
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZeolites in catalysis: sustainable synthesis and its impact on properties and applications
Hongwei
Zhang
 ,
Ismail bin
Samsudin
,
Stephan
Jaenicke
and
Gaik-Khuan
Chuah
*
,
Ismail bin
Samsudin
,
Stephan
Jaenicke
and
Gaik-Khuan
Chuah
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, Kent Ridge, 117543 Singapore. E-mail: chmcgk@nus.edu.sg
First published on 29th August 2022
Abstract
Zeolites are versatile catalysts not only for large scale petrochemical processes but also in applications related to fine chemicals synthesis, biomass conversion and CO2 utilization. Introduction of mesopores and heteroatoms opens up additional applications. Heteroatoms in the crystalline framework create active sites where the diversity in the coordination offers tunability in acidity and redox properties. Their framework locations within the unit cell and their open/closed nature greatly influence the catalytic ability. Depending on the synthesis conditions, the properties of the pore walls can vary from hydrophobic to hydrophilic which affects the confinement of reactant and solvent molecules within the well-ordered pores. This mini-review traces the synthesis of heteroatom zeolites from traditional solvent-based methods to the more sustainable routes of post-synthesis and mechanochemistry. The properties of the zeolites formed by the different methods are compared and the impact on selected catalytic reactions is discussed.
 Hongwei Zhang | Hongwei Zhang received his M.Sc. degree in Materials Science and Engineering from Tsinghua University in 2014 and PhD degree in Chemistry from the National University of Singapore (NUS) in 2019. He then took up the position of research fellow in NUS, under the supervision of Prof. Gaik-Khuan Chuah. and Prof. Stephan Jaenicke. His research interests focus on heterogeneous catalysis in biomass utilization and green chemistry. |
 Ismail bin Samsudin | Ismail Samsudin studied Chemistry at the National University of Singapore (NUS) and obtained his M.Sc. degree in 2019. He is currently doing his PhD studies in the group of Prof. Gaik-Khuan Chuah at NUS. His research interests are in catalysts for green and sustainable chemistry and materials chemistry. |
 Stephan Jaenicke | Stephan Jaenicke obtained his Ph.D. in 1981 at the Technical University of Karlsruhe (Germany). After postdoctoral work at Michigan State University (1982–1983) and the Fritz Haber Institute of the Max Planck Society (1984–1988), he joined the National University of Singapore in 1989 where he worked on the development of new catalysts for sustainable processes. |
 Gaik-Khuan Chuah | Gaik-Khuan Chuah obtained her PhD in Chemistry from Texas A&M University, USA in 1987. Her PhD thesis on catalysis and surface reactivity studied by pulsed field desorption mass spectrometry was carried out at the Fritz-Haber Institute, Berlin. She is presently an associate professor at the Department of Chemistry, National University of Singapore. Her research interests in heterogeneous catalysts and materials include solventless synthesis, zirconium compounds, transformation of biomass to chemicals and cascade reaction in fine chemicals synthesis. |
1. Introduction
Porous materials are ubiquitous in our daily lives. Covering a wide range of pore sizes from micropores (diameter, d < 2 nm), mesopores (2 < d < 50 nm) to macropores (d > 50 nm), these materials are versatile and indispensable in applications such as ion exchangers, gas separators, catalysts, for energy storage, in sensors, and many more.1–3 Pore size is an important parameter that affects not only the physico-chemical properties of the material but also the properties of molecules inside the pores. Zeolites are microporous crystalline materials with defined pore geometry. The traditional aluminosilicate zeolites have the general formula Mx/n[(AlO2)x(SiO2)y]·mH2O, where M indicates an exchangeable charge-compensating cation and n refers to the charge of this cation. Their structural characteristics, such as high surface area, pore channel systems, accessible void space, ion-exchange sites, adjustable acidity, and high thermal stability make zeolites viable catalysts. Since the 1950s when Al-rich Y-type zeolites were introduced for fluid catalytic cracking of heavy oil fractions, various zeolites have been developed for the synthesis of fine chemicals, utilization of biomass and as environmental catalysts.4 Replacing the framework aluminium with other heteroatoms such as boron, gallium, or iron can modify their chemical properties, in turn directing the activity and selectivity of reactions. Particularly interesting materials are obtained when Si4+ is partially substituted with other M4+ ions.5 Substitution of titanium into silicalite, an MFI zeolite with 10-ring structure, formed TS-1 which showed unique activity for the oxidation of organic substrates with hydrogen peroxide.6,7 Consequently, these materials have attracted continuing attention in both academic research and industrial applications.8 As solid acids, zeolites can be regarded as low-waste and green compared to conventional Brønsted and Lewis acid catalysts.9There are two sides to the small pore sizes of zeolites. On the one hand, it can be limiting when bulky molecules are of interest. On the other hand, the regular crystalline micropores confer these materials uniquely different properties from those of amorphous aluminosilicates. The surface curvature of the internal walls results in non-covalent van der Waals interactions between the framework and molecules located in the intracrystalline space, different from interactions with a flat surface.10 These supramolecular interactions are the basis of confinement effects which govern the sorption, shape selectivity, diffusivity and catalytic behaviour of microporous zeolites. As first pointed out by Derouane,11 molecules with cross-sections that match closely with the micropore channels have enhanced diffusivity as the probability of transverse motions becomes negligible. For this reason, molecules that are smaller than the pore window encounter higher energy barriers and lower diffusion rates. The advent of the ordered mesoporous M41S (MCM-41) silica materials, first reported in 1992, widened the scope for larger molecules.12–14 Experimental results suggest that confinement effects exist in these materials as well.15,16 For example, neutron diffraction studies showed that methanol in the pores of MCM-41 was highly ordered with a higher density than that of bulk methanol.17,18 However, it was subsequently discovered that these materials lack the acidity and thermal stability of zeolites because of their non-crystalline nature. They have proven to be more useful as supports with their precise pore sizes and high surface areas. Nevertheless, it is possible to modify the pore walls of the mesoporous silicates; attaching acidic or basic groups, grafting of metal complexes, isomorphous substitution of heteroatoms and immobilization of enzymes can convert these materials into highly specific catalysts.19–21
The preparation of zeolites and ordered mesoporous materials is unfortunately not very eco-friendly. This is largely due to the usage of organic templates as structure-directing agents to yield the desired framework topology and pore size. Various ionic and non-ionic surfactants such as quaternary ammonium salts and triblock polyoxyalkene copolymers are used as structure directors in addition to bulk chemicals such as silica, alumina and sodium hydroxide. While non-ionic surfactants can be recovered by extraction due to their weaker assembly forces, the ionic surfactants cannot be recycled but need to be removed by heat treatment, resulting in carbon dioxide and nitrous oxides emissions. Environmental adversities brought about by unsustainable practices have turned attention to low waste eco-friendly processes. A comprehensive review by Meng and Xiao covers various routes to green synthesis of zeolites including elimination of templates and solvents as well as microwave-assisted synthesis.22 In particular, the use of mechanochemistry in synthesis is attractive given its inherent opportunities for cleaner synthesis. In this mini-review, we focus on sustainable routes for the synthesis of heteroatom-incorporated zeolites and the impact that modifications of porosity, acidity and hydrophobicity have on their catalytic properties.
2. Synthesis of microporous zeolites
Zeolites are conventionally synthesized using hydrothermal methods with structure-directing agents (SDA) under alkaline conditions where the hydroxide ion acts as mineralizer. Procedures that minimize the usage of water as a solvent and reduce or eliminate the organic auxiliaries are sought after to minimize effluents which would have to be treated. Dry gel conversion (DGC) or steam-assisted synthesis (SAC) are promising strategies. Xu et al. developed this novel technique to synthesize ZSM-5 from amorphous aluminosilicate gels in a vapour of ethylenediamine, triethylamine, and water.23 The starting materials are first formed into a gel, dried, and placed in a specially designed sample holder separated from the solvent and SDA liquid phase at the bottom of the autoclave. Crystallization of the gel occurs upon contact with vapors of the solvent and SDA.24 Significantly lower amounts of SDA are required as they can participate in the crystallization process by absorbing into the gel. In the SAC method, only steam and not the SDA-containing vapour is supplied from the gas phase.25 For both DGC and SAC, less waste is generated than by the conventional hydrothermal route.26,27 The crystallization time could also be shortened, e.g., the synthesis of a CHA-type zeolite, SSZ-13, was reduced from several days to 6 h without seeding or pre-aging at the same crystallization temperature of 160 °C (Fig. 1a and b).28 | ||
| Fig. 1 SEM images of (a) steam-assisted SSZ-13 formed in 7 h and (b) conventional SSZ-13 formed after 4 days. Reprinted from ref. 28. Copyright (2020) with permission from Elsevier. (c) Hydrothermal HF-synthesized and (d) post-synthesized Zr-Beta. Reprinted from ref. 36. Copyright (2015) with permission from Elsevier. (e) Solvent-free Al-Beta and (f) solvent-free ZSM-5 zeolite. Reprinted with permission from ref. 56. Copyright (2014) American Chemical Society. | ||
Flanigen and Patton were the first to report the use of fluoride as mineralizer for the synthesis of zeolites.29 This method was further developed by Guth and Kessler.30,31 The zeolites formed with the fluoride route are of larger particle size with few defects compared to those synthesized under alkaline conditions. Drawbacks of the fluoride route include high concentrations of SDA and fluoride, typically, SDA/Si and HF/Si of 0.5, long crystallization times and lower yields of 40–60% compared to 85–90% for the alkaline synthesis.32 However, the acidic conditions of the fluoride synthesis make it possible to incorporate ions that would otherwise precipitate out under alkaline conditions. Corma's group were the first to report the synthesis of Al-free Ti- and Sn-Beta zeolites using hydrogen fluoride as the mineralizer and tetraethylorthosilicate (TEOS) as the silica source.33–35 Zr-Beta zeolites have been similarly synthesized.36–39 The amount of heteroatoms that can be incorporated into the zeolite framework is generally limited to a few atom-%, even with prolonged crystallization times of several days to weeks. The corrosive and toxic nature of fluorides, especially hydrogen fluoride, poses problems for commercialization due to safety considerations and associated costs. The development of strategies for fluoride-free synthesis is challenging.40 For siliceous zeolites, there are currently less than 25 structures that can be synthesized under hydrothermal or dry gel conditions without fluoride.41
Dzwigaj et al. developed a two-step post-treatment synthesis method to obtain various heteroatom-incorporated zeolites.42,43 In this top-down method, the pre-formed zeolite is first treated with acid to remove the framework aluminium atoms. The dealuminated zeolite is rich in silanol nests and exposure to the heteroatoms by various means such as wet impregnation, solid-state mixing, refluxing or gas–solid adsorption, can cause their incorporation into the vacated tetrahedral (T) sites.36,44–46 A distinct advantage is that the synthesis duration is much shorter because the zeolite framework is already formed. In addition, more heteroatoms can be introduced than with direct hydrothermal synthesis as the number of available T-sites vacancies depends on the aluminium content of the parent zeolite. Our group compared the pros and cons of Zr-Beta zeolites synthesized by HF-assisted hydrothermal synthesis and the two-step post-synthesis method starting from a nanocrystalline Al-Beta sample (Fig. 1c and d).36 The Zr-Beta zeolites obtained by the post-synthesis method had a higher density of acid sites with a wider spread of acid strength due to the many remaining silanol nests and partially hydrolysed framework zirconium.
