
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacing single-atom catalysis with continuous-flow organic electrosynthesis
Mark A.
Bajada
 a,
Jesús
Sanjosé-Orduna
b,
Giovanni
Di Liberto
c,
Sergio
Tosoni
c,
Gianfranco
Pacchioni
c,
Timothy
Noël
b and
Gianvito
Vilé
*a
a,
Jesús
Sanjosé-Orduna
b,
Giovanni
Di Liberto
c,
Sergio
Tosoni
c,
Gianfranco
Pacchioni
c,
Timothy
Noël
b and
Gianvito
Vilé
*a
aDepartment of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milano, Italy. E-mail: gianvito.vile@polimi.it
bFlow Chemistry Group, van’t Hoff Institute for Molecular Sciences, Universiteit van Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
cDepartment of Materials Science, Università di Milano Bicocca, via R. Cozzi 55, 20125 Milano, Italy
First published on 28th April 2022
Abstract
The global warming crisis has sparked a series of environmentally cautious trends in chemistry, allowing us to rethink the way we conduct our synthesis, and to incorporate more earth-abundant materials in our catalyst design. “Single-atom catalysis” has recently appeared on the catalytic spectrum, and has truly merged the benefits that homogeneous and heterogeneous analogues have to offer. Further still, the possibility to activate these catalysts by means of a suitable electric potential could pave the way for a true integration of diverse synthetic methodologies and renewable electricity. Despite their esteemed benefits, single-atom electrocatalysts are still limited to the energy sector (hydrogen evolution reaction, oxygen reduction, etc.) and numerous examples in the literature still invoke the use of precious metals (Pd, Pt, Ir, etc.). Additionally, batch electroreactors are employed, which limit the intensification of such processes. It is of paramount importance that the field continues to grow in a more sustainable direction, seeking new ventures into the space of organic electrosynthesis and flow electroreactor technologies. In this piece, we discuss some of the progress being made with earth abundant homogeneous and heterogeneous electrocatalysts and flow electrochemistry, within the context of organic electrosynthesis, and highlight the prospects of alternatively utilizing single-atom catalysts for such applications.
 Mark A. Bajada | Mark A. Bajada holds a BSc (with honours) in chemistry and physics from the University of Malta and a MPhil in energy engineering from the University of Cambridge. He received his PhD from the University of Cambridge with a thesis on fuel-generating electrolysers and continuous-flow photoreactors, conducted under the supervision of Prof. Erwin Reisner, and funded through the Endeavour Scholarship. In 2021, he was awarded a Marie Skłodowska-Curie individual fellowship to join the Vilé Lab at Politecnico di Milano and research novel and sustainable single-atom catalysts for photo- and electrosynthetic applications. |
 Jesús Sanjosé-Orduna | Jesús Sanjosé-Orduna studied organic chemistry at the University of Barcelona and pursued his PhD at the Institute of Chemical Research of Catalonia, under the supervision of Dr. Mónica H. Pérez-Temprano, studying cobalt-catalyzed C–H transformations. He has received the 2019 CAS Future Leaders award and the Josep Castells award. After a short postdoctoral stay in the Martin group (at ICIQ), he joined the Noël Research Group at the University of Amsterdam, where he is currently developing innovative chemical transformations employing flow chemistry. In 2022, he has been awarded a Marie Skłodowska-Curie postdoctoral fellowship. |
 From left to right: Gianfranco Pacchioni, Sergio Tosoni and Giovanni Di Liberto | Gianfranco Pacchioni is active in the field of theoretical heterogeneous catalysis. Prior to joining the University of Milano-Bicocca, he worked at the IBM Almaden Research Center, the TU Munich, and the Fritz-Haber Institute of the Max Planck Society, among others. In Milan, he is Professor of Chemistry and has been Vice-Rector for Research and Director of the Department of Materials Science. He is co-author of >550 papers and has given >500 invited talks. He received several awards (Humboldt Award, Pascal Medal, etc.) and is Fellow of the Accademia Nazionale dei Lincei, the Academia Europaea, and the European Academy of Sciences. Sergio Tosoni works on the computational description of surfaces and interfaces. He obtained his PhD from the University of Turin in 2007. Afterwards, he worked as postdoctoral researcher at the Humboldt University in Berlin and the University of Barcelona. He joined the University of Milano-Bicocca in 2016 and was promoted to associate professor in 2022. Giovanni Di Liberto works on theoretical modelling of nanomaterials for photo- and electro-catalysis. He received a PhD in Theoretical Chemistry from University of Milan and he has been visiting scientist at University of Barcelona in 2019. Since 2021, he is an assistant professor at the University of Milano-Bicocca. For his research, he has received several awards by the Italian Society of Chemistry. |
 Timothy Noël | Timothy Noël currently holds a position as Full Professor at the University of Amsterdam, where he holds the chair in Flow Chemistry. His research interest ranges from organic chemistry to chemical engineering and encompass more specifically flow chemistry, organic synthesis, and synthetic catalytic methodology development. His work received several awards, including the DECHEMA prize (2017), the Hoogewerff Jongerenprijs (2019), the IUPAC-ThalesNano prize in flow chemistry and microfluidics (2020) and the KNCV Gold Medal (2021). He serves as editor-in-chief of Journal of Flow Chemistry. |
 Gianvito Vilé | Gianvito Vilé is a tenure-track assistant professor of Chemical Engineering at Politecnico di Milano. He received his PhD from ETH Zurich and, from 2016 to 2019, was Lab Head in Chemistry Technologies at Idorsia Pharmaceuticals. His research focuses on understanding the structure and reactivity of single-atom catalysts, and designing sustainable chemical processes. He has received the ETH medal, the Dimistris N. Chorafas Award, and the Felder Award for his research. In addition, he has been selected among the emerging early-career researchers by several journals of the American Chemical Society, Royal Society of Chemistry, and Institute of Physics. |
1. The quest for greener and more sustainable catalysts
One of the greatest challenges we are facing as a society is the development and wider implementation of alternative synthetic technologies, coupled with more sustainable and “energy smart” chemical manufacturing processes. Within this framework, catalysis engineering is recognized as the key enabling technology that has a substantial impact on the manufacturing sector. It can contribute to the Green Deal agenda, by driving reactions along selective molecular pathways to reduce waste, unlock clean energy sources, and preventing pollution. Hence, the nanoscale design of better catalytic methods is an essential step in our communal challenge of greening chemical manufacturing.In 1913, the development of iron-based catalysts in the Haber–Bosch process represented an industrial landmark innovation that enabled the synthesis of ammonia from N2 and H2. This laid the foundation for the development of industrial chemistry, which in turn has enabled the production of a wide variety of bulk and fine chemicals. Catalysts are used today in almost 90% of all chemical processes, and are essential for the production of bulk and fine chemicals. In particular, bulk, or so-called commodity, chemicals are manufactured on a large scale in order to satisfy global demand, while fine chemicals are produced in more limited quantities (<1000 tonnes per year).1 Fine chemicals are inherently more expensive than bulk chemicals, and their cost typically reflects the number of synthetic steps involved in their manufacture along with their commercial performance. These useful building blocks affect everyday life through combinations with other chemicals or substrates, to generate, for example, biofuels, plastics, fertilizers, active pharmaceutical ingredients (APIs), or food additives.
It is undeniable however that the traditional chemical industry is accompanied by a serious waste of resources and heavy pollution of water, air, and soil. Chemical manufacturing remains today energy intensive and requires a large amount of fossil fuel-supplied heat (responsible for around 26% of the world energy demand) to drive thermochemical processes.2 It is essential, but hugely challenging, that we transform the chemical industry, making it capable of producing various products to meet our daily needs while avoiding any possible negative impact on the environment. These challenges have led to an explosion in catalyst innovation, to engineer the action of solid catalysts at the atomic scale. In this context, single-atom catalysts (SACs) have recently emerged as an exciting new approach to catalyst design (Fig. 1). Due to the high surface free energy of single atoms, individual metal atoms are generally stabilised or confined by means of a heterogeneous support system with high surface area and abundant anchoring sites to prevent aggregation during synthesis and catalytic processes.3 The term “confining” means that the single metal atoms can be stabilised by the lattice or the coordination environment of the 2D materials, via strong covalent bonds. However, the SACs exhibits unsaturated coordination features, which is what gives rise to their active sites and what effectively allows the binding of reaction intermediates and facilitates the catalytic reaction pathway.4 Because of their atomic-scale design, these materials thus represent the ideal paradigm of site isolation,5 and have been hailed as the limit of heterogeneous catalysis in terms of economy and optimal use of the metal phase.6
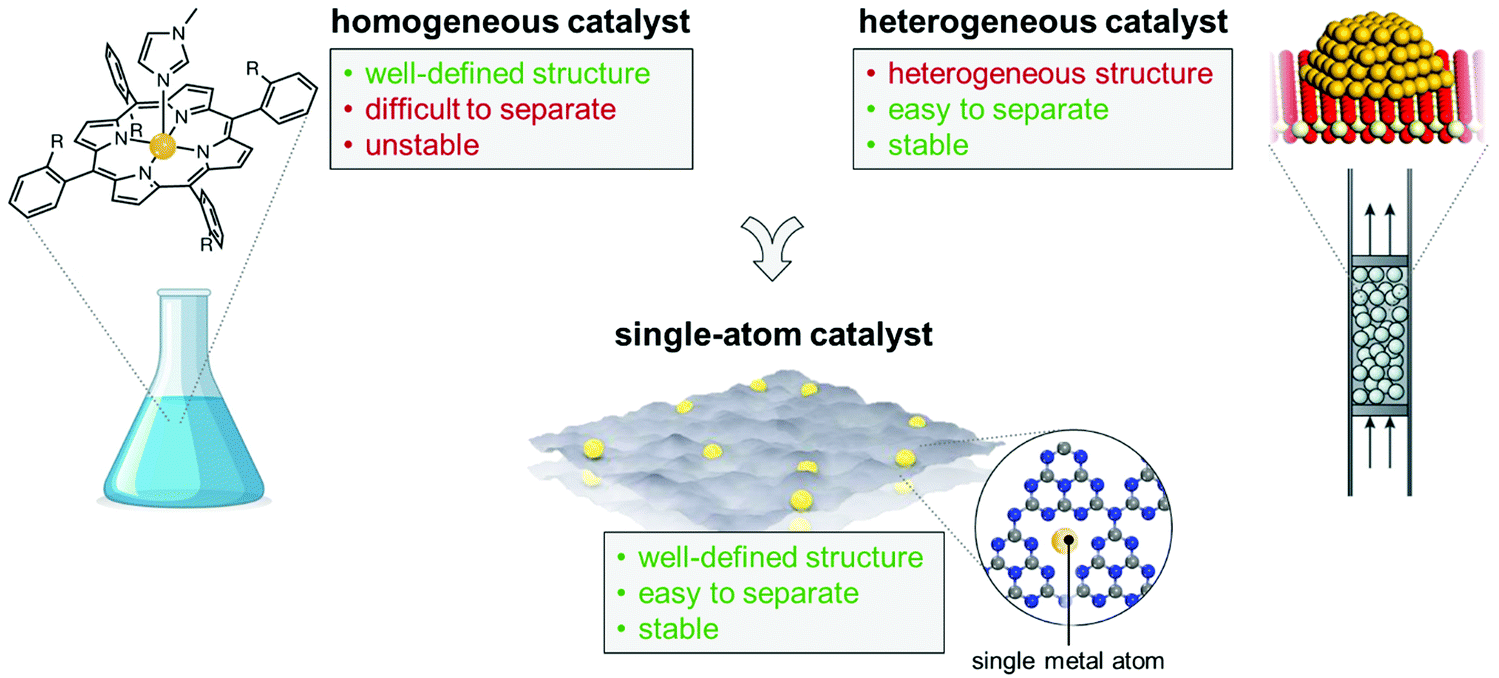 | ||
| Fig. 1 SACs incorporate numerous beneficial features of homogeneous and heterogeneous catalysts, as exemplified in the figure. | ||
One of the earliest SACs was reported in 1999, featuring atomically dispersed Pt species on MgO.7 Tested in propane combustion, the material was as active as the reference catalyst made of MgO-stabilized Pt nanoparticles. The new catalyst, however, was not stable and the difficulty to precisely characterize the metal atoms, together with the obvious catalyst restructuring under reaction conditions, left open questions on the true nature of the “active site”. A breakthrough came in 2003 with a report from Flytzani-Stephanopoulos and co-workers.8 Using a model catalyst, the group provided evidence that cationic Au3+ or Pt2+ species (and not the metal nanoparticles) on CeO2 were responsible for the activity observed in the water-gas shift reaction.
They assumed, in particular, that Au3+ ions substituted Ce4+ ions, triggering the concomitant formation of oxygen vacancies to balance charge. The correctness of this assumption was demonstrated a few years later, when the role of oxygen vacancies on CeO2 was deciphered.9 Wilson, Lee, and co-workers reported atomically dispersed Pd species on the surface of mesoporous Al2O3 for the selective oxidation of allylic alcohols to produce intermediates of pharmaceutical relevance.10 In this work, strong evidence for the presence of single atom came from extended X-ray absorption fine structure (EXAFS) spectroscopy, which gave bond lengths consistent with Pd–O and not with Pd–Pd. In 2011, Zhang and co-workers reported that single Pt atoms deposited on FeOx were active for CO oxidation.11 The group also combined, for the first time, the use of high-angle annular dark-field scanning transmission electron microscopy, EXAFS, and computational techniques to elucidate both the bonding between the single Pt atoms and FeOx carrier along with the catalytic mechanism involved. Finally, they also suggested the term “single-atom catalysis”, modifying the initial definition (“single-site catalysis”) given by Sir John Meurig Thomas,12 and further defining these materials as “supported heterogeneous metal catalyst exclusively containing isolated monometallic active sites on a surface, possibly entrapped in the cavities of porous structures”.
Since the definition of SACs in 2011,13 this class of materials have been applied within a broad range of reactions, including energy-related electrochemical processes, exhibiting superior performance to their cluster or nanoparticle analogues.14–17 Their ability to achieve 100% efficiency of atomic utilisation is particularly advantageous in the field of large-scale renewable energy conversion18–20 from a resource and atom economic perspective, for lowering the consumption of (precious) metals during material preparation, and for effectively minimising wasted bulk catalytic material. A strong metal support interaction was thought to be critical to preventing aggregation of single atoms on the surface. Remarkable has been the effort of Diebold and Parkinson in understanding the surface chemistry of a single-atom catalyst using a model Fe3O4(001) surface.21 Surface science techniques have revealed that CO adsorption strength at single metal sites differs from the respective metal surfaces and supported clusters. The authors proved in this manner that, under surface science conditions, charge transfer into the support modifies the d states of the metal and the strength of the metal–CO bond.21 Such an endeavour, which has been recently validated experimentally,22–26 has led to the hypothesis that charge transfer occurs in single-atom catalysis, making these materials heterogenized forms of homogeneous catalysts and bridging the two fundamental pillars of catalysis science.5,27 Indeed, as illustrated in Fig. 2A, there has been an exponential increase in the number of publications that utilize SACs since 2014, giving rise to a highly active and diverse research field that continues at the present time. Landmark publications and milestones reached within the SAC community are summarised in Fig. 2B.
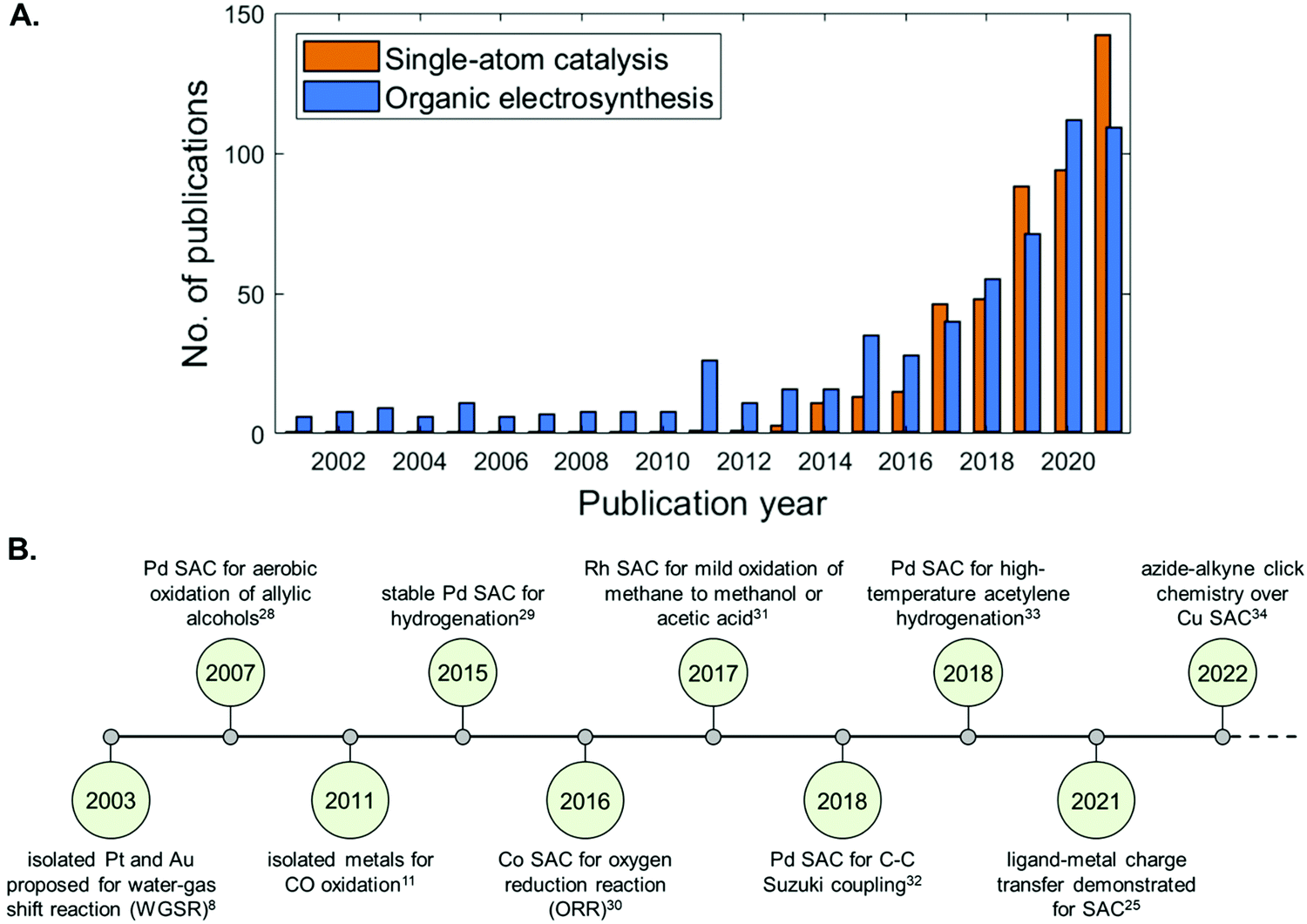 | ||
| Fig. 2 (A) Bar chart of the papers published per year for “single-atom catalysts” (applications and fundamental studies), and for “organic electrosynthesis” (electrosynthetic and electrochemical studies) generated using the Web of Science database. (B) Timeline showing key milestones in the development and application of SACs.8,11,25,28–34 | ||
Work on energy-related single-atom electrocatalysts has been pivotal to the advancement of this class of materials. Pt-based materials remain the most active catalysts toward the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER), but their scarcity and high-cost call for alternative catalysts based on non-precious transition metals. Tour and co-workers first reported the use of non-precious metal-based SACs for electrocatalytic HER,35 in which they demonstrated that atomic cobalt on N-doped graphene was highly active towards this transformation in both acidic and alkaline media. The electrocatalytic CO2 reduction reaction (CO2RR) enables the conversion of CO2 into value-added chemicals and fuels.36 Recently, first-row transition metals (e.g., Mn, Fe, Co, Ni, Cu) atomically dispersed in graphene layers have been explored as CO2RR electrocatalysts in aqueous solution with CO as the dominant product, and Ni-based ones are the most widely studied among different atomic metal catalysts.37
2. Beyond small molecule activation: SAC-driven organic electrosynthesis
To-date, most SACs have been studied for electrochemical energy-related reactions or traditional thermochemical organic chemistry, but rarely within the context of forefront organic synthesis of APIs and other fine chemicals featuring more than one carbon atom. However, the transfer of green chemistry technologies into industry also requires their wide, effective, and economically viable implementation in less explored industrial areas. Over the past years, the field of preparative organic methods has experienced increased attention to the adoption of new and viable technologies. Indeed, as demonstrated in Fig. 2A, there has been an exponential increase in the number of publications that utilize electrosynthetic methods since 2012, giving rise to a diverse and highly active field of research that continues at the present time. This development has been also encouraged by the uptake of green chemistry concepts by the organic synthesis community and by the definition of new “metrics”, such as the E(nvironmental)-factor, which is defined as the ratio between the mass of waste generated and the mass of product synthesized, and which assess how environmentally friendly or harmful a chemical process is.38 By showing that the synthesis of organic compounds generate 10 times higher waste than the energy and oil-and-gas industry (due to solvent losses and the large amounts of downstream waste generated from the use of toxic reductants and oxidants during pharmaceutical synthesis),39 these metrics have led to the re-evaluation of many practices in the fine chemical and pharmaceutical industries, and encouraged a reduced use of hazardous substances in the synthesis and production of various chemical goods.40 In this context, the renaissance of electrosynthesis has offered a viable alternative to traditional thermochemical approaches, and has shown the potential to improve the sustainability of chemical manufacturing. The range of examples exploiting electrocatalytic steps in organic synthesis is significantly smaller in volume than thermochemical analogues.41–44 However, it has been demonstrated that there is substantial market potential to implement electrosynthetic routes as an alternative to traditional thermochemical approaches, for the sustainable production of organic chemicals, such as is the case with the Simons fluorination process,45 the Monsanto adiponitrile process,46 and electrochemical deuteration methods.47On a more fundamental level, electrochemistry uses an electrical current to drive, otherwise endergonic, chemical reactions. This electricity is supplied to an electrochemical cell, which consists of two complementary electrodes and a suitable electrolyte.48 The two electrodes, anode and cathode, are responsible for the oxidation and reduction processes, respectively. The electrode material is crucial, and a judicious choice is required to steer and maximize the chemical outcome of the process (see Section 7).49,50 The electrolyte is usually a highly-charged solution that acts as a link between the two redox events that take place at the electrode surfaces. The selection of a proper solvent/electrolyte combination will be very important for closing the overall circuit in a productive manner, and consequently will have an impact in the overall efficiency (see Section 6).51 In other words, the presence of an electrolyte minimizes the Ohmic drop of the electrochemical cell and is thus important to increase the energy efficiency of the cell. Overall, electrosynthesis can replace the need for oxidising or reducing chemical agents used in conventional thermochemical processes, which typically lead to the generation of wasteful and toxic by-products. The use of an electric current as a renewable, affordable, and safe source of electrons to develop chemical transformations can be considered ideal from the vantage point of green chemistry.52
Progress has also been made to expand the list of existing organic electrosynthetic reactions for large-scale applications. An electrochemical allylic C–H oxidation has been recently developed by Bristol Myers Squibb and the Baran group.53 The elimination of toxic reagents (such as chromium or selenium) and precious metals, and the use of carbon graphite rods and vitreous carbon electrodes, implied that the process was suitable for scale-up. Industrial applicability was demonstrated in a 100 g scale batch electroreactor, with the conversion of α-pinene (1) to verbenone (2, Scheme 1A), and the synthesis of sterol 5 and its acetate 6 (Scheme 1B). Additionally, a process greenness score (PGS) of 56% was obtained, >50% improvement in the PGS compared with the CrO3-mediated oxidation of acetate 6 (32%). In 2019, a desirable building block for the preparation of drug compounds like citiolone and ersteine was prepared by BASF AG, using a large-scale electrochemical reduction protocol to convert DL-homocysteine to thiolactone 8 (Scheme 2).54 Their alternative electrochemical procedure was developed and conducted on a 200 kg scale, through the use of a carbon cathode coated in a lead/bismuth alloy, to produce compound 8 with 45% yield. Smaller scale protocols, with the potential of large-scale operation, are also being devised within the area of electrochemical reductive coupling using Ni catalysts. Recently, Pfizer, in collaboration with the Weix group at the University of Wisconsin-Madison, reported such a system, featuring a homogeneous Ni-catalysed coupling reaction between aryl bromides and alkyl bromides in a divided electrochemical cell. These mechanisms alleviate the production of stoichiometric metal waste,55 but further work is required to move beyond the use of elements such as nickel and lead, which have environmental consequences in their own right.
