
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using molecular oxygen and Fe–N/C heterogeneous catalysts to achieve Mukaiyama epoxidations via in situ produced organic peroxy acids and acylperoxy radicals†
Mengjun
Gong
 ,
Yanjun
Guo
,
Daniel
Malko
,
Javier
Rubio-Garcia
,
Jack M.S.
Dawson
,
George J. P.
Britovsek
and
Anthony
Kucernak
*
,
Yanjun
Guo
,
Daniel
Malko
,
Javier
Rubio-Garcia
,
Jack M.S.
Dawson
,
George J. P.
Britovsek
and
Anthony
Kucernak
*
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, London W12 0BZ, UK. E-mail: Anthony@imperial.ac.uk
First published on 24th March 2022
Abstract
Under mild conditions of room temperature and pressure, and using either pure oxygen or air, aldehydes are converted using a heterogeneous Fe–N/C catalyst to produce the corresponding organic peroxy acid and acylperoxy radicals, which forms the epoxide from cyclohexene with high yield (91% for isobutyraldehyde in O2). Real-time monitoring of the rate of oxygen consumption and the electrochemical potential of the Fe–N/C catalyst has been used to study the formation of the peroxy acid and subsequent catalytic epoxidation of cyclohexene. Using isobutyraldehyde, it is shown that the aldehyde and the iron-based carbon catalyst (Fe–N/C) are involved in the rate determining step. Addition of a radical scavenger increases the induction time showing that radicals are initiated by the reaction between the aldehyde and the catalyst. Furthermore, UV-vis spectroscopy with 2,2′-azino-di-(3-ethylbenzthiazoline sulfonic acid) (ABTS) proved the in situ formation of peroxy acid. In the presence of cyclohexene, the peroxy acid leads to the corresponding epoxide with high yield. Monitoring the open circuit potential (OCP) and oxygen flow concurrently follows the production of the peroxy acid. The epoxidation reaction can take place only when the increase in open circuit potential is greater than 0.14 V, suggesting an in situ direct link between the relative oxidative strength of the peroxy acid and the likelihood of epoxidation.
Introduction
Oxidation plays an important role in both electrochemical processes and organic synthesis, with molecular oxygen being one of the most desirable oxidants due to its abundance and sustainability. However, the reaction with molecular oxygen is kinetically sluggish because of the triplet ground state; therefore, either harsh reaction conditions or a catalyst is required to activate oxygen. Several studies involving oxidation of organic compounds with molecular oxygen have been performed including oxidation of alkenes,1,2 allylic oxidation3,4 and epoxidation.5–7 Organic peroxides are produced at the rate of about 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes p.a. with ¼ of the total associated with peroxy carboxylic acids and their esters.8 Peroxy carboxylic acids are used in disinfection, and bleaching, but also as oxidising agents in Bayer–Villiger oxidation and epoxidations.8 Epoxidation is a significant chemical process since epoxides are a versatile organic intermediate in the production of diols such as glycol and 1,3-propanediol as well as epoxy resins.9 Hence, epoxides are used as the starting materials in various industries such as pharmaceuticals and cosmetics etc., which leads to its high demand.10,11 For example, the capacity for ethylene oxide production was 34.5 million tons globally in 2006.12 Industrially, ethylene oxide is prepared by epoxidation with oxygen and silver is used as the catalyst.12 Although this method is well-developed for ethylene oxide, there are two main problems: (1) precious metal use as catalyst; (2) this method is not applicable to other alkenes.
000 tonnes p.a. with ¼ of the total associated with peroxy carboxylic acids and their esters.8 Peroxy carboxylic acids are used in disinfection, and bleaching, but also as oxidising agents in Bayer–Villiger oxidation and epoxidations.8 Epoxidation is a significant chemical process since epoxides are a versatile organic intermediate in the production of diols such as glycol and 1,3-propanediol as well as epoxy resins.9 Hence, epoxides are used as the starting materials in various industries such as pharmaceuticals and cosmetics etc., which leads to its high demand.10,11 For example, the capacity for ethylene oxide production was 34.5 million tons globally in 2006.12 Industrially, ethylene oxide is prepared by epoxidation with oxygen and silver is used as the catalyst.12 Although this method is well-developed for ethylene oxide, there are two main problems: (1) precious metal use as catalyst; (2) this method is not applicable to other alkenes.
In 1983, Groves et al. found that 5,10,15,20-tetraphenyl-21H,23H-porphyrin iron(III) chloride (FeTPPCl) is an efficient catalyst for the epoxidation of cyclohexadiene with molecular oxygen with a the yield of 93%.13 Mukaiyama and co-workers have studied the epoxidation with molecular oxygen using an aldehyde as co-reductant and they showed that not only the Fe(III)-based catalyst14 but also Ni(II)15,16 and Mn(III) complexes17 are efficient catalysts for this epoxidation, leading to the epoxidation reaction with oxygen and aldehyde being known as the Mukaiyama epoxidation. Furthermore, Nam et al. proved that metal cyclams are also suitable catalysts for Mukaiyama epoxidation.18 Several possible mechanisms are proposed, and they all agreed that different oxidising agents are formed during the epoxidation; therefore, the reaction has different pathways.19,20
Although all of the Mukaiyama epoxidations mentioned above have very high yields, the catalyst used is homogenous, which makes the process unfavourable for industrial application due to the difficulties in the separation of the catalyst. Therefore, a heterogeneous catalytic Mukaiyama epoxidation is attractive. In 2004, Brown et al. used a manganese porphyrin-based metal–organic framework as catalyst, but the solvent used was dichloromethane, which is prohibited in industrial use.21 We have proved that a nitrogen doped carbon-based iron catalyst (Fe–N/C), which showed a high reactivity for the oxygen reduction reaction in fuel cells,22 is also able to catalyse the Mukaiyama epoxidation with a high yield (91%) under very moderate conditions of room temperature and pressure.23 In this epoxidation, acetonitrile is used as solvent and the Fe–N/C is produced through a simple synthetic step using low-cost starting materials. We also tried N/C and Co–N/C with a similar structure to Fe–N/C, but the yield of epoxide is only 40% and 59% respectively.23 In other words, the Fe centre plays a very important role in this heterogeneous catalysis reaction. However, the mechanism of this heterogeneous catalytic epoxidation and the extent of side reactions have not been significantly studied, knowledge of which would be useful to further improve the reaction. Therefore, we decided to study this epoxidation further with Fe–N/C, which shows a promising activity for the Mulaiyama epoxidation reaction. In this paper, we determine the rate equation of this heterogeneous epoxidation to understand the rate-limiting step. We also introduce a range of new experimental approaches to study this reaction including the on-line measurement of oxygen consumption and electrochemical approaches to characterise the system in situ. We also prove that the role of the Fe–N/C catalysts is to activate the aldehyde with molecular oxygen and this activation produces peroxy carboxylic acids and acylperoxy radicals, which are powerful oxidants for epoxidation. Hence, catalytic oxidation of the aldehyde with oxygen by Fe–N/C is a promising process for in situ production of peroxy acids.