Using mechanochemistry in lieu of solvents opens the door to a viable sustainable synthesis of zeolites. The addition of energy through grinding promises high yields, shorter synthesis time, significant reduction of wastes together with simplified processing which avoids the hazards of high pressure inherent to liquid phase hydrothermal synthesis.47,48 The effectiveness of mechanochemistry has been attributed to the simultaneous combination of shear forces, mechanical pressure, and high instantaneous local temperatures resulting in structural transformations via lower energy reaction paths than conventional wet synthesis.49 However, mechanochemical synthesis of zeolites typically requires a thermal step in a closed system in addition to the input of mechanical energy. This is in contrast to organic materials such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and zeolitic imidazolate frameworks (ZIFs), where the porous structures form by purely mechanical activation of the precursors. According to Majano et al.,49 the mechanical activation produces a chemically stressed composite which during subsequent hydrothermal treatment undergoes an irreversible structural reorganization to the zeolite. The key properties of zeolite synthesis by hydrothermal, post-synthesis and mechanochemical methods are summarized in Table 1.
| Hydrothermal synthesis | Post-synthesis | Mechanochemistry/solvent-free | |
|---|---|---|---|
| Procedure | Hydrothermal treatment of reactive gels in either alkaline media (high pH) or fluoride-containing media (low pH) at temperatures between ∼80 and 200 °C. A structure directing agent is frequently required | Al-zeolites (typically from commercial sources) are dealuminated using concentrated acids, followed by incorporation of the desired heteroatoms into vacant T-sites, e.g., by wet or solid state impregnation | Mechanical mixing of the solid reactants followed by treatment in autoclave at temperatures ∼80–200 °C |
| Advantages | Many zeolite types can be formed. Well-documented and verified synthesis procedures | Short synthesis time | Essentially solvent-free; only source of water is from hydrated salts |
| No structure directing agent required as presynthesized zeolites are used | |||
| Minimal wastes generated | |||
| Various incorporated heteroatoms | Safe reaction conditions with low autoclave pressure | ||
| Adjustable concentration of heteroatoms | |||
| High yields of zeolites | Maximizes usage of autoclave space with high yields of zeolites | ||
| Disadvantages | Solvent intensive | Large amounts of concentrated acid pose safety and disposal concerns | Insufficient information on the scope of the method; only a limited number of structures have been reported so far |
| Long crystallization time especially for siliceous zeolites | |||
| Textural properties dependent on the precursor zeolite | |||
| Limited incorporation of heteroatoms with synthesis time extending to weeks | |||
| Zeolite framework contains high concentration of defect sites | |||
| Variable zeolite yields | Generally hydrophilic with low water tolerance | ||
Yamamoto et al. reported the mechanochemical activation of titanium oxide and silica by ball milling to form an amorphous composite of the well-dispersed oxides which by hydrothermal treatment in the presence of the appropriate SDA and water could be transformed into titanosilicate TS-1.50 Inexpensive bulk oxides can be used as starting materials together with the cheaper SDA salts such as tetrapropylammonium bromide instead of the hydroxide. This route was also used successfully to synthesize a number of Beta and silicalite-1 zeolites containing Sn, Mn, Zn and Fe.51–54 Xiao and co-workers further simplified this method with a solvent-free protocol utilizing only the water present in the hydrated reactants. Hydrated sodium silicate or hydrated silica, the relevant organic templates and sources of heteroatoms in the form of simple salts or oxides were ground together followed by heating at 80–180 °C in a closed system for 24–72 h.55 Thermal treatment under the prevailing alkaline conditions caused crystallization. Several types of zeolites (MFI, SOD, MOR, BEA and FAU) were obtained in excellent yields in the pure silica form as well as containing heteroatoms (Al, Fe, Ga and B). Xiao's group further showed that the structure-directing agents can be replaced by seeds, and they successfully synthesized several types of zeolites using this method (Fig. 1e and f).56 The zeolites obtained by this route comprised well-formed particles of high crystallinity, similar to those formed by the hydrothermal route.
Mechanical activation of the substrates can be achieved by manual grinding with mortar and pestle or may require using a ball mill. For instance, in the solvent-free synthesis of SSZ-13 by Pashkova et al., a planetary mill was necessary to activate the reagents as manual grinding only formed an amorphous product.57 Nada et al. found that grinding the solid raw materials for a sufficiently long time caused water to be released from the hydrated starting materials, forming a paste which transformed into crystalline ZSM-5 after thermal treatment.58 The presence of trace amounts of water is a necessary condition for the solvent-free synthesis of zeolites.55,56 Wu et al. proposed that water serves as a “catalyst” in the zeolite crystallization, being consumed in the hydration of silica species and reformed in the subsequent condensation step.59 The use of completely anhydrous reactants has also been reported and a number of structures (MFI, BEA*, EUO, and TON) could be obtained including the incorporation of heteroatoms such as B and Fe.60 In all these syntheses, NH4F was used, which reacted chemically with the silica. Upon simple grinding at room temperature, (NH4)2SiF6 was formed together with transformation of Q4 silica to Q3 species.
3. Porous structure adjustment and hierarchical zeolites
The nanosized pore diameters (<2 nm) in zeolites often pose challenges for reactions involving bulky molecules due to hindered mass transfer and inaccessibility to active sites located inside the pore channels.61 Significant research efforts have been devoted to address this by introducing mesopores. Such hierarchical structures can be obtained by either a top-down or a bottom-up approach (Fig. 2).62–64 The bottom-up approach is an in situ synthetic strategy using soft surfactants or hard templates to engineer microporous and mesoporous domains in the zeolite.65–68 Many of the early efforts used a mixed template of micelle-forming surfactant molecules and small molecule structure-directing agents to simultaneously direct the formation of mesoporous and microporous structures in one material. This dual-templating process often leads to physical mixtures of the amorphous mesoporous material and microporous crystalline zeolite due to phase separation of the different templating systems.69,70 To overcome this, molecules with both the mesopore templating function and the SDA functionality have been synthesized to form mesoporous zeolites with various topologies.66,71,72 These dual-function templates are surfactant-based as the supermolecular self-assembly of the surfactants is necessary for the formation of periodically ordered mesostructures. Notwithstanding this, it remains difficult to obtain long-range order of both micropores and mesopores within the material as the ordered mesostructure disrupts the continuity of the three-dimensional zeolitic framework. | ||
| Fig. 2 “Bottom-up” and “top-down” approaches to mesoporous zeolites. Reprinted from ref. 65. Copyright (2013) with permission from John Wiley and Sons. | ||
Zhu et al. used the cationic polymer, polydiallyldimethylammonium chloride (PDADMA), to form highly mesoporous single-crystalline zeolite Beta.73 The polymer contains abundant quaternary ammonium groups that serve as the structure-directing agent for zeolite formation. As it lacks the hydrophobic segments, it does not self-assemble to form regular structures, but functions more as a porogen, giving rise to disordered mesopores. Mesopores with diameters of 4 to 10 nm could be obtained by varying the molecular weight of the polymer. PDADMA is an attractive template for synthesis of hierarchical zeolites, being commercially available and inexpensive, compared to specially designed templates that have to be synthesized. PDADMA was also used as a flocculating agent to aid in the association of in situ-formed nanosized Beta crystallites into easily recoverable mesoporous aggregates.74 The pore size could be adjusted from 40 to 400 nm by changing the amount of polymer in the synthesis gel. In a more environmentally friendly approach to hierarchical zeolites, nanosized Beta zeolite was first prepared from highly concentrated precursor gels, followed by condensation into a mesoporous network under steam-assisted hydrothermal conditions without any porogen.67 The mesoporosity arises from the intraparticle voids and the material has been reported to be stable against mechanical and thermal treatments. In this hierarchical structure, the microporosity of the zeolite Beta is fully retained with additional contribution to the porosity from mesopores.
Top-down routes comprise post-synthetic treatment of previously formed zeolites by leaching of framework atoms, commonly through treatment with acid or base, steaming and surfactant-templating.75,76 The efficiency of desilication depends on the zeolite morphology and the framework type, Si/Al ratio, and treatment conditions of alkalinity, time and temperature.77 Although desilication using NaOH or Na2CO3 generates intracrystalline mesoporosity, it is accompanied by a substantial loss of material and microporosity. While the reagents used are inexpensive, the material loss and the additional wastewater generated have to be considered if hierarchical zeolites are to be synthesized in a sustainable and environmentally friendly manner. A review by Verboekend and Pérez-Ramírez discusses measures to increase reactor productivity, recycle the waste streams, recover the organic compounds instead of their combustion, and minimization of separation efforts.78 If the desilication is performed in the presence of surfactants such as cetyltrimethyl ammonium salts, hierarchical zeolites with ordered mesoporosity can be obtained with less loss of material.79,80 It is postulated that under basic conditions, cleavage of Si–O–Si bonds occurs, creating some flexibility in the crystalline structure and also negatively charged sites in the zeolite framework. Surfactant cations are attracted to these sites and undergo self-assembly to micelles within the zeolite crystal. Rearrangement of the crystal structure around the micelles then forms the mesopores. Recycling of the surfactants is a necessary step to achieve sustainability. In an adaptation to a solvent-free process, Huang et al. created meso–/macropores in Silicalite-1 by mechanochemical grinding with tetrapropylammonium bromide and ammonium fluoride.81 Diffusion of the fluoride ions into the zeolite channels selectively etches areas that had structural defects, forming mesopores which could be made to coalesce into macropores by heating to 180 °C (Fig. 3). The size of the pores could be tuned by adjusting the heating duration and by using other quaternary ammonium salts. By incorporating an impregnation step prior to the grinding, Pt and Co nanoparticles could be encapsulated in the pores. The introduction of mesoporosity in zeolites is an effective way to influence the diffusion properties of these microporous solids. Previous work has concentrated on Al-containing zeolites and more investigations into other heteroatom zeolites would be needed.