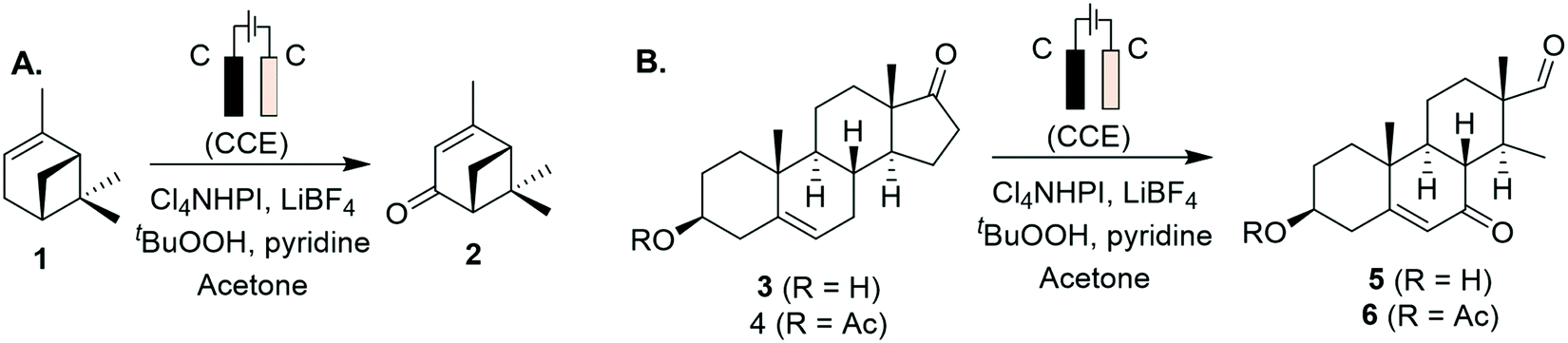 | ||
| Scheme 1 Electrochemical allylic oxidation on a 100 g scale, using inexpensive graphite plate electrodes in an open-air beaker, and LiBF4 as supporting electrolyte: (A) α-pinene to verbenone conversion, (B) sterol product and its acetate (CCE: constant current electrolysis). | ||
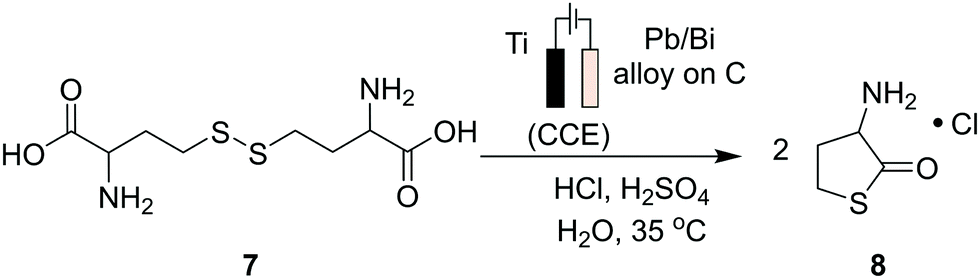 | ||
| Scheme 2 A large-scale electrochemical reduction protocol to synthesize DL-homocysteine thiolactone hydrochloride from DL-homocysteine. This product is a useful pharmaceutical building block required for the preparation of drug compounds such as citiolone and Ersteine. | ||
As shown from these examples, organic electrosynthesis can offer several advantages: conventional chemical oxidizing or reducing agents can be replaced by electric current in which electrons are used as “green” reagents for redox-based chemistry.56 This makes this method an environmentally friendly synthetic tool that is well aligned with the principles outlined in the Green Agenda for organic synthesis.38,57–59 The other contributing factor to the broader adoption of electrosynthetic methods is the drastic rise in the deployment of wind turbines and photovoltaics (Fig. 3), which have allowed us to tap into renewable sources of energy, and corresponding drop in the price of renewable electricity.60 Integrating the energy and chemical sectors would also be beneficial for continually promoting the dispatchability and penetration of renewables on an even broader scale, as excess renewable power could be directed to and stored in the form of a stable, chemical bond, such as in the electrolysis of water to generate H2 fuel.61–64
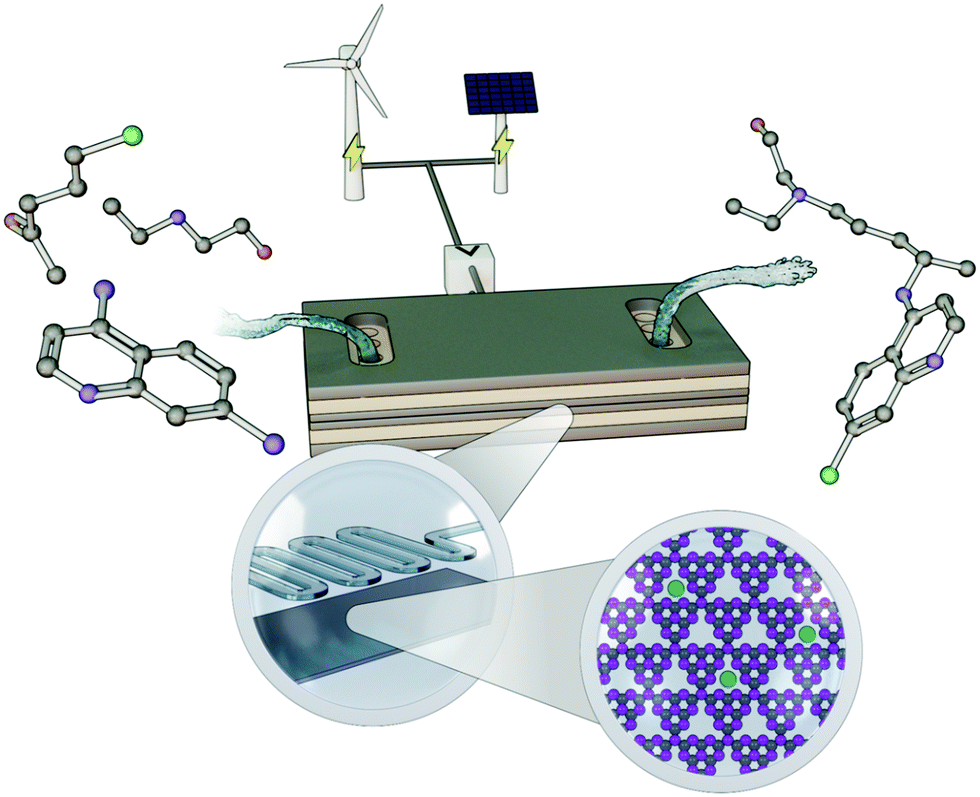 | ||
| Fig. 3 Schematic showing the electrocatalytic conversion of simpler building block molecules into value-added active pharmaceutical ingredients (in this case, hydroxychloroquine), using a single-atom catalyst incorporated within a microflow device and powered through renewable electricity. | ||
The use of SACs for the great potential of electrifying chemistry has been emphasised in the literature, with special importance on designing, developing, and optimizing suitable single-atom electrocatalysts for clean energy conversion reactions.37,65–67 However, little consideration has been given with regards to the application of those design principles to drive targeted organic transformations in a strategically planned electrocatalytic manner. As illustrated in Fig. 2A both the field of “SAC” and the field of “organic electrosynthesis” are substantially growing, but a review merging these complementary fields has, to-date, not been formulated. Promoting the integration of these two sectors would allow to encompass a more comprehensive portfolio of applicable, green chemistries, including both those for bulk and fine chemical production pathways.
3. Targeting five classes of reactions for SAC-based organic electrosynthesis
Five dominant reaction types are typically invoked in organic and pharmaceutical synthesis, and represent more than 60% of the reactions used in these fields.68 These include amide formation, cross-coupling reaction, amine tert-butyloxycarbonyl (Boc)-deprotection, electrophilic reactions with amines, and enantioselective hydrogenation (Fig. 4). These reactions are widely used in synthesis, and predominately require redox conditions via the addition of terminal reductants or oxidants, and are already catalysed over transition metals. The development of sustainable organic electrosynthetic processes and the implementation of single-atom catalysts in the aforementioned reactions could thus have a transformative impact due to the dominant or recurring nature of certain key reactive steps.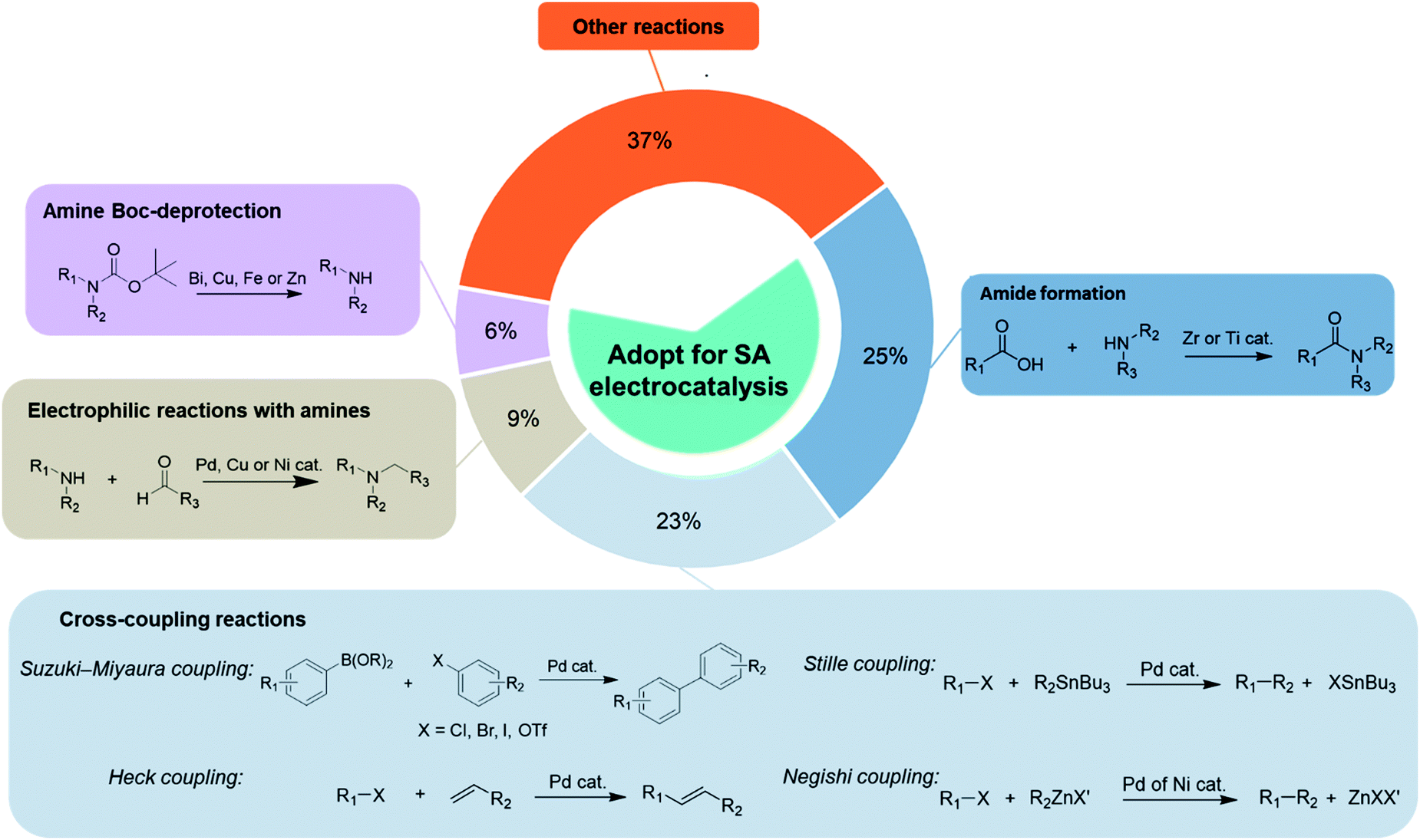 | ||
| Fig. 4 The percentages of the most frequently used reaction types (amide formation, cross-coupling reactions, amine Boc-deprotection and electrophilic reactions with amines) which dominate contemporary practice to produce compounds in fine chemistry. These present a clear reaction space that can be targeted with single-atom electrocatalysis. Data sourced from ref. 30, 83 and 84. | ||
Amide formation protocols are either mediated by a stoichiometric reagent or a catalyst. In the case of the former, typical methods for amide bond formation employ stoichiometric quantities of activating reagents such as 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), thionyl chloride or n-propylphosphonic acid anhydride (T3P),69 which lead to the production of large quantities of waste. Vis-à-vis the catalytic route, the most successful catalysts to date include group(IV) metal salts70–73 and boron based systems.74–81 Cp2ZrCl2 and ZrCl4 are reported to be more effective catalysts in relation to Ti(OiPr)4, and have been applied to a wider range of substrates, albeit the fact that the titanium catalyst is an inexpensive bulk chemical.71 The reactions have also been tested on a 20 mmol scale, demonstrating improved efficiency. Boron compounds constitute the other important class of amidation catalysts. One of the main concerns however is the potential reproductive toxicity of boric acid,82 which will inevitably result from boron-based catalyst breakdown. Commercially available borate esters offer significant improvements in reactivity and scope over boric acid,78 and many pharmaceutically relevant amides can be prepared such as those derived from poorly nucleophilic anilines, heterocyclic compounds, and amino acids.81 These reactions can be scaled up to access multigram quantities of amide with high efficiency. The most reported amidation catalysts within this class are in fact boronic acids, and range from simple examples (phenyl, n-butyl) to more complex functionalized systems.76,77,80
Cross-couplings are a set of different reactions where two fragments are joined together with the aid of a metal catalyst (e.g., Pd). These reactions (i.e., Heck, Kumada, Stille, Negishi, Suzuki–Miyaura, Hiyama, to name a few) use organohalides and other surrogates as starting materials, and are essential tools in synthetic chemistry for the construction of many organic and pharmaceutical compounds. Over the past two decades, the Suzuki–Miyaura coupling reaction, catalysed through the Pd0/PdII cycle, has become arguably one of the most efficient methods for the construction of biaryl or substituted aromatic moieties.85 Most early work for this coupling reaction was conducted using triarylphosphines as supporting ligand for the homogeneous palladium phase. However, more recently, the application of new ligands has dramatically improved the efficiency and selectivity attainable in such cross-coupling reactions. From the available list of ligands for such cross-coupling reactions, bulky dialkylbiaryl86,87 and trialkylphosphines88 remain the most widely used, followed by N-heterocyclic carbenes (NHCs).89 The range of substrates for Suzuki–Miyaura coupling reactions based on these ligands include aryl bromides, aryl triflates, unactivated aryl chlorides, aryl tosylates, a variety of heteroaryl systems as well as very hindered substrate combinations.85
Many biologically active substructures and APIs incorporate nitrogen atoms, hence C–N bond formation is an important area within the pharmaceutical arena. The Buchwald–Hartwig amination, featuring the transition-metal-catalysed cross-coupling of electrophilic aryl halides with amines,90 has become an elegant and resourceful method for creating such bonds. These reactions typically involve palladium, copper, or nickel centres, decorated with sophisticated and expensive ligands. In addition, they usually require the use of a stoichiometric base and high reaction temperatures.91 These ligands were first described by Buchwald for Pd-catalyzed cross-coupling in 1998.92 Since then, additional research has led to the development of a versatile group of structurally related ligands that have been shown to yield highly active catalysts for a range of reactions.93–101 For the case of the Pd-catalyzed amination, the ligand BrettPhos (9, Fig. 5A) for example was shown to be the most active dialkylbiaryl-based phosphine system for the selective arylation of primary amines.102,103 Another breakthrough was made with the ligand RuPhos (10, Fig. 5B), which was found to be useful for the arylation of secondary amines.104,105 Tertiary amines on the other hand, can be synthesised via the Cu-catalysed electrophilic amination of diorganozinc reagents with O-acyl hydroxylamine.106
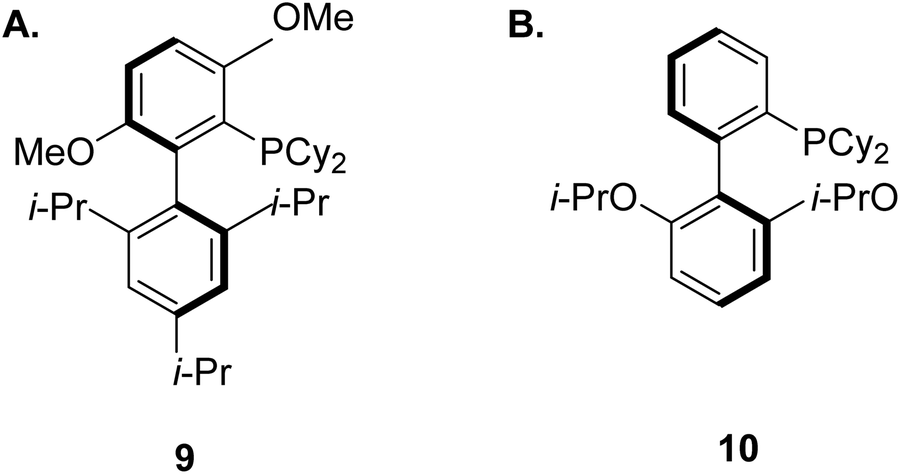 | ||
| Fig. 5 Chemical structures for two homogenous ligands: (A) BrettPhos, and (B) RuPhos. | ||
The Boc group is one of the most widely used amino protecting groups, and the most common procedures for its removal require an excess of an organic acid such as TFA,107 or mineral acids such as sulfuric,108 hydrochloric,109 and phosphoric acid110 on large scale. However, catalytic methods are also known, and they include the use of bismuth(III) bromide,111 copper(II) triflate,112 iron(III) salts,113 and zinc-hydroxyapatite.114 The list is non-exhaustive but sheds some light on the versatility of the applicability of metal systems for catalytic Boc-deprotection.
