
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle- and double-bridged PNP ligands in chromium-catalysed ethylene oligomerisation†
Quintin
Lo
a,
Dominic
Pye
a,
Sami
Gesslbauer
a,
Ying
Sim
b,
Felipe
García
 bc,
Andrew J. P.
White
a and
George J. P.
Britovsek
*a
bc,
Andrew J. P.
White
a and
George J. P.
Britovsek
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, 82 Wood Lane, London, W12 0BZ, UK. E-mail: g.britovsek@imperial.ac.uk
bDivision of Chemistry & Biological Chemistry, NTU, 21 Nanyang Link, 637371, Singapore
cDepartamento de Química Orgánica e Inorgánica, Facultad de Química, Universidad de Oviedo, Julián Claveria 8, 33006 Oviedo, Spain
First published on 31st May 2022
Abstract
Several PNP-type diphosphine ligands have been synthesised and characterised, featuring a single or a double N-bridge between the P-donor atoms. PNP ligands 1 and 2 containing diazaphospholane donors have been prepared and reaction with [CrCl3(thf)3] results in coordination in a bidentate fashion to give dinuclear complexes [(1)CrCl3]2 and [(2)CrCl3]2 which have been characterised by scXRD analysis. In situ prepared catalysts using ligands 1 and 2 provide good activities and selectivities for the tri- and tetramerisation of ethylene reaching 35% 1-hexene and 61% 1-octene at 5400 g g−1 per Cr per h in the case of 1, and 42% 1-hexene and 55% 1-octene at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g g−1 (Cr) h−1 in the case of 2, comparable to standard iPrN(PPh2)2-type ligands under similar conditions. Chromium-catalysed ethylene oligomerisations with a doubly N-bridged cyclodiphosphazane ligand (4) result in a Schulz–Flory distribution of α-olefins with relatively low α values of 0.42 and 0.52. Computational studies using DFT on mononuclear chromium complexes of ligands 1 and 2 have shown that the binding of ethylene is favoured in these complexes compared to the benchmark PNP ligand iPrN(PPh2)2 and that the oligomerisation mechanism involves both single and double ethylene insertions.
000 g g−1 (Cr) h−1 in the case of 2, comparable to standard iPrN(PPh2)2-type ligands under similar conditions. Chromium-catalysed ethylene oligomerisations with a doubly N-bridged cyclodiphosphazane ligand (4) result in a Schulz–Flory distribution of α-olefins with relatively low α values of 0.42 and 0.52. Computational studies using DFT on mononuclear chromium complexes of ligands 1 and 2 have shown that the binding of ethylene is favoured in these complexes compared to the benchmark PNP ligand iPrN(PPh2)2 and that the oligomerisation mechanism involves both single and double ethylene insertions.
Introduction
Linear alpha olefins (LAOs, or 1-alkenes) are important building blocks for the chemical industry that find extensive use for example as co-monomers in olefin polymerisation and as intermediates to detergents, lubricants, and plasticisers.1–3 Global consumption of LAOs is currently more than 6 million tonnes per annum and this increases annually.4 LAOs such as 1-hexene and 1-octene are high value products used predominantly as co-monomers in polyethylene production (LLDPE). As a result, there is much attention focused on selective ethylene trimerisation and tetramerisation catalysed by metal complexes that can afford these valuable intermediates in high yield.1,5–9 A great deal of experimental and theoretical studies have been carried out on the Cr/PNP/MAO class of catalysts (PNP = diphosphinoamine, MAO = methylaluminoxane), first reported for selective trimerization by Wass and co-workers.10 These PNP ligands of type A in Fig. 1 are also capable of high tetramerisation selectivity and have been developed into a commercial process by Sasol.11–14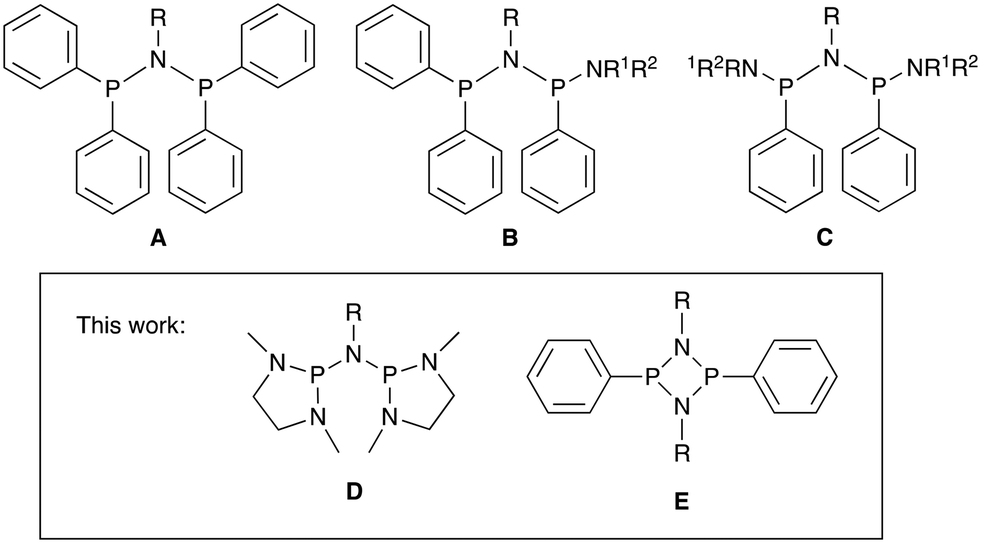 | ||
| Fig. 1 PNP-type ligand classes A–E used for chromium-catalysed ethylene oligomerisation. | ||