Experimental
Catalyst preparation
The Fe–N/C catalyst was synthesised similarly as reported by Malko.24 9.00 g (56.9 mmol) of 1,5-diaminonaphthalene (97%, Sigma-Aldrich) and 320 mg of iron(II) chloride tetrahydrrate (≥99.0%, Honeywell) were mixed in 2 L of ethanol (absolute, VWR). 100 mL of 0.394 mol dm−3 ammonium persulfate (≥98%, Sigma-Aldrich) solution in DI water (MilliQ 18.2 MΩ) was added and stirred at room temperature for 24 h. The solvent was removed under reduced pressure and dried in an oven overnight. The dried powder was pyrolysed in an alumina boat in a quartz tube furnace (Carbolite) at 950 °C for 8 h with a heating rate of 7 °C·min−1, under a constant flow of N2 (N5.5, Research Grade, BO). Uncoordinated iron was removed from the catalyst by leaching in 0.5 M H2SO4 (95–98%, VWR) for 16 h under reflux. The sample was filtered and washed with DI water. The dried sample was further pyrolysed at 900 °C for 5 h under the same condition as previously described. Approximately 6 g of Fe–N/C catalyst was collected after this pyrolysis. For the poisoned catalyst, the catalyst was ground manually and immersed into 0.125 M sodium nitrite solution for 15 min. Then the catalyst was washed with DI water for three times to remove the residual solute. Finally, the poisoned catalyst was dried in the oven at 80 °C under vacuum overnight. Full characterisation of the catalyst has been provided in our previous studies.22,23Production of peroxy carboxylic acid and subsequent epoxidation reaction
Detailed methods and setup are given in the ESI† section 1. All experiments were operated at 25 ± 2 °C under a pure oxygen atmosphere unless otherwise stated. Less than 1 g of catalyst per 1 dm3 of solvent (dried acetonitrile) was used for all reactions to make sure the catalyst was saturated, and the mixtures were sonicated to obtain a homogeneous dispersion. The molar ratio of cyclohexene (99%, Sigma Aldrich) and aldehyde was 1:2 unless stated otherwise. Leak test was done before injection of any reactants to make sure that the digital mass flow meter (15 mln min−1 O2, Bronkhorst Flow Meter) can accurately monitor the rate of reaction and the oxygen flow rate was accurately recorded. The reaction was run until the oxygen flow decreased to zero. Aliquots were taken from the reaction mixture by using a syringe filter to remove the heterogeneous catalyst and the filtered solutions were analysed by gas chromatography. More details are displayed in ESI† section 2.
Electrochemical measurement
A solution of tetrabutylammonium tetrafluoroborate in dried acetonitrile (0.1 M) was used as solvent in order to increase the conductivity of the reaction mixture. All other conditions were the same as for the epoxidation reaction. More details are shown in ESI† section 9 and 10. For open circuit potential (OCP) measurements, Ag/AgCl is used as reference electrode and glassy carbon as counter electrode; the OCP was measured using a Gamry Ref 600 Potentiostat/Galvanostat. For cyclic voltammetry measurements, a Rotating Ring Disk Electrode (Pine Instruments, model AFE6R1AU having a mirror polished glassy carbon as disk and rotator model AFMSRCE) was used and the catalyst was deposited on the glassy carbon disk unless otherwise stated. A three-compartment electrochemical glass cell was used; glassy carbon was used as the counter electrode. A potentiostat (Autolab, model PGSTAT20) was used for current and potential control during the cyclic voltammetry measurements and all the potentials were reported against the Fc/Fc+ couple.Safety considerations
Peroxy carboxylic acids may undergo rapid decomposition at high concentrations and temperatures – for instance 40% peracetic acid in acetic acid/water has a self-accelerating decomposition temperature of >60 °C.8 In our experiments we have kept the concentration of aldehyde low (≤0.2 M) to limit the maximum concentration of peroxy carboxylic acid produced.Discussion
1. Kinetic studies
In order to estimate the reaction order for the epoxidation reaction of cyclohexene using isobutyraldehyde as a co-reductant (Scheme 1), the reaction was carried out in the apparatus as displayed in ESI†Fig. 1 at a temperature of 25 °C under pure oxygen (except where noted) at atmospheric pressure. The design of the apparatus allows the instantaneous measurement of the oxygen consumption rate as the flow of oxygen through the digital mass flow meter into the reaction flask. | ||
| Scheme 1 Balanced epoxidation of cyclohexene with molecular oxygen and isobutylaldehyde. | ||