Finally, enantioselective hydrogenations of enamines and imines represent another highly important reaction pathway for the synthesis of a variety of biologically active molecules, and are often incorporated within the industrial production of chiral drugs and agrochemicals.115 The chiral catalyst used for the hydrogenation of imines and enamines is the primary determinant vis-à-vis the reactivity and enantioselectivity of the reaction. To-date, a broad range of these catalysts have been developed for such purposes, providing the desired chiral amines or their derivatives with good to excellent enantioselectivities. Chiral catalysts for imine hydrogenation are in fact predominantly based on iridium, although examples incorporating rhodium or ruthenium do exist.116 The most notable example is the chiral Ir-Xyliphos catalyst, featuring a chiral ferrocenyl-based diphosphine Josiphos-type ligand, which has been used in the enantioselective synthesis of the herbicide (S)-Metolachlor (Scheme 3).117 Numerous studies on chiral rhodium catalysts based on either diphosphine ligands or monophosphorus ligands have been published in the past couple of decades. In 1996, Burk et al.118 made a breakthrough in enantioselective hydrogenation of enamides via Rh-based catalysts featuring the diphosphine ligands DuPhos and BPE, and a wide range of these substrates were hydrogenated to N-acetyl amines with high enantioselectivities, up to 98% enantioselective excess (ee).
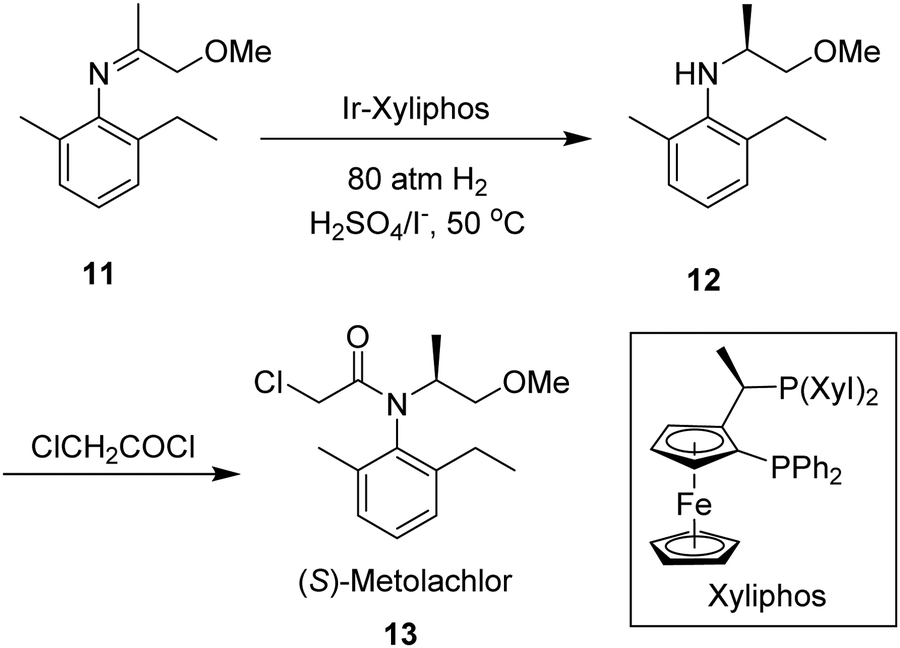 | ||
| Scheme 3 Enantioselective synthesis of (S)-metolachlor, using an Iridium-based chiral catalyst. Note: Xyl = 3,5-xylene. | ||
4. Why SACs can be applied for organic synthesis
Focusing on the class of reactions identified above, in this section we will delve into some key examples which predominately highlight metal-based electrocatalytic applications for organic synthesis, critically discussing limitations of both the homogeneous and heterogeneous perspectives. In this context, we shed light on how single-atom catalysts emerge from the ashes of these two pillars, and can provide unprecedented opportunities for the development of green organic synthesis protocols.Early pioneering reports from Kolbe119 and Shono120 amongst others focused on direct electrolysis methods, where the electro-reaction is predominantly governed by bond cleavage of the weakest link. However, over the past decade, there has been a strong push from the academic organic community to further explore the realm of preparative electrosynthetic methods.121–123 The merger of transition metal catalysis with electrocatalysis – coined metallaelectrocatalysis – has given a unique opportunity to move from substrate engineering to catalyst design, thus extending the electro-reaction past the domain of simple dissociation energy control.124–126 In most cases, homogeneous catalysts comprising of organometallic complexes, metal salts, enzymes, inorganic, and organic species that are solubilized in the same phase as the reactants, are employed.127 Nonetheless, separating homogeneous catalysts from the reaction mixture requires expensive and tedious purification steps. Heterogeneous catalysts have been proposed as future materials for all of the reactions described above. These involve distinct phases for the catalyst (a solid) and the reactants (which are liquid or gaseous) and are preferred since they are stable and easy to separate from the reaction mixture.
Until recently, electrochemical synthesis was conducted over atomically-precise molecular systems, but was limited to the use of costly platinum group metal (PGM) electrocatalysts, and the application of earth-abundant 3d metal catalysts for such purposes had been elusive.128 Earth-abundant 3d metals, being more cost-effective and generally less toxic in comparison with their noble 4d and 5d homologues,129 provide an invaluable platform for resource economical, metallaelectrocatalysed organic synthesis, and 3d transition metals such as Mn,130 Fe,131 Co,132 and Ni,133 present an attractive alternative for intensified applications. For example, anodic C–H activation has been catalysed by Ni and Co, while Mn and Fe are increasingly being employed. Ni complexes are most frequently applied within an electroreductive reaction pathway, although a variety of 3d block metals, such as Co and Zn, are also being implemented as efficient electrocatalysts.134
Co-catalysed metallaelectrochemical C–H activation studies were initiated by the Ackermann group in 2018,135 where they reported a Co(OAc)2 electrocatalytic system featuring the C–H/N–H annulation of alkynes at ambient temperature (Scheme 4). Other metal salts attempted (Mn, Cu, Au, Ir and Pd amongst others) proved to be ineffective, thus highlighting the importance of Co for this electrocatalytic application. Their mechanistic understanding of the system involves the initial formation of the catalytically active CoIII carboxylate species by anodic oxidation of CoI or CoII carboxylate. Carboxylate-assisted C–H activation of benzamides, migratory insertion of the alkyne compound, and a reductive elimination step, affords both the desired product and the CoI species. The latter is converted back to the catalytically active CoIII carboxylate complex through anodic oxidation.
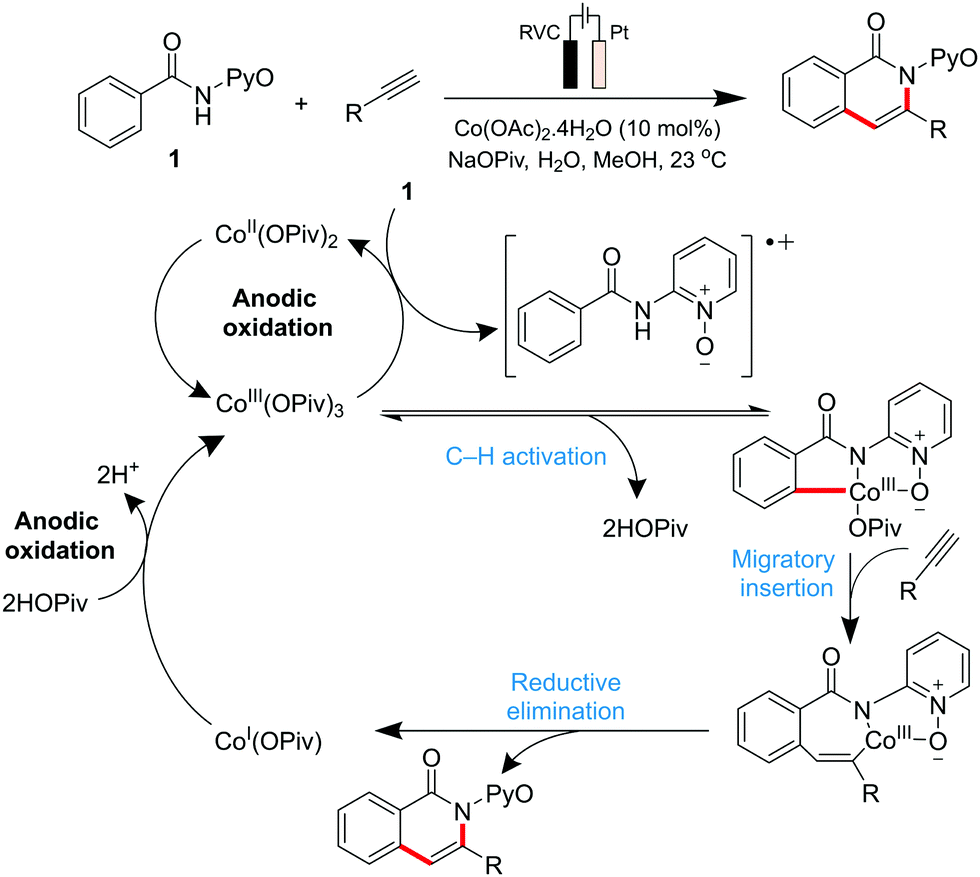 | ||
| Scheme 4 Proposed cobalt-catalytic cycle for C–H/N–H activation. Note: RVC = reticulated vitreous carbon. | ||
The same group also proposed a Fe electrocatalysed route for the regioselective ortho C–H arylation of amides (Scheme 5).136 Typically, the Fe-catalysed oxidative C–H transformation requires superstoichiometric amounts of expensive 1,2-dichloroisobutane as the sacrificial reagent, which in their case was replaced with electricity as an environmentally-benign oxidant. An electrooxidative Fe(II/III/I) catalytic scheme was described for the system, based on experimental, spectroscopic, and DFT mechanistic studies. The Fe-electrocatalytic cycle is thus believed to commence via an organometallic C–H cleavage, followed by anodic single-electron-transfer oxidation and transmetallation of the FeII intermediate to generate a five-membered FeIII species. The latter furnishes the arylated product and the key FeI intermediate via reductive elimination. The catalytically active FeII complex is finally regenerated at the anode-electrolyte interface. Their metallaelectrocatalysis strategy also proved to be versatile vis-à-vis the central metal redox centre whereby the authors substituted Fe for Mn in the C–H arylation scheme, thus demonstrating the use of electricity and first-row, earth-abundant transition metal catalysis for targeting such organic transformations.
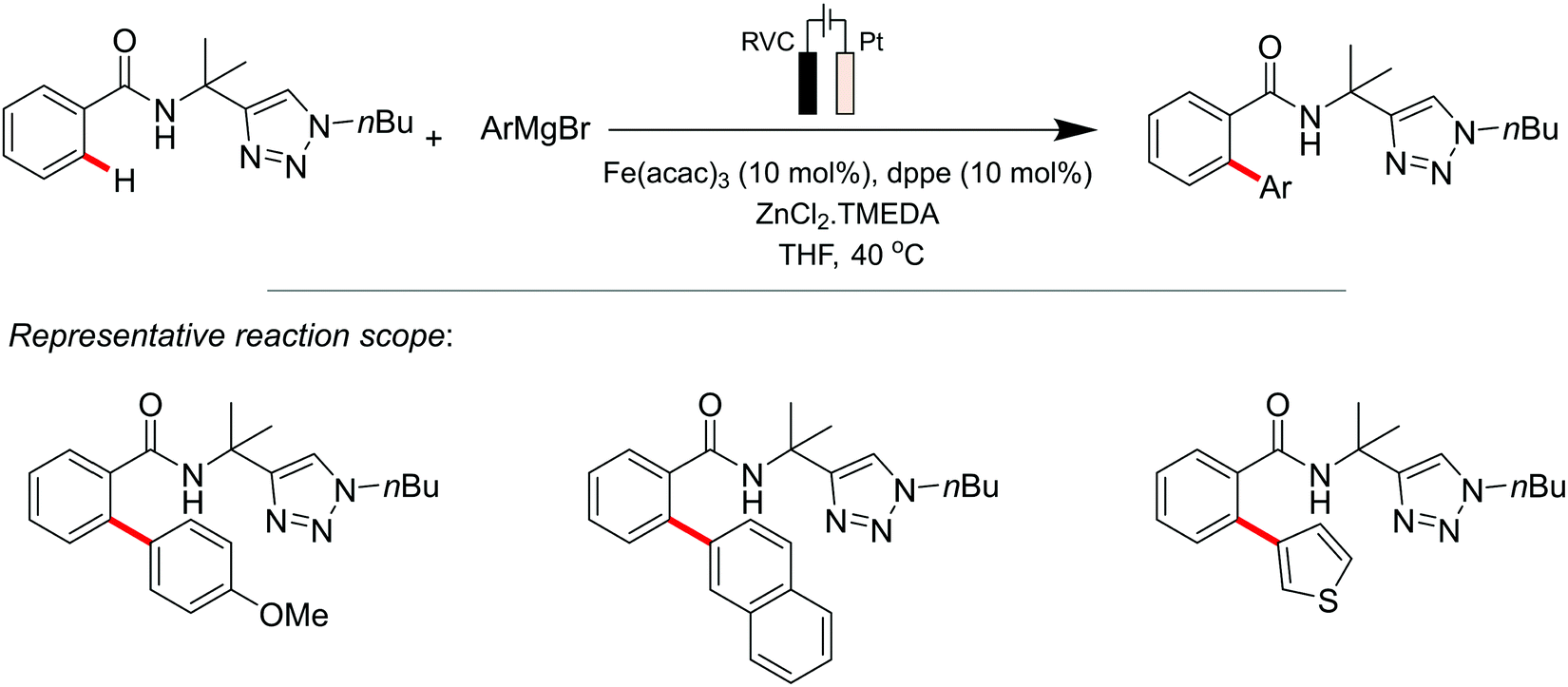 | ||
| Scheme 5 Fe-electrocatalyzed C–H arylation. | ||
Sevov et al.137 reported the Heck coupling with aryl halides and alkenes, replacing the precious-metal Pd with an earth-abundant Ni electrocatalyst for this electroreductive transformation (Scheme 6). Despite parallels in reactivity between Ni and Pd complexes,138 Ni-catalyzed Heck reactions of aryl halides are uncommon, but this seminal report by Sevov highlights the utilisation of electrochemistry to accomplish this rare, Ni-catalysed, transformation. For their catalyst design, an additive (cyclohexanone) was required to convert the redox inert NiII aryl complex to a redox active NiII species. The latter could then subsequently undergo single-electron reduction at the cathode to form a low-valent NiI complex that reacts rapidly with unactivated alkenes, via a migratory insertion and β-H elimination pathway, to yield the Heck coupled product. Their data suggested the occurrence of a stabilising interaction between the electron-deficient additive and the reduced NiI intermediate, allowing the reaction to procced at a mild potential, thereby preventing degradation of the aryl bromide.
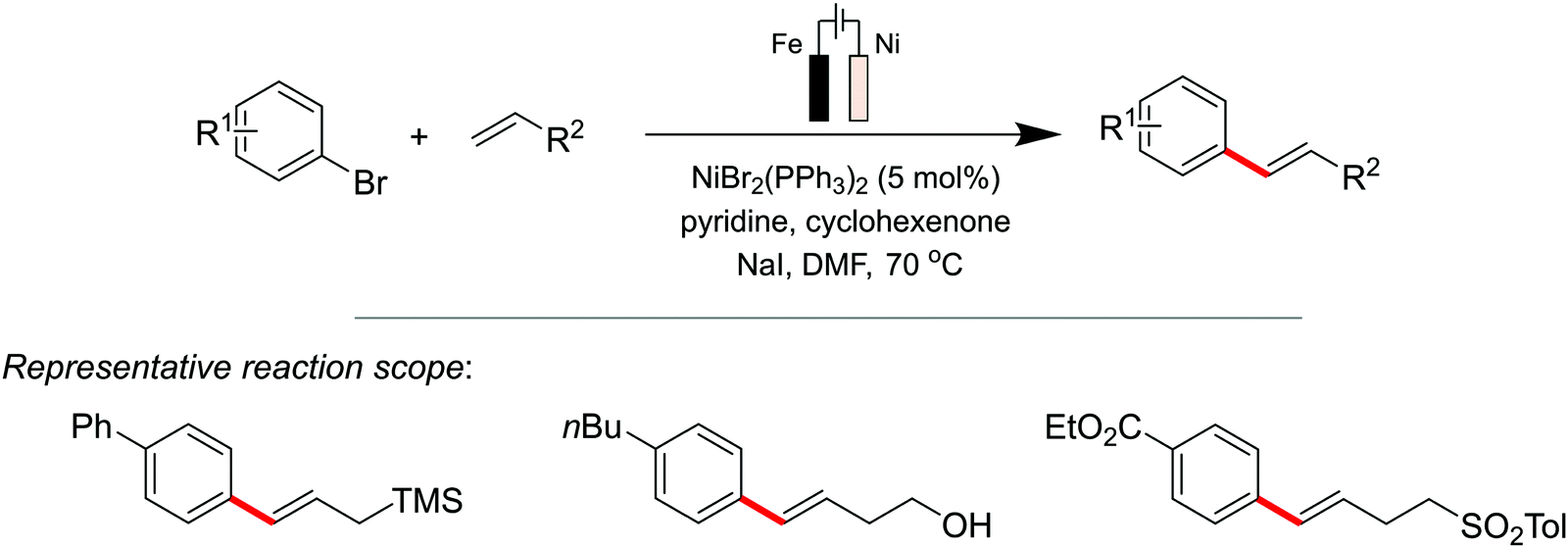 | ||
| Scheme 6 Nickel-catalyzed electrochemical Mizoroki–Heck coupling reaction, where cyclohexenone was employed as an additive to transform the redox inert NiII aryl complex to a redox active NiII species. | ||
These are but a few examples to demonstrate the electrocatalytic versatility of homogeneous metal catalysts. As mentioned, the metal can take the form of a salt or a neutral complex within the reaction mixture, and these studies depict the variation in catalyst design for various targeted transformation. They also shed light on the ability to utilise earth-abundant 3d metals for green applications. The homogeneous catalysts presented herein exhibit unparalleled catalytic activity and selectivity at mild conditions, well-defined active sites and tunable coordination environments,127 which allows for the rational design and synthesis of high-performance catalysts based on the established structure–property correlations, and the possibility to describe their reactivity with atomic precision. In a practical sense however, homogeneous catalysts are limited by low stability and a typically complex separation and recovery process, thereby impeding their application within an industrial context.139 Furthermore, Pt is typically used as one of the electrode materials, increasing the overall cost of the process, or a sacrificial anode is employed, further complicating the reaction process and leading to the additional generation of wasteful by-products. In several cases, the actual role of the additives employed is not always clear, and their exact function vis-à-vis the net reaction is ambiguous. In an analogous manner to the homogeneous catalyst itself, their presence tends to complicate the process and lead to additional steps in the purification stage.
For most of the reactions explored within the realm of electrochemical organic synthesis, non-targeted, commercial first-generation electrodes (such as carbon rod, platinum, and reticulated vitreous carbon) are applied as the current collectors. The advancements made using well-designed reaction-specified electrodes in improving the catalytic activity for energy conversion heterogeneously catalysed reactions, such as water splitting, nitrogen reduction reactions, and CO2RR,140–142 is further testament to the importance of electrode design for improving the rate of catalysis. This sheds light on the huge gap between the design of electrode materials and the requirements for efficient and sustainable organic electrosynthesis. Examples involving the use of modified electrodes for heterogeneous catalysis are indeed quite scarce in the literature, but we envision that this particular area of research will gain traction soon. Some key designs have indeed been envisioned, albeit for organic transformations which lie within or outside the five dominant reactions listed earlier. Nonetheless, they are included herein in order to present the latest advancements made within this sector.
