
Additive-free selective methylation of secondary amines with formic acid over a Pd/In2O3 catalyst†
Caroline
Genre
 *,
Idir
Benaissa
,
Timothé
Godou
,
Mathieu
Pinault
and
Thibault
Cantat
*
*,
Idir
Benaissa
,
Timothé
Godou
,
Mathieu
Pinault
and
Thibault
Cantat
*
CEA, CNRS, NIMBE, Université Paris-Saclay, 91191 Gif-sur-Yvette, France. E-mail: caroline.genre@cea.fr; thibault.cantat@cea.fr
First published on 8th December 2021
Abstract
Formic acid is used as the sole carbon and hydrogen source in the methylation of aromatic and aliphatic amines to methylamines. The reaction proceeds via a formylation/transfer hydrogenation pathway over a solid Pd/In2O3 catalyst without the need for any additive.
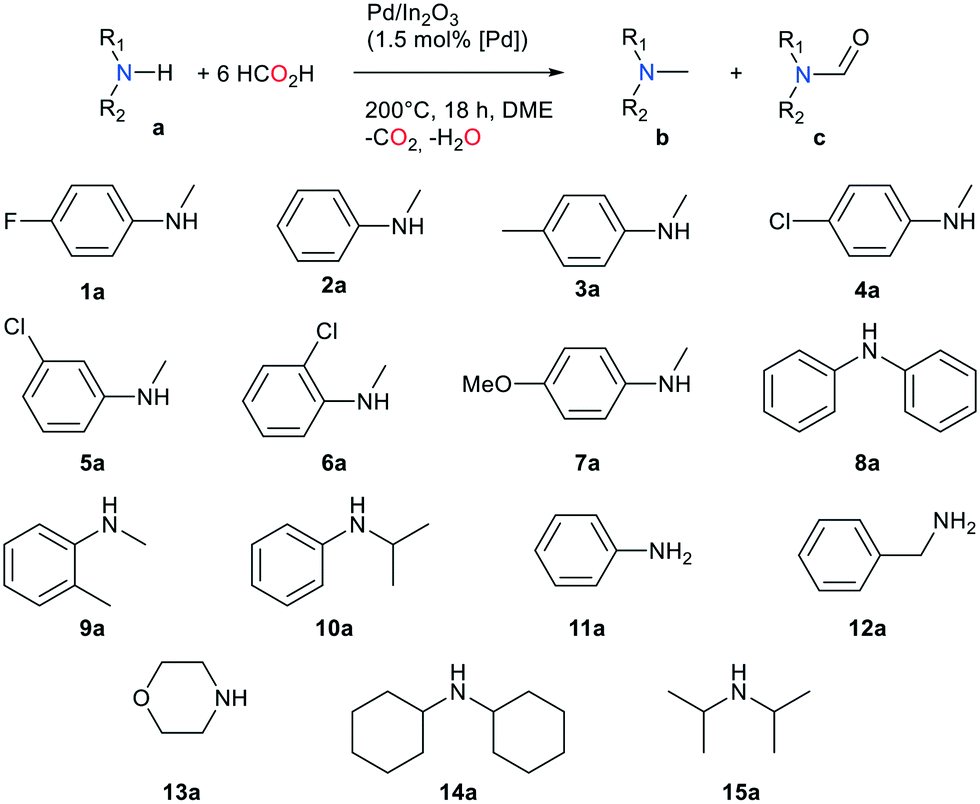
Methylamines are widely utilized compounds in both fine and bulk chemical industries. They are used as intermediates for the synthesis of agrochemicals, polymers, dyes and pharmaceuticals and are valuable organic building blocks.1 Methylation of an amine in a drug molecule can have a strong effect on its lipophilicity and potency.2 For these reasons, the development of methylation methods continues to attract the interest of chemists. The most common methods for synthesizing methylamines involve stoichiometric quantities of highly toxic methylating agents such as methyl iodide, formaldehyde, diazomethane or dimethylsulfate and generate abundant waste.3 In the quest for greener, more sustainable reactions, catalytic methods utilizing renewable C1 sources (CO2, HCOOH (FA), dimethyl carbonate) associated with reductants (silanes, boranes, H2) or additives (acids, salts) have been developed.4 Examples of such catalytic systems involve organometallic complexes of ruthenium,5 zinc,6 rhodium7 or iron.8 Amines were successfully methylated by CO2/H2 over solid catalysts as well, such as CuAlOx,9 Pt–MoOx/TiO2,10 PdGa/TiO2,11 Re/TiO2,12 or Cu/TiO2.13 These systems, however, require a high pressure of H2. This issue can be solved with the use of liquid FA instead of a mix of gases, as it can act as both a carbon source and a reductant. Moreover, a now mature technology has been developed to promote its synthesis from the 2-electron reduction of CO2 in an electrochemical cell over electro-catalysts, making FA a renewable reagent.14 A few heterogeneous catalysts have been described for the methylation of amines with FA. Kim and co-workers reported the successful additive-free methylation of aromatic amines with FA catalyzed by a supported PdAg alloy in 2015.15 In 2016, Zhu et al. achieved this reaction over a Pd/C catalyst, albeit using hydrosilanes as reductants.16 In order to expand the range of possible catalysts for this reaction, we sought to exploit the association of a catalyst known for its CO2 reduction activity and metal species active in reduction processes. We report here on the additive-free, one pot methylation of a variety of amines with an easily synthesized, water and air resistant catalyst. To our delight, we observed that Pd supported on indium oxide is active in the FA-mediated methylation of amines with yields up to 99%.