A metallacyclic mechanism is generally invoked for chromium-catalysed trimerisation and tetramerisation reactions.14–18 Detailed mechanistic studies have been hampered due to the paramagnetic nature of both the pre-catalyst and the active species, and many questions still remain regarding the finer details of the reaction mechanism and the exact nature of the active species, despite considerable spectroscopic and computational efforts in recent years.19–26 Our previous experimental and theoretical studies on PNP chromium catalysts has led to a mechanistic model which invokes a competition between mono-ethylene and bis-ethylene coordinated chromacyclopentane complexes, leading to both single and double ethylene insertion pathways (Scheme 1).27,28 1-Hexene results from a mono-ethylene intermediate and a bis-ethylene complex leads to 1-octene, with only a minor contribution from the dashed pathway in Scheme 1. This model is able to explain the preferred formation of 1-octene over 1-hexene in tetramerisation catalysts, especially at higher pressures, and also accounts for the formation of various alkyl cyclopentane and alkane side products (see Scheme 1). Additionally, we have expanded the mono- versus bis-ethylene model to other non-PNP catalyst systems to show how metallacycles can lead to alternating LAO distributions where α-olefins formed from an odd number of ethylene units (n = 1, 3, 5, etc.) are significantly less abundant than the even-numbered oligomers such as 1-butene (n = 2), 1-octene (n = 4), and 1-dodecene (n = 6).4,29,30
 | ||
| Scheme 1 Ethylene oligomerisation via a metalacyclic mechanism. | ||
Many attempts using a variety of bidentate ligand sets have been made to improve selectivity in ethylene oligomerisation.6,31–33 While 1-hexene selectivities of >95% are generally easily obtained, only modest improvements in 1-octene selectivity have been achieved so far and there appears to be an upper limit of approximately 70–80% selectivity for 1-octene.13 We are interested in the role of the ligand in this oligomerisation process, especially why so many different and diverse ligand structures give similar results in terms of selectivity and what the requirements for improvements in selectivity are. Most PNP ligands investigated so far contain aryl substituents at the phosphorus donors. Some exceptions are dialkyl phosphine derivatives,34–36 phospholyl derivatives,37 and the amino-substituted derivatives B and C in Fig. 1 reported by Rosenthal and co-workers.38,39
The first step in the oligomerisation process is an oxidative cyclisation of two ethylene monomers by a low valent chromium(I) species to form a chromium(III) cyclopentane complex (Scheme 1).25 According to our previous computational studies, the next step, which is key for high selectivity to 1-octene over 1-hexene, is the coordination of two more ethylene units and a double insertion in the Cr–C bond of the metallacyclopentane complex, without elimination of 1-hexene during this process. Considering that the binding of ethylene to a metal centre relies heavily on back donation from the metal, we reasoned that an increase in the Lewis basicity of the phosphorus donors of the PNP ligand should increase the ethylene affinity of the chromium centre and thus increase selectivity towards 1-octene. Furthermore, it has been shown that the 1-octene/1-hexene ratio is sensitive to ligand sterics and bulky ligands tend to favour 1-hexene formation.1,33
Inspired by Verkade's classic superbasic phosphatranes,40,41 and the basic properties of amino-substituted phosphines P(NR2)3,42,43 we have prepared two new PNP ligands with amine rather than the phenyl substituents. The first class involves ligands of type D featuring sterically unencumbered diazaphospholane donors, and the second type E involves a doubly N-bridged cyclodiphosphazane unit (Fig. 1). The coordination chemistry of PNP ligands of type D has been little explored so far.44 We have investigated the coordination chemistry with [CrCl3(THF)3] and their application in ethylene oligomerisation. Computational studies have been carried out using DFT to study the effect of these ligands on the binding of ethylene and subsequent barriers to insertion and product formation.