Zhang adopted a bi-functional Ni2P nanosheet electrode, but extended the anode reaction to include the selective semi-dehydrogenation of tetrahydro- to dihydroisoquinolines,143 while Grätzel and Hu used a robust photoanode, haematite, to conduct C–H/N–H coupling of anisole with pyrazole in a photoelectrochemical setting.144 Manthiram and co-workers developed an electrochemical method to epoxidize alkene substrates at room temperature and ambient pressure.145 The catalytic system incorporated a carbon electrode coated with monodisperse manganese oxide nanoparticles, while water was the sole oxygen atom source.
Reisner et al. modified an indium tin oxide electrode with a silatrane-functionalised TEMPO electrocatalyst to yield a hybrid anode, for the selective oxidation of glycerol to glyceraldehyde, which was concomitantly coupled with CO2RR in the cathodic compartment (Fig. 6A).146 Wang and co-workers successfully identified the optimal geometrical sites in Co3O4 spinel oxide for the electrochemical oxidation of hydroxymethylfurfural (HMF),147 and showed that CoII tetrahedral sites were capable of chemical adsorption for acidic organic molecules while CoIII octahedral sites play a key role in the oxidation pathway of HMF (Fig. 6B). Based on these findings, the rational design of a specifically tailored mixed spinel oxide (CuCo2O4) was accomplished, to yield an anodic system with an enhanced electrocatalytic activity for HMF electrooxidation relative to Co3O4.
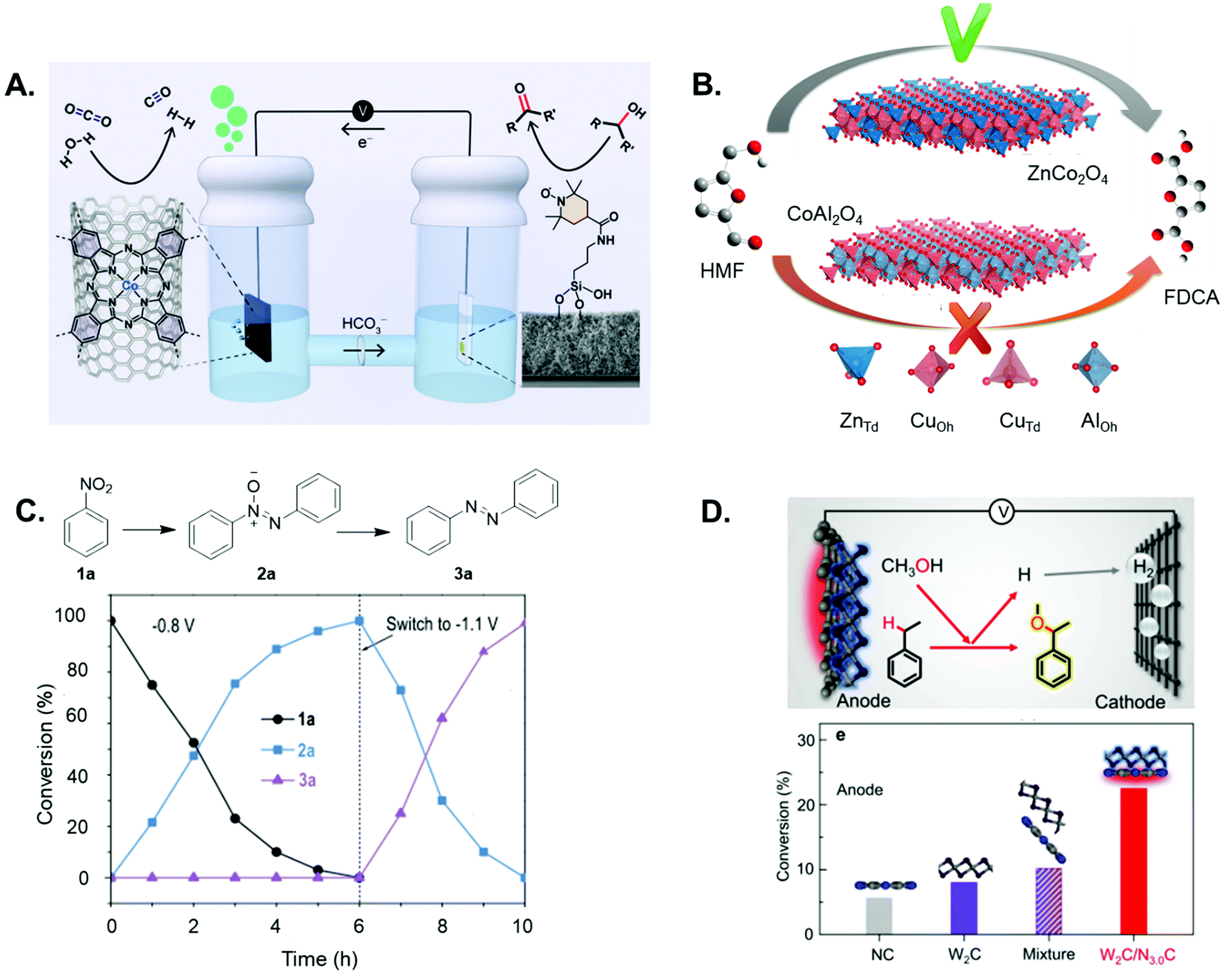 | ||
| Fig. 6 (A) Coupled alcohol oxidation and CO2 reduction electrolyzer, featuring a mesoporous indium tin oxide scaffold modified with a silatrane-anchored TEMPO electrocatalyst to yield a hybrid anode for selective alcohol oxidation (right-hand compartment), and a polymeric cobalt phthalocyanine CO2 reduction electrocatalyst, deposited onto porous carbon paper to yield the cathode (left-hand compartment) (reprinted with permission from ref. 146). (B) Scheme to show the relative performance for electrochemical HMF oxidation to the targeted product on spinel oxides, whereby tetrahedral and octahedral sites in Co3O4 were substituted by Zn2+ and Al3+ to form ZnCo2O4 and CoAl2O4, respectively (reprinted with permission from ref. 147). (C) Time-dependent conversion plots for the cobalt phosphide electrocatalysed formation of azobenzene, which can be efficiently synthesized through a one-pot two-step procedure, involving first the formation of azoxy products at a less negative bias (−0.8 V vs. Ag/AgCl) and the subsequent reduction to azo at more negative potentials (−1.1 V vs. Ag/AgCl) (figure adapted from ref. 149). D. Schematic illustration of the coupled electrolyzer, including a highly coupled W2C/N3.0C dyad as the anode for the electrocatalytic alkoxylation of ethylbenzene with methanol, and a Ti cathode for H2 production (top); conversions of ethylbenzene on the W2C/N3.0C anode and control anodes, featuring the same amount of a bare NC sample, W2C catalyst, or a mechanical mixture of the two components (figure adapted from ref. 155). | ||
To support green research efforts to explore the synthesis of deuterated pharmaceuticals, the Zhang group also pioneered an electrocatalytic strategy for the one-pot halogenation–deuterodehalogenation of C–H to C–D bonds, without isolating the halide precursors.148 Here, they employed noble-metal-free Cu nanowire arrays as a low-cost cathode to electrocatalyse the reductive deuteration of halides, using D2O as the deuterium source under ambient conditions. The same group engineered a CoP nanosheet cathode for the electrocatalytic reduction of nitro substrates to yield azoxy-, azo- and amino-aromatics,149 highlighting a more sustainable route to generate compounds used in, for example, dyestuff production.150 The authors describe how the selectivity can be tuned by varying the applied potential/s, in order to modulate which nitro reduction product is generated at the cathode (Fig. 6C). A two-electrode electrolyser featuring the CoP cathode and a Ni2P anode was then assembled, to couple two synthetically useful half-reactions – the anodic oxidation of aliphatic amines to nitriles, and nitrobenzene reduction to azoxybenzene.
Sun and co-workers reported a heterogeneous electrocatalytic system comprised of monodisperse CuPd nanoparticles, for the selective cross-coupling of alkyl halides and allylic halides to form C–C hydrocarbons in aqueous solution and at room temperature.151 Yu et al. report an electrolyser featuring a nanostructured NiFe oxide anode and a NiFe nitride cathode, synthesized from NiFe layered double hydroxide nanosheet arrays on three-dimensional Ni foams, for the purposes of conducting glucose oxidation and HER, respectively.152 The coupled system demonstrated a high yield for glucaric acid in the anodic compartment, a “top value added compound” derived from biomass, owing to the fact that it is a key intermediate for the production of synthetic, biodegradable polymers.153,154 Electron-deficient W2C nanocrystal-based electrodes for the highly efficient electrochemical alkoxylation of C–H bonds were developed by Li et al.155 They highlighted the importance of the modified physicochemical properties of electrode materials in boosting additive-free C–H activation reactions, and focused on the alkoxylation of ethylbenzene with methanol on the electron-deficient W2C nanocrystal-based anode (Fig. 6D). The authors were thus able to tune the electron density of W2C nanocrystals by constructing Schottky heterojunctions with nitrogen-doped carbons, so as to promote preferred adsorption of benzylic C–H bonds of ethylbenzene on the electrode surface. This greatly facilitated subsequent C–H activation, which is the rate-limiting step in the overall transformation. The efficient oxidative reaction was simultaneously coupled with HER at the cathodic side.
These heterogeneous catalysts are highly appealing owing to their high stability, easy separation and recyclability, and facile immobilisation for continuous flow (electro)catalytic applications. However, their broad distribution of particle sizes and surface characteristics leads to ambiguity vis-à-vis the role of catalysis and the exact nature of the active crystal facets, edges, and corners which give rise to such effects. This makes it difficult to deduce accurate structure–function relationships which facilitate the fundamental mechanistic understanding and rational design of active sites with tailored activity and selectivity.17 The combination of varying size and surface features also implies that overall atom efficiency with regards to metal utilisation is low, and certain atoms present on the surface may actually be inert or trigger undesired side reactions.139 These disadvantages push for a foundational rethink in the type of catalytic systems for organic electrochemistry, and make SACs an ideal candidate for such applications.
5. Engineering SACs for organic electrosynthesis
SACs can offer unprecedented opportunities for rational catalysis engineering (Fig. 1). The aspect of resource economy intrinsic in SACs is an encompassing term to consider,156 as it deals not only with the energy source used to synthesize the desired chemical feedstocks, but also delves into aspects related to the sustainability of the catalysts employed. There is thus a high incentive to replace precious metal with earth-abundant catalysts, and to atomically tailor the material so as to maximise both the reactivity of the active sites and the utilisation of metallic components.157 SA-based electrocatalysts can be tailored to selectively conduct targeted organic transformations. At a suitable electric potential, these materials can become activated and will oxidize or reduce reactants to generate reactive intermediates that can then undergo subsequent reaction(s), with no permanent chemical changes to the material itself.158,159The properties of the support materials (e.g., composition and nanostructure) can impact the coordination environment and hence, the mechanism of the electrocatalytic reaction under observation.67 The presence of foreign atoms, such as N or B, at certain sites of the support, may tune the electronic properties of the composite material, and enhance catalysis in that way. In effect, noble metal-free nanocarbon materials featuring M–N–C moieties show an electrocatalytic activity comparable with (and often exceeding that of) Pt-based materials for ORR.160
Carbon-based materials have widely been adopted as metal-free supports in the fabrication of SACs for electrocatalytic applications. This is principally due to electrical conductivity, high specific surface area, tunable porous structure, and versatility for chemical functionalisation.37,65,161,162 An array of supports featuring carbon nanotubes, graphene, graphdiyne, graphitic carbon nitride (g-CNx), and porous carbon substrates derived from metal organic frameworks (MOFs) or polymers,3,163–173 have been developed using a variety of synthetic methodologies. Generally, pyrolysis and wet chemistry techniques have been most commonly employed for the preparation of SACs based on such carbon substrates. Wet chemistry methods do not require any special equipment and are typically easy to operate, hence their potential for the large-scale production of SACs. The impregnation approach is one such example of this synthetic route, and relies on the chelation between metal centres and N, O, or S coordination sites in carbon-based materials. For instance, Vilé et al.29 synthesized Pd SACs supported on mesoporous graphitic carbon nitride (mpg-CNx) by mixing PdCl2 and NaCl in a mpg-CNx dispersion under magnetic stirring and sonication, followed by NaBH4 reduction (Fig. 7A). This was primarily ascribed to the strong coordination between the metal centres and the N coordination sites of mpg-CNx. The procedure has also been used to prepare other catalysts, such as Pt-g-CNx, Ru-g-CNx, and Ni-g-CNx SACs.26,174,175
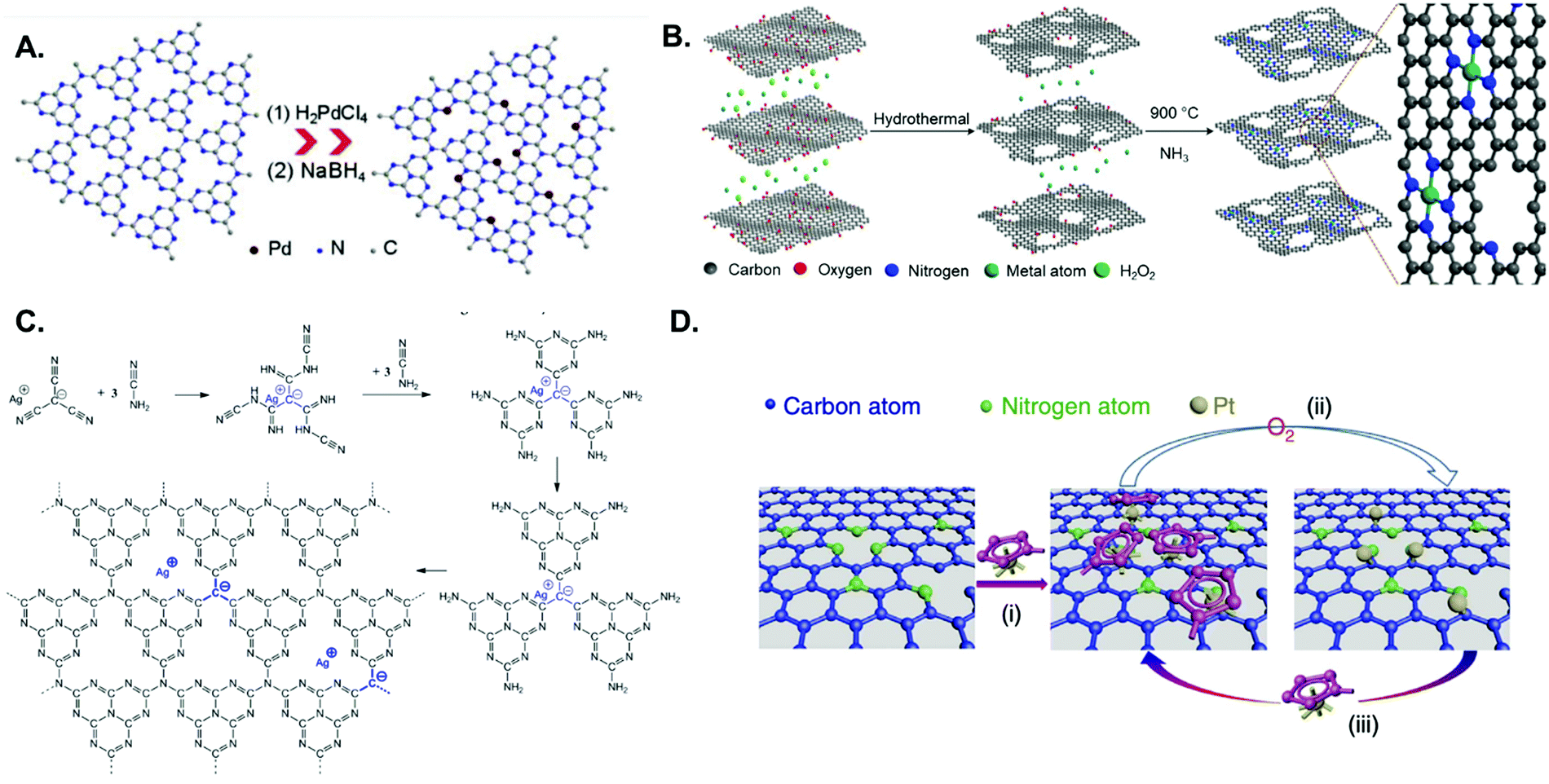 | ||
| Fig. 7 Examples of metal SACs supported on carbon materials, prepared through various means: (A) impregnation; strongly anchored and isolated Pd species in the host cavities of the mpg-CNx framework, resulting from stirring, sonication, and NaBH4 reduction of the corresponding metal salt in the presence of mpg-CNx (adapted from ref. 187). (B) Pyrolysis; preparation of single metal atoms embedded in nitrogen-doped holey graphene frameworks with MN4C4 moieties, in which the precursors are metal ions adsorbed on a 3D graphene hydrogel and the pyrolytic step is performed under an NH3 atmosphere (reprinted with permission from ref. 176). (C) Copolymerisation method; Ag-CNx derived from silver tricynomethanide and cyanamide (reprinted with permission from ref. 178). (D) ALD; schematic illustration of the Pt ALD mechanism on nitrogen-doped graphene nanosheets, in which the Pt precursor (MeCpPtMe3) first reacts with the N-dopant sites (process (i)). This is followed by O2 exposure, in which the Pt precursor on the nanosheets is completely oxidized, creating a Pt containing monolayer (process (ii)). The new adsorbed oxygen layer that forms on the Pt surface provides functional groups for the next ALD cycle (process (iii)) (reprinted with permission from ref. 184). | ||
Pyrolysis has been used rather extensively in the preparation of SACs by thermal decomposition of select precursors, such as carbon matrices and MOFs, at elevated temperatures under a controlled atmosphere (e.g., N2, NH3, Ar or H2). Fig. 7B illustrates the synthesis of SACs from a carbon matrix and features single metal atoms embedded in a nitrogen-doped holey graphene framework (M-NHGF, M = Ni, Co and Fe).176 The two-step process involves the initial creation of a metal ion-containing porous hydrogel, obtained by hydrothermal treatment of a graphene oxide (GO) solution with metal salts and H2O2, followed by pyrolysis at elevated temperatures under an NH3/Ar atmosphere. The NH3 gas acts as the reducing agent and nitrogen dopant source, thus serving a dual function in the procedure. In the final products, MN4 moieties are formed and embedded within the graphene lattice. MOFs are another interesting SAC precursor, on account of their well-defined porous structure and highly ordered arrangement of organic linkers and metal nodes. Li and coworkers for instance showed that during pyrolysis of a Co-, Zn-containing bimetallic MOF (BMOF), the organic linkers were converted into N-doped porous carbon while single Co atoms were formed after pyrolysis and embedded in the N-doped carbon matrix.177
The copolymerisation method and atomic layer deposition (ALD) have also been utilized, albeit to a lesser extent, for the preparation of SAC materials. In the copolymerisation method, metal single atoms are introduced into the g-CNx network during the synthesis of the carbon nitride material itself (in a one-step reaction), rather than post-synthetically as in the case of the impregnation method. For instance, Chen et al. created a single-atom material featuring silver atoms dispersed within a carbon nitride polymer (Fig. 7C).178 Silver tricyanomethanide was used as a reactive co-monomer during templated carbon nitride synthesis to introduce both negative charges and silver atoms to the system. ALD is a deposition technique based on the sequential use of a gas phase chemical reaction. Due to the ability to control the amount of material that is deposited, it has been used for the synthesis of nanoparticles, nanoclusters, and even single atoms.179–186 For example, the Sun and Botton groups were able to tailor the ALD cycle numbers, in order to fabricate Pt SACs, Pt nanoclusters, and Pt nanoparticles on supported graphene surfaces, using (methylcyclopentadienyl)-trimethylplatinum as the Pt source (Fig. 7D).179,181,184
An in-depth understanding of the phenomena occurring at the catalyst surface with atomic resolution and their micro-kinetic consequences at the reactor scale is also of paramount relevance for the rational design of SACs. This comprehension remains however complex in the presence of SACs. For example, four types of supports are typically used for single-atom catalytic applications, and include: (i) bulk metals and alloys, (ii) nanoparticles, (iii) metal-nonmetal compounds (e.g., doped/defective oxides and chalcogenides), and (iv) carbon-based materials.188 Among these, carbonaceous supports, such as carbon-nitride structures (e.g., Cu–CNx)189 or metal-organic frameworks built on metal-porphyrin structural motifs,190 are particularly interesting candidates for modelling applications. The local coordination of a supported metal single atom determines the reactivity, and hence the activity of the catalyst. For instance, as shown in Fig. 8, the local coordination of N-dopants and SACs determine (along with the chemical nature of the single-atom species) the activity of SAC/N-doped graphene systems for HER.191 Therefore, the identification of the active site and the rationalization of the electronic properties is a critical aspect to be addressed. Single atoms supported on such materials are less computationally demanding than conventional oxides, given their smaller size and corresponding number of possible conformations, which makes it affordable to screen various metal atoms on a single support, or a specific metal atom supported on different scaffolds. For instance, a computational screening over several metal–porphyrin adducts showed how cobalt could in principle fulfil CO2 electroreduction at a rather small overpotential (0.2–0.3 V).190 Even more promising results were reported for a non-noble SAC, namely Fe, anchored to a graphdiyne support,192 where the reaction free energy profile for the CO2 electroreduction to CH4 and CO2 carbonylation to CH3CH2OH envisages a small ΔG, and thus, the need for only a moderate applied potential.
