Experimental and numerical investigation of NO oxidation on Pt/Al2O3- and NOx storage on Pt/BaO/Al2O3-catalysts
Received
24th March 2022
, Accepted 19th May 2022
First published on 23rd May 2022
Abstract
Planar laser-induced fluorescence with laser synchronized flow control is employed as a non-invasive in situ technique to investigate a NOx storage catalyst, especially to grant a deeper insight into the reaction dynamics and its interaction with mass transfer. In addition to visualizing the spatial and temporal NO evolution over a Pt/BaO/Al2O3 catalyst, the spatially resolved NO distribution over a Pt/Al2O3 catalyst in the steady-state is measured to understand the oxidation of NO to NO2 as a precursor step of NOx storage. The experimental results are compared with corresponding numerical simulations using transient one- and two-dimensional reactor simulations with detailed surface reaction mechanisms. The thermodynamic equilibrium for NO oxidation over Pt/Al2O3 approaches between 623 K and 723 K, as a reduced conversion is observed at a higher temperature. The NOx storage on the Pt/BaO/Al2O3 catalyst decreases with time, which is partly due to the reduced storage capacity, but also strongly limited by the NO oxidation rate.
1. Introduction
NOx abatement technologies are needed for all combustion systems using air as the oxidizer due to the stringent requirements of emission control. NOx storage catalysts (NSCs) are one of the common concepts for NOx removal, especially for engines operating under lean conditions.1,2 Aside from the catalyst support, typical NSCs consist of noble metals, e.g., Pt, for the NO oxidation under lean conditions and the reduction of stored NOx under rich conditions, and alkaline or alkaline-earth metal oxides as the NOx storage material.3–5 Numerous studies have been conducted experimentally and numerically to understand the mechanism of NOx storage, and to further optimize the design and the control of the exhaust aftertreatment. Y. Nagai et al. conducted spatiotemporally resolved surface species measurements on an NSR catalyst to reveal the cause of the NOx spike emission.6 L. Lietti, E. Tronconi and their co-workers7–9 explored the insights into the NOx storage/reduction mechanism with various experiments, including both gas-phase and surface species measurements. They claimed that NO2 is adsorbed onto BaO to form only nitrates via a disproportionation reaction, while NO uptake under lean conditions has two parallel routes. The first route is through NO oxidation to NO2 on Pt, followed by NO2 adsorption on BaO. The second occurs through the formation of surface nitrites that evolve into nitrates, which can be promoted by the presence of Pt. P. J. Schmitz and R. J. Baird10 agreed on the nitrite formation from the direct NO adsorption on BaO, but they believed that as an intermediate, nitrite is also formed during the NO2 uptake. Models ranging from global kinetic11–13 to micro kinetic schemes14–18 have been proposed to simulate the NOx storage process. Olsson et al. have conducted a series of studies, starting from NO oxidation and NOx storage mechanism on Pt/BaO/Al2O3 with 20 reactions,14 then extending to sulphur deactivation,15,19 investigating the influence of CO2 and H2O,16 and the regeneration mechanism.17,18 Rankovic et al.20,21 optimized the mechanism by comparing simulations with experimental data from various systems with a wide range of conditions. In the microkinetic model by Larson et al.,22 the direct adsorption of NO2 on BaO–O was excluded, instead Ba(NO3)2 formation was only initiated by the adsorption of NO2 on BaO.
Although the kinetic models for NOx storage catalysts have been examined in a wide range of systems, the gas-phase experimental data used for the model validation is limited to end-of-pipe gas concentrations for most of the literature. Shwan et al. from Olsson's group18 validated their model with the intra-catalyst concentrations measured by SpaciMS, a gas sampling technique with multiple capillary probes. The same technique has also been used to study the impact on NH3 and N2O selectivity for the NOx storage/reduction catalysts.23 However, since an invasive method is applied, flow distortion due to the probes cannot be avoided.24 Furthermore, only 1D concentration profiles in flow direction with limited spatial resolution can be obtained, and the temporal broadening caused by the diffusion in between the sampling position and the mass spectrometer affects the direct comparison to the simulations.
In combustion research, non-invasive laser-based techniques such as planar laser-induced fluorescence (PLIF) and Raman spectroscopy have been used for decades to obtain spatially and temporally resolved gas-phase species distributions.25–27 In the last 15 years, such techniques have been applied in the field of heterogeneous catalysis as well. Mantzaras et al. conducted several pioneering studies with PLIF and Raman spectroscopy to investigate the catalyst-assisted combustion in microchannels.28,29 In our group, NO-PLIF measurements were performed in a parallel–wall channel reactor to investigate NO reduction by H2 on Pt-based diesel oxidation catalysts.30 In the same setup, the reaction mechanism of HCHO oxidation on Pt proposed by Torkashvand et al.31 was experimentally evaluated32 based on in situ two-dimensional HCHO distributions measured by HCHO–PLIF. These PLIF investigations allow a better understanding of the interaction of reaction kinetics with the convective and diffusive mass transfer processes in the flow field of the catalytic channel of typical monolithic converters. Quantitative data has been provided in studies mentioned above, as PLIF image averaging could be easily applied for those steady-state processes to increase the signal-to-noise ratio (SNR). However, controlling the SNR is challenging for transient processes. There were a few attempts in the past. Visualization of HCHO formation during catalytic combustion of CH3OH on Pt under transient conditions was reported by Kang et al. using only qualitative data from single-shot measurements.33 Zetterberg et al. demonstrated the spatial and temporal evolution of CO and CO2 on Pt or Pd with both single-shot and time-averaged images.34,35 However, images averaged from multiple consecutive laser shots degrade the temporal resolution, depending on the repetition rate of the laser, from sub-seconds to a few seconds or more, which significantly limits the application in catalytic processes with rapid variations in gas phase composition. To maintain high SNRs at high temporal resolution, recently we demonstrated the potential of phase-locked PLIF to quantitatively determine gas-phase species concentration distributions by averaging the images over several repetitive cycles.36 Therefore, in this study, phase-locked PLIF imaging is further used to obtain spatially and temporally resolved two-dimensional maps of absolute NO concentration distributions for kinetic model evaluation.
The objective of this work is to further develop the NOx storage reaction kinetics by comparing numerical and experimental data with spatial and temporal resolutions. First, NO oxidation on Pt/Al2O3 is investigated in both a packed bed reactor for light-off measurements and a parallel–wall channel configuration for steady-state NO-PLIF measurements. Phase-locked PLIF is further employed to obtain the spatially and temporally resolved NO distributions over a Pt/BaO/Al2O3 catalyst-coated plate in the parallel–wall channel at different temperatures and flow conditions. Corresponding simulations with detailed surface reaction mechanisms are conducted using the one-dimensional packed bed reactor code and the two-dimensional channel flow code of DETCHEM software package.37–40
2. Experimental methodology
2.1. Catalyst preparation
Catalyst powder.
After 5 h of calcination at 973 K in static air, γ-Al2O3 (Puralox, SASOL Germany GmbH) is used as support material for the powder catalyst samples. The Pt/Al2O3 catalyst sample with a target noble metal loading of 1 wt% is prepared by incipient wetness impregnation (IWI) using (NH3)4Pt(NO3)2 (VWR, purity >99.9%) as a precursor that is dissolved in purified water (ROTIPURAN®Ultra, Carl Roth GmbH + Co. KG). After drying the powder for 2 h at 353 K, it is calcined in static air for 5 h at 823 K. Similarly, Pt/Ba/Al2O3 is prepared by stepwise impregnation, analogous to a procedure previously described by Casapu et al.,41 using an aqueous solution of Ba(CH3COO)2 (Merck KGaA) as Ba precursor and an aqueous solution of (NH3)4Pt(NO3)2, as Pt precursor. After each step, the powder is dried at 353 K for 2 h and subsequently calcined at 823 K for 5 h in static air.
Catalytically coated plate for PLIF measurements.
Planar plates with a length of 25 mm, a width of 18 mm and a thickness of 1 mm are cut from a monolithic cordierite substrate (NGK, 3.0 mil, 600 cpsi). Afterwards, plates are coated with an aqueous slurry, which contains catalyst powder and AlO(OH) (Dispersal P2, SASOL) in a weight ratio of 9 to 1 and which is made analogous to the procedure previously described by Karinshak et al.42 The aqueous suspension containing about 50 mg of catalyst powder and AlO(OH) is applied onto the cordierite plate by using a syringe. Subsequently, the plate is turned and tilted to ensure a homogenous distribution of the slurry. After drying in static air, the coated plates are calcined for 5 h at 823 K in static air, ensuring good adhesion of the washcoat on the cordierite substrate. The catalytically coated plate is placed in a groove of a ceramic holder to form a 2 mm high parallel wall channel.
2.2. Catalyst characterization
The target catalyst composition is confirmed by elemental analysis. After degassing the catalyst sample for 2 h at 573 K, N2-physisorption using a Belsorp mini II (MicrotracBEL) and subsequent evaluation according to the BET method provides information on the specific surface area and pore volume of the catalyst powder. Assuming a CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt adsorption stoichiometry of 1:1,43 CO-chemisorption measurements of the catalyst powder are conducted in a continuous flow reactor setup equipped with an X-Stream Enhanced XEGK compact gas analyzer (Emerson Electric Co.) for precise quantification of CO and CO2 in order to obtain the noble metal dispersion. Analogous to the procedure described earlier by our group,44–46 Pt/Al2O3 was subject to a temperature programmed desorption of pre-adsorbed CO, whereas the reactor loaded with Pt/BaO/Al2O3 was cooled to 196 K by means of a dry ice/iso-propanol bath prior to saturation with CO and measurement of the CO uptake, which is supposed to suppress CO–BaO interactions and an incorporation of CO into the BaO structure.
:Pt adsorption stoichiometry of 1:1,43 CO-chemisorption measurements of the catalyst powder are conducted in a continuous flow reactor setup equipped with an X-Stream Enhanced XEGK compact gas analyzer (Emerson Electric Co.) for precise quantification of CO and CO2 in order to obtain the noble metal dispersion. Analogous to the procedure described earlier by our group,44–46 Pt/Al2O3 was subject to a temperature programmed desorption of pre-adsorbed CO, whereas the reactor loaded with Pt/BaO/Al2O3 was cooled to 196 K by means of a dry ice/iso-propanol bath prior to saturation with CO and measurement of the CO uptake, which is supposed to suppress CO–BaO interactions and an incorporation of CO into the BaO structure.
The number of BaO sites on the coated catalyst plate was estimated by assuming that all NOx will eventually form Ba(NO)3 if the storage phase is sufficiently long. Therefore, the number of BaO sites is two times the total amount of adsorbed NOx. The latter can be calculated by integrating the difference of inlet and outlet NOx concentrations until the difference becomes zero. Prior to the BaO site measurements, the catalyst is first reduced in 5 vol% H2/N2 at 673 K for 1 h, and then oxidized in 5 vol% O2/N2 at 723 K for 1 h. During the storage phase, 400 ppm NO and 1 vol% O2 balanced by N2 flows into the reactor at 723 K for over 4 hours, until at the outlet the sum of the NO and NO2 concentrations equals the NO concentration at the inlet.
Finally, the plates are cut in the middle, sputtered with gold and scanned along their entire length by means of scanning electron microscopy (SEM) using an FEI, Quanta 250 ESEM (FEG). Analysis of the obtained images using the program package Fiji47 allowed to determine the washcoat thickness, hereby confirming a homogeneous distribution of the catalyst washcoat on the plate.
2.3. Packed bed reactor for kinetic measurement of the powder catalyst
The activity measurements of the Pt/Al2O3 powder catalyst are conducted in a packed bed reactor, which consists of a quartz tube with an inner diameter of 8 mm filled with 300 mg of 1 wt% Pt/Al2O3 catalyst powder over a length of about 1.5 cm. The quartz tube is heated uniformly by a furnace with a PID controller (Eurotherm). The temperature ramp of 3 K min−1 for the light-off cycle ranges from 473 K to 723 K and a Fourier-transform infrared spectrometer (FTIR, Multigas MG2030, MKS) measures the outlet gas components from the reactor or the inlet gas components via a bypass. Details about the fixed bed reactor have been described in previous studies.48,49 To ensure that all light-off cycles start with an identical initial condition, the catalysts are first oxidized at 723 K for 0.5 h in 10 vol% O2 in N2. Subsequently, a degreening at the same temperature for 1 h in 1000 ppm NO, 10 vol% O2 in N2 is conducted.
2.4. PLIF measurement over catalystically coated plate
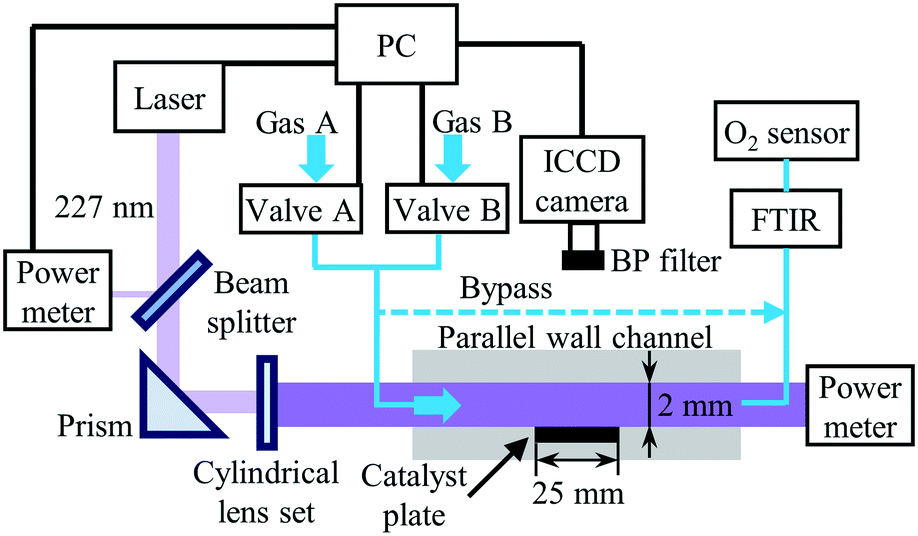
The experimental setup for the PLIF measurements in the parallel–wall channel with catalystically coated plate, as described in detail in our previous studies,30,32,36 is illustrated in Fig. 1. The optically accessible reactor with an inner channel of 2 mm in height, 18 mm in depth and 150 mm in length is used. The catalyst-coated plate (18 mm × 25 mm) is placed in an indentation located in the middle of the channel. Heating coils (HORST) with insulation layers are wrapped around the quartz tube and the end caps are heated with heating cartridges (HORST). The temperature of the reactor is controlled by a PID controller (Eurotherm) between room temperature and up to 723 K. A Fourier-transform infrared spectrometer (FTIR) (Series 2000, MKS) and an O2 sensor (AO2040, ABB) are connected to the exhaust of the reactor to monitor the NO, NO2 and O2 concentrations at the outlet and can be alternatively switched to the bypass connection for measuring the inlet concentrations.