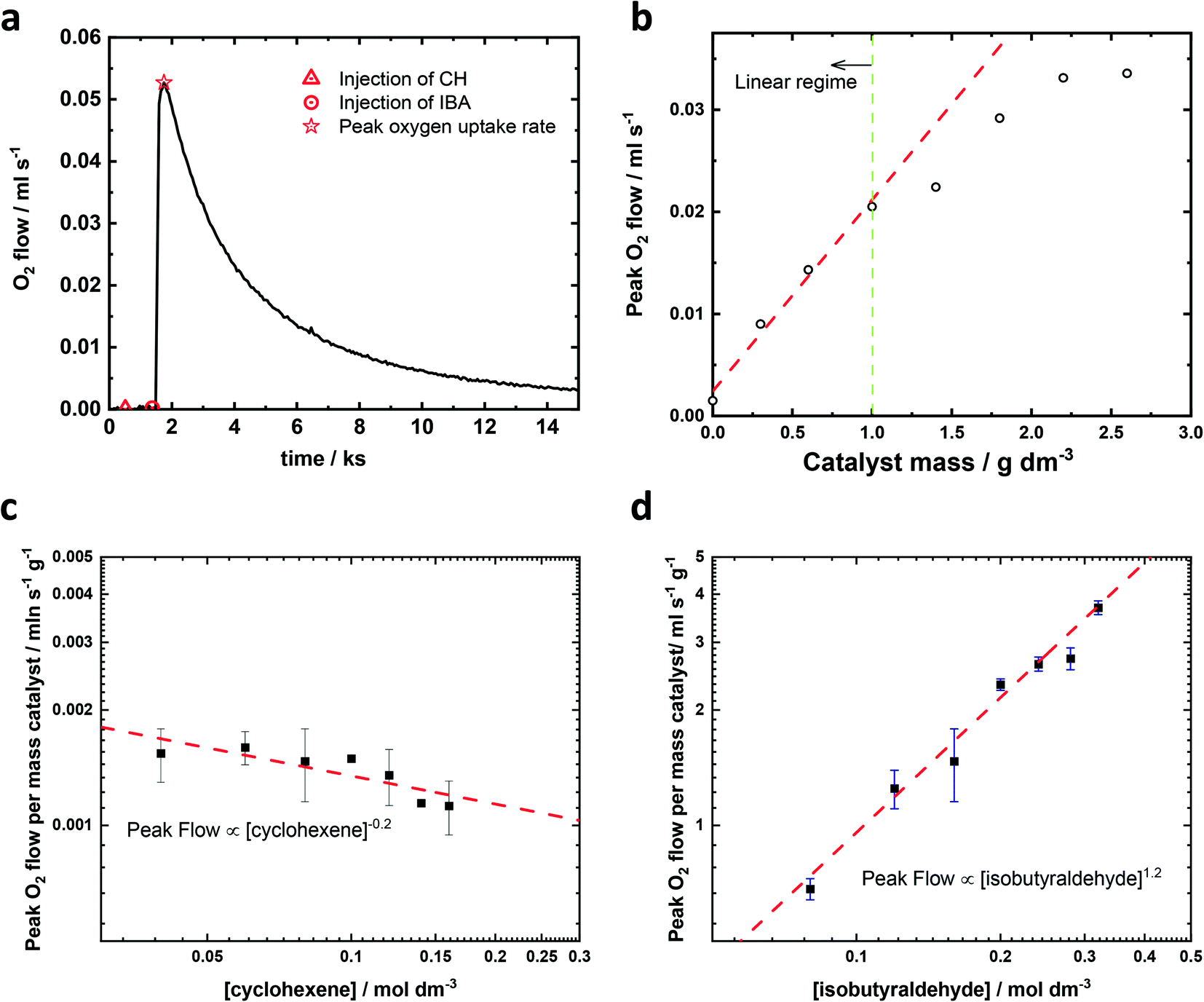 | ||
| Fig. 1 Analysis of kinetics (as measured by peak O2 consumption rate) of the epoxidation of cyclohexene using Fe–N/C catalyst in the presence of isobutyraldehyde (a) oxygen uptake profile during the epoxidation reaction showing the cyclohexene and isobutyraldehyde additions along with the peak oxygen consumption rate; (b) catalyst dosing plot showing the peak oxygen consumption rate versus the amount of catalyst in the reaction mixture; red trendline linear region of catalyst undersaturation; (c) kinetic reaction order plot for cyclohexene with 4 mmol of isobutyraldehyde; (d) kinetic reaction order plot for isobutyraldehyde with 2 mmol of inhibitor-free cyclohexene. All reactions were carried out at 25 ± 2 °C under pure oxygen and 6 mg of Fe–N/C catalyst in 25 cm3 of anhydrous acetonitrile (0.24 gFe–N/C catalyst dm−3acetonitrile) were used for (a), (c) and (d). | ||
A typical oxygen transient is shown in Fig. 1a, in which the oxygen flowrate can be treated as oxygen consumption rate.‡ There is no flow of oxygen into the reaction flask when only catalyst, or catalyst plus cyclohexene are present in the acetonitrile solvent. However, once the aldehyde is added, there is a rapid increase in the flow of oxygen into the reaction flask. This flow (i.e. the rate of oxygen consumption) increases and reaches a peak around 0.5 ks after the injection of the aldehyde, and then decays away with t1/2 ∼2.5 ks, reaching close to zero after 15 ks. In the absence of cyclohexene, the total oxygen consumption is half the number of moles of aldehyde added to the flask, suggesting that the aldehyde reacts with oxygen quantitatively (see ESI† section 3 for further information). Herein, the maximum instantaneous oxygen consumption rate was selected as a proxy for the initial rate of reaction because it represents the maximum rate of reaction as measured by oxygen consumption. The influence of catalyst and reactant concentration on the reaction rate were studied under conditions in which all other variables were kept constant. The catalyst studies, shown in Fig. 1b, reveal that when the amount of catalyst was less than 1 gFe–N/C catalyst dm−3acetonitrile, the catalyst was saturated, i.e. increasing the amount of catalyst increased the reaction rate by the same proportion. At higher amounts of catalyst, the peak oxygen consumption levelled off, which means the reactant transport (probably oxygen) to the catalyst becomes rate limiting, and hence the catalyst is not saturated. All subsequent experiments were performed with the catalyst amount within the linear region (less than 1 gFe–N/C catalyst dm−3acetonitrile).
The peak flow of oxygen during the epoxidation of cyclohexene is 1st order in catalyst (Fig. 1b), −0.2th order in cyclohexene (Fig. 1c), 0th order with respect to the concentration of oxygen (Fig. 3a) and an order of 1.2 was determined for isobutyraldehyde (Fig. 1d). A zero reaction order for cyclohexene and first reaction order with respect to the catalyst have been reported in the epoxidation reaction catalysed by metal porphyrin complexes. For example, studies on the olefin epoxidation with hydrogen peroxide and ferric chloride or iron(III) tetrakis(pentafluorophenyl) porphyrin catalysts also observed that the reaction rate was independent of the concentration of the olefin,25,26 whereas the reaction order of Mn(III) tetraphenylporphyrin chloride (Mn(TPP)Cl) or phthalocyanine iron(III) chloride in the epoxidation of olefins with molecular oxygen was reported to be close to one.27,28 These biomimetic catalysts and haem Cytochrome P-450 could form active metal-oxo species.27 Due to the similarity in structure, Fe–N/C may also produce metal-oxo species during the epoxidation reaction. For the reaction order in aldehyde, a number of previous papers have reported a reaction order for isobutyraldehyde close to one.20,29 However, in the studies reported by Takeuchi,30 Zhou28 and Nam,19 the reaction order for isobutyraldehyde was approximately 3/2. There is no doubt that the larger reaction order in isobutyraldehyde should reveal a more important role it plays in the epoxidation and based on our results, the reaction order of isobutyraldehyde is between 1 and 3/2. Epoxidations under reduced partial pressure of oxygen have also been carried out and the initial rate is the same as the rate under pure oxygen as shown in Fig. 3a. Hence, we can write the observed rate equation associated with the maximum rate of oxygen consumption as shown in eqn (1).
 | (1) |
Eqn (1) indicates that only catalyst and isobutyraldehyde are involved in the rate determining step (RDS); therefore, the aldehyde is activated by the catalyst and the active intermediate epoxidises the alkene in the following step as shown in Scheme 2.
 | ||
| Scheme 2 Possible reaction steps in cyclohexene epoxidation. | ||
Takeuchi has suggested that the non-catalytic oxidation of isobutyraldehyde involves the formation of peroxy acid, which is a well-known epoxidation reagent.30 The epoxidation with peroxy acid is stereospecific; however, in our previous study, the epoxidation of cis-stilbene produced trans-epoxides as a major product, suggesting that another active intermediate is present in this epoxidation reaction.23 Nam and Wentzel proposed that, for a transition metal catalysed epoxidation reaction, the acylperoxy radical (RCOOO˙), which is formed through the oxidation of aldehyde, is a possible oxidising agent for epoxidation.19,20 The nature of the radical based mechanism of this reaction was confirmed by the addition of the radical scavenger butylated hydroxytoluene (BHT). Rodrigues et al. stated that the addition of BHT could efficiently react with radicals formed by the oxidation of aldehyde.31 Upon adding 0.02 wt% of BHT into the reaction mixture, the induction time of the reaction significantly increased to 2 h (details for the epoxidation reaction with a radical inhibitor are shown in ESI† section 4). Hence, increasing the induction time suggests that the epoxidation of cyclohexene with aldehyde is a radical based reaction.
2. Mechanistic studies
As mentioned above, peroxy acid, acylperoxy radicals and Fe-oxo species are three possible intermediates involved in the oxidation of cyclohexene. The Fe-oxo species are formed on the surface of the catalyst and in order to ascertain the potential involvement of Fe-oxo species in this epoxidation reaction, we used NO poisoned Fe–N/C catalyst. Poisoning of the Fe–N/C catalyst by NO is reversible and has been used to count the number of active Fe sites in this catalyst. The interaction with NO (and NO2−) does not irreversibly destroy the active site, but instead deactivates it towards oxygen binding.24,32 In these experiments we used poisoning by exposure to nitrite, which will strongly combine with the iron metal centre in this Fe–N/C catalyst. Using the poisoned catalyst did not decrease the yield of epoxide significantly (more information shown in the ESI† Section 6). This result implies that the main active intermediate is not formed at the iron centre. In other words, an Fe-oxo species is not a key intermediate in this heterogeneous Mukaiyama epoxidation. Cyclohexene is oxidised by the oxidation products generated from isobutyraldehyde and the main role of the catalyst is associated with the initiation of the aldehyde chain reaction to form the oxidising species, the acylperoxy radical and peroxy acid.Although peroxy acids and acylperoxy radicals are difficult to detect using common methods because of their relative low stability, the detection of peroxy acid is possible using ABTS (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt), which forms a green coloured intermediate upon reaction.33,34 As shown in Fig. 2a, the coloured products can be used to determine the amount of peroxy acid generated in situ, due to the oxidation of colourless ABTS to green ABTS+˙, which absorbs at several wavelengths (415 nm, 650 nm, 732 nm and 820 nm) as determined by UV-vis spectroscopy.33 The peak heights showed a linear relationship with the concentration of the peroxy acid, which was calibrated using meta-chloroperoxybenzoic acid (more details shown in ESI† section 5). Because the acylperoxy radical is the starting material for the formation of peroxy acid, this experiment confirms the formation of not only the peroxy acid but also the acylperoxy radicals. The peak height increases with reaction time of epoxidation, suggesting that the concentration of peroxy acid increases during the reaction. As peroxy acids are well-known oxidising agents for epoxidation, the subsequent epoxidation reaction of cyclohexene with peroxy acid is inevitable.35
 | ||
| Fig. 2 Analysis of the reaction intermediate using UV-vis spectra of ABTS doped reaction aliquots. (a) UV-vis spectra of 25-fold diluted samples which contain ABTS, KI and an aliquot taken from the epoxidation of cyclohexene with isobutyraldehyde in acetonitrile at different reaction times; (b) the amount of peroxide generated at different reaction times with and without the addition of cyclohexene according to the peak at 415 nm. All reactions were carried out with 3 mg of Fe–N/C catalyst in 10 mL of anhydrous acetonitrile (0.3 gFe–N/C catalyst dm−3acetonitrile), 1 mmol of inhibitor-free cyclohexene and 2 mmol of isobutyraldehyde under pure oxygen at 25 ± 2 °C. | ||
Fig. 2b shows that the concentration of peroxy acid increases over time for reactions both with and without cyclohexene. In the absence of cyclohexene, the amount of peroxy acid present in the reaction mixture reaches approximately 75% of the initial concentration of isobutyraldehyde. Hence Fe–N/C is an effective catalyst to oxidise isobutyraldehyde into peroxy acid by molecular oxygen. However, with the addition of cyclohexene, the amount of peroxy acid decreases significantly and is generally less than 20% of the initial concentration of isobutyraldehyde. This implies that the concentration of acylperoxy radicals decreases when cyclohexene is added. In the first 11 ks, the total oxygen consumption decreases from 4 mmol to 3 mmol, which suggests that the lower amount of peroxy acid is due to the slower rate of isobutyraldehyde activation, rather than epoxidation with peroxy acid (more information shown in the ESI† section 3). Moreover, the oxygen flow peak decreases significantly with the presence of cyclohexene as shown in the ESI† section 3. Several metal-free nitrogen-doped carbon catalysts have been used for the selective allylic oxidation of cyclohexene with molecular oxygen, and the proposed mechanism suggests that cyclohexene will be attracted to the nitrogen-doped carbon catalyst by π-interaction.36–38 Therefore, the addition of cyclohexene is able to block active sites of the catalyst and reduce the rate of acylperoxy radical formation and hence, the concentration of peroxy acid decreases when cyclohexene is added. The −0.2th order with respect to the concentration of cyclohexene also supports that the addition of cyclohexene will block some active sites which used to activate the isobutyraldehyde.