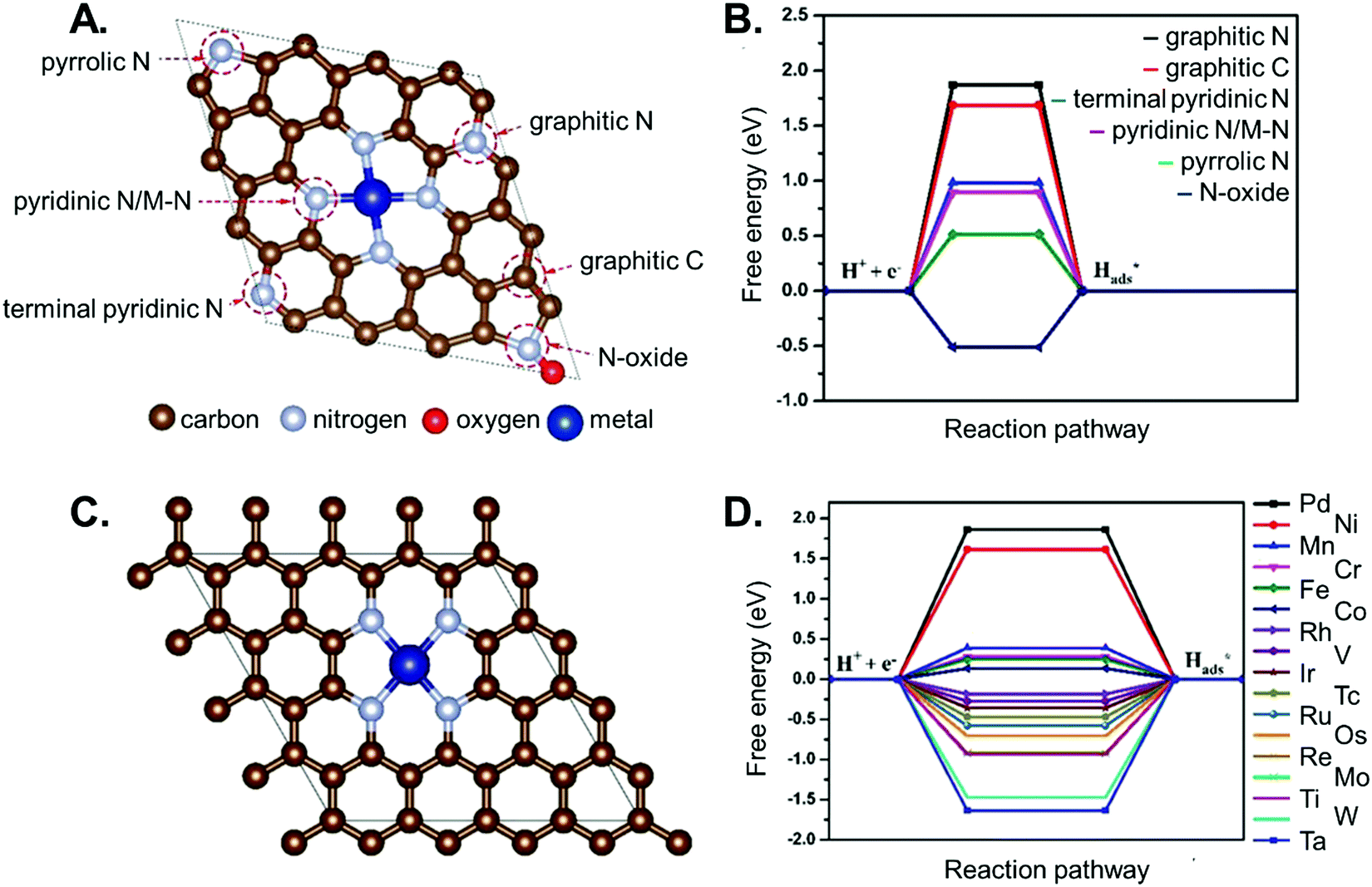 | ||
| Fig. 8 Hydrogen adsorption free energy on different metal single atoms supported on N-doped graphene. Panel A shows possible metal binding sites in nitrogen doped graphene. Panel B reports the calculated Gibbs free energy of H adsorption on the different sites. Panel C shows a metal atom coordinated by four nitrogen dopant atoms, and panel D reports the corresponding calculated Gibbs free energy for the adsorption of a hydrogen atom on top of different metals. Figure adapted from ref. 191. | ||
In most of the examples discussed in the literature, theoretical modelling focuses on the coordination, activation, and eventually the reaction of small molecules, such as hydrogen, methane, ethane, CO2, and so on. However, nowadays, DFT can treat much more complex molecular substrates, up to some hundredths of atoms, such as organic moieties. Nevertheless, when modelling a heterogeneous catalytic process, one must consider that the overall computational burden does not only scale with the physical size of the system (namely, N3, where N is the number of atoms included in the structure) but also with the number of possible configurations that a reactant molecule may assume on the catalyst's surface. The latter factor grows steadily with the increasing structural complexity of a more or less bulky organic molecule, where rotation around the axis of σ bonds and any other form of structural isomerism contribute to the complexity of the problem. It is thus appealing to adopt a reductionist approach, relating the properties of complex systems to their simpler components: hydrogen/oxygen evolution as a key to redox chemistry, CH4 activation as model system for C–H bond cleavage and so on.
In spite of a remarkable, and steadily growing, number of publications featuring computational studies of SAC applications in thermal and (to a smaller extent) electrocatalysis, more work is necessary toward a full understanding of the complex structure–activity relationships of SACs, involving both the chemical reactivity of the single atoms, their (often not univocally defined) coordination to the support, and the role of the environment. This poses severe challenges from the point of view of the level of theory, the adequacy of the structural models and the number of variables at stake. Nevertheless, we foresee that theoretical research will be performed in this area. It is thus an exciting moment to conduct studies aiming to find novel and efficient catalysts, along with catalytic conditions, for greening synthetic chemistry.
6. Emerging trends in continuous-flow organic electrosynthesis
In stark contrast with classical chemical reactors which have been employed for several decades, where the setup characteristics (i.e., round-bottom flask or similar glass vessels) has a minor impact on the outcome of the transformation, organic electrosynthetic transformations are very sensitive towards the design of the reactor (Fig. 9).193 Consequently, understanding the effect that every component of the setup has in the electrochemical reaction is of paramount importance towards developing an efficient and reproducible chemical process. The main features that must be taken into account during the design of electrochemical reactors are not only the catalyst type, but also the cell and separator, transport phenomena (including fluid dynamic regime), supporting electrolyte, and choice of electrode. Once these parameters have been studied and defined, electrochemistry is, undoubtfully, in a privileged position to develop new environmentally benign chemical transformations, using electricity as the greenest source of electrons imaginable, replacing toxic, expensive, and dangerous reducing/oxidizing agents.194 | ||
| Fig. 9 Synthetic chemistry has largely remained unchanged in the past centuries, dominated by the use of batch reactors. This has also been a standard in electrochemistry. | ||
An electrochemical reaction is composed of two half-reactions (reduction and oxidation), and in the basic (batch-type) setup, the anode and cathode share the same compartment/chamber, i.e., an undivided cell. However, a divided cell must be employed when high energy intermediates can prematurely react at the other electrode, giving rise to deleterious reaction pathways. In this case, the anodic and cathodic chambers have to be separated using for example, a salt bridge, a glass frit, a porous ceramic, a porous polymer sheet, or an ion-selective (cation or anion) exchange membrane.195 Hence, the selection of either a divided or undivided microreactor is one of the most important decisions to be made, influenced by the targeted reaction and catalyst type.
In both the divided and undivided cases, batch electrochemical cells tend to suffer from a lack of design specificity; for instance, scaling up the volume would lead to an increase in the relative distance between the two electrodes (the interelectrode gap, d), giving rise to a higher ohmic drop (iRu). This drop is a result of the resistance encountered by the flow of charge (i) through the solution, and can be represented by:
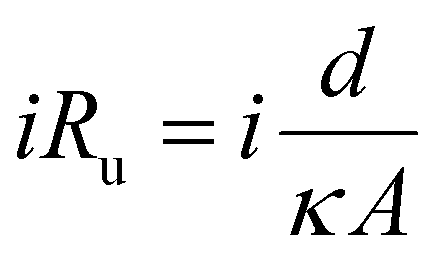 | (1) |
One of the most effective techniques to circumvent these issues is through the implementation of continuous-flow microreactors (Fig. 10A), as supported through comparative techno-economic analysis (TEA) studies performed on batch and flow processes.198–200 Such devices provide a high surface-to-volume (S-to-V) ratio for the catalytic layer; this also implies a narrow d value (below 1 mm) and thus, lower ohmic drop.201 Consequently, such short distances allow to reduce the solution conductivity, and therefore minimise the amount of supporting electrolyte, rendering a greener overall methodology. The undivided setup can be used as a quintessential example to highlight the construction of a flow electrochemical microreactor with a “parallel plate” geometry (Fig. 10B). Such a reactor can be made with either a metal body (e.g., stainless steel),202 or from polymer resins by using additive manufacturing technology (e.g., 3D printing).203 The latter route offers the possibility for rapid reactor prototyping at lower costs, and enables the user to easily customise the dimensions and features of the reactor body in accordance with the specifications of the reaction. The large electrode area (ranging from 15 to 50 cm2 each) allows to develop fast and reliable electrochemistry and enables scalability. The two electrodes are separated by a thin elastomer polymer (e.g., PTFE or FEP) gasket, in which a reaction channel is carved. The distance between the electrodes is governed by the thickness of the film (typically 100–500 μm), and the internal volume of the microreactor. The polymer spacer also serves as a sealant between the two electrodes. In addition to this, solid polymer electrolytes (SPE, also known as a membrane-electrode assembly or MEA) provide an alternative to these systems, where the supporting electrolyte can also be avoided, due to the fact that the separator also functions as an ion exchange membrane, thus avoiding the wasteful processing steps of having to separate and recycle the supporting electrolyte of typical electrosynthetic methodologies.204,205
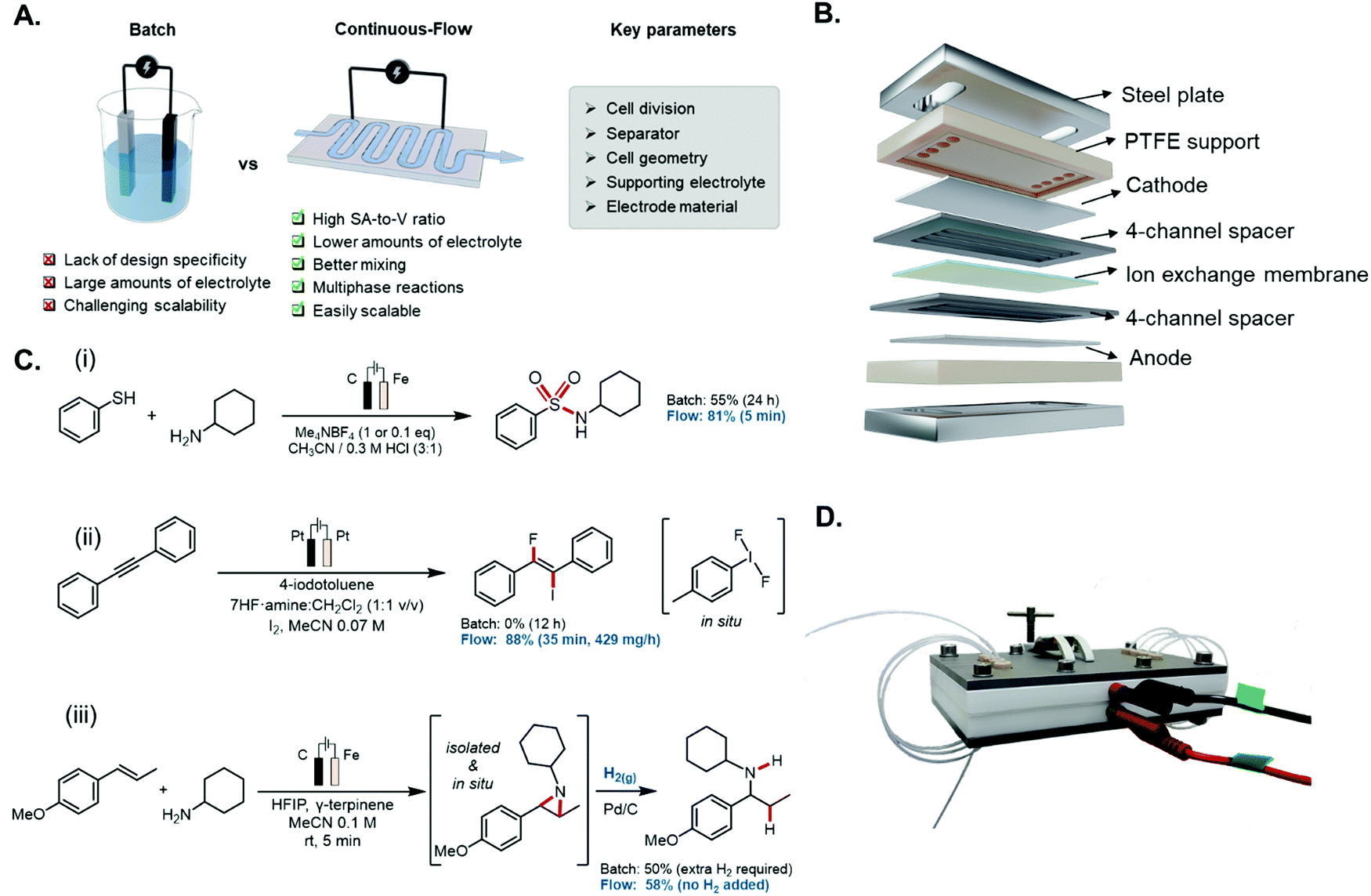 | ||
| Fig. 10 (A) Advantages of electrochemical flow cells over traditional batch cells. (B) Exploded schematic of a divided-cell parallel plate flow microreactor. (C) Selected examples of electrochemical flow microreactor applications versus analogous batch reactions: (i) electrochemical synthesis of sulfonamides; (ii) iodofluorination of alkynes with electrochemically generated (difluoroiodo)arenes; (iii) two-step biphasic hydroamination of in situ generated aziridines. (D) Image of an assembled flow electroreactor. | ||
Flow cells based on the “parallel plate” geometry can be operated in two ways: (i) by recirculating the reactant solution between the cell and a reservoir, or (ii) in a single pass of reactant, through the electrochemical cell. A single pass of the reagents has the advantage that the chemicals are only briefly exposed to the electrochemical conditions resulting in less by-product formation.195 In the past few years, several research groups have taken advantage of all the benefits that microfluidic systems can provide in electrochemistry to develop more efficient transformations, such as lower amounts of supporting electrolyte, faster reaction times and chemoselective processes, even in complex multiphasic systems. For instance, Noël et al. applied this technology for the electrosynthesis of sulfonamides via oxidative coupling of thiols and amines.206 They could reduce the amount of supporting electrolyte Me4NBF4 from 1 equivalent in batch (d = 1 cm) to 0.1 equivalents in flow (d = 250 μm), and also the reaction time could be shortened, from 24 h in batch to only 5 min in flow (Fig. 10C(i)). In another example by Yoshida et al., the anodic methoxylation of several organic compounds was carried out in an electrochemical reactor with an even narrower interelectrode gap (d = 75 μm).207 This allowed the researchers to avoid completely the addition of supporting electrolyte while still obtaining high conversions and yields. Wirth and co-workers surmounted the batch limitations associated with the synthesis of unstable, toxic (difluoroiodo)arenes. Exploiting an in situ generated hypervalent iodine via anodic oxidation in flow, they could develop a range of different chemical transformations, such as fluorocyclisations, di- and monofluorinations, ring contractions and iodofluorination of alkynes. Microfluidic technologies allowed to develop electrochemical transformations that were unsuccessful in batch due to the instability of the difluoroiodo arenes (Fig. 10C(ii)).208
Another prevailing scenario when developing electrochemical reactions is the involvement of multiphasic mixtures. Among the different immiscible phases combinations, one of the most common systems in electrochemistry are gas-liquid reactions, where reduction of H+ takes place at the cathode, giving rise to H2 gas as a by-product. This gas evolution can result into complex scenarios such as non-uniform local current densities alongside the reactor or formation of gas layers which can lead to a substantial rise in ohmic resistance.209 On the other hand, the formation of these gas bubbles can increase dramatically the mass transport phenomena, taking advantage of the toroidal vortices generated between two liquid segments.210 In sharp contrast with batch setups, where this area is poorly defined, microfluidic setups present an exceptional S-to-V ratio. Therefore, continuous-flow technologies present an excellent opportunity to optimize multiphasic reactions. Noël and collaborators used this for the hydrogenation of electrochemically generated aziridines.211 Anodic oxidation of alkenes and amines in a microflow electrocell gave rise to a wide variety of aziridines, with concomitant formation of hydrogen gas bubbles. By connecting in-line a packed-bed reactor filled with Pd/C to the electrochemical flow reactor, they were able to consume all the gas generated in the first step to ultimately obtain the corresponding hydroaminated products, without using additional hydrogen gas (Fig. 10C(iii)).
Compatibility with scale-up is an important concept for any given chemical transformation that is to be adapted from laboratory scale to pilot and production scale. In this regard, electrocatalytic transformations are surface dependant, so classical strategies, such as enlarging the dimensions of the reactor, fall short due to the concomitant increase in the interelectrode gap. Therefore, industrial electrochemical reactors consist almost exclusively of flow cells and numbering-up approaches.212,213 In these systems, a narrow interelectrode gap can be preserved, and by using stacked modular units a high specific area with a low cell voltage can be achieved.
Thanks to the benefits of flow microreactors in electrochemistry and the unique qualities offered by this technology, significant progress has been made in the field of large-scale organic synthesis.214 In 2019, Baran, Minter, Neurock and collaborators targeted the challenge of performing a reductive electroreduction of aromatic rings in a chemoselective, safe and scalable fashion, hence updating the classical Birch reduction conditions (Scheme 7A).215 Initial investigations allowed them to find proper reaction conditions in batch, avoiding strongly reductive alkali metals and using a cheap, nontoxic proton source. The 0.1 mmol scale developed in batch could be increased in a safe and sustainable manner to a 100 g scale, making use of a cheap and robust modular flow setup. Also, Weber et al. in collaboration with GlaxoSmithKline reported the hydrogenation of nitroarenes, nitriles and unsaturated aldehydes using a continuous-flow electrocatalytic system (Scheme 7B).216 Inspired by redox flow batteries, they successfully developed three sizes of reactors with rectangular catalyst cavities from 5 cm2 to 100 cm3 and a turbulent flow regime. Using two 27 mL reactors in parallel, they could produce a combined stream of 2.4 g h−1 of the reduced nitroarene into the corresponding aniline. Given that the reaction kinetics were close to first order, a linear extrapolation of those results indicated that a 20 kg y−1 productivity could be obtained, and up to 500 kg y−1 if an assembly of 50 reactors in parallel would be used.