 | ||
| Fig. 3 TEM images of Silicalite-1 after grinding with TPABr–NH4F at (a) room temperature for 15 h, (b) 453 K for 1 h, (c) 453 K for 15 h, (d) 453 K for 48 h. STEM images and EDS maps of (e and f) Pt@silicalite-1 and (g and h) Co-silicalite-1 samples. Reproduced from ref. 81 with permission from the Royal Society of Chemistry. | ||
4. Properties of zeolites
4.1 Tailoring acidity by heteroatom substitution
The acidity of zeolites can be tuned by replacing the Si4+ with atoms of other elements without affecting the framework nature.82 Aluminium-containing zeolites are well-known solid acid catalysts used industrially in cracking and isomerization of hydrocarbons. The substitution of Si4+ by Al3+ or other ions with lower charges, e.g., Fe3+, Ga3+, Mg2+, introduces negative charges to the framework which must be charge balanced by extra-framework cationic species (Scheme 1). If the cation is a proton, the hydroxyl group bridging an aluminium and silicon forms a Brønsted acid site. The nature of aluminium Lewis acidity is less well-defined and a recent review by van Bokhoven and coworkers categorized them into framework Al (AlF), framework-associated Al (AlFA), and extra-framework Al (AlEF).83 Several types of extra-framework aluminium were identified, including cationic and neutral species such as Al3+, Al(OH)2+, Al(OH)2+, AlOOH, Al(OH)3, and Al2O3. In a study of commercial ultrastabilized Y zeolites using 27Al NMR spectroscopy, it was established that cationic extra-framework Al caused perturbation of the Brønsted acid sites, which could be correlated with the rate of propane cracking.84 However, Batool et al. found that the introduction of extra-framework aluminium into zeolite Y by ion exchange did not affect the Brønsted acidity although the number of Lewis acid sites was significantly increased.85 Zhao et al. attempted to determine how aluminium occupancy at different lattice sites in the unit cell of Beta zeolite affects the Brønsted acidity.86 By dealuminating Al-rich H-Beta (Si/Al 7), a series of samples were prepared, which were characterized by 27Al MAS/MQMAS and 31P MAS NMR with trimethylphosphine oxide as molecular probe, together with DFT calculations. The strongest Brønsted acid sites were ascribed to the synergistic interaction between Lewis and Brønsted sites, aluminium occupying the T1 sites was associated with medium strength, and the weakest Brønsted acid sites originate from Al at T6 sites. The removal of one framework aluminium from 5- and 6-rings that originally contained two Al3+ ions increased the Brønsted acid strength of the dealuminated H-Beta. | ||
| Scheme 1 Hosting of (a) tri- (b) tetra- and (c) pentavalent heteroatoms in the zeolitic framework. Reprinted from ref. 82. Copyright (2014) with permission from Elsevier. | ||
In contrast to aluminium or other M3+ ions, the isomorphous substitution of Si4+ by other M(IV) heteroatoms, e.g., Ti, Sn, Hf, Zr, does not generate charge imbalance in the framework. Brønsted acidity is generally absent although some weak Brønsted acid sites may arise due to differences in electronegativity of Si4+ and M4+.87 Instead, the M(IV) atoms form isolated Lewis acid sites with properties that depend on the type, site occupation, concentration and state of hydration. From EXAFS studies, Bare et al. found that Sn does not randomly insert into the Beta-zeolite structure but rather, occupies T5 and T6 sites on the 6-ring.88 The substitution of Sn in pairs on opposite sides of the 6-ring was postulated to be the basis for the uniform catalytic activity and high selectivity in the Baeyer–Villiger oxidation of ketones into lactones and esters.
The non-random substitution of heteroatoms was supported by periodic density functional theory calculations by Yang et al.89 However, their calculations for Ti-, Sn- and Zr-Beta zeolites showed that substitution at the T2 site was most favoured and T7 the least (Fig. 4). The stability trend at the other seven distinct T sites differed, depending on the type of heteroatom. The non-random substitutions are due to local flexibility of the framework and structural perturbations reflected in the changes of the θ(O–T–O) bond angles. The adsorption of water was used to probe the Lewis acidity of the heteroatom zeolites. Both Sn- and Zr-Beta have higher Lewis acidity than Ti-Beta. Incorporation of a second heteroatom in the unit cell causes obvious alterations to the θ(O–T–O) bond angles and is more difficult than the substitution of the first heteroatom. In Sn-Beta, stable structures containing dual Sn were identified at neighbouring T2 sites. In contrast, the formation of paired sites was absent in the case of Ti and Zr framework substitution. The presence of paired framework Sn sites substantially enhances the Lewis acidity of Sn-Beta and is postulated to be responsible for its higher catalytic activity in the conversion of sugar to methyl lactate compared to the other two zeolites.90
 | ||
| Fig. 4 (a) Structural representation of BEA zeolite, with the nine crystallographically distinct T sites indicated. (b) Relative energies of the nine different T sites in Ti-, Sn-, and Zr-BEA zeolites. (c) Two 6-membered rings in BEA zeolite that share the T2 and T2b substituted sites. Reprinted with permission from ref. 89. Copyright (2013) American Chemical Society. | ||
The incorporation of divalent and pentavalent ions has been less frequently reported. Mg-Beta formed by the post-synthesis method has both Lewis and Brønsted acidity which correlated with the amount of Mg incorporated.91 A mechanochemical route was used to synthesize MFI-type zincosilicates where a mixture of fumed silica and zinc oxide was intensely ball-milled before adding more silica and SDA followed by hydrothermal synthesis.52 The mechanochemical step was essential to the successful synthesis as hand mixing of the silica/zinc oxide mixture resulted in low crystallinity of the zincosilicate together with zinc oxide and extraframework Zn. In this way, it was possible to obtain zinc zeolites essentially free of aluminum in contrast to the preparation by conventional seed-assisted hydrothermal routes. Vanadium-containing MFI zeolites could be prepared by hydrothermal synthesis under alkaline conditions.43,92 The V5+ ions were associated with Si–OH hydroxyl nests. XAFS studies showed that the V5+ is tetrahedrally coordinated with three V–OSi single bonds and a shorter V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond.93 From IR studies using different probe molecules such as CD3CN, pyridine and tert-butyl cyanide on a V-MFI sample (Si/V 545), it was concluded that strong Lewis acid sites are associated with V5+ inside the zeolitic pores whereas the silanol groups on the external surface contribute to weak Lewis acidity.43
O double bond.93 From IR studies using different probe molecules such as CD3CN, pyridine and tert-butyl cyanide on a V-MFI sample (Si/V 545), it was concluded that strong Lewis acid sites are associated with V5+ inside the zeolitic pores whereas the silanol groups on the external surface contribute to weak Lewis acidity.43
Recently, it has been questioned whether observed shifts in spectroscopic properties of adsorbed probe molecules do indeed correlate with a conventional acidity scale.94–96 Calculations indicate that the deprotonation energies for zeolites with different crystalline structures are close to each other, which implies that the intrinsic acidity is independent of the zeolite structure or the location of the site within the framework.97 However, Boronat and Corma posit that confinement effects can influence the interaction of probe molecules with Bronsted acid sites.94 Weak bases, such as CO, form neutral OH–CO adducts but spatial constraints due to the void space may distort the linear configuration and weaken the interaction. This is counteracted by increased dispersion forces if the adduct fits tightly into the space, enhancing its stabilization. Hence, the correlation between shift in CO frequency and acid strength of zeolite may be distorted. When strong bases, e.g., NH3 or pyridine are used, the overall acidity depends on deprotonation energy of the zeolite, the proton affinity of the base, and the stabilization of the zeolite–BH+ ion pair by surrounding framework oxygen atoms. The 31P-NMR shift of adsorbed trimethylphosphine oxide (TMPO) is widely used to determine the Brønsted acid strength of zeolites, but it has recently been shown that confinement effects and the formation of protonated TMPO dimers affect the chemical shifts.95 Only by limiting the TMPO loadings (P/Al < 0.4) can the 31P chemical shift be correlated to the Brønsted acid strength and also used to distinguish between Lewis and Brønsted acid sites.96 Generally, measurements based on adsorption of probe molecules can reveal the heterogeneity of acid sites but cannot determine their intrinsic acidity. Therefore, theoretical calculations for specific conformations are required to supplement the experimental results.
4.2 Open/closed nature of incorporated heteroatom sites
The isomorphous incorporation of the metal atom (M) in the zeolite framework can result in two types of sites, classified as closed or open sites. This phenomenon has been primarily studied on Sn-containing Beta zeolites. In the closed sites, there are four M–O–Si bonds whereas in open sites, the metal atom is partially hydrolyzed with three M–O–Si and one M–OH group (Fig. 5).98–100 Interconversion between the sites occurs through hydrolysis/dehydration so that the hydrolyzed-open sites cannot be detected by spectroscopic techniques that involve sample pretreatment at high temperatures. Instead, the open sites which are detected in this case are sites which cannot transform into closed configurations upon heating. It is suggested that these “defect-open” sites are preferentially located at stacking fault grain boundaries.99 They comprise (HO)–M–(OSi)3, similar to the open sites formed by hydrolysis of closed sites. However, the compensating Si–OH groups are sufficiently distant so that they do not revert to closed sites upon heat treatment. The two types of open sites and the transformation of open into closed sites were identified in Sn-Beta using one and two-dimensional proton-detected 1H/119Sn correlation solid-state NMR spectroscopy.101 Kolyagin et al. used 119Sn MAS NMR to identify different Sn sites in fully dehydrated Sn-Beta zeolite (Si/Sn 200) synthesized by the fluoride hydrothermal route.102 Applying a combination of Carr–Purcell–Meiboom–Gill (CPMG) pulse sequences, they obtained NMR spectra with three sets of narrow peaks assigned to isomorphously substituted Sn sites in defect-free framework positions (Fig. 6). A broad signal at −443 ppm attributed to Sn atoms in proximity to a terminal or internal Si–OH group. Of the nine non-equivalent T sites, the T5, T6 and T7 had the highest population for Sn. No open sites could be observed due to the dehydration pretreatment. | ||
| Fig. 5 Structurally distinct configurations of Sn Lewis acid sites in zeolites. Reprinted with permission from ref. 99. Copyright (2019) American Chemical Society. | ||
 | ||
| Fig. 6 (a) 119Sn DP-CPMG MAS NMR spectrum of dehydrated 119Sn-BEA. (b) Assignment of signals to three groups of non-equivalent T-sites based on the existing theoretical predictions: (I) T9, T4, and T3; (II) T2, T1, and T8; and (III) T7, T5, and T6 and broad signal at −443.9 ppm due to Sn with neighbouring defects. Reprinted with permission from ref. 102. Copyright (2018) American Chemical Society. | ||
Open and closed sites have different reactivities in chemical reactions.98,103–106 Sushkevich et al. observed a linear correlation between the relative amount of open sites and the activity for the conversion of ethanol to butadiene over different Zr-Beta catalysts.107 In contrast, the number of closed sites did not show any obvious correlation with the activity. The catalytic efficiency of the open sites was attributed to their higher acid strength and better steric accessibility compared to closed sites. Open sites in Sn-Beta zeolite were found to be the active sites for the isomerization of glucose into fructose and epimerization of glucose to mannose.108 It is proposed that the presence of nearby Si–OH groups in the defect-open sites is responsible for the high isomerization activity. However, closed Sn sites appear to be involved in ethanol dehydration, perhaps because they can interconvert between closed and open configurations.
The group of Román-Leshkov used 15N pyridine as probe to study the interaction with metal centres by MAS NMR and thereby assess the acidity of a number of Beta zeolites.109 The isotropic 15N chemical shift correlated linearly with the Mulliken electronegativity of the metal atoms in the order Ti < Hf < Zr < Nb < Ta < Sn < H+ (Fig. 7). As the signal intensity is proportional to the number of spins, this technique can be used to quantify the framework content for any metal in a Beta zeolite. However, more recently, it was observed that very small extraframework SnOx clusters with sizes of 0.5–2 nm confined within the pores or cages of zeolites had similar Lewis acidity as framework Sn.110 The Lewis acidity rapidly decreases for larger SnOx particles beyond 2 nm.