Results and discussion
The synthesis of the diphosphinoamines 1 and 2, adapted from a procedure published by Scherer and Wokulat, involved reaction of the chlorodiaminophosphine precursor with the appropriate amine (see eqn (1) (Scheme 2) and ESI† for details).45 The products were purified by distillation and analysis by 31P NMR showed a singlet at 112.3 ppm for the N-propyl ligand 1 and at 109.6 ppm for the N-isopropyl ligand 2, close to the value of 105.4 ppm reported for a similar N-methyl derivative.46 However, for both compounds 1 and 2, additional small signals were seen in the NMR spectra. In the 31P NMR spectrum of ligand 1, two additional singlets were present at 115.6 ppm (∼6%) and at 122.0 ppm (∼2%), and in the case of ligand 2 at 115.6 ppm (∼10%) and at 122.0 ppm (∼3%). These by-products could not be removed by repeated distillation. VT 31P NMR studies for both 1 and 2 over the temperature range from −80 to 100 °C showed no change in the ratio between the three signals, confirming there is no equilibrium between these three species (see Fig. S14 and S15†). A rearrangement of PNP to PPN ligands was first considered, similar to that seen for other PNP and PNPN ligands,47–52 but this was not consistent with the singlets observed in the 31P NMR spectra. The signal at 115.6 ppm, which was common to both 1 and 2, was identified as the P–P bridged diphospholane compound [(CH2)2N(CH3)2P]2 (3) which was prepared by independent synthesis through reduction of 2-chloro-1,3-diazaphospholane with magnesium (eqn (2) (Scheme 2)).53,54 We therefore propose that a redistribution takes place during the distillation process, whereby two PNP ligands react to give a P–P bridged diphospholane and a PNNP diphosphinohydrazine as shown in eqn (3). The mechanism for this process has not been investigated but may involve radical intermediates as diphospholanes are known to undergo homolytic P–P bond cleavage,55 and form stable phosphinyl radicals with sterically hindered N-substituents.53,54,56 The cyclodiphosphazane 4 was synthesised by reacting the chloro precursor [ClP(μ-NtBu)]2 (ref. 57) with phenyl lithium (eqn (4) (Scheme 2)). Spectroscopic analysis by 31P NMR showed a single isomer at 155.4 ppm, which we presume is the thermodynamically favoured cis isomer.58 | ||
| Scheme 2 Synthesis and redistribution reactions of PNP ligands. | ||
The addition of 1 and 2 to [CrCl3(THF)3] in toluene led to the formation of blue solutions from which dark blue PNP chromium(III) chloride complexes [(1)CrCl3]2 and [(2)CrCl3]2 could be isolated cleanly in good yields (eqn (5)). Analysis of the paramagnetic complexes by single crystal XRD showed the formation of chloro-bridged dinuclear complexes [(1)CrCl3]2 and [(2)CrCl3]2, similar to other PNP chromium trichloride complexes (Fig. 2).51,59 Little structural difference is seen in the n-propyl versus the isopropyl complex. The axial Cr–P bond lengths are all significantly longer than the equatorial ones (cf. 2.5015(11) Å vs. 2.4134(10) Å for complex [(2)CrCl3]2), which is also seen in similar complexes containing the R-PNP ligand A (Fig. 1).51 The equatorial Cr–P bonds in [(1)CrCl3]2 and [(2)CrCl3]2 are both shorter than in the iPr-PNP chromium complex,51cf. 2.450(5) Å vs. 2.4187(8) Å and 2.4134(10) Å, presumable a consequence of the increased basicity at phosphorous and the reduced steric hindrance. The reaction of 4 with [CrCl3(THF)3] in toluene resulted in a dark purple solid product after evaporation of the solvent, but no complex could be isolated, most likely due to the formation of a coordination polymer. Previous studies on the coordination behaviour of type E ligands have shown that these cyclodiphosphazanes normally act either as monodentate or as bridging ligands between two metals, but never as bidentate ligands coordinating to a single metal.60,61
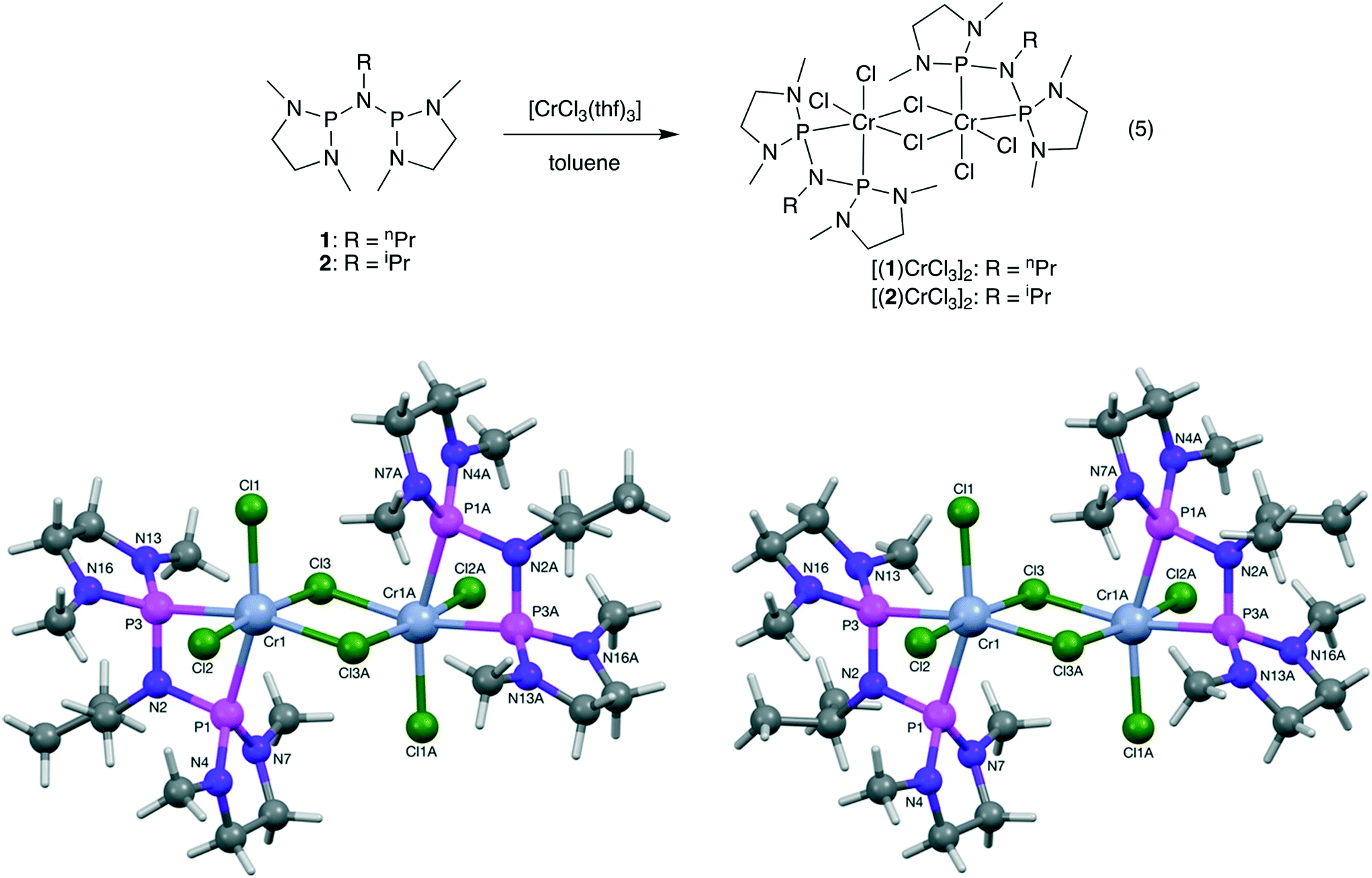 | ||
| Fig. 2 Synthesis and molecular structures of complexes [(1)CrCl3]2 and [(2)CrCl3]2. | ||