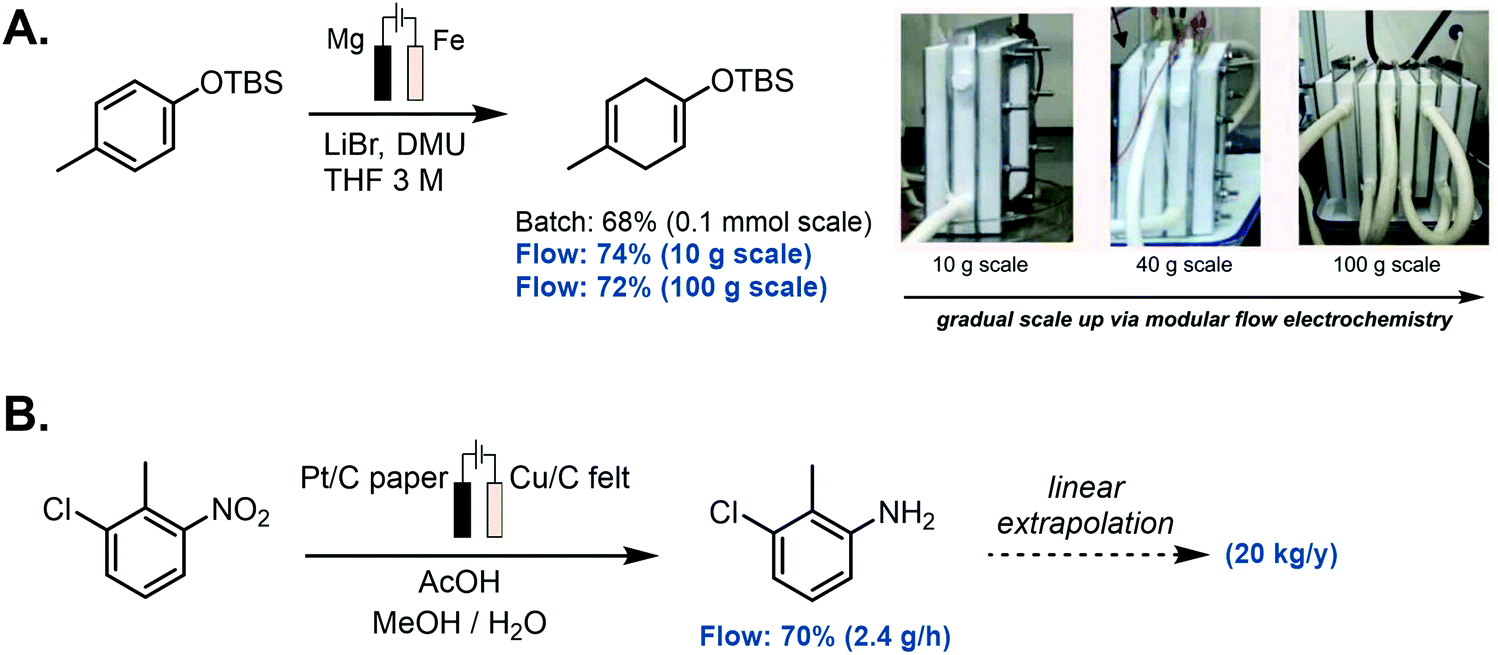 | ||
| Scheme 7 Selected examples of scalable electrochemical reactors in flow: (A) modular scale-up of Birch-type reduction, (B) electrochemical reduction of relevant nitroarenes. | ||
7. Combining SACs with continuous-flow reactor technologies
One of the most critical components in an electrochemical cell are the electrodes. Hence, particular attention is needed when selecting the type of electrode material, the morphology of the electrode, in addition to the incorporation of either an immobilised or a homogeneous electrocatalyst. Most electrodes consist of a single conductive material, although electrocatalytic coatings can also be used for the case of heterogeneous or hybrid electrocatalysts.49 For heterogeneous electrocatalysts, substrates need to be transported from the bulk to the active surface; hence, the high S-to-V ratio afforded by continuous-flow microreactors over batch systems is beneficial from a mass transport and kinetic perspective too.217 Immobilisation of electrocatalytic species in the flow reactor assists with downstream processing (catalyst separation, as required when using homogeneous electrocatalysts, is avoided), and hence facilitates reusability and easier integration within a multistep synthetic process.218As an example, Atobe et al. found out that selecting the appropriate material for the working electrode was crucial in the electrocatalytic hydrocarboxylation of imines in flow.219 They observed a clear relationship between the over potential of the material of the cathode with the yield of the reaction, with glassy carbon being the more efficient material, followed by graphite, platinum, and silver. Wirth and collaborators discovered that the material of the counter electrode was also very important for the outcome of an intramolecular cyclisation of N-centered radicals when translated to a flow set up.203 In their report, they observed that when using a graphite cathode, no reaction was taking place. Interestingly, when switching to nickel, an increase of the yield to 60% was detected, which could be further improved by using a platinum cathode, with a yield of over 90%. In this example, the anode material did not have any impact, so it could be chosen for other reasons apart from its reactivity.
Ideally, the electrode material should be inexpensive, non-toxic, highly stable under the effect of different voltages, current densities, temperatures, solvents, and pressures. Lower resistivities are also highly desired, in order to reduce the ohmic drop, which would require higher cell potentials, giving rise to undesired heating of the system due to the excess energy input. Although this might not have a large impact at the laboratory scale, it might limit the choice of materials for industrial applications.49
In terms of electron transfer processes, the electrode can either be simply inert, acting only as a source/sink of electrons, or it can be modified with an electrocatalyst to yield a heterogeneous electrocatalytic material. Hybrid electrodes, featuring an immobilised molecular electrocatalyst, or single-atom electrocatalysts present two classes of ‘electrode materials’. Both these classes are examples of ‘supported electrocatalysis’, in which the electrons flow from the electrode to the active site to drive the catalysis, through the application of an applied potential that is more positive or negative (anodic or cathodic process respectively) than the onset potential of the catalyst.
The immobilization of molecular catalysts onto surfaces provides a variety of advantages over homogeneous systems. A large portion of the homogeneous catalyst is present in the bulk solution, and thus, since the catalyst is only active while in the diffusion layer close to the electrode surface, is inactive at any given time. Having solubilized catalysts also complicates downstream processing of the post-reaction mixture, since additional treatment is required to recover the catalytic components, and the issue is all the more relevant as we shift towards electrosynthetic organic transformations which typically occur in the liquid phase. Having the catalyst localized on the electrode surface is also beneficial for maintaining direct electronic communication and efficient connectivity between the electrode and the catalyst, implying that the latter is constantly undergoing catalytic turnover under the application of a suitable bias, and is not impeded by mass transfer limitations.220 Metrics used to assess catalyst performance, in terms of system stability and rate of product generation, include the turnover number (TON) and turnover frequency (TOF).221 These quantities become more meaningful for benchmarking purposes vis-à-vis immobilized catalysts, since as mentioned, for the majority of an experiment, catalyst molecules in a homogeneous system are inactive within the bulk solution.222 In contrast to homogeneous systems, product isolation and catalyst recyclability are greatly facilitated through the immobilization procedure, which in turn promotes the integration of such systems within continuous flow electrochemical devices and more industrially relevant processes. Another benefit provided by immobilization is that it has allowed water-insoluble catalysts to be used under aqueous conditions. More generally, this implies that catalyst solubility in any reaction medium is no longer a requirement. Reports have indeed emerged where the catalyst was not operable in homogeneous aqueous conditions but was able to demonstrate catalytic activity within the same aqueous medium once immobilized on the electrode surface.223,224 In light of these aforementioned advantages, SACs, which combine the single active site properties of homogenous molecular catalysts with the immobilized properties of heterogeneous systems, also provide a highly suitable platform for electrocatalytic studies.
The choice of electrode material can help minimize undesirable effects such as electron transfer impedance towards the catalyst, high capacitive currents, side-reactions, and instability under catalysis conditions. Carbon-based electrodes, which include a large variety of carbon allotropes such as glassy carbon and carbon nanotubes (CNTs),225 are commonly used for the fabrication of molecular catalyst-modified electrodes. CNTs for instance, have a high active surface area and conductivity, good mechanical stability, and wide operational electrochemical window, making them a suitable candidate for such applications.
Covalent and non-covalent strategies could be applied in order to modify the electrode surface and anchor a molecular catalyst. For the former technique, derivatization of the sp2 carbon surface through strongly oxidizing conditions, succeeded by a chemical reaction between the modified surface and the specific function on the molecular catalyst, allows for the formation of a covalent linker between the electrode material and catalytic species. Examples of chemically or electrochemically generated derivations on the sp2 surface include ketones, quinones, or carboxylic acid groups (Fig. 11A).226–229 For example, carboxylic acid sites allow for grafting of compounds onto the carbon surface through formation of ester or amide bonds.230,231 However, the defects in the carbon structure induced through oxidizing the surface may lead to the degradation of electrode material, in terms of its conductivity or mechanical stability. The reduction of aryl diazonium compounds on sp2 carbon (Fig. 11B) and the 1,3-dipolar cycloaddition of azomethine ylides (Fig. 11C) provide more benign means to modify carbon-based electrodes, and have been applied to glassy carbon, carbon fibers, or highly oriented pyrolytic graphite,232,233 and CNTs or ordered mesoporous carbon surfaces,234–236 respectively. Noncovalent strategies have also been utilized to modify carbon electrodes without altering their intrinsic properties via grafting procedures. Derivatization of π-conjugated carbon materials with polyaromatic compounds via supramolecular π–π interactions (π-stacking) has been frequently adopted, with the pyrene moiety being one of the most popular functionalities used to anchor catalysts to CNTs and graphene (Fig. 11D).237–241
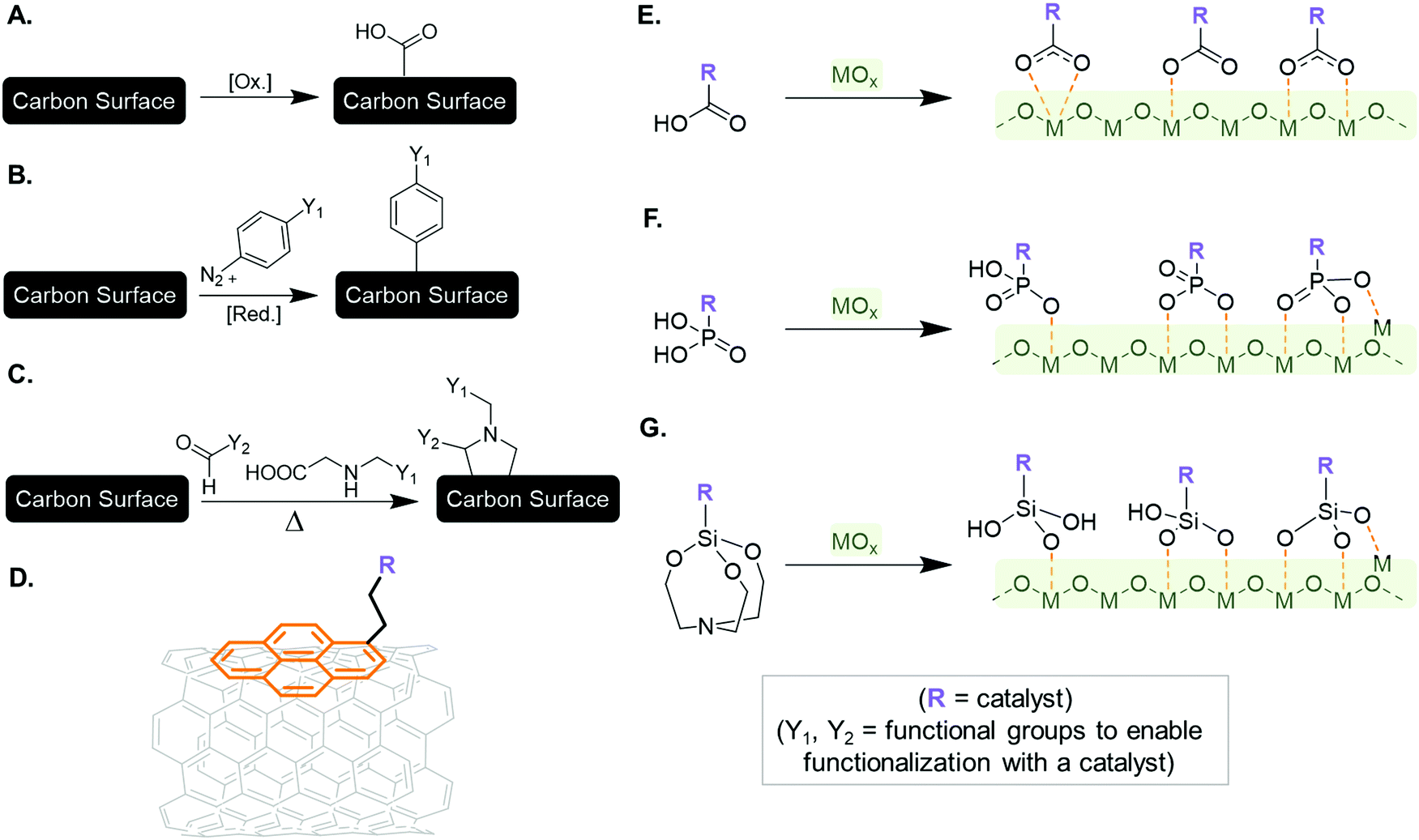 | ||
| Fig. 11 Key examples of frequently used methods to covalently introduce functional groups onto sp2 carbon surfaces, that subsequently enable functionalization with a catalyst: (A) chemical or electrochemical surface oxidation, (B) chemical or electrochemical reduction of in situ generated diazonium salts, and (C) 1,3-dipolar cycloaddition of azomethine ylides. (D) Schematic highlighting the noncovalent modification of a π-conjugated carbon surface with a polyaromatic moiety, enabling π-stacking. Surface binding motifs of some of the various anchors used for grafting catalysts onto metal oxide (MOx) surfaces: (E) carboxylate moieties through a chelating, unidentate, or bridging binding mode; (F) phosphonate anchors through mono-, bi-, and tridentate motifs; (G) silatrane anchor and corresponding silane functions through a mono-, bi-, or tridentate binding mode. Note: carboxylate and phosphonate groups are believed to undergo a condensation reaction with the hydroxy groups at the surface of the MOx, forming an ester-type linkage with the metal; the reaction of the silatrane following a nucleophilic attack from the hydroxy group of the MOx surface leads to the formation of a strong silyl ether-type surface bonding.247 Figure adapted from ref. 159. | ||
Distinct to carbon-based electrodes are the metal and metal oxide-based substrates. Many metallic electrodes exhibit catalytic activity in the absence of any additional molecular catalyst, and in many cases have been incorporated in flow devices for applications in, for instance, CO2 electroreduction (e.g., Cu and Ag).242 Metal electrodes however often present flat surfaces that prevent high loading of the molecular catalyst, and for this reason, metal oxides present an interesting alternative. These materials can be nanostructured to introduce a higher surface area-to-volume ratio which consequently allows for an increased loading of catalytic species.243–246 Additional benefits include their facile synthesis, using low-cost solution processing techniques, and their ability to exhibit different electronic properties, as demonstrated by the metallic behaviour of indium tin oxide (ITO) and the semiconducting properties of TiO2.
A number of chemical functionalities have been employed for anchoring and grafting molecular catalysts to metal oxide surfaces, via the formation of chemical bonds to the metal of the metal oxide surface (Fig. 11E–G),248,249 including carboxylic acids,250,251 phosphonic acids,252–254 silatranes,247,255–258 hydroxamic acids,259–261 acetylacetonate anchors,262 and catechol anchors.263 As per the case of metal electrodes, metal oxide and non-oxide materials can also display active catalytic surfaces. For example, well-known catalysts for the OER (to oxidize water to O2) include the formation of an electrodeposited cobalt–phosphate film,264 and the synthesis of NiO nanoparticles using a solvothermal approach followed by spin-coating or dropcasting on fluorine-doped tin oxide glass to form thin films.265
The list of covalent and noncovalent strategies provided herein, for anchoring catalysts on carbon and non-carbon substrates, along with fabrication techniques for metal oxide and nonoxide electrodes is inexhaustive, and interested readers are referred to other reviews more closely focused on this subject matter.159,266,267
Single-atom electrocatalysts represent the other class of ‘supported electrocatalysts’, which differ from their thermal analogues in that the isolated and anchored metal atoms need to be confined on highly conductive scaffolds, such as heteroatom-doped carbonaceous supports.5 Members of the M–N–C class of materials (where, for example, M = Co, Fe, Ni or Cu) are considered some of the most promising candidates to replace Pt in electrochemical reactions such as the ORR.268 Electrode fabrication techniques for single-atom electrocatalysts (both precious and non-precious SACs) involve the direct formation of the active centers on the aforementioned conductive substrate. This could be through, for example, pyrolysis,130 electrodeposition (potential cycling),269 or impregnation methods,270 as previously highlighted.
8. Identifying the cell potential for SAC-based organic electrosynthesis
DFT may provide an estimate of the redox potential for a continuous-flow electrocatalytic reaction employing SACs. The characterization of a chemical reaction by means of computational tools starts by investigating whether the reaction is endergonic or exergonic. This is done by comparing the Gibbs free energies, ΔG, of the initial state of the chemical process, defined as the sum of the Gibbs free energies of all reactants, and that of the final state, analogously defined as the sum of the Gibbs free energies of all products (eqn (2)):| Reactant1 + Reactant2 + … + ReactantN → Product1 + Product2 + … + ProductN |
 | (2) |
We discuss the practical case of H2 adsorption on a catalytic surface, given its importance in many reactions, including hydrogenations29 and reductive aminations.271 The catalytic active site (*) may be either a metal site, a cation or anion site at an oxide surface, a supported metal nanocluster, or a supported metal single-atom. In this case, the energy of the initial state of the process is calculated as shown in eqn (3):
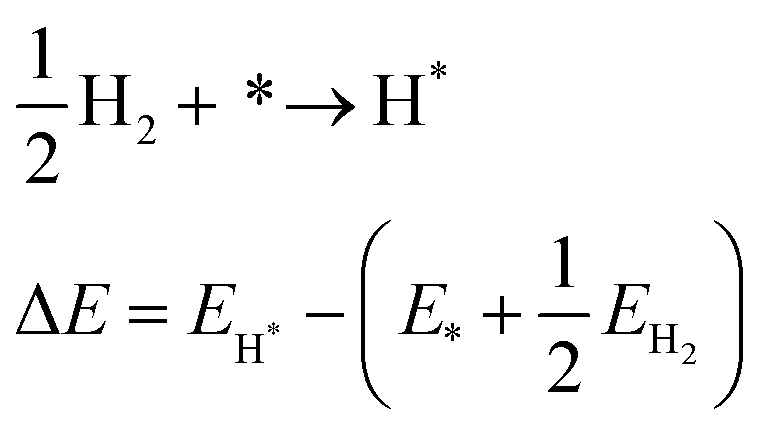 | (3) |
Hence, ΔG can be obtained from ΔE by including two additional and relevant terms, the zero point-energy correction ΔEZPE, and the entropy variation ΔS (eqn (4)):
| ΔG = ΔE + ΔEZPE − TΔS | (4) |
Then, the entropy contribution must be considered, which is very relevant when gaseous species are involved in the chemical reaction, such as hydrogenation, reductive amination, and CO2 carbonylation, to name a few. At 0 K, the entropy contribution is vibrational, while at higher temperatures one should account for translational and rotational contributions too.283,284 In accordance with eqn (3), one can approximate the entropy of the reaction to be (eqn (5)):
 | (5) |
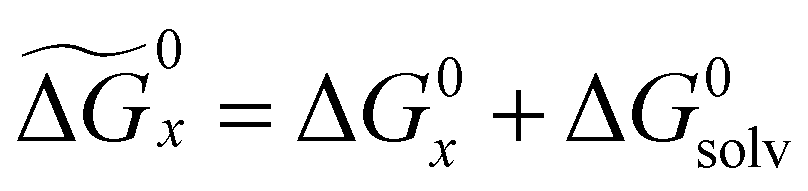 .
.
It is important to note that all the aforementioned contributions are related to the thermodynamic aspects of a reaction, i.e., the energy balance between reactants and products. In reality, the kinetic aspects should be also considered. Any chemical reaction involves an initial induction state where some chemical bonds within the reactants are broken. This leads the system from a thermodynamic minimum state (the reactants) to a transient non-equilibrium state (transition state). The reaction then evolves to a new equilibrium state (the products) through the formation of new chemical bonds. While the free energy difference between reactant and products tells if the reaction is exergonic or endergonic, the energy difference between the transition state and the reactant provides the activation energy, a measure of the barrier to overcome for the reaction to proceed. The picture becomes complicated for multistep processes, where several intermediate stages and possible reaction paths should be considered. However, given a reaction mechanism, one can evaluate the free energy of each reaction step, and hence provide estimates of the required thermal or electrical energy. For instance, the hydrogen adsorption on a catalyst is just the first stage of the reductive amination of benzonitrile (PhCN), which occurs via adsorption of two H2 molecules, causing the reduction of a –CN group to –CH2NH2.271 The calculated Gibbs free energy landscape for this reaction is reported in Fig. 12. The reaction PhCN + 2H2 → PhCH2NH2 is clearly a complex multistep process, where one can identify the molecular H2 adsorption on the catalyst, followed by its transfer to PhCN. Then, a second hydrogen atom is adsorbed on the catalyst and transferred to PhCHNH*, forming and releasing the product, PhCH2NH2, and regenerating the catalyst.