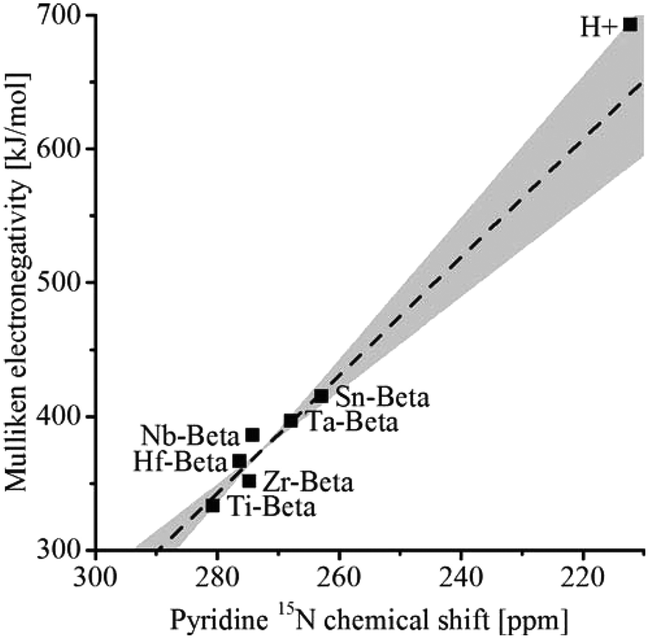 | ||
| Fig. 7 Experimental Mulliken electronegativity vs. pyridine 15N MAS NMR chemical shift for heteroatom Beta zeolites. Reprinted with permission from ref. 109. Copyright (2016) American Chemical Society. | ||
4.3 Hydrophobic effect in zeolites
Hydrophobicity within the zeolitic pores affects rates of reactions through preferential adsorption of molecules or alteration of free energy barriers.111 One of the determinants of hydrophilicity–hydrophobicity in zeolites is the presence of trivalent heteroatoms, commonly Al3+. The selectivity for adsorption of water decreases with smaller aluminium content signifying an increase in hydrophobicity.112,113 Other factors are the crystallinity of the zeolite and the concentration of T-site defects which affect the amount of terminal silanol groups. Highly hydrophobic zeolites typically have few T-site vacancies and high crystallinity. Post-synthesis incorporation of heteroatoms forms zeolites that are more hydrophilic than those directly synthesized by the fluoride route. Zr-Beta obtained by the two-step post-synthesis protocol contains a considerable concentration of T-site vacancies, making it susceptible to poisoning by water although the poisoning can be reversed by calcination.36 In contrast, Zr-Beta synthesized via the HF-assisted hydrothermal method was hydrophobic and exhibited high water tolerance and stability when used as catalyst for the cyclisation of citronellal and in MPV reductions.36,38 Di Iorio et al. investigated the hydrophobic effect on Sn-Beta zeolites prepared in neutral to slightly acidic fluoride-containing medium and by post-synthesis grafting of dealuminated Beta.111,114 They found that hydrophobic Sn-Beta-F had a 10-times higher turnover frequency than Sn-Beta-OH for the transfer hydrogenation of cyclohexanone (Fig. 8). The IR spectra showed that the solvent, 2-butanol, rearranged into networks of ordered 2-butanol dimers in the hydrophobic Sn-Beta-F, whereas it formed liquid-like 2-butanol oligomers in the hydrophilic Sn-Beta-OH zeolite. This rate enhancement was attributed to a greater stabilization of the transition state for hydrogen transfer caused by changes in the adsorption enthalpies and entropies.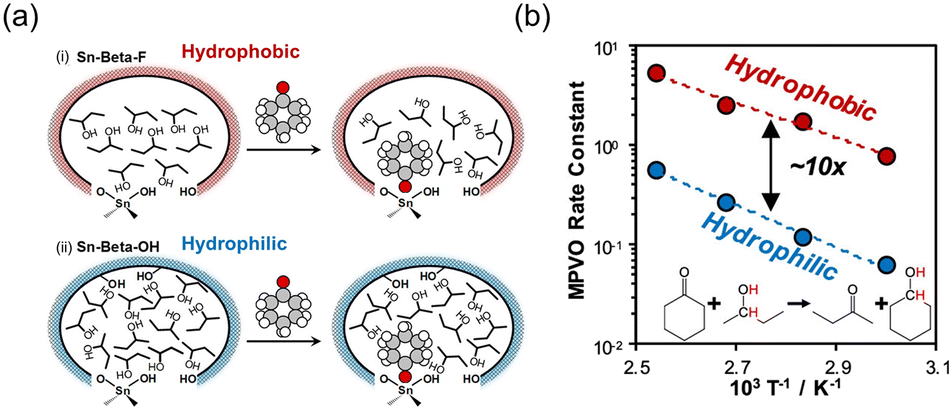 | ||
| Fig. 8 (a) Adsorption of cyclohexanone within the 2-butanol filled pores of hydrophobic Sn-Beta-F and hydrophilic Sn-Beta-OH and (b) differences in the rate constant for MPV of cyclohexanone. Reprinted with permission from ref. 111. Copyright (2020) American Chemical Society. | ||
Differences in the polarity of the zeolite pores can also arise as a consequence of the structure of the zeolite.115 Zr-HY was found to be more hydrophobic than Zr-Beta when both materials were prepared with the same Si/Zr of ∼50. The IR spectra showed that 2-pentanol adsorbed on Zr-HY forms predominantly dimers and oligomers (∼3430 cm−1) but for Zr-Beta, an absorption band at ∼3204 cm−1 indicates hydrogen bonding between silanol groups and the alcohol (Fig. 9). In Zr-HY, a low density of surface OH groups limits their interaction with the alcohol, so that there is a preference for intermolecular hydrogen bonding between the alcohol molecules instead. These results are corroborated by thermogravimetry measurements of water loss from the two zeolites, where significantly less water is bound in Zr-HY than in Zr-Beta.
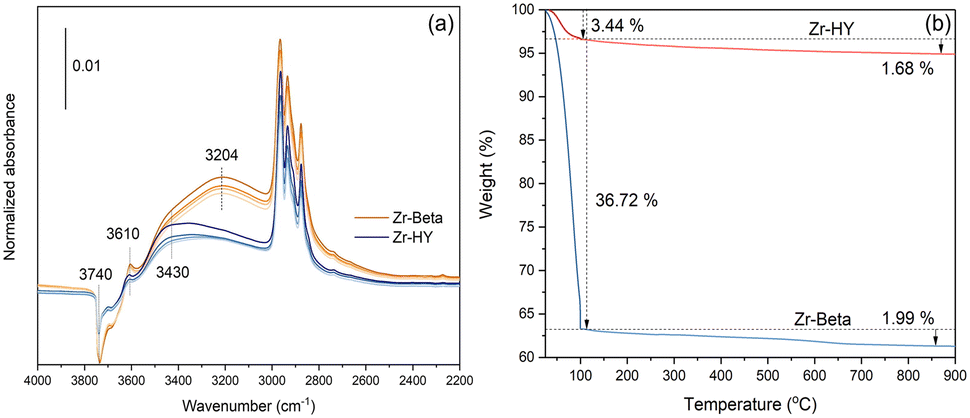 | ||
| Fig. 9 (a) Difference IR spectra of 2-pentanol on Zr-HY (blue) and Zr-Beta (orange) under dynamic evacuation at 0.05, 0.5, 20, and 50 min (top to bottom) and (b) TGA of Zr-HY and Zr-Beta. Reprinted from ref. 115. Copyright (2021) with permission from Elsevier. | ||
Besides intrinsic tuning by heteroatom or zeolite type, the surface can be modified with organosilanes to alter the hydrophobicity.116 However, the active sites can be easily covered during silanization and it is necessary to strike a fine balance to achieve optimum catalytic activity. Resasco's group found that organosilanes with short alkyl chains are able to reach all the defects and small pockets in the zeolite.117 Therefore, they were most effective in preventing structural collapse when the zeolites were exposed to liquid water at high temperatures. On the other hand, use of the long alkyl chain octadecyltrichlorosilane in HY zeolites enabled a high rate of reaction for the acetalization of glycerol to solketal.118 This was traced to emulsion formation between the immiscible reactants, glycerol and acetone, promoted by the bound organic phase, and easier access of both reaction partners to the acid sites on the zeolite catalyst.
5. Application as heterogeneous catalysts
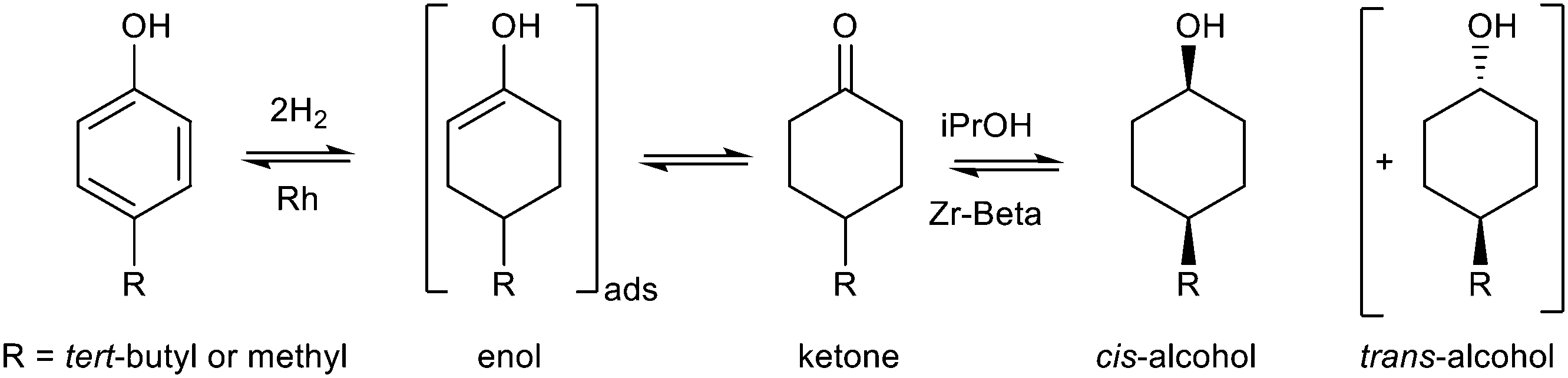
For more than half a century, zeolites have been used in refining and the petrochemical industry for fluid catalytic cracking (FCC) and isomerization and also in environmental remediation, nuclear waste management, and groundwater decontamination.8 The application of zeolites in fine chemical synthesis is extremely promising. The most interesting catalysts are based on zeolites that contain metals other than Al in the framework. The well-defined porous structure, controllable acidity and the variable nature of the active sites offer unique opportunities to fine tune their catalytic properties. The group of Corma established Sn-Beta as an excellent catalyst for Baeyer–Villiger oxidation of ketones to esters and lactones by H2O2. The isolated Sn sites in the zeolite framework form the Lewis acid sites that preferentially activate the carbonyl group rather than H2O2 and this is critical for the high selectivity to lactones.35 When a different heteroatom, titanium, is incorporated in the zeolite, a catalyst active for the epoxidation of olefins with aqueous H2O2 is obtained instead.34,119 Besides the nature of the incorporated heteroatom, the type of zeolite and its synthesis mode determine the performance of the catalyst. Blasco et al. compared fluoride-synthesized Ti-Beta(F), alkali-synthesized Ti-Beta(OH) and TS-1 as catalysts for the epoxidation of 1-hexene.34 TS-1 had the highest turnover number of the three catalysts with methanol as the solvent but the lowest in acetonitrile. The activities of Ti-Beta(F) and Ti-Beta(OH) were similar in both solvents but the more hydrophobic Ti-Beta(F) had a higher selectivity in the use of H2O2. The influence of the hydrophobic character was demonstrated by the epoxidation of oleic acid where Ti-Beta(F) is more active than Ti-Beta(OH). As Ti-Beta(OH) is more hydrophilic, the oleic acid adsorbs strongly through the polar head group, making it more difficult to oxidize the double bond in the molecule.Substituted Beta zeolites are efficient catalysts for Meerwein–Ponndorf–Verley (MPV) reduction of aldehydes and ketones using secondary alcohols as hydrogen donors and for the Oppenauer oxidation of alcohols by carbonyl oxidants. The benefits of using solid zeolites for the transfer hydrogenation instead of the conventional homogeneous aluminium alkoxide catalysts are easy workup and catalyst recovery. The transfer hydrogenation provides a greener solution to reduction under milder conditions without needing high hydrogen pressures. Zirconium and tin are especially active for the MPV reactions.39,120 Their high chemoselectivity for the reduction of the carbonyl group has been employed to convert α,β-unsaturated aldehydes into the corresponding unsaturated alcohols, e.g., cinnamaldehyde is reduced to cinnamyl alcohol with >98% selectivity over Zr-Beta.37 In the MPV reduction of 4-tert-butylcyclohexanone using hydrophobic Zr-Beta, only cis-4-tert-butylcyclohexanol was obtained.121 The activity and selectivity of Zr-Beta for this reaction outperforms that of Al- and Sn-Beta. The high stereoselectivity of the reduction is due to steric constraints which force the reaction to proceed via the less bulky transition state that can align along the channel wall.