In2O3 was recently shown to be a highly efficient and specific catalyst for the hydrogenation of CO2 to methanol.17 In a first attempt, we tested whether this oxide showed activity for the methylation of p-fluoro-N-methylaniline (1a) with FA (Table 1, entry 2). The catalytic reaction was conducted in a sealed autoclave under atmospheric pressure. A low p-fluoro-N,N-dimethylaniline (1b) yield of about 11% was observed, as well as a 74% yield of p-fluoro-N-methylformanilide (1c). It thus appears that In2O3 is capable of methylating 1a to 1b with FA but the activity remains low. For this reason, In2O3-based catalysts doped with metals known for their hydrogenation properties (Pd, Pt, Ru) were synthesized and tested. The results are listed in Table 1. The catalysts were synthesized by an impregnation technique followed by calcination of the resulting powder at 400 °C for 3 h under air. Pre-reducing the catalysts (20 bar H2, 200 °C, 1 h) did not improve their activity nor their selectivity, therefore they were used as such. GC analysis of the atmosphere in the autoclave at the end of the reaction showed the presence of significant quantities of H2 and CO2 (Fig. S1†). We thus postulate that in situ reduction of the catalysts was achieved from H2 resulting from the decomposition of FA over the catalyst.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Conv. (%) | Yield 1b (%) | Yield 1c (%) |
| a Reaction conditions: 1a (5 mmol), catalyst 1.5 mol%, FA (60 mmol), DME (10 mL), 200 °C, 18 h. Yields determined by GC/MS using isooctane as an internal standard, after calibration. | ||||
| 1 | None | 97 | Traces | 96 |
| 2 | In2O3 | 98 | 11 | 74 |
| 3 | Pd/In2O3 | 100 | 94 | 4 |
| 4 | Pd/C | 88 | 9 | 72 |
| 5 | Pt/In2O3 | 85 | 30 | 51 |
| 6 | Pt/C | 58 | 5 | 47 |
| 7 | Ru/In2O3 | 82 | 14 | 59 |
| 8 | Ru/C | 75 | 8 | 44 |
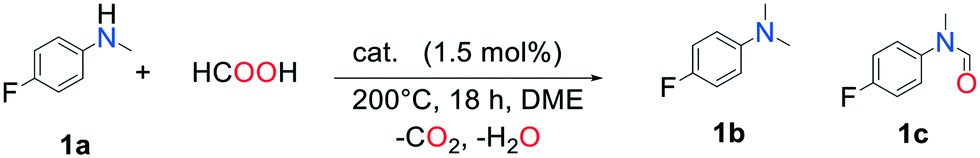
4-Fluoro-N-methylaniline was retained as a benchmark substrate. Among the various catalysts tested, Pd/In2O3 afforded the best activity for N-methylation (yield: 94%). Remarkably, Pd/In2O3 showed better activity than both In2O3 and Pd/C, indicating that a positive synergistic effect occurs between the oxidic support and the Pd catalytic species. The same observation can be made with Ru and Pt containing catalysts (entries 5–8).