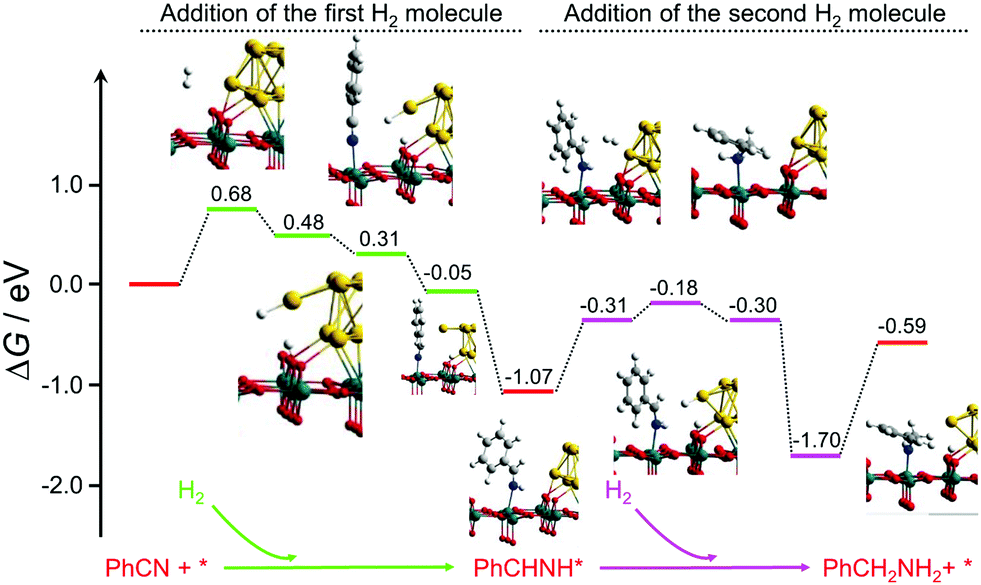 | ||
| Fig. 12 Calculated Gibbs free energy diagram for the reductive amination of benzonitrile to benzylic amine. Figure adapted from ref. 271. | ||
Based on the energy profile, one can estimate the Gibbs free energy of the whole catalytic process (in eV or the equivalent kJ mol−1). To determine the equivalent electrochemical conditions, ΔG0reaction can be associated to a required thermodynamic potential under experimental conditions, E0, by means of eqn (6):
| ΔG0reaction = −n × F × E0 | (6) |
| η = |Eappl − E0| | (7) |
It is clear that under working conditions, the catalyst should bind neither too strongly nor too weakly to the reaction intermediate, leading to an overpotential close to 0 eV. In practice, when modelling a particular electrocatalyst, it is essential to understand both E0 and η, as both parameters will dictate the applied potential needed to sustain the electrochemical reaction of interest. This assists in designing catalysts with sustainable performances over the whole electrocatalytic cycle, and also in unraveling the rationale behind the potentials observed in electrosynthetic experiments. Current DFT methods provide estimates of redox potentials with an error of around 200 mV,292 tacitly considered the limit for a reasonable estimation. It is important to highlight that the computational tools described above, even though developed on classical catalytic systems such as metals or metal oxide surfaces, allow one to study catalytic and electrocatalytic processes on any type of heterogeneous catalysts, including those based on SACs.
9. Concluding remarks and learning outcomes
Ever growing concerns regarding the degradation of our environment and the negative impacts associated with increased levels of CO2 in our atmosphere has fuelled the need for a new generation of greener and more sustainable chemical manufacturing technologies. The field of homogeneous metallaelectrocatalysis has grown significantly in recent years, and the case studies presented herein shed light on the versatility and uniqueness that this field of research has to offer within the context of catalytic systems based on earth-abundant components. However, the downside of homogeneous metallaelectrocatalysts lies in their multi-component characteristics, as they tend to have a number of solubilised catalysts and additives which complicates downstream processing, increases running costs, and hampers scalability. Analogously for heterogeneous electrocatalysts, research efforts are being placed on using well-designed reaction-specified electrodes for improving the catalytic activity of organic redox transformations, and in deducing structure–function properties of the novel materials for such applications. Here, the disadvantages lie in the limited extent of the organic electrosynthetic reactions which have been probed via such techniques (as traditionally, such electrocatalysts have been adopted for energy-related reactions), and the large amount of electrosynthetic reactions which still rely on non-targeted, commercial first-generation electrodes.We have discussed five key reaction types (amide formation, cross-coupling reaction, amine tert-butyloxycarbonyl (Boc)-deprotection, electrophilic reactions with amines, and enantioselective hydrogenation) which dominate contemporary practice to produce compounds in fine chemistry (more than 60% of the reactions). These present a clear reaction space that can be targeted with single-atom electrocatalysis. There are therefore many opportunities to develop catalysts to conduct organic transformations and generate pharmaceutically relevant compounds. SACs combine the merits of the two aforementioned catalysis pillars, and possess well-defined active centres, leading to unique opportunities for the rational design of new catalysts with high activities, selectivities, and stabilities. Their atomically dispersed nature also allows for complete metal utilisation, facilitating the resource economics associated with catalyst development and fabrication. Nonetheless, SAC applications within the electrocatalytic sphere have mostly been reserved for energy-related applications, and studies have precluded the use of such materials within the organic electrosynthetic field.
One of the key technologies that is coupled with the growth of electrosynthetic methods are continuous-flow micro- and electroreactors. Such progressions are enabling diverse chemical transformations by solving the main problems related with batch electrochemical systems, including mass transfer limitations, ohmic drop, scalability, and selectivity. In that sense, understanding the impact that the different parameters of a flow cell can have on the overall reaction outcome is of important to develop efficient electrochemical transformations. Doing so, several research groups have found many useful applications of electrochemical cells in flow, such as fast, chemoselective reactions, in situ generation of highly active reaction catalysts or multiphase reaction conditions, all scenarios that proved to be difficult to work under batch conditions. On top of that, and given the modularity of these systems, large-scale reactions are also within reach, as numbering-approaches allow to increase the scale in a cheap, robust, safe, and sustainable fashion. In addition to that, computational fluid dynamic293 and machine learning-assisted models294 are also being applied to further understand the different physical processes occurring in the electrochemical cells, such as hydrodynamics, mass transport, heat transfer and current distribution, in an effort to streamline the choice of suitable reaction conditions. DFT and computational modelling can be utilised to predict the overpotential requirements of SAC-based electrosynthetic transformations, providing a mechanistic understanding and facilitating the deduction of structure–function correlations, from which more efficient and optimal single-atom electrocatalysts can be rationally designed and fabricated.
In the coming years, we envision that earth-abundant metal SACs will find new applications within the context of organic electrosynthesis, as researchers shift their focus to explore a vast array of applicable chemistries with these novel and unique catalysts. In order to ensure that this vision becomes a reality, computational techniques will complement experimental planning to allow for a deep understanding of the mechanistic aspects of a catalytic reaction at the subatomic level, as provided by accurate quantum chemical simulations. This will provide added value toward the design of reaction-specific, single-atom electrocatalysts and boost interest toward the implementation of machine-learning approaches to the problems related to catalysis.295–301 Continuous-flow electroreactors will also play a pivotal role in this transition, due to the numerous advantages offered over their batch counterparts, providing further industrial relevance and scale-up possibilities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Marie Skłodowska-Curie Fellowship from the European Commission H2020 (M. A. B., 101031710, SSEFR), the Italian Ministry of Education, University and Research (MIUR) through PRIN Project 20179337R7 and grant Dipartimenti di Eccellenza-2017 “Materials for Energy” (G. L., S. T., G. P.), and COST (European Cooperation in Science and Technology) Action 18234 (G. L., S. T., G. P.).References
- M. Monai, M. Gambino, S. Wannakao and B. M. Weckhuysen, Chem. Soc. Rev., 2021, 50, 11503–11529 RSC.
- M. J. Orella, Y. Román-Leshkov and F. R. Brushett, Curr. Opin. Chem. Eng., 2018, 20, 159–167 CrossRef.
- R. Qin, P. Liu, G. Fu and N. Zheng, Small Methods, 2018, 2, 1700286 CrossRef.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- J. M. Thomas, Nature, 2015, 525, 325–326 CrossRef CAS PubMed.
- K. Asakura, H. Nagahiro, N. Ichikuni and Y. Iwasawa, Appl. Catal., A, 1999, 188, 313–324 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- G. Vilé, S. Colussi, F. Krumeich, A. Trovarelli and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2014, 53, 12069–12072 CrossRef PubMed.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456–6482 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- X. Su, X. F. Yang, Y. Huang, B. Liu and T. Zhang, Acc. Chem. Res., 2019, 52, 656–664 CrossRef CAS PubMed.
- Y. Cheng, S. Yang, S. P. Jiang and S. Wang, Small Methods, 2019, 3, 1800440 CrossRef.
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC.
- J. Hulva, M. Meier, R. Bliem, Z. Jakub, F. Kraushofer, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Science, 2021, 371, 375–379 CrossRef CAS PubMed.
- N. Daelman, M. Capdevila-Cortada and N. López, Nat. Mater., 2019, 18, 1215–1221 CrossRef CAS PubMed.
- H. V. Thang and G. Pacchioni, ChemCatChem, 2020, 12, 2595–2604 CrossRef CAS.
- J. Resasco, L. Derita, S. Dai, J. P. Chada, M. Xu, X. Yan, J. Finzel, S. Hanukovich, A. S. Hoffman, G. W. Graham, S. R. Bare, X. Pan and P. Christopher, J. Am. Chem. Soc., 2020, 142, 169–184 CrossRef CAS PubMed.
- J. Liu, Y. Zou, D. Cruz, A. Savateev, M. Antonietti and G. Vilé, ACS Appl. Mater. Interfaces, 2021, 13, 25858–25867 CrossRef CAS PubMed.
- G. Vilé, P. Sharma, M. Nachtegaal, F. Tollini, D. Moscatelli, A. Sroka-Bartnicka, O. Tomanec, M. Petr, J. Filip, I. S. Pieta, R. Zbořil and M. B. Gawande, Sol. RRL, 2021, 5, 2100176 CrossRef.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard, G. Vilé and J. Pérez-Ramírez, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- S. Wei, A. Li, J. C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W. C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- G. Vilé, G. Di Liberto, S. Tosoni, A. Sivo, V. Ruta, M. Nachtegaal, A. H. Clark, S. Agnoli, Y. Zou, A. Savateev, M. Antonietti and G. Pacchioni, ACS Catal., 2022, 12, 2947–2958 CrossRef.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. Dong Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- H. Fei, J. Dong, D. Chen, T. Hu, X. Duan, I. Shakir, Y. Huang and X. Duan, Chem. Soc. Rev., 2019, 48, 5207–5241 RSC.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed.
- G. G. Botte, Electrochem. Soc. Interface, 2014, 23, 49 CrossRef.
- D. E. Blanco and M. A. Modestino, Trends Chem., 2019, 1, 8–10 CrossRef CAS.
- M. P. Grotheer, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000, vol. 9, p. 618 Search PubMed.
- D. Pletcher and F. C. Walsh, Industrial Electrochemistry, Blackie Academic & Professional, London, 2nd edn, 1993 Search PubMed.
- J. H. Simons, J. Electrochem. Soc., 1949, 95, 47–52 CrossRef CAS.
- D. E. Danly, J. Electrochem. Soc., 1984, 131, 435C–442C CrossRef.
- P. L. Norcott, Chem. Commun., 2022, 58, 2944–2953 RSC.
- S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750–14759 CrossRef CAS PubMed.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- J. Rein, J. R. Annand, M. K. Wismer, J. Fu, J. C. Siu, A. Klapars, N. A. Strotman, D. Kalyani, D. Lehnherr and S. Lin, ACS Cent. Sci., 2021, 7, 1347–1355 CrossRef CAS PubMed.
- H. O. House, E. Feng and N. P. Peet, J. Org. Chem., 1971, 36, 2371–2375 CrossRef CAS.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS PubMed.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC.
- R. J. Perkins, A. J. Hughes, D. J. Weix and E. C. Hansen, Org. Process Res. Dev., 2019, 23, 1746–1751 CrossRef CAS.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- H. J. Schäfer, C. R. Chim., 2011, 14, 745–765 CrossRef.
- R. Service, Can the world make the chemicals it needs without oil? https://www.sciencemag.org/news/2019/09/can-world-make-chemicals-it-needs-without-oil.
- D. T. Mah, Electrochem. Soc. Interface, 2014, 23, 47 CrossRef CAS.
- Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10–14 CrossRef.
- K. M. van Geem, V. V. Galvita and G. B. Marin, Science, 2019, 364, 734–735 CrossRef CAS PubMed.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- B. Lu, Q. Liu and S. Chen, ACS Catal., 2020, 10, 7584–7618 CrossRef CAS.
- H. Chen, X. Liang, Y. Liu, X. Ai, T. Asefa and X. Zou, Adv. Mater., 2020, 32, 2002435 CrossRef PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- C. L. Allen, A. R. Chhatwal and J. M. J. Williams, Chem. Commun., 2012, 48, 666–668 RSC.
- H. Lundberg, F. Tinnis and H. Adolfsson, Chem. – Eur. J., 2012, 18, 3822–3826 CrossRef CAS PubMed.
- H. Lundberg, F. Tinnis and H. Adolfsson, Synlett, 2012, 2201–2204 CAS.
- H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271–3277 CrossRef CAS.
- K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS PubMed.
- T. Maki, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 1431–1434 CrossRef CAS PubMed.
- K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Green Chem., 2008, 10, 124–134 RSC.
- S. Fatemi, N. Gernigon and D. G. Hall, Green Chem., 2015, 17, 4016–4028 RSC.
- M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed.
- H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 571–577 CrossRef CAS PubMed.
- K. Wang, Y. Lu and K. Ishihara, Chem. Commun., 2018, 54, 5410–5413 RSC.
- M. T. Sabatini, V. Karaluka, R. M. Lanigan, L. T. Boulton, M. Badland and T. D. Sheppard, Chem. – Eur. J., 2018, 24, 7033–7043 CrossRef CAS PubMed.
- J. A. Moore, Reprod. Toxicol., 1997, 11, 123–160 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- S. Harkal, F. Rataboul, A. Zapf, C. Fuhrmann, T. Riermeier, A. Monsees and M. Beller, Adv. Synth. Catal., 2004, 346, 1742–1748 CrossRef CAS.
- C. M. So, C. P. Lau and F. Y. Kwong, Org. Lett., 2007, 9, 2795–2798 CrossRef CAS PubMed.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
- E. A. B. Kantchev, C. J. O’Brien and M. G. Organ, Palladium complexes of N-heterocyclic carbenes as catalysts for cross-coupling reactions – A synthetic chemist's perspective, 2007, vol. 46 Search PubMed.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- M. Corpet and C. Gosmini, Synthesis, 2014, 2258–2271 CAS.
- D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722–9723 CrossRef CAS.
- F. Rataboul, A. Zapf, R. Jackstell, S. Harkal, T. Riermeier, A. Monsees, U. Dingerdissen and M. Beller, Chem. – Eur. J., 2004, 10, 2983–2990 CrossRef CAS PubMed.
- Q. Dai, W. Gao, D. Liu, L. M. Kapes and X. Zhang, J. Org. Chem., 2006, 71, 3928–3934 CrossRef CAS PubMed.
- R. A. Singer, M. Doré, J. E. Sieser and M. A. Berliner, Tetrahedron Lett., 2006, 47, 3727–3731 CrossRef CAS.
- N. Schwarz, A. Tillack, K. Alex, I. A. Sayyed, R. Jackstell and M. Beller, Tetrahedron Lett., 2007, 48, 2897–2900 CrossRef CAS.
- S. Doherty, J. G. Knight, C. H. Smyth and G. A. Jorgenson, Adv. Synth. Catal., 2008, 350, 1801–1806 CrossRef CAS.
- C. M. So, Z. Zhou, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 6402–6406 CrossRef CAS PubMed.
- G. J. Withbroe, R. A. Singer and J. E. Sieser, Org. Process Res. Dev., 2008, 12, 480–489 CrossRef CAS.
- K. Suzuki, Y. Hori and T. Kobayashi, Adv. Synth. Catal., 2008, 350, 652–656 CrossRef CAS.
- R. Pratap, D. Parrish, P. Gunda, D. Venkataraman and M. K. Lakshman, J. Am. Chem. Soc., 2009, 131, 12240–12249 CrossRef CAS PubMed.
- B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS PubMed.
- D. Maiti, B. P. Fors, J. L. Henderson, Y. Nakamura and S. L. Buchwald, Chem. Sci., 2011, 2, 57–68 RSC.
- M. D. Charles, P. Schultz and S. L. Buchwald, Org. Lett., 2005, 7, 3965–3968 CrossRef CAS PubMed.
- B. P. Fors, N. R. Davis and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 5766–5768 CrossRef CAS PubMed.
- A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680–5681 CrossRef CAS PubMed.
- T. W. Greene and P. G. M. Wuts, Protective groups in Organic Synthesis, John Wiley & Sons, 3rd edn, 1999 Search PubMed.
- P. Strazzolini, N. Misuri and P. Polese, Tetrahedron Lett., 2005, 46, 2075–2078 CrossRef CAS.
- D. S. Coffey, M. K. N. Hawk, S. W. Pedersen, S. J. Ghera, P. G. Marler, P. N. Dodson and M. L. Lytle, Org. Process Res. Dev., 2004, 8, 945–947 CrossRef CAS.
- B. Li, M. Berliner, R. Buzon, C. K. F. Chiu, S. T. Colgan, T. Kaneko, N. Keene, W. Kissel, T. Le, K. R. Leeman, B. Marquez, R. Morris, L. Newell, S. Wunderwald, M. Witt, J. Weaver, Z. Zhang and Z. Zhang, J. Org. Chem., 2006, 71, 9045–9050 CrossRef CAS PubMed.
- J. Zheng, B. Yin, W. Huang, X. Li, H. Yao, Z. Liu, J. Zhang and S. Jiang, Tetrahedron Lett., 2009, 50, 5094–5097 CrossRef CAS.
- V. Evans, M. F. Mahon and R. L. Webster, Tetrahedron, 2014, 70, 7593–7597 CrossRef CAS.
- J. M. López-Soria, S. J. Pérez, J. N. Hernández, M. A. Ramírez, V. S. Martín and J. I. Padrón, RSC Adv., 2015, 5, 6647–6651 RSC.
- E. Langille, C. S. Bottaro and A. Drouin, J. Flow Chem., 2020, 10, 377–387 CrossRef CAS.
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, eds., Comprehensive Asymmetric Catalysis, Springer, Berlin, 2004 Search PubMed.
- J. H. Xie, S. F. Zhu and Q. L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed.
- H.-U. Blaser, R. Hanreich, H.-D. Schneider, F. Spindler and B. Steinacher, Asymmetric Catalysis on Industrial Scale, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 1999, vol. 53, pp. 55–70 Search PubMed.
- M. J. Burk, Y. M. Wang and J. R. Lee, J. Am. Chem. Soc., 1996, 118, 5142–5143 CrossRef CAS.
- H. Kolbe, J. Prakt. Chem., 1847, 41, 137–139 CrossRef.
- T. Shono, Tetrahedron, 1984, 40, 811–850 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- C. Ma, P. Fang and T. S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS.
- S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CAS.
- L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed.
- Y. Xia, C. T. Campbell, B. Roldan Cuenya and M. Mavrikakis, Chem. Rev., 2021, 121, 563–566 CrossRef CAS PubMed.
- N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
- J. Feng, H. Gao, L. Zheng, Z. Chen, S. Zeng, C. Jiang, H. Dong, L. Liu, S. Zhang and X. Zhang, Nat. Commun., 2020, 11, 4341 CrossRef PubMed.
- A. Li, S. A. Nicolae, M. Qiao, K. Preuss, P. A. Szilágyi, A. Moores and M. M. Titirici, ChemCatChem, 2019, 11, 3602–3625 CrossRef CAS.
- T. Sun, S. Zhao, W. Chen, D. Zhai, J. Dong, Y. Wang, S. Zhang, A. Han, L. Gu, R. Yu, X. Wen, H. Ren, L. Xu, C. Chen, Q. Peng, D. Wang and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12692–12697 CrossRef CAS PubMed.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383–2387 CrossRef CAS PubMed.
- C. Zhu, M. Stangier, J. C. A. Oliveira, L. Massignan and L. Ackermann, Chem. – Eur. J., 2019, 25, 16382–16389 CrossRef CAS PubMed.
- B. R. Walker and C. S. Sevov, ACS Catal., 2019, 9, 7197–7203 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- A. Corma, P. Concepción, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. López-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Nat. Chem., 2013, 5, 775–781 CrossRef CAS PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- Z. H. Xue, S. N. Zhang, Y. X. Lin, H. Su, G. Y. Zhai, J. T. Han, Q. Y. Yu, X. H. Li, M. Antonietti and J. S. Chen, J. Am. Chem. Soc., 2019, 141, 14976–14980 CrossRef CAS PubMed.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12014–12017 CrossRef CAS PubMed.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed.
- K. Jin, J. H. Maalouf, N. Lazouski, N. Corbin, D. Yang and K. Manthiram, J. Am. Chem. Soc., 2019, 141, 6413–6418 CrossRef CAS PubMed.
- M. A. Bajada, S. Roy, J. Warnan, K. Abdiaziz, A. Wagner, M. M. Roessler and E. Reisner, Angew. Chem., Int. Ed., 2020, 59, 15633–15641 CrossRef CAS PubMed.