Cascade transformations, either with multiple catalysts in one pot or with multiple active sites present on one catalyst, provide the ability to form products with high selectivity and suppression of side reactions.122 The advantages of this strategy are high atom economy, reduced waste generation and savings in time required to work up each step. The redox functionality can be added to the zeolite. For example, Zr-Beta was loaded with rhodium to introduce gas phase hydrogenation capability. Applied to the cascade hydrogenation of 4-alkylphenols, the designed Rh/Zr-Beta first catalysed the hydrogenation of the phenol ring to the intermediate, 4-alkylcyclohexanone, which could be reduced via the highly stereoselective MPV reduction over zirconium Lewis acid sites to give the cis-alcohol (Table 2, entry 1). Even with a low metal loading of 0.5% Rh on Zr-beta, 4-tert-butylphenol, and p-cresol were hydrogenated to the corresponding cis-alcohols with 95 and 89% stereoselectivity, respectively, under mild reaction conditions (80 °C, 0.5 MPa H2).123 The synthesis of menthol from citral, abundantly available as the main component of lemongrass oil, requires three steps: selective hydrogenation of citral to citronellal, followed by cyclization of citronellal to isopulegols and hydrogenation (Table 2, entry 2).124 The cyclization step creates two additional chiral centres and therefore results in eight stereoisomers of which (−)-isopulegol is the most desired isomer as it can be hydrogenated to (−)-menthol which has a refreshing peppermint odour and exerts a cooling effect. Although the cyclization occurs easily over Lewis acid catalysts, Zr-Beta catalyzes the reaction with diastereoselectivity for isopulegol >93%. This is comparable to the industrial process where ZnBr2 is used in stoichiometric amounts, with the associated problems of disposal on work-up. With bifunctional catalysts containing nickel or rhodium supported on Zr-Beta, citral could be converted in one pot directly into menthols with yields of 87–89% and an excellent diastereoselectivity of 94% for the desired (±)-menthol.125 Using two separate catalysts, Zr-Beta and nano dispersed Ni on an MCM-41 support, was equally effective and had the advantage that the rates of each step could be independently varied by adjusting the amount of the appropriate catalyst. By increasing the hydrogen pressure from 0.2 MPa initially to 2 MPa towards the end of the reaction, the over-reduction of citral and citronellal to undesired by-products was minimized and the yield of menthols could be increased to 95% of which 94% were (±)-menthol.
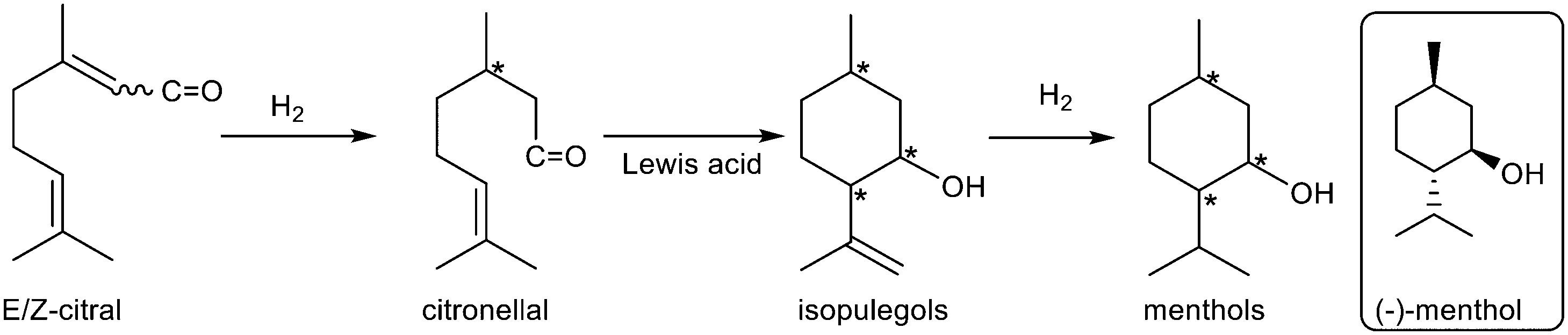


Anethole is an important ingredient in many flavors, fragrances and pharmaceutical formulations.115,126 It can be synthesized via sequential hydrogen transfer reduction and dehydration of 4′-methoxypropiophenone (Table 2, entry 3). Zr-HY zeolite (Si/Zr 50) is an extremely active catalyst for this synthesis. The reaction rate was 4–7 times faster than over mesoporous Zr-MSU-3 or Zr-Beta. This has been attributed to its accessible pore sizes, the abundance of the stronger Lewis acid sites required for dehydration activity, and a hydrophobic surface that aids in the adsorption of the organic reactants and the desorption of water.
Interest in sustainable production of energy and chemicals has spurred attention to biomass and CO2 utilization.127 Zeolite catalysts are important in the transformation of carbohydrates to various platform chemicals. Despite challenges due to their microporosity and limited stability in the liquid phase, they have potentials due to their Brønsted and Lewis acidity.122 As many of the carbohydrate reactions occur in water, hydrophobic zeolites are ideal due to their intrinsic repulsion of water. Glucose is the most abundant monosaccharide but before it can be converted into other compounds, it is necessary to isomerize it to the more reactive fructose (Scheme 2). While this can be readily achieved by high temperature treatment of glucose with a base,128 sustainable chemocatalytic processes are desirable. Sn-Beta can catalyse the glucose to fructose isomerization in aqueous media with high activity and selectivity.129 Mechanistic studies revealed that glucose binds to the Lewis acid centre followed by an intramolecular hydride transfer between C-1 and C-2 carbons to form the acyclic fructose.130,131 Despite some suppression of Lewis acidity in the presence of water, the framework Sn atoms, presumably open sites, were sufficiently active to mediate the reaction. The isomerization of glucose to fructose can also be catalyzed by extraframework SnO2 located within the pore channel.132 The SnO2 acts as a base catalyst by abstracting a proton at the C-2 carbon to form an enolate intermediate. Changing the solvent from water to methanol results in the epimerization of glucose to mannose rather than isomerization. The epimerization occurs via an intramolecular carbon shift between the C-1 and C-2 positions catalysed by framework Sn atoms in Sn-Beta.
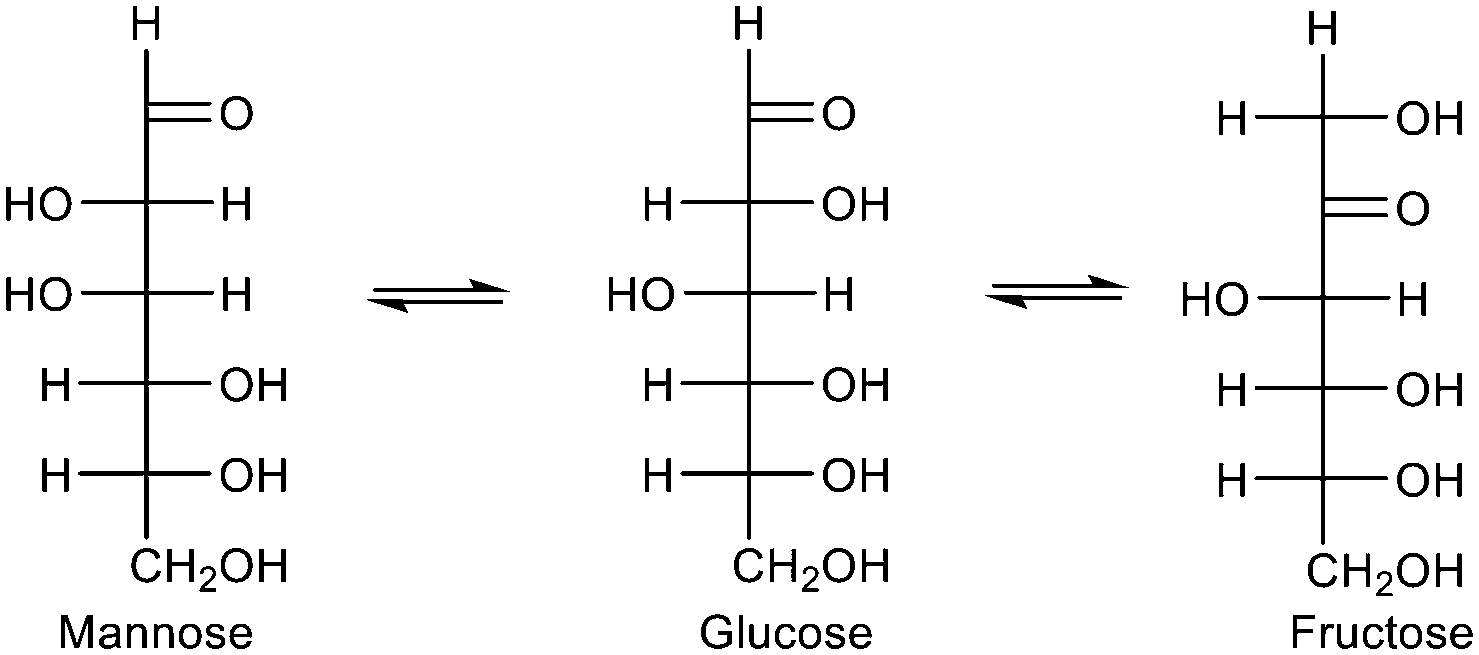 | ||
| Scheme 2 Isomerization of glucose to fructose and epimerization to mannose. | ||
Lactic acid is a promising carbohydrate-derived molecule which is extensively used in the food industry as a monomer for biodegradable plastics and for bulk chemicals such as acrylic acid.133 Currently, its production is by enzymatic fermentation but zeolites and mesoporous silica isomorphously substituted with Sn have been reported to be active catalysts for the reaction.90,133,134 Lactic acid and its esters have been synthesized from glucose, fructose and sucrose using Sn-Beta.135 At high temperatures (140–160 °C) in methanol, the sugars undergo a rate-determining retro aldol reaction to form two trioses which are converted to racemic methyl lactate through sequential dehydration and methanol addition, followed by a 1,2-hydride shift (Table 2, entry 4). When sucrose was the substrate, a substantially higher yield of methyl lactate, 68%, was obtained than from fructose or glucose (43–44%). The Sn-substituted medium-pore zeolite MWW is another active and selective catalyst for the conversion of sugars to methyl lactate and lactic acid.134 With this catalyst, the yields of methyl lactate from sucrose are only slightly higher than from glucose and fructose, which was attributed to a fast methanolysis of sucrose over Sn-MWW.