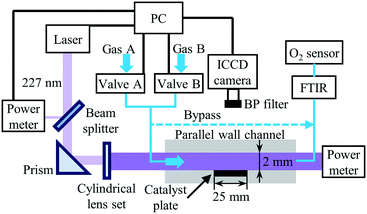 |
| | Fig. 1 Schematic of the experimental setup for phase-locked NO-PLIF measurements. Reproduced from ref. 36 with permission. Copyright 2020 by S. Wan et al. | |
For NO-PLIF, the P2(12.5) + Q12(12.5) line in the NO A2Σ+ − X2Π (0,0) band is excited by a 226.668 nm laser,30,36 which is generated by a frequency-doubled Nd:YAG (Quanta-Ray Pro, Spectra-Physics) pumped dye laser (PrecisionScan, Sirah) sum-frequency mixed with an additional third harmonic output of the Nd:YAG laser. The output laser beam, about 6 mJ per pulse, is split into two beams by a quartz plate serving as a beam splitter. One beam with ∼4% of the total power is sent to a power meter to monitor the pulse energy fluctuation. The other beam is shaped into a ∼0.3 mm thin laser sheet by a set of cylindrical lenses and sent to the reactor. In all measurements, the PLIF laser sheet is placed in the middle of the 18 mm wide channel to minimize the influence of the front and backside quartz boundaries on the results and to closely approximate the 2D flow field in the simulations. The NO fluorescence is collected through a band pass-filter (BP) at 248 nm (FWHM = 10 nm, peak transmission 15%, LaVision) and a UV objective lens (f = 100 mm, f/2.8, LaVision) with a 75 mm extension tube, and finally captured by an image-intensified charge-coupled device (ICCD) camera (IRO 25 & Imager QE, LaVision). The spatial resolution in this study is ∼22 μm per pixel.
An embedded programmable timing unit (PTU X, LaVision) generates the trigger pulses with a repetition frequency of 9.6 Hz for synchronizing the hardware of the PLIF system (laser, camera, power meter, etc.). The oxidation of NO over Pt is investigated in steady-state. For each condition, 500 images are taken for averaging. To perform the phase-locked PLIF for the transient measurement of the NO storage over Pt/BaO/Al2O3, the PTU also sends triggers to control the gas flow. The synchronization scheme is identical to our previous study.36 Briefly, the switching of the gas feed is realized via two 3/2-way solenoid valves (Bürkert), and controlled by the PTU as well as a programmable logic controller (SIMATIC, Siemens). All flow rates given in the manuscript refer to the normal temperature and pressure (293.15 K, 1 atm). To enhance the SNR in this study, all images for transient measurements are averaged phase-locked images over ten repeated cycles. Before each recording, the catalyst is first reduced by flowing 1 vol% H2/N2 for 15 min and subsequently another 5 min with 1 vol% O2/N2, to ensure that all cycles start with the identical status for the catalyst. With the experimental setup in this study, the SNR for a single-shot NO PLIF image at 400 ppm NO, 1% O2, and N2 at 623 K is about 5. Averaging over 10 images increases the SNR to a value of 10 or higher. Note that the averaging is done over phase-locked images, i.e., at identical times after switching the input gas streams. The variation between individual measurements is smaller or comparable to the noise in the NO frames. Only at times of very slow concentration changes was averaging also performed over short time intervals, where it does not affect the temporal gradients.
Because all measurements are conducted with the laser power in the linear regime, in principle collisional quenching has to be considered. To get rid of the spatial variations in quenching of the NO fluorescence, an excess of oxygen representing the dominant quencher is adjusted, to keep its concentration approximately constant. As a consequence, the systematic error caused by the local quenching fluctuations is less than 5% in this study. The absolute NO concentration under reactive conditions is calculated by comparing the PLIF image of the reactive flow to a reference image that was taken under non-reactive conditions. The same PLIF calibration method has been described in previous studies.32,36 The local NO concentration C(x,y) in the target image is given by
| | 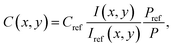 | (1) |
where
I is the fluorescence signal intensity at each pixel,
P is the laser pulse energy, and the subscript ref stands for the reference image.
Cref is determined by the settings of the mass flow controllers and monitored by the FTIR.
3. Modeling approach and numerical simulation
The experiments of NO oxidation on Pt/Al2O3 and NOx storage on Pt/BaO/Al2O3, are modeled and numerically simulated by computer code of DETCHEM software package.37–40 Detailed surface reactions are used for modeling the chemical kinetics in both cases.
3.1. Packed bed reactor
The light-off experiment with the Pt/Al2O3 catalyst powder is simulated with a 1D model of a packed bed reactor, DETCHEMPBR, based on the continuum modeling approach under steady-state and isothermal conditions. To simplify the continuum and species balance equations, it is assumed that there are no radial variations in the flow properties.| | 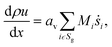 | (2) |
where, ρ is the fluid phase density, x is the axial coordinate, u is the axial superficial velocity, av is the particle area to reactor volume ratio, ṡi is the surface phase reaction rate, Mi is the molar mass of species, Sg is the set of gas-phase species.| | 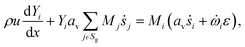 | (3) |
where Yi is mass fraction of species, ![[small omega, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e15a.gif) i is fluid phase reaction rate (not considered in this paper) and ε is bed porosity. It should be noted that for catalytic gas-phase reactions the right-hand term in eqn (2) vanishes at steady-state.
i is fluid phase reaction rate (not considered in this paper) and ε is bed porosity. It should be noted that for catalytic gas-phase reactions the right-hand term in eqn (2) vanishes at steady-state.
The approximated resistance to species mass transport between the composition at reacting catalyst surface and the mean composition is improved with the consideration of external fluid–solid mass transfer coefficient by following relationship equations.
| | | Miṡi = ki,fs(ρi,sYi,s − ρi,fYi,f), | (4) |
where
kfs is fluid–solid mass transfer coefficient.
DETCHEMPBR uses various empirical correlations, as a function of the dimensionless flow number, to calculate fluid–solid heat and mass transfer. Wakao and Kagei50 proposed an empirical equation for the average fluid–solid heat transfer in a packed bed:
| | | Nu = 2 + 1.1Pr1/3Re0.6, | (5) |
where Nu, Pr and Re are the Nusselt, Prandtl and Reynolds number, respectively. The fluid–solid heat transfer coefficient (
hfs) is calculated using the Nusselt number (Nu):
| | 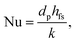 | (6) |
where
dP is the volume-based particle diameter, and
k is thermal conductivity. Based on the heat and mass transfer analogy for dimensionless numbers, the fluid–solid mass transfer can be calculated using the Sherwood number Sh:
| | | Sh = 2 + 1.1Sc1/3Re0.6, | (7) |
where Sc is the Schmidt number. Moreover, the fluid–solid mass transfer coefficient (
kfs) is determined by the Sherwood number (Sh):
| | 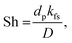 | (8) |
where
D is effective mass diffusivity. Furthermore, the flow numbers Re, Pr, Sc are defined as follows,
| | 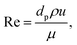 | (9) |
| | 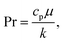 | (10) |
| | 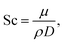 | (11) |
where
μ is fluid viscosity, and
cp is specific heat capacity.
3.2. Channel reactor for PLIF measurements
For the reactive flows in the parallel–wall channel, the code DETCHEMCHANNEL37 is used for the steady-state simulation of NO oxidation on Pt/Al2O3. Below listed represents, the governing equations based on boundary layer approximation used in the parallel–wall channel simulations, total continuity, horizontal momentum (in streamwise direction), vertical momentum (in wall-normal direction), and species continuity respectively.| | 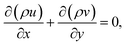 | (12) |
| | 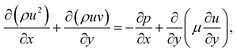 | (13) |
| | 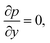 | (14) |
| | 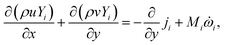 | (15) |
The vertical flux ji can be calculated using following conditions,| | 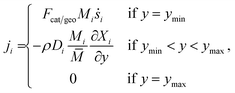 | (16) |
where x is the coordinate for the streamwise direction, y is the coordinate for the wall-normal direction, u and v are the horizontal and the vertical component of velocity, respectively, M is the molar mass, Di and Xi are the mixture-average diffusion coefficient and the mole fraction of species i, respectively. The Peclet number, Pe, for the flow with the lowest flow rate in this study is approximately 150. Therefore, the influence of the counter-current diffusion may be considered rather small.
In the simulation, all walls are modeled with non-slip boundary conditions with the same wall-temperature as those measured in the experiments. The inlet gas temperature is the same as the wall temperature for each case. The flow velocity and the gas components at the inlet are determined according to the experimental conditions. For the transient simulation of NOx storage, the PLIF measured NO inlet concentration profiles including the concentration fluctuations at the rising and falling edges are used as the inlet boundary condition. Constant O2 concentration and flow velocity are assumed in the simulation, despite the possible instability caused by the valve switching in the experiments.
Furthermore, DETCHEMRESERVOIR37–39 is used for the transient simulation of NOx storage on Pt/BaO/Al2O3. The single channel simulation DETCHEMCHANNEL with storage effect is achieved with DETCHEMRESERVOIR which works as a transient wrapper. For present simulation, it is assumed that the time scale for storage reactions is greater than the residence time in the gas phase. The transient species equation, for surface species involved in storage reaction, solved by DETCHEMRESERVOIR is given below.
| | 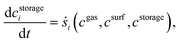 | (17) |
where
cgas is concentration of gas phase species,
csurf is concentration of non-storage surface species and
cstorage is concentration of storage species. The simulation is run iteratively. At each time step, DETCHEM
RESERVOIR calls DETCHEM
CHANNEL. Since the reaction rate of the NO oxidation on Pt is always orders of magnitude higher than that of the storage reaction, a quasi steady-state simulation of single channel is performed for the calculation of concentration profiles of gas-phase species, while keeping the concentrations of storage species fixed. The locally resolved gas-phase concentrations of a single channel are transferred back to the reservoir model followed by a time integration step for the transient species equation,
eqn (17).
To account for the actual active catalytic surface into account, the ratio of catalytic to geometric surface area, Fcat/geo, is used as
| | 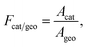 | (18) |
where
Acat is the catalytic active area, and
Ageo is the geometric area.
37 For Pt sites,
Acat is calculated as
| | 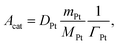 | (19) |
where
ΓPt is the surface-site density of Pt,
DPt is the dispersion of platinum measured by CO chemisorption,
MPt is the molar mass of Pt, and
mPt is the amount of Pt loading. For BaO sites,
| | 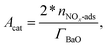 | (20) |
where
ΓBaO is the surface-site density of BaO and
nNOx-ads is the amount of adsorbed NO
x, calculated by integrating the difference of the inlet and outlet NO
x concentration until the difference reduces to zero.
Ageo for the catalyst plate is calculated as 18 mm (width) × 25 mm (length). In this study, the
Fcat/geo for Pt on the Pt/Al
2O
3 coated plate is calculated to be 45. The
Fcat/geo for Pt and BaO on the Pt/BaO/Al
2O
3 coated plate are 112 and 60, respectively.
3.3. Reaction mechanism
Homogenous gas-phase reactions can be neglected, because no change of NO or O2 concentration is detected in an inert channel in the temperature range examined in this study. The surface reaction model for NOx storage on Pt/BaO/Al2O3 contains two parts: the fast catalytic reactions of NO oxidation on Pt and the slow storage reactions of NOx on BaO. Detailed surface reaction mechanisms using the mean-field approximation are applied. The surface phase reaction rate of surface species i, ṡi, is calculated as| | 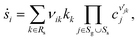 | (21) |
where Rs is the set of surface reactions, Ss is the set of all surface species, νik are the stoichiometric coefficients, 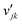 are the reaction orders of the species. The rate coefficient k is determined by a modified Arrhenius expression
are the reaction orders of the species. The rate coefficient k is determined by a modified Arrhenius expression| | 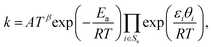 | (22) |
where the pre-exponential factor A, the temperature exponent β, the activation energy Ea and the coverage dependence parameter εi are specified in Table 1 for each reaction. θi is the surface coverage of surface species i. For adsorption reactions, competitive adsorption is considered on both Pt and BaO sites. The rate coefficient is specified in terms of sticking coefficient S, as shown in eqn (23).| | 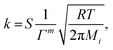 | (23) |
Here, m is the stoichiometric coefficient of the adsorbing surface species and Mi is the molar mass of the adsorbed gas-phase species. The sticking coefficient S has an “Arrhenius-like” form as follows:| | 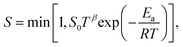 | (24) |
where the initial sticking coefficient S0 is given in Table 1.
Table 1 Reactions in the surface mechanism and the corresponding kinetic parameters
| No. |
Reaction |
A (cm, mol, s) or S0 (*) |
β
|
E
a (kJ mol−1) |
ε
i
(kJ mol−1) |
Denotes coverage dependency on O–Pt.
On Ba(NO3)2.