Ethylene oligomerisation
The PNP ligands were used in combination with [CrCl3(THF)3] as catalysts for ethylene oligomerisation, using MAO as the co-catalyst (Table 1). The catalyst systems prepared from ligands 1 and 2 show good activities and selectivities for the formation of 1-hexene and 1-octene at room temperature and 20 bar ethylene pressure, but are less active than the benchmark catalyst containing the iPrN(PPh2)2 ligand under similar conditions (run 10). The selectivity for 1-octene is slightly better for ligand 1 compared to 2, but both are lower than iPrN(PPh2)2. The selectivity to 1-octene improves with ethylene pressure and decreases somewhat with temperature from 20 to 50 °C. Overall, the product spectrum for 1 and 2 looks comparable to that obtained with the iPrN(PPh2)2 ligand, with less C10 and C10+ side-products being obtained, but more solid PE formation. A comparison between the in situ prepared catalyst system with the preformed complex [(2)CrCl3]2 under the same conditions showed a similar productivity, but a shift in the product distribution to favour the formation of 1-hexene. The origin of this shift is unclear at this stage.| # | Cat.a | P bar | Temp. °C | Olig. g | PE yield g | Prod. g g−1 (Cr) h−1 | C6b mol% | C8 mol% | C10c mol% | C10+ mol% | α |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: catalyst prepared in situ from 20 μmol PNP ligand and 20 μmol [CrCl3(THF)3]; MAO 500 equiv.; 100 ml toluene; 60 min. b C6 denotes 1-hexene and C8 denotes 1-octene. c C10 includes 1-decene, internal and branched decenes. d Catalyst prepared in situ from 5 μmol PNP ligand 4 and 5 μmol [CrCl3(THF)3]; MAO 500 equiv.; 100 ml toluene; 60 min. e Data taken from ref. 27; conditions: 10 μmol iPrN(PPh2)2 (A) and 10 μmol [CrCl3(THF)3]; MAO 300 equiv.; 100 ml toluene; 30 min. | |||||||||||
| 1 | 1/Cr | 3 | 21 | 0.32 | 0.81 | 1088 | 66.0 | 25.4 | 1.8 | 0 | |
| 2 | 1/Cr | 20 | 21 | 3.97 | 1.68 | 5432 | 34.9 | 61.4 | 0.6 | 0.6 | |
| 3 | 1/Cr | 20 | 50 | 8.39 | 1.40 | 9413 | 39.3 | 57.8 | 0.8 | 0.3 | |
| 4 | 2/Cr | 3 | 21 | 0.45 | 1.47 | 1842 | 43.1 | 35.5 | 0.5 | 0 | |
| 5 | 2/Cr | 20 | 21 | 15.9 | 2.10 | 17307 |
42.2 | 55.2 | 0.8 | 0.2 | |
| 6 | 2/Cr | 20 | 50 | 16.8 | 1.30 | 18076 |
43.1 | 53.8 | 0.8 | 0.4 | |
| 7 | [(2)CrCl3]2 | 20 | 22 | 15.8 | 1.12 | 16269 |
76.1 | 21.2 | 1.4 | 0.4 | |
| 8 | 4/Cr | 10 | 24 | 19.0 | 1.21 | 19432 |
30.2 | 15.8 | 8.6 | 9.8 | 0.52 |
| 9 | 4/Crd | 20 | 24 | 5.87 | 0.22 | 23423 |
32.9 | 15.8 | 7.2 | 6.3 | 0.42 |
| 10 | A/Cre | 20 | 30 | 6.46 | 0.41 | 26441 |
19.5 | 67.5 | 5.4 | 7.6 | |
Interestingly, oligomerisation experiments with the cyclodiphosphazane ligand 4 yielded a Schulz–Flory distributions of 1-alkenes, with α values of 0.52 at 10 bar and 0.42 at 20 bar (less catalyst was used in the latter case). These results indicate that the binding of ligand 4 in the active catalyst is likely to be different compared to the iPrN(PPh2)2 ligand. As discussed previously, these ligands normally act either as monodentate or as bridging ligands between two metals.60,61
Computational studies
The PNP ligands 1 and 2 form, upon reaction with [CrCl3(THF)3], dinuclear chloro-bridged chromium(III) complexes. Upon activation with MAO, these PNP/Cr catalysts have shown up to 61% selectivity for 1-octene in the oligomerisation of ethylene. While the catalytic activity is somewhat lower compared to the benchmark iPrN(PPh2)2 catalyst, it is remarkable that the product spectrum obtained with these ligands 1 and 2 is not significantly different, despite the very different ligand properties in terms of steric requirements and basicity. In order to understand these electronic and steric differences, we have carried out a DFT investigation, using ligand 2. The same method (M06L) and basis sets were employed that we have used previously for other PNP ligands so that results can be directly compared (see ESI†).27,28,62,63Upon addition of MAO to the Cr(III) precursor