Pd/In2O3 displays higher activity than the Pt and Ru containing analogues (100% conversion vs. 85 and 82%, respectively), and higher selectivity towards 1b (94% yield vs. 30 and 14% respectively). The reaction conditions for the methylation of various amines were therefore optimized with this catalyst (see Table S1†). When the reaction is conducted at 150 °C, there is total conversion of 1a but the yield in 1b is limited to 22% after 12 h (entry 2). The results are much better at 200 °C, with an 85% yield in the desired product. This is consistent with reports showing that In2O3 is more active for CO2 hydrogenation above this temperature.17 Screening different solvents revealed that DME is the best suited for the methylation reaction (entry 1), while yields fall when the reaction is conducted in hexane (entry 3). With THF, significant degradation of the solvent is observed, rendering its use impractical (entry 4).
A 12 h reaction time with 4-fluoro-N-methylaniline as a substrate leads to a 100% conversion with a 85% yield of 1b (Table S1,† entry 1), which increased to 94% after 18 h (entry 5). Longer reaction times did not provide better yields (entry 6).
The quantity of FA relative to the substrate was also optimized. Tests were made with 2, 4, 8 and 12 equivalents of FA (entries 5 and 7–9). The best conversion (100% vs. 58, 73, and 93%, respectively) and 1b yield (94% vs. 6, 16 and 23% respectively) were obtained with 12 eq. FA (entry 5). This stoichiometry was thus selected for further catalytic reactions.
The Pd/In2O3 catalyst is a brown powder that turns to black after it is reduced, and turns back to brown after it is annealed again, as would be the case in a typical recycling experiment. SEM-EDX mappings of a fresh catalyst sample show that Pd, In, and O are spread uniformly on a micrometric scale (Fig. S3†). XPS spectra of the samples were recorded before and after reaction with a substrate, as well as after annealing under air of a spent catalyst sample. They confirmed the presence of In, Pd and O in the samples. In the as-prepared state, the Pd3d signal at 336.7 eV is ascribed to PdO (Fig. S4†).18 The In3d5/2 signal was detected at 444.1 eV, characteristic of In2O3 (Fig. S5†).19 A shift of the Pd3d signal to 335.6 eV is observed for the reacted catalyst sample, which according to literature corresponds to elemental Pd.18 After annealing of the spent catalyst, the Pd signal appeared at 337.3 eV, indicating a re-oxidation of palladium. In the reacted catalyst, the In 3d5/2 signal was recorded at 444.9 eV and the peak became broader, signaling that while indium was not significantly reduced during the reaction, another chemical environment is present, possibly In(OH)3. After annealing of the spent catalyst, the In signal was detected at 444.5 eV and the peak became narrower again, signaling a return to In2O3. The atomic concentration of Pd of the near surface region was 16.7% for the as-prepared catalyst and decreased to 4.1% in the spent catalyst (Table S2†). It retained a similar value (4.2%) over annealing (400 °C, 3 h) of the spent catalyst.
HR-TEM images of samples of the fresh, reacted and recycled catalyst were recorded, as well as EDX spectra. The fresh Pd/In2O3 materials is made of objects varying in size from about 20 to 50 nm aggregated into bigger objects (100–500 nm) (Fig. S6a†). The bigger objects seem to result from the aggregation of two different kinds of material (Fig. S6b†). Close-ups of zones containing both types of material, (zones 1 and 2 on Fig. S6b†) were recorded, and EDX analysis of both zones were carried-out. The material in zone 1 is highly crystalline, with the planes clearly visible (Fig. S6c†). The EDX spectrum shows a high concentration of palladium in the material (Fig. S6d†). A close up of zone 2 shows that the material is crystalline as well (Fig. S6e†). EDX showed a high concentration of indium and no palladium in this zone (Fig. S6f†). We can deduct from these studies that fresh Pd/In2O3 catalyst is likely made of particles of indium oxide decorated with palladium oxide. This is consistent with the catalyst synthesis method, where indium oxide is suspended in a solution containing Pd2+ ions before solvent evaporation and subsequent annealing under air.