According to eqn (22)–(24), rate coefficient 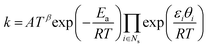 or or 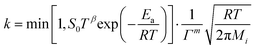 . .
|
| NO oxidation on Pt12 |
| (S1) |
O2 + 2Pt <=> 2O–Pt |
9.515 × 10−2* |
0.009 |
0.0 |
|
| 6.804 × 1021 |
−0.035 |
229.3 |
114.4a |
| (S2) |
NO + Pt <=> NO–Pt |
6.465 × 10−1* |
0.016 |
0.2 |
|
| 3.885 × 1012 |
−0.065 |
79.9 |
|
| (S3) |
NO2 + Pt <=> NO2–Pt |
9.997 × 10−1* |
−0.009 |
−0.2 |
|
| 2.472 × 1012 |
0.141 |
61.2 |
|
| (S4) |
N2O + Pt <=> N2O–Pt |
2.610 × 10−2* |
−0.017 |
−0.1 |
|
| 1.083 × 1010 |
0.070 |
1.0 |
|
| (S5) |
NO + O–Pt <=> NO2–Pt |
3.493 × 1010 |
0.215 |
111.3 |
57.2a |
| 5.229 × 1012 |
−0.215 |
115.5 |
|
| (S6) |
NO–Pt + O–Pt <=> NO2–Pt + Pt |
2.766 × 1016 |
−0.075 |
133.3 |
57.2a |
| 1.682 × 1017 |
0.075 |
57.7 |
|
| (S7) |
NO–Pt + N–Pt <=> N2O–Pt + Pt |
8.417 × 1020 |
0.070 |
91.1 |
|
| 3.415 × 1024 |
−0.070 |
132.9 |
|
| (S8) |
NO–Pt + Pt <=> N–Pt + O–Pt |
4.206 × 1020 |
0.070 |
108.0 |
|
| 1.184 × 1021 |
−0.070 |
122.3 |
57.2a |
| (S9) |
2N–Pt => N2 + 2Pt |
3.700 × 1021 |
0 |
113.9 |
|
| NOx storage on BaO14 |
| (S10) |
O2 + 2BaO <=> 2BaO–O |
1.116 × 10−6* |
−0.019 |
58.0 |
|
| 1.549 × 1018 |
0.035 |
208.9 |
|
| (S11) |
NO2 + BaO <=> BaO–NO2 |
3.936 × 10−5* |
0.017 |
37.0 |
|
| 1.017 × 109 |
−0.069 |
168.2 |
|
| (S12) |
NO + BaO–O <=> BaO–NO2 |
4.010 × 10−6* |
0 |
0.940 |
|
| 1.979 × 107 |
0.001 |
113.3 |
|
| (S13) |
NO2 + BaO–O <=> BaO–NO3 |
2.800 × 10−6* |
0 |
1.0 |
|
| 1.000 × 108 |
0 |
130.0 |
|
| (S14) |
NO2 + BaO–NO3 <=> Ba(NO3)2 |
1.000 × 1014 |
0 |
19.0 |
|
| 1.000 × 108 |
0 |
250.0 |
55.6b |
The model for NO oxidation on Pt consists 17 elementary-step-like reactions on a platinum-based catalyst among 5 gas-phase species and 6 surface adsorbed species. The mechanism is based on our previously published mechanism,12 originally much larger and developed for modeling NSC in the presence of CO, CO2 and light hydrocarbons. We removed all “C”-containing reactions and species from the mechanism for this study. The mechanism focuses on two main reaction pathways towards NO oxidation, which is the reaction of NO as a surface species as well as an Eley-Rideal based reaction with NO directly reacting with pre-adsorbed oxygen. The kinetic parameters of the NO oxidation mechanism on Pt are modified to match the light-off curves measured in the packed bed reactor, and then directly applied to simulate the NO oxidation in the parallel–wall channel. The microkinetic mechanism exhibits thermodynamic consistency.40
The NOx storage mechanism in this study relies on the reaction scheme proposed by Olsson et al.,14 containing five bidirectional reactions for NOx storage, as listed in Table 1. After combining the NOx storage mechanism on BaO with the NO oxidation mechanism on Pt, only the pre-exponential factors for the steps of NOx storage on BaO have been adapted to match the experimental data. The activation energies, including the dependence of the activation energy on the coverages of some species, remain, and only are adapted for thermodynamic consistency. In previous literature, the reported pre-exponential factors either have unit that is coupled with the catalyst loading, e.g. mol s−1 kg−1 catalyst,14,16,17 or must be scaled according to the loading.15,18,20–22 In this study, the effect of catalyst loading is included in the ratio of catalytic to geometric surface area, Fcat/geo, therefore, the pre-exponential factors are independent of the loading, which allows it to be directly adapted to other cases.
4. Results and discussion
4.1. Catalyst characterization
Table 2 summarizes the results of the powder catalyst characterization, which reveals a decrease of the specific surface area from more than 170 m2 g−1 for the Pt/Al2O3 catalyst samples to 126 m2 g−1 for the Pt/BaO/Al2O3 sample. Similarly, the addition of barium results in a decrease of the pore volume from 0.48 mL g−1 to 0.36 mL g−1. Moreover, elemental analysis confirms the target catalyst composition for all catalyst samples. Based on CO-chemisorption measurements, the noble metal dispersion is determined as 23% for Pt/Al2O3 and 58% for Pt/BaO/Al2O3. By analysis of SEM images that were taken along the entire length of the catalyst plate and by taking a generous estimation of thickness deviations into account, an average washcoat thickness of approximately 50 ± 5 μm is determined (Fig. 2). An estimation of the internal and external diffusion shows that the effect of internal diffusion in this thin catalyst coating is neglectable.
Table 2 Catalyst properties as determined by N2-physisorption, CO-chemisorption and elemental analysis
| Catalyst |
Pt/Al2O3 |
Pt/BaO/Al2O3 |
| BET surface area (m2 g−1) |
175 |
126 |
| Pore volume (mL g−1) |
0.48 |
0.36 |
| Pt dispersion (%) |
23 |
58 |
| Pt loading (wt%) |
0.918 |
0.912 |
| Ba loading (wt%) |
— |
15.7 |
 |
| | Fig. 2 Representative image of substrate and washcoat as obtained by means of scanning electron microscopy. | |
4.2. NO oxidation on Pt/Al2O3
NO oxidation on Pt/Al2O3 is investigated with both a light-off measurement in a packed bed reaction and with a steady-state PLIF measurement in a parallel wall channel. The experimental conditions are listed in Table 3. The mechanism is based on reactions for the conversion of NO, which is discussed in the original work by Koop et al.12 It contains pathways with the Langmuir–Hinshelwood and Eley–Rideal reaction type, contributing both to the oxidation of NO. Initial activation energies of both reaction types were kept constant to 133 kJ mol−1 and 111 kJ mol−1 respectively. However, additional coverage dependencies of adsorbed O species were introduced and changed to match measured NO oxidation activities within the packed bed experiments. For this reason, pre-exponential factors also differ compared to the initial published mechanism, which contained further coverage dependencies such as CO and H adsorbed species.
Table 3 Experimental conditions for NO oxidation on Pt/Al2O3
| Reactor type |
Temperature |
NO (ppm) |
O2 (%) |
| Packed bed reactor |
Transient, 3 K min−1 |
1000 |
10 |
| Packed bed reactor |
Transient, 3 K min−1 |
400 |
5 |
| Parallel wall channel |
Steady-state, every 50 K |
400 |
1 |
| Parallel wall channel |
Steady-state, every 50 K |
150 |
1 |
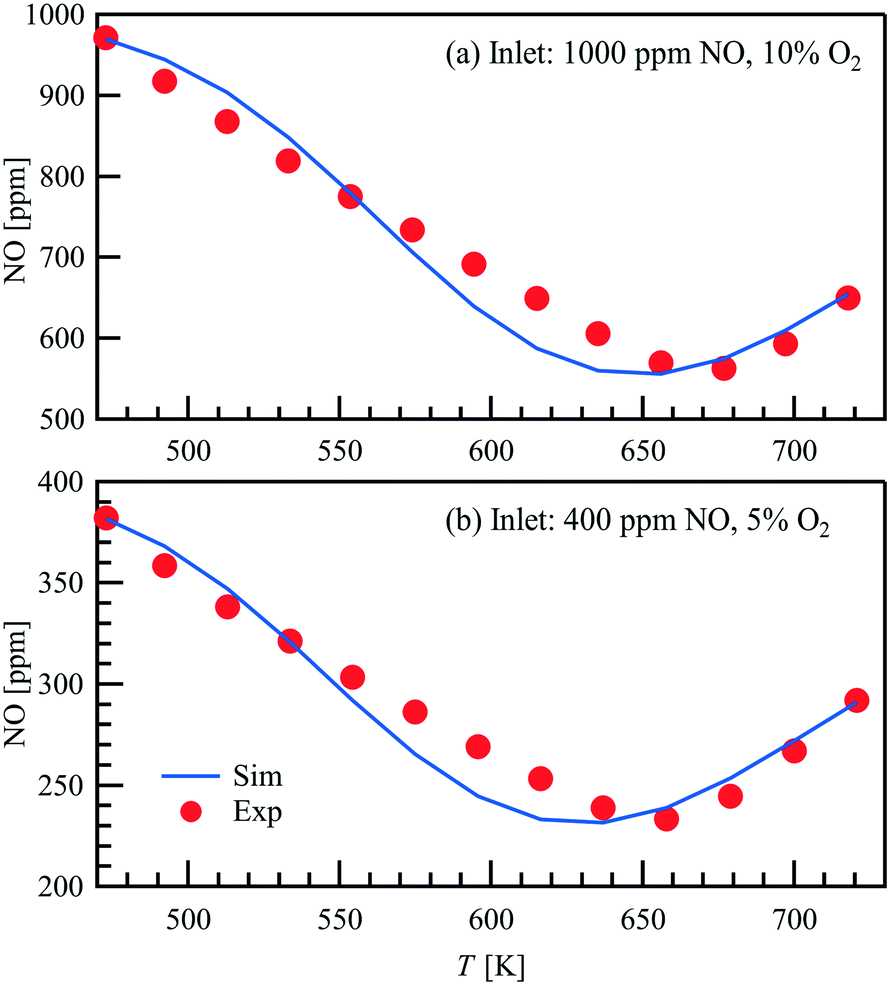
Light-off measurements are conducted in a packed bed with a temperature ramp of 3 K min−1 in a steady gas flow of 1 L min−1. Two different gas atmospheres with varying NO concentration are used. The results of the light-off measurements are shown in Fig. 3, where the NO outlet concentration obtained by FTIR is plotted as a function of temperature. The maximum conversion of the 1000 ppm NO inlet concentration in 10 vol% O2 is around 43.8% at a temperature of 677 K, whereas for an inlet concentration of 400 ppm NO in 5 vol% O2 the peak is shifted towards a lower temperature of 658 K and a similar conversion of 41.6%. Both trends are represented in good agreement within the thermodynamic consistent simulations.
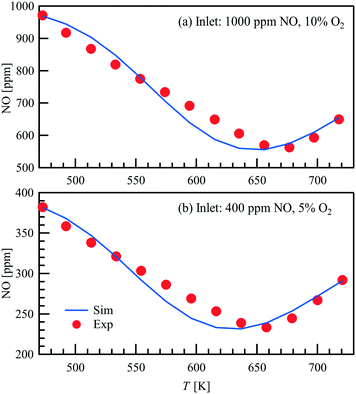 |
| | Fig. 3 Comparison of the FTIR measured and simulated NO outlet concentrations in a light-off measurement of the 1wt% Pt/Al2O3 powder catalyst. | |
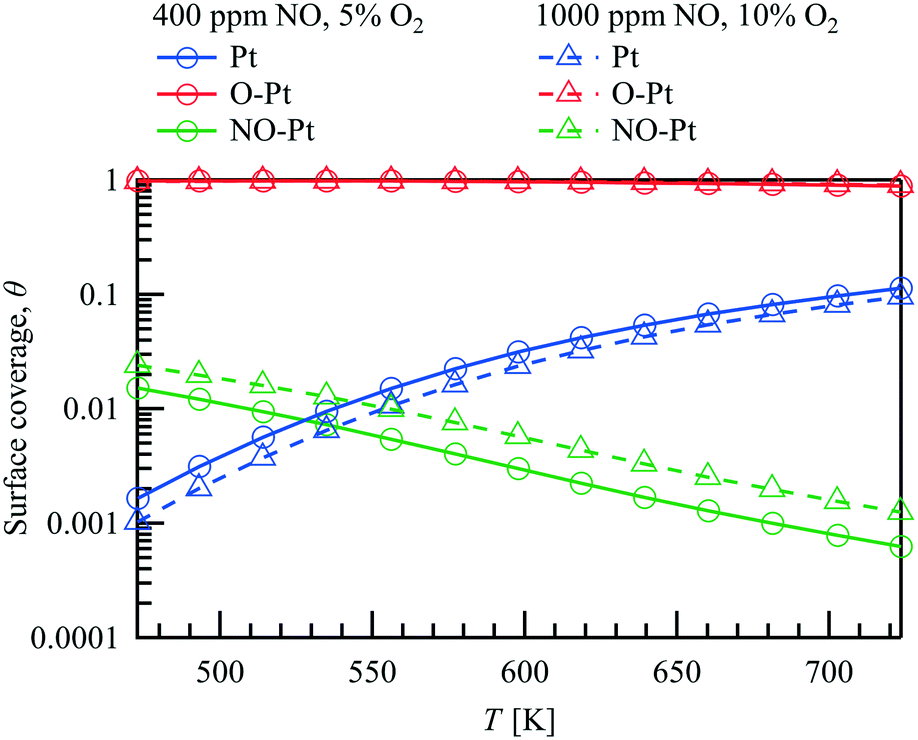
The simulation shows that with respect to the major surface species only minor variations of the surface coverages occur along the catalyst. Therefore, an averaged surface coverage of each species along the catalyst is calculated and shown in Fig. 4 to demonstrate the influence of temperature on the surface coverage at ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) = 1 L min−1 within the packed bed experiment under the two conditions examined. Due to the excess of oxygen, the adsorbed oxygen is the most abundant species at all conditions tested. As the temperature increases from 473 K to 723 K, the shift of the adsorption–desorption-equilibrium results in a small decrease of θO–Pt from approximately 0.97 to 0.90 for both cases although the O2 concentration differs with 5% and 10% respectively. Besides that, θPt increases with increasing temperature, whereas with higher NO concentration at the inlet (1000 vs. 400 ppm), θNO–Pt increases slightly as depicted in Fig. 4. Both Langmuir–Hinshelwood (LH) and Eley–Rideal (ER) reaction types have been included in the NO oxidation reaction pathway. Despite the high oxygen surface coverage, NO adsorption on Pt is orders of magnitude faster than NO adsorption on O–Pt originating from a lower activation energy. However, the combination reaction of NO–Pt and O–Pt limits the NO oxidation rate in the Langmuir–Hinshelwood reaction pathway. The simulations show that the NO oxidation rate is comparable between the LH and ER reaction paths for all conditions tested, with a larger contribution found for the ER path at higher temperatures.
= 1 L min−1 within the packed bed experiment under the two conditions examined. Due to the excess of oxygen, the adsorbed oxygen is the most abundant species at all conditions tested. As the temperature increases from 473 K to 723 K, the shift of the adsorption–desorption-equilibrium results in a small decrease of θO–Pt from approximately 0.97 to 0.90 for both cases although the O2 concentration differs with 5% and 10% respectively. Besides that, θPt increases with increasing temperature, whereas with higher NO concentration at the inlet (1000 vs. 400 ppm), θNO–Pt increases slightly as depicted in Fig. 4. Both Langmuir–Hinshelwood (LH) and Eley–Rideal (ER) reaction types have been included in the NO oxidation reaction pathway. Despite the high oxygen surface coverage, NO adsorption on Pt is orders of magnitude faster than NO adsorption on O–Pt originating from a lower activation energy. However, the combination reaction of NO–Pt and O–Pt limits the NO oxidation rate in the Langmuir–Hinshelwood reaction pathway. The simulations show that the NO oxidation rate is comparable between the LH and ER reaction paths for all conditions tested, with a larger contribution found for the ER path at higher temperatures.