complex containing ligand 2, reduction to a PNP-Cr(I) species is believed to occur as a result of halide/alkyl exchange and abstraction reactions, combined with reductive elimination. Our previous studies have indicated that a Cr(I)–Cr(III) cycle is more likely than a Cr(II)–Cr(IV) cycle.62 Thus, the reaction pathway starts from the bis(ethylene)complex Cr(I) complex [(2)Cr(C2H4)2]+ (I), (Fig. 3), which is generally believed to be the active species in these reactions.25 Complex I undergoes oxidative coupling viaTSI-II to the metallacyclopentane complex II, with a barrier of +9.3 kcal mol−1, smaller but similar in magnitude as 11.9 kcal mol−1 for the MeN(PPh2)2/Cr system.63 More likely in this case, coordination of a further ethylene monomer to complex I to give [(2)Cr(C2H4)3]+ (III) takes place first, which is most stable in the quartet spin state (4III) as shown by the blue route (the PNP ligand has been omitted for clarity). The sextet spin state was also calculated for I and III, but these were found to be higher in energy by +0.5 and +6.2 kcal mol−1, respectively. There are a number of interesting differences between ligand 2 when comparing the energies with the previously calculated MeN(PPh2)2 ligand, using the same DFT method and basis sets.63 The coordination of the third ethylene to give 4III leads to a stabilisation of −10.4 kcal mol−1 relative to the bis(ethylene) starting complex, which is significantly greater than the stabilisation of less that 1 kcal mol−1 calculated for [(MeN(PPh2)2)Cr(C2H4)3]+.63 The increased affinity of the metal centre to bind ethylene is likely due to the basicity of the phosphine donors in ligand 2 and possibly also due to the reduced steric hindrance. Oxidative coupling of two ethylene moieties in 4III leads to the metallacyclopentane complex IVviaTSIII-IV with a barrier of 9.6 kcal mol−1 (blue route). Intermediate IV either undergoes ethylene insertion viaTSIV-V (+10.7 kcal mol−1) to the metallacycloheptane species V, or binds another ethylene to give the bis(ethylene) metallacyclopentane complex VII. This coordination of a second ethylene is favourable by −3.6 kcal mol−1, compared to +1.3 kcal mol−1 for the MeN(PPh2)2/Cr system.63 The β-H transfer process from intermediate V has a modest barrier of approximately 10 kcal mol−1, which leads to the intermediate Cr(I)–hexene complex VI that ultimately releases 1-hexene.
 | ||
| Fig. 3 Reaction profile with Gibbs free energies (kcal mol−1) for the single ethylene insertion pathway of the activated chromium complex containing the n-propyl PNP ligand 2. All chromium species are cations. (M06L/BS1//M06L/BS2, 298 K). | ||
The metallacyclopentane intermediate VII with two coordinated ethylene units is stabilised by −3.6 kcal mol−1 relative to the mono(ethylene) complex IV. At higher ethylene pressures, VII will likely be the dominant intermediate, leading to the formation of 1-octene via a double ethylene insertion pathway (Fig. 4). The first ethylene insertion from VII to intermediate VIII occurs viaTSVII-VIII with a modest barrier of +8.5 kcal mol−1 (cf. +10.9 kcal mol−1 for MeN(PPh2)2/Cr).63 Note that this barrier is lower than for the mono(ethylene) complex IV (+10.7 kcal mol−1). Due to the increased flexibility of the metallacycloheptane ring in VIII and the various coordination modes of the coordinated ethylene ligand, this intermediate exists as a number of energetically closely related isomers with different agostic interactions, the lowest of which is found at −38.7 kcal mol−1. The second ethylene insertion shows a slightly lower barrier (+7.9 kcal mol−1) than the first, and leads to the chromacyclononane complex IX, which can undergo β-H transfer viaTSIX-X to produce the Cr(I)–octene complex X and eventually 1-octene. Complexes VIII, IX and X as well as the TS states exist as various isomers due to different agostic interactions and the most likely energetic pathway is indicated in red in Fig. 4.
