
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed synthesis of mixed anhydrides via carbonylative telomerization†
Kevin
Hares
 ,
Dennis
Vogelsang
,
Charlotte S.
Wernsdörfer
,
Dennis
Panke
,
Dieter
Vogt
and
Thomas
Seidensticker
*
,
Dennis
Vogelsang
,
Charlotte S.
Wernsdörfer
,
Dennis
Panke
,
Dieter
Vogt
and
Thomas
Seidensticker
*
TU Dortmund University, Department for Biochemical and Chemical Engineering, Laboratory of Industrial Chemistry, Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de; Tel: +49 231 755 2310
First published on 2nd May 2022
Abstract
For the first time, mixed carboxylic anhydrides were accessed directly via homogeneous palladium catalysis from 1,3-butadiene and carboxylic acids. Under carbonylative telomerization conditions, the respective mixed 3,8-nonadienoic anhydrides are formed in a single reaction step with yields of up to 82%. These very reactive mixed anhydrides can then be used for consecutive reactions in a one-pot manner and selectively transfer the newly formed unsaturated C9 unit. Possible changes in the proposed mechanism were discussed and in a first example, the mixed anhydrides were utilized to form amides.
Introduction
Due to the rising demand for energy, developing more efficient ways to convert basic feedstock into novel products has become a critical challenge. Hence, the direct use of simple building blocks, such as 1,3-butadiene and carbon monoxide, is of high interest. One of the most relevant tools enabling such efficient conversions is catalysis and particularly homogeneous catalysis.1 Compared to heterogeneous catalysis, it offers mainly higher activity due to the lack of mass transfer limitations.1 Additionally, the catalytic mechanism can be studied more easily, and its detailed understanding enables the development of new reactions and more sophisticated reaction designs.1 This possibility offers excellent potential for addressing the challenges mentioned above and synthesizing complex molecules in as few steps as possible.In 1971, Billups discovered in collaboration with Union Carbide Corporation the homogeneously catalyzed carbonylative telomerization (Fig. 1).2 They investigated the carbonylation of dienes and it was reported that halide-free palladium catalysts, such as Pd(acac)2, give ethyl nona-3,8-dienoate from 1,3-butadiene and ethanol in the presence of carbon monoxide and PPh3 as ligand.2 They showed that the ester is directly formed from 1,3-butadiene and not by carbonylation of the ether, the telomerization product of 1,3-butadiene and ethanol.2 This multi-component reaction was later named “carboxytelomerization” due to the newly formed carboxylate group. Remarkably, four molecules react in a single catalytic cycle to form the final product, mediated by a single catalyst complex (Fig. 1), which offers a highly efficient and atom-economic 2 + 2 + 1 + X synthesis route starting from basic chemicals.2,3
 | ||
| Fig. 1 Pd-Catalyzed carbonylative telomerization of 1,3-butadiene. | ||
Eight years later, in 1979, Knifton published a more comprehensive study on the Pd-catalyzed carboxytelomerization.3 With monodentate phosphine ligands, high yields and selectivities were achieved in N-heterocyclic or tertiary arylamine solvents, presumably due to increased catalyst stability. Until then, the reaction was limited to 1,3-butadiene and short-chain aliphatic alcohols as substrates.2–5 In 2018, Vogelsang et al. were able to connect the carboxytelomerization to an additional carbonylation. They developed a protocol for synthesizing a linear, terminal C10-diester from 1,3-butadiene, methanol and carbon monoxide in a one-pot manner.6 Following this “rediscovery” of carbonylative telomerization, several other papers were published, exploring the scope of that particular reaction for the first time. Carboxytelomerization converts the renewable β-myrcene in quantitative yields into highly branched esters, with best results in the absence of additional solvents,7 which resembles the first diene used, other than 1,3-butadiene. Furthermore, Vogelsang et al. reported the conversion of amines under carbonylative telomerization conditions for the first time.8 Inspired by the resulting products, this reaction was named amidotelomerization. It offers a one-step synthesis for unsaturated C9-amides, otherwise synthesized in a multistep reaction associated with high waste production.8,9 All these examples show the high potential of carbonylative telomerization reactions to produce unsaturated C9-esters and their derivatives. However, some nucleophiles remain challenging to convert efficiently. On the one hand, phenols and tertiary alcohols are less active in carbonylative telomerization and result in low yields. On the other hand, amines required a high catalyst loading for their conversion in carbonylative telomerization.7,8,10
In the same year, Sauthier and co-workers showed the positive influence of carboxylic acids on carboxytelomerization. They proposed a cationic palladium complex (Fig. 2, right) that plays a critical role in converting phenols, as it allows soft nucleophiles such as phenols to cleave the product from the palladium catalyst more efficiently, resulting in higher yields.10 In 2019, they applied their system to different di- and polyols and different bio-based alcohols.11 Recently, similar results were achieved by Vogelsang et al., who also synthesized di- and triesters but under acid-free conditions. Additionally, they were able to control the degree of esterification via control over the 1,3-butadiene concentration.12 These di- and triesters can be used as synthetic fats, lubricants or polymer precursors.