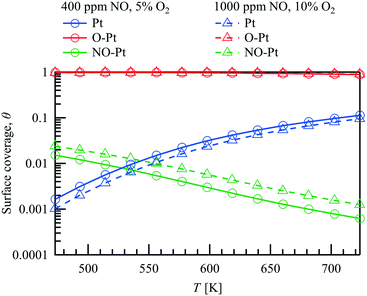 |
| | Fig. 4 Predicted surface coverage for NO oxidation on 1wt% Pt/Al2O3 catalyst in the packed bed reactor. | |
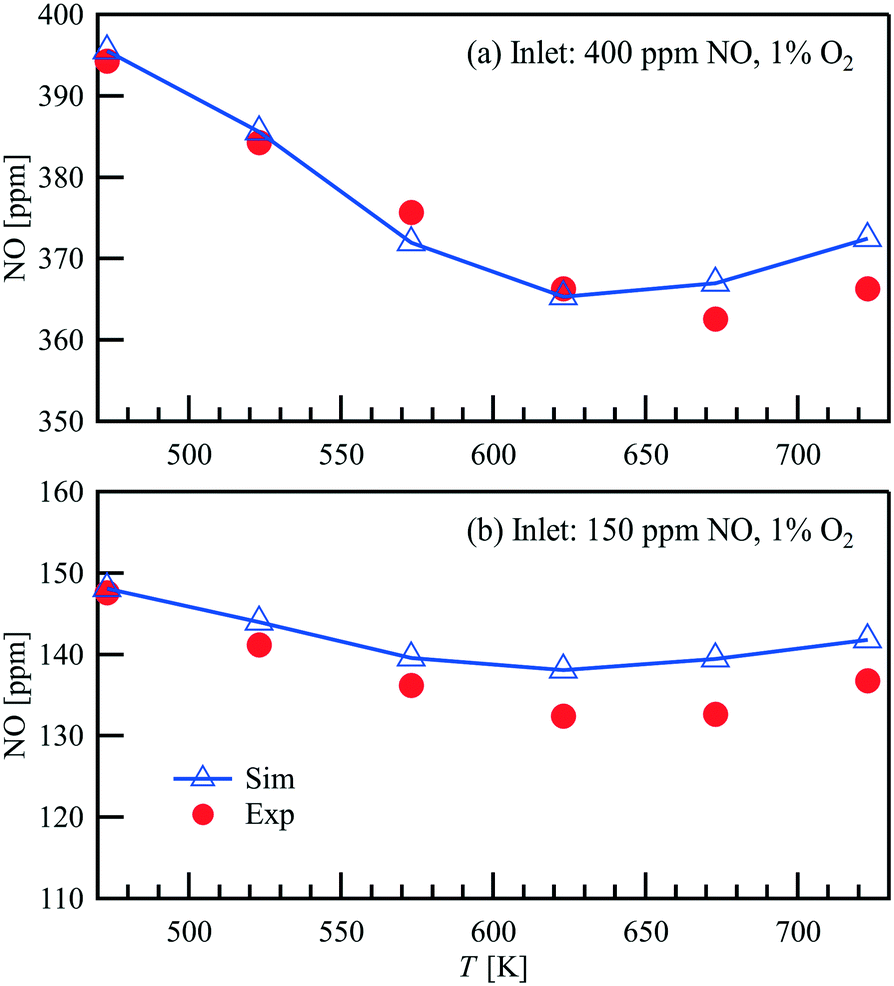
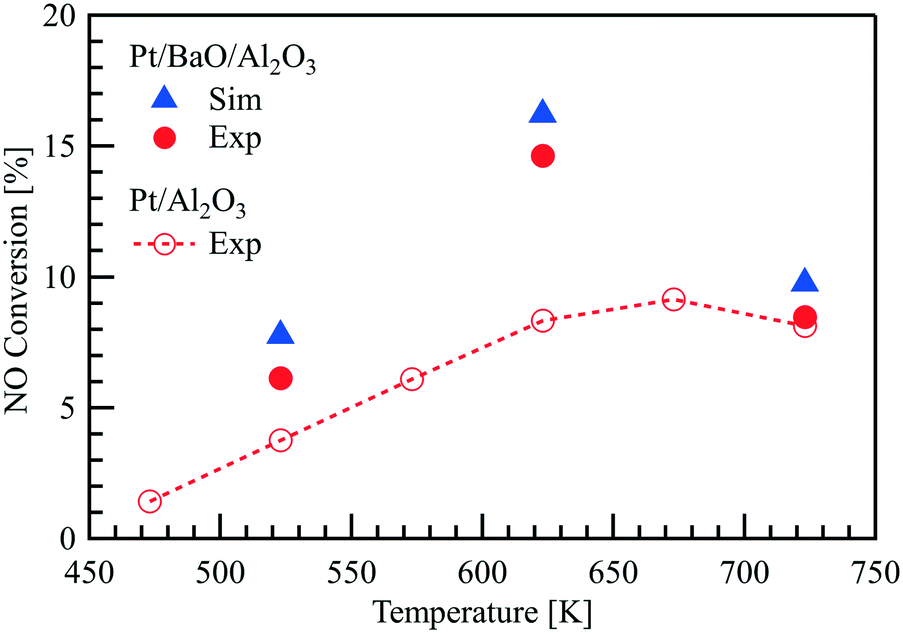
Steady-state NO oxidation over Pt in the parallel–wall channel is performed with an inlet flow rate of 1 L min−1, inlet NO concentrations of 150 and 400 ppm, and excess O2 (1 vol%). Fig. 5 compares the outlet NO concentration measured with FTIR and the simulated results as a function of temperature of the catalyst. Similar as shown in the light-off powder reactor measurements, the lowest outlet NO concentrations are observed at 623–673 K, indicating that the reaction converges/approaches the thermodynamic equilibrium. A good agreement is also found between the experimental data and the simulated results in the parallel wall channel, showing the NO oxidation mechanism in this study can well predict the NO conversion in different reactors under various flow conditions.
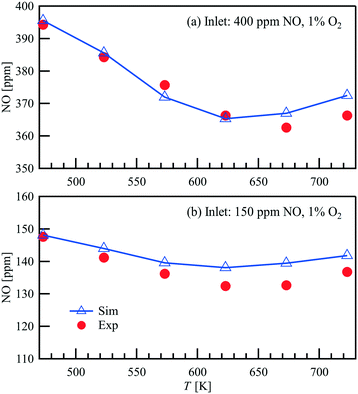 |
| | Fig. 5 Comparison of the measured and simulated NO outlet concentrations in a parallel wall channel for NO oxidation on a 1wt% Pt/Al2O3 catalyst coated plate at a flow rate of 1 L min−1. | |
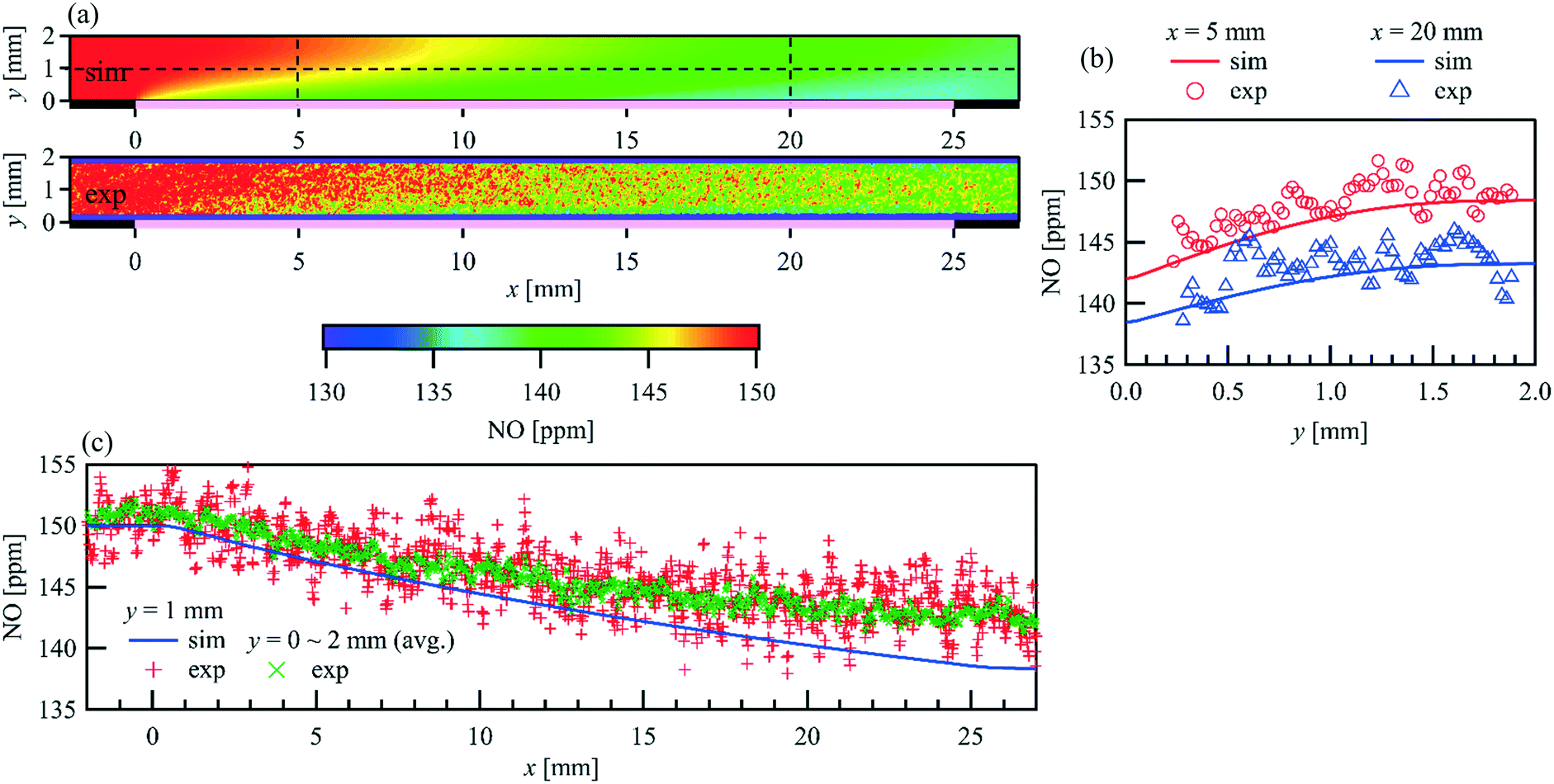
The spatial distribution of species over the catalyst is influenced by the combined effects of surface kinetics and, as a consequence, of diffusive and convective mass transfer. Since the flow is laminar, the modeling of diffusive and convective mass transfer is well established. Therefore, the spatial distribution of the species in the gas phase is a good indicator of the local surface reaction rate. Fig. 6(a) shows the comparison of the PLIF measured and simulated two-dimensional NO distribution at an inlet NO concentration of 150 ppm, an inlet flow rate of 1 L min−1, and a catalyst temperature of 723 K (Reynolds number, Re = 63.4). Wall normal NO concentration profiles at x = 5 and 20 mm and streamwise profiles at y =1 mm as well as a profile for the averaged NO concentration of the full channel height are given in Fig. 6(b) and (c), respectively. A uniform and constant NO concentration is observed upstream of the catalyst (x < 0 mm), which is identical to the concentration at the inlet, as no gas-phase reaction occurs under the given conditions. The NO concentration decreases in the flow direction above the catalyst plate, and a lower NO concentration is always observed closer to the catalyst plate, indicating the adsorption and/or conversion of NO at the catalytic surface, and the NO diffusion towards it. A good fitting indicates that the applied NO oxidation mechanism predicts the reaction along the entire catalyst plate quite well.
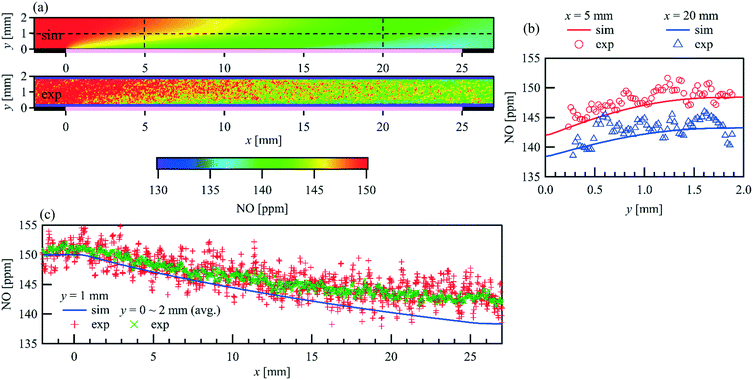 |
| | Fig. 6 Comparison of the PLIF measured and simulated 2D spatially resolved NO concertation within a parallel wall channel for NO oxidation over a 1wt% Pt/Al2O3 catalyst coated plate. = 1 L min−1, T = 723 K, Re = 63.4, inlet: 150 ppm NO, 1% O2, balance N2. (a) 2D map (pink line: catalytic plate, x = 0 to 20 mm); (b) profiles in wall-normal direction at x = 5 and 20 mm; (c) profiles in streamwise direction. | |
4.3. NOx storage on Pt/BaO/Al2O3
The NOx storage model is based on the combination of the fast catalytic reactions of NO oxidation on Pt and the slow storage reactions of NOx on BaO. The NOx storage process is transient as the coverages of the storage species change over time. When the active sites of the storage medium are fully occupied, the process approaches steady-state and the species concentrations are determined only by the fast NO oxidation over Pt. Hence, NO oxidation over Pt/BaO/Al2O3 can be measured independent of storage reactions when the steady-state is reached with = 1 L min−1 and 400 ppm NO, 1 vol% O2. Fig. 7 compares the NO conversion in the channel reactor on Pt/Al2O3 with the Pt/BaO/Al2O3 catalyst in steady-state. Despite the same Pt loading, higher NO conversions are observed for the Pt/BaO/Al2O3 catalyst, which can be attributed to a higher platinum dispersion: the Pt dispersion was found to be 23% for Pt/Al2O3 and 58% for Pt/BaO/Al2O3, respectively (see Table 2). The influence of temperature on NO oxidation is similar for both catalysts. After NOx storage on Pt/BaO/Al2O3 reaches the steady-state, the highest NO conversion is found at 623 K, implicating Pt in the Pt/BaO/Al2O3 catalyst performs the best NO oxidation to NO2 at this temperature. The increased Pt dispersion is reflected by the increased Fcat/geo, according to eqn (18) and (19). The simulation also well captures the steady-state NO conversion on Pt/BaO/Al2O3.