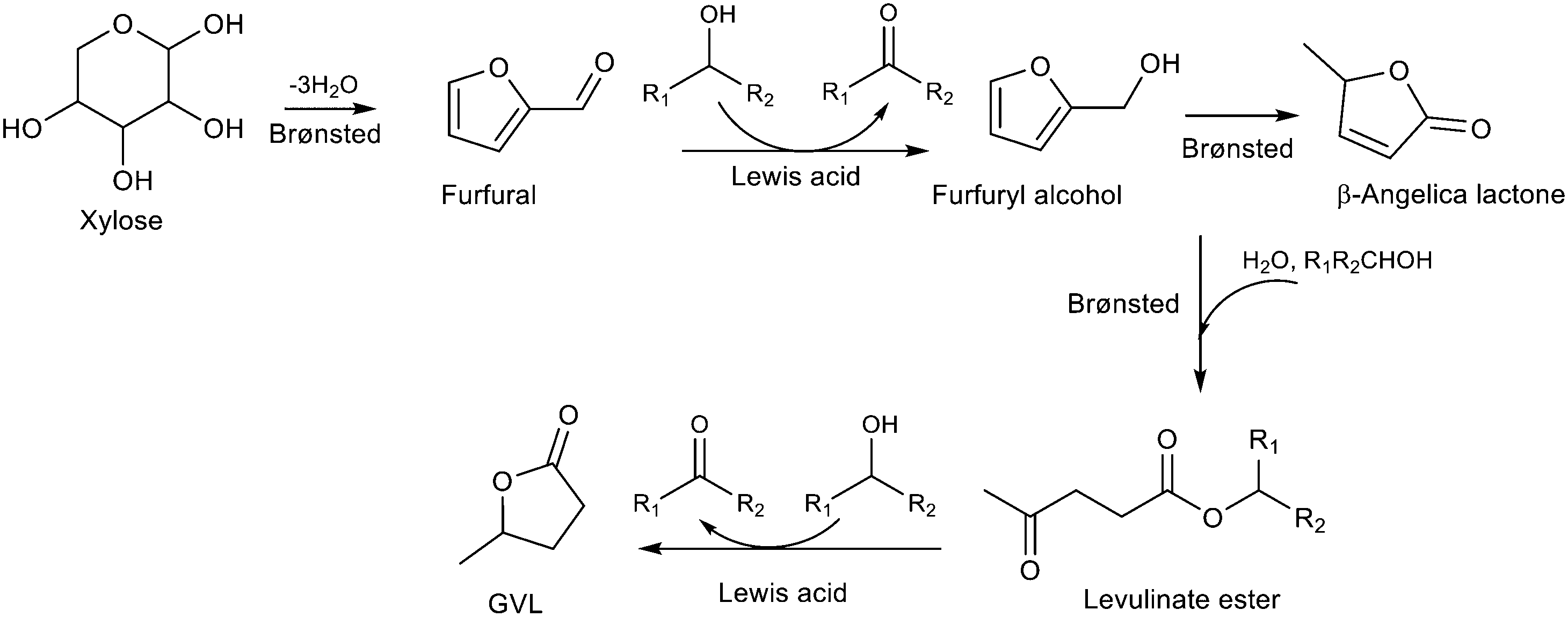
γ-Valerolactone (GVL) has been identified as a platform molecule which can be used as a renewable feedstock in the production of both energy and chemicals.136,137 GVL is produced by hydrogenation of levulinic acid, obtained from cellulose and sugars. While gas phase hydrogenation can be used, the MPV reduction offers an alternative approach with secondary alcohols as hydrogen donors. Zr-Beta (Si/Zr-100) proved to be a highly efficient and stable catalyst for this reaction; at 250 °C in a continuous flow reactor with WHSV of 0.64 h−1, the GVL yield remained steady at >99% for 87 h.138 The space–time-yield of 0.46 molGVLgZr−1 h−1 is comparable with that from high pressure gas phase hydrogenation using precious metal catalysts. Furfural and xylose as starting materials for GVL have been less studied as the transformation involves a multistep cascade of dehydration, (transfer) hydrogenation, etherification, elimination/addition, ring-opening and lactonization (Table 2, entry 5). These reactions require both Lewis and Brønsted acid sites. To meet this requirement, Hernández et al. prepared Zr–Al-Beta by the post-synthesis method.139 The Al/Zr ratio was tuned to optimize the Brønsted to Lewis acidity for the conversion of xylose to GVL. A zeolite with 5.7 wt.% Zr and Al/Zr molar ratio of 0.22 gave 35 mole% GVL after 48 h at 190 °C in 2-propanol. Much higher GVL yields can be obtained when using furfural as the starting material. Bui et al. used a combination of Zr-Beta and Al-MFI nanosheets in a one-pot batch reactor with 2-butanol as hydrogen source for the MPV reduction step.140 Yields of close to 80% were obtained at 120 °C after 48 h. Our group found that a physical mixture of Zr-HY and Al-HY was a highly efficient catalytic system with ∼85% GVL yield after only 5 h at 120 °C.141 The Zr-HY (Si/Zr 20) catalyst was more active than Zr-Beta for the MPV reduction of furfural and levulinate ester with 2-pentanol. This was attributed to the larger pore size and stronger Lewis acidic strength compared to Zr-Beta with comparable Zr content. The Brønsted acid steps proceeded differently over Al-HY and Al-Beta. Al-HY preferentially catalyzed the ring opening of furfuryl alcohol to levulinate ester whereas Al-Beta was more selective to β-angelica lactone. This reaction path requires a subsequent transesterification with an alcohol to form the levulinate ester. A side reaction of etherification between furfural and the solvent was more predominant over Al-Beta than Al-HY zeolite. Using Al-HY with a high Al content (Si/Al 6) where the Brønsted acid strength is weaker significantly reduced the etherification side products.
CO2 utilization is another reaction which attracts increasing attention in the context of global warming and a circular economy.142 Bacariza et al. recently studied the application of nickel on zeolites for the hydrogenation of CO2 to methane.143 The type of zeolite framework and compensating ions influence the metal dispersion and the basicity of the zeolites. Water, a byproduct of methanation, can inhibit the reaction because it competes with CO2 for adsorption sites but this can be addressed by increasing the silica content in the zeolite. Hydrophobic USY zeolites with high Si/Al ratios as supports for nickel gave significantly improved methane yields and longer catalyst lifetime. The hydrogenation of CO2 over Cu@Na-Beta catalyst in a fixed bed reactor at 300 °C and 2.1 MPa yielded exclusively ethanol.144 The catalyst was prepared in a two-step process where commercial Na-Beta was first treated with alkali to generate mesopores, followed by introduction of CuO. The resultant solid was subjected to dry gel conversion. Such a procedure is claimed to form irregularly-shaped copper nanoparticles of 2–5 nm with a high density of step sites. The surrounding zeolite framework constrains the CO2 reaction to the Cu sites and block by-product formation.
6. Conclusions and outlook
High surface area and porosity are desired properties for heterogeneous catalysts. Zeolites are outstanding candidates of porous materials with unique performance in the fields of petroleum industry and fine chemicals production. Post-synthetic treatment of zeolites offers a chance to modify the textural properties and customize them for optimum performance. Reduction of particle size and introduction of mesopores facilitate diffusion of reactants to active sites in the porous channels. Modifying the zeolites by incorporating heteroatoms other than Si, Al and P in the silicate framework forms new Brønsted or Lewis acid sites. Framework-incorporated Ti, Sn and Zr zeolites are excellent catalysts for many reactions such as epoxidation, isomerization, cyclization, and transfer hydrogenation.It is challenging to introduce heterometals into the zeolitic framework due to precipitation of many metal ions as hydroxides under the highly basic synthesis conditions necessary for crystallization. Therefore, the fluoride-assisted hydrothermal method is often used due to the excellent mineralization ability of fluoride ions in neutral to slightly acidic conditions. The amount of metal ions that can be incorporated is limited and higher concentrations require a significant increase in the crystallization times of up to several weeks. Furthermore, the use of fluoride ions, particularly in the form of HF, poses safety concerns for handling on an industrial scale. Another strategy is the two-step post-synthesis method using already-synthesized zeolites as starting material for heteroatom incorporation. This typically requires a dealumination step forming silanol nests which are coordination sites for the heteroatoms. These heteroatoms can be introduced by wet or dry impregnation. Zeolites with high heteroatom substitution can be obtained within a few days. Because of the remaining silanol groups, these zeolites are generally more hydrophilic than those formed using the fluoride-assisted hydrothermal method.
Mechanochemistry-based routes provide simpler and faster access to high quality heteroatom-containing zeolites. The input of mechanical energy varies from manual grinding to more intensive ball milling, depending on the type of zeolite and reactants. The zeolites obtained by mechanochemical-based routes share several similarities with those synthesized by the fluoride-assisted hydrothermal method, e.g., crystallite size, low defect concentration and hydrophobic nature. In applications as solid acid catalysts, hydrophobicity of the pore walls affects the concentration of organic reactants in the pores and the extent to which water can bind to the surface. Hydrophobicity is particularly important in dehydration reactions, which are often encountered in biomass valorization. Mechanochemistry-based protocols offer several attractive characteristics and should be added to the toolbox for green and sustainable zeolite synthesis. The advantages include (i) the possibility of using readily available precursors instead of costly exotic ones (ii) smaller concentrations of organic SDA or even complete elimination (iii) solventless nature with intrinsic lower pressures during crystallization (iv) minimal waste generation and (v) high space–time yield.
The site specificity and intrinsic acidity of the incorporated atoms play defining roles in the catalytic activity. Spectroscopic characterization for open/closed sites and the determination of the precise location of the heteroatom within the unit cell have given insights into the role of different active sites. Further development of analytical techniques with in situ capabilities will be invaluable in advancing our understanding of the underlying chemical mechanisms in zeolite crystallization and site-activity correlations. In this minireview, selected applications of the zeolite-based catalysts from single step transformation to complex one-pot cascade reactions, and in the valorization of CO2 and renewable biomass sources to fine chemicals have been discussed. They illustrate the tremendous potential for developing sustainable processes using zeolite catalysts. With its many advantages, it is to be anticipated that mechanochemistry will play an increasingly important role in the synthesis and design of zeolites.