A sample of the reacted catalyst was analyzed with similar methods. The reacted catalyst displays nanostructures with a 5–10 nm size aggregated into bigger objects (Fig. S7a†). The material is crystalline with the planes clearly visible. The material is much more homogeneous in nature than the fresh catalyst. EDX spectra of various zones of a number of different particles all confirmed the presence of both metals in every point of the sample (Fig. S7b†). These results are consistent with observations reported by Neumann et al., who described the formation of intermetallic In–Pd particles, as well as diffusion of Pd into the bulk upon exposure of similar Pd/In2O3 samples to reducing conditions at 200 °C.19 These observations, combined with the homogenization of the catalyst material most likely account for the decrease of the concentration of palladium observed by XPS.
A sample of reacted catalyst that was subsequently re-oxidized by annealing under air was observed with HR-TEM as well. The morphology is the same as with the reacted catalyst, with nanostructures of ca. 10 nm aggregated into bigger objects (Fig. S8a†). The material is still crystalline, and quite homogeneous. EDX analyses in various points of the sample showed that confirmed the presence of both In and Pd everywhere (Fig. S8b†). This is consistent with the stable atomic concentration of Pd in the samples detected with XPS.
In order to ensure that there is no contribution from Pd possibly leached from the solid catalyst, our benchmark reaction was carried out with pure In2O3 powder in the liquid phase recovered from a previous run of the reaction, but only extremely limited activity for amine methylation could be observed (3%).
All these observations allow us to propose that Pd in the catalyst is reduced by either FA or H2 (produced upon FA decomposition), and that metallic Pd on In oxide and/or In–Pd intermetallic species are the active catalytic species.
Recycling experiments were carried out with 1a as a substrate (Fig. 1). After each run, the recovered catalyst was annealed under air at 400 °C for 3 h. Full conversion and excellent specificity were retained in a second and third runs of the catalyst. In a fourth run, specificity decreased to 85% of the desired product.
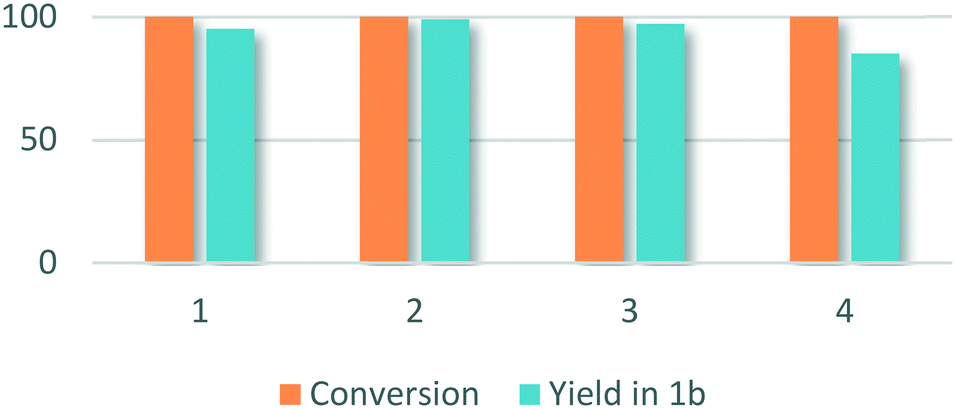 | ||
| Fig. 1 Conversion and yield in 1b (%) for methylation of 4-fluoro-N-methylaniline over Pd/In2O3 after 1, 2, 3 and 4 runs. | ||
