
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo step activation of Ru-PN3P pincer catalysts for CO2 hydrogenation†‡§
Alex S.
Tossaint¶
 a,
Christophe
Rebreyend¶
a,
Vivek
Sinha
a,
Manuela
Weber
b,
Stefano
Canossa
c,
Evgeny A.
Pidko
*a and
Georgy A.
Filonenko
*a
a,
Christophe
Rebreyend¶
a,
Vivek
Sinha
a,
Manuela
Weber
b,
Stefano
Canossa
c,
Evgeny A.
Pidko
*a and
Georgy A.
Filonenko
*a
aInorganic Systems Engineering Group, Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, Van der Maasweg 9, 2629 HZ, Delft, The Netherlands. E-mail: e.a.pidko@tudelft.nl; g.a.filonenko@tudelft.nl
bFreie Universität Berlin, Institut für Chemie und Biochemie, Anorganische Chemie, Fabreckstrasse 34/36, D-14195 Berlin, Dahlem, Germany
cMax Planck Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany
First published on 21st March 2022
Abstract
Activation of homogeneous catalysts is an important step in ensuring efficient operation of any catalytic system as a whole. For the majority of pincer catalysts, the activation step leans heavily on the metal ligand cooperative chemistry that allows these complexes to react with small molecule substrates and engage in catalytic transformations. While the majority of such catalysts require a single activation event to become cooperative, herein we report an exception to this trend. Specifically, we demonstrate that a Ru-PN3P aminopyridine pincer catalyst, which lacks conventional reactivity with hydrogen upon typical one-fold activation, can exhibit this reactivity when a sequential two-step activation is performed. The resulting anionic complexes readily activate molecular hydrogen and react further with CO2 showing the previously unknown reactivity that is critical for CO2 hydrogenation catalysts. While active in CO2 hydrogenation, Ru-PN3Ps are significantly more efficient in hydrogenation of bicarbonates – a likely consequence of the chemistry of these pincers requiring formation of anionic complexes for hydrogen activation.
Introduction
Chemical conversion of CO2 is a potent strategy for the production of C1 building blocks and small molecules that can store hydrogen and, by extension, chemical energy.1 Among others, formic acid (FA) was highlighted as a C1 hydrogen storage medium2,3 due to the ease of its dehydrogenation – a reaction allowing stored hydrogen to be recovered rapidly. Production of formic acid from CO2 is a catalytic hydrogenation process, and late transition metal complexes (Fig. 1) have been the most common and potent catalysts for CO2 hydrogenation; among the rich state of the art, iridium and ruthenium complexes hold the performance records to date.4–10 Most commonly, formate salts rather than pure formic acid are produced in homogeneous CO2 