- Y. Lu, C. L. Dong, Y. C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- C. Liu, S. Han, M. Li, X. Chong and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18527–18531 CrossRef CAS PubMed.
- X. Chong, C. Liu, Y. Huang, C. Huang and B. Zhang, Natl. Sci. Rev., 2020, 7, 285–295 CrossRef CAS PubMed.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed.
- Z. Yin, H. Pang, X. Guo, H. Lin, M. Muzzio, M. Shen, K. Wei, C. Yu, P. Williard and S. Sun, Angew. Chem., Int. Ed., 2020, 59, 15933–15936 CrossRef CAS PubMed.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G. W. Huber and H.-Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- T. Mehtiö, M. Toivari, M. G. Wiebe, A. Harlin, M. Penttilä and A. Koivula, Crit. Rev. Biotechnol., 2016, 36, 904–916 CrossRef PubMed.
- X. Lin, S.-N. Zhang, D. Xu, J.-J. Zhang, Y.-X. Lin, G.-Y. Zhai, H. Su, Z.-H. Xue, X. Liu, M. Antonietti, J.-S. Chen and X.-H. Li, Nat. Commun., 2021, 12, 3882 CrossRef CAS PubMed.
- P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC.
- S. Ding, M. J. Hülsey, J. Pérez-Ramírez and N. Yan, Joule, 2019, 3, 2897–2929 CrossRef CAS.
- A. Dey, T. B. Gunnoe and V. R. Stamenkovic, ACS Catal., 2020, 10, 13156–13158 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- X. Zhang, S. Zhang, Y. Yang, L. Wang, Z. Mu, H. Zhu, X. Zhu, H. Xing, H. Xia, B. Huang, J. Li, S. Guo and E. Wang, Adv. Mater., 2020, 32, 1906905 CrossRef CAS PubMed.
- Z. Liang, C. Qu, D. Xia, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 57, 9604–9633 CrossRef CAS PubMed.
- J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu and G. Wang, Small Methods, 2019, 3, 1900159 CrossRef.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H. T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang, C. He, X. Ouyang, M. Gu, J. Liu and X. Sun, Energy Environ. Sci., 2019, 12, 1000–1007 RSC.
- Z. Zhang, Y. Chen, L. Zhou, C. Chen, Z. Han, B. Zhang, Q. Wu, L. Yang, L. Du, Y. Bu, P. Wang, X. Wang, H. Yang and Z. Hu, Nat. Commun., 2019, 10, 1657 CrossRef PubMed.
- M. Tavakkoli, N. Holmberg, R. Kronberg, H. Jiang, J. Sainio, E. I. Kauppinen, T. Kallio and K. Laasonen, ACS Catal., 2017, 7, 3121–3130 CrossRef CAS.
- X. P. Yin, H. J. Wang, S. F. Tang, X. L. Lu, M. Shu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2018, 57, 9382–9386 CrossRef CAS PubMed.
- J. Lai, A. Nsabimana, R. Luque and G. Xu, Joule, 2018, 2, 76–93 CrossRef CAS.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef PubMed.
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef.
- H. Zhang, P. An, W. Zhou, B. Y. Guan, P. Zhang, J. Dong and X. W. (David) Lou, Sci. Adv., 2018, 4, eaao6657 CrossRef PubMed.
- Y. Qu, B. Chen, Z. Li, X. Duan, L. Wang, Y. Lin, T. Yuan, F. Zhou, Y. Hu, Z. Yang, C. Zhao, J. Wang, C. Zhao, Y. Hu, G. Wu, Q. Zhang, Q. Xu, B. Liu, P. Gao, R. You, W. Huang, L. Zheng, L. Gu, Y. Wu and Y. Li, J. Am. Chem. Soc., 2019, 141, 4505–4509 CrossRef CAS PubMed.
- S. Chen, N. Zhang, C. W. Narváez Villarrubia, X. Huang, L. Xie, X. Wang, X. Kong, H. Xu, G. Wu, J. Zeng and H. L. Wang, Nano Energy, 2019, 66, 104164 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- Y. Peng, B. Lu, L. Chen, N. Wang, J. E. Lu, Y. Ping and S. Chen, J. Mater. Chem. A, 2017, 5, 18261–18269 RSC.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- Z. Chen, S. Pronkin, T. P. Fellinger, K. Kailasam, G. Vilé, D. Albani, F. Krumeich, R. Leary, J. Barnard, J. M. Thomas, J. Pérez-Ramírez, M. Antonietti and D. Dontsova, ACS Nano, 2016, 10, 3166–3175 CrossRef CAS PubMed.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T. K. Sham and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- J. Lu, J. W. Elam and P. C. Stair, Acc. Chem. Res., 2013, 46, 1806–1815 CrossRef CAS PubMed.
- S. Stambula, N. Gauquelin, M. Bugnet, S. Gorantla, S. Turner, S. Sun, J. Liu, G. Zhang, X. Sun and G. A. Botton, J. Phys. Chem. C, 2014, 118, 3890–3900 CrossRef CAS.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed.
- Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. B. Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef CAS PubMed.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- X. Huang, Y. Xia, Y. Cao, X. Zheng, H. Pan, J. Zhu, C. Ma, H. Wang, J. Li, R. You, S. Wei, W. Huang and J. Lu, Nano Res., 2017, 10, 1302–1312 CrossRef CAS.
- C. Wang, X. K. Gu, H. Yan, Y. Lin, J. Li, D. Liu, W. X. Li and J. Lu, ACS Catal., 2017, 7, 887–891 CrossRef CAS.
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 CrossRef PubMed.
- Z. Tian, C. Priest and L. Chen, Adv. Theory Simulations, 2018, 1, 1800004 CrossRef.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- S. Wannakao, W. Jumpathong and K. Kongpatpanich, Inorg. Chem., 2017, 56, 7200–7209 CrossRef CAS PubMed.
- M. D. Hossain, Z. Liu, M. Zhuang, X. Yan, G. Xu, C. A. Gadre, A. Tyagi, I. H. Abidi, C. Sun, H. Wong, A. Guda, Y. Hao, X. Pan, K. Amine and Z. Luo, Adv. Energy Mater., 2019, 9, 1803689 CrossRef.
- X. Liu, Z. Wang, Y. Tian and J. Zhao, J. Phys. Chem. C, 2020, 124, 3722–3730 CrossRef CAS.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS PubMed.
- D. K. Leahy, E. M. Simmons, V. Hung, J. T. Sweeney, W. F. Fleming and M. Miller, Green Chem., 2017, 19, 5163–5171 RSC.
- T. Noël, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858–2869 CrossRef PubMed.
- P. Desir, B. Saha and D. G. Vlachos, Energy Environ. Sci., 2019, 12, 2463–2475 RSC.
- A. Somoza-Tornos, O. J. Guerra, A. M. Crow, W. A. Smith and B. M. Hodge, iScience, 2021, 24, 102813 CrossRef CAS PubMed.
- S. Bagi, S. Yuan, S. Rojas-Buzo, Y. Shao-Horn and Y. Román-Leshkov, Green Chem., 2021, 23, 9982–9991 RSC.
- M. Atobe, H. Tateno and Y. Matsumura, Chem. Rev., 2018, 118, 4541–4572 CrossRef CAS PubMed.
- G. Laudadio, W. de Smet, L. Struik, Y. Cao and T. Noël, J. Flow Chem., 2018, 8, 157–165 CrossRef PubMed.
- A. A. Folgueiras-Amador, K. Philipps, S. Guilbaud, J. Poelakker and T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 15446–15450 CrossRef CAS PubMed.
- J. Jörissen, Electrochim. Acta, 1996, 41, 553–562 CrossRef.
- V. Montiel, A. Sáez, E. Expósito, V. García-García and A. Aldaz, Electrochem. Commun., 2010, 12, 118–121 CrossRef CAS.
- G. Laudadio, E. Barmpoutsis, C. Schotten, L. Struik, S. Govaerts, D. L. Browne and T. Noël, J. Am. Chem. Soc., 2019, 141, 5664–5668 CrossRef CAS PubMed.
- R. Horcajada, M. Okajima, S. Suga and J. I. Yoshida, Chem. Commun., 2005, 1303–1305 RSC.
- B. Winterson, T. Rennigholtz and T. Wirth, Chem. Sci., 2021, 12, 9053–9059 RSC.
- Y. Cao, C. Soares, N. Padoin and T. Noël, Chem. Eng. J., 2021, 406, 126811 CrossRef CAS.
- Y. Cao, N. Padoin, C. Soares and T. Noël, Chem. Eng. J., 2022, 427, 131443 CrossRef CAS.
- M. Ošeka, G. Laudadio, N. P. van Leest, M. Dyga, A. D. A. Bartolomeu, L. J. Gooßen, B. de Bruin, K. T. de Oliveira and T. Noël, Chem, 2021, 7, 255–266 Search PubMed.
- J. Zhang, K. Wang, A. R. Teixeira, K. F. Jensen and G. Luo, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 285–305 CrossRef PubMed.
- Z. Dong, Z. Wen, F. Zhao, S. Kuhn and T. Noël, Chem. Eng. Sci. X, 2021, 10, 100097 CAS.
- K. Lovato, P. S. Fier and K. M. Maloney, Nat. Rev. Chem., 2021, 5, 546–563 CrossRef CAS.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838–845 CrossRef CAS PubMed.
- J. D. Egbert, E. C. Thomsen, S. A. O’Neill-Slawecki, D. M. Mans, D. C. Leitch, L. J. Edwards, C. E. Wade and R. S. Weber, Org. Process Res. Dev., 2019, 23, 1803–1812 CrossRef CAS.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- Y. Qu, C. Tsuneishi, H. Tateno, Y. Matsumura and M. Atobe, React. Chem. Eng., 2017, 2, 871–875 RSC.
- J.-M. Saveant, Chem. Rev., 2008, 108, 2348–2378 CrossRef CAS PubMed.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- C. D. Windle and E. Reisner, Chimia, 2015, 69, 435–441 CrossRef CAS PubMed.
- E. S. Andreiadis, P. A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pécaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef CAS PubMed.
- B. Reuillard, K. H. Ly, T. E. Rosser, M. F. Kuehnel, I. Zebger and E. Reisner, J. Am. Chem. Soc., 2017, 139, 14425–14435 CrossRef CAS PubMed.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- A. D. Jannakoudakis, P. D. Jannakoudakis, E. Theodoridou and J. O. Besenhard, J. Appl. Electrochem., 1990, 20, 619–624 CrossRef CAS.
- M. L. Bowers and B. A. Yenser, Anal. Chim. Acta, 1991, 243, 43–53 CrossRef CAS.
- Y. Xing, L. Li, C. C. Chusuei and R. V. Hull, Langmuir, 2005, 21, 4185–4190 CrossRef CAS PubMed.
- K. J. Ziegler, Z. Gu, H. Peng, E. L. Flor, R. H. Hauge and R. E. Smalley, J. Am. Chem. Soc., 2005, 127, 1541–1547 CrossRef CAS PubMed.
- A. Gromov, S. Dittmer, J. Svensson, O. A. Nerushev, S. A. Perez-García, L. Licea-Jiménez, R. Rychwalski and E. E. B. Campbell, J. Mater. Chem., 2005, 15, 3334–3339 RSC.
- C. H. Andersson and H. Grennberg, Eur. J. Org. Chem., 2009, 4421–4428 CrossRef CAS.
- M. Delamar, R. Hitmi, J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1992, 114, 5883–5884 CrossRef CAS.
- P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J. M. Savéant, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS.
- M. Melchionna and M. Prato, Nanocarbon-Inorg. Hybrids Next Gener. Compos. Sustain. Energy Appl., 2014, 124, 43–70 Search PubMed.
- N. Tagmatarchis and M. Prato, J. Mater. Chem., 2004, 437–439 RSC.
- X. Wang, D. Jiang and S. Dai, Chem. Mater., 2008, 20, 4800–4802 CrossRef CAS.
- Y.-L. Zhao and J. F. Stoddart, Acc. Chem. Res., 2009, 42, 1161–1171 CrossRef CAS PubMed.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2015, 44, 6425–6433 RSC.
- M. T. De Groot and M. T. M. Koper, Phys. Chem. Chem. Phys., 2008, 10, 1023–1031 RSC.
- H. L. Smith, R. L. Usala, E. W. McQueen and J. I. Goldsmith, Langmuir, 2010, 26, 3342–3349 CrossRef CAS PubMed.
- J. A. Mann and W. R. Dichtel, ACS Nano, 2013, 7, 7193–7199 CrossRef CAS PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- Z. Chen, J. J. Concepcion, J. F. Hull, P. G. Hoertz and T. J. Meyer, Dalton Trans., 2010, 39, 6950 RSC.
- P. G. Hoertz, Z. Chen, C. A. Kent and T. J. Meyer, Inorg. Chem., 2010, 49, 8179–8181 CrossRef CAS PubMed.
- S. Morra, F. Valetti, S. J. Sadeghi, P. W. King, T. Meyer and G. Gilardi, Chem. Commun., 2011, 47, 10566–10568 RSC.
- T. E. Rosser, M. A. Gross, Y.-H. Lai and E. Reisner, Chem. Sci., 2016, 7, 4024–4035 RSC.
- B. J. Brennan, M. J. Llansola Portolés, P. A. Liddell, T. A. Moore, A. L. Moore and D. Gust, Phys. Chem. Chem. Phys., 2013, 15, 16605–16614 RSC.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CrossRef CAS PubMed.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS PubMed.
- R. Ernstorfer, L. Gundlach, S. Felber, W. Storck, R. Eichberger and F. Willig, J. Phys. Chem. B, 2006, 110, 25383–25391 CrossRef CAS PubMed.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. E. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389–2392 RSC.
- P. H. Mutin, G. Guerrero and A. Vioux, J. Mater. Chem., 2005, 15, 3761–3768 RSC.
- L. Wang, D. L. Ashford, D. W. Thompson, T. J. Meyer and J. M. Papanikolas, J. Phys. Chem. C, 2013, 117, 24250–24258 CrossRef CAS.
- Y. Gao, L. Zhang, X. Ding and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 12008–12013 RSC.
- B. J. Brennan, A. E. Keirstead, P. A. Liddell, S. A. Vail, T. A. Moore, A. L. Moore and D. Gust, Nanotechnology, 2009, 20, 505203 CrossRef PubMed.
- K. L. Materna, B. J. Brennan and G. W. Brudvig, Dalton Trans., 2015, 44, 20312–20315 RSC.
- K. L. Materna, B. Rudshteyn, B. J. Brennan, M. H. Kane, A. J. Bloomfield, D. L. Huang, D. Y. Shopov, V. S. Batista, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2016, 6, 5371–5377 CrossRef CAS.
- K. L. Materna, J. Jiang, R. H. Crabtree and G. W. Brudvig, ACS Appl. Mater. Interfaces, 2019, 11, 5602–5609 CrossRef CAS PubMed.
- W. R. McNamara, R. C. Snoeberger, G. Li, C. Richter, L. J. Allen, R. L. Milot, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig and V. S. Batista, Energy Environ. Sci., 2009, 2, 1173–1175 RSC.
- W. R. McNamara, R. L. Milot, H. E. Song, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, G. W. Brudvig and R. H. Crabtree, Energy Environ. Sci., 2010, 3, 917–923 RSC.
- T. P. Brewster, S. J. Konezny, S. W. Sheehan, L. A. Martini, C. A. Schmuttenmaer, V. S. Batista and R. H. Crabtree, Inorg. Chem., 2013, 52, 6752–6764 CrossRef CAS PubMed.
- L. A. Martini, G. F. Moore, R. L. Milot, L. Z. Cai, S. W. Sheehan, C. A. Schmuttenmaer, G. W. Brudvig and R. H. Crabtree, J. Phys. Chem. C, 2013, 117, 14526–14533 CrossRef CAS.
- C. R. Rice, M. D. Ward, M. K. Nazeeruddin and M. Grätzel, New J. Chem., 2000, 24, 651–652 RSC.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109–114 RSC.
- K. Fominykh, J. M. Feckl, J. Sicklinger, M. Döblinger, S. Böcklein, J. Ziegler, L. Peter, J. Rathousky, E. W. Scheidt, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2014, 24, 3123–3129 CrossRef CAS.
- R. M. Bullock, A. K. Das and A. M. Appel, Chem. – Eur. J., 2017, 23, 7626–7641 CrossRef CAS PubMed.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed.
- L. Zhang, L. Han, H. Liu, X. Liu and J. Luo, Angew. Chem., Int. Ed., 2017, 56, 13694–13698 CrossRef CAS PubMed.
- J. Kim, C.-W. Roh, S. K. Sahoo, S. Yang, J. Bae, J. W. Han and H. Lee, Adv. Energy Mater., 2018, 8, 1701476 CrossRef.
- R. Coeck, J. Meeprasert, G. Li, T. Altantzis, S. Bals, E. A. Pidko and D. E. De Vos, ACS Catal., 2021, 11, 7672–7684 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- S. Yoo, E. Aprà, X. C. Zeng and S. S. Xantheas, J. Phys. Chem. Lett., 2010, 1, 3122–3127 CrossRef CAS.
- X. Wang and J. M. Bowman, J. Chem. Theory Comput., 2013, 9, 901–908 CrossRef CAS PubMed.
- F. Gabas, G. Di Liberto, R. Conte and M. Ceotto, Chem. Sci., 2018, 9, 7894–7901 RSC.
- T. Halverson and B. Poirier, J. Phys. Chem. A, 2015, 119, 12417–12433 CrossRef CAS PubMed.
- A. Pandey and B. Poirier, J. Phys. Chem. Lett., 2020, 11, 6468–6474 CrossRef CAS PubMed.
- J. Neugebauer and B. A. Hess, J. Chem. Phys., 2003, 118, 7215–7225 CrossRef CAS.
- V. Barone, J. Chem. Phys., 2005, 122, 014108 CrossRef PubMed.
- M. Ceriotti, W. Fang, P. G. Kusalik, R. H. McKenzie, A. Michaelides, M. A. Morales and T. E. Markland, Chem. Rev., 2016, 116, 7529–7550 CrossRef CAS PubMed.
- X. Ma, G. Di Liberto, R. Conte, W. L. Hase and M. Ceotto, J. Chem. Phys., 2018, 149, 164113 CrossRef PubMed.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- P. Pracht and S. Grimme, Chem. Sci., 2021, 12, 6551–6568 RSC.
- J. A. Gauthier, C. F. Dickens, L. D. Chen, A. D. Doyle and J. K. Nørskov, J. Phys. Chem. C, 2017, 121, 11455–11463 CrossRef CAS.
- Z. Da, He, S. Hanselman, Y. X. Chen, M. T. M. Koper and F. Calle-Vallejo, J. Phys. Chem. Lett., 2017, 8, 2243–2246 CrossRef PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- Q. Zhang and A. Asthagiri, Catal. Today, 2019, 323, 35–43 CrossRef CAS.
- F. Calle-Vallejo, R. F. De Morais, F. Illas, D. Loffreda and P. Sautet, J. Phys. Chem. C, 2019, 123, 5578–5582 CrossRef CAS.
- A. Bouzid, P. Gono and A. Pasquarello, J. Catal., 2019, 375, 135–139 CrossRef CAS.
- Á. Valdés, Z.-W. Qu, G.-J. Kroes, J. Rossmeisl and J. K. Nørskov, J. Phys. Chem. C, 2008, 112, 9872–9879 CrossRef.
- M. H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407–7412 CrossRef CAS.
- F. F. Rivera, T. Pérez, L. F. Castañeda and J. L. Nava, Chem. Eng. Sci., 2021, 239, 116622 CrossRef CAS.
- E. Sato, M. Fujii, H. Tanaka, K. Mitsudo, M. Kondo, S. Takizawa, H. Sasai, T. Washio, K. Ishikawa and S. Suga, J. Org. Chem., 2021, 86, 16035–16044 CrossRef CAS PubMed.
- G. A. Landrum, J. E. Penzotti and S. Putta, Meas. Sci. Technol., 2005, 16, 270–277 CrossRef CAS.
- N. Fey, Chem. Cent. J., 2015, 9, 38 CrossRef PubMed.
- Z. W. Ulissi, A. J. Medford, T. Bligaard and J. K. Nørskov, Nat. Commun., 2017, 8, 14621 CrossRef PubMed.
- J. R. Kitchin, Nat. Catal., 2018, 1, 230–232 CrossRef.
- P. Schlexer Lamoureux, K. T. Winther, J. A. Garrido Torres, V. Streibel, M. Zhao, M. Bajdich, F. Abild-Pedersen and T. Bligaard, ChemCatChem, 2019, 11, 3581–3601 CrossRef CAS.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Science, 2019, 363, eaau5631 CrossRef CAS PubMed.
- T. Toyao, Z. Maeno, S. Takakusagi, T. Kamachi, I. Takigawa and K. I. Shimizu, ACS Catal., 2020, 10, 2260–2297 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |