
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sustainable route for the synthesis of alkyl arylacetates via halogen and base free carbonylation of benzyl acetates†
Roberto
Sole
 *ab,
Sofia
Toldo
a,
Marco
Bortoluzzi
a and
Valentina
Beghetto
ab
*ab,
Sofia
Toldo
a,
Marco
Bortoluzzi
a and
Valentina
Beghetto
ab
aDipartimento di Scienze Molecolari e Nanosistemi, Università Ca' Foscari Venezia, Via Torino 155, 30172 (VE), Italy. E-mail: roberto.sole@unive.it
bConsorzio Interuniversitario per le Reattività Chimiche e la Catalisi (CIRCC), via C. Ulpiani 27, 70126 Bari, Italy
First published on 23rd May 2022
Abstract
The unprecedented palladium catalysed carbonylation of benzyl acetate derivatives herein presented allowed a broad library of alkyl arylacetates in excellent yields to be obtained. Notably, no use of halogen or base additives was required to perform the reaction. Furthermore, all reactions were accomplished at carbon monoxide ambient pressure, increasing the sustainable feature of the protocol reported. The binary system palladium acetate (2 mol%)/DPPF (4 mol%) (DPPF = 1,1′-bis(diphenylphosphino)ferrocene) was found to be the best performing catalyst at 130 °C and 18 hours for different carbonylative transformations, such as alkoxycarbonylation in the presence of several alcohols and hydroxycarbonylation of benzyl acetates. The synthesis of the substrates was achieved by simply treating benzyl alcohol with cheap, safe and non-toxic isopropenyl acetate (IPAc). IPAc is an emerging acetylating agent which allows acetates under mild reaction conditions to be obtained, producing only acetone as the by-product. Thus, the whole process is completely halogen free. Finally, a set of control experiments and DFT studies led to the identification of a plausible reaction pathway.
Introduction
In the context of a green transition towards a modern society with minimal environmental impact, the appeal of new solutions enhancing sustainable synthesis of fine chemicals is rapidly increasing.1,2 In this scenario, alkyl arylacetates and their derivatives are a prominent example, as they play a pivotal role in the framework of pharmaceuticals, agrochemicals and valuable building blocks for fine chemicals production (Fig. 1). These are generally prepared via a multi-step synthesis starting from benzyl halides and making use of harmful and toxic reagents such as sodium cyanide and strong acids (Scheme 1a). Thus, environmentally-friendly alternatives are required to tackle the synthesis of such valuable compounds in a more sustainable way.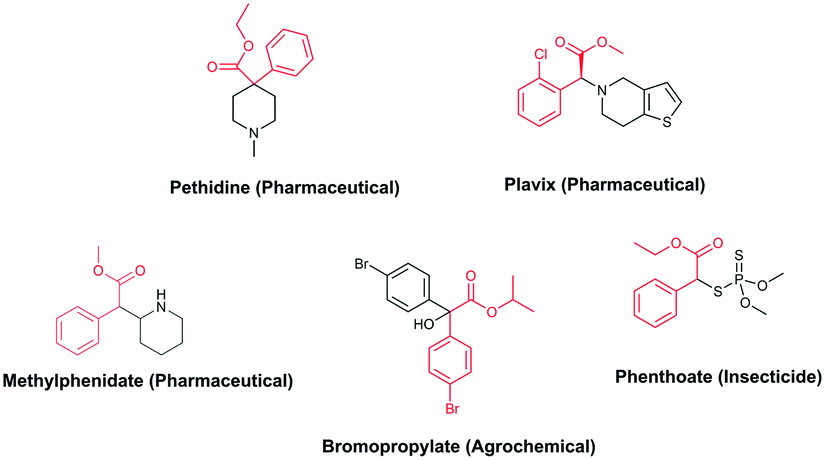 | ||
| Fig. 1 Fine chemicals containing the alkyl arylacetate moiety employed as pharmaceuticals, an agrochemical and an insecticide. | ||
 | ||
| Scheme 1 Known procedures for the synthesis of alkyl arylacetates (a–c), the oxidative addition step (d) and the target reaction of this work (e). | ||
In this light, the peculiar properties of carbon monoxide suggest the possibility of employing this C1-building block for the carbonylation of a variety of organic substrates by insertion of CO into σ-bonds.3 Since the pioneering report of Heck and co-workers in 1974,4 palladium catalysed alkoxycarbonylation of benzyl halides has been recognised as an efficient option to carry out the synthesis of alkyl arylacetates (Scheme 1b).5 Generally, the reaction takes place in the presence of stoichiometric amounts of a base necessary to neutralize the acid formed during the reaction, at temperatures between 60 and 140 °C and 5–60 bar of CO pressure.6
One might say that benzyl halides are industrially produced from the corresponding benzyl alcohols, hence an ideal way to optimize this process, in terms of economic and environmental impact, would be the direct use of benzyl alcohols. Few studies were carried out in the past,7 but several drawbacks related to limited substrate scope, low product yields and hazardous reagents used limited the upgrade of these protocols. In fact, most of the procedures required the use of halogen acids (HCl, HI, HBr) or halogen containing salts (KI, KBr) to in situ convert benzyl alcohols into the corresponding benzyl halides (Scheme 1c), because the oxidative addition of a C(sp3)–OH group to Pd(0) species is difficult (Scheme 1d, left part).5c,8 Therefore, it is generally accepted that alcohols must be activated to promote the oxidative addition to palladium (Scheme 1d, right part).
In view of sustainability, the dependence on strong mineral acids causes several concerns related to the generation of massive amounts of waste. Furthermore, corrosion of plants is also considered a major drawback when strong acids are employed.9
Beyond the use of benzyl halides or halogen additives, further sustainable alternatives could be implemented for the synthesis of alkyl arylacetates. Specifically, we were interested in exploring the possibility of exploiting cheap and easily accessible activated benzyl alcohols in order to reduce the load of additives employed.
To this aim, we reckoned interesting the selection of benzyl acetates as a class of activated benzyl alcohols, readily available, economically sustainable and easily prepared at the laboratory or industrial scale.9 In this framework, isopropenyl acetate (IPAc) recently emerged as a non-toxic, cheap and commercially available compound able to acetylate alcohols10 and amines,11 providing the desired acetylated derivative and acetone as the by-product, due to tautomerization of propen-2-ol. Thus, no halogen salt or strong mineral acid is involved.
Surprisingly, the carbonylation of benzyl acetates has never been reported in the past. This lack of reports could derive from the fact that intermediate acetato-complexes of palladium often undergo a back reaction to give starting acetates, especially in the presence of bases,12 thus reducing the possibility of achieving high selectivity and yield towards the carbonylated products. As a matter of fact, in the early 1980s Yamamoto and co-workers reported a complex prepared from Pd(PCy3)2 (Cy = cyclohexyl) and allyl acetate which, upon addition of CO, gave back allyl acetate.13
Later, Murahashi and co-workers reported the carbonylation of allyl acetates although in the presence of NaBr. The reactions proceeded via formation of π-allylpalladium complexes, which allow facile CO insertion into the Pd–allyl bond.12a
Summing up, given our interest in palladium catalysed carbonylation reactions14 and sustainability,15 we realized that the identification of a halogen-free synthetic strategy for the synthesis of alkyl arylacetates from benzyl acetates could represent a valuable alternative to industrial practices adopted to date. With this in mind, the unprecedented carbonylation of benzyl acetates is herein discussed (Scheme 1e).
Results and discussion
Carbonylation of benzyl acetate
To test the feasibility of the reaction and the role of the acetate group, the carbonylation of benzyl acetate (BzAc) was initially chosen as the target reaction. Thus, a preliminary set of experiments was carried out to optimize the reaction conditions in terms of palladium precursor, phosphine ligand, CO pressure, temperature and reaction times (Table 1). The selectivity towards methyl phenylacetate (MeAcPh) was calculated versus the formation of benzyl alcohol (BzOH) which may be obtained by transesterification of BzAc with MeOH in the presence of acetic acid.16 We started by selecting the conditions generally adopted in the literature for the carbonylation of benzyl halides,5–6i.e. PCO = 20 bar, T = 135 °C and a reaction time of 18 hours. Preliminarily, an experiment carried out without Pd allowed the assessment that the reaction does not proceed in the absence of a metal catalyst (entry 1, Table 1). Next, a catalytic system based on Pd(TFA)2 (TFA = trifluoroacetate) (2 mol%) and DPPP (1,3-bis(diphenylphosphino)propane, 4 mol%) was chosen to start evaluating the influence of a palladium catalyst, while maintaining the reaction conditions tested in the previous experiment (entry 2, Table 1). The addition of halogen salts or bases was avoided. Gratifyingly, it was possible to recover methyl phenylacetate albeit with modest yield and selectivity. According to the literature the use of at least 2 equivalents of phosphine is required to form in situ Pd(0) species.17 Experiments reported in the ESI† (Table S1) on the influence of palladium/phosphine ratio confirmed this trend. To the best of our knowledge, only one paper concerning the halogen and base free methoxycarbonylation of C(sp3)–OR bonds is present in the literature. In 1995 Yamamoto reported halogen and base free carbonylation of allyl formates to yield β,γ-unsaturated carboxylic acids.18 In this case a π-allylpalladium complex facilitated the CO insertion in the Pd–allyl bond. Encouraged by this result, we established that further experiments could proceed in the absence of any additive. Therefore, different palladium precursors were screened in order to reach higher conversion of BzAc and higher selectivity towards MeAcPh (entries 3–7, Table 1). The use of bis(acetonitrile)dichloropalladium (entry 3, Table 1) led to very unsatisfactory conversions of BzAc, similarly to dichloro(1,5-cyclooctadiene)palladium (entry 4, Table 1) and palladium chloride (entry 5, Table 1). Tris(dibenzylideneacetone)dipalladium (entry 6, Table 1) was also tested, as it was revealed to perform well for the carbonylation of benzylamines at low CO pressure.19 However, minimal conversion of BzAc was observed. Finally, the combination of palladium acetate with DPPP showed the best performance, since 92% conversion of BzAc and 70% yield MeAcPh (entry 7, Table 1) were achieved. Thus, PdAc2 was selected as a palladium precursor and different phosphine ligands were evaluated (see Fig. 2) in order to increase the selectivity towards MeAcPh.|
|
|||||
|---|---|---|---|---|---|
| Entrya | [Pd] | Ligand | Conv.b (%) | Yield MeAcPhb (%) | Yield BzOHb (%) |
| Reaction conditions: a BzAc = 1 mmol, [Pd] = 2 mol%, ligand = 4 mol%, MeOH = 1 mL, PCO = 20 bar, time = 18 h, T = 130 °C. b Conversion of BzAc, and yields of MeAcPh and BzOH were calculated by GC analysis using mesitylene as an internal standard. c Ligand = 8 mol%. d P CO = 1 bar. e Time = 6 h. f T = 100 °C. g [Pd] = 1 mol%, ligand = 2 mol%. h Addition of Cs2CO3 (2 eq.). | |||||
| 1 | — | — | n.d. | n.d. | n.d. |
| 2 | Pd(TFA)2 | DPPP | 46 | 21 | 25 |
| 3 | Pd(CH3CN)2Cl2 | DPPP | 12 | 2 | 9 |
| 4 | Pd(COD)Cl2 | DPPP | 33 | 5 | 28 |
| 5 | PdCl2 | DPPP | 45 | 12 | 33 |
| 6 | Pd(dba)2 | DPPP | 3 | n.d. | n.d. |
| 7 | PdAc2 | DPPP | 92 | 70 | 22 |
| 8 | PdAc2 | DPPE | 75 | 15 | 60 |
| 9 | PdAc2 | DPPB | 80 | 77 | 3 |
| 10 | PdAc2 | DPPF | 97 | 99 | n.d. |
| 11c | PdAc2 | PPh2PyMe | 3 | n.d. | 3 |
| 12c | PdAc2 | PPh3 | 10 | 3 | n.d. |
| 13 | PdAc2 | DCPE | 22 | 5 | 12 |
| 14d | PdAc2 | DPPF | >99 | 98 | n.d. |
| 15d,e | PdAc2 | DPPF | 60 | 59 | n.d. |
| 16d,f | PdAc2 | DPPF | 3 | n.d. | n.d. |
| 17d,g | PdAc2 | DPPF | 80 | 79 | 1 |
| 18d,h | PdAc2 | DPPF | 99 | 3 | 98 |
| 19 | [Pd(allyl)(DPPF)]ClO4 | DPPF | 99 | 25 | 74 |

 | ||
| Fig. 2 Phosphine ligands evaluated in this study for carbonylation of BzAc. Natural bite angle (β) taken from ref. 16. | ||
Based on the effects of phosphine bite angles observed by Amadio et al. for the oxidative carbonylation of alcohols,20 we screened a set of phosphines covering bite angle values in the 78.1–99.1° range. It is in fact known that when catalysts bearing chelating diphosphine ligands are used, the natural bite angle defined only by the ligand backbone is a key parameter since it may alter considerably the electronic and steric properties and therefore the reactivity of the metal centre, with effects of paramount importance in the reductive elimination step.21
It is generally accepted that wider bite angles promote a rapid reductive elimination step, whilst smaller bite angles usually lead to the formation of stable cis Pd(II) complexes which slow down the reaction.22 Considering DPPP with a bite angle of 86.2°, we initially tested a diphosphine with a lower bite angle, such as bis(diphenylphosphino)ethane (DPPE, βn = 78.1°) (entry 8, Table 1). As expected, the conversion of BzAc decreased while the selectivity shifted to BzOH. On the other hand, increasing the bite angle of the phosphine by using bis(diphenylphosphino)butane (DPPB, βn = 98.7°) allowed the selectivity towards MeAcPh (entry 9, Table 1) to be increased, but not the conversion of BzAc. 1,1′-Bis(diphenylphosphino)ferrocene (DPPF) with a bite angle of 99.1° was found to perform the best resulting in quantitative conversion of BzAc and 97% yield of MeAcPh (entry 10, Table 1). The overall conversion and the selectivity are therefore not linearly related to the bite angle, which probably does not affect only one of the steps of the catalytic cycle. For instance, it is likely that the stability of the in situ formed Pd(0) phosphine complexes plays a role in the competition between carbonylation and hydrolysis.23
For the sake of completeness, monophosphines such as triphenylphosphine (PPh3) and diphenyl-(6-methyl-pyridin-2-yl)phosphine (PPh2PyMe) were also evaluated, since they found large application in carbonylation chemistry as ligands. However, the use of monophosphines was detrimental for the catalyst activity (entries 11 and 12, Table 1). Moreover, highly basic bis(dicyclohexylphosphino)ethane (DCPE, entry 13) gave only 22% conversion of BzOH. Subsequent experiments, performed in the presence of PdAc2/DPPF, aimed to evaluate the influence of CO pressure, time, temperature and catalyst loading. Notably, the quantitative conversion of BzAc was achieved by decreasing PCO up to 1 bar, along with total selectivity towards MeAcPh (entry 14, Table 1). In contrast, when PCO was increased to 30 and 40 bar, an unfavourable effect was observed (see Table S2 in the ESI†). It is likely that CO competes with the substrate in the coordination sphere of palladium, a behaviour which is not unusual in carbonylation chemistry due to the π-acidic nature of CO.5c,8 The reduction of the reaction times to 6 h did not allow full conversions (entry 15, Table 1) to be reached. A similar result was obtained by reducing the temperature to 100 °C (entry 16, Table 1). Reducing the catalyst loading to 1 mol% allowed a 79% yield of MeAcPh (entry 17, Table 1) to be achieved. The carbonylation of BzAc under standard conditions was also performed in the presence of 2 equivalents of Cs2CO3 (entry 18, Table 1) to evaluate if the influence of a base could be beneficial to the reaction. The reaction produced BzOH as the major product (98%) due to the base promoted hydrolysis of the acetate group.
Finally, in accordance with what was stated in the Introduction section, the use of an allylpalladium complex as a precursor was investigated, employing freshly synthesised Pd(η3-C3H5)(DPPF)]ClO4 (see the ESI†). Despite the good activity in terms of conversion, a high yield of BzOH was detected, while only 25% yield of MeAcPh was achieved (entry 19, Table 1).
Summing up, the best performing catalytic system was found to be composed of PdAc2 at 2 mol% and DPPF at 4 mol%. As a matter of fact, Pd(II)/DPPF catalytic systems proved to be efficient in a variety of homogeneous reactions,24 such as cross-coupling reactions,25 aryl halide amination,26 hydroamination of alkynes,27 and Heck reactions.28 It is also worth mentioning that Pd[(η3-C3H5)(cod)]BF4/DPPF was found to be an efficient catalytic system for the alkylation of malonate derivatives with benzyl acetates, as reported by Kuwano and coworkers.29 Furthermore, Gusev et al. demonstrated that Pd(II) complexes combined with DPPF could also be used for ethene methoxycarbonylation.30 Similarly, Beller and co-workers reported the use of DPPF as a stabilizing ligand in the carbonylation of chloroarenes, carried out at 145 °C and 1 bar of CO pressure with [PdCl2(PhCN)2] as a precursor and in the presence of 3 equivalents of Na2CO3 as a base.31
Methoxycarbonylation of benzyl acetates
As far as the synthesis of the substrates is concerned, the most common way to synthesize benzyl acetates is the acid catalysed transesterification reaction of benzyl alcohol or benzyl halides in the presence of acetic acid. However, this procedure presents several drawbacks in terms of hazardous, toxic, and corrosive chemicals used and unsafe reaction conditions. Alternatively, acetylation of hydroxyl groups can be performed using acetyl chloride or acetic anhydride along with stoichiometric amounts of a base.10 Also in this case, issues regarding sustainability are significantly present. To improve the sustainability of the synthesis of alkyl arylacetates, all the acetate substrates reported in this study were prepared by treating the corresponding benzyl alcohol with IPAc and a catalytic amount of Cs2CO3 as a base without the use of any other solvent. All the substrates were prepared in high yields (>90%) and easily purified by removal of the volatiles under reduced pressure. All the products were characterized by means of 1H and 13C NMR spectroscopy (see the ESI†). The excellent yields achieved with all substrates tested demonstrate that the system shows outstanding tolerance to different functional groups. On considering the subsequent carbonylation reactions with optimized reaction conditions in hand, we tested the robustness of the optimized catalytic system towards the methoxycarbonylation of a wide range of substrates (see Table 2). The influence of different groups in the p-position (entries 2–7, Table 2) was evaluated. The presence of electron-withdrawing groups (entries 2–4, Table 2) allowed the corresponding carbonylated products in excellent yields to be obtained. The same trend was displayed by electron-donating groups, since 4-methoxybenzyl acetate (entry 5, Table 2) and 4-methylbenzyl acetate (entry 6, Table 2) were converted to methyl (4-methoxyphenyl)acetate and methyl 2-(2-tolyl)acetate respectively in 95% and 89% isolated yields. Also the presence of the thioether group in the p-position had no influence on the activity of the catalytic system formed by PdAc2/DPPF (entry 7, Table 2). Subsequently, we examined if the presence of more than one group on the aromatic ring could decrease the activity of the system (entry 8, Table 2). Notably, 3,4-dimethoxybenzyl acetate was converted with 95% yield into the corresponding carbonylated product. It is worth mentioning that 3,4-dimethoxybenzyl acetate is obtained by acetylation of 3,4-dimethoxybenzyl alcohol, a natural compound derived from lignin.32 The effect of various groups in meta and ortho-positions was further investigated (entries 9–12, Table 2). While meta substituted benzyl acetates still gave very good conversions in the corresponding carbonylated products, ortho-alkyl benzyl acetates were converted with slightly minor efficiency, probably due to steric effects, which are not significantly present in meta and para isomers.33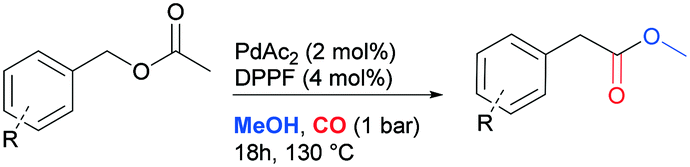




















Alkoxycarbonylation of benzyl acetates
The palladium catalysed alkoxycarbonylation reaction in the presence of higher molecular weight or sterically hindered alcohols may be very difficult to achieve, since steric effects often limit the reaction.12cThus, to further explore the potential of this study, BzAc was carbonylated in the presence of various alcohols (entries 2–6, Table 3). To our delight, the use of ethanol, isopropanol, tert-butanol, and n-butanol afforded the desired alkyl arylacetates in excellent yields.








Notably, even when a diol such as ethylene glycol was used, a 91% yield of 2-hydroxyethyl phenylacetate was achieved (entry 6, Table 3). No trace of the diester was observed by GC-MS and NMR analysis, similarly to what was reported by Sauthier et al. for the alkoxycarbonylation of α-chloro ketones.34
Hydroxycarbonylation of benzyl acetate
Phenylacetic acid plays a pivotal role in the context of fragrances, pharmaceuticals, and polymer synthesis. Several studies reported the carbonylative transformation of aryl halides into phenylacetic acid via palladium catalysed hydrocarbonylation. However, there is no example of an additive-free carbonylation to achieve the synthesis of this key compound. In our study, the carbonylation of BzAc with water as a nucleophile allowed phenylacetic acid in 71% isolated yield (entry 7, Table 3) to be obtained.Control experiments
In order to gain further insights on this new reaction, a set of control experiments was run.First, the formation of acetic acid was evaluated, which should be formed in the final step of the catalytic cycle. To test this, methanol, BzAc and p-chlorobenzyl alcohol (BzClOH) were added into the same reactor at 5 bar of CO pressure.
BzClOH should be initially converted to 4-chlorobenzyl acetate (BzClAc) by the generated acetic acid and then to methyl (4-chlorophenyl)acetate MeAcPhCl by methoxycarbonylation, in a tandem-type reaction (Scheme 2a). To our delight, both MeAcPh and MeAcPhCl were isolated in excellent yields after 18 h. Furthermore, traces of the BzClAc intermediate formed during the reaction were detected by GC-MS analysis (m/z [M–CH3CO2]+ = 125).
 | ||
| Scheme 2 Control experiments. Standard conditions: PdAc2 = 2 mol%, DPPF = 4 mol%, MeOH = 1 mL, 18 h, 130 °C, PCO = 1 bar. a) Tandem carbonylation of BzAc and BzClOH. b) Direct carbonylation of BzOH in the presence of acetic acid. | ||
This experiment calls for testing the direct methoxycarbonylation of benzyl alcohol in the presence of acetic acid as an activating agent, thus avoiding pre-activation of substrates. Gratifyingly, the reaction still led to MeAcPh in high yield (78%) under standard reaction conditions. In this way it was confirmed that acetic acid promoted the carbonylation of benzylic substrates. Furthermore, this method opens up the way to a green, sustainable and efficient method for the carbonylation of benzylic alcohols in the absence of base and halogen sources.
Mechanistic studies
The largest part of mechanistic studies on carbonylation of C(sp3)–X bonds generally refers to the carbonylation of aryl halides.35 Nevertheless, despite the effort of many research groups with long-term expertise in this field, critical steps in the catalytic cycle remain ambiguous and hard to define.According to the literature, carbonylation mechanisms commonly involve well-defined steps, i.e. i) formation of Pd(0) species; ii) oxidative addition of the substrate; iii) insertion of CO into the Pd–substrate bond; iv) interaction of the alcohol with the metal centre; v) reductive elimination of the product with simultaneous regeneration of the catalyst (see Fig. 3.1).
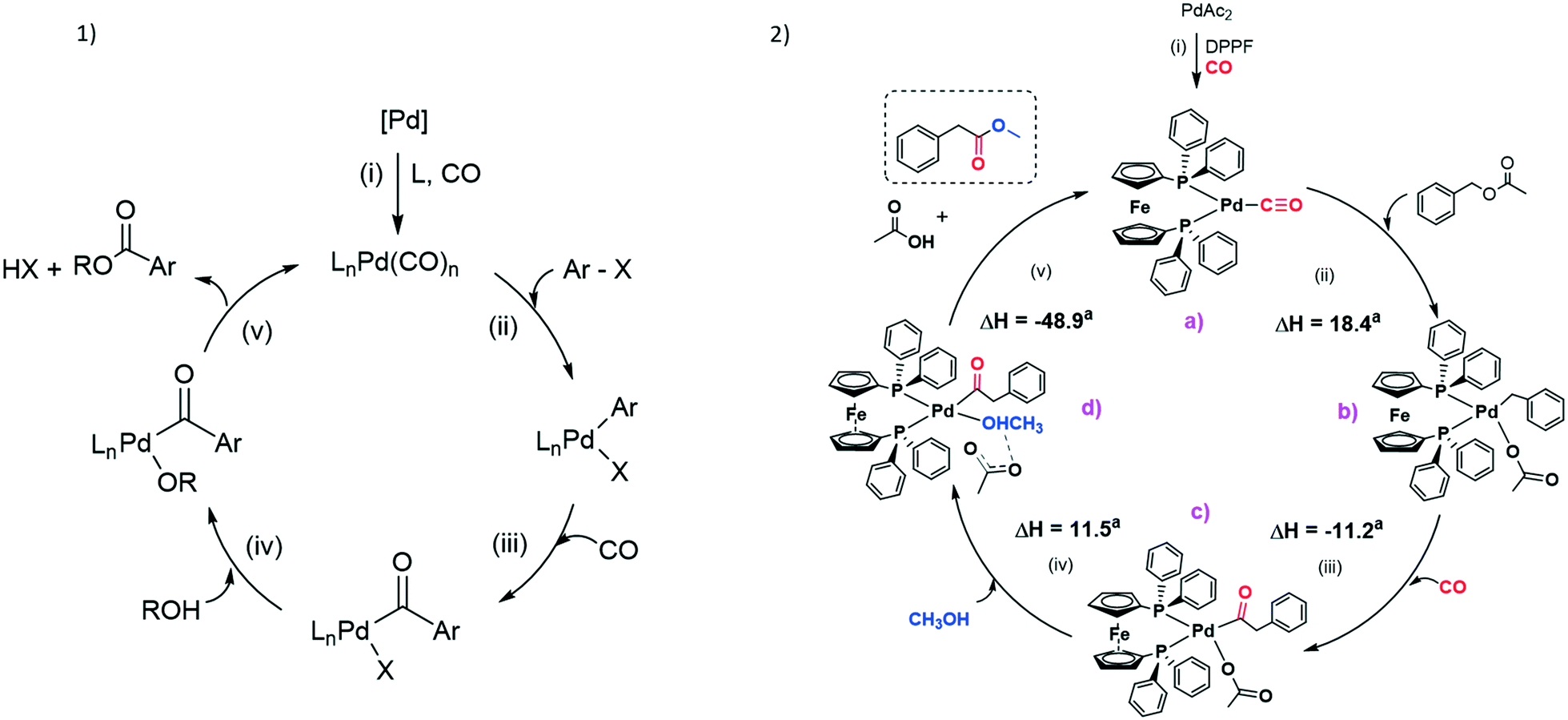 | ||
| Fig. 3 1) General mechanism of Pd-catalysed alkoxycarbonylation of aryl halides (X = Cl, Br, I). 2) Plausible catalytic cycle of the Pd-catalysed methoxycarbonylation of benzyl acetate. See the ESI† (Fig. S6) for DFT optimized geometries. aGibbs energy variations in kcal mol−1. | ||
As far as the formation of Pd(0) complexes is concerned, studies are inclined to accept that carbonyl adducts are likely to be involved in the oxidation step rather than L–Pd(0)–L complexes. To verify this hypothesis, we tried to react Pd(DPPF)2 (ref. 36) with BzAc, but no oxidative addition occurred. A similar result was reported by Amatore et al. trying to react Pd(DPPF)2 with phenyl iodide.37
Next, [PdAc2(DPPF)] was synthesized by replicating a procedure reported by Gusev et al. (see section 3.2 in the ESI†).30 This complex was catalytically active, as MeAcPh was obtained in 68% yield. Addition of an equivalent of DPPF slightly improved the activity of the catalyst, since MeAcPh was provided with 89% yield. These results are of much interest in terms of mechanistic consideration, since they allow confirmation that species with only one DPPF ligand are likely to be more reactive as starting precursors.
Unfortunately, further reaction of Pd(DPPF)2 with CO in the effort to isolate active species failed.
Nevertheless, possible intermediate species in the catalysed methoxycarbonylation of BzAc were investigated by means of DFT calculations. Considering the reaction conditions and the experiments aforementioned, the Pd(0) complex [Pd(CO)(DPPF)] depicted in Fig. 3.2 (a) should be a reasonable catalytic precursor. The reaction [Pd(CO)(DPPF)] + CO → [Pd(CO)2(DPPF)] has computed Gibbs energy variation close to zero (calculated value of −1.4 kcal mol−1, see Fig. S3†). It is likely that the coordinatively saturated dicarbonyl complex is catalytically inactive, therefore the increase of catalytic activity on lowering PCO is probably due to a greater concentration of the tricoordinated monocarbonyl complex in the reaction mixture. Similarly, Quesnel et al. considered the monocarbonyl complex (tBu3P)Pd(CO) as the active Pd(0) complex for the Pd-catalysed carbonylation of aryl chlorides.35d The presence of one carbonyl ligand in the Pd(0) coordination sphere was supported by the high Gibbs energy variation computed for the reaction [Pd(CO)(DPPF)] + MeOH → [Pd(MeOH)(DPPF)] + CO, 25.2 kcal mol−1 (Fig. S3†).
It is likely that the reaction starts with the oxidative addition of BzAc, affording a Pd(II) complex. The complex [Pd(CH2Ph)(Ac)(DPPF)] (Ac = acetate) sketched in Fig. 3.2 (b) is more stable than the alternative possible oxidative addition product, [Pd(CH2Ph)(CO)(DPPF)][Ac]: [Pd(CH2Ph)(Ac)(DPPF)] + CO → [Pd(CH2Ph)(CO)(DPPF)][Ac], ΔG = 13.6 kcal mol−1 (Fig. S4†). The oxidative addition is however associated with an increase of the Gibbs energy variation with respect to the reactants (ΔG = 18.4 kcal mol−1), in part compensated by the subsequent exergonic insertion of CO in the Pd–C bond, with the formation of [Pd(COCH2Ph)(Ac)(DPPF)], (c) in Fig. 3.2 (ΔG of the insertion step −11.2 kcal mol−1). The reaction of [Pd(CH2Ph)(Ac)(DPPF)] with methanol before the CO insertion was also considered (Fig. S4†), but the high Gibbs energy variation of the reaction [Pd(CH2Ph)(Ac)(DPPF)] + MeOH → [Pd(CH2Ph)(OMe)(DPPF)] + HAc, 14.0 kcal mol−1, indicates the CO insertion in the Pd–C bond as a more probable step. The compound [Pd(COCH2Ph)(Ac)(DPPF)] (c) can then react with methanol, and it was found that in the most stable ground-state geometry the MeOH molecule is coordinated to Pd(II) (Pd–OC(O)Me = 2.109 Å) and forms a hydrogen bond with acetate (MeO–H 1.331 Å, H–OC(O)Me 1.098 Å). The formula of the intermediate (d) in Fig. 3.2 is therefore [Pd(COCH2Ph)(MeOH–Ac)(DPPF)]. The complete deprotonation of methanol by acetate to obtain a methoxy-complex and acetic acid is less energetically favourable by about 5.9 kcal mol−1 (Fig. S5†). The reaction of (c) with methanol is associated with a positive Gibbs energy variation (ΔG = 11.5 kcal mol−1), but it is possibly driven by the large excess of methanol in the reaction mixture. It is likely that the weakly coordinated acetate in (d) supports the reductive elimination of methyl phenylacetate with the formation of acetic acid, and the coordination of CO restores the catalytic precursor. The last step of the reaction is accompanied by a strongly negative Gibbs energy variation, ΔG = −48.9 kcal mol−1.
Unfortunately, we were unable to localize the transition steps connecting the optimized intermediate species, therefore the pathway here proposed must be considered with great caution. The high relative Gibbs energies of the intermediates related to the oxidative addition of BzAc and to the coordination of MeOH however suggest that these steps are probably critical for the catalytic activity. The DFT-optimized structures are shown in Fig. S6.† The computed energy profile shown in Fig. S7,† obtained at a lower computational level with respect to the other data reported, indicates the breaking of one of the Pd–P bonds as possibly involved in the initial oxidative addition of BzAc to [Pd(CO)(DPPF)].
The role of the bidentate phosphine in the catalytic cycle deserves further investigation, even if recent computational studies revealed that the electronic influence of DPPF towards Pd centres should be comparable to that of DPPP.38 The variation of catalytic activity on changing the chelating P-donor ligand is therefore probably due mainly to the different bite angle. The localization of (3, −1) bond critical points (b.c.p.) for the optimized DPPF complexes did not highlight particular interactions between the Pd centre and the ferrocenyl moiety. Selected examples are provided in Fig. S8.†
Conclusions
In summary, we herein proposed a sustainable and straightforward method for the synthesis of alkyl arylacetates, a class of key compounds in organic synthesis.Carbonylative transformations of benzyl acetates have never been investigated before. Nevertheless, they have been found to proceed smoothly at atmospheric CO pressure, in the absence of any halogen or base additive, a rare example in carbonylation chemistry. Among the various in situ catalytic systems tested, PdAc2 (2 mol%)/DPPF (4 mol%) displayed the best performance and allowed a plethora of carbonylated products to be obtained showing excellent tolerance towards substituted aryl acetates and different alcohols.
Looking at the whole picture, the syntheses of alkyl arylacetates were carried out from benzyl alcohols avoiding the use of halogen containing salt or strong acids, but employing IPAc as a safe acylating agent. In this framework, it was demonstrated that the direct carbonylation of benzyl alcohol in the presence of acetic acid allowed MeAcPh with 78% yield to be obtained. This method opens up the way to obtain alkyl arylacetates directly from benzyl alcohols, thus avoiding expensive pre-activation steps.
Furthermore, preliminary experimental and computational studies indicated the key role of CO in generating the catalytic active species and suggested a non-innocent role of coordinated acetate in the steps preceding the reductive elimination of the product. Further mechanistic investigations are however currently ongoing to reveal the pivotal role of the acetate group in carbonylation chemistry.
Experimental
General
Commercial solvents (Aldrich) were purified as described in the literature.39 Reagents were commercially available compounds and were used as received unless otherwise stated. PdAc2, Pd(COD)Cl2, PdCl2 and Pd(dba)2 were provided by Sigma Aldrich. Pd(TFA)2 was provided by Strem Chemicals, while Pd(CH3CN)2Cl2 was prepared by following a literature procedure.40DPPP, DPPE, DPPB, PPh3 and DPCE were provided by Sigma Aldrich. DPPF was purchased from Strem Chemicals while PPh2PyMe was synthesized according to the literature.41
Benzyl acetate and p-methyl benzyl acetate were purchased from Sigma Aldrich. All other starting substrates employed for the substrate scope were synthesized (see the ESI†).
High purity CO was obtained from SIAD. The 1H and 13C NMR spectra were recorded on a Bruker AVANCE 300 spectrometer operating at 300.1 and 75.44 MHz, respectively. Gas–liquid chromatography (GLC) analyses were performed on an Agilent 6850 gas chromatograph; gas chromatography-mass spectrometry (GC-MS) analyses were performed on a HP 5890 series II gas chromatograph interfaced to an HP 5971 quadrupole mass detector.
Computational details
Preliminary geometry optimizations were carried out using the PBEh-3c method,42 which is a reparametrized version of PBE0 (with 42% HF exchange) that uses a split-valence double-zeta polarized basis set (def2-mSVP) for light atoms and the def2/J auxiliary basis set. 28 electrons of Pd are included in relativistic ECPs.43 The method adds three corrections that consider dispersion, basis set superposition and other basis set incompleteness effects.44 The C-PCM implicit solvation model was added to PBEh-3c calculations, considering methanol as a continuous medium.45 Calculations were performed with the ORCA 4.0.1.2 software.46 Further calculations were carried out with the Gaussian 16 software,47 using the previously described basis set and C-PCM solvation model in combination with the hybrid PBE1PBE DFT functional.48 The “restricted” approach was used in all the cases. The stationary points were characterized by IR simulation (harmonic approximation), from which zero-point vibrational energies and thermal corrections were obtained. Gibbs free energy values were calculated for T = 403 K and P = 7.8 atm.49 AIM analyses were carried out using MultiWFN, version 3.5.50 Potential energy surface scans were carried out in vacuo with the EDF2 hybrid functional51 in combination with the 6-31G* basis set for light atoms and the LANL2DZ basis set for Pd.52 The software used was Spartan 16.53Cartesian coordinates of the DFT-optimized structures are collected in a separate .xyz file including selected computed data.
Synthesis of acetates
In a 50 mL round bottom flask benzyl alcohol (5 mmol) and Cs2CO3 (2.5 mmol) were added into 10 mL of isopropenyl acetate. The heterogeneous mixture obtained was allowed to stir overnight at 50 °C. Afterwards, the reaction mixture was cooled down to room temperature and solids were removed by filtration. Residues were concentrated at reduced pressure to yield the corresponding acetate, without further purification.Carbonylation experiments
The carbonylation experiments were carried out in a magnetically stirred stainless steel autoclave (total volume 150 ml) connected to a thermostatic bath in order to keep the reaction temperature constant within ±1 °C. The palladium precursor, the ligand and solid substrates were weighed in air in a 4 mL vial, which was then closed with a PTFE/rubber septum pierced with a needle and purged with nitrogen. Afterwards, the solvent, mesitylene and the liquid reagent were added in this order. The vials were then placed in the pre-purged autoclave along with a PTFE support to prevent the vial from moving (see the ESI†). As an example, the experimental procedure for the methoxycarbonylation of benzyl acetate (entry 1, Table 2) is reported. Under an inert atmosphere, in a 4 mL vial equipped with a small magnetic bar Pd(OAc)2 (5 mol%, 0.05 mmol) and DPPF (10 mol%, 0.1 mmol) were introduced. Then, after closing the vial with a PTFE/rubber septum pierced with a needle and purged with nitrogen, MeOH (1 mL), mesitylene (0.5 mmol) and benzyl acetate (1 mmol) were added, obtaining a reddish solution. Afterwards, the vial was placed in a pre-purged 150 mL autoclave and 1 atm of CO was added. The autoclave was then heated at 130 °C and kept under constant magnetic stirring. After 18 h, the autoclave was cooled to room temperature and the residual gas was carefully vented off. The raw reaction mixture was filtered over SiO2 and then analysed by GLC to determine the substrate conversion and product composition. The reaction crudes were purified by flash chromatography (silica gel, 60 Å, 70–230 mesh, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethyl acetate and n-hexane) to give methyl 2-phenylacetate.
:1 mixture of ethyl acetate and n-hexane) to give methyl 2-phenylacetate.
Characterization of products
Author contributions
Roberto Sole: conceptualization; data curation; investigation; supervision; project administration; writing – original draft; writing – review & editing. Sofia Toldo: formal analysis, investigation. Marco Bortoluzzi: conceptualization; investigation; writing – review & editing. Valentina Beghetto: supervision; funding acquisition; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Università Ca′ Foscari and CINECA are gratefully acknowledged for financial support (ADIR 2020) and availability of computing resources (COLUMN21 project 2021). All the authors wish to thank Prof. Alberto Scrivanti for essential discussion and inspiration.References
- (a) European Commission, The European Green Deal, 2019 Search PubMed; (b) European commission, Horizon Europe. Work Programme 2021–2022, 2021 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- T. Nasr Allah, L. Ponsard, E. Nicolas and T. Cantat, Green Chem., 2021, 23, 723–739 RSC.
- A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318–3326 CrossRef CAS.
- (a) X.-F. Fu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 5122–5150 RSC; (b) R. Sang, Y. Hu, R. Razzaq, R. Jackstell, R. Franke and M. Beller, Org. Chem. Front., 2021, 8, 799–811 RSC; (c) L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X. F. Wu, ACS Catal., 2014, 4, 2977–2989 CrossRef CAS.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- (a) G. Cavinato and L. Toniolo, J. Mol. Catal., 1993, 78, 131–142 CrossRef CAS; (b) G. Papadogianakis, L. Maat and R. A. Sheldon, J. Mol. Catal. A: Chem., 1997, 116, 179–190 CrossRef CAS; (c) G. Papadogianakis, L. Maat and R. A. Sheldon, J. Chem. Soc., Chem. Commun., 1994, 2659–2660 RSC; (d) J. Duan, J. Jiang, J. Gong, Q. Fan and D. Jiang, J. Mol. Catal. A: Chem., 2000, 159, 89–96 CrossRef CAS; (e) Y. S. Lin and A. Yamamoto, Tetrahedron Lett., 1997, 38, 3747–3750 CrossRef CAS; (f) S. Sabater, M. Menche, T. Ghosh, S. Krieg, S. L. Ruck, R. Paciello, A. Schafer, P. Comba, A. S. K. Hashmi and T. Shaub, Organometallics, 2020, 39, 870–880 CrossRef CAS; (g) Y. Li, Z. Wang and X.-F. Wu, Green Chem., 2018, 20, 969–972 RSC.
- (a) G. Zanti and D. Peeters, Eur. J. Inorg. Chem., 2009, 3904–3911 CrossRef CAS; (b) Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788–10799 CrossRef CAS PubMed.
- P. Kalck, C. Le Berre and P. Serp, Coord. Chem. Rev., 2020, 402, 213078 CrossRef CAS.
- (a) A. Temperini, L. Minuti, T. Morini, O. Rosati and F. Piazzolla, Tetrahedron Lett., 2017, 58, 4051–4053 CrossRef CAS; (b) D. Rigo, R. Calmanti, A. Perosa and M. Selva, Green Chem., 2020, 22, 5487–5496 RSC.
- R. Pelagalli, I. Chiarotto, M. Feroci and S. Vecchio, Green Chem., 2012, 14, 2251–2255 RSC.
- (a) S. Murahashi, Y. Imada, Y. Taniguchi and S. Higashiura, Tetrahedron Lett., 1988, 29, 4945–4948 CrossRef CAS; (b) S. Murahashi, Y. Imada, Y. Taniguchi and S. Higashiura, J. Org. Chem., 1993, 58, 1538–1545 CrossRef CAS; (c) R. Sole, A. Scrivanti, M. Bertoldini, V. Beghetto and M. Alam, Mol. Catal., 2020, 496, 111179 CrossRef CAS.
- T. Yamamoto, O. Saito and A. Yamamoto, J. Am. Chem. Soc., 1981, 103, 5600–5602 CrossRef CAS.
- (a) R. Sole, A. Scrivanti, Md. M. Alam and V. Beghetto, Appl. Organomet. Chem., 2021, e6391 CAS; (b) R. Sole, M. Bortoluzzi, A. Spannenberg, S. Tin, V. Beghetto and J. G. de Vries, Dalton Trans., 2019, 48, 13580–13588 RSC; (c) A. Scrivanti, R. Sole, M. Bortoluzzi, V. Beghetto, N. Bardella and A. Dolmella, Inorg. Chim. Acta, 2019, 498, 119129 CrossRef CAS.
- (a) V. Beghetto, R. Sole, C. Buranello, M. Al-abkal and M. Facchin, Materials, 2021, 14, 4782 CrossRef CAS PubMed; (b) V. Beghetto, V. Gatto, S. Conca, N. Bardella, C. Buranello, G. Gasparetto and R. Sole, Carbohydr. Polym., 2020, 249, 116810 CrossRef CAS PubMed; (c) V. Beghetto, N. Bardella, R. Samiolo, V. Gatto, S. Conca, R. Sole, G. Molin, A. Gattolin and N. Ongaro, J. Cleaner Prod., 2021, 318, 128627 CrossRef CAS; (d) R. Sole, V. Gatto, S. Conca, N. Bardella, A. Morandini and V. Beghetto, Molecules, 2021, 26, 191 CrossRef CAS PubMed; (e) R. Sole, L. Agostinis, S. Conca, V. Gatto, N. Bardella, A. Morandini, C. Buranello and V. Beghetto, Synthesis, 2021, 53, 1672–1682 CrossRef CAS; (f) V. Ferraro, R. Sole, M. Bortoluzzi and V. Beghetto, Appl. Organomet. Chem., 2021, 1–10 Search PubMed.
- J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS.
- C. Amatore, A. Jutand and A. Thuilliez, Organometallics, 2001, 20, 3241–3249 CrossRef CAS.
- A. Yamamoto, Bull. Chem. Soc. Jpn., 1995, 68, 433–446 CrossRef CAS.
- Y. Li, Z. Wang and X. F. Wu, ACS Catal., 2018, 8, 738–741 CrossRef CAS.
- E. Amadio, Z. Freixa, P. W. N. M. Van Leeuwen and L. Toniolo, Catal. Sci. Technol., 2015, 5, 2856–2864 RSC.
- (a) M. N. Birkholz, Z. Freixa and P. Van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC; (b) P. W. N. M. Van Leeuwen and Z. Freixa, in Modern Carbonylation Methods, ed. L. Kollàr, Wiley-VCH Verlag, Weinheim, Germany, 2008, pp. 1–25 Search PubMed.
- (a) Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1980 Search PubMed; (b) J. E. Marcone and K. J. Moloy, J. Am. Chem. Soc., 1998, 120, 8527 CrossRef CAS; (c) M. Zuideveld, B. H. G. Swennenhuis, M. D. K. Boele, Y. Guari, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz, L. Spek and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 2002, 2308 RSC.
- M. Portnoy and D. Milstein, Organometallics, 1993, 12, 1665 CrossRef CAS.
- T. J. Colacot, Platinum Met. Rev., 2001, 45, 22 CAS.
- (a) M. Ogasawara, K. Yoshida and T. Hayashi, Organometallics, 2000, 19, 1567 CrossRef CAS; (b) B. E. Bosch, I. Brummer, K. Kunz, G. Erker, R. Frolich and S. Kotila, Organometallics, 2000, 19, 1255 CrossRef CAS.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 7217 CrossRef CAS.
- T. E. Muller, M. Berger, M. Grosche, E. Herdtweck and F. P. Schmidtchen, Organometallics, 2001, 20, 4384 CrossRef.
- A. L. Boyes, I. R. Butler and S. C. Quayle, Tetrahedron Lett., 1998, 39, 7763 CrossRef CAS.
- R. Kuwano, Y. Kondo and Y. Matsuyama, J. Am. Chem. Soc., 2003, 125, 12104–12105 CrossRef CAS PubMed.
- O. V. Gusev, A. M. Kalsin, M. G. Peterleitner, P. V. Petrovskii and K. A. Lyssenko, Organometallics, 2002, 21, 3637–3649 CrossRef CAS.
- W. Mägerlein, A. F. Indolese and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 2856–2859 CrossRef.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- V. V. Grushin and H. Alper, Organometallics, 1993, 12, 3846–3850 CrossRef CAS.
- B. Wahl, H. Bonin, A. Mortreux, S. Giboulot, F. Liron, G. Poli and M. Sauthier, Adv. Synth. Catal., 2012, 354, 3105–3114 CrossRef CAS.
- (a) J. X. Xu and X. F. Wu, Org. Lett., 2018, 20, 5938–5941 CrossRef CAS PubMed; (b) A. G. Sergeev, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2008, 130, 15549–15563 CrossRef CAS PubMed; (c) F. M. Miloserdov, C. L. McMullin, M. M. Belmonte, J. Benet-Buchholz, V. I. Bakhmutov, S. A. Macgregor and V. V. Grushin, Organometallics, 2014, 33, 736–752 CrossRef CAS; (d) J. S. Quesnel, S. Moncho, K. E. O. Ylijoki, G. M. Torres, E. N. Brothers, A. A. Bengali and B. A. Arndtsen, Chem. – Eur. J., 2016, 22, 15107–15118 CrossRef CAS PubMed; (e) G. M. Torres, J. S. Quesnel, D. Bijou and B. A. Arndtsen, J. Am. Chem. Soc., 2016, 138, 7315–7324 CrossRef CAS PubMed; (f) J. Y. Wang, A. E. Strom and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 7979–7993 CrossRef CAS PubMed.
- Z.-G. Fang, P. M. N. Low, S.-C. Ng and T. S. A. Hor, J. Organomet. Chem., 1994, 483, 17–20 CrossRef CAS.
- C. Amatore, G. Broeker, A. Jutand and F. Khalil, J. Am. Chem. Soc., 1997, 119, 5176–5185 CrossRef CAS.
- T. R. Kégl, N. Pálinkás, L. Kollár and T. Kégl, Molecules, 2018, 23, 3176 CrossRef PubMed.
- Purification of Laboratory Chemicals, ed. W. L. F. Armarego, Elsevier Science Publishers B.V., Amsterdam, The Netherlands, 8th edn, 2017 Search PubMed.
- J. C. Carretero and R. G. Arrayás, Encycl. Reagents Org. Synth., John Wiley and Sons Ltd, Chichester, UK, 2008 Search PubMed.
- K. Zhao and T. P. Loh, Chem. – Eur. J., 2014, 20, 16764–16772 CrossRef CAS PubMed.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, J. Chem. Phys., 2015, 143, 054107 CrossRef PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS; (c) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- (a) H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (c) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed; (b) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- (a) F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS; (b) F. Neese, WIREs Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, USA, 2019 Search PubMed.
- (a) M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS; (b) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- C. Y. Lin, M. W. George and P. M. W. Gill, Aust. J. Chem., 2004, 57, 365–370 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- (a) Spartan '16, Build 2.0.3, Wavefunction Inc., Irvine CA, USA, 2016 Search PubMed; (b) Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio Jr., H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Steck, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00203e |
| This journal is © The Royal Society of Chemistry 2022 |