 | ||
| Fig. 4 Reaction profile with Gibbs free energies (kcal mol−1) for the double ethylene insertion pathway of the activated chromium complex containing the isopropyl PNP ligand 2. All chromium species are cations (M06L/BS1//M06L/BS2, 298 K). | ||
Overall, the energetics of single and double ethylene insertion pathways in Fig. 3 and 4 are very comparable to those previously calculated for MeN(PPh2)2/Cr and related systems.63 The increased basicity of ligand 2 leads to stronger ethylene coordination and a stabilisation of ethylene bound species such as III and VII. This however has not yet resulted in an increased overall yield of 1-octene.
Multiple attempts have been made to prepare metallacyclic chromium PNP complexes. The alkylation reaction of [(iPrN(PPh2)2)CrCl3]2 with Li2C4H8 in Et2O invariably failed and attempts to prepare Cr(II) PNP complexes by reacting PNP ligands with [CrCl2(thf)2] in THF also failed, as did reductions of [(iPrN(PPh2)2)CrCl3]2 with a range of reducing agents such as Al/AlCl3, AlMe3, or KC8 in toluene, or nBuLi, PhMgBr, NaHBEt3 or Mg in Et2O. Starting from the dinuclear tetra(1,4-butanediyl) Cr(II) precursor complex [Li(OEt2)]4[{Cr(C4H8)2}2] (6),64,65 prepared here from [CrCl2(thf)2] and Li2C4H8 in Et2O, and reaction with various PNP ligands also failed to generate any isolable products. A new and more accurate XRD analysis for [Li(OEt2)]4[{Cr(C4H8)2}2] (6) has been included in the ESI.†
In conclusion, we have shown that Lewis basic PNP ligands of type D in Fig. 1 afford active chromium-based oligomerisation catalysts upon activation with MAO, with selectivities for 1-octene and 1-hexene that are comparable to the classic iPrN(PPh2)2/Cr catalyst system. A doubly-bridged PNP ligand of type E resulted in a mixture of linear α-olefins that follow a Schulz–Flory distribution. Computational studies have shown that ethylene binding to the chromium centre is favoured, most likely as a result of the increased basicity of the PNP ligand and reduced steric hindrance, but the experimental oligomerisation studies have not shown an improved selectivity for 1-octene, which appears to be generally limited to approximately 80%. Further studies are needed in order to deepen our understanding of these industrially important oligomerisation systems.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the EPSRC and the UK Catalysis Hub for funding (Grant: EP/K014668/1). F. G. also thanks the support of Fundación para el Fomento en Asturias de la Investigación Científica Aplicada y la Tecnología (FICYT) through the Margarita Salas Senior Program (AYUD/2021/59709).References
- O. L. Sydora, Organometallics, 2019, 38, 997–1010 CrossRef CAS.
- P.-A. R. Breuil, L. Magna and H. Olivier-Bourbigou, Catal. Lett., 2014, 145, 173–192 CrossRef.
- W. Keim, Angew. Chem., Int. Ed., 2013, 52, 12492–12496 CrossRef CAS PubMed.
- C. T. Young, R. von Goetze, A. K. Tomov, F. Zaccaria and G. J. P. Britovsek, Top. Catal., 2020, 63, 294–318 CrossRef CAS.
- T. Agapie, Coord. Chem. Rev., 2011, 255, 861–880 CrossRef CAS.
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed.
- P. W. N. M. van Leeuwen, N. D. Clément and M. J.-L. Tschan, Coord. Chem. Rev., 2011, 1499–1517 CrossRef CAS.
- K. A. Alferov, G. P. Belov and Y. Meng, Appl. Catal., A, 2017, 542, 71–124 CrossRef CAS.
- D. F. Wass, Dalton Trans., 2007, 816–819 RSC.
- A. Carter, S. A. Cohen, N. A. Cooley, A. Murphy, J. Scutt and D. F. Wass, Chem. Commun., 2002, 858–859 RSC.
- A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuinness, D. H. Morgan, A. Neveling, S. Otto, M. Overett, A. M. Slawin, P. Wasserscheid and S. Kuhlmann, J. Am. Chem. Soc., 2004, 126, 14712–14713 CrossRef CAS PubMed.
- S. Kuhlmann, C. Paetz, C. Hägele, K. Blann, R. Walsh, J. T. Dixon, J. Scholz, M. Haumann and P. Wasserscheid, J. Catal., 2009, 262, 83–91 CrossRef CAS.
- R. Walsh, D. H. Morgan, A. Bollmann and J. T. Dixon, Appl. Catal., A, 2006, 306, 184–191 CrossRef CAS.
- M. J. Overett, K. Blann, A. Bollmann, J. T. Dixon, D. Haasbroek, E. Killian, H. Maumela, D. S. McGuinness and D. H. Morgan, J. Am. Chem. Soc., 2005, 127, 10723–10730 CrossRef CAS PubMed.
- T. Agapie, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2007, 129, 14281–14295 CrossRef CAS PubMed.
- T. Agapie, S. J. Schofer, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2004, 126, 1304–1305 CrossRef CAS PubMed.
- J. R. Briggs, J. Chem. Soc., Chem. Commun., 1989, 674–675 RSC.
- J. Petit, L. Magna and N. Mézailles, Coord. Chem. Rev., 2022, 450, 214227 CrossRef CAS.
- B. Venderbosch, L. A. Wolzak, J.-P. H. Oudsen, B. de Bruin, T. J. Korstanje and M. Tromp, Catal. Sci. Technol., 2020, 10, 6212–6222 RSC.
- N. A. Hirscher, C. H. Arnett, P. H. Oyala and T. Agapie, Organometallics, 2020, 39, 4420–4429 CrossRef CAS.
- B. Venderbosch, J. P. H. Oudsen, D. J. Martin, B. de Bruin, T. J. Korstanje and M. Tromp, ChemCatChem, 2019, 12, 881–892 CrossRef.
- J. Rabeah, M. Bauer, W. Baumann, A. E. C. McConnell, W. F. Gabrielli, P. B. Webb, D. Selent and A. Brückner, ACS Catal., 2013, 3, 95–102 CrossRef CAS.
- D.-H. Kwon, S. M. Maley, J. C. Stanley, O. L. Sydora, S. M. Bischof and D. H. Ess, ACS Catal., 2020, 10, 9674–9683 CrossRef CAS.
- T. Gunasekara, J. Kim, A. Preston, D. K. Steelman, G. A. Medvedev, W. N. Delgass, O. L. Sydora, J. M. Caruthers and M. M. Abu-Omar, ACS Catal., 2018, 8, 6810–6819 CrossRef CAS.
- S. Chabbra, D. M. Smith, N. L. Bell, A. J. B. Watson, M. Buhl, D. J. Cole-Hamilton and B. E. Bode, Sci. Adv., 2020, 6, eabd7057 CrossRef CAS PubMed.
- I. Y. Skobelev, V. N. Panchenko, O. Y. Lyakin, K. P. Bryliakov, V. A. Zakharov and E. P. Talsi, Organometallics, 2010, 29, 2943–2950 CrossRef CAS.
- G. J. P. Britovsek, D. S. McGuinness, T. S. Wierenga and C. T. Young, ACS Catal., 2015, 5, 4152–4166 CrossRef CAS.
- G. J. P. Britovsek, D. S. McGuinness and A. K. Tomov, Catal. Sci. Technol., 2016, 6, 8234–8241 RSC.
- G. J. P. Britovsek, R. Malinowski, D. S. McGuinness, J. D. Nobbs, A. K. Tomov, A. W. Wadsley and C. T. Young, ACS Catal., 2015, 5, 6922–6925 CrossRef CAS.
- A. K. Tomov, J. D. Nobbs, J. J. Chirinos, P. K. Saini, R. Malinowski, S. K. Y. Ho, C. T. Young, D. S. McGuinness, A. J. P. White, M. R. J. Elsegood and G. J. P. Britovsek, Organometallics, 2016, 36, 510–522 CrossRef.
- J. H. Lee, J. W. Baek, D. G. Lee, J. H. Ko, D. G. Lee, K. S. Cho, J. W. Lee and B. Y. Lee, Catalysts, 2021, 11, 1122–1136 CrossRef CAS.
- H. Lee, Y. Joe and H. Park, Catal. Commun., 2019, 121, 15–18 CrossRef CAS.
- J. E. Radcliffe, A. S. Batsanov, D. M. Smith, J. A. Scott, P. W. Dyer and M. J. Hanton, ACS Catal., 2015, 5, 7095–7098 CrossRef CAS.
- F. Alam, H. Fan, C. Dong, J. Zhang, J. Ma, Y. Chen and T. Jiang, J. Catal., 2021, 404, 163–173 CrossRef CAS.
- S. D. Boelter, D. R. Davies, K. A. Milbrandt, D. R. Wilson, M. Wiltzius, M. S. Rosen and J. Klosin, Organometallics, 2020, 39, 967–975 CrossRef CAS.
- S. D. Boelter, D. R. Davies, P. Margl, K. A. Milbrandt, D. Mort, B. A. Vanchura, D. R. Wilson, M. Wiltzius, M. S. Rosen and J. Klosin, Organometallics, 2020, 39, 976–987 CrossRef CAS.
- T. E. Stennett, T. W. Hey, L. T. Ball, S. R. Flynn, J. E. Radcliffe, C. L. McMullin, R. L. Wingad and D. F. Wass, ChemCatChem, 2013, 5, 2946–2954 CrossRef CAS.