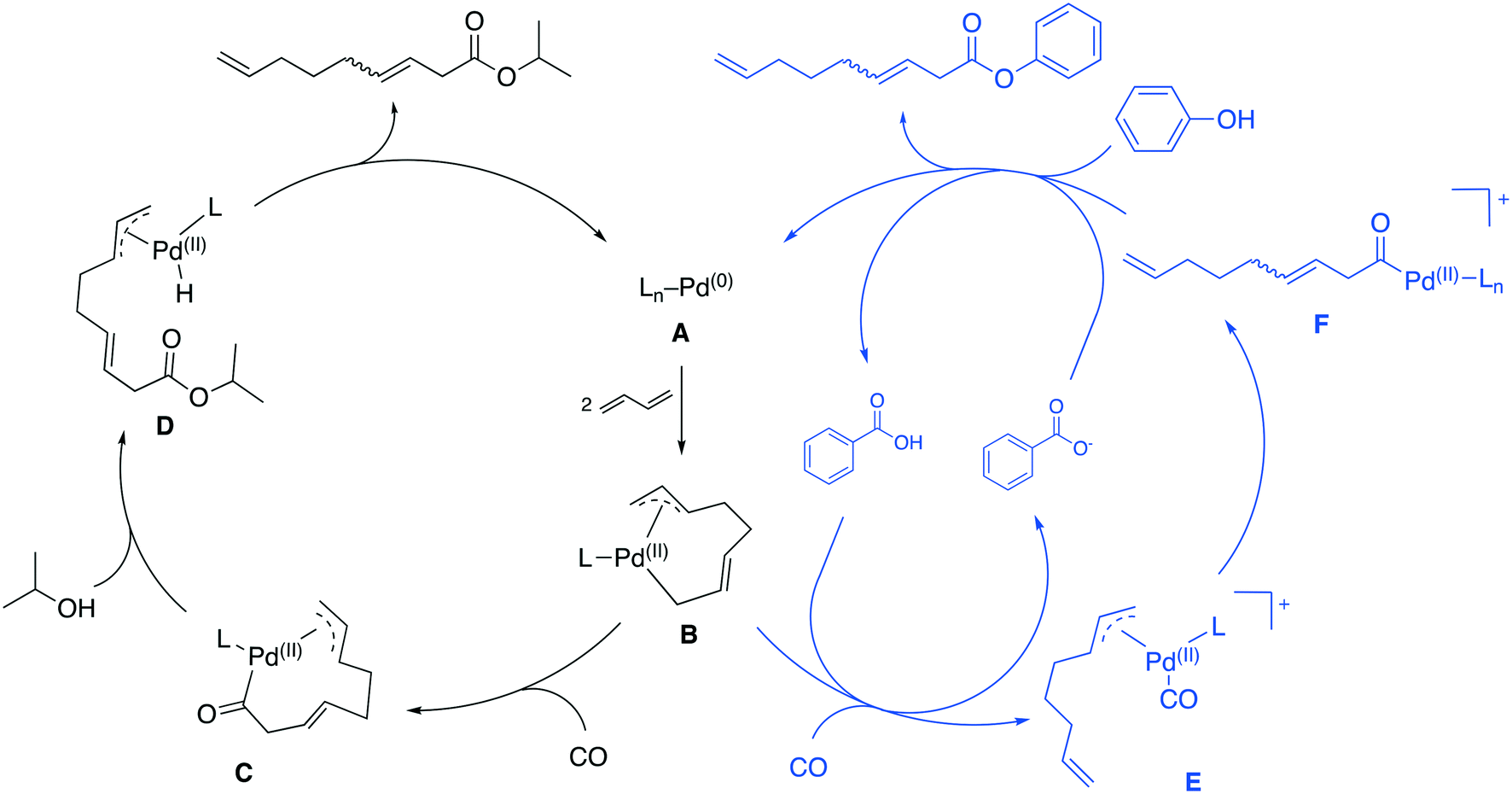 | ||
| Fig. 2 Catalytic cycles proposed for the carboxytelomerization proposed by Knifton (left),3 and Sauthier (right).10 | ||
So far, two different reaction mechanisms have been proposed for the carbonylative telomerization. The first was proposed by Billups2 and was later refined by Knifton (Fig. 2, left).3
Similar to the mechanism of a “simple” telomerization reaction, the first step represents an oxidative coupling of two 1,3-budadiene molecules forming the allyl complex B. Insertion of CO into the σ-allyl C–Pd bond forms the acyl complex C. This species is then attacked by the alcohol nucleophile to form the intermediate D, which collapses via a reductive elimination into the desired ester product and the active catalyst complex A.3
Sauthier et al. proposed a different mechanism for the carbonylative telomerization of phenols (Fig. 2, right cycle).10 Phenols result in low yields under common carbonylative telomerization conditions. The key for this transformation was the addition of benzoic acid. Therefore, a cationic Pd-complex was proposed. Due to its positive charge, the complex supposedly becomes more electrophilic increasing the interaction with the phenol nucleophile. In their mechanism, the complex B is protonated by the acid and forms the cationic complex E upon coordination of CO. The CO then undergoes a migratory insertion into the η3-allyl C–Pd bond forming F. In the last step, the phenol cleaves the ester from the Pd-complex, while restoring the active catalyst A as well as a proton.10
Within our longtime interest in developing novel and sustainable catalytic synthesis strategies, we set ourselves the goal to further expand the valuable synthesis tool carbonylative telomerization. In this context, the implementation of new nucleophiles is of particular interest since entirely new classes of compounds would be accessible for further applications. Here, it is of interest to consider the closely related telomerization reaction.13,14 The telomerization is complemented by its carbonylative counterparts for many nucleophiles (Fig. 3), such as alcohols and amines. However, for carboxylic acids, which are well-known nucleophiles for telomerization (Fig. 3),15–19 the carbonylative counterpart is not yet described in the literature. Thus, four substrate molecules under carbonylative telomerization conditions, including the carboxylic acids, would be transformed into mixed unsaturated C9-anhydrides in a single reaction step in the presence of a palladium catalyst. Such a synthesis method is particularly interesting, since mixed carboxylic anhydrides are highly reactive intermediates with a broad range of applications. Surprisingly, best to our knowledge, no transition metal-catalyzed synthesis for mixed carboxylic acid anhydrides from dienes was reported until now.
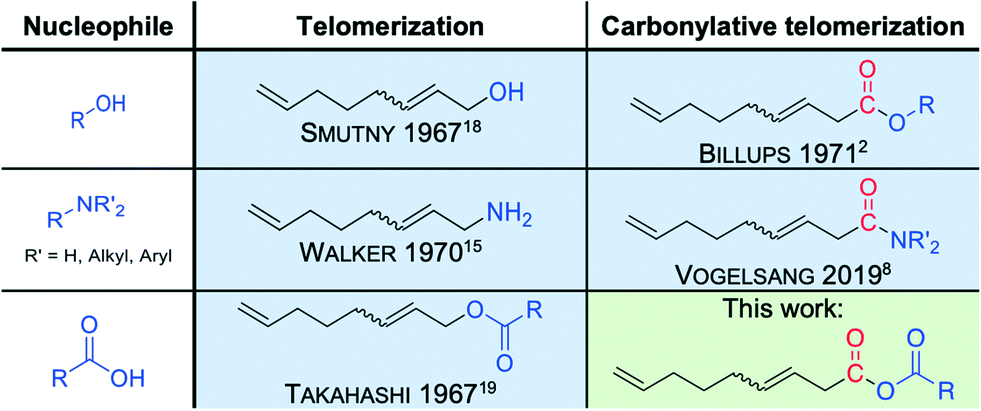 | ||
| Fig. 3 Accessible products from 1,3-butadiene and different nucleophiles under telomerization (left) and carbonylative telomerization (right) conditions, respectively. | ||
Mixed carboxylic acid anhydrides are often used to activate otherwise weakly reactive carboxylic acids. A prominent example for the application of mixed anhydrides is the Yamaguchi esterification, which utilizes 2,3,6-trichlorobenzoic acid chloride to form a mixed anhydride with the carboxylic acid to be activated. The anhydride can react with an alcohol to form the desired ester in high yield and selectivity in a second reaction step.20 A major drawback of such syntheses, and the synthesis of mixed anhydrides in general, is the necessity of acid chlorides as substrates, which is associated with the production of stoichiometric amounts of waste products, i.e. salts.21,22
Herein, we present a novel Pd-catalyzed synthesis of mixed carboxylic acid anhydrides via carbonylative telomerization of 1,3-butadiene and carboxylic acids. Additionally, the corresponding anhydrides are implemented in consecutive reactions.
Experimental section
Carbonylative telomerization
First, the acid was weighed into the autoclave's 20 mL stainless steel vessel. The reactor was closed, then evacuated and purged with argon. A solution containing Pd(OAc)2 (6.74 mg, 0.03 mmol, 1 mol%) and PCy2Ph (17.3 mg, 0.06 mmol, 2 mol%), 3 mL dry dioxane and the internal standard n-decane (75 mg, 0.527 mmol) was prepared. Pd(OAc)2 and PCy2Ph were weighed into a 20 mL headspace vial, sealed with a crimp cap, and then alternately evacuated and purged with argon. The corresponding amount of solvent and internal standard was added via syringe. The mixture was placed in an ultrasonic bath for homogenization and then transferred to the reactor via syringe under argon counterflow. 1,3-Butadiene was condensed under auto-pressure into a PTFE tube. Afterwards, the tube was connected to the reactor, and 1,3-butadiene was transferred into the reactor. The correct amount was adjusted via differential weighing. The reactor was pressurized with carbon monoxide, placed in a heating block, and magnetically stirred for 20 h.After 20 h, the reactor was cooled on water/ice, depressurized and purged with argon. Piperidine (1032 mg, 12 mmol, 4 eq.) was added to the reaction mixture via syringe under argon counterflow. The solution was stirred for 5 h at 50 °C. Then it was cooled, and the solvent was removed under reduced pressure. The residue was dissolved in 10 mL dichloromethane and extracted with 20 mL diluted sulfuric acid (5 v%). The aqueous phase was then extracted twice with 10 mL dichloromethane. The combined organic phases were dried with MgSO4 and concentrated under reduced pressure. The residue was dissolved in 15 mL diethyl ether and extracted with 30 mL of a 0.5 M NaOH solution. The aqueous phase was extracted two more times with 20 mL diethyl ether. The combined organic phases were dried with MgSO4. Two spoons of celite were added, and the mixture was concentrated under reduced pressure until dry. The resulting powder was further purified using a 40 g “Flash Pure” cartridge (ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cyclohexane, 1% to 10% in 2 CV, 10% until eluted).
:cyclohexane, 1% to 10% in 2 CV, 10% until eluted).
Exclusion of alternative pathways
3,8-Nonadienoic acid (467 mg, 3 mmol, 1 eq.) was weighed into a 20 mL stainless steel reactor. The reactor was closed, then evacuated and purged with argon. The palladium precursor Pd(OAc)2 (6.74 mg, 0.03 mmol, 1 mol%) and PCy2Ph (17.3 mg, 0.06 mmol, 2 mol%) were weighed into a 20 mL headspace vial and sealed with a crimp cap. The vial was then alternately evacuated and purged with argon while connected to a Schlenk-line via a cannula. The corresponding amount of dry dioxane (3 mL) was added to the vial via syringe. The mixture was placed in an ultrasonic bath until a uniform solution was observed. Then the solution was transferred to the reactor via syringe under argon counterflow. Piperidine (1032 mg, 3 mmol, 4 eq.) and 75 mg of the internal standard n-decane were added to the reactor via syringe under argon counterflow. The reactor was stirred for 5 h at 50 °C. Afterwards, the reaction was stopped by an ice bath, and the reaction mixture was analyzed via GC–FID.Results and discussion
The experiments were initially carried out employing 1,3-butadiene in combination with glacial acetic acid 1a as model substrates, as the latter is a well-known nucleophile in the related telomerization reaction.15,17,18 Carbonylative telomerization conditions (Fig. 4) were adapted from our previous studies,6 and applied using a 20 mL stainless steel autoclave with magnetic stirring. The catalyst was in situ generated from Pd(OAc)2 as the precursor and the ligand tri-n-butyl phosphine.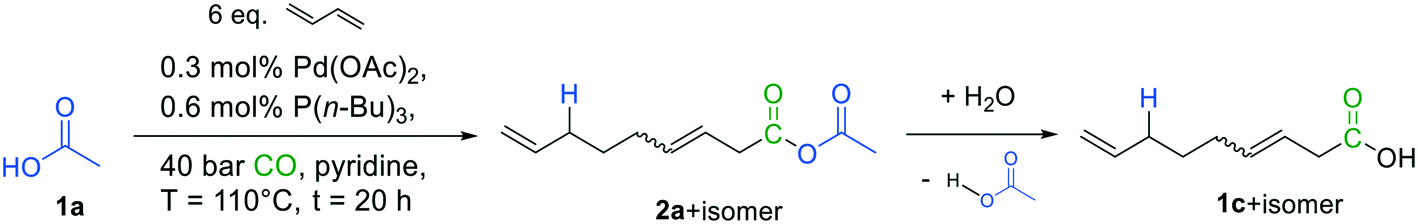 | ||
| Fig. 4 Initial reaction conditions for carbonylative telomerization of 1,3-butadiene with acetic acid. | ||
The reaction was examined via GC–MS (see ESI†). To our delight, analysis of the spectra indicated the presence of the desired mixed anhydride 2a beside one main isomer, presumably its branched isomer. Additionally, two derivatives of 3,8-nonadienoic acid (1c + isomer) were detected, which may have formed by hydrolysis of the mixed anhydrides, underlining the expected high reactivity of the formed product. Consequently, isolation of 2avia standard purification methods such as chromatography is rather tricky and complicates its precise quantification.
However, this high reactivity allows easy derivatization into more stable compounds, quantified with more precision. Hence, carbonylative telomerization was repeated with acetic acid, but after depressurizing the reactor, butyl alcohol 3a was added as the nucleophile (Fig. 5) before the analysis. The corresponding esters could be readily detected via GC–MS and were quantified by GC–FID.
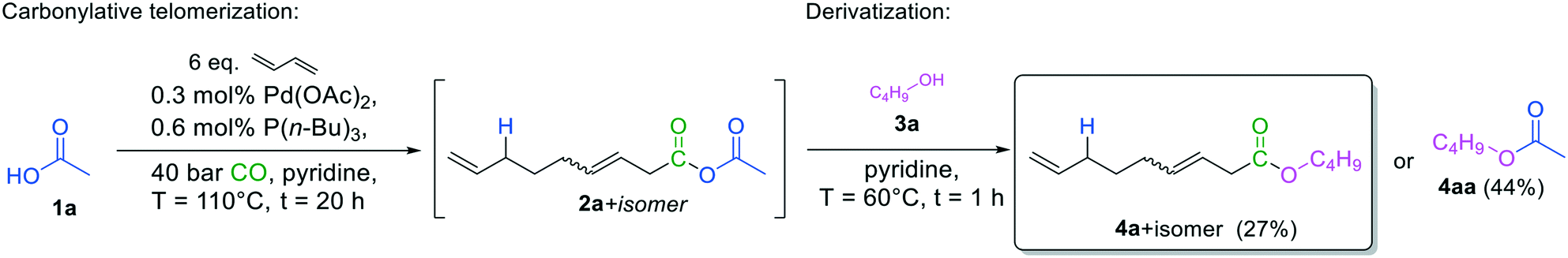 | ||
| Fig. 5 Synthesis of butyl nona-3,8-dienoate 4a by carbonylation telomerization of 1,3-butadiene with acetic acid and subsequent derivatization. | ||
Under these non-optimized conditions, however, yields were not satisfactory yet. The corresponding acetate 4aa, resulting from a nucleophilic attack of the alcohol on the acetate side of the anhydride, was the main product. Since more straightforward and effective acylating agents are available, it is not favorable to transfer the acetic acid side of the mixed anhydride to the nucleophile. Instead, transfer of the 3,8-nonadienoic acid part of the anhydride to the nucleophile is desired. In this way, it is possible to produce valuable unsaturated products that can be readily functionalized further due to their terminal double bond. Moreover, if the transfer of the 3,8-nonadienoic acid part of the anhydride to the nucleophile is selective, the initial acid 1a is reformed, offering potential reuse.
The relatively low selectivity can be explained by the nature of the formed mixed anhydride 2a: the acid functionalities in acetic acid and nonadienoic acid have a similar pKa-value and nucleophilicity since both are aliphatic acids. High selectivity may be achieved if one of the acid groups is a significantly better leaving group. For instance, acids having a lower pKa-value are usually better leaving groups. Another option would be the use of resonance stabilized acids such as benzylic acid derivatives. A well-known reaction making use of this concept is the Yamaguchi esterification.20 The reaction results in rapid and highly selective formation of esters from carboxylic acids by activation through a mixed anhydride. In this case, electron-poor benzoic acid derivatives are used to increase the reactivity and selectivity drastically. Benzoates are resonance stabilized; therefore, they are better leaving groups than non-aromatic acid anions. Hence, benzoic acid (1b) was used, providing a more stable leaving group, aiming for higher selectivity.
Improvement and validation of the system with benzoic acid (1b)
Benzoic acid (1b) was chosen as the nucleophile and was tested under carbonylative telomerization conditions (Fig. 6).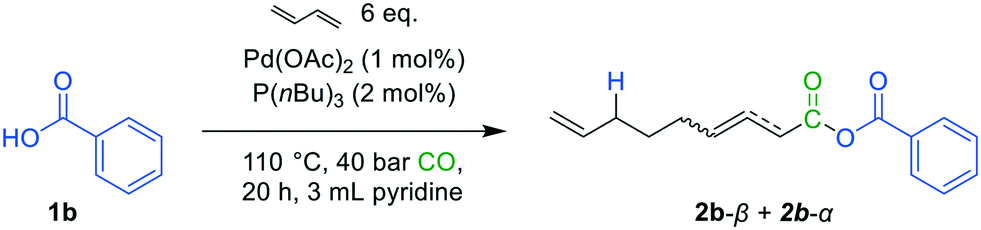 | ||
| Fig. 6 Carbonylative telomerization of 1,3-butadiene with benzoic acid (1b). | ||
GC–MS analysis revealed the presence of the mixed carboxylic anhydride. A small amount of the product could be separated via chromatography with an isolated yield of 6% using dry solvents. The presence of anhydrides was confirmed by FT-IR (see in ESI†), indicating the success of the reaction. NMR spectra of the product revealed the isomerization of the β-unsaturated product 2b-β to the α-unsaturated isomer of 2b-α (see in ESI†). This isomer seems to be more stable, presumably due to the conjugated double-bond and therefore could be isolated. The formation of the conjugated isomer of 2b could be promoted by the acidic reaction environment, which can lead to Pd–hydride species. These species are known for their isomerization activity.23–25
The corresponding symmetrical anhydrides, i.e. benzoic acid anhydride and nonadienoic acid anhydride, respectively, were detected during GC-analysis. Presumably, the highly reactive mixed anhydrides undergo disproportionation reactions under the high temperatures in the GC injecting block, yielding an equilibrium of the two symmetrical and the mixed anhydride (Fig. 7).26–28 We strongly assume that symmetrical anhydrides are not formed during carbonylation telomerization. However, this must be excluded for precise quantification, and a different analysis protocol must be developed.
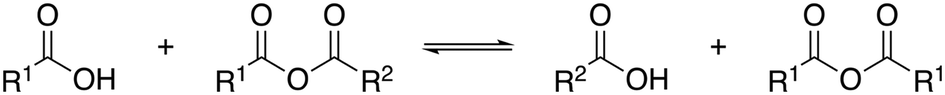 | ||
| Fig. 7 Disproportionation reaction of a mixed anhydride to the corresponding symmetrical anhydrides.26 | ||
For quantitative analysis of mixed anhydrides formed via carbonylative telomerization under our initial conditions, a nucleophile was added to the reaction mixture after stopping (ice bath and depressurization) (Fig. 8). Next, amines were added for derivatization, which are more nucleophilic compared to the initially applied alcohols. The reaction mixture was analyzed afterwards via GC–FID, and the corresponding amides were quantified. After a short screening of potent amines (see in ESI†) and the corresponding reaction conditions for the derivatization step, piperidine (3b) was chosen as a suitable candidate resulting in the highest total amide yield of 67% after a reaction time of 5 h. An excess of 4 equivalents of 3b was applied to ensure a complete amidation of the mixed anhydrides. Additionally, dioxane was adapted as the solvent form Sauthier et al. since it already had proven to be beneficial for carbonylative telomerization in the presence of acids and will avoid pyridinium salt formation.10 This sequential reaction set up was used in the following to optimize the carbonylative telomerization of benzoic acid (1b) regarding the amount of 1,3-butadiene, the ligand, the CO pressure and the temperature (Fig. 8).
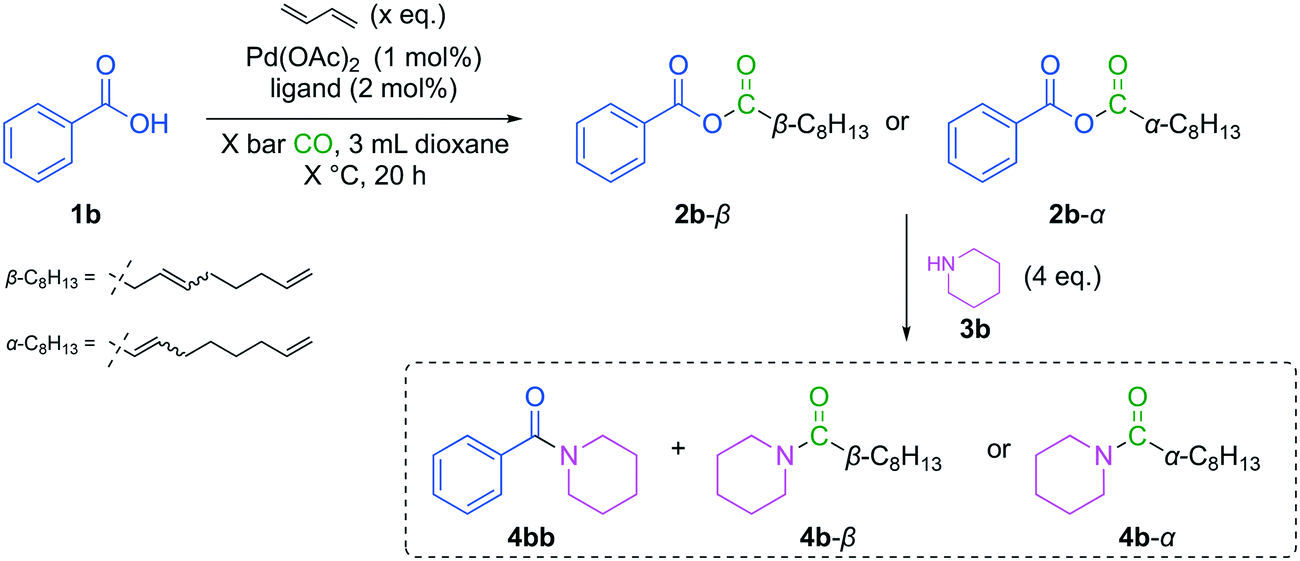 | ||
| Fig. 8 Carbonylative telomerization of 1,3-butadiene with benzoic acid (1b) for the synthesis of mixed anhydrides (2b) prior to optimization and subsequent derivatization yielding amides for quantification. | ||
It is important to exclude other reaction pathways towards the analyzed amides, as this would lead to false quantification of the mixed anhydrides. We tested whether amides 4b and 4bb may be formed by direct reactions between the acid and the amine (Fig. 9) to validate the chosen analysis method consisting of derivatization and quantification of the resulting amides. 3,8-Nonadienoic acid (1c) was included since it can be a by-product of the side reaction towards 4bb.
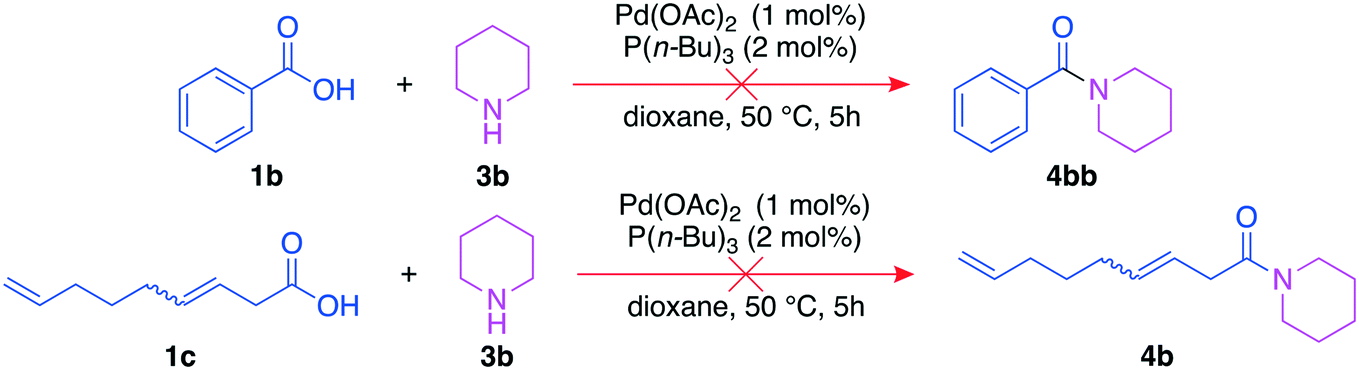 | ||
| Fig. 9 Exclusion experiments with benzoic acid 1b and 3,8-nonadienoic acid 1c. | ||
In both cases, no amides were detected. Hence, all amides from the derivatization step result from the reaction of the mixed anhydride and the amine. Their combined yield can thus give precise information about the yield of the mixed anhydride formed via carbonylative telomerization.
Optimization via DoE
Having an established protocol for quantifying carbonylative telomerization in hand (Fig. 8), this set-up was used to optimize the reaction conditions via a design of experiments (DoE) towards an increased anhydride yield.Previous investigations on carbonylative telomerization reactions have revealed significant parameters, such as temperature, CO pressure and the excess of 1,3-butadiene. These factors were varied in the ranges stated in Table 1, taking other research on these reactions into account. Additionally, the ligand plays a crucial role regarding reaction yield. Knifton has initially shown that basic monodentate phosphines resulted in good to excellent yields, and Vogelsang et al. have shown the positive effects of large Tolman angles.3,7 Therefore, P(n-Bu)3, PPh3 and PCy2Ph were chosen for this investigation; all other parameters were kept constant (see Fig. 8).
| Factor | Temperature [°C] | CO pressure [bar] | 1,3-Butadiene eq. [–] | Ligand (Tolman angle) |
|---|---|---|---|---|
| Minimum | 80 | 20 | 4 | P(n-Bu)3 (132°) |
| Middle | 100 | 30 | 6 | PPh3 (148°) |
| Maximum | 120 | 40 | 9 | PCy2Ph (164°) |
| Optimized | 93 | 20 | 7.9 | PCy2Ph (164°) |
The sum of the detected amides after derivatization was considered the anhydride yield, hence the output for the DoE. A central composite face-centered design was chosen using the optimization software Modde©, which resulted in a total of 27 experiments, each carried out as duplicates (see in ESI†). The data was evaluated, and the model had to be changed for better results to enhance the accuracy of the prediction. The calculations revealed that the CO pressure was not a significant factor in the given ranges. It was thus excluded from further calculations. Therefore, the pressure was set to 20 bar, as it represents the mildest reaction condition.
The calculations made by Modde© applied to the resulting dataset suggested a reaction temperature of 93 °C, and excess of 7.9 eq. 1,3-butadiene and PCy2Ph as the ligand. The predicted yield was calculated to be 76%. These results were verified by the corresponding experiments using the optimized reaction conditions, which resulted in good reproducibility since the duplicates show high similarity (Table 2, entry 1 and 2). The yield distribution of the derivatized products under optimized reaction conditions showed good reproducibility (Table 2). The sum of amides, in this case, equals the yield for the desired mixed anhydride intermediates 2b-β and 2b-α, showing an average yield of the mixed anhydride of 80%, which is 4% higher than predicted. Moreover, the desired amides 4b-β and 4b-α account for over 70% of the total amide yield. Consequently, the mixed anhydride favors the transfer of the aliphatic acyl rest (i.e. 3,8-nonadienyl) to the amide yielding linear and branched C9-amides. The optimization was carried out using calculated response factors using an established estimation method.29 Afterwards, the products were isolated to confirm the calculated results (Table 2, entry 3).
| Entry | Predicted yield [%] | ∑ Y [%] | Y (4b-β) [%] | Y (4b-α) [%] | Y (4bb) [%] |
|---|---|---|---|---|---|
| Conditions: carbonylative telomerization: 3 mmol benzoic acid, 23.7 mmol (7.9 eq.) 1,3-butadiene, 1 mol% Pd(OAc)2, 2 mol% PCy2Ph, 3 mL dioxane, 20 bar CO, 93 °C, 20 h, 600 rpm. Internal standard 75 mg decane; derivatization: 12 mmol piperidine, 50 °C, 5 h, 600 rpm. Yields (Y) were determined via GC-FID analysis using n-decane as an internal standard and calculated with respect to the carboxylic acid. a Isolated yield. | |||||
| 1 | 76 | 79 | 36 | 20 | 23 |
| 2 | 76 | 82 | 37 | 22 | 23 |
| 3a | 76 | 81 | 37 | 20 | 24 |
Implementation of optimized conditions
Afterwards, the optimized conditions were applied to a small scope investigation (Fig. 10). Since the electronic nature of benzoic acid derivatives is mainly influenced by substituents on the aromatic ring, different electron-withdrawing and donating groups were tested (Table 3). In this investigation, two effects must be considered. First, the nucleophilicity of the acid influences the reactivity in carbonylative telomerization. And second, the reactivity of the resulting anhydride is again influenced by the nature of the benzoic acid. An electron-deficient acid is an excellent leaving group. If the anhydride consists of two very different acid groups in pKa, it becomes more reactive. The electron-rich acid is transferred preferentially in a consecutive reaction, as seen in Yamaguchi esterification.20 Since the requirements for both reaction parts are opposing, a trade-off has to be found with high reactivity in carbonylative telomerization and satisfactory selectivity of the consecutive reaction.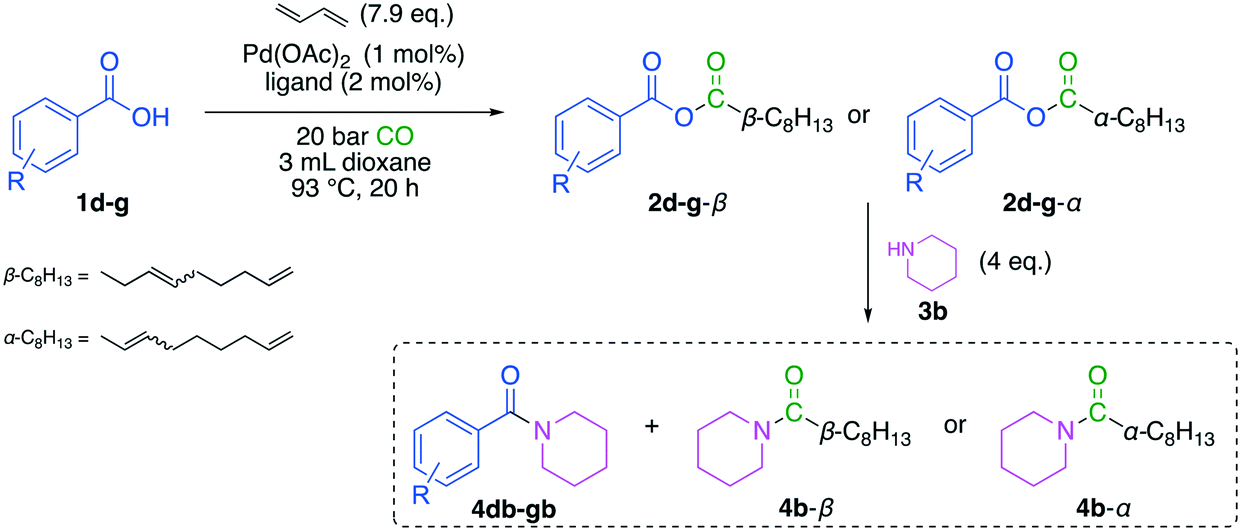 | ||
| Fig. 10 Applied substituted benzoic acid derivates. | ||
| Entry | R (acid) | pKa (ref. 30) | ∑ yield [%] | Y (4b-β) [%] | Y (4b-α) [%] | Y (4db–gb) [%] |
|---|---|---|---|---|---|---|
| Conditions: carbonylative telomerization: 3 mmol acid, 24 mmol 1,3-butadiene, 1 mol% Pd(OAc)2, 2 mol% PCy2Ph, 3 mL dioxane, 20 bar CO, 93 °C, 20 h, 600 rpm. Internal standard 75 mg decane; derivatization: 12 mmol piperidine, 50 °C, 5 h, 600 rpm. Isolated yield (Y) related to acid. a 0.5 mol% Pd(OAc)2, 1 mol% PCy2Ph; derivatization: 12 mmol piperidine, 50 °C, 5 h, 600 rpm. Isolated yield (Y) related to acid. b Not isolated. | ||||||
| 1 | m-Me (1d) | 4.28 | 85 | 39 | 22 | 24 |
| 2 | p-OMe (1e) | 4.49 | 91 | 40 | 26 | 25 |
| 3 | p-OMea (1e) | 4.49 | 71 | 36 | 17 | 18 |
| 4 | p-Cl (1f) | 3.98 | 74 | 36 | 37 | Tracesb |
| 5 | p-NO2 (1g) | 3.42 | — | Tracesb | Tracesb | — |
| Entry | ∑ yield [%] | Y (4b-β) [%] | Y (4b-α) [%] | Y (4bb) [%] | X (3b) [%] |
|---|---|---|---|---|---|
| Conditions: carbonylative telomerization: 3 mmol benzoic acid, 23.5 mmol 1,3-butadiene, 1 mol% Pd(OAc)2, 2 mol% PCy2Ph, 3 mL dioxane, 20 bar CO, 93 °C, 20 h, 600 rpm. Internal standard 75 mg decane; derivatization: 2.26 mmol piperidine, 50 °C, 5 h, 600 rpm. Yield (Y) and conversion (X) related to amine determined via GC-analysis with decane as internal standard. | |||||
| 1 | 77 | 36 | 22 | 19 | 100 |
The highest yield of 91% can be achieved using anisic acid (1e). Due to the +−M-effect, anisic acid is electron-rich acid and therefore has a higher pKa value. More electron-rich acids appear to be more reactive in carbonylative telomerization, showing higher overall anhydride yield. The anhydride yield is reduced with a lower pKa value. In contrast, the selectivity of 4b increases with a lower pKa value. As mentioned before, electron-poor acids are better leaving groups and are more stable in their anionic form. All tested acids have a lower pKa value than 3,8-nonadienoic acid (1c), which should be similar to nonanoic acid (was “pelargonic acid”), i.e. 4.96.30 Hence, the 3,8-nonadienoic rest is preferably transferred to the nucleophile. This effect increases with larger differences in pKa values and results in higher selectivity with a lower pKa. For 1f, it was impossible to isolate the arylamide 4fb, resulting in the highest selectivity for 4b. Although the overall activity of 1f is lower compared to the more electron rich acids, the selectivity towards the unsaturated C9-amides is high. This offers a way to selective pass the newly formed unsaturated C9-chain along to the consecutive product (Table 3, entry 4), since p-Cl-benzoate is the better leaving group. In comparison with the anhydride 2e a selectivity of only 72% was achieved in the amidation reaction. For 1g, only traces of the products could be observed, indicating low activity in carbonylative telomerization. These results can be explained with the lower nucleophilicity of electron poor carboxylic acids. Mayr et al. reported a nucleophilicity scale, showing p-nitro benzoate to be significant less nucleophilic than the benzoate ion.31 The lower nucleophilicity renders the acid les reactive for the carbonylative telomerization, thus forming less of the corresponding anhydride. This shows a compromise has to be found, between a high reactivity in carbonylative telomerization resulting from electron rich carboxylic acids and an ideal anhydride for consecutive reaction built with electron poor carboxylic acids.20 Additionally, the electronic properties of the acid have an influence on the formation of the α-unsaturated isomer 4b-α and 2d–g-4b-α, respectively. More electron-withdrawing groups in para-position of the acid result in a higher degree of isomerization and result in equal amounts of 4b-α and 4b-β (Table 3, entry 4), which is in contrast to more electron-rich acids, which result in 2:1 ratio of 4b-α and 4b-β. This aligns with the results found by Sauthier et al. for the carbonylative telomerization of phenols in the presence of catalytic amounts of benzoic acid.10 This effect could also be emphasized, by the higher acidity of the p-Cl substituted benzoic acid, which could also lead to more Pd–H species forming and thus more isomerization.
Mixed carboxylic anhydrides are mainly utilized to form consecutive products, such as the here implemented amidation. However, a large excess of the nucleophile is typically not desirable. Previously, an excess of piperidine (3b) was introduced to the system to guarantee full conversion of the mixed anhydrides for quantification. Since carbonylative telomerization as the first step is now optimized, further investigations for the derivatization step were carried out. A sub-stoichiometric amount, i.e. 0.75 eq., of piperidine was applied. This results in a 1.1 eq. excess of the anhydride (Table 4).
The total yield of the desired product reaches 77%, and 23% of the piperidine is converted to other by-products. These by-products can be assigned to the formation of ammonium salts since unreacted acid readily reacts with piperidine to form piperidyl benzoate.
Proposed reaction mechanism
The best results in the carbonylative telomerization with carboxylic acids were achieved with the bulky, basic phosphine PCy2Ph. Such donor phosphine ligands are reported to push the equilibrium of the η3-allyl into a σ-allyl complex (B)32 and an insertion of CO is preferred in this position, thus the carbonyl Pd complex C is formed, analogous to the proposed mechanism by Knifton (Fig. 11).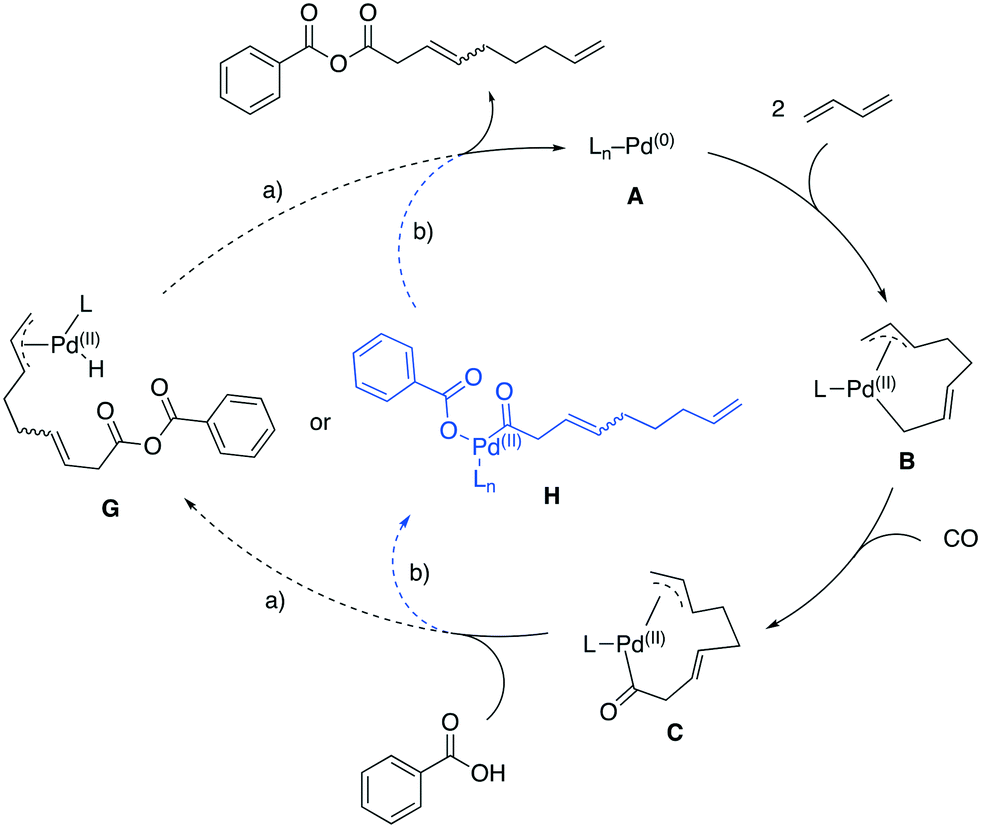 | ||
| Fig. 11 Possible reaction mechanisms for carbonylative telomerization of 1,3-butadiene with benzoic acid. | ||
Two pathways for the interaction of the carboxylic acid are feasible pathways. On the one hand, the reaction continues the mechanism proposed by Knifton and forms a Pd–H while the carboxylate inserts in the Pd carbonyl bond (Fig. 11, path a)),3 resulting in complex G. A reductive elimination of the allyl and the hydride forms the product and closes the catalytic cycle by releasing A. On the other hand, the acid could first protonate the remaining Pd–allyl and the resulting carboxylate could coordinate to the complex replacing the former allyl (Fig. 11, path b)). Again, a reductive elimination of the product forms the desired anhydride product and A. Both proposed pathways would proceed without a cationic catalyst species, since the insertion of CO into the σ-allyl–Pd bond is more likely compared to the migratory-insertion into a η3-allyl–Pd bond.
Conclusion
A novel, palladium-catalyzed synthesis for mixed carboxylic acid anhydrides was developed. For the first time, it was possible to generate mixed anhydrides catalytically from bulk chemicals, namely 1,3-butadiene, CO, and benzoic acid derivatives. In addition, a quantification protocol was developed using amines for quantitative derivatization in combination with standard GC-analysis. It was possible to optimize the conditions for the anhydride synthesis using this protocol and yields up to 82% were achieved with the model substrate benzoic acid.A small investigation of the scope of the reaction was carried out, including different benzoic acid derivatives. The experiments have shown the importance of the nature of the acid, as more electron-rich benzoic acid derivatives achieved higher total yields of up to 94% due to their higher nucleophilicity. In contrast, p-Cl-benzoic acid, which is less electron rich, resulted in only 74% yield in carbonylative telomerization, but the consecutive amidation was highly selective due to the better leaving group p-Cl-benzoate. Further investigations of the scope, including different dienes, are currently in progress, as well as other applications of the resulting anhydrides. Additionally, the mechanism must be studied in more detail, as the currently accepted proposed mechanism has to be updated for the conversion of carboxylic acids.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) − TRR 63 “Integrierte chemische Prozesse in flüssigen Mehrphasensystemen” (Teilprojekt A11) − 56091768. Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) − TRR 63 “Integrated Chemical Processes in Liquid Multiphase Systems” (subproject A11) − 56091768. We thank Umicore for the generous donation of the Pd(OAc)2 used in this work.References
- A. Behr and P. Neubert, Applied homogeneous catalysis, Wiley-VCH Verlag, Weinheim, Germany, 2012 Search PubMed.
- W. E. Billups, W. E. Walker and T. C. Shields, J. Chem. Soc. D, 1971, 1067–1068 RSC.
- J. F. Knifton, J. Catal., 1979, 60, 27–40 CrossRef CAS.
- J. Tsuji, Y. Mori and M. Hara, Tetrahedron, 1972, 28, 3721–3725 CrossRef CAS.
- M. Romanelli and G. Romanelli, Preparation of alkadienoic acid esters, US Pat., 3780074, 1973, 1–5 Search PubMed.
- D. Vogelsang, B. A. Raumann, K. Hares and A. J. Vorholt, Chem. – Eur. J., 2018, 24, 2264–2269 CrossRef CAS PubMed.
- D. Vogelsang, M. Dittmar, T. Seidensticker and A. J. Vorholt, Catal. Sci. Technol., 2018, 8, 4332–4337 RSC.
- D. Vogelsang, J. Vondran and A. J. Vorholt, J. Catal., 2018, 365, 24–28 CrossRef CAS.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- C. Dumont, F. Belva, R. M. Gauvin and M. Sauthier, ChemSusChem, 2018, 11, 3917–3922 CrossRef CAS PubMed.
- E. Wilson, C. Dumont, M. Drelon, I. Suisse, C. Penverne and M. Sauthier, ChemSusChem, 2019, 12, 2457–2461 CAS.
- D. Vogelsang, J. Vondran, K. Hares, K. Schäfer, T. Seidensticker and A. J. Vorholt, Adv. Synth. Catal., 2020, 362, 679–687 CrossRef CAS.
- A. Behr, M. Becker, T. Beckmann, L. Johnen, J. Leschinski and S. Reyer, Angew. Chem., Int. Ed., 2009, 48, 3598–3614 CrossRef CAS PubMed.
- T. A. Faßbach, A. J. Vorholt and W. Leitner, ChemCatChem, 2019, 11, 1153–1166 CrossRef.
- W. E. Walker, R. M. Manyik, K. E. Atkins and M. L. Farmer, Tetrahedron Lett., 1970, 11, 3817–3820 CrossRef.
- E. J. Smutiny, Ester Production, US Pat., 3407224, 1968 Search PubMed.
- D. Rose and H. Lepper, J. Organomet. Chem., 1973, 49, 473–476 CrossRef CAS.
- E. J. Smutny, J. Am. Chem. Soc., 1967, 89, 6793–6794 CrossRef CAS.
- S. Takahashi, T. Shibano and N. Hagihara, Tetrahedron Lett., 1967, 8, 2451–2453 CrossRef.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef CAS.
- A. R. Hajipour and G. Mazloumi, Synth. Commun., 2002, 32, 23–30 CrossRef CAS.
- M. Pasha and S. Rizwana, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2005, 44, 420–421 Search PubMed.
- X. Wang and L. K. Woo, J. Mol. Catal. A: Chem., 1998, 130, 171–176 CrossRef CAS.
- A. L. Kocen, M. Brookhart and O. Daugulis, Chem. Commun., 2017, 53, 10010–10013 RSC.
- P. Mamone, M. F. Grünberg, A. Fromm, B. A. Khan and L. J. Gooßen, Org. Lett., 2012, 14, 3716–3719 CrossRef CAS PubMed.
- J. Peydecastaing, C. Vaca-Garcia and E. Borredon, Chromatographia, 2008, 68, 685–688 CrossRef CAS.
- J. Peydecastaing, C. Vaca-Garcia and E. Borredon, Eur. J. Lipid Sci. Technol., 2009, 111, 723–729 CrossRef CAS.
- K. Kikukawa, K. Kono, K. Nagira, F. Wada and T. Matsuda, J. Org. Chem., 1981, 46, 4413–4416 CrossRef CAS.
- A. D. Jorgensen, K. C. Picel and V. C. Stamoudis, Anal. Chem., 1990, 62, 683–689 CrossRef CAS.
- D. R. Lide and W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, 90th edn, 2010 Search PubMed.
- H. F. Schaller, A. A. Tishkov, X. Feng and H. Mayr, J. Am. Chem. Soc., 2008, 130, 3012–3022 CrossRef CAS PubMed.
- P. W. Jolly, Angew. Chem., Int. Ed. Engl., 1985, 24, 283–295 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00486k |
| This journal is © The Royal Society of Chemistry 2022 |