The methylation of a variety of primary and secondary amines was carried out to explore the potential of this novel catalytic transformation (Table 2). Using 12 equivalents of FA, the methylation of secondary aromatic amines (entries 1–7) is efficient, with yields ranging between 70 and 93% (18 h, 200 °C, 1.5 mol% [Pd]). The catalytic system tolerates a variety of substituents on the aromatic ring. Interestingly, no hydrogenation of the aromatic cycle was observed, as can be the case with other Pd containing catalysts during hydrogenation of aromatic species including amides.20 With aniline and N-methylaniline, coupling by-products, mostly 4,4′-methylenebis(N,N-dimethylaniline), were observed, which explains the low yield in product 11b. The same kind of coupling was observed with 5a and 10a, yielding 4,4′-methylenebis(3-chloro-N,N-dimethylaniline) and 4,4′-methylenebis(N-isopropyl-N-methylaniline) as side products. The catalytic system displayed lower activity with aliphatic amines (entries 9–13). The best yield was obtained with morpholine (60%). With substrate 12a, secondary products such as N,N-diethylbenzylamine and N-isopropylbenzylamine are observed, driving down the yield in desired product 12b.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Conv. (%) | Yield b (%) | Yield c (%) |
| a 4.5 mol% catalyst. b Side-product: 4,4′-methylenebis(3-chloro-N,N-dimethylaniline). c Side-product: methylenebis(N-isopropyl-N-methylaniline). d N-Methylaniline. e Mix of various products including starting material and ethylated products. f N-Methylbenzylamine. g Mix of N-benzylformamide, N-benzyl-N-methylformamide, N-benzyl-N-formylformamide. | ||||
| 1 | 1a | 100 | 94 | Traces |
| 2 | 2a | 100 | 84 | 8 |
| 3 | 3a | 100 | 90 | Traces |
| 4a | 4a | 100 | 76 | 24 |
| 5 | 5a | 100 | 77 | Tracesb |
| 6 | 6a | 95 | 70 | Traces |
| 7 | 7a | 100 | 78 | 22 |
| 8 | 8a | 100 | 99 | Traces |
| 9 | 9a | 100 | 82 | 16 |
| 10 | 10a | 100 | 80 | Tracesc |
| 11 | 11a | 89 | 29 (12d) | 20 |
| 12 | 12a | 88e | 40 (1f) | 20g |
| 13 | 13a | 100 | 60 | 38 |
| 14 | 14a | 98 | 31 | 65 |
| 15 | 15a | 100 | 25 | 73 |
With aromatic amines, products of methylation of the aromatic cycle were observed via Friedel–Crafts reaction: the reaction of N,N-dimethylaniline with FA over Pd/In2O3 (DME, 200 °C, 18 h) yielded 15% of N,N,4-trimethylaniline, 52% of 4,4′-methylenebis(N,N-dimethylaniline), and a mixture of various methylated derivatives of the latter (Scheme 1). This is further indication that the reaction time must be chosen accurately to avoid over-methylation.
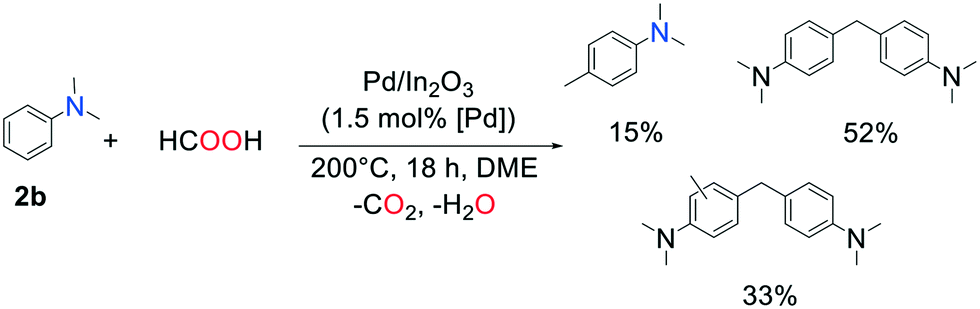 | ||
| Scheme 1 Reaction of N,N-dimethylaniline with FA over Pd/In2O3. | ||
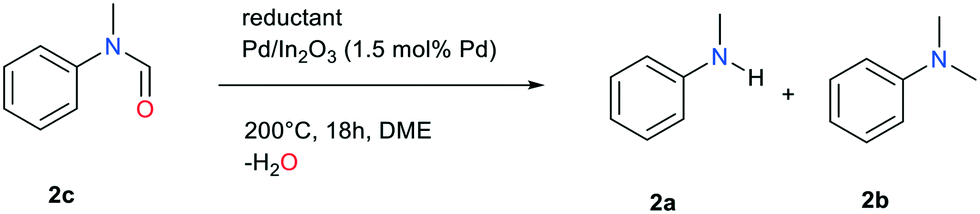
In light of these results, and in order to enhance the scope of this catalytic transformation, we investigated possible reaction pathways and intermediates. Based on the species detected at the end of the reaction, it quite plausible that the methylation of the NH function involves the formation of a formamide intermediate, which is subsequently reduced.