Based on these results, a proposed mechanism is shown in Scheme 3. In the initiation step, isobutyraldehyde is activated by the Fe–N/C catalyst to form an acyl radical, which is the rate determining step. The radical interacts with oxygen to form the acylperoxy radical. In the absence of cyclohexene, the acylperoxy radical will further interact with isobutyraldehyde to produce a peroxy acid and another acyl radical. The peroxy acid will oxidise the aldehyde to isobutyric acid. For the termination step, one of the possible termination products is α-diketone, produced by the dimerisation of acyl radicals. Due to the instability, the α-diketone will be oxidised to two carboxylic acids by oxygen.39 Hence, isobutyric acid will be the main product in the absence of cyclohexene. When cyclohexene is added, the acylperoxy radical is an oxidising agent to produce the epoxide, which is labelled as radical epoxidation. The epoxidation by acylperoxy radicals will generate an alkyl radical and Russell termination was proven by GC analysis of an aliquot from the epoxidation reaction with isobutyraldehyde where isopropanol and acetone were detected.40 A competitive step for the radical epoxidation is that the peroxy acid is able to oxidise cyclohexene to form the epoxide, which is a molecular epoxidation. The peroxy acid will oxidise another aldehyde to form a carboxylic acid as by-product.
 | ||
| Scheme 3 Possible mechanism for epoxidation of cyclohexene with isobutyraldehyde as a co-reductant [grey: colour shows the prospective termination product]. | ||
It is difficult to determine under the conditions of the reaction (room temperature and pressure), which of the two reaction pathways dominates: either the epoxidation with acylperoxy radical or the epoxidation with peroxy acid. A kinetic study on the rate of epoxide formation might be able to discern between these two pathways because the molecular reaction with peroxy acid tends to be more sluggish than the radical reaction due to the high reactivity of radicals. Isobutyraldehyde, a hindered aldehyde, and n-hexanal, a linear aldehyde, were chosen for the kinetic study since the oxygen uptake behaviour for epoxidation with these two aldehydes was significantly different, as shown in Fig. 3a. It can be seen that the concentration of oxygen does not affect the initial rate, which proves the zero order with respect to oxygen in eqn (1). There was a clear oxygen uptake peak when isobutyraldehyde was added, no matter whether the reaction was carried out under pure oxygen atmosphere or under reduced oxygen partial pressure (20 vol% O2). However, the oxygen uptake was more steady when n-hexanal was used and the oxygen flow profile did not change majorly. The yield of epoxide increased with the concentration of oxygen, which has a greater influence on the epoxidation with isobutyraldehyde. This suggests that the structure of the aldehyde potentially determines the route of epoxidation. The concentration of epoxide at different reaction times is shown in Fig. 3b and the reactant and product distribution with time are displayed in the ESI† section 7. When n-hexanal was used, the rate of cyclohexene oxide formation was much slower compared to when isobutyraldehyde was used. For the reaction with isobutyraldehyde, 3.8 ks were needed to convert 20% of cyclohexene, whereas 12 ks were required for n-hexanal to produce the same amount of epoxide. Hence, this suggests that epoxidation with a hindered aldehyde could potentially be dominated by the radical pathway and with a linear aldehyde the molecular reaction is preferred. However, it is difficult to make any firm conclusions whether these two different routes take place simultaneously or not.
 | ||
| Fig. 3 Analysis of the epoxidation reaction with different aldehydes: (a) oxygen uptake profile during the epoxidation reaction with isobutyraldehyde and n-hexanal under different concentration of oxygen (the molar ratio of cyclohexene to aldehyde is 1:2); (b) the conversion of cyclohexene to cyclohexene epoxide with time under 20 vol% of oxygen (the molar ratio of cyclohexene to aldehyde is 1:2). All reactions were taken at 25 ± 2 °C, 6 mg of Fe–N/C catalyst and 25 cm3 of anhydrous acetonitrile (0.24 gFe–N/C catalyst dm−3acetonitrile). | ||
The benefits of this catalytic epoxidation are that oxygen is a desirable oxidant with high abundance and the co-reductants used are not dangerous or harmful for the environment. Several Mukaiyama epoxidations used dichloromethane as solvent,21,28,40 which is labelled as hazardous in the CHEM21 selection guide.41 The solvent we used is acetonitrile and it is more environmentally friendly than dichloromethane. Furthermore, for similar epoxidations most of the reactions require high temperature or pressure, for example 80 °C and 10 bar of oxygen.37,38,42 Also the selectivity to epoxide is relatively low compared with ours.37,38,42 Since pure oxygen is used, the yield of epoxide is 91% even under moderate room temperature and pressure conditions. Finally, it is a heterogeneous catalyst and it can be easily recycled and reused for at least 5 runs.23 A comparison between different catalysts and our own is displayed in a table in ESI† Section 8. We also tried to use reduced partial pressure of oxygen since the oxygen is a strong oxidising agent and potentially will cause severe fire, and the use of pure oxygen involves extra cost and complexity (Table 1). Therefore, synthetic air (20 vol% O2), was used. However, the yield of epoxide decreased for all aldehydes except n-hexanal, which also suggests the mechanism of epoxidation with n-hexanal is different compared with isobutyraldehyde. When n-hexanal is used, the Russell termination will produce n-pentanal as by product and it can be further activated and promote epoxidation. A better understanding of mechanism will be helpful to find out the most efficient co-reductant under pure oxygen and air conditions.