hydrogenation. One of the earliest examples of a potent catalyst for CO2 hydrogenation was reported by Nozaki and co-workers in 2009 who demonstrated that an iridium complex with a lutidine based PNP-pincer ligand could operate with exceptional turnover frequency (TOF) and turnover number (TON) values, reaching 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and 3500000, respectively (Fig. 1).11 Our group has reported a highly active ruthenium PNP-pincer catalyst for the reversible hydrogenation of CO2 in organic media, with TOF and TON values of 1100000 and 206000, respectively.12,13 We have recently reported the use of this ruthenium PNP-pincer complex for bicarbonate hydrogenation in biphasic water–toluene mixtures.14 Non-symmetrical Ru-PNN pincers have also been reported to produce formates efficiently.15 Finally, a wide range of early transition metal pincers have been developed in the recent decade16,17 with some showing outstanding catalytic activity in FA dehydrogenation.18,19
000 h−1 and 3500000, respectively (Fig. 1).11 Our group has reported a highly active ruthenium PNP-pincer catalyst for the reversible hydrogenation of CO2 in organic media, with TOF and TON values of 1100000 and 206000, respectively.12,13 We have recently reported the use of this ruthenium PNP-pincer complex for bicarbonate hydrogenation in biphasic water–toluene mixtures.14 Non-symmetrical Ru-PNN pincers have also been reported to produce formates efficiently.15 Finally, a wide range of early transition metal pincers have been developed in the recent decade16,17 with some showing outstanding catalytic activity in FA dehydrogenation.18,19
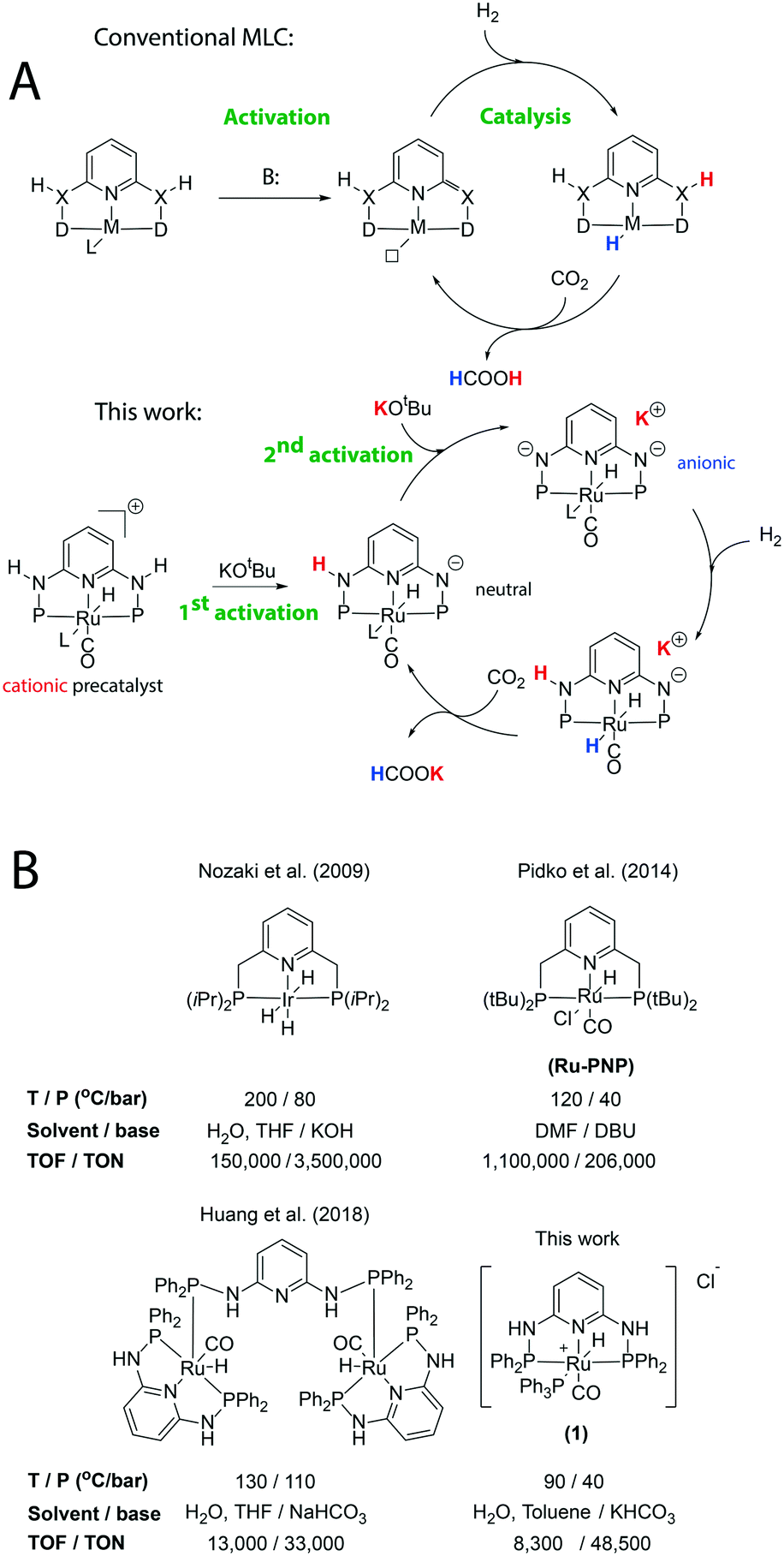 | ||
| Fig. 1 Activation and MLC chemistry of common pincer catalysts in CO2 hydrogenation compared to Ru-PN3P (A) and examples of noble metal pincer catalysts for this process (B). | ||
In this work we focus specifically on pincer catalysts – a class that has heavily influenced the CO2 hydrogenation field. Evolving from the pioneering research that started nearly 30 years ago,20 various pincer ligands are commonplace in several fields of catalysis, especially those involving hydrogen activation and hydride transfer to polar and unsaturated substrates.21–24 One of the common explanations of such utility of pincer complexes in catalysis is the ability of some of them to engage in metal–ligand cooperative (MLC) transformations, wherein both the metal and ligand functional groups participate in substrate binding, activation, or conversion (Fig. 1). While the role and involvement of MLC is often debated and is likely to remain controversial, cooperative chemistry is often involved in catalyst activation and, thus, remains integral to catalysis at large.