Reaction of N-methylformanilide 2c with 12 equivalents of FA in our chosen conditions indeed provided N,N-dimethylaniline with an excellent 94% yield. A possible pathway is thus the conversion of the starting amine into the corresponding formamide, followed by either direct or transfer hydrogenation.
During the reaction, the pressure inside the autoclave increased to 30 bar. Gas chromatography analysis of the autoclave atmosphere confirms the presence of H2 and CO2, likely resulting from FA decomposition over the catalyst. It is thus possible that either FA or/and H2 are responsible for formamide (transfer)-hydrogenation. To test these hypotheses, the hydrogenation of N-methylformanilide was conducted in our preferred conditions with FA at 200 °C and under a H2 and a H2/CO2 atmosphere. At 160 °C, conversion was extremely low, regardless the nature of the reductant, indicating that reduction of the amide is the rate determining step in the methylation of amines. A >99% conversion and excellent 94% yield of N,N-dimethylaniline were obtained with FA as a reductant (Table 3, entry 1) at 200 °C. In contrast, the direct hydrogenation (30 bar H2) of N-methylformanilide 2c provided a mixture of N-methylaniline 2a and N,N-dimethylaniline 2b with a 50 to 60% yield depending on the conditions. This is consistent with recent results published by Sorribes et al.,21 whereas only traces of the de-methylated product were observed with FA (Table 3, entries 2, 3). A higher H2 pressure (50 bar) did not significantly modify the results, and a CO2 pressure only had a marginal impact (Table 3, entries 3, 4). This leads us to propose that FA, and not H2 issued from its decomposition, is the principal reductant in the methylation of amines over Pd/In2O3 (Fig. 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reductant | Pressure (bar) | Conv. (%) | Yield 2a (%) | Yield 2b (%) |
| a Reaction conditions: 1a (5 mmol), Pd/In2O3 80 mg, DME (10 mL), 200 °C, 18 h. Yields determined by GC/MS using isooctane as an internal standard, after calibration. | |||||
| 1 | FA (12 eq.) | — | >99 | Traces | 94 |
| 2 | H2 | 30 | >99 | 32 | 60 |
| 3 | H2 | 50 | >99 | 50 | 49 |
| 4 | H2/CO2 | 30/30 | >99 | 56 | 42 |
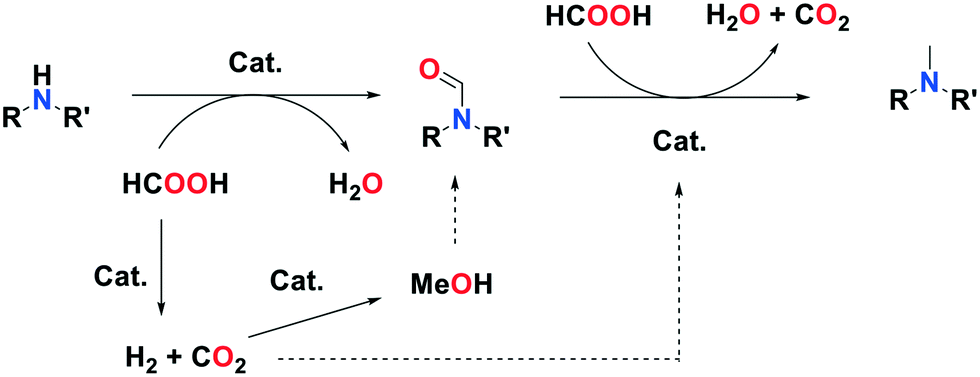 | ||
| Fig. 2 Proposed mechanism for the methylation of amines by FA. | ||
Methanol was observed in the reaction medium, as could be anticipated from the fact that In2O3 catalyzes the hydrogenation of CO2 to methanol. The use of 4 equivalents of methanol as a methylating agent was tested with N-methylaniline in our preferred conditions. The reaction yielded a moderate 22% yield of methylated product (conversion: 30%). It is thus possible that methanol issued from CO2 hydrogenation is a minor contributor to amine methylation as well.