- U. Rosenthal, ChemCatChem, 2019, 12, 41–52 CrossRef.
- S. Härzschel, F. E. Kühn, A. Wöhl, W. Müller, M. H. Al-Hazmi, A. M. Alqahtani, B. H. Müller, N. Peulecke and U. Rosenthal, Catal. Sci. Technol., 2015, 5, 1678–1682 RSC.
- P. B. Kisanga, J. G. Verkade and R. Schwesinger, J. Org. Chem., 2000, 65, 5431–5432 CrossRef CAS PubMed.
- C. Lensink, S. K. Xi, L. M. Daniels and J. G. Verkade, J. Am. Chem. Soc., 1989, 111, 3478–3479 CrossRef CAS.
- R. F. Weitkamp, B. Neumann, H. G. Stammler and B. Hoge, Chem. – Eur. J., 2021, 27, 10807–10825 CrossRef CAS PubMed.
- I. A. Koppel, R. Schwesinger, T. Breuer, P. Burk, K. Herodes, I. Koppel, I. Leito and M. Mishima, J. Phys. Chem. A, 2001, 105, 9575–9586 CrossRef CAS.
- A. Tarassoli, H. J. Chen, M. L. Thompson, V. S. Allured, R. C. Haltiwanger and A. D. Norman, Inorg. Chem., 1986, 25, 4152–4157 CrossRef CAS.
- O. J. Scherer and J. Wokulat, Z. Anorg. Allg. Chem., 1968, 361, 296–305 CrossRef CAS.
- H. Nöth and W. Storch, Chem. Ber., 1977, 110, 2607–2623 CrossRef.
- R. Keat, L. Manojlović-Muir, K. W. Muir and D. S. Rycroft, J. Chem. Soc., Dalton Trans., 1981, 2192–2198 RSC.
- Z. Fei, N. Biricik, D. Zhao, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2004, 43, 2228–2230 CrossRef CAS PubMed.
- D. S. McGuinness, M. Overett, R. P. Tooze, K. Blann, J. T. Dixon and A. M. Z. Slawin, Organometallics, 2007, 26, 1108–1111 CrossRef CAS.
- M. F. Haddow, J. Jaltai, M. Hanton, P. G. Pringle, L. E. Rush, H. A. Sparkes and C. H. Woodall, Dalton Trans., 2016, 45, 2294–2307 RSC.
- A. M. Lifschitz, N. A. Hirscher, H. B. Lee, J. A. Buss and T. Agapie, Organometallics, 2017, 36, 1640–1648 CrossRef CAS.
- S. Peitz, N. Peulecke, B. R. Aluri, B. H. Muller, A. Spannenberg, U. Rosenthal, M. H. Al-Hazmi, F. M. Mosa, A. Wohl and W. Muller, Chem. – Eur. J., 2010, 16, 12127–12132 CrossRef CAS PubMed.
- R. Edge, R. J. Less, E. J. McInnes, K. Muther, V. Naseri, J. M. Rawson and D. S. Wright, Chem. Commun., 2009, 1691–1693 RSC.
- O. Puntigam, D. Förster, N. A. Giffin, S. Burck, J. Bender, F. Ehret, A. D. Hendsbee, M. Nieger, J. D. Masuda and D. Gudat, Eur. J. Inorg. Chem., 2013, 2013, 2041–2050 CrossRef CAS.
- O. Puntigam, I. Hajdók, M. Nieger, M. Niemeyer, S. Strobel and D. Gudat, Z. Anorg. Allg. Chem., 2011, 637, 988–994 CrossRef CAS.
- N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850 CrossRef CAS PubMed.
- K. W. Muir, J. Chem. Soc., Dalton Trans., 1975, 259 RSC.
- R. Keat, D. S. Rycroft and D. G. Thompson, J. Chem. Soc., Dalton Trans., 1980, 321 RSC.
- P. R. Elowe, C. McCann, P. G. Pringle, S. K. Spitzmesser and J. E. Bercaw, Organometallics, 2006, 25, 5255–5260 CrossRef CAS.
- M. M. Siddiqui, J. T. Mague and M. S. Balakrishna, J. Organomet. Chem., 2015, 794, 81–87 CrossRef CAS.
- M. S. Balakrishna, J. Organomet. Chem., 2010, 695, 925–936 CrossRef CAS.
- D. S. McGuinness, B. Chan, G. J. P. Britovsek and B. F. Yates, Aust. J. Chem., 2014, 67, 1481–1490 CrossRef CAS.
- G. J. Britovsek and D. S. McGuinness, Chem. – Eur. J., 2016, 22, 16891–16896 CrossRef CAS PubMed.
- J. Krausse and G. Schödl, J. Organomet. Chem., 1971, 27, 59–67 CrossRef CAS.
- R. Fischer, H. Görls, S. Krieck and M. Westerhausen, Z. Anorg. Allg. Chem., 2017, 643, 1276–1294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic procedures, NMR, details for DFT calculations and XRD analysis. All computational results are collected in the repository. CCDC 2108180, 2108181 and 2115885. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cy00550f |
| This journal is © The Royal Society of Chemistry 2022 |