Typical pincer ligands are activated by deprotonation of methylene or amine groups to engage in MLC (Fig. 1); however, other pincers are also utilized in Ru-catalysed CO2 hydrogenation. Among these are 2,6-diaminopyridine based pincers, which were extensively explored for early metal catalysis by Kirchner and co-workers.25 Huang and co-workers successfully utilized these ligands in Ru-catalysed CO2 hydrogenation.26 Having the advantage of ease of preparation and elevated stability towards oxidation, these PN3P pincer ligands were reported to form binuclear ruthenium complexes (Fig. 1) while a mononuclear deprotonated Ru-PN3P species, formed during catalyst activation, was suggested to be the active component of the catalytic mixture.26 While the state of the art PNP pincers resemble PN3Ps structurally, the latter lack the same level of mechanistic description. Specifically, Ru-PN3Ps are not known to activate molecular hydrogen and show no stoichiometric reactivity with carbon dioxide, posing a question of which active species operate in catalysis and which precatalyst transformations furnish them.
We aimed to uncover the origin of the catalytic activity of Ru aminophosphine pincers. Having investigated their stoichiometric reactivity, we have discovered an unusual chemistry of these pincers originating from the high acidity of the PN3P ligand and its ability to readily undergo multiple deprotonations. We demonstrated that Ru-PN3Ps can engage in MLC transformations immediately relevant for CO2 hydrogenation; they can split H2 heterolytically and engage in hydride transfer reactions. However, they require a two-fold consecutive activation for this chemistry to manifest.
Results and discussion
In order to study the activation and catalytic behaviour of Ru-PN3P pincers we aimed to isolate the representative mononuclear complexes. Although complexation of the N,N′-bis(diphenylphosphino)-2,6-diaminopyridine ligand (Fig. 2, PN3P, L1) was reported to yield dinuclear complexes upon reaction with RuHCl(CO)(PPh3)3, we consistently observed the formation of mononuclear species 1 upon complexation, marking a difference from previous reports25 (Fig. 2). The reaction of ligand L1 with the ruthenium precursor in benzene at 80 °C led to the formation of this cationic complex sparingly soluble in benzene. The 1H NMR spectrum of 1 features hydride resonance as a doublet of triplets (δ = 7.41 ppm, 2JHP = 88.7 Hz, 22.4 Hz in DMSO-d6) with a coupling pattern consistent with the presence of two equivalent phosphine donors of the PN3P ligand and a PPh3 ligand bound trans to the hydride as shown in Fig. 2. The IR spectrum of 1 in the solid state features a strong band at 1932 cm−1 confirming the retention of the carbonyl ligand, and the suggested structure of 1 was further confirmed using single crystal X-ray diffraction (Fig. 2). The cationic complex 1 adopts a distorted octahedral geometry in the solid state with a chloride counterion located in the outer sphere of the complex. The presence of a PPh3 ligand in 1 resulted in the breaking of the molecular symmetry along the pincer ligand plane that was reflected in the 1H NMR spectra as inequivalency of the phenyl rings of the pincer ligand that produce two distinct sets of resonances (see S1 in the ESI‡). | ||
| Fig. 2 (A) Synthesis and single deprotonation of complex 1; (B) solid state structures of complexes 1 and 2. See S4 in the ESI‡ for details. | ||
The Ru-PN3P complex 1 is an active CO2 hydrogenation catalyst. It can operate in a fully organic medium but remains inferior to the reference Ru-PNP catalyst (Table 1, entries 1 and 2). Specifically, in a DMF solvent containing a DBU base promoter under 40 bar pressure of an equimolar H2/CO2 gas mixture, 1 provides a 68% formate yield compared to the 93% produced by the reference RuPNP complex. Aiming to improve the performance of 1, we investigated its activation and stoichiometric reactivity.
| Catalysta | Solvent | Base | P(H2:CO2) |
TON | Yield |
|---|---|---|---|---|---|
|
a RuPNP: 0.107 μmol, 1:0.13 μmol. Conditions: 2 mL DMF or 2 mL of 1:1 mixture with water; base: 5 mmol KHCO3 or 2.23 mmol DBU, reaction time – 16 h at 90 °C and indicated pressure.
|
|||||
| RuPNP | DMF | DBU | 20:20 |
19400 |
93 |
| 1 | DMF | DBU | 20:20 |
11800 |
68 |
| 1 | DMF | DBU | 39:1 |
13000 |
75 |
| RuPNP | H2O/toluene | KHCO3 | 40:0 |
27700 |
66 |
| 1 | H2O/toluene | KHCO3 | 40:0 |
25800 |
71 |
| 1 | H2O/THF | KHCO3 | 40:0 |
21600 |
60 |
| 1 | H2O/dioxane | KHCO3 | 40:0 |
25800 |
73 |
Reaction with strong bases like KOtBu is the most common way of activating Ru pincers. While Ru-PNP pincers form five-coordinate species upon deprotonation, we expected 1 to form a six-coordinate complex due to the presence of a PPh3 ligand in this complex (Fig. 1A). Indeed, upon reaction with 1 equivalent of KOtBu, complex 1 transforms into a neutral species 2 (Fig. 2) characterized by a marginal shift of the hydride resonances in 1H NMR to 7.78 ppm. The PPh3 ligand in complex 2 is retained as evidenced by the doublet of triplets pattern (2JHP = 92.8 Hz, 22.9 Hz) nearly identical to that of 1 (see S1 in the ESI‡).
Compared to its methylene bridged analogue, complex 1 is significantly more acidic and susceptible to deprotonation. For comparison, the pKa of the Ru-PNP complex is calculated to be 33.9 and a value of 22.1 was calculated for complex 1. The presence of a PPh3 ligand has a marginal effect on the ligand acidity as the pKa values computed for the neutral analogue of 1 were similar (pKa = 23.6 vs. 22.1 for 1, see S3 in the ESI‡). As a result, complex 1 also reacts with weaker non-nucleophilic bases, e.g., DBU, to produce 2 as one of the major products (Fig. S39‡). In addition, complex 1 can also react with aqueous KOH solutions in THF producing singly deprotonated 2, which is stable in the presence of water as long as excess base is present (Fig. S41 and S42‡), indicating the significantly higher acidity of the PN3P ligand compared to that of PNP.
The one-fold activated neutral complex 2 that was previously identified by Huang and co-workers26 who produced it upon deprotonation of a dinuclear Ru complex is shown in Fig. 1. As 2 is similar to the activated Ru-PNP complexes structurally, the same reactivity was expected for this complex and the authors proposed that 2 was an active component of the catalytic cycle in CO2 hydrogenation and would participate in heterolytic H2 splitting. This would produce neutral Ru dihydrido complexes active in the generation of formates upon contact with CO2 in complete similarity to the mechanistic proposals made by Pidko13 and Sanford15 for related Ru-PNP and PNN pincer complexes. Complex 2, however, does not react with hydrogen under pressures up to 3 bar upon prolonged exposure, nor does it react with carbon dioxide under these conditions, marking a difference in reactivity with other Ru pincers for which MLC activation of both H2 and CO2 is well known.
The absence of reactivity with CO2 pointed to the fact that the hydride in 2 might have limited involvement in catalytic CO2 hydrogenation and be a resting or an off-cycle state rather than an active species. To verify this suggestion, we started a search for alternative transformation pathways that would render Ru-PN3Ps active in hydrogenation of CO2. Analysing the computed pKa data for 1, we noted that even relatively weak bases can promote deprotonation of 1 and the high acidity of the remaining NH group protons in PN3P suggests the feasibility of double deprotonation in this complex. Our calculations estimate the pKa for the second deprotonation to be 26.6, thus accessible for alkoxide bases like KOtBu.
Although fairly uncommon, double deprotonation of methylene bridged pincers has been documented in the literature. Studies by Milstein and co-workers have shown that double deprotonation of methylene-bridged PNP Pd(II) and Pt(II) complexes leads to anionic pincers – a rare example considering the lack of strong π-acceptor ligands that lower the electron density at the metal centre.27 Similarly, Ni-PNP pincers were doubly deprotonated and, in this form, exhibited ligand centred addition of CO2 leading to anionic carboxylate complexes.28 Both observations suggest that anionic complexes formed via double deprotonation of PNP ligands might exhibit an unusual ligand-centred reactivity towards CO2, a suggestion recently confirmed by Saouma and co-workers who disclosed a precedent of anionic Mn-PNP pincers engaging in ligand centred reactivity with CO2.29
We found that two-fold deprotonation of 1 with KOtBu base quantitatively converts it to a new complex 3 (Fig. 3A). In tetrahydrofuran, this complex retains the PPh3 ligand which is rapidly replaced in the presence of traces of DMSO or in neat DMSO-d6 solvent. In DMSO, 3 has a triplet resonance in the hydride region of the 1H NMR spectrum significantly shifted upfield to −10.84 ppm, indicative of PPh3 dissociation. IR spectroscopy reveals the presence of a carbonyl ligand in 3 (1910 cm−1), suggesting that 3 is an anionic complex existing in solution as an ion pair with a complex anion charge compensated by an alkali metal cation. Complex 3 does not readily crystallize; however, its DMSO analogue 3DMSO can be obtained in crystalline form. The solid state structure of this complex (Fig. 3B) confirms the presence of a potassium cation arranged close to one of the ligand side arms in the solid state – an arrangement very similar to that reported by Milstein and co-workers for anionic Ni-PNP complexes.28
 | ||
| Fig. 3 (A) Synthesis and interconversion of doubly deprotonated complex 3 and its solid state structure (B); hydride region of the 1H NMR spectra (C) and carbonyl region of the IR spectra (D) for associated complexes. | ||
We found that small amounts of 3 can also be formed in the reaction of 1 with DBU base (Fig. S39‡), suggesting that even relatively weak bases can produce detectable quantities of the doubly deprotonated complex. Surprisingly, unlike the unreactive singly deprotonated 2, complex 3 readily engages in small molecule activation and exhibits MLC behaviour typical for other Ru pincers. In particular, exposure of 3 generated in situ to hydrogen at room temperature leads to the generation of dihydrido species 4 at 3 bar pressure with the loss of the PPh3 ligand. Formation of 4 in THF solvent proceeds at room temperature in the course of several minutes, while in DMSO, hydrogen dissociation over 3DMSO was not observed, likely as a consequence of low H2 solubility and a high concentration of the coordinating solvent. Importantly, under identical conditions, hydrogen addition to Ru-PNP pincers proceeds instantly,30 indicating apparently lower energetic barriers for H2 splitting in this pincer compared to complex 3. We also noted that the presence of a small amount of free butoxide base was necessary to produce complex 4 which did not form when sub-stoichiometric amounts of KOtBu, e.g., 1.95 equivalents compared to 1, were used to generate complex 3. This observation triggered a theoretical examination of Ru-PN3Ps and their reactivity patterns (see section S3 in the ESI‡).
According to DFT calculations, the singly deprotonated complex 2 indeed has a high energetic barrier for H2 dissociation of up to 113 kJ mol−1. Deprotonation of 2 that generates the anionic complex 3 opens the possibility of base-assisted H2 activation that proceeds with a significantly lower barrier. When hydrogen activation is assisted by KOtBu, the computed barrier was found to be 20 kJ mol−1, with a further decrease to 6 kJ mol−1 if the butoxide anion is considered as an assisting base. Surprisingly, direct dissociation of H2 over 3 is computed to proceed with a high barrier of 108 kJ mol−1 which indicates the necessity of base promotion being involved in catalysis. These results confirm our experimental studies indicating no formation of dihydride 4 in the absence of free KOtBu base.