In conclusion, we have developed a simple, additive-free procedure for the N-methylation of amines with FA as a carbon and hydrogen source, by using a water and air resistant Pd/In2O3 catalyst, without the need for an additional reductant. Aromatic amines are methylated with good yields and no reduction of the aromatic cycle is observed.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a grant from Fondation de la Maison de la Chimie (Paris, France). We acknowledge CEA, CNRS, and the European Research Council (ERC Consolidator Grant Agreement no. 818260) for financial support. The authors thank Dr Jocelyne Leroy (Université Paris-Saclay, CEA, CNRS, NIMBE – Gif-sur-Yvette, France) for the XPS measurements and Dr Éric Larquet (CIMEX, École Polytechnique, Palaiseau, France) for the HR-TEM imaging.References
- K. Weissermehl and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, 4th edn, 2003 Search PubMed.
- H.-G. Cheng, M. Pu, G. Kundu and F. Schoenebeck, Org. Lett., 2020, 22, 331–334 CrossRef CAS PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, John Wiley & Sons Inc, 5th edn, 2001 Search PubMed.
- (a) Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077–1086 CrossRef CAS; (b) J. R. Cabrero-Antonino, R. Adam and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 12820–12838 CrossRef CAS PubMed; (c) J. R. Cabrero-Antonino, R. Adam, V. Papa and M. Beller, Nat. Commun., 2020, 11, 3893 CrossRef PubMed.
- (a) S. Savourey, G. Lefèvre, J.-C. Berthet and T. Cantat, Chem. Commun., 2014, 50, 14033–14036 RSC; (b) Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed; (c) Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed; (d) K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed; (e) J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Catal. Sci. Technol., 2016, 6, 7956–7966 RSC.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- R. H. Lam, C. M. A. McQueen, I. Pernik, R. T. McBurney, A. F. Hill and B. A. Messerle, Green Chem., 2019, 21, 538–549 RSC.
- X. Frogneux, O. Jacquet and T. Cantat, Catal. Sci. Technol., 2014, 4, 1529–1533 RSC.
- X. Cui, X. Dai, Y. Zhang, Y. Deng and F. Shi, Chem. Sci., 2014, 5, 649–655 RSC.
- K. Kon, S. M. A. H. Siddiki, W. Onodera and K.-i. Shimizu, Chem. – Eur. J., 2014, 20, 6264–6267 CrossRef CAS PubMed.
- X. Su, W. Lin, H. Cheng, C. Zhang, Y. Li, T. Liu, B. Zhang, Q. Wu, X. Yu and F. Zhao, RSC Adv., 2016, 6, 103650–103656 RSC.
- T. Toyao, S. M. A. H. Siddiki, Y. Morita, T. Kamachi, A. S. Touchy, W. Onodera, K. Kon, S. Furukawa, H. Ariga, K. Asakura, K. Yoshizawa and K.-i. Shimizu, Chem. – Eur. J., 2017, 23, 14848–14859 CrossRef CAS PubMed.
- K. Liu, Z. Zhao, W. Lin, Q. Liu, Q. Wu, R. Shi, C. Zhang, H. Cheng, M. Arai and F. Zhao, ChemCatChem, 2019, 11, 3919–3926 CrossRef CAS.
- S. C. Perry, P.-k. Leung, L. Wang and C. Ponce de León, Curr. Opin. Electrochem., 2020, 20, 88–98 CrossRef CAS.
- A. K. Singh, Y.-H. Hwang and D.-P. Kim, NPG Asia Mater., 2015, 7, e222 CrossRef CAS.
- L. Zhu, L.-S. Wang, B. Li, W. Li and B. Fu, Catal. Sci. Technol., 2016, 6, 6172–6176 RSC.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- M. Brun, A. Berthet and J. C. Bertolini, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 55–60 CrossRef CAS.
- M. Neumann, D. Teschner, A. Knop-Gericke, W. Reschetilowski and M. Armbrüster, J. Catal., 2016, 340, 49–59 CrossRef CAS.
- M. Stein and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 2231–2234 CrossRef CAS.
- I. Sorribes, S. C. S. Lemos, S. Martín, A. Mayoral, R. C. Lima and J. Andrés, Catal. Sci. Technol., 2019, 9, 6965–6976 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01626a |
| This journal is © The Royal Society of Chemistry 2022 |