Author contributions
Hongwei Zhang: writing – original draft. Ismail bin Samsudin: -data curation. Stephan Jaenicke: writing – review & editing. Gaik-Khuan Chuah: conceptualization, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge financial support from the Ministry of Education (ARC Tier 1 grants R-143-000-667-114 and A-0004107-00-00) administered by the Faculty of Science, National University of Singapore.References
- A. H. Chowdhury, N. Salam, R. Debnath, S. M. Islam and T. Saha, in Nanomaterials Synthesis, ed. Y. Beeran Pottathara, S. Thomas, N. Kalarikkal, Y. Grohens and V. Kokol, Elsevier, 2019, ch. 8, pp. 265–294 Search PubMed.
- G. S. Day, H. F. Drake, H.-C. Zhou and M. R. Ryder, Commun. Chem., 2021, 4, 114 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- N. E. R. Zimmermann and M. Haranczyk, Cryst. Growth Des., 2016, 16, 3043–3048 CrossRef CAS.
- G. Yang and L. Zhou, Sci. Rep., 2017, 7, 16113 CrossRef PubMed.
- M. G. Clerici and P. Ingallina, J. Catal., 1993, 140, 71–83 CrossRef CAS.
- M. Taramasso, G. Perego and B. Notari, US Pat., 4410501, 1983 Search PubMed.
- R. Millini and G. Bellussi, in Zeolites in Catalysis: Properties and Applications, The Royal Society of Chemistry, 2017, ch. 1, pp. 1–36 Search PubMed.
- H. van Bekkum and H. W. Kouwenhoven, Zeolite manual for the organic chemist, Mijnbestseller, NL, 2014 Search PubMed.
- P. Ferri, C. Li, R. Millán, J. Martínez-Triguero, M. Moliner, M. Boronat and A. Corma, Angew. Chem., Int. Ed., 2020, 59, 19708–19715 CrossRef CAS PubMed.
- E. G. Derouane, J. Mol. Catal. A: Chem., 1998, 134, 29–45 CrossRef CAS.
- J. C. Vartuli, K. D. Schmitt, C. T. Kresge, W. J. Roth, M. E. Leonowicz, S. B. McCullen, S. D. Hellring, J. S. Beck, J. L. Schlenker, D. H. Olson and E. W. Sheppard, in Stud. Surf. Sci. Catal., ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Hölderich, Elsevier, 1994, pp. 53–60 Search PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- S. Kittaka, S. Takahara, H. Matsumoto, Y. Wada, T. J. Satoh and T. Yamaguchi, J. Chem. Phys., 2013, 138, 204714 CrossRef PubMed.
- F. Goettmann and C. Sanchez, J. Mater. Chem., 2007, 17, 24–30 RSC.
- T. Takamuku, H. Maruyama, S. Kittaka, S. Takahara and T. Yamaguchi, J. Phys. Chem. B, 2005, 109, 892–899 CrossRef CAS PubMed.
- R. Guégan, D. Morineau and C. Alba-Simionesco, Chem. Phys., 2005, 317, 236–244 CrossRef.
- Z. Zhou and M. Hartmann, Chem. Soc. Rev., 2013, 42, 3894–3912 RSC.
- Z. Wu and D. Zhao, Chem. Commun., 2011, 47, 3332–3338 RSC.
- U. Ciesla and F. Schüth, Microporous Mesoporous Mater., 1999, 27, 131–149 CrossRef CAS.
- X. Meng and F.-S. Xiao, Chem. Rev., 2014, 114, 1521–1543 CrossRef CAS PubMed.
- W. Xu, J. Dong, J. Li, J. Li and F. Wu, J. Chem. Soc., Chem. Commun., 1990, 755–756 RSC.
- M.-H. Kim, H.-X. Li and M. E. Davis, Microporous Mater., 1993, 1, 191–200 CrossRef CAS.
- M. Matsukata, M. Ogura, T. Osaki, P. R. H. P. Rao, M. Nomura and E. Kikuchi, Top. Catal., 1999, 9, 77–92 CrossRef CAS.
- Z. Kang, X. Zhang, H. Liu, J. Qiu and K. L. Yeung, Chem. Eng. J., 2013, 218, 425–432 CrossRef CAS.
- P. S. Niphadkar, M. S. Kotwal, S. S. Deshpande, V. V. Bokade and P. N. Joshi, Mater. Chem. Phys., 2009, 114, 344–349 CrossRef CAS.
- L. Zeng, Z. Yu, Z. Sun, Y. Han, Y. Xu, J. Wu, Z. Liang and Z. Wang, Microporous Mesoporous Mater., 2020, 293, 109789 CrossRef CAS.
- E. M. Flanigen and R. L. Patton, US Pat., 4073865, 1978 Search PubMed.
- H. Kessler, J. Patarin and C. Schott-Darie, in Stud. Surf. Sci. Catal., ed. J. C. Jansen, M. Stöcker, H. G. Karge and J. Weitkamp, Elsevier, 1994, pp. 75–113 Search PubMed.
- J. Guth, H. Kessler, P. Caullet, J. Hazm, A. Merrouche and J. Patarin, in Proceedings of the 9th International Zeolite Conference, ed. R. V. Ballmoos, J. B. Higgins and M. M. J. Treacy, Butterworth-Heinemann, Montreal, 1993, pp. 215–222 Search PubMed.
- S. I. Zones, S.-J. Hwang, S. Elomari, I. Ogino, M. E. Davis and A. W. Burton, C. R. Chim., 2005, 8, 267–282 CrossRef CAS.
- A. Corma, M. E. Domine and S. Valencia, J. Catal., 2003, 215, 294–304 CrossRef CAS.
- T. Blasco, M. A. Camblor, A. Corma, P. Esteve, J. M. Guil, A. Martínez, J. A. Perdigón-Melón and S. Valencia, J. Phys. Chem. B, 1998, 102, 75–88 CrossRef CAS.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef CAS PubMed.
- J. Wang, K. Okumura, S. Jaenicke and G.-K. Chuah, Appl. Catal., A, 2015, 493, 112–120 CrossRef CAS.
- Y. Zhu, G.-K. Chuah and S. Jaenicke, J. Catal., 2006, 241, 25–33 CrossRef CAS.
- Z. Yongzhong, N. Yuntong, S. Jaenicke and G.-K. Chuah, J. Catal., 2005, 229, 404–413 CrossRef.
- Y. Zhu, G. Chuah and S. Jaenicke, J. Catal., 2004, 227, 1–10 CrossRef CAS.
- Y. Shinno, K. Iyoki, K. Ohara, Y. Yanaba, Y. Naraki, T. Okubo and T. Wakihara, Angew. Chem., Int. Ed., 2020, 59, 20099–20103 CrossRef CAS PubMed.
- V. Vattipalli, A. M. Paracha, W. Hu, H. Chen and W. Fan, Angew. Chem., Int. Ed., 2018, 57, 3607–3611 CrossRef CAS PubMed.
- S. Dzwigaj and T. Shishido, J. Phys. Chem. C, 2008, 112, 5803–5809 CrossRef CAS.
- S. Dzwigaj, M. J. Peltre, P. Massiani, A. Davidson, M. Che, S. Dzwigaj, P. Massiani, T. Sen and S. Sivasanker, Chem. Commun., 1998, 87–88 RSC.
- J. Dijkmans, J. Demol, K. Houthoofd, S. Huang, Y. Pontikes and B. Sels, J. Catal., 2015, 330, 545–557 CrossRef CAS.
- J. Dijkmans, D. Gabriëls, M. Dusselier, F. de Clippel, P. Vanelderen, K. Houthoofd, A. Malfliet, Y. Pontikes and B. F. Sels, Green Chem., 2013, 15, 2777–2785 RSC.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- D. N. Rainer and R. E. Morris, Dalton Trans., 2021, 50, 8995–9009 RSC.
- R. E. Morris and S. L. James, Angew. Chem., Int. Ed., 2013, 52, 2163–2165 CrossRef CAS PubMed.
- G. Majano, L. Borchardt, S. Mitchell, V. Valtchev and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2014, 194, 106–114 CrossRef CAS.
- K. Yamamoto, S. E. Borjas García and A. Muramatsu, Microporous Mesoporous Mater., 2007, 101, 90–96 CrossRef CAS.
- M. Yabushita, H. Kobayashi, R. Osuga, M. Nakaya, M. Matsubara, S. Maki, K. Kanie and A. Muramatsu, Ind. Eng. Chem. Res., 2021, 60, 2079–2088 CrossRef CAS.
- P. Hu, K. Iyoki, H. Yamada, Y. Yanaba, K. Ohara, N. Katada and T. Wakihara, Microporous Mesoporous Mater., 2019, 288, 109594 CrossRef CAS.
- T. Iida, A. Takagaki, S. Kohara, T. Okubo and T. Wakihara, ChemNanoMat, 2015, 1, 155–158 CrossRef CAS.
- T. Iida, M. Sato, C. Numako, A. Nakahira, S. Kohara, T. Okubo and T. Wakihara, J. Mater. Chem. A, 2015, 3, 6215–6222 RSC.
- L. Ren, Q. Wu, C. Yang, L. Zhu, C. Li, P. Zhang, H. Zhang, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 15173–15176 CrossRef CAS PubMed.
- Q. Wu, X. Wang, G. Qi, Q. Guo, S. Pan, X. Meng, J. Xu, F. Deng, F. Fan, Z. Feng, C. Li, S. Maurer, U. Müller and F.-S. Xiao, J. Am. Chem. Soc., 2014, 136, 4019–4025 CrossRef CAS PubMed.
- V. Pashkova, K. Mlekodaj, P. Klein, L. Brabec, R. Zouzelka, J. Rathousky, V. Tokarova and J. Dedecek, Chem. – Eur. J., 2019, 25, 12068–12073 CrossRef CAS PubMed.
- M. H. Nada, E. G. Gillan and S. C. Larsen, Microporous Mesoporous Mater., 2019, 276, 23–28 CrossRef CAS.
- Q. Wu, X. Meng, X. Gao and F.-S. Xiao, Acc. Chem. Res., 2018, 51, 1396–1403 CrossRef CAS PubMed.
- Q. Wu, X. Liu, L. Zhu, L. Ding, P. Gao, X. Wang, S. Pan, C. Bian, X. Meng, J. Xu, F. Deng, S. Maurer, U. Müller and F.-S. Xiao, J. Am. Chem. Soc., 2015, 137, 1052–1055 CrossRef CAS PubMed.
- M. Hartmann, A. G. Machoke and W. Schwieger, Chem. Soc. Rev., 2016, 45, 3313–3330 RSC.
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC.
- A. Feliczak-Guzik, Microporous Mesoporous Mater., 2018, 259, 33–45 CrossRef CAS.
- X. Jia, W. Khan, Z. Wu, J. Choi and A. C. K. Yip, Adv. Powder Technol., 2019, 30, 467–484 CrossRef CAS.
- K. Li, J. Valla and J. Garcia-Martinez, ChemCatChem, 2014, 6, 46–66 CrossRef CAS.
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, J. Messinger Robert, F. Chmelka Bradley and R. Ryoo, Science, 2011, 333, 328–332 CrossRef CAS PubMed.
- K. Möller, B. Yilmaz, R. M. Jacubinas, U. Müller and T. Bein, J. Am. Chem. Soc., 2011, 133, 5284–5295 CrossRef PubMed.