Unlike a singly activated complex 2, dihydride 4 is a competent hydride transfer reagent. Exposure of Ru-PN3P dihydride 4 to CO2 results in a rapid reaction with the carbon dioxide substrate followed by the precipitation of a potassium formate product. Complex 4 upon this transformation is converted into a singly deprotonated complex 2, thus formally closing the catalytic cycle. This reactivity highlights an unusual role of double deprotonation in Ru-PN3P; apart from enabling heterolytic H2 splitting, it allows rapid elimination of the formate product as potassium salt. We observed no formate-bound Ru species in our experiments in contrast to common Ru dihydride pincers that produce formate complexes upon hydride addition to CO2.15,30 This reactivity implies that Ru-PN3P continuously utilizes both basic sites with one of them required for the product liberation. This, in turn, renders complex 2 a likely resting state in catalysis promoted by dihydride 4.
Aiming to confirm that anionic dihydride 4 is the likely active state of the catalyst, we attempted a catalytic turnover using singly activated complex 2 as a starting compound (see S1 in the ESI‡). Under 3 bar pressure of an equimolar H2/CO2 gas mixture, complex 2 requires over 16 hours to produce detectable amounts of formate compared to several seconds when the reaction is performed using dihydride 4.
Building on the reactivity studies indicating that H2 dissociation over Ru-PN3P might be the rate limiting step, we aimed to improve the performance of 1 in catalysis by increasing the H2 partial pressure. In a H2/CO2 atmosphere mixed at a 39/1 ratio, catalyst 1 allows for a 75% yield to be reached, indicating that higher H2 concentrations maintained during catalysis are beneficial for the conversion (Table 1). Considering the anionic nature of complex 3 that is active in H2 activation, we assumed that ionic bicarbonate should be preferred over neutral DBU as a base. In fact, we found a significant improvement in the performance of 1 in a biphasic medium with KHCO3 as the base and reagent. At 90 °C under 40 bar H2 pressure, catalyst 1 provided >25000 turnovers, on a par with the Ru-PNP reference catalyst in various solvent combinations (Table 1).
Considering the low rate of H2 activation observed in the reactivity studies, our further experiments in biphasic medium were aimed at tracking hydrogenation yields under varying H2 pressures. The latter, together with the reaction temperature, were found to strongly affect the formate yields (Fig. 4). At both 65 °C and 90 °C, the increase of H2 pressure from 5 to 40 bar led to a monotonic formate yield increase from ca. 15 to 75%. Notably, this dependence is significantly less pronounced for the RuPNP reference catalyst14 that is known to activate hydrogen via the MLC mechanism directly.20 Finally, to estimate the kinetic performance of 1, we performed scale-up experiments combining the previous insights into its reactivity, namely, the preference for elevated hydrogen pressure and biphasic reaction medium and the necessity to produce the doubly deprotonated species 3 for hydrogen activation. The latter was promoted by increasing the potassium bicarbonate loading two-fold to 10 M which ensures saturation of the aqueous layer with base during catalysis. We obtained a bicarbonate conversion of 68%, corresponding to >48500 turnovers produced with an initial TOF of 8300 h−1 (see S2 in the ESI‡). Compared to dinuclear Ru-PN3P catalysts (Fig. 1), this presents a nearly 50% improvement in the reaction rate and a three-fold improvement in productivity per mol Ru despite the significantly milder conditions used in this study.
 | ||
| Fig. 4 Formate yield in biphasic KHCO3 hydrogenation as a function of the reaction temperature and hydrogen pressure. Conditions: catalyst 1 0.14 μmol, 1 mL water, 1 mL toluene, 5 mmol KHCO3, reaction time – 16 h at the indicated temperature and pressure. | ||
Conclusions
In summary, this work reports on the catalysis and highly unusual metal–ligand cooperative chemistry of diaminopyridine-based Ru pincer catalysts. While the initial survey of their reactivity could lead to their dismissal as non-cooperative, we demonstrate that two-fold consecutive deprotonation can unlock their new bifunctional reactivity. When activated two-fold, they are active in hydrogen dissociation in a fashion similar to conventional pincer complexes. The main consequence of this chemistry is a potential for Ru-PN3Ps to operate outside of conventional CO2 hydrogenation mechanisms. While the latter revolve around the transformations of neutral complexes, our data suggest that Ru-PN3P can operate in a neutral-anionic manifold, where continuous transition between the two states is necessary to engage both substrates of CO2 hydrogenation. Furthermore, the anionic nature of the active dihydride complex leads to a strikingly different mechanism of the formate product liberation that utilizes the basic site at the active catalyst rather than the external base as in the case of common CO2 hydrogenation catalysts. These features render Ru-PN3P one of the few pincer catalysts where metal–ligand cooperation, manifested in an unusual form, appears to be the main driving force behind catalytic performance.Author contributions
A. S. T. and C. R.: experiments and writing; V. S.: pKa analysis; M. W.: X-ray data collection and analysis; S. C.: refinement and analysis of X-ray data; E. A. P.: DFT calculations, resources, supervision, and writing; G. A. F.: project management, writing, and spectroscopy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The use of the national computer facilities in this research was subsidized by NWO Domain Science. The authors acknowledge Nitto Denko Corporation for supporting this work.Notes and references
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 RSC.
- S. Enthaler, ChemSusChem, 2008, 1, 801–804 CrossRef CAS PubMed.
- F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef PubMed.
- R. Kanega, M. Z. Ertem, N. Onishi, D. J. Szalda, E. Fujita and Y. Himeda, Organometallics, 2020, 39, 1519–1531 CrossRef CAS.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- B. Chen, M. Dong, S. Liu, Z. Xie, J. Yang, S. Li, Y. Wang, J. Du, H. Liu and B. Han, ACS Catal., 2020, 10, 8557–8566 CrossRef CAS.
- S. A. Burgess, K. Grubel, A. M. Appel, E. S. Wiedner and J. C. Linehan, Inorg. Chem., 2017, 56, 8580–8589 CrossRef CAS PubMed.
- S. Oldenhof, J. I. van der Vlugt and J. N. H. Reek, Catal. Sci. Technol., 2016, 6, 404–408 RSC.
- Y. M. Badiei, W.-H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS.
- G. A. Filonenko, E. J. M. Hensen and E. A. Pidko, Catal. Sci. Technol., 2014, 4, 3474–3485 RSC.
- C. Rebreyend, E. A. Pidko and G. A. Filonenko, Green Chem., 2021, 23, 8848–8852 RSC.
- C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- F. Bertini, M. Glatz, N. Gorgas, B. Stoger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Chem. Sci., 2017, 8, 5024–5029 RSC.
- A. Z. Spentzos, C. L. Barnes and W. H. Bernskoetter, Inorg. Chem., 2016, 55, 8225–8233 CrossRef CAS PubMed.
- Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- Y. Himeda, in CO2 Hydrogenation Catalysis, V. C. H. Wiley, 2021 Search PubMed.
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- M. L. Clarke and M. B. Widegren, Homogeneous Hydrogenation Non-Precious Catal., 2019, 111–140 Search PubMed.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef CAS PubMed.
- C. Guan, Y. Pan, E. P. L. Ang, J. Hu, C. Yao, M.-H. Huang, H. Li, Z. Lai and K.-W. Huang, Green Chem., 2018, 20, 4201–4205 RSC.
- M. Feller, E. Ben-Ari, M. A. Iron, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, L. Konstantinovski and D. Milstein, Inorg. Chem., 2010, 49, 1615–1625 CrossRef CAS PubMed.
- M. Vogt, O. Rivada-Wheelaghan, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, Organometallics, 2013, 32, 300–308 CrossRef CAS.
- K. Schlenker, E. G. Christensen, A. A. Zhanserkeev, G. R. McDonald, E. L. Yang, K. T. Lutz, R. P. Steele, R. T. Van der Linden and C. T. Saouma, ACS Catal., 2021, 11, 8358–8369 CrossRef CAS.
- G. A. Filonenko, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2013, 3, 2522–2526 CrossRef CAS.
Footnotes |
| † A dataset deposited under DOI: 10.4121/19190237 contains raw spectral data and machine-readable DFT data. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2133066, 2133067, and 2125682. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cy00485b |
| § In memory of Yehoshoa Ben-David. |
| ¶ A. S. T. and C. R. contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2022 |