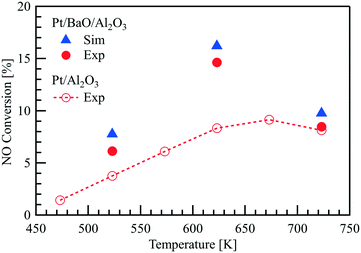 |
| | Fig. 7 Steady-state NO conversion over a Pt/Al2O3 catalyst coated plate and over a Pt/BaO/Al2O3 catalyst coated plate in the parallel wall channel with an inlet gas mixture of 400 ppm NO, 1% O2 in N2 at = 1 L min−1. | |
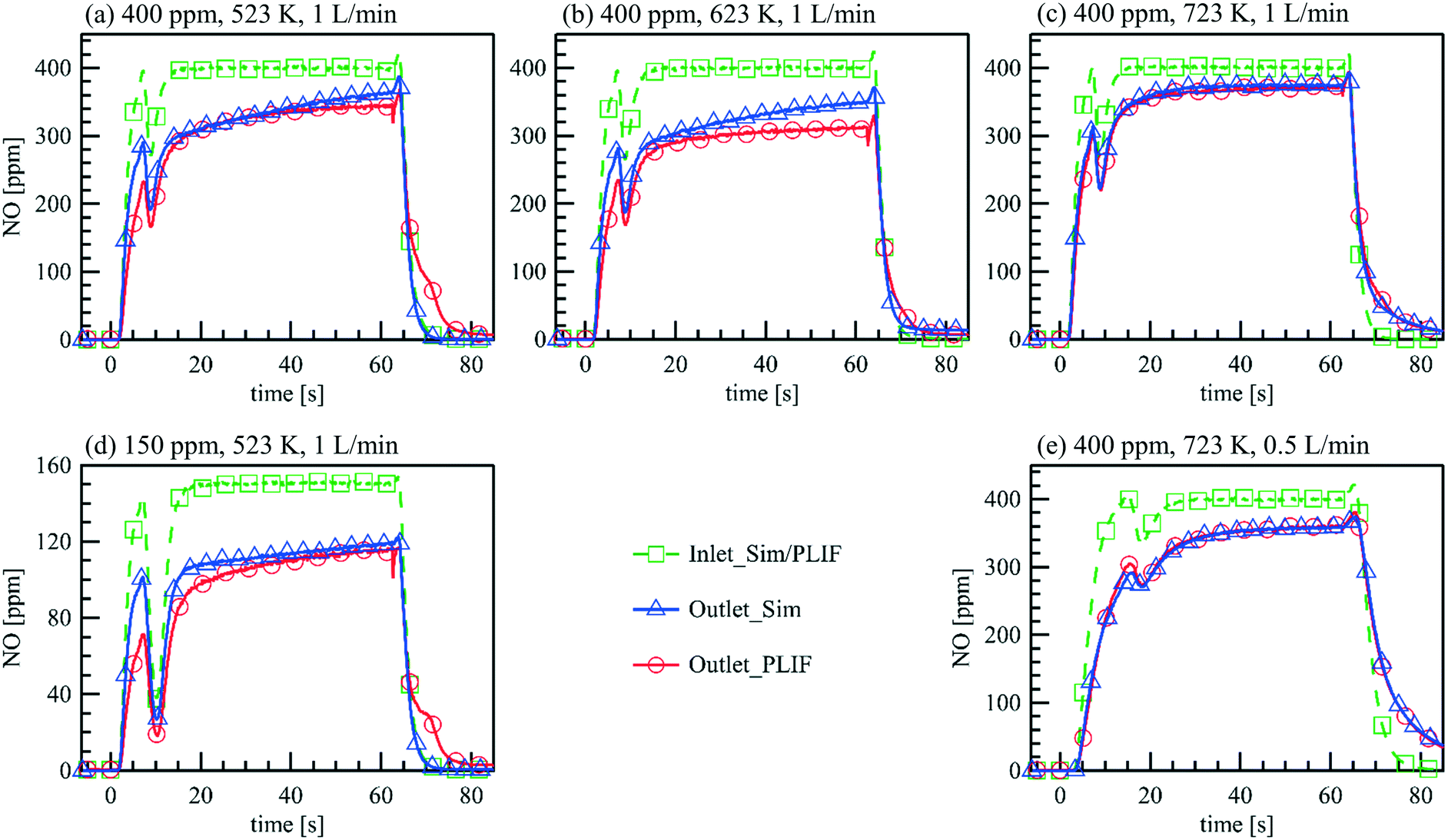
Transient measurements of NOx storage on Pt/BaO/Al2O3 are conducted in the parallel wall channel. The experimental conditions are listed in Table 4. Fig. 8 compares the measured and calculated NO inlet and outlet concentrations during the NOx storage cycle. Before the cycle starts, the Pt/BaO/Al2O3 catalyst is first reduced by 1 vol% H2/N2 for 15 min, and then continuously exposed to 1 vol% O2/N2 for over 5 min. At t = 0 s, 150/400 ppm NO is added to the inlet feed additionally, and at t = 62.5 s, the feeding gas is switched back to the mixture without NO. The PLIF data are the averaged NO concentrations at the upstream side towards the gas inlet and the downstream side towards the outlet of the catalytic plate. Overall, NO concentrations at the inlet and the outlet measured by PLIF and FTIR (not shown) are in accordance with each other and differ only in the rise and fall time when the NO feed is turned on or off.36 The latter is due to a larger volume in the FTIR cell and diffusion in the connection pipe towards the cell. As an in situ method, PLIF responds instantaneously to concentration changes inside the channel. At a flow rate of 1 L min−1, the first NO signals are observed at t = 1.7 s upstream of the catalyst plate, corresponding to the line length and volume upstream of the catalyst, and rapidly increase to a constant level within 14 s, which is equal to the input concentration. Such steep temporal gradients in the NO concentration changes cannot be monitored by averaging consecutive PLIF images. The fluctuation in the concentration profiles at t = ∼7 s in the rising edge and at t = ∼64 s in the falling edge are caused by the valve switching which induces instabilities of the mass flow controllers. The time-resolved NO inlet concentration measured via PLIF with the fluctuation due to valve switching is used as the actual inlet condition for the simulation. The simulation is able to predict the NO concentration development with reasonable accuracy for all tested conditions, e.g., temperatures, flow rates, inlet NO concentrations. Both the experimental and numerical results show that during the NO storage period, the NO concentration difference between the inlet and the outlet decreases over time for all cases. An increasing concentration of desorbed NO2 is detected by the FTIR at the outlet, but no other nitrogen oxides, which indicate that the NOx storage efficiency decreases over time due to the reduced storage capacity.
Table 4 Experimental conditions for NOx storage on Pt/BaO/Al2O3
| Flow rate (L min−1) |
Temperature (K) |
NO (ppm) in storage phase |
O2 (%) |
| 1 |
523 |
400 |
1 |
| 1 |
623 |
400 |
1 |
| 1 |
723 |
400 |
1 |
| 1 |
523 |
150 |
1 |
| 0.5 |
723 |
400 |
1 |
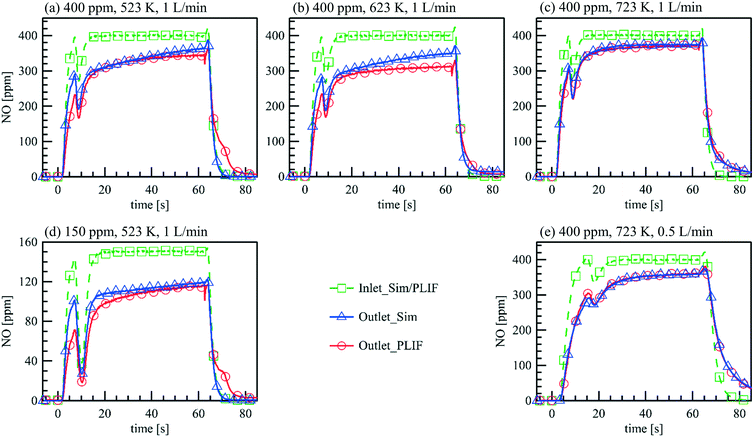 |
| | Fig. 8 Comparison of the measured and simulated NO inlet and outlet concentrations during the NOx storage cycle. (a)–(c) Inlet: 400 ppm NO, 1% O2, balance N2 (t = 0–62.5 s), = 1 L min−1, (a) T = 523 K, (b) T = 623 K, (c) T = 723 K; (d) inlet: 150 ppm NO, 1% O2, balance N2 (t = 0–62.5 s), = 1 L min−1, T = 523 K; (e) inlet: 400 ppm NO, 1% O2, balance N2 (t = 0–62.5 s), = 0.5 L min−1, T = 723 K. | |
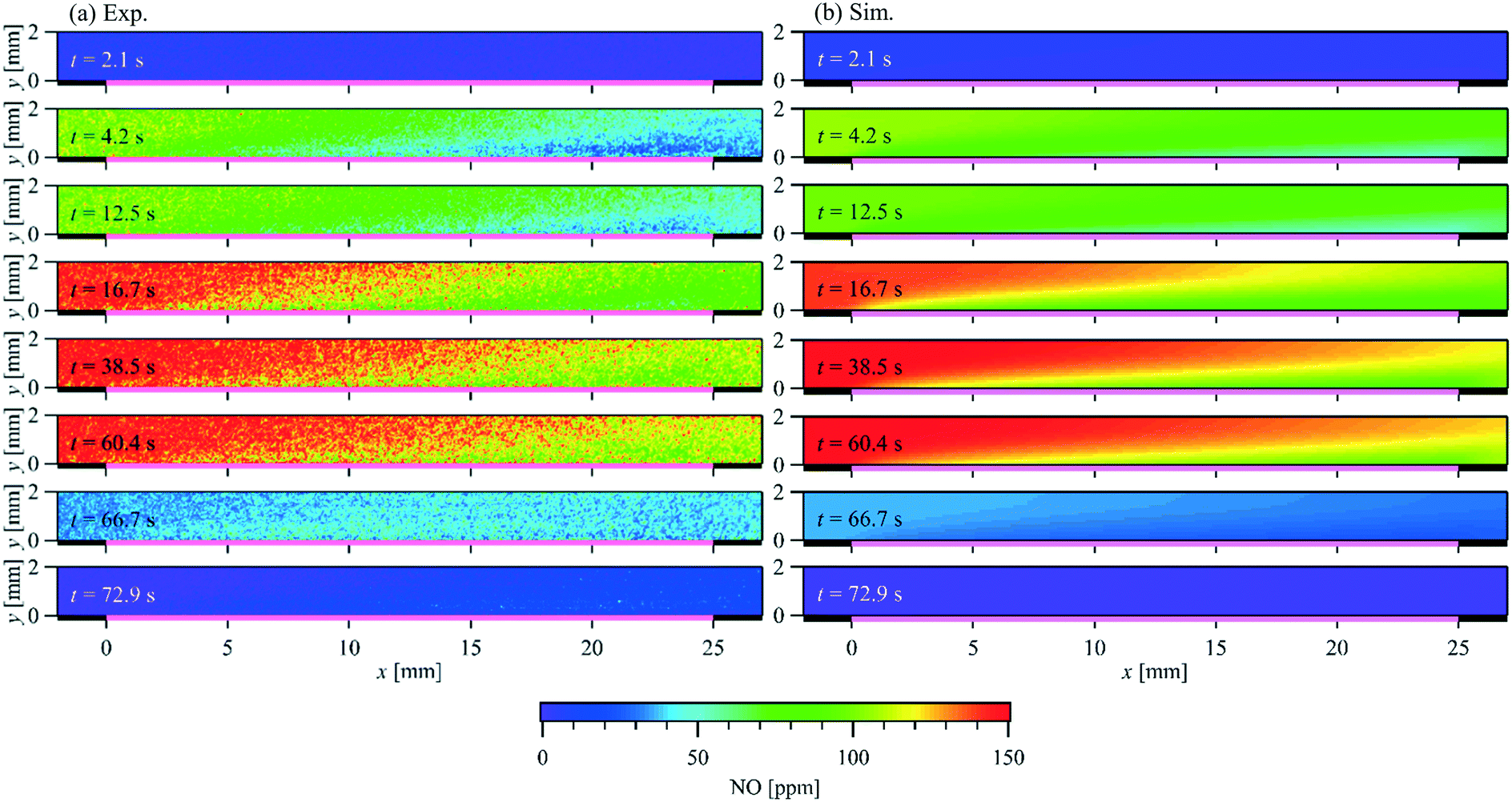
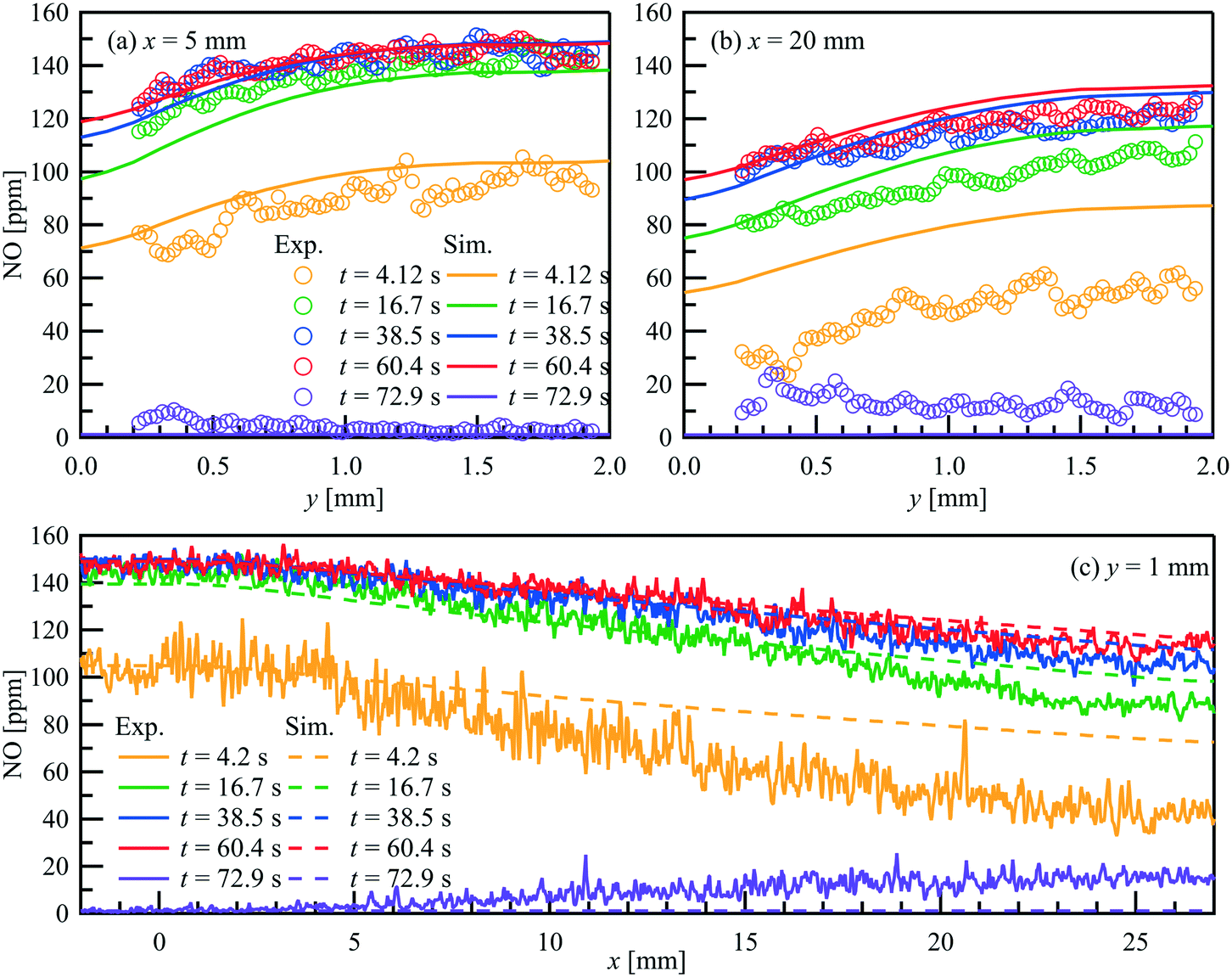
Comparison of the exhaust gas concentrations offers only the validation of the overall reaction rate. To verify the local catalytic activity and its interaction with the flow field, a spatially resolved comparison is necessary. Fig. 9 compares the PLIF measured and simulated 2D NO-concentration maps over time at T = 523 K and = 1 L min−1 (Re = 79.0) as an example. The corresponding NO inlet and outlet concentrations are plotted in Fig. 8(d). The PLIF images show the mean NO concentration at the indicated time phase resolved averaged over 10 storage cycles. The simulation captures the general development of the NO distribution over the catalyst plate. NO concentration gradients are observed in both streamwise and wall-normal directions, due to the interaction of catalytic surface reactions with the NO diffusion and convection in wall-normal and streamwise directions, respectively. Fig. 10 plots both the experimental measured and numerical calculated NO profiles at x = 5 and 20 mm in wall-normal direction (Fig. 10(a) and (b)) and at y = 1 mm in streamwise direction (Fig. 10(c)). Due to the small concentration changes between t = 15 and 62.5 s, a combination of phase-locked and time averaging is used to further improve the signal-to-noise ratio for experimental data.
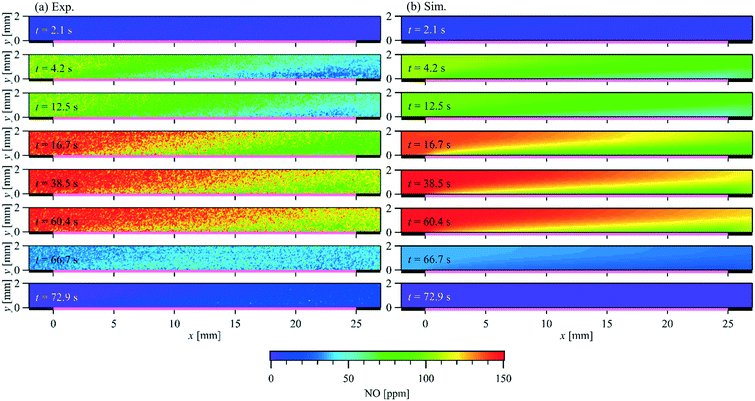 |
| | Fig. 9 Comparison of the PLIF measured and simulated 2D spatially and temporally resolved NO concentration within a parallel wall channel for NO storage over a Pt/BaO/Al2O3 catalyst coated plate. = 1 L min−1, T = 523 K, Re = 79.0, inlet: 150 ppm NO, 1% O2, balance N2 (t = 0–62.5 s). | |
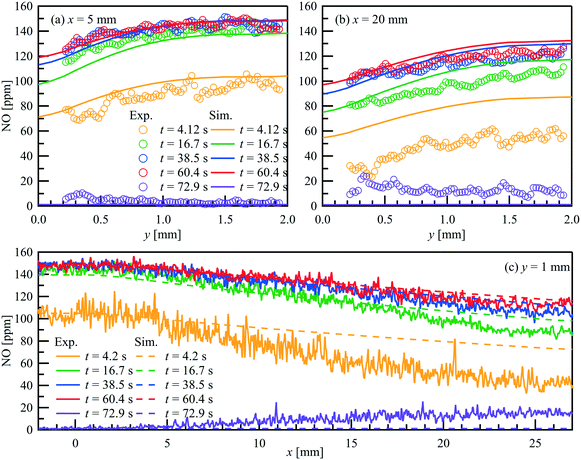 |
| | Fig. 10 Comparison of the PLIF measured and simulated NO concertation profiles within a parallel wall channel for NO storage over a Pt/BaO/Al2O3 catalyst coated plate at selected position. = 1 L min−1, T = 523 K, inlet: 150 ppm NO, 1% O2, balance N2 (t = 0–62.5 s). (a) x = 5 mm; (b) x = 20 mm; (c) y = 1 mm. | |
In each case, 9 consecutive laser shots (repetition rate 9.6 Hz) were averaged over 10 storage cycles. During the NO storage phase (from 0 to 62.5 s), from both experimental and numerical results, lower NO concentrations are found just above the catalytic plate than at the top of the channel, showing NO is consumed at the catalytic surface. Since the NO consumption rate equals the NO mass flux in the wall-normal direction at the surface, the changes of the NO concentration profiles reflect the changes of surface coverages. A discrepancy can be seen at t = 4.2 s downstream. At this stage, it is unclear whether it is caused by a modest underestimation of the activity that accumulated in the streamwise direction during the mass transportation or the unconsidered flow instability due to the valve switching. Therefore, the focus is given to the period after the flow is stabilized. From t = 15 s to 62.5 s, both, the experimental and the numerical results show, that despite the same NO feeding concentration at inlet, the NO concentration gradients in both the flow and wall normal directions decrease with time, as does the total storage capacity, and converge toward the profiles under steady-state conditions. At the same time, the reactive flow profiles upstream (x = 5 mm) reach steady-state faster than downstream (x = 20 mm). Numerical models must account for such local and temporal changes in surface coverage and thus reaction rates, which is not the case for global reaction models, for example. After the NO input stream is turned off (at t = 62.5 s), the situation reverses and the NO concentration is slightly higher at the catalyst surface than further away from the surface (see purple curves in Fig. 10(a) and (b)) as NO desorbs and diffuses away from the catalyst plate. Although such desorption has also been confirmed by the FTIR measured outlet NO profiles, it is not yet correctly reflected in the current simulation for the low temperature (523 K).
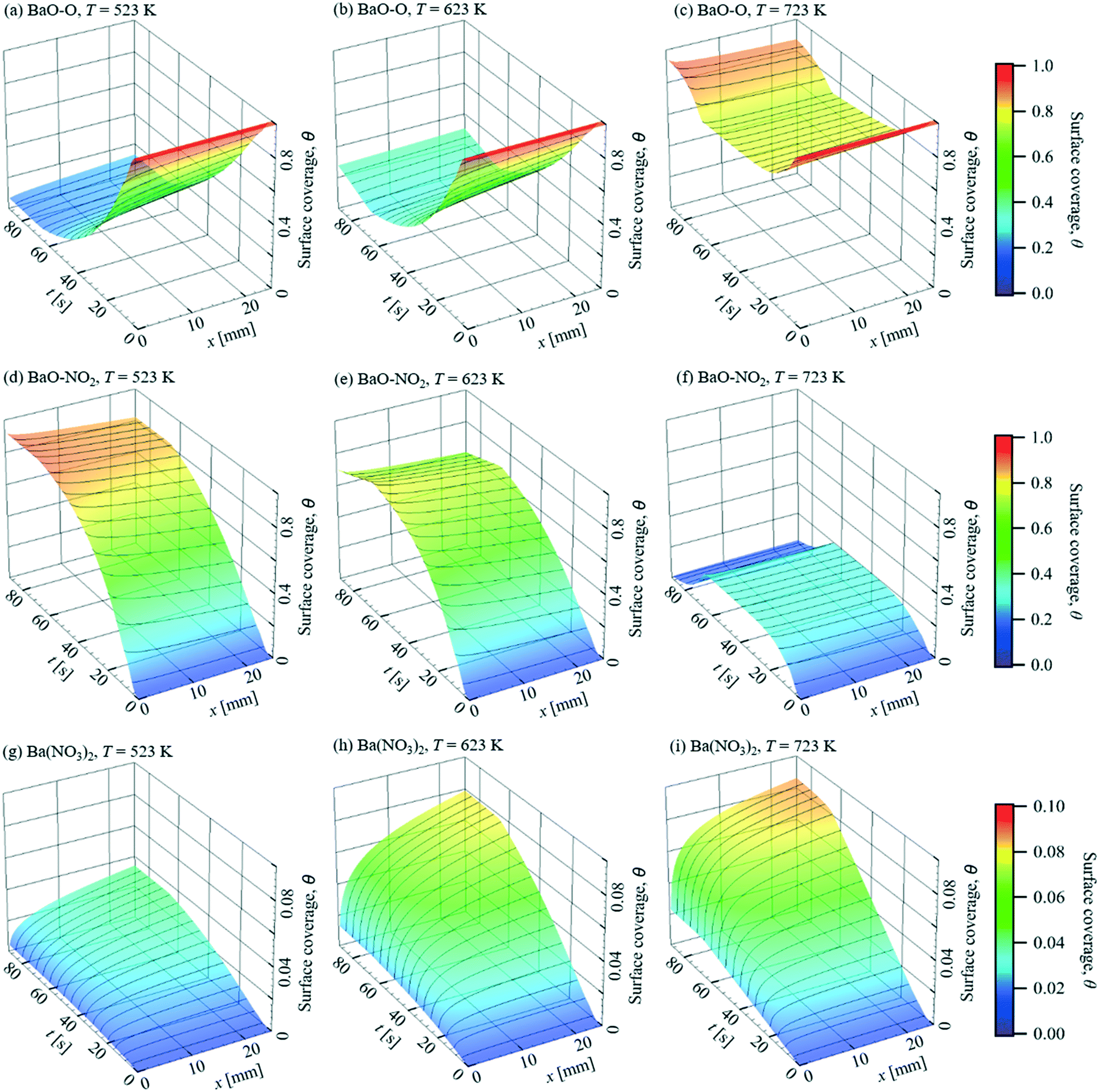
The simulated surface coverages, θ, plotted in Fig. 11, show the changes of the storage capacity in a more direct way. Due to the continuous dosing of 1 vol% O2, the surface is initially mainly covered by BaO–O. Therefore, the main reaction paths under excess oxygen are NO(g) → BaO–NO2 and NO2(g) → BaO–NO3 → Ba(NO3)2, which are triggered by NO and NO2 adsorption on BaO–O, respectively. At T = 523 K and 623 K and with the onset of the storage phase, the surface coverage of BaO–O decreases rapidly and is mainly replaced by BaO–NO2 due to the higher NO concentration compared to NO2, which is accompanied by a decreased storage capacity. In the meantime, due to the higher NO concentration upstream, θBaO–NO2 decreases in streamwise direction, and a higher storage capacity remains downstream. At T = 723 K, more uniform surface coverages of BaO–O and Ba–NO2 are found along the catalyst. Although θBaO–O decreases over time under these conditions during the storage phase as well, it remains larger than 70% because the reaction rate of NO desorption from BaO–O increases with temperature substantially faster than the adsorption reaction. The faster NO desorption is also noticeable by comparing the θBaO–O at t = 60.4 s and 93.8 s. At T = 723 K, after the NO stream has been stopped, θBaO–O rises again to ∼0.9, and θBaO–NO2 decreases to about 10−2, due to the NO desorption. In contrast, θBaO–NO2 remains over 0.8 and 0.6 at T = 523 K and 623 K, respectively, as little NO is desorbed. NO2 from the oxidation reaction of NO on Pt is stored as Ba(NO3)2. Due to the higher energy barrier, Ea = 250 kJ mol−1, almost no Ba(NO3)2 decomposition happens on the surface. Comparing between T = 523 K and 623 K, much higher coverage of Ba(NO3)2 is found at the higher temperature throughout the entire cycle, which is attributed to the higher oxidation rate of NO on Pt and faster NO2 adsorption. However, as the NO oxidation on Pt is limited by the thermodynamic equilibrium around T = 623 K, despite the increased NO2 adsorption rate, no obvious improvement of Ba(NO3)2 formation is found at higher temperatures.
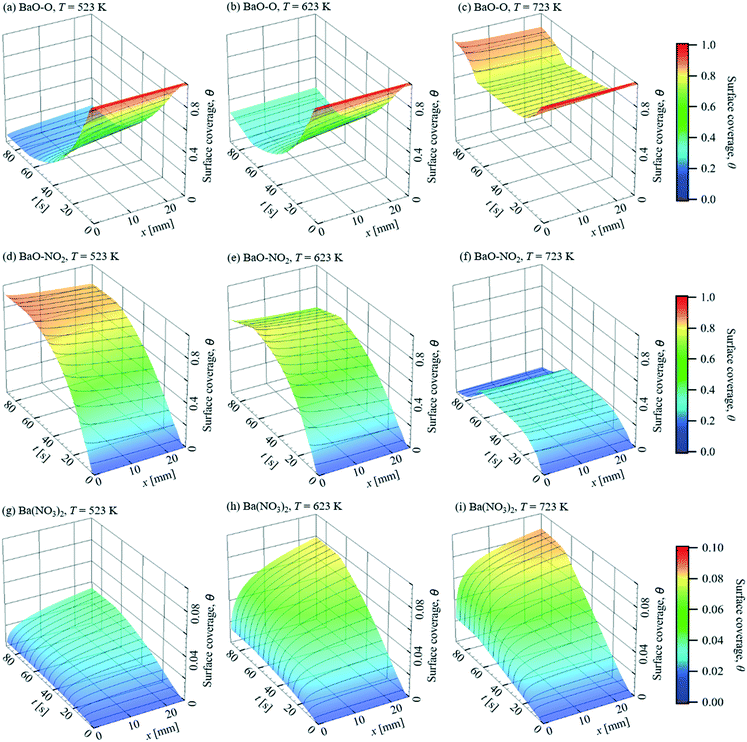 |
| | Fig. 11 Predicted surface coverage, θ, for NO storage over a Pt/BaO/Al2O3 catalyst coated plate. = 1 L min−1, inlet: 400 ppm NO, 1% O2, balance N2 (t = 0–62.5 s). | |
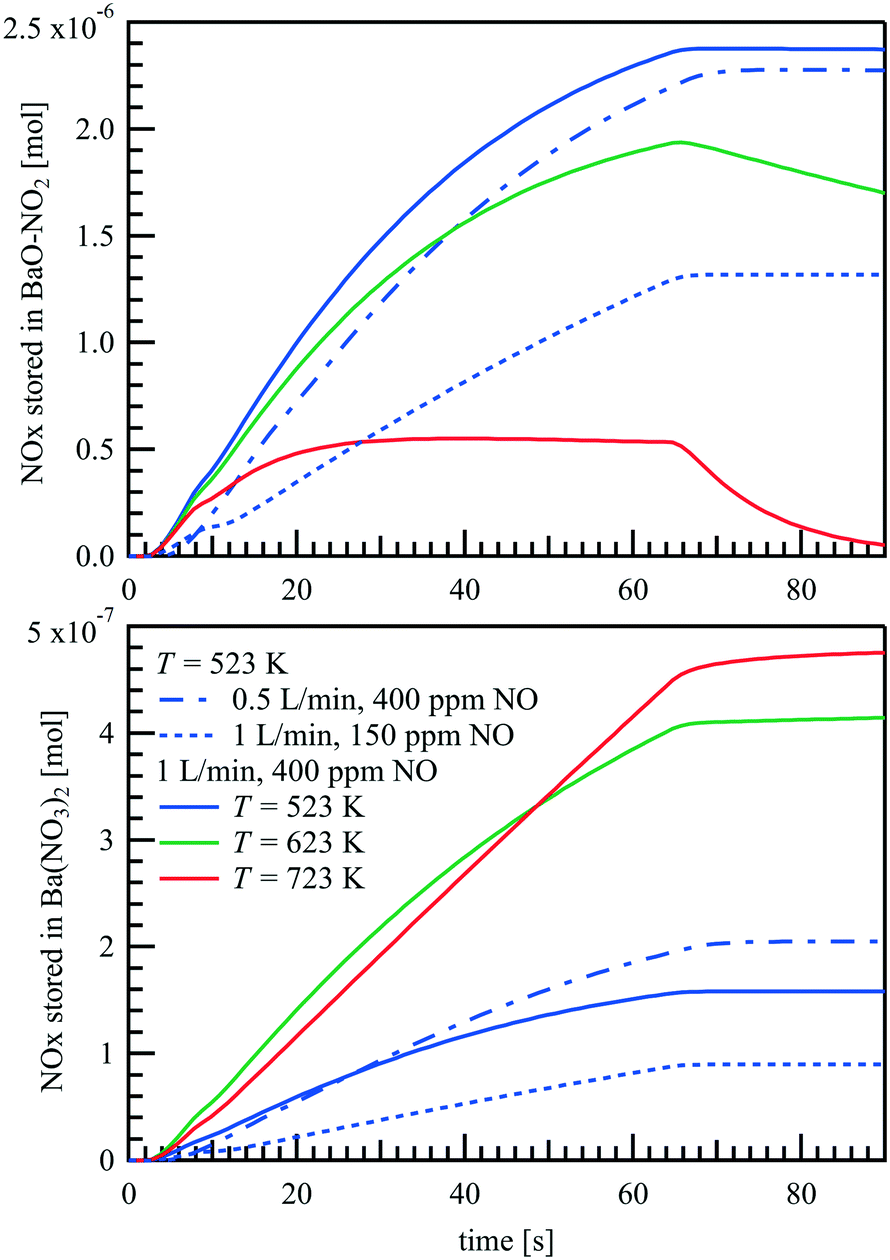
The amounts of NOx stored as BaO–NO2 and Ba(NO3)2 are plotted in Fig. 12 as a function of time. For T = 523 K and 623 K, NOx is mainly stored as BaO–NO2, which is not a stable species at higher temperatures. Therefore, at T = 723 K, during the storage phase, almost no further accumulation of BaO–NO2 is found after t = 20 s, but only an increase of Ba(NO3)2. Since the Ba(NO3)2 formation rate is limited by the NO oxidation rate on Pt, the NO concentration at the outlet changes only slightly after t = 20 s and at T = 723 K, as shown in Fig. 8(c). Compared to the formation of BaO–NO2, the formation of Ba(NO3)2 is almost non-reversible. The simulation shows that the surface is completely covered with Ba(NO3)2 after a long period of NO supply (not shown here). However, limited by the NO oxidation rate, such processes take about 2 hours at 723 K with = 1 L min−1 and 400 ppm NO at the inlet, and take even longer at lower temperatures, as the slower NO desorption rate has to be taken into consideration.
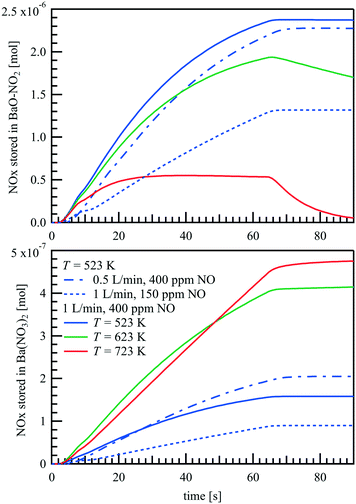 |
| | Fig. 12 Predicted amount of stored NOx as BaO–NO2 and Ba(NO3)2 during a NOx storage cycle over a Pt/BaO/Al2O3 catalyst coated plate. | |
Fig. 12 shows the NO storage at T = 523 K with lower inlet NO concentration and slower flow rate as well. When the NO concentration is lowered from 400 to 150 ppm by more than 60%, the total amount of NOx stored does not decrease by the same amount, but by less than 50%. That is, the effective storage efficiency is somewhat higher at lower NO inlet concentrations, presumably because the storage capacity is not reached as quickly locally. When the inlet flow rate decreases from 1 L min−1 to 0.5 L min−1, the integral amount of NO added over the same time-period is halved whereas the residence time doubles. At the end of the storage phase, however, the amount of BaO–NO at a flow rate of 0.5 L min−1 is still over 95% of the amount at a flow rate of 1 L min−1. Moreover, with the prolonged residence time, the higher conversion of NO to NO2 on Pt site leads to even a higher amount of Ba(NO3)2 at the slower flow rate.
5. Conclusions
In this study, NOx storage on Pt/BaO/Al2O3 catalysts has been investigated numerically and experimentally. A thermodynamically consistent reaction mechanism for NOx storage is developed by combining the fast catalytic reactions of NO oxidation on Pt and the slow storage reactions of NOx on BaO. The kinetic parameters are based on literature values and fine-tuned to match the experimental data in our present study under various conditions.
Light-off measurements in a packed bed reactor with catalyst powder and spatially resolved NO distribution measurements over a catalystically coated plate are conducted to investigate the NO oxidation to NO2 on Pt as a precursor step for the NOx storage. Both measurements show that NO conversion on Pt above approximately 650 K is limited by the reaction equilibrium of the reversible reaction. The previously published Koop et al.'s automotive exhaust gases abatement model12 is tailored to a mechanism of NO oxidation on Pt. Using the 1D packed bed code, DETCHEMPBR, which is presented in this study, and comparing with the experimental data, the kinetic data of the 17-reaction-scheme is thermodynamically consistent and is adapted to match the light-off curve. The NO oxidation mechanism is subsequently employed in parallel–wall channel simulations with DETCHEMCHANNEL, which predicts 2D NO distribution over the Pt/Al2O3 catalyst plate measured by the PLIF technique. This mechanism is then implemented as sub-steps in the NOx storage mechanism.
The spatial and temporal development of the absolute NO concentration distributions over a NOx storage catalyst is obtained with in situ phase-locked PLIF. Decreasing storage capacity with time is reflected by decreasing concentration gradients that rapidly converge to steady-state conditions locally. The NOx storage model on BaO is based on Olsson et al.'s mechanism14 and combined with the NO oxidation mechanism on Pt. Only the pre-exponential factors for the steps NOx storage on BaO are further tuned to fit the simulated results of DETCHEMRESERVOIR to the experimental data. A reasonable agreement is found under various conditions, e.g., temperatures, flow rates and inlet gas concentrations. It is shown that the NOx storage efficiency over a Pt/BaO/Al2O3 catalyst highly depends on the temperature, the storage capacity, the flow field, and the NO oxidation rate on Pt. At T = 523 K and 623 K, during the 62.5 s storage phase with NO/O2/N2 mixture, NO is mainly stored as BaO–NO2, which is unstable at higher temperatures and strongly diminishes the NOx storage efficiency at T = 723 K. On one hand, only a small amount of NO reacts to form BaO–NO2 due to the high NO desorption rate, and on the other hand, the formation of Ba(NO3)2 is limited due to the low conversion of NO to NO2.
The study shows that a micro kinetic model established with experimental data by kinetic data from packed bed reactor measurements using a catalyst powder can be incorporated into more complex flow simulations in which convective and conductive transport processes are interacting with the kinetics. Furthermore, the micro kinetic model also works under transient conditions of a NSR catalyst.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was founded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project #237267381 (TRR 150) and #426888090 (SFB 1441), concerning the PLIF setup and the experimental and kinetic modeling studies. Steinbeis GmbH & Co KG für Technologietransfer (STZ 240 Reactive Flow) provided the cost-free license of the software tool DETCHEM. The authors thank Dr. T. Bergfeldt (IAM-AWP, KIT) for elemental analysis.
References
-
O. Deutschmann and A. G. Konstandopoulos, Catalytic technology for soot and gaseous pollution control, in Handbook of Combustion: Online, 2010, vol. 15, pp. 465–509 Search PubMed.
- B. Choi, K. Lee and G. Son, Int. J. Automot. Technol., 2020, 21(6), 1597–1618 CrossRef.
- N. Takahashi, H. Shinjoh, T. Iijima, T. Suzuki, K. Yamazaki, K. Yokota, H. Suzuki, N. Miyoshi, S. I. Matsumoto and T. Tanizawa, Catal. Today, 1996, 27, 63–69 CrossRef CAS.
- S. Roy and A. Baiker, Chem. Rev., 2009, 109, 4054–4091 CrossRef CAS PubMed.
- G. Liu and P. X. Gao, Catal. Sci. Technol., 2011, 1(4), 552–568 RSC.
- Y. Nagai, A. Kato, M. Iwasaki and K. Kishita, Catal. Sci. Technol., 2019, 9(5), 1103–1107 RSC.
- T. Selleri, F. Gramigni, I. Nova, E. Tronconi, S. Dieterich, M. Weibel and V. Schmeisser, Catal. Sci. Technol., 2018, 8(9), 2467–2476 RSC.
- L. Lietti, M. Daturi, V. Blasin-Aubé, G. Ghiotti, F. Prinetto and P. Forzatti, ChemCatChem, 2012, 4, 55–58 CrossRef CAS.
- F. Prinetto, G. Ghiotti, I. Nova, L. Castoldi, L. Lietti, E. Tronconi and P. Forzatti, Phys. Chem. Chem. Phys., 2003, 5, 4428–4434 RSC.
- P. J. Schmitz and R. J. Baird, J. Phys. Chem. B, 2002, 106, 4172–4180 CrossRef CAS.
- D. Bhatia, R. D. Clayton, M. P. Harold and V. Balakotaiah, Catal. Today, 2009, 147, S250–S256 CrossRef CAS.
- J. Koop and O. Deutschmann, Appl. Catal., B, 2009, 91, 47–58 CrossRef CAS.
- P. Kočí, F. Plát, J. Štěpánek, Š. Bártová, M. Marek, M. Kubíček, V. Schmeißer, D. Chatterjee and M. Weibel, Catal. Today, 2009, 147, S257–S264 CrossRef.
- L. Olsson, H. Persson, E. Fridell, M. Skoglundh and B. Andersson, J. Phys. Chem. B, 2001, 105, 6895–6906 CrossRef CAS.
- J. Dawody, M. Skoglundh, L. Olsson and E. Fridell, Appl. Catal., B, 2007, 70, 179–188 CrossRef CAS.
- A. Lindholm, N. W. Currier, J. Li, A. Yezerets and L. Olsson, J. Catal., 2008, 258, 273–288 CrossRef CAS.
- L. Olsson, E. Fridell, M. Skoglundh and B. Andersson, Catal. Today, 2002, 73, 263–270 CrossRef CAS.
- S. Shwan, W. Partridge, J. S. Choi and L. Olsson, Appl. Catal., B, 2014, 147, 1028–1041 CrossRef CAS.
- Z. Say, O. Mihai, M. Tohumeken, K. E. Ercan, L. Olsson and E. Ozensoy, Catal. Sci. Technol., 2017, 7(1), 133–144 RSC.
- N. Rankovic, A. Nicolle and P. D. Costa, J. Phys. Chem. C, 2010, 114, 7102–7111 CrossRef CAS.
- N. Rankovic, A. Nicolle, D. Berthout and P. D. Costa, Catal. Commun., 2010, 12, 54–57 CrossRef CAS.
- R. S. Larson, V. K. Chakravarthy, J. A. Pihl and C. S. Daw, Chem. Eng. J., 2012, 189, 134–147 CrossRef.
- J. S. Choi, W. P. Partridge, J. A. Pihl, M. Y. Kim, P. Kočí and C. S. Daw, Catal. Today, 2012, 184, 20–26 CrossRef CAS.
- M. Hettel, C. Diehm, B. Torkashvand and O. Deutschmann, Catal. Today, 2013, 216, 2–10 CrossRef CAS.
- G. Kychakoff, R. D. Howe, R. K. Hanson and J. C. McDaniel, Appl. Opt., 1982, 21, 3225–3227 CrossRef CAS PubMed.
- R. K. Hanson, J. M. Seitzman and P. H. Paul, Appl. Phys. B., 1990, 50, 441–454 CrossRef.
-
N. Ebersohl, T. Klos, R. Suntz and H. Bockhorn, 27th Symposium (International) on Combustion, 1998, vol. 27, pp. 997–1005 Search PubMed.
- R. Sui, J. Mantzaras and R. Bombach, Proc. Combust. Inst., 2017, 36, 4313–4320 CrossRef CAS.
- J. Mantzaras, Prog. Energy Combust. Sci., 2019, 70, 169–211 CrossRef.
- A. Zellner, R. Suntz and O. Deutschmann, Angew. Chem., Int. Ed., 2015, 54, 2653–2655 CrossRef CAS PubMed.
- B. Torkashvand, L. Maier, P. Lott, T. Schedlbauer, J.-D. Grunwaldt and O. Deutschmann, Top. Catal., 2018, 62, 206–213 CrossRef.
- S. Wan, B. Torkashvand, T. Haeber, R. Suntz and O. Deutschmann, Appl. Catal., B, 2020, 264, 118473 CrossRef.
- W. Kang, O. Fujita and K. Ito, J. Energy Resour. Technol., 1996, 118, 82–87 CrossRef.
- J. Zetterberg, S. Blomberg, J. Gustafson, J. Evertsson, J. Zhou, E. C. Adams, P. A. Carlsson, M. Aldén and E. Lundgren, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- S. Blomberg, J. Zhou, J. Gustafson, J. Zetterberg and E. Lundgren, J. Phys.: Condens. Matter, 2016, 28, 453002 CrossRef PubMed.
- S. Wan, Y. Guo, T. Haeber, R. Suntz and O. Deutschmann, ChemPhysChem, 2020, 21, 2497 CrossRef CAS PubMed.
- O. Deutschmann, Catal. Lett., 2015, 145, 272–289 CrossRef CAS.
- J. Koop and O. Deutschmann, SAE [Tech. Pap.], 2007, 2007-01-1142 Search PubMed.
- R. Schwiedernoch, S. Tischer, C. Correa and O. Deutschmann, Chem. Eng. Sci., 2003, 58, 633–642 CrossRef CAS.
- K. Herrera Delgado, H. Stotz, L. Maier, S. Tischer, A. Zellner and O. Deutschmann, Catalysts, 2015, 5, 871–904 CrossRef.
- M. Casapu, J. D. Grunwaldt, M. Maciejewski, M. Wittrock, U. Göbel and A. Baiker, Appl. Catal., B, 2006, 63, 232–242 CrossRef CAS.
- K. A. Karinshak, P. Lott, M. P. Harold and O. Deutschmann, In situ activation of bimetallic Pd-Pt Methane oxidation catalysts, ChemCatChem, 2020, 12, 3712–3720 CrossRef CAS.
-
G. Bergeret and P. Gallezot, Particle size and dispersion measurements, in Handbook of heterogeneous catalysis, ed. G. Bergeret, P. Gallezot, G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, pp. 738–765 Search PubMed.
- C. Karakaya and O. Deutschmann, Appl. Catal., A, 2012, 445-446, 221–230 CrossRef CAS.
- D. Chan, A. Gremminger and O. Deutschmann, Top. Catal., 2013, 56, 293–297 CrossRef CAS.
- D. Chan, S. Tischer, J. Heck, C. Diehm and O. Deutschmann, Appl. Catal., B, 2014, 156-157, 153–165 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- K. Kevin, P. Lott, H. Stotz, L. Maier and O. Deutschmann, Catalysts, 2020, 10, 922 CrossRef.
- M. Borchers, K. Keller, P. Lott and O. Deutschmann, Ind. Eng. Chem. Res., 2021, 60, 6613–6626 CrossRef CAS.
-
N. Wakao and S. Kagei, Heat and Mass Transfer in Packed Beds, Taylor & Francis, 1982 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Kevin
Keller
b,
Patrick
Lott
a,
Kevin
Keller
b,
Patrick
Lott
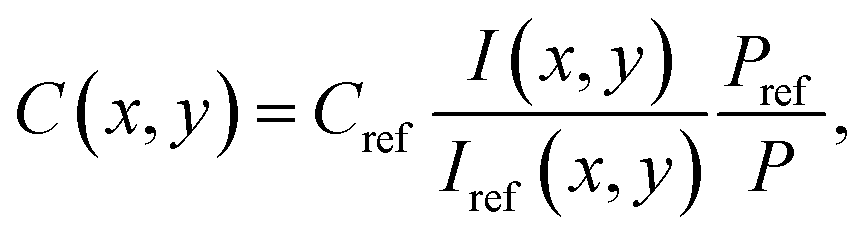
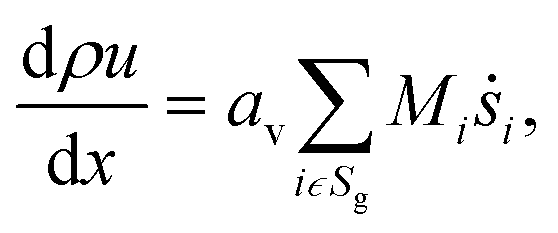
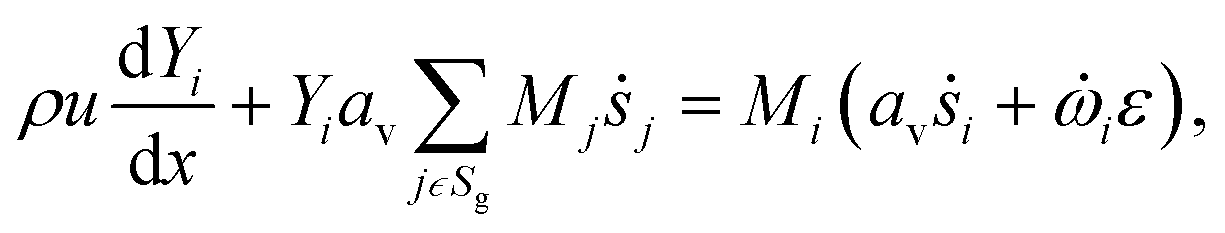
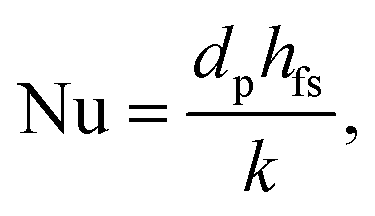
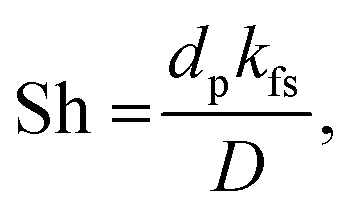
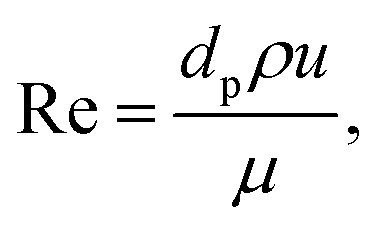
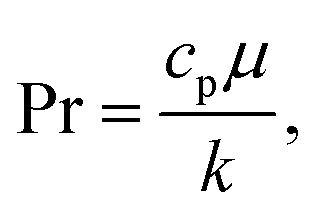
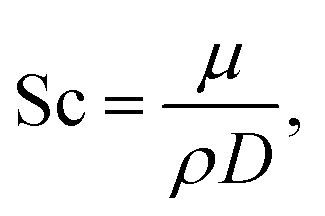
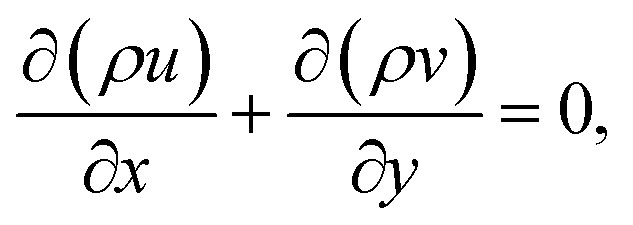
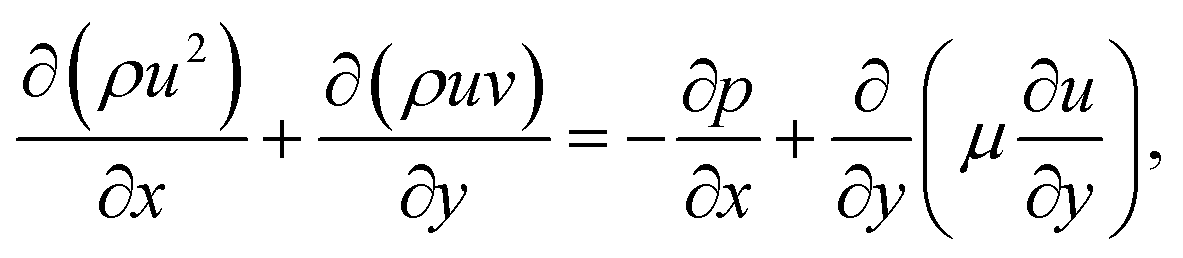
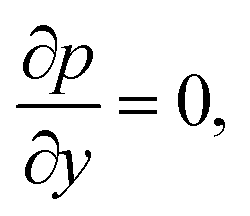
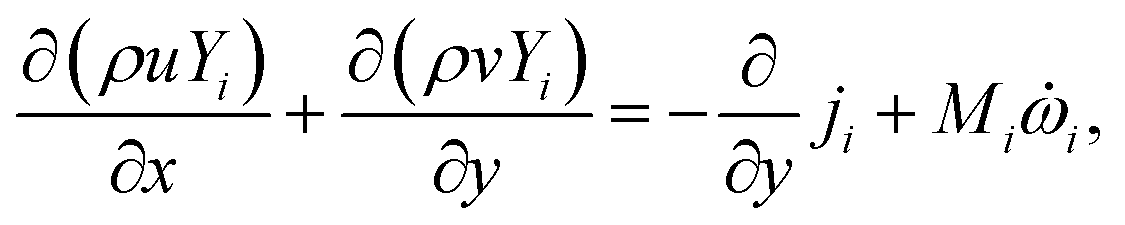
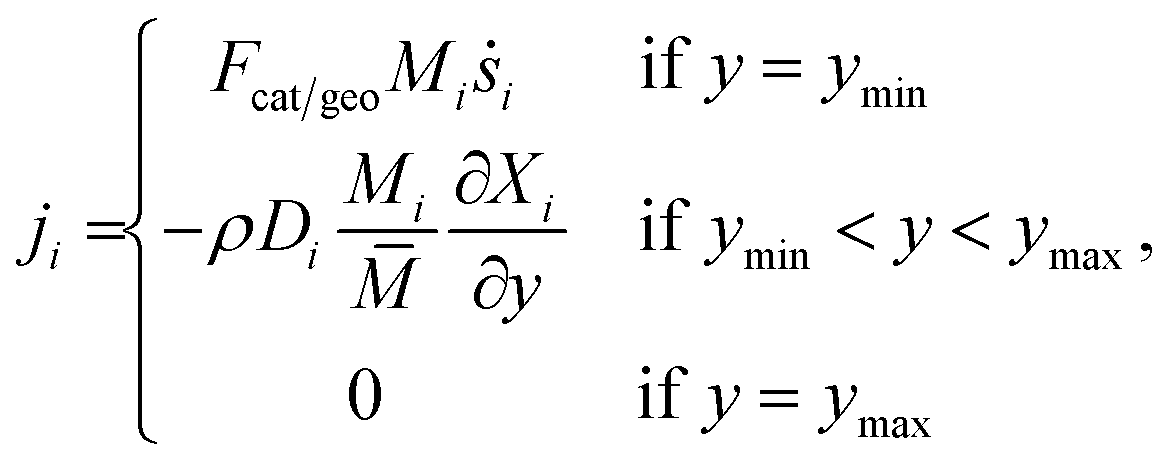
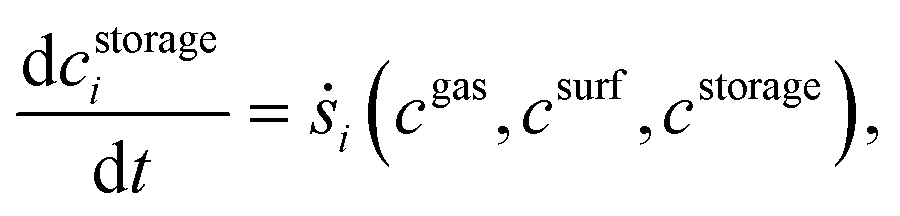
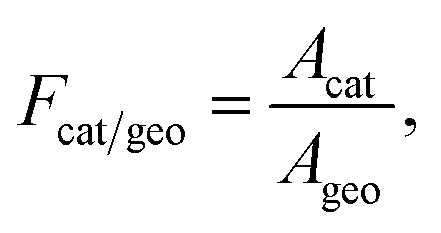
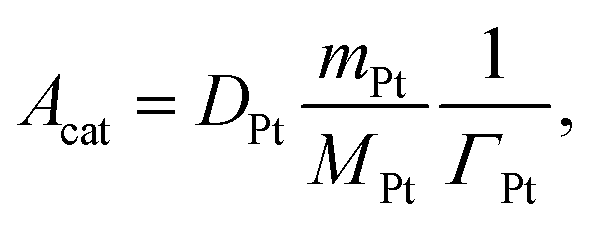
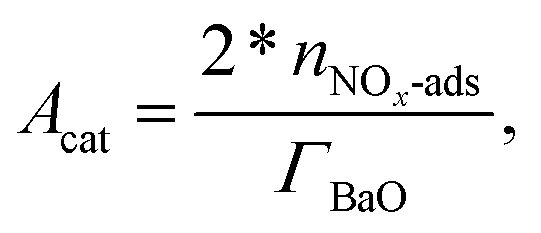
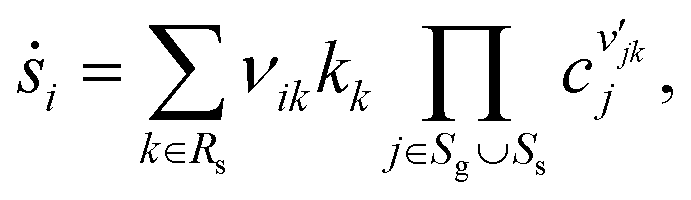
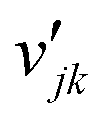 are the reaction orders of the species. The rate coefficient k is determined by a modified Arrhenius expression
are the reaction orders of the species. The rate coefficient k is determined by a modified Arrhenius expression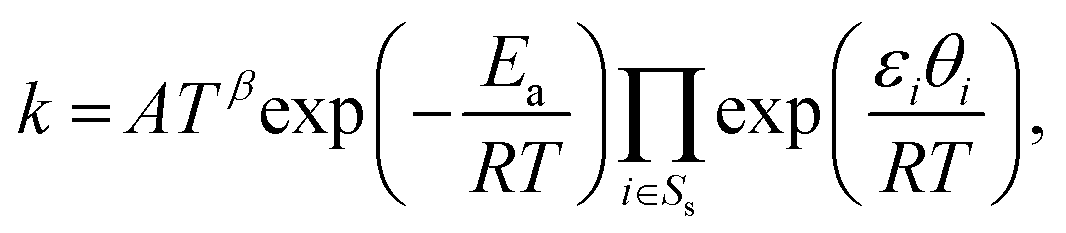
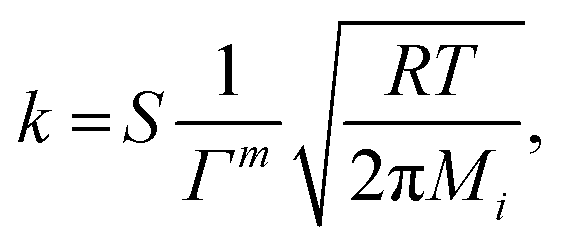
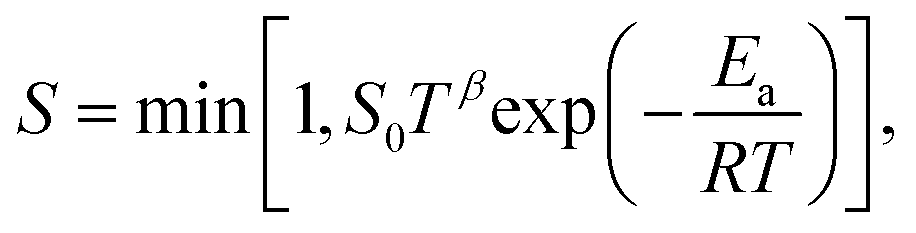
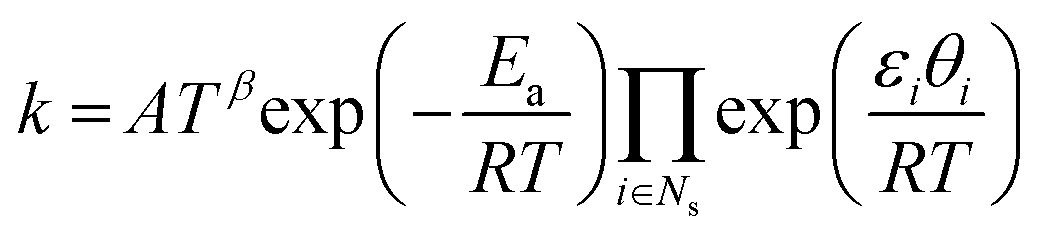 or
or 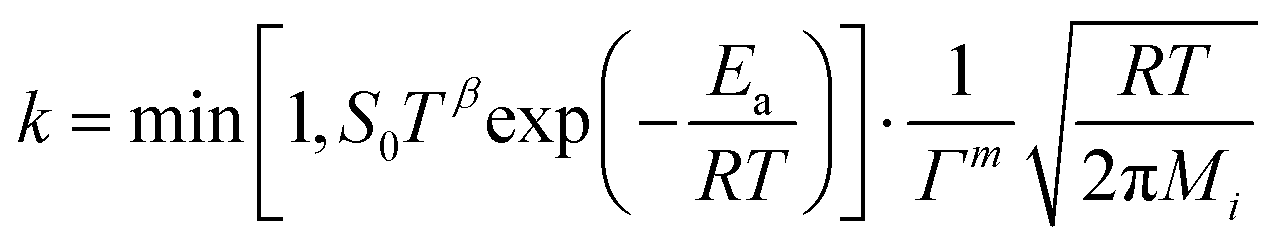 .
.