- J.-B. Koo, N. Jiang, S. Saravanamurugan, M. Bejblová, Z. Musilová, J. Čejka and S.-E. Park, J. Catal., 2010, 276, 327–334 CrossRef CAS.
- A. Karlsson, M. Stöcker and R. Schmidt, Microporous Mesoporous Mater., 1999, 27, 181–192 CrossRef CAS.
- K. R. Kloetstra, H. W. Zandbergen, J. C. Jansen and H. van Bekkum, Microporous Mater., 1996, 6, 287–293 CrossRef CAS.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718–723 CrossRef CAS PubMed.
- J. Zhu, Y. Zhu, L. Zhu, M. Rigutto, A. van der Made, C. Yang, S. Pan, L. Wang, L. Zhu, Y. Jin, Q. Sun, Q. Wu, X. Meng, D. Zhang, Y. Han, J. Li, Y. Chu, A. Zheng, S. Qiu, X. Zheng and F.-S. Xiao, J. Am. Chem. Soc., 2014, 136, 2503–2510 CrossRef CAS PubMed.
- K. Möller, B. Yilmaz, U. Müller and T. Bein, Chem. Mater., 2011, 23, 4301–4310 CrossRef.
- A. H. Janssen, A. J. Koster and K. P. de Jong, J. Phys. Chem. B, 2002, 106, 11905–11909 CrossRef CAS.
- A. H. Janssen, A. J. Koster and K. P. de Jong, Angew. Chem., Int. Ed., 2001, 40, 1102–1104 CrossRef CAS PubMed.
- D. Verboekend and J. Pérez-Ramírez, Catal. Sci. Technol., 2011, 1, 879–890 RSC.
- D. Verboekend and J. Pérez-Ramírez, ChemSusChem, 2014, 7, 753–764 CrossRef CAS PubMed.
- A. Sachse and J. García-Martínez, Chem. Mater., 2017, 29, 3827–3853 CrossRef CAS.
- J. García-Martínez, M. Johnson, J. Valla, K. Li and J. Y. Ying, Catal. Sci. Technol., 2012, 2, 987–994 RSC.
- J. Huang, Y. Fan, G. Zhang and Y. Ma, RSC Adv., 2020, 10, 13583–13590 RSC.
- J. A. van Bokhoven and C. Lamberti, Coord. Chem. Rev., 2014, 277-278, 275–290 CrossRef CAS.
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, Nat. Mater., 2020, 19, 1047–1056 CrossRef CAS PubMed.
- S. M. T. Almutairi, B. Mezari, G. A. Filonenko, P. C. M. M. Magusin, M. S. Rigutto, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2013, 5, 452–466 CrossRef CAS.
- S. R. Batool, V. L. Sushkevich and J. A. van Bokhoven, J. Catal., 2022, 408, 24–35 CrossRef CAS.
- R. Zhao, Z. Zhao, S. Li and W. Zhang, J. Phys. Chem. Lett., 2017, 8, 2323–2327 CrossRef CAS PubMed.
- M. Shamzhy, M. Opanasenko, P. Concepción and A. Martínez, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- S. R. Bare, S. D. Kelly, W. Sinkler, J. J. Low, F. S. Modica, S. Valencia, A. Corma and L. T. Nemeth, J. Am. Chem. Soc., 2005, 127, 12924–12932 CrossRef CAS PubMed.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Phys. Chem. C, 2013, 117, 3976–3986 CrossRef CAS.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- H. Zhang, L. Zhong, I. Bin Samsudin, K. Okumura, H.-R. Tan, S. Li, S. Jaenicke and G.-K. Chuah, J. Catal., 2022, 405, 489–498 CrossRef CAS.
- T. Sen, P. R. Rajamohanan, S. Ganapathy and S. Sivasanker, J. Catal., 1996, 163, 354–364 CrossRef CAS.
- S. Higashimoto, S. G. Zhang, H. Yamashita, Y. Matsumura, Y. Souma and M. Anpo, Chem. Lett., 1997, 26, 1127–1128 CrossRef.
- M. Boronat and A. Corma, ACS Catal., 2019, 9, 1539–1548 CrossRef CAS PubMed.
- C. Bornes, M. Fischer, J. A. Amelse, C. F. G. C. Geraldes, J. Rocha and L. Mafra, J. Am. Chem. Soc., 2021, 143, 13616–13623 CrossRef CAS PubMed.
- Y. Wang, S. Xin, Y. Chu, J. Xu, G. Qi, Q. Wang, Q. Xia and F. Deng, J. Phys. Chem. C, 2021, 125, 9497–9506 CrossRef CAS.
- M. Brändle and J. Sauer, J. Am. Chem. Soc., 1998, 120, 1556–1570 CrossRef.
- M. Boronat, P. Concepción, A. Corma, M. Renz and S. Valencia, J. Catal., 2005, 234, 111–118 CrossRef CAS.
- J. S. Bates, B. C. Bukowski, J. W. Harris, J. Greeley and R. Gounder, ACS Catal., 2019, 9, 6146–6168 CrossRef CAS.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef CAS.
- G. Qi, Q. Wang, J. Xu, Q. Wu, C. Wang, X. Zhao, X. Meng, F. Xiao and F. Deng, Commun. Chem., 2018, 1, 22 CrossRef.
- Y. G. Kolyagin, A. V. Yakimov, S. Tolborg, P. N. R. Vennestrøm and I. I. Ivanova, J. Phys. Chem. Lett., 2018, 9, 3738–3743 CrossRef CAS PubMed.
- G. Li, E. A. Pidko and E. J. M. Hensen, Catal. Sci. Technol., 2014, 4, 2241–2250 RSC.
- D. Gleeson, G. Sankar, C. R. A. Catlow, J. M. Thomas, G. Spanó, S. Bordiga, A. Zecchina and C. Lamberti, Phys. Chem. Chem. Phys., 2000, 2, 4812–4817 RSC.
- B. A. Johnson, J. R. Di Iorio and Y. Román-Leshkov, J. Catal., 2021, 404, 607–619 CrossRef CAS.
- A. Rodríguez-Fernández, J. R. Di Iorio, C. Paris, M. Boronat, A. Corma, Y. Román-Leshkov and M. Moliner, Chem. Sci., 2020, 11, 10225–10235 RSC.
- V. L. Sushkevich, D. Palagin and I. I. Ivanova, ACS Catal., 2015, 5, 4833–4836 CrossRef CAS.
- R. Bermejo-Deval, M. Orazov, R. Gounder, S.-J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288–2297 CrossRef CAS.
- W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. Román-Leshkov, J. Phys. Chem. C, 2016, 120, 28533–28544 CrossRef CAS PubMed.
- W. Dai, Q. Lei, G. Wu, N. Guan, M. Hunger and L. Li, ACS Catal., 2020, 10, 14135–14146 CrossRef CAS.
- J. R. Di Iorio, B. A. Johnson and Y. Román-Leshkov, J. Am. Chem. Soc., 2020, 142, 19379–19392 CrossRef CAS PubMed.
- J. N. Kondo, M. Iizuka, K. Domen and F. Wakabayashi, Langmuir, 1997, 13, 747–750 CrossRef CAS.
- S. Namba, N. Hosonuma and T. Yashima, J. Catal., 1981, 72, 16–20 CrossRef CAS.
- B. A. Johnson, J. R. Di Iorio and Y. Román-Leshkov, Acc. Mater. Res., 2021, 2, 1033–1046 CrossRef CAS.
- H. Zhang, Z. J. Quek, S. Jaenicke and G.-K. Chuah, J. Catal., 2021, 400, 50–61 CrossRef CAS.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F.-S. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- P. A. Zapata, Y. Huang, M. A. Gonzalez-Borja and D. E. Resasco, J. Catal., 2013, 308, 82–97 CrossRef CAS.
- M. S. Rahaman, T. K. Phung, M. A. Hossain, E. Chowdhury, S. Tulaphol, S. B. Lalvani, M. O'Toole, G. A. Willing, J. B. Jasinski, M. Crocker and N. Sathitsuksanoh, Appl. Catal., A, 2020, 592, 117369 CrossRef CAS.
- M. Boronat, A. Corma, M. Renz and P. M. Viruela, Chem. – Eur. J., 2006, 12, 7067–7077 CrossRef CAS PubMed.
- A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194–3195 CrossRef CAS PubMed.
- Y. Zhu, G. Chuah and S. Jaenicke, Chem. Commun., 2003, 2734–2735 RSC.
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- Y. Nie, S. Jaenicke, H. van Bekkum and G.-K. Chuah, J. Catal., 2007, 246, 223–231 CrossRef CAS.
- Y. Nie, G.-K. Chuah and S. Jaenicke, Chem. Commun., 2006, 790–792 RSC.
- Y. Nie, S. Jaenicke and G.-K. Chuah, Chem. – Eur. J., 2009, 15, 1991–1999 CrossRef CAS PubMed.
- H. Zhang, C. L.-F. Lim, M. Zaki, S. Jaenicke and G. K. Chuah, ChemSusChem, 2018, 11, 3007–3017 CrossRef CAS PubMed.
- A. Corma, ChemSusChem, 2020, 13, 6054–6055 CAS.
- J. C. Speck, in Advances in Carbohydrate Chemistry, ed. M. L. Wolfrom, Academic Press, 1958, pp. 63–103 Search PubMed.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S.-J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS PubMed.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef PubMed.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- Q. Guo, F. Fan, E. A. Pidko, W. N. P. van der Graaff, Z. Feng, C. Li and E. J. M. Hensen, ChemSusChem, 2013, 6, 1352–1356 CrossRef CAS PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602 CrossRef CAS PubMed.
- K. Yan, C. Jarvis, J. Gu and Y. Yan, Renewable Sustainable Energy Rev., 2015, 51, 986–997 CrossRef CAS.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- J. Wang, S. Jaenicke and G.-K. Chuah, RSC Adv., 2014, 4, 13481–13489 RSC.
- B. Hernández, J. Iglesias, G. Morales, M. Paniagua, C. López-Aguado, J. L. García Fierro, P. Wolf, I. Hermans and J. A. Melero, Green Chem., 2016, 18, 5777–5781 RSC.
- L. Bui, H. Luo, W. R. Gunther and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2013, 52, 8022–8025 CrossRef CAS PubMed.
- H. Zhang, W. Yang, I. I. Roslan, S. Jaenicke and G.-K. Chuah, J. Catal., 2019, 375, 56–67 CrossRef CAS.
- S. Saeidi, S. Najari, V. Hessel, K. Wilson, F. J. Keil, P. Concepción, S. L. Suib and A. E. Rodrigues, Prog. Energy Combust. Sci., 2021, 85, 100905 CrossRef.
- M. C. Bacariza, M. Maleval, I. Graça, J. M. Lopes and C. Henriques, Microporous Mesoporous Mater., 2019, 274, 102–112 CrossRef CAS.
- L. Ding, T. Shi, J. Gu, Y. Cui, Z. Zhang, C. Yang, T. Chen, M. Lin, P. Wang, N. Xue, L. Peng, X. Guo, Y. Zhu, Z. Chen and W. Ding, Chem, 2020, 6, 2673–2689 CAS.
| This journal is © The Royal Society of Chemistry 2022 |