| Aldehyde | [O2]/vol% | Selectivity of epoxide/% | Conversion of aldehyde/% | Yield/% |
|---|---|---|---|---|
| Conditions: 2 equivalents of aldehyde to cyclohexene, 1 atm O2/air, room temperature in acetonitrile. | ||||
| Isobutyraldehyde | 100 | 99 | 91 | 91 |
| 20 | 37 | 68 | 32 | |
| Butyraldehyde | 100 | 79 | 74 | 69 |
| 20 | 33 | 68 | 24 | |
| n-Hexanal | 100 | 65 | 86 | 50 |
| 20 | 76 | 81 | 40 | |
3. Electrochemical analysis
In order to understand the role of the catalyst and the impact of reactants and products on the epoxidation reaction, we have tried several electrochemical measurements, including open circuit potential and cyclic voltammetry. In these experiments it is necessary to add a supporting electrolyte in order to perform the electrochemical measurements, (0.1 M tetrabutylammonium tetrafluoroborate). We confirmed that adding the electrolyte did not influence the epoxidation reaction since the oxygen uptake profiles were identical.(1) Open circuit potential change in the presence of different aldehydes
Seven aldehydes were used as co-reductant for the epoxidation of cyclohexene while monitoring the open circuit potential (OCP) and the oxygen flow rate at the same time. The experimental setup is shown in Fig. 4a and a high impedance potentiostat was used as a voltmeter to record the open circuit potential while measuring the oxygen flow rate. The graphs of OCP and oxygen flow versus time for epoxidation of cyclohexene with isobutyraldehyde are shown in Fig. 4b and the graphs of epoxidation with other aldehydes are displayed in ESI† section 9. | ||
| Fig. 4 Analysis of open circuit potential (OCP) of epoxidation of cyclohexene using Fe–N/C catalyst in the presence of aldehyde (a) experimental setup of the epoxidation reaction with monitoring the oxygen flow and open circuit potential simultaneously (b) oxygen uptake profile and open circuit potential (OCP) during the epoxidation reaction showing the injection of cyclohexene and isobutyraldehyde; (c) different aldehydes investigated in this section; (d) the relationship between the change in OCP of epoxidation with different aldehydes and oxygen consumption. All experiments were performed in 0.1 M solution of tetrabutylammonium tetrafluoroborate in dried acetonitrile for Fe–N/C catalyst and the ratio of cyclohexene to aldehyde is 1:2; reference electrode: Ag/AgCl; working electrode: glassy carbon. | ||
The OCP increased significantly when isobutyraldehyde was injected, which implies a strong oxidising agent is produced. Compared to the reported different aldehyde reactivities,23 the potential difference on addition of aldehyde also correlates with an increase in oxygen consumption and conversion. A higher OCP value can be interpreted as a more oxidative environment, which is generated by the active intermediate produced. As can be seen in Fig. 4d, the aldehyde which had a greater increase in OCP has a faster reaction rate and greater conversion due to the presence of the oxidising products. Only when the change in OCP is greater than 0.139 V is a significant epoxidation reaction observed. Furthermore, a linear relationship between the change in OCP and the molar ratio of oxygen consumption to aldehyde was observed. Herein, most tested aldehydes are efficient co-reductants, except benzaldehyde and m-CBA. Theoretically, m-CBA should be a good co-reductant because m-CBA will form m-CPBA during autoxidation with oxygen, which is one of the frequently used oxidising agents for epoxidation. Also benzaldehyde is light sensitive and could be easily decomposed to acyl radicals under light.43 However, none of these could be autoxidised with oxygen by the Fe–N/C catalyst. This might be caused by the presence of the α-aromatic unit in the aldehyde because the benzene ring will be strongly attracted to the nitrogen doped carbon catalyst and block active sites of the catalyst.44 However, PPA could be activated by the Fe–N/C catalyst while presenting a benzene ring in the β position, because the aldehyde group could rotate and approach to the active sites even though the benzene ring is attracted to the catalyst surface. Moreover, the change in OCP also suggests that those two co-reductants do not work in this epoxidation. Hence, the OCP measurements can be helpful to determine a suitable co-reductant and could potentially be applied to other oxidation reactions to allow in situ analysis and monitoring of complex catalytic systems.
(2) Cyclic voltammetry of the cyclohexene epoxidation reaction system with isobutylaldehyde as co-reductant
Although the increase in OCP proved the formation of an oxidising species when the aldehyde interacts with oxygen and Fe–N/C catalyst, this epoxidation is still not fully understood. Therefore, several cyclic voltammetric experiments were performed to investigate the impact on the addition of catalyst under conditions which mimic the conditions of the epoxidation reaction. Fig. 5a shows a comparison of cyclic voltammetry under different solution compositions to show how the electrochemical reaction changes with the addition of reactants and possible products. (cyclic voltammetry without catalyst is shown in the ESI† Fig. S8). A schematic graph of the experimental setup is displayed in Fig. 5b. Two different systems were tested: one with deposited Fe–N/C catalyst on the glassy carbon disk electrode in a three-electrode electrochemical cell; the other with dispersed Fe–N/C catalyst in solution, which is more similar to the conditions of the epoxidation reaction as shown in Fig. 4a. This electrochemical approach allows direct probing of the local environment and electrochemistry of the reaction. In the absence of alkene and aldehyde, one moderately reversible peak was seen centred at −1.3 V (vs. Fc), which is associated with the reduction of oxygen to the superoxide anion radical.45 When alkene was included in the solution, this reduction peak hardly changed, suggesting that the rate of reaction of the superoxide anion with the alkene is slow. In contrast, when aldehyde was added to the solution, the re-oxidation peak of superoxide anion disappeared. This indicates that superoxide was removed from the solution by reaction with aldehyde. Electrogenerated superoxide anion radical has been reported as a strong oxidant, which is able to oxidise a primary alcohol to a carboxylic acid and also a secondary alcohol to a ketone.46,47 A possible reaction is shown below: | ||
| Fig. 5 Cyclic voltammetry of epoxidation of cyclohexene with isobutyraldehyde and Fe–N/C catalysts. (a) The CV of systems with different compositions using either a working electrode composed of Fe–N/C catalyst or Fe–N/C catalyst dispersed in solution and a glassy carbon working electrode; (b) the setup of a three-electrode electrochemical cell with dispersed catalyst and deposited catalyst on rotating disk electrode [CH: cyclohexene, IBA: isobutyraldehyde, IBC: isobutyric acid, CO: cyclohexene oxide] all experiments were performed in 0.1 M solution of tetrabutylammonium tetrafluoroborate in dried acetonitrile for Fe–N/C catalyst using a rotating disk electrode setup unless stated. | ||

Conclusion
We have proved that the rate of the epoxidation of cyclohexene with molecular oxygen using a heterogeneous Fe–N/C catalyst at room temperature and pressure is 1st order with respect to the Fe–N/C catalyst, −0.2th order in cyclohexene, 0th order in oxygen, and 1.2th order in isobutyraldehyde. Only Fe–N/C catalyst and isobutyraldehyde are involved in the rate-determining step and the presence of cyclohexene will block the active site for activation of isobutyraldehyde. Also, the formation of peroxy acid and radicals during the activation of isobutyraldehyde are confirmed. We suggest a possible reaction mechanism with two potential oxidising agents: (a) acylperoxy radical; (b) peroxy acid. The structure of the aldehyde is the key factor determining whether the reaction is radical-based or molecular-based. Under room temperature and pressure, the yield of epoxide is 91% when isobutyraldehyde and pure oxygen is used. The yield of epoxide decreased when reduced partial pressure of oxygen is used. Moreover, another improvement, which can be done in the future, is to increase the macroporous and mesoporous area. Although the catalyst is a porous catalyst and has a large surface area, most of the porosity is microporous comprising around 80% of total surface area. Therefore, the accessibility of aldehyde to the active sites will be limited due to size constraints, and diffusional limitations through the tortuous catalyst structure. For further improvements, increasing the mesoporous and macroporous surface area should be performed to make sure the large organic molecule can easily access the active sites. Furthermore, we have used electrochemical measurements to analyse the organic reaction. We found a linear relationship between the change in OCP and the ratio of oxygen consumption to aldehyde and the epoxidation reaction takes place only when the change in OCP is greater than 0.139 V. Aldehydes with an α-phenyl ring are not suitable as co-reductant due to the block of active sites. Finally, cyclic voltammetry results suggest that isobutyraldehyde reacts with superoxide rapidly due to the disappearance of the reduction peak at −1.3 V (vs. Fc).Associated content
The following material is provided in the ESI:† experimental setup use for measurement of oxygen consumption; gas chromatography analysis of products; oxygen consumption transients without cyclohexene; effect of injecting inhibitor while monitoring the OCP; calibration of UV-vis detection of peroxy carboxylic acids with m-CPBA; Epoxidation reaction with nitrite poisoned Fe–N/C catalyst; concentration of reactants and product at different reaction time; OCP measurement with different aldehyde; cyclic voltammetry of epoxidation reaction.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors would like to thank the U.K. Engineering and Physical Sciences research council under project EP/J016454/1 for financial assistance. Data supporting this study and used in the production of the figures in this paper are openly available from Zenodo at https://doi.org/10.5281/zenodo.6384017.References
- D. E. Hamilton, R. S. Drago and A. Zombeck, Mechanistic studies on the cobalt(II) Schiff base catalyzed oxidation of olefins by O2, J. Am. Chem. Soc., 1987, 109(2), 374–379 CrossRef CAS.
- A. Zombeck, D. E. Hamilton and R. S. Drago, Novel catalytic oxidations of terminal olefins by cobalt(II)-Schiff base complexes, J. Am. Chem. Soc., 1982, 104(24), 6782–6784 CrossRef CAS.
- A. Hanyu, Y. Sakurai, S. Fujibayashi, S. Sakaguchi and Y. Ishii, Selective aerobic oxidation of isophorone catalyzed by molybdovanadophosphate supported on carbon (NPMo V/C), Tetrahedron Lett., 1997, 38(32), 5659–5662 CrossRef CAS.
- R.-M. Wang, C.-J. Hao, Y.-P. Wang and S.-B. Li, Amino acid Schiff base complex catalyst for effective oxidation of olefins with molecular oxygen, J. Mol. Catal. A: Chem., 1999, 147(1–2), 173–178 CrossRef CAS.
- R. I. Kureshy, N. H. Khan, S. H. R. Abdi, P. Iyer and A. K. Bhatt, Aerobic, enantioselective epoxidation of non-functionalized olefins catalyzed by Ni(II) chiral Schiff base complexes, J. Mol. Catal. A: Chem., 1998, 130(1–2), 41–50 CrossRef CAS.
- S. Takagi, E. Takahashi, T. K. Miyamoto and Y. Sasaki, A New Iron (III) Porphyrin System for Olefin Epoxidation Catalysts, Chem. Lett., 1986, 15(8), 1275–1278 CrossRef.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Recent advances in transition metal catalyzed oxidation of organic substrates with molecular oxygen, Chem. Rev., 2005, 105(6), 2329–2363 CrossRef CAS PubMed.
- A. Uhl, M. Bitzer, H. Wolf, D. Hermann, S. Gutewort, M. Völkl and I. Nagl, Peroxy Compounds, Organic, Ullmann's Encyclopedia of Industrial Chemistry, 2018, pp. 1–45 Search PubMed.
- G. Sienel, R. Rieth and K. T. Rowbottom, Epoxides, In Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Recent developments on the epoxidation of alkenes using hydrogen peroxide as an oxidant, Green Chem., 2003, 5(1), 1–7 RSC.
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, The Production of Propene Oxide: Catalytic Processes and Recent Developments, Ind. Eng. Chem. Res., 2006, 45(10), 3447–3459 CrossRef CAS.
- S. Rebsdat and D. Mayer, Ethylene Oxide, In Ullmann's Encyclopedia of Industrial Chemistry, 2001 Search PubMed.
- J. T. Groves and T. E. Nemo, Epoxidation reactions catalyzed by iron porphyrins, Oxygen transfer from iodosylbenzene, J. Am. Chem. Soc., 1983, 105(18), 5786–5791 CrossRef CAS.
- P. Mastrorilli and C. F. Nobile, Catalytic activity of a polymerizable tris(β-ketoesterate)iron(III) complex towards the oxidation of organic substrates, Tetrahedron Lett., 1994, 35(24), 4193–4196 CrossRef CAS.
- T. Yamada, T. Takai, O. Rhode and T. Mukaiyama, Highly Efficient Method for Epoxidation of Olefins with Molecular Oxygen and Aldehydes Catalyzed by Nickel(II) Complexes, Chem. Lett., 1991, 20(1), 1–4 CrossRef.
- R. Irie, Y. Ito and T. Katsuki, Catalytic epoxidation with molecular oxygen using nickel complex, Tetrahedron Lett., 1991, 32(47), 6891–6894 CrossRef CAS.
- T. Yamada, K. Imagawa, T. Nagata and T. Mukaiyama, Enantioselective Epoxidation of Unfunctionalized Olefins with Molecular Oxygen and Aldehyde Catalyzed by Optically Active Manganese(III) Complexes, Chem. Lett., 1992, 21(11), 2231–2234 CrossRef.
- W. Nam, R. Ho and J. S. Valentine, Iron-cyclam complexes as catalysts for the epoxidation of olefins by 30% aqueous hydrogen peroxide in acetonitrile and methanol, J. Am. Chem. Soc., 1991, 113(18), 7052–7054 CrossRef CAS.
- W. Nam, H. J. Kim, S. H. Kim, R. Y. Ho and J. S. Valentine, Metal Complex-Catalyzed Epoxidation of Olefins by Dioxygen with Co-Oxidation of Aldehydes, A Mechanistic Study, Inorg. Chem., 1996, 35(4), 1045–1049 CrossRef CAS PubMed.
- B. B. Wentzel, P. A. Gosling, M. C. Feiters and R. J. M. Nolte, Mechanistic studies on the epoxidation of alkenes with molecular oxygen and aldehydes catalysed by transition metal–β-diketonate complexes, J. Chem. Soc., Dalton Trans., 1998,(13), 2241–2246 RSC.
- J. W. Brown, Q. T. Nguyen, T. Otto, N. N. Jarenwattananon, S. Glöggler and L.-S. Bouchard, Epoxidation of alkenes with molecular oxygen catalyzed by a manganese porphyrin-based metal–organic framework, Catal. Commun., 2015, 59, 50–54 CrossRef CAS.
- D. Malko, T. Lopes, E. Symianakis and A. R. Kucernak, The intriguing poison tolerance of non-precious metal oxygen reduction reaction (ORR) catalysts, J. Mater. Chem. A, 2016, 4(1), 142–152 RSC.
- D. Malko, Y. Guo, P. Jones, G. Britovsek and A. Kucernak, Heterogeneous iron containing carbon catalyst (Fe-N/C) for epoxidation with molecular oxygen, J. Catal., 2019, 370, 357–363 CrossRef CAS.
- D. Malko, A. Kucernak and T. Lopes, Performance of Fe-N/C Oxygen Reduction Electrocatalysts toward NO2(−), NO, and NH2OH Electroreduction: From Fundamental Insights into the Active Center to a New Method for Environmental Nitrite Destruction, J. Am. Chem. Soc., 2016, 138(49), 16056–16068 CrossRef CAS PubMed.
- H. Sugimoto, L. Spencer and D. T. Sawyer, Ferric chloride-catalyzed activation of hydrogen peroxide for the demethylation of N,N-dimethylaniline, the epoxidation of olefins, and the oxidative cleavage of vicinal diols in acetonitrile: a reaction mimic for cytochrome P-450, Proc. Natl. Acad. Sci. U. S. A., 1987, 84(7), 1731–1733 CrossRef CAS PubMed.
- N. A. Stephenson and A. T. Bell, Mechanistic insights into iron porphyrin-catalyzed olefin epoxidation by hydrogen peroxide: Factors controlling activity and selectivity, J. Mol. Catal. A: Chem., 2007, 275(1–2), 54–62 CrossRef CAS.
- N. Safari and F. Bahadoran, Cytochrome P-450 model reactions: a kinetic study of epoxidation of alkenes by iron phthalocyanine, J. Mol. Catal. A: Chem., 2001, 171(1–2), 115–121 CrossRef CAS.
- X. Zhou and H. Ji, Biomimetic kinetics and mechanism of cyclohexene epoxidation catalyzed by metalloporphyrins, Chem. Eng. J., 2010, 156(2), 411–417 CrossRef CAS.
- G. Emig, T. Haeberle, W. Höss and O. Watzenberger, Kinetic analysis of the oxidation of isobutyraldehyde in liquid phase, Chem. Eng. Technol., 1988, 11(1), 120–126 CrossRef.
- T. Takeuchi, T. Cho, H. Yanai and T. Osa, Non-Catalyzed Oxidation of Aldehydes in Benzene, J. Jpn. Pet. Inst., 1977, 20(3), 243–248 CrossRef CAS.
- C. A. B. Rodrigues, M. N. de Matos, B. M. H. Guerreiro, A. M. L. Gonçalves, C. C. Romão and C. A. M. Afonso, Water as efficient medium for mild decarbonylation of tertiary aldehydes, Tetrahedron Lett., 2011, 52(22), 2803–2807 CrossRef CAS.
- D. Malko, A. Kucernak and T. Lopes, In situ electrochemical quantification of active sites in Fe-N/C non-precious metal catalysts, Nat. Commun., 2016, 7, 13285 CrossRef CAS PubMed.
- U. Pinkernell, H.-J. Lüke and U. Karst, Selective Photometric Determination of Peroxycarboxylic Acids in the Presence of Hydrogen Peroxide, Analyst, 1997, 122(6), 567–571 RSC.
- A. El-Agamey and D. J. McGarvey, Acyl/aroylperoxyl radicals: a comparative study of the reactivity of peroxyl radicals resulting from the α-cleavage of ketones, Phys. Chem. Chem. Phys., 2002, 4(9), 1611–1617 RSC.
- T. W. Findley, D. Swern and J. T. Scanlan, Epoxidation of Unsaturated Fatty Materials with Peracetic Acid in Glacial Acetic Acid Solution, J. Am. Chem. Soc., 1945, 67(3), 412–414 CrossRef CAS.
- X.-H. Li, X. Wang and M. Antonietti, Solvent-Free and Metal-Free Oxidation of Toluene Using O2 and g-C3N4 with Nanopores: Nanostructure Boosts the Catalytic Selectivity, ACS Catal., 2012, 2(10), 2082–2086 CrossRef CAS.
- Y. Cao, H. Yu, F. Peng and H. Wang, Selective Allylic Oxidation of Cyclohexene Catalyzed by Nitrogen-Doped Carbon Nanotubes, ACS Catal., 2014, 4(5), 1617–1625 CrossRef CAS.
- I. M. Denekamp, M. Antens, T. K. Slot and G. Rothenberg, Selective Catalytic Oxidation of Cyclohexene with Molecular Oxygen: Radical Versus Nonradical Pathways, ChemCatChem, 2018, 10(5), 1035–1041 CrossRef CAS PubMed.
- G. E. Gream, J. C. Paice and C. C. R. Ramsay, Photochemistry of α-diketones. II. Some photo-oxidation reactions, Aust. J. Chem., 1969, 22(6), 1229–1247 CrossRef CAS.
- B. B. Wentzel, P. L. Alsters, M. C. Feiters and R. J. Nolte, Mechanistic studies on the Mukaiyama epoxidation, J. Org. Chem., 2004, 69(10), 3453–3464 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, CHEM21 selection guide of classical- and less classical-solvents, Green Chem., 2016, 18(1), 288–296 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Tools and techniques for solvent selection: green solvent selection guides, Sustainable Chem. Processes, 2016, 4(7), 1–24 Search PubMed.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chemistry of Acyl Radicals, Chem. Rev., 1999, 99(8), 1991–2070 CrossRef CAS PubMed.
- Y. Cao, H. Yu, H. Wang and F. Peng, Solvent effect on the allylic oxidation of cyclohexene catalyzed by nitrogen doped carbon nanotubes, Catal. Commun., 2017, 88, 99–103 CrossRef CAS.
- P. S. Singh and D. H. Evans, Study of the electrochemical reduction of dioxygen in acetonitrile in the presence of weak acids, J. Phys. Chem. B, 2006, 110(1), 637–644 CrossRef CAS PubMed.
- M. Singh and R. A. Misra, Electrogenerated Superoxide Initiated Oxidation with Oxygen: A Convenient Method for the Conversion of Secondary Alcohols to Ketones, Synthesis, 1989, 1989(05), 403–404 CrossRef.
- M. Singh, K. N. Singh, S. Dwivedi and R. A. Misra, Superoxide (O2-·) - Initiated Oxidation of Primary Alcohols to Carboxylic Acids, Synthesis, 1991, 1991(04), 291–293 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00356b |
| ‡ During the measurements, the total oxygen consumption was 4-fold the total free space in the flask indicating that no gases were significantly produced during the course of the reaction. |
| This journal is © The Royal Society of Chemistry 2022 |