
Organoboron molecules and polymers for organic solar cell applications
Junhui
Miao
a,
Yinghui
Wang
 ab,
Jun
Liu
*a and
Lixiang
Wang
a
ab,
Jun
Liu
*a and
Lixiang
Wang
a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: liujun@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei 230026, P. R. China
First published on 1st December 2021
Abstract
Organic solar cells (OSCs) are emerging as a new photovoltaic technology with the great advantages of low cost, light-weight, flexibility and semi-transparency. They are promising for portable energy-conversion products and building-integrated photovoltaics. Organoboron chemistry offers an important toolbox to design novel organic/polymer optoelectronic materials and to tune their optoelectronic properties for OSC applications. At present, organoboron small molecules and polymers have become an important class of organic photovoltaic materials. Power conversion efficiencies (PCEs) of 16% and 14% have been realized with organoboron polymer electron donors and electron acceptors, respectively. In this review, we summarize the research progress in various kinds of organoboron photovoltaic materials for OSC applications, including organoboron small molecular electron donors, organoboron small molecular electron acceptors, organoboron polymer electron donors and organoboron polymer electron acceptors. This review also discusses how to tune their opto-electronic properties and active layer morphology for enhancing OSC device performance. We also offer our insight into the opportunities and challenges in improving the OSC device performance of organoboron photovoltaic materials.
 Junhui Miao | Junhui Miao received his PhD degree from Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences (CAS), in 2020. Now he is an assistant researcher in Professor Jun Liu's group in State Key Laboratory of Polymer Physics and Chemistry at CIAC. His research interest is focused on organic photovoltaic materials. |
 Yinghui Wang | Yinghui Wang received her BS degree from Zhengzhou University (China) in 2018. She is now a PhD student under the supervision of Prof. Jun Liu, in CIAC, CAS. Her research topic is organoboron polymer for organic solar cell applications. |
 Jun Liu | Jun Liu received his PhD degree in Polymer Physics and Chemistry in 2007 from CIAC, CAS. After postdoctoral training at the University of Würzburg (Germany), University of California, Los Angeles (USA), and Case Western Reserve University (USA) for five years, he joined CIAC as a principal investigator in 2013. His main research interests are organic/polymer opto-electronic materials and polymer solar cells. |
 Lixiang Wang | Lixiang Wang received his PhD degree from CIAC, CAS, in 1989. During 1994–1997, he was a postdoctoral researcher at Max Planck Institute for Polymer Research (Germany) and University of Massachusetts Amherst (USA). Since 1997, he has been a principal investigator in CIAC. His main research interests focus on solution-processable dendrimers/polymers for organic light-emitting diodes, chemical sensors and polymer solar cells. |
1. Introduction
Solar cells are an important kind of renewable clean energy technology to address the energy and environmental issues mankind is facing today.1–4 Among various kinds of solar cells, organic solar cells (OSCs) have the advantages of flexibility, light-weight, semi-transparency and low cost.5–14 These advantages make OSCs promising in portable, wearable power supplies and building integrated photovoltaics.15–23 Recently, great progress has been made in the field of OSCs. Small-area single-junction OSC devices with a power conversion efficiency (PCE) exceeding 18% or with a lifetime of over twenty years have been demonstrated in labs.24–27 Large-area OSC modules have also been fabricated by vacuum-deposition or roll-to-roll processing. Provided that three key factors, efficiency, stability and cost, can be synergistically addressed, OSCs will be used in commercial applications.OSCs use a pair of small molecular/polymer electron donors and electron acceptors as photoactive layers. The development of electron donor and electron acceptor materials plays a key role in the OSC device performance improvement. In 1986, Tang et al. reported the pioneering work on OSCs which used double layers (Fig. 1a) of an electron donor and an electron acceptor and gave a PCE of ∼1%.28 In 1995, Heeger et al. developed bulk heterojunction (BHJ) OSCs (Fig. 1b) using a mixture of an electron donor and an electron acceptor as the photoactive layer.29 Since then, the widely used electron acceptors have been fullerene derivatives.30,31 Materials chemists have made attempts to develop small molecular electron donors or polymer electron donors to match fullerene electron acceptors to enhance the PCE of OSCs.32–41 From 2015 to date, owing to the emergence and optimization of non-fullerene electron acceptors, the PCE of OSCs has been enhanced from ca. 10% to more than 18%. Many high-efficiency non-fullerene electron acceptors have been developed, including small molecular electron acceptors and polymer electron acceptors.42–62
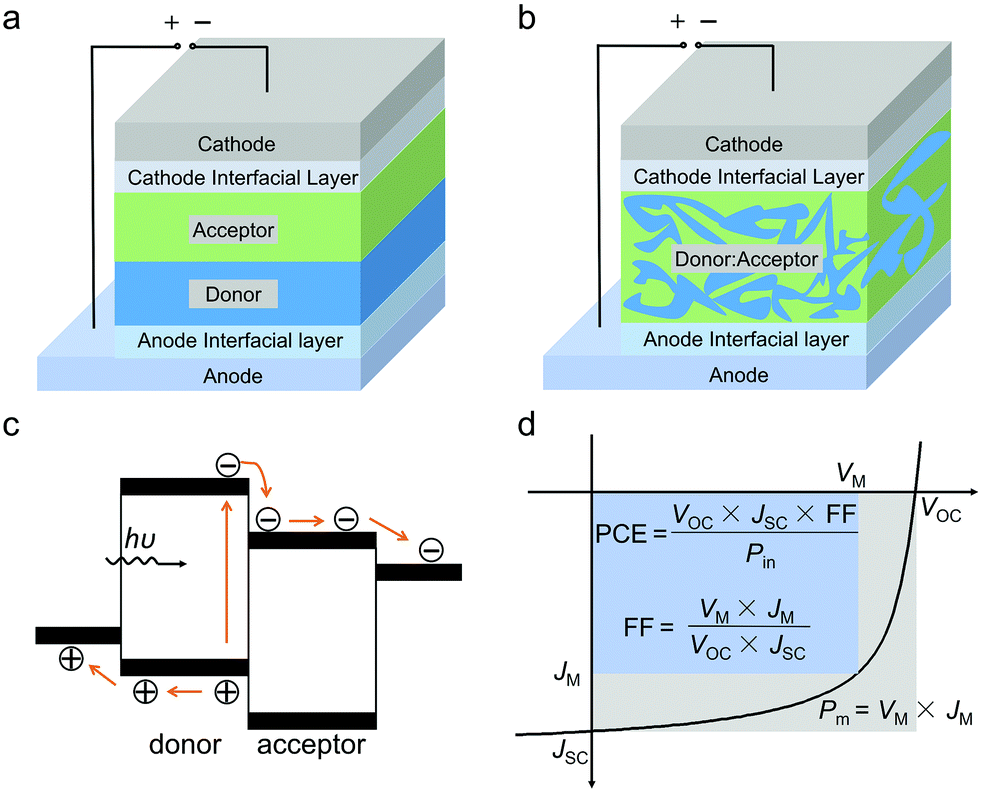 | ||
| Fig. 1 Schematic illustration of the device structure of (a) two-layer heterojunction and (b) bulk heterojunction OSCs. (c) Schematic illustration of the empirical working principle of OSCs. (d) A typical current density–voltage (J–V) curve of OSC devices and the meaning of JSC, VOC, FF and PCE. | ||
Organoboron chemistry offers a toolbox not only to design new organic opto-electronic materials, but also to tune their optoelectronic properties towards excellent device performance.63–74 In the past decade, a large amount of organoboron small molecule or polymer electron donors/acceptors have been developed and used in OSC devices.75–78 High PCEs and excellent OSC device stability have been demonstrated with these organoboron photovoltaic materials. Therefore, organoboron molecules and polymers have become an important class of materials for OSCs.
The aim of this review is to discuss various kinds of organoboron photovoltaic materials for OSC applications, including organoboron small molecular electron donors, organoboron small molecular electron acceptors, organoboron polymer electron donors and organoboron polymer electron acceptors. We discuss not only how to design novel organoboron photovoltaic materials, but also how to tune their opto-electronic properties and active layer morphology for enhanced OSC device performance. In this review, we start with the research background, i.e. introduction to OSC devices, requirement of electron donor/acceptor materials, and principle of organoboron chemistry to tune opto-electronic properties. Then, we discuss the various kinds of organoboron photovoltaic materials. Finally, we provide an outlook on the opportunities and challenges in improving the OSC device performance of organoboron photovoltaic materials.
2. Research background
2.1 A brief introduction to OSCs
Fig. 1a and b show the schematic illustration of bilayer and BHJ OSC device structures, respectively. A pair of electron donors and electron acceptors with a bilayer structure or single layer structure are used as active layers, which are sandwiched between the anode and the cathode. Fig. 1c shows a schematic illustration of the empirical working principle of OSCs. Under solar light irradiation, the active layer absorbs photons and generates excitons (electron/hole pairs). The excitons diffuse to the donor/acceptor interfaces and dissociate to separated electrons and holes. The electrons and holes are transported in the active layer and are collected by the cathode and the anode, respectively. With these processes of light absorption, exciton diffusion/dissociation, electron/hole transportation and electron/hole collection, solar light energy can be converted to electrical energy.The device performance of OSCs can be expressed by the current density–voltage (J–V) curve under simulated solar light irradiation (Fig. 1d). The main parameter for evaluating photovoltaic performance is power conversion efficiency (PCE), which is defined as the ratio between the maximum power output density (Pm) and the incident light input power (Pin). The Pm of the cell is determined as the product of open-circuit voltage (VOC), short-circuit current (JSC), and fill factor (FF). Therefore, the PCE can be written as the following equation: PCE = VOC × JSC × FF/Pin. For OSCs, VOC mainly depends on the difference between the highest occupied molecular orbital (HOMO) energy level of the electron donor and the lowest unoccupied molecular orbital (LUMO) energy level of the electron acceptor. In addition, charge carrier recombination and electronic coupling between the donor and the acceptor also affect VOC. JSC is determined by light absorption of the active layer, diffusion and dissociation of excitons, transport and collection of charge carriers, etc. FF reflects the ratio of charge extraction and charge recombination and is also affected by multiple factors.
2.2 Requirements of electron donor/acceptor materials
According to the working principle of OSC devices, to obtain excellent OSC device performance, electron donor and electron acceptor materials should meet multiple requirements. At first, to effectively harvest sunlight, electron donor materials and electron acceptor materials in active layers should have wide absorption spectra and large absorption coefficients. Secondly, HOMO and LUMO energy levels of electron donor materials and electron acceptor materials greatly affect the exciton dissociation process and to a large extent determine the voltage output of OSCs. Therefore, donor and acceptor materials should have appropriate LUMO/HOMO energy level alignment. Thirdly, to improve charge transport in active layers, the donor and acceptor materials should have high hole mobilities and high electron mobilities, respectively. Fourthly, to improve exciton dissociation at the donor/acceptor interfaces and provide pathways for electron/hole transportation, donor materials and acceptor materials should form bis-continuous network phase separation morphology with a phase domain size of ca. 10–30 nm. It is challenging to develop an electron donor or acceptor material that fulfills all these requirements. Materials chemists always have to carefully optimize the chemical structures of electron donor or acceptor materials to fulfill all these requirements for high OSC device efficiency.2.3 Organoboron chemistry as a toolbox for molecular design
The boron element, neighboring the carbon element in the periodic table of elements, has the atomic number of 5. The boron atom has a vacant 2p orbital, leading to strong Lewis acidity and high propensity to coordinate with Lewis bases (Fig. 2). In organic molecules and polymers, the boron atom adopts either sp2 hybridization (tri-coordination) or sp3 hybridization (tetra-coordination) (Fig. 2). In tri-coordinated organoboron π-conjugated compounds, the empty 2p orbital of the boron atom forms p–π* conjugation with the π system, leading to a slightly downshifted LUMO energy level.68,73 Therefore, tri-coordinated organoboron molecules or building blocks are always used in electron donors with optimized LUMO/HOMO energy levels. For tetra-coordinated organoboron π-conjugated compounds, the empty 2p orbital of the boron atom (Lewis acidic) coordinates with the lone electron pairs of some atoms, such as oxygen atom and nitrogen atom (Lewis basic).79–82 Since coordination bonds of B ← N and B ← O have strong electron withdrawing capability, many tetra-coordinated organoboron compounds or polymers exhibit very low-lying LUMO energy levels. As a result, the majority of tetra-coordinated organoboron molecules or building blocks are used in electron acceptors with deep LUMO/HOMO energy levels. | ||
| Fig. 2 Illustration of a tri-coordinated organoboron compound and a tetra-coordinated organoboron compound. | ||
Many tri-coordinated and tetra-coordinated organoboron small molecules and polymers with excellent opto-electronic properties are used for organic opto-electronic device applications.72,83–91 Generally, two paradigms have been adopted to develop organoboron photovoltaic materials. The first one is to modify the chemical structures of some well-known organoboron compounds, e.g. 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) and boron subphthalocyanine (SubPc), for optimized opto-electronic properties. BODIPY and SubPc derivatives have already been intensively studied before OSCs attracted wide-spread attention. Materials chemists utilize molecular design to tune their opto-electronic properties to meet the requirement of electron donors or electron acceptors for excellent OSC device performance. The other paradigm is to design brand-new organoboron molecules or building blocks for OSC application. A typical example is double B ← N bridged bipyridine (BNBP), which is used as an electron-deficient building block to design polymer electron acceptors and small molecular electron acceptors.76 BNBP was firstly developed aiming at OSC application by Liu et al. in 2016.
An important issue for organoboron molecules or polymers for OSC application is their chemical stability. Both tri-coordinated organoboron compounds and tetra-coordinated organoboron compounds are highly labile upon Lewis base. For tri-coordinated organoboron compounds, the vacant 2p orbital of the boron atom may be attacked by some molecules containing oxygen or nitrogen atoms with lone electron pairs, e.g. H2O, alcohol, triethylamine, etc. Normally, large steric hindrance or a rigid π-conjugated skeleton can be used to substantially enhance the chemical stability of tri-coordinated organoboron compounds. For tetra-coordinated organoboron compounds, coordination bonds of B ← N and B ← O are sometimes inadequately strong. For example, the intermolecular B ← N bond is reversible, and the intramolecular B ← N bond is irreversible but the bond energy is only ca. 30 kJ mol−1. A portion of tetra-coordinated organoboron compounds is not stable against moisture or polar solvents. Therefore, for both tri-coordinated and tetra-coordinated organoboron compounds to be used in OSCs, their chemical structures must be carefully designed to ensure adequate chemical stability.
3. Organoboron small molecules for OSCs
Both organic small molecules and conjugated polymers can be used as electron donors or electron acceptors in OSCs. The small molecules have the merits of explicit chemical structures, high purity of materials and easy tuning of phase separation morphology. In comparison, the conjugated polymers have the advantages of excellent mechanical properties and morphological stability. Empirically, the combination of small molecular electron acceptors and polymer electron donors affords optimal OSC device efficiency, and the combination of polymer electron donors and polymer acceptors gives the best OSC device stability. In this part, we will introduce organoboron small molecular electron donors and electron acceptors. The general strategy to design small molecular organoboron photovoltaic materials is to modify the chemical structures of already-known organoboron compounds, e.g. BODIPY and SubPc, to tune their opto-electronic properties.3.1 BODIPY-based small molecular electron donors
BODIPY (Fig. 3a) is a well-known dye with strong and tunable absorption in the visible and near infrared (NIR) region, a high molar extinction coefficient and a high fluorescence quantum yield, as well as excellent photostability and chemical stability.92–101 BODIPY was discovered by Kreuzer and Treibs in 1968.78,102–108 In 2009, Roncali and coworkers pioneered the use of BODIPY derivatives as electron donors in OSCs.109 In the past decade, a large family of BODIPY derivatives have been synthesized for application in OSCs.92,93 In this review, due to the limited space, we are not able to discuss all the BODIPY-based organic photovoltaic materials. For more comprehensive and more detailed information, the readers are recommended to refer to some recent excellent reviews on BODIPY derivatives for organic opto-electronic device applications.78,92,93,106,108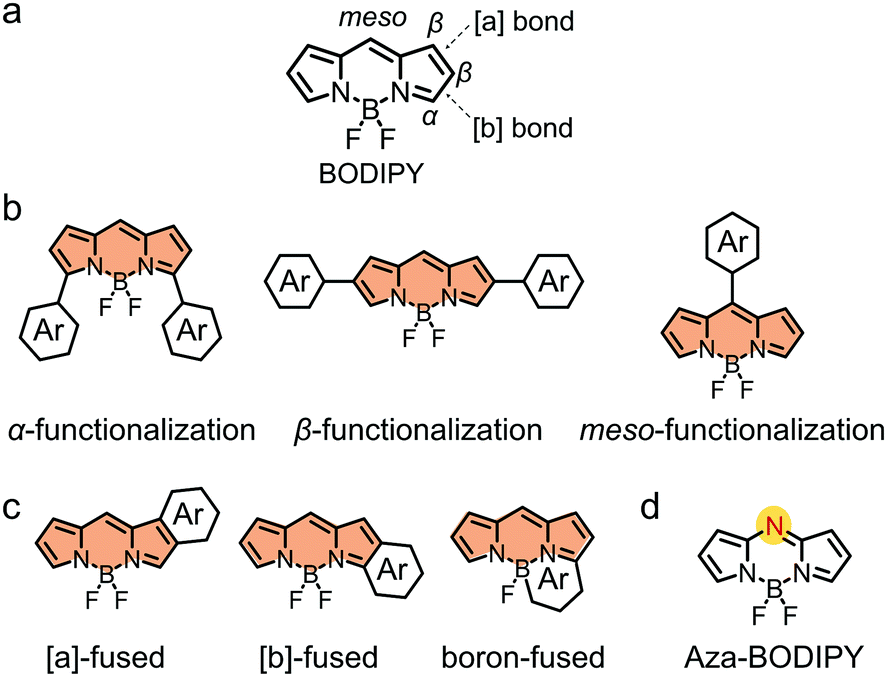 | ||
| Fig. 3 (a) Chemical structure of unmodified BODIPY. (b) Functionalization of BODIPY at the α-, β- or meso-positions. (c) Fusing the BODIPY core with aromatic rings. (d) Substitution of the meso-carbon atom by a nitrogen atom. | ||
Unmodified BODIPY shows an absorption spectrum peaking at ca. 503 nm with a bandgap of ca. 2.4 eV. The opto-electronic properties of BODIPY must be tuned by chemical modification to meet the requirements of electron donors for high-performance OSCs. As shown in Fig. 3, in view of the chemical structure of BODIPY, three strategies can be used to functionalize BODIPY:110–112 (i) functionalization of BODIPY at the α-, β- or meso-positions; (ii) fusing the BODIPY core with aromatic rings to extend π-conjugation; and (iii) substitution of the meso-carbon atom by a nitrogen atom to afford aza-BODIPYs. Following the three strategies and aiming at optimized opto-electronic properties, BODIPY has been functionalized with π-conjugated units, other dye building blocks or electron-donating/withdrawing substituents. The resulting BODIPY-based small molecular electron donors exhibit excellent photovoltaic performance in OSC devices.113,114
Sobral et al. reported a series of BODIPY derivatives (M1–M6) with various aromatic groups at the meso-positions (Fig. 4a).126 The meso-substitution aromatic groups were nearly perpendicular to the BODIPY plane and consequently were not effectively conjugated with the BODIPY skeleton (Fig. 4b). Therefore, M1–M6 exhibited similar LUMO/HOMO energy levels and absorption spectra (Fig. 4c and d). Solution-processed OSCs were fabricated using M1–M6 as the electron donors and [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) as the electron acceptor Table 1. The device based on M6 exhibited a low PCE of 0.47%, which might result from the non-optimal active layer morphology and the low charge carrier mobility. An effective approach to alleviate the steric hindrance effect of meso-substitution is to use π-linkers between the BODIPY skeleton and meso-substitution. Sharma et al. employed ethynyl as the π-linker and attached the bulky 2,7-tert-butyl-carbazole moiety to the meso-position of BODIPY to synthesize M7 (Fig. 4e).127 Due to the acetylene connection between BODIPY and the carbazole substituent, M7 showed a strong donor–acceptor (D–A) character. The absorption spectrum of M7 in solution covered from 450 nm to 650 nm, and a shoulder peak appeared at around 590 nm. From solution to film, the absorption spectrum became broad and redshifted, which may be due to the strong intermolecular interaction. M7 exhibited a narrow bandgap with the onset absorption wavelength exceeding 700 nm. Moreover, this compound adopted a highly planar configuration and showed high crystallinity in thin film, which improved the hole mobility. The optimized OSC devices based on M7:[6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) showed a PCE of 5.05% with a VOC of 0.90 V, a JSC of 10.20 mA cm−2, and an FF of 0.55.
 | ||
| Fig. 4 (a) Chemical structures of BODIPY dyes M1–M6 functionalized at meso-positions. (b) Oak Ridge Thermal Ellipsoid Plot (ORTEP) drawing of the molecular structure with ellipsoids drawn at the 50% probability level of the M5 structure. (c) Electronic distribution of LUMO and HOMO of M5. (d) Normalized absorption spectra of M1–M6 (BDP1–BDP6) in solution. (e) Chemical structure of M7. Panels (b–d) were reproduced from ref. 126 with permission from Elsevier Ltd, copyright 2019. | ||
| Small molecules | HOMO/LUMO (eV/eV) | E optg (eV) | Device structure | V OC (V) | J SC (mA cm−2) | FF | PCEa (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The maximum PCE value of the corresponding device. | ||||||||
| M1 | −5.35/−3.08 | 2.28 | ITO/PEDOT:PSS/M1:PC61BM/LiF/Al | 0.54 | 1.93 | 0.26 | 0.31 | 126 |
| M2 | −5.41/−3.12 | 2.25 | — | — | — | — | — | 126 |
| M3 | −5.50/−3.29 | 2.19 | ITO/PEDOT:PSS/M3:PC61BM/LiF/Al | 0.42 | 0.09 | 0.26 | 0.01 | 126 |
| M4 | −5.27/−2.99 | 2.25 | ITO/PEDOT:PSS/M4:PC61BM/LiF/Al | 0.53 | 0.49 | 0.30 | 0.09 | 126 |
| M5 | −5.39/−3.10 | 2.26 | ITO/PEDOT:PSS/M5:PC61BM/LiF/Al | 0.62 | 1.55 | 0.27 | 0.30 | 126 |
| M6 | −5.35/−3.08 | 2.27 | ITO/PEDOT:PSS/M6:PC61BM/LiF/Al | 0.67 | 2.27 | 0.27 | 0.47 | 126 |
| M7 | −5.48/−3.44 | 1.72 | ITO/PEDOT:PSS/M7:PC71BM/Al | 0.90 | 10.20 | 0.55 | 5.05 | 127 |
| M8 | −5.56/−3.75 | 1.70 | ITO/PEDOT:PSS/M8:PC71BM/Al | 0.75 | 4.14 | 0.44 | 1.34 | 109 |
| M9 | −5.69/−3.66 | 1.95 | ITO/PEDOT:PSS/M9:PC71BM/Al | 0.80 | 4.43 | 0.34 | 1.17 | 109 |
| M10 | −5.32/−3.86 | 1.45 | ITO/PEDOT:PSS/M10:PC61BM/Al | 0.7 | 14.3 | 0.47 | 4.7 | 129 |
| M11 | −5.06/−3.73 | 1.41 | ITO/PEDOT:PSS/M11:PC71BM/Ca/Al | 0.74 | 11.28 | 0.38 | 3.13 | 140 |
| M12 | −5.02/−3.64 | 1.40 | ITO/PEDOT:PSS/M12:PC71BM/Ca/Al | 0.67 | 6.80 | 0.34 | 1.56 | 140 |
| M13 | −5.23/−3.72 | 1.60 | ITO/PEDOT:PSS/M13:PC71BM/PFN/Al | 0.97 | 10.55 | 0.47 | 4.75 | 141 |
| M14 | −5.06/−3.60 | 1.46 | ITO/PEDOT:PSS/M14:PC71BM/Ca/Al | 0.7 | 12.98 | 0.62 | 5.61 | 142 |
| M15 | −4.93/−3.28 | — | ITO/PEDOT:PSS/M15:PC71BM/Ca/Al | 0.77 | 13.79 | 0.67 | 7.2 | 143 |
| M16 | −5.36/−3.79 | 1.44 | ITO/PEDOT:PSS/M16:PC71BM/PFN/Al | 0.95 | 14.32 | 0.67 | 8.98 | 144 |
| M17 | −5.62/−3.52 | 1.84 | ITO/PEDOT:PSS/M17:PC71BM/Al | 0.90 | 10.48 | 0.56 | 5.29 | 145 |
| M18 | −4.95/−3.50 | — | ITO/PEDOT:PSS/P3HT:IC70BM:M18/Ca/Al | 0.87 | 7.0 | 0.71 | 4.3 | 152 |
| M19 | −5.45/−3.87 | 1.37 | ITO/MH250:W2(hpp)4/C70/M19:C60/BPAPF/BPAPF:NDP9/NDP9/Al | 0.89 | 6.1 | 0.45 | 2.5 | 153 |
| M20 | −5.36/−3.80 | 1.35 | ITO/MH250:W2(hpp)4/C70/M20:C60/BPAPF/BPAPF:NDP9/NDP9/Al | 0.85 | 9.9 | 0.54 | 4.6 | 153 |
| M21 | −5.23/−3.78 | 1.32 | ITO/MH250:W2(hpp)4/C70/M21:C60/BPAPF/BPAPF:NDP9/NDP9/Al | 0.73 | 13.3 | 0.63 | 6.1 | 153 |
| M22 | −5.68/−4.01 | 1.62 | ITO/C60/M22/PV-TPD/p-PV-TPD/p-ZnPc/Au | 0.92 | 3.0 | 0.33 | 0.8 | 157 |
| M23 | −5.22/−3.65 | 1.48 | ITO/C60/M23/PV-TPD/p-PV-TPD/p-ZnPc/Au | 0.65 | 2.4 | 0.65 | 1.1 | 157 |
| M24 | −5.13/−3.40 | — | ITO/MH250:W2(hpp)4/C60/M24:C60/M24/BF-DPB/BF-DPB:NDP9/NDP9/Al | 0.77 | 10.4 | 0.57 | 4.5 | 162 |
| M25 | −5.40/−3.79 | 1.73 | ITO/ZnO/P3HT:M25/MoO3/Ag | 0.65 | 3.09 | 0.60 | 1.21 | 163 |
| M26 | −5.16/−3.82 | 1.54 | ITO/ZnO/P3HT:M26/MoO3/Ag | 0.62 | 3.90 | 0.63 | 1.51 | 163 |
| M27 | −5.14/−3.74 | 1.47 | ITO/ZnO/P3HT:M27/MoO3/Ag | 0.57 | 3.28 | 0.63 | 1.18 | 163 |
| M28 | −5.36/−3.79 | 1.50 | ITO/PEDOT:PSS/PTB7-Th:p-DTS(FBTTh2)2:M28/Ca/Al | 0.76 | 7.03 | 0.53 | 2.84 | 164 |
α-Functionalization of BODIPY is used to design small molecular electron donors with small bandgap and NIR absorption. In the first report of BODIPY-based small molecular electron donors by Roncali et al., the BODIPY core was functionalized with phenylene-vinyl at the two α-positions (M8 and M9 in Fig. 5a). Preliminary OSC device results show a PCE of 1.34% for M8 and 1.17% for M9.109,128 Ziessel and coworkers developed M10 by attaching two vinyl-bithiophene substituents to the α-positions of BODIPY (Fig. 5b).129 The single-crystal structure of M10 indicated that the vinyl-bithiophene skeletons were coplanar with the BODIPY core (Fig. 5c). Moreover, the vinyl-bithiophene skeletons are well conjugated with the BODIPY core, leading to a narrow bandgap of 1.45 eV with an absorption maximum wavelength of 780 nm. In solid state, M10 molecules organized in three dimensions with strong intermolecular interactions, favoring short distances between neighboring molecules (Fig. 5d). As a result, M10 showed excellent charge transporting property, i.e. both hole and electron mobilities in the order of 10−3 cm2 V−1 s−1 in ambipolar organic field-effect transistors (OFETs). The OSC device with the M10:PC61BM blend as the active layer showed a broad external quantum efficiency (EQE) response covering from 350 nm to ca. 900 nm. The device exhibited a VOC of 0.7 V, a JSC of 14.3 mA cm−2, and an FF of 0.47, leading to a PCE of 4.7%.
 | ||
| Fig. 5 (a) Chemical structures of BODIPY derivatives M8 and M9 functionalized at α-positions. (b) Chemical structure of BODIPY derivative M10. (c) ORTEP view of the single crystal of compound M10. (d) Packing view down the ac direction of M10. Panels (c) and (d) were reproduced from ref. 129 with permission from the American Chemical Society, copyright 2012. | ||
α-Functionalized BODIPY can be used as the A terminals in acceptor–donor–acceptor (A–D–A) type small molecular electron donors.130–139 This molecular design motif can be used to tune the active layer morphology of OSCs for improved device performance. Zhan et al. synthesized M11 (Fig. 6),140 in which two α-functionalized BODIPY units were connected to a terthiophene unit. M11 showed a similar absorption spectrum and LUMO/HOMO energy levels to those of its monomeric derivative M12. When blended with PC71BM in thin film, the monomeric M12 lost its crystalline structure but the dimeric M11 could maintain its ordered packing. This was due to the expanded π-conjugated backbone and the steric-pairing effect of the twisted configuration of M11. The OSC device based on the blend of M11 and PC71BM exhibited a PCE of 3.13% and a high hole/electron mobility of 1.16 × 10−3 cm2 V−1 s−1/0.90 × 10−3 cm2 V−1 s−1. Zhu et al. synthesized M13 by end-capping a dialkylthienyl flanked benzo[1,2-b:4,5-b′]dithiophene (BDT) core with two BODIPY units (Fig. 6).141M13 exhibited a HOMO energy level of −5.23 eV, a LUMO energy level of −3.72 eV and an onset absorption wavelength at 774 nm. With a blend of M13 and PC71BM as the active layer, the OSC device achieved a PCE of 4.75%. The good OSC device performance of M13 was assigned to the optimal phase separation morphology with a small domain size in the active layer.
 | ||
| Fig. 6 Chemical structures of M11–M13 functionalized at the α-positions of BODIPY. | ||
β-Funtionalized BODIPY can be used to develop small molecular electron donors with panchromatic properties, i.e. those that have ultra-wide absorption spectra. Xu et al. developed M14 by functionalizing a BODIPY core at the β-position with two BDT terminals via two alkynyl linkages (Fig. 7a).142 Although the BDT units were co-planar with the BODIPY core, the BDT units were not well conjugated with the BODIPY core. This was evidenced by the delocalized HOMO over the whole skeleton and the localized LUMO on the BODIPY core unit of M14. An interesting property of M14 was its ultra-wide absorption spectrum in thin film, which effectively covered from 300 nm to 800 nm. This wide absorption spectrum indicated strong sunlight harvesting capability and was desirable for solar cell applications. Using M14 as the electron donor and PC71BM as the electron acceptor, the authors demonstrated OSC devices with a PCE of 5.61%. Another example of BODIPY-based electron donors with wide absorption spectra is M15, which is reported by Singh and coworkers.143 In M15, BODIPY acted as the core and was functionalized at the β-position, two dithiafulvalene acted as the terminals, and the furan unit acted as the bridging unit (Fig. 7b). M15 showed a wide absorption spectrum in film ranging from 300 nm to 800 nm with three absorption peaks at 376 nm, 458 nm and 627 nm. The optimized OSC device based on the M15:PC71BM blend showed a high PCE of 7.2% together with a high EQE value of 88.1% at 371 nm.
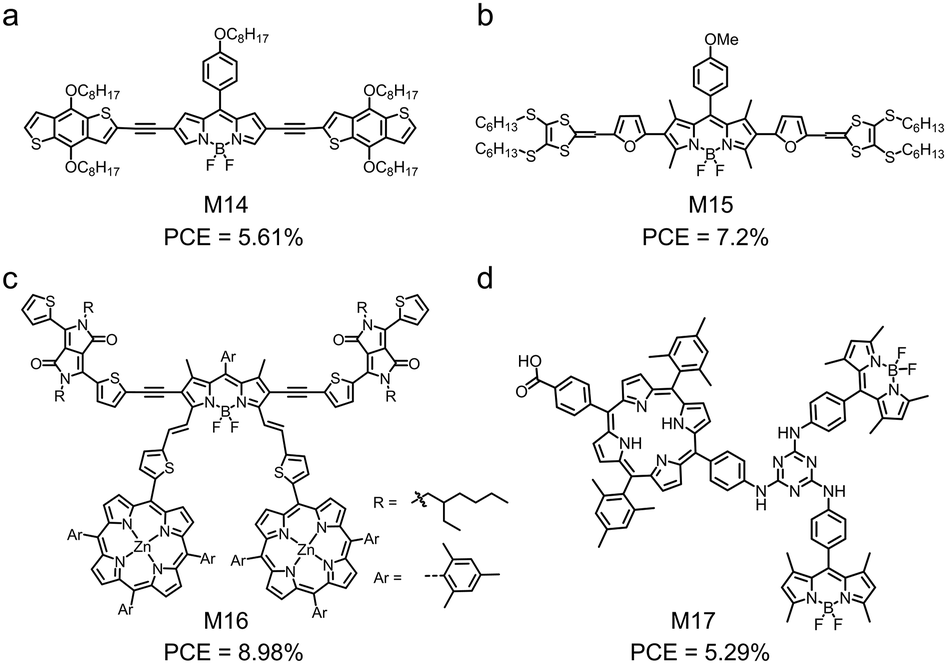 | ||
| Fig. 7 Chemical structures of M14–M17 functionalized at the β-positions of BODIPY. | ||
An interesting approach to design small molecular electron donors is to link BODIPY and other low-bandgap dyes for wide absorption spectra. Gros et al. designed and synthesized M16 by introducing two porphyrin units at the α-positions and attaching two dithienyl-diketopyrrolopyrrole (Th-DPP) units to the β-positions of BODIPY (Fig. 7c).144 The absorption spectra of BODIPY, porphyrin and Th-DPP were complementary, leading to the broad absorption spectrum of M16 covering from 350 nm to 850 nm. The OSC device based on the M16:PC71BM active layer showed a remarkable PCE of 8.98% with a JSC of 14.32 mA cm−2, a VOC of 0.95 V and an FF of 0.67. Moreover, the device showed a small photon energy loss of 0.5 eV due to the small LUMO offset between M16 and PC71BM. Sharma et al. designed a triad of porphyrin and BODIPY bridged by 1,3,5-triazine (M17) (Fig. 7d).145 Its absorption spectrum showed five peaks at 422 nm, 552 nm, 596 nm, 652 nm and 502 nm. The first four peaks were assigned to porphyrin and the last one peak was attributed to BODIPY. The OSC device based on M17 and PC71BM showed a PCE of 5.29% with a JSC of 10.48 mA cm−2.
Annulated BODIPYs always show a small bandgap, redshifted absorption spectra and strong sunlight harvesting capability. For example, M18,152 in which a BODIPY core was fused with two benzene rings at the [a]-bond (Fig. 8a), showed a narrow bandgap with the absorption spectrum peaking at 733 nm. M18 could be used as the third component to be added to the poly(3-hexylthiophene) (P3HT)/indene-C70 bisadduct (IC70BA) active layer to improve the sunlight absorption of the OSC devices. As a result, upon the addiction of M18, the JSC of the device was increased from 6.3 mA cm−2 to 7.0 mA cm−2 and the PCE was improved from 3.7% to 4.3%. Leo et al. developed a series of furan-fused BODIPY derivatives (M19–M21) with trifluoromethyl (CF3) on the meso-carbon atom and various substituents at the peripheral phenyl groups (Fig. 8a).153 The substituents on the phenyl groups greatly affected molecular packing in solid state of these furan-fused BODIPY derivatives (Fig. 8b). M19 showed a slipped face-to-face stacking with a mean plane distance of 3.8 Å. In M20 with the methyl substituents, two parallel molecules were arranged as a repeating unit in a tilted ladder-type arrangement. M21 with the methoxy substituents exhibited a brickwork-type arrangement. It seemed that M19 and M21 adopted J-aggregation and M20 adopted H-aggregation. Their absorption spectra in thin film spanned from 500 nm to 900 nm, and the redshift and blueshift of absorption spectra might be attributed to the J-/H-aggregation of the annulated BODIPY in solid state (Fig. 8c). From M19 to M21, HOMO energy levels continuously upshifted from −5.45 eV to −5.23 eV, and LUMO energy levels upshifted from −3.87 eV to −3.78 eV. The upshifted HOMO/LUMO energy levels were due to the electron-donating properties of the methyl and methoxyl groups. Vacuum-deposited OSC devices were fabricated using M19–M21 as the electron donors and C60 as the electron acceptor. The PCEs of the single-junction OSC devices were 2.5% for M19, 4.6% for M20 and 6.1% for M21 (Fig. 8d). The poor device performance of the M19-based OSC was mainly due to the low-lying LUMO energy level of M19, which could not provide sufficient driving force for photo-induced charge separation. The multi-junction OSC device with M21 as the NIR absorber showed a PCE as high as 9.9%.
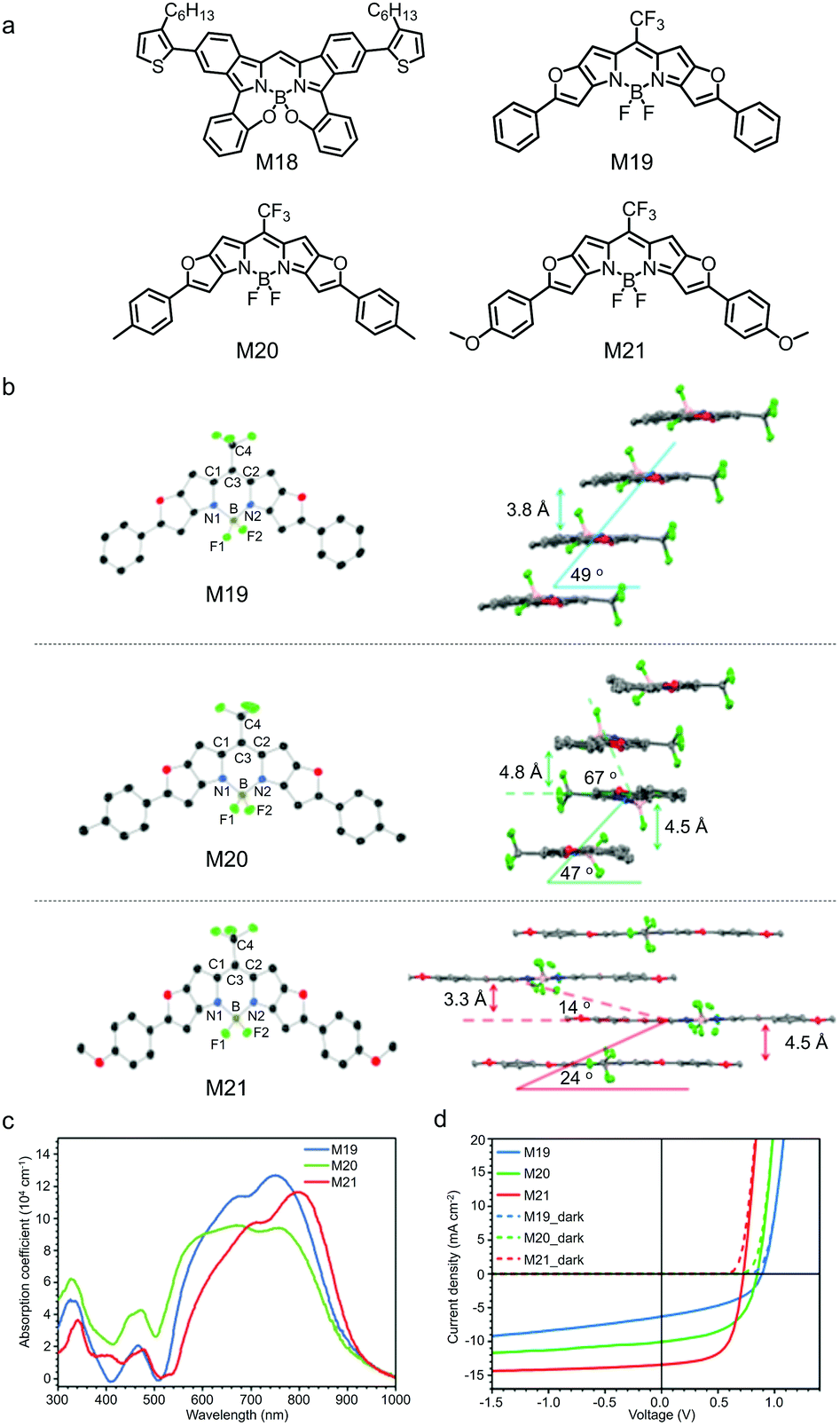 | ||
| Fig. 8 (a) Chemical structures of BODIPY derivatives M18–M21 annulated at the [a]-bond or the [b]-bond. (b) ORTEP plots (left) and packing configurations (right) of M19–M21; hydrogen atoms are omitted. (c) Absorption spectra of M19–M21 in film. (d) J–V curves in the dark and under AM 1.5G illumination of the single-junction OSCs using M19–M21 as the electron donors. Panels (b)–(d) were reproduced from ref. 153 with permission from the American Chemical Society, copyright 2017. | ||
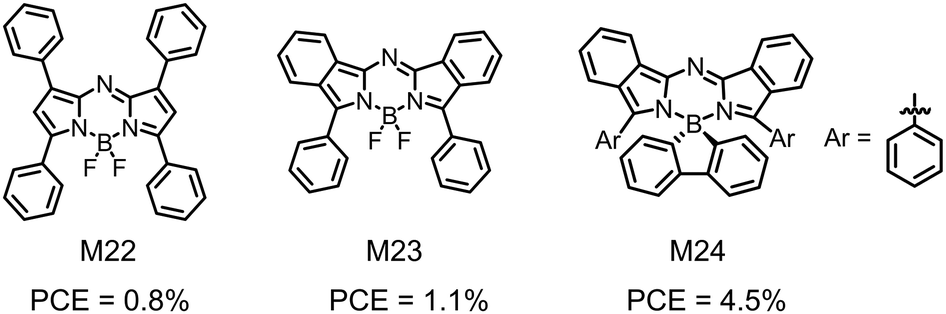 | ||
| Fig. 9 Chemical structures of aza-BODIPY derivatives M22–M24. | ||
In summary, the rich chemistry and facile synthesis enable the development of a large family of BODIPY-based small molecular electron donors. After careful chemical structure optimization and OSC device optimization, a PCE of about 10% has been demonstrated with these BODIPY-based small molecular electron donors. A striking feature of this class of organic photovoltaic materials is their strong sunlight harvesting capability. Both narrow bandgap with strong NIR absorption and panchromatic property with wide absorption spectra have been demonstrated with BODIPY-based small molecular electron donors. It is worth noting that most of the above OSC device results are obtained with fullerene derivatives as the electron acceptors. Great efforts should be devoted to the matching of BODIPY-based small molecular electron donors and non-fullerene electron acceptors. In view of the excellent opto-electronic properties of BODIPY-based small molecules, large room for PCE enhancement of BODIPY-based small molecular electron donors can be anticipated, provided that high-efficiency non-fullerene electron acceptors are used in these OSC devices.
3.2 BODIPY-based small molecular electron acceptors
In view of the low LUMO energy levels of BODIPY, materials chemists have attempted to design BODIPY-based small molecular electron acceptors for OSCs. These compounds always use some π-conjugated linkers to connect two BODIPY units as the terminals. Fig. 10 shows several examples. However, their device performance is not satisfactory possibly due to the unfavorable charge transport and the non-optimal active layer morphology. Thayumanavan et al. reported a series of A–D–A type compounds (M25–M27), in which BODIPY as the A unit was linked at the meso-position by various electron donating units (BDT, cyclopenta-[2,1-b:3,4-b]dithiophene (CPDT) and dithieno [3,2-b:2′,3′-d]pyrrole (DTP)) (Fig. 10).163 These compounds exhibited strong visible absorption with the extinction coefficient on the order of 104 M−1 cm−1 and low-lying LUMO energy levels of lower than −3.7 eV. They showed pure n-type characteristics with electron mobilities on the order of 10−5 cm2 V−1 s−1. The low electron mobilities of M25–M27 might be due to the limited intermolecular interactions, caused by the skeletal twist between the thiophene and the meso-position linked BODIPY. Inverted OSC devices with M25, M26 or M27 as the electron acceptor and P3HT as the electron donor showed a PCE of 1.51%. The limited device performance may be attributed to the noneffective charge transport. Zhan et al. used the DPP unit as the core and two BODIPY units as the endcapper, and linked them with two ethynyl bridges to design M28 (Fig. 10).164 The ethynyl bridges not only enhanced the planarity, but also played an important role in π-electron delocalization of the molecule.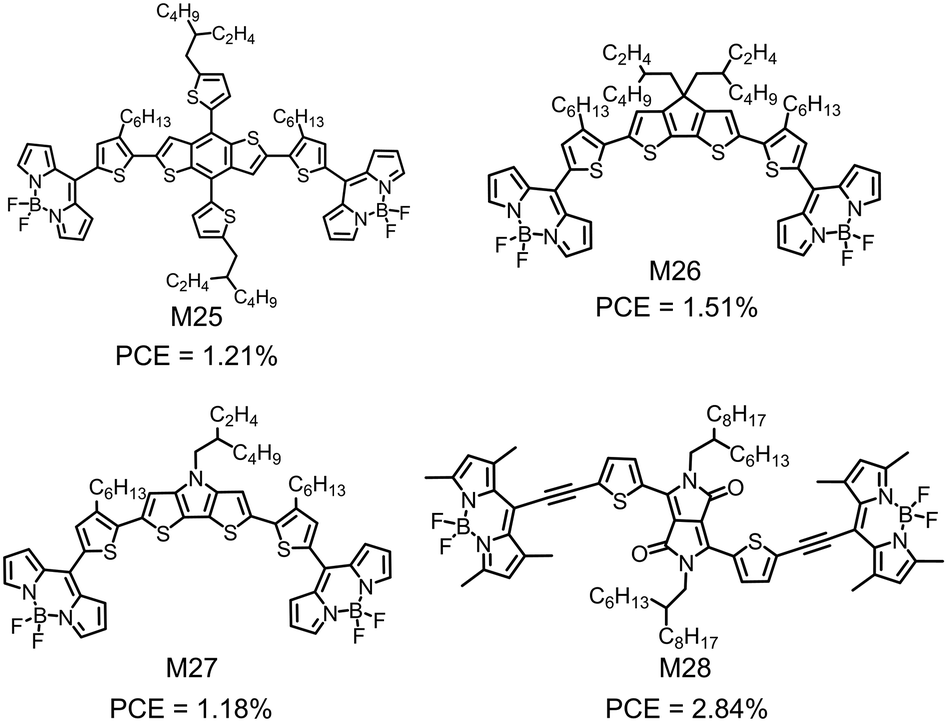 | ||
| Fig. 10 Chemical structures of BODIPY-based electron acceptors M25–M28. | ||
M28 showed a wide absorption spectrum covering the ultraviolet-visible-near infrared (UV-vis-NIR) region with a low bandgap of 1.5 eV. The OSC device based on M28 as the electron acceptor showed a PCE of 2.84% (Fig. 17).
3.3 SubPc-based small molecular electron donors
Boron subphthalocyanine (SubPc, see Fig. 11) is a 14 π-electron heteroaromatic molecule, in which a tetracoordinated boron atom is surrounded by three N-fused diiminoisoindole units.77,165–167 SubPc can be synthesized with high yield by the cyclotrimerization reaction of phthalonitrile precursors with boron trichloride (BCl3). Unmodified SubPc has strong absorption in the wavelength range of 450–620 nm with a bandgap of ca. 2.1 eV. Its opto-electronic properties can be tuned by functionalization at the axial position and peripheral position. SubPc has a C3-symmetric cone-shaped geometry. The nonplanar configuration of SubPc is beneficial not only for vacuum sublimation deposition, but also for good phase separation morphology when blended with other organic opto-electronic materials and processed by spin-coating. SubPc and its derivatives are always used as electron donors in OSCs, while carefully modified SubPc derivatives can also work as electron acceptors Table 2. Unmodified SubPc is always used in vacuum-deposited OSC devices, while modified SubPc with enhanced solubility can be used to fabricate solution-processed OSC devices. | ||
| Fig. 11 Chemical structures, functionalized positions and 3D-geometry of SubPc. The 3D-geometry of SubPc was reproduced from ref. 165 with permission from the American Chemical Society, copyright 2012. | ||
| Small molecules | HOMO/LUMO (eV/eV) | E optg (eV) | Device structure | V OC (V) | J SC (mA cm−2) | FF | PCEa (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The maximum PCE value of the corresponding device. | ||||||||
| SubPc | −5.6/−3.6 | 2.07 | ITO/SubPc/C60/BCP/Al | 0.97 | 3.36 | 0.57 | 2.1 | 168 |
| ITO/mCP/SubPc/C60/BCP/Al | 1.09 | 7.87 | 0.59 | 5.08 | 171 | |||
| ITO/MoO3/SubPc:C70/BCP/Al | 1.02 | 12.90 | 0.41 | 5.4 | 172 | |||
| ITO/MoOx/Tc/SubPc/BCP/Al | 1.24 | 4.01 | 0.58 | 2.89 | 191 | |||
| SubNc | −5.4/−3.6 | 1.71 | ITO/PEDOT:PSS/SubNc/C60/BCP/Al | 0.55 | 5.59 | 0.49 | 1.47 | 182 |
| ITO/MoO3/SubNc:PC71BM/BCP/Al | 0.92 | 10.10 | 0.43 | 4.0 | 183 | |||
| ITO/PEDOT:PSS/α-6T/SubNc/BCP/Al | 0.94 | 12.04 | 0.54 | 6.02 | 199 | |||
| ITO/PEDOT:PSS/α-6T/SubNc/SubPc/BCP/Al | 0.96 | 14.55 | 0.61 | 8.40 | 199 | |||
| M29 | −5.06/— | — | ITO/PEDOT:PSS/M29/C60/BCP/Al | 0.73 | 3.72 | 0.51 | 1.39 | 184 |
| M30 | −4.81/— | — | ITO/PEDOT:PSS/M30/C60/BCP/Al | 0.63 | 3.30 | 0.34 | 0.70 | 184 |
| M31 | −5.6/−3.4 | 2.22 | ITO/MoOx/SubPc/M31/BCP/Al | 1.10 | 0.83 | 0.22 | 0.19 | 192 |
| M32 | −5.8/−3.6 | 2.24 | ITO/MoOx/SubPc/M32/BCP/Al | 1.22 | 1.56 | 0.43 | 0.80 | 192 |
| M33 | −5.8/−3.7 | 2.17 | ITO/MoOx/SubPc/M33/BCP/Al | 1.31 | 3.53 | 0.58 | 2.68 | 192 |
| ITO/ZnO/PTB7-Th:M33/MoOx/Ag | 0.77 | 10.7 | 0.48 | 4.0 | 201 | |||
| M34 | —/−3.54 | — | ITO/MoO3/SubNc/M34/BCP:C60/Ag | 1.08 | 4.83 | 0.48 | 2.44 | 193 |
| M35 | —/−3.71 | — | ITO/MoO3/SubNc/M35/BCP:C60/Ag | 0.89 | 6.18 | 0.43 | 2.36 | 193 |
| M36 | —/−3.85 | — | ITO/PEDOT:PSS/DIP/SubNc/M36/BCP:Yb | 0.75 | 8.55 | 0.53 | 3.31 | 193 |
| M37 | −5.7/−3.6 | — | ITO/MoO3/SubNc/M37/BCP/Al | 1.06 | 2.06 | 0.43 | 0.94 | 195 |
| M38 | — | — | ITO/PEDOT:PSS/α-6T/M38/BCP/Ag | 1.17 | 4.83 | 0.49 | 2.77 | 198 |
| M39 | — | — | ITO/PEDOT:PSS/α-6T/M39/BCP/Ag | 1.21 | 4.40 | 0.47 | 2.51 | 198 |
| M40 | −6.00/−3.84 | 2.16 | ITO/ZnO/PTB7-Th:M40/MoOx/Ag | 0.74 | 3.6 | 0.40 | 1.1 | 201 |
| M41 | −6.02/−3.86 | 2.16 | ITO/ZnO/PTB7-Th:M41/MoOx/Ag | 0.81 | 5.3 | 0.41 | 1.8 | 201 |
| M42 | −6.08/−3.93 | 2.15 | ITO/ZnO/PTB7-Th:M42/MoOx/Ag | 0.50 | 2.1 | 0.45 | 0.5 | 201 |
| M43 | −5.69/−3.98 | 1.82 | ITO/ZnO/PBDB-T:M43/MoO3/Al | 0.74 | 5.18 | 0.46 | 1.78 | 202 |
| M44 | −5.71/−3.83 | 1.88 | ITO/ZnO/PBDB-T:M44/MoO3/Al | 0.89 | 9.18 | 0.55 | 4.53 | 202 |
| M45 | −5.60/−3.90 | 1.68 | ITO/PEDOT:PSS/PBDB-T:M45/Ca/Al | 0.87 | 10.06 | 0.54 | 4.69 | 203 |
| M46 | −5.63/−4.06 | 1.66 | ITO/PEDOT:PSS/PBDB-T:M46/Ca/Al | 0.77 | 10.51 | 0.47 | 3.80 | 203 |
| M47 | −5.82/−3.91 | 1.93 | ITO/ZnO/PM6:M47/MoO3/Ag | 0.84 | 9.69 | 0.60 | 4.92 | 204 |
| M48 | −5.39/−3.81 | 1.74 | ITO/ZnO/PM6:M48/MoO3/Ag | 0.97 | 13.97 | 0.55 | 7.41 | 205 |
In 2006, Thompson and co-workers pioneered the development of OSCs based on SubPc.168 They used SubPc as the electron donor and fullerene (C60) as the electron acceptor to fabricate bilayer OSC devices by vacuum deposition (Fig. 12a and b). Because of the deep HOMO energy level (−5.6 eV) of SubPc, the SubPc/C60-based OSC device exhibited a high VOC of 0.97 V, a JSC of 3.36 mA cm−2 and a PCE of 2.1%. This vacuum deposited SubPc/C60 bilayer OSC device allows in-depth investigation of the fundamental physics of organic photovoltaics. Device physicists have used SubPc/C60 bilayer OSC devices as a test-bed to study the energy transfer mechanism, photoinduced charge transfer, interface energetics, charge recombination dynamics, charge collection efficiency, etc. of OSC devices. For a comprehensive review of the physics of SubPc/C60 bilayer OSC devices, the readers are recommended to refer to some excellent reviews.77,165,167
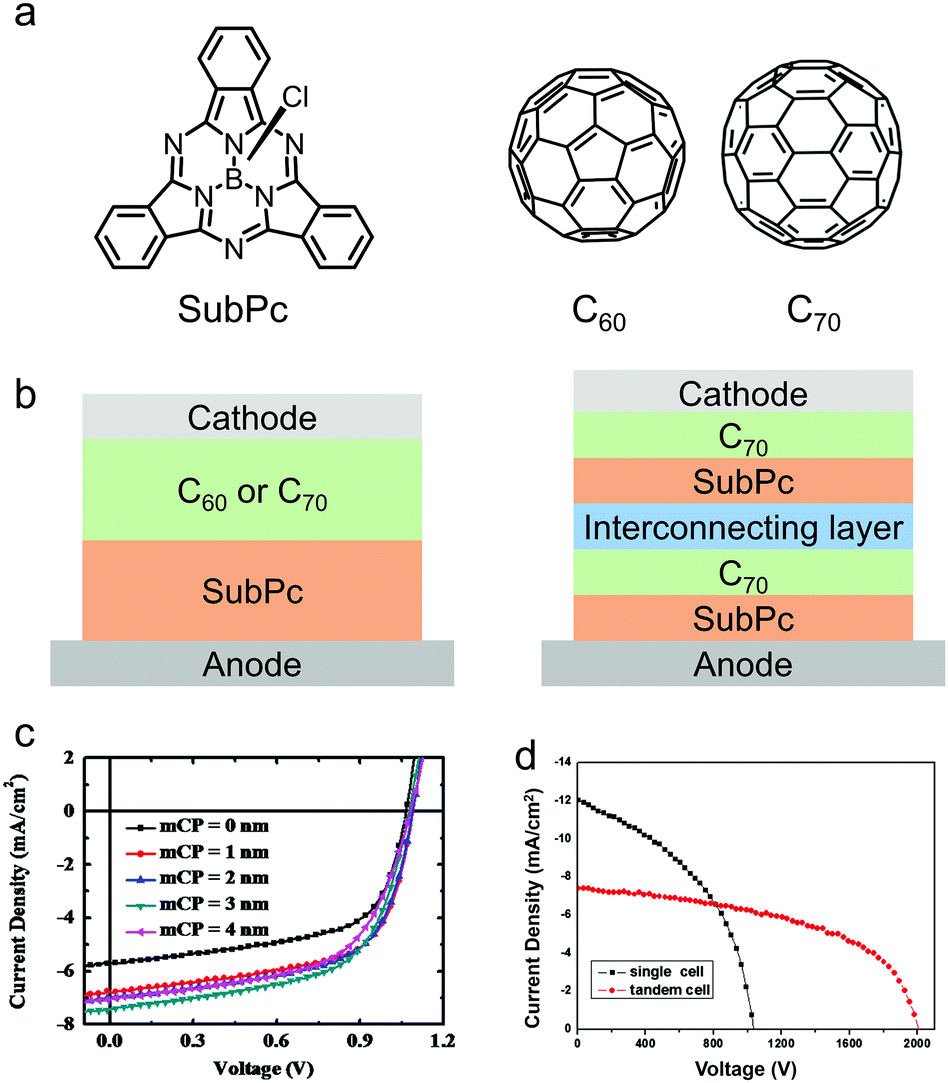 | ||
| Fig. 12 (a) Chemical structures of the electron donor SubPc and the electron acceptors C60 and C70. (b) Device architectures of vacuum-deposited planar heterojunction (PHJ) OSCs. (c) J–V curves of the devices based on the active layer of SubPc:C60 with different anode interfacial layer (mCP) thickness. (d) J–V curves of the single device and tandem device based on the active layer of SubPc:C70. Panel (c) was reproduced from ref. 171 with permission from Elsevier, copyright 2013. Panel (d) was reproduced from ref. 173 with permission from Elsevier, copyright 2014. | ||
Great efforts have been made to improve the PCE of SubPc-based vacuum-deposited OSC devices.169,170 The deep HOMO energy level of SubPc leads to an energy barrier for hole extraction from the active layers to the indium tin oxide (ITO) anode. Therefore, the device performance of SubPc/C60 can be improved by modification of the ITO surface to minimize the energy barrier. Lin et al. introduced a thin layer of N,N-dicarbazolyl-3,5-benzene (mCP) between the ITO anode and the SubPc layer.171 The mCP layer not only improved hole extraction, but also acted as an electron blocking layer and reduced the leakage current of the OSC device. As a result, all the three key parameters, VOC, JSC and FF, of the device increased, leading to a greatly enhanced PCE of 5.1% (Fig. 12c). A drawback of C60 is the weak light absorption capability, i.e. a small extinction coefficient in the visible range. C70 has a larger extinction coefficient and a red-shifted absorption spectrum compared to those of C60, thereby has stronger sunlight absorption capability. Holmes et al. utilized C70 as the electron acceptor and SubPc as the electron donor, and fabricated a vacuum-deposited OSC with a continuously graded heterojunction layer.172 The device exhibited a significantly improved JSC of 12.90 mA cm−2, which was due to the improved sunlight harvesting capability and the increased donor/acceptor interfacial area in the active layers. A PCE of 5.4% was obtained, which was much higher than that of the C60-based device. Furthermore, by stacking two SubPc:C70 layers with an interconnecting layer inserted, a tandem OSC device was made and a high PCE of 7.66% was demonstrated (Fig. 12d).173 These results indicate that unmodified SubPc is an excellent electron donor for vacuum-deposited OSCs.174–178
Compared with vacuum deposition, solution processing techniques such as. spin-coating and blade-coating are low-cost processing techniques to fabricate OSCs. The cone-shaped geometry of SubPc derivatives prevents compact molecular stacking and makes them readily soluble in organic solvents. Therefore, SubPc derivatives can also be solution-processed to fabricate OSCs. A typical example is chloroboron(III) subnaphthalocyanine (SubNc), which is comprised of three N-fused benzoisoindole units and an axial ligand around a boron core (Fig. 13a).179,180 Compared with SubPc, SubNc has extended π-conjugation and consequently shows an upshifted HOMO energy level and a red-shifted absorption spectrum.181 SubNc exhibits strong light absorption spanning from 550 nm to 750 nm. Ma et al. initially adopted solution processing to fabricate bilayer OSC devices based on SubNc. The SubNc layer was spin-coated with the chlorobenzene solution and the C60 layer was vacuum-deposited.182 Hong et al. reported OSC devices using a blend of SubNc and PC71BM as the active layer, which was spin-coated with their blend solution in 1,2-dichlorobenzene (Fig. 13b).183 The device exhibited a high JSC of 10.1 mA cm−2 and a PCE of 4.0%.
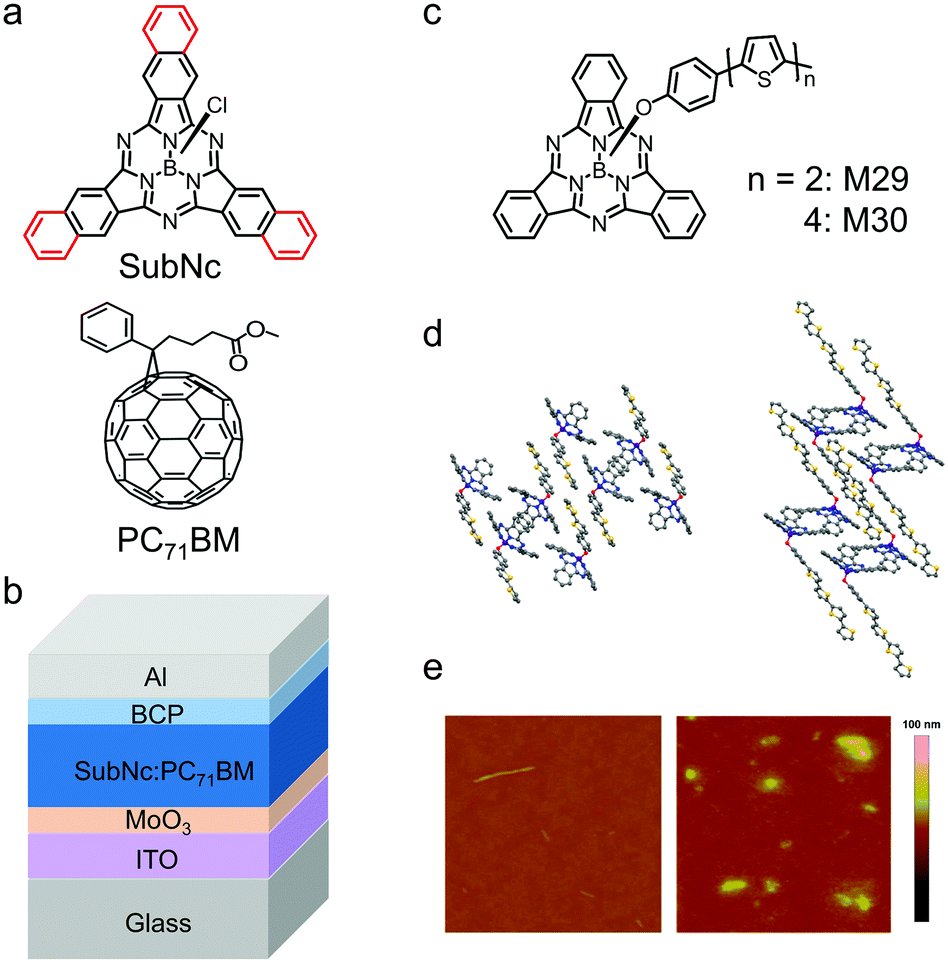 | ||
| Fig. 13 (a) Chemical structures of the electron donor SubNc and the electron acceptor PC71BM. (b) Device architecture of the SubNc:PC71BM bulk heterojunction (BHJ) device for solution-processed OSCs. (c) Chemical structures of M29 and M30. (d) View of the crystal packing of M29 (left) and M30 (right). (e) Atomic force microscopy (AFM) images of M29 (left) and M30 (right). Panels (d) and (e) were reproduced from ref. 184 with permission from the American Chemical Society, copyright 2010. | ||
An interesting approach to develop a solution-processed SubPc-based electron donor is to introduce bithiophene or quaterthiophene as the axial ligand in SubPc with phenoxy as the linker. The chemical structures of the resulting two compounds, M29 and M30, are shown in Fig. 13c and their molecular packing in crystals is provided in Fig. 13d.184 The oligothiophene axial ligands of M29 and M30 provided the driving force for ordered molecular stacking and thus enhanced crystallization. Moreover, the longer the oligothiophene ligand of the linkage, the stronger the crystallization tendency of the compounds. In the films, M30 with quaterthiophene as the axial ligand was more crystalline than M29 with bithiophene as the axial ligand. The atomic force microscopy (AFM) height images of the two films are shown in Fig. 13e. The surface of the M29 film was smooth with a root-mean-square (RMS) roughness of about 0.5 nm. The surface of the M30 film displayed some aggregates with a relatively high RMS roughness of 1.4 nm, which could be attributed to the crystallization of M30. Bilayer OSCs were fabricated with the spin-coated M29 or M30 layer and the vacuum-evaporated C60 layer and the preliminary PCE was 1.39%.
3.4 SubPc-based small molecular electron acceptors
The LUMO/HOMO energy levels of unsubstituted SubPc are about −3.6 eV/−5.6 eV and it is initially used as the electron donor.168 When the peripheral or axial positions of SubPc are modified with electron-deficient groups, the resulting SubPc derivatives will show downshifted HOMO/LUMO energy levels and can be used as electron acceptors in OSCs.185–190 That is, SubPc derivatives can be converted from electron donors to electron acceptors by substitution with electron-deficient groups.The feasibility of SubPc as an electron acceptor for OSCs was firstly demonstrated by Jones et al. They fabricated a vacuum-deposited OSC with tetracene (Tc) as the electron donor and SubPc as the electron acceptor.191 The device showed a high VOC of 1.24 V and a PCE of 2.89%. Although SubPc was typically used as an electron donor, the compound could also work as an electron acceptor provided that the offset between the donor and the acceptor was sufficient to dissociate charges efficiently. The authors introduced different types and numbers of halogen to the peripheral positions of SubPc and obtained three halogenated-SubPc (M31–M33, see Fig. 14).192 Compared with M31 with three F substituents at the peripheral position, the increase in the number of F atoms in M32 significantly reduced the LUMO energy level from −3.4 eV to −3.6 eV. The replacement of F atoms in M32 with Cl atoms further reduced the LUMO energy level to −3.7 eV of M33, the LUMO energy level was further reduced to −3.7 eV, which was derived from the stronger electron-withdrawing property of the Cl atom compared to the F atom. The low-lying LUMO energy levels made the halogenated-SubPc compounds work as electron acceptors to match unsubstituted SubPc as the electron donor. Among the three halogenated-SubPc compounds, M33 with the lowest LUMO energy level of −3.7 eV exhibited the best OSC device performance. The device based on the SubPc:M33 active layer showed a PCE of 2.68% with a VOC of 1.31 V, a JSC of 3.53 mA cm−2 and an FF of 0.58.
 | ||
| Fig. 14 Chemical structures of SubPc derivatives with substitution at the peripheral and axial positions. | ||
A main reason for the low JSC of the SubPc:halogenated-SubPc device was the overlap of the absorption spectra between the electron donor and the electron acceptor. Device efficiency enhancement was achieved by selecting electron donors with complementary absorption spectra. Cnops et al. used four peripherally substituted SubPc derivatives (M33–M36) as the electron acceptors.193 The electron-deficient character of the substituents (F, Cl and cyano group) could lower the frontier orbital energies of M33–M36 in different degrees, resulting in LUMO energy levels ranging from −3.54 eV to −3.85 eV. They chose small-bandgap donor materials and fabricated vacuum-evaporated PHJ OSC devices. The devices gave a large photocurrent because of the improved sunlight absorption. After elegant device optimization, the OSC device based on SubNc:M33 showed a PCE of 6.86% with a VOC over 1.0 V and a JSC of 10.1 mA cm−2. The high VOC of SubPc-based OSC devices can be used to construct tandem OSCs with extremely high VOC to power a typical battery for energy storage. Jones et al. selected two sub-cell D/A pairs of SubPc/C60 (VOC = 1.1 V) and SubPc/M33 (VOC = 1.3 V) to fabricate multi-junction OSC devices.194 The resulting single-stack tandem OSC device showed a PCE of 3.4% and a VOC of 4.54 V. Moreover, an ultra-high VOC of over 7 V was achieved with 6-heterojunction. The devices could be used to charge a micro-energy battery under low intensity white light or monochromatic LED illumination.
An alternative approach to design SubPc-based electron acceptors is axial substitution with electron-withdrawing groups. Bender et al. developed a SubPc derivative (M37) with pentafluorophenoxy functionalization in the axial position.195 The pentafluorophenoxy functionalization led to downshift of both HOMO and LUMO energy levels by about 0.1 eV. M37 could be used as either an electron donor or an electron acceptor in PHJ OSC devices. The authors also investigated the outdoor performance and stability of axially functionalized SubPc derivatives as electron acceptors.196,197 They used phenoxy, phenyl and chloro-functionalized SubPcs (M38, M39 and SubPc) as electron acceptors and α-sexithiophene (α-6T) as an electron donor to construct PHJ OSC devices,198 which were encapsulated and tested under outdoor conditions. The axial functionalization groups greatly affected the device lifetime. The OSC device based on SubPc with the chlorine substituent exhibited the highest JSC and PCE under passive load and took far longer to degrade than the device based on M38 with the phenoxy substituent and the device based on M39 with the phenyl substituent. The T80 (the operation time at which the OPV reaches 80% of its original PCE) of SubPc is more than 80 h of irradiation, while the T50 (the operation time at which the OPV reaches 50% of its original PCE) of M38 and M39 is only 39 h and 22 h, respectively. Compared with M38 and M39, chloro-SubPc showed the highest outdoor device stability, which is attributed to the best intrinsic stability and the slowest degradation rate of chloro-SubPc. A possible reason for the faster degradation rates of M38 and M39 is that they are more susceptible to hydrolysis, compared with chloro-SubPc. These studies have shown that the functional groups at the axial positions of SubPcs have an important impact on their stability, and minor chemical modification may greatly affect their stability.
SubNc with extended π-conjugation and a redshifted absorption spectrum can also work as an electron acceptor in OSCs. Cnops et al. used SubNc as the electron acceptor and α-6T as the electron donor to make a PHJ OSC device, which showed a high PCE of 6.0% with a VOC of 0.94 V, a JSC of 12.04 mA cm−2 and an FF of 0.54.199 To further improve the sunlight absorption of the OSC device, the authors developed a three-layer PHJ architecture comprising both SubNc and SubPc as separated acceptor layers and α-6T as the donor layer (Fig. 15a).199 In this device, excitons generated in the SubPc acceptor (outer layer) could be transferred to the smaller-bandgap SubNc acceptor (middle layer) by long-range Förster energy transfer, and then dissociated at the α-6T/SubNc interface (Fig. 15b). SubNc functioned not only as an energy acceptor for excitons generated in the SubPc layer, but also as a charge acceptor at the interface of α-6T/SubNc. SubNc, SubPc and α-6T all contributed to the generation of photocurrent, leading to the increase from 12.04 mA cm−2 for the α-6T/SubNc-based device to 14.55 mA cm−2 for the α-6T/SubNc/SubPc-based device. Surprisingly, this three-layer device exhibited a record-high PCE of 8.4% with a high VOC close to 1 V and a wide EQE spectrum covering from 400 nm to 720 nm (Fig. 15c–e).
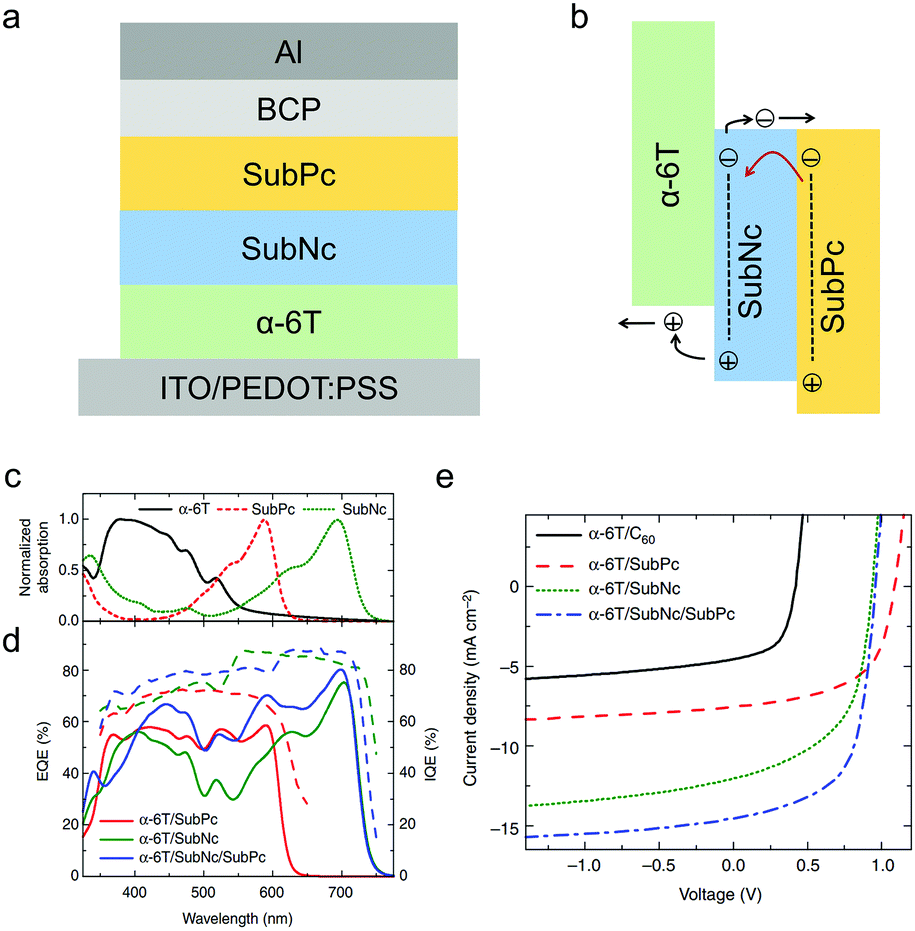 | ||
| Fig. 15 (a) The device structure of the three-layer PHJ OSC based on α-6T, SubPc and SubNc. (b) Illustration of the two-step exciton dissociation process of the active layer. (c) The absorption spectra of α-6T, SubPc and SubNc. (d) The external quantum efficiency (EQE) and internal quantum efficiency (IQE) spectra of the two-layer or three-layer PHJ OSC devices. (e) J–V curves of the two-layer or three-layer PHJ OSC devices. Panels (c–e) were reproduced from ref. 199 with permission from Springer Nature, copyright 2014. | ||
SubPc derivatives with enhanced solubility had been used in solution-processed OSCs and excellent photovoltaic performance had been demonstrated.200 Janssen et al. reported a series of chlorinated SubPcs (M33, M40–M42) bearing different axial substituents, including chlorine and substituted phenoxy groups (Fig. 14 and 16).201 Modification of axial substituents had little effect on the absorption spectra and LUMO/HOMO energy levels of the compounds. The optical bandgaps of these compounds were almost identical, and the LUMO/HOMO energy levels only spanned from −3.84 eV/−6.00 eV for M40 to −3.93 eV/−6.08 eV for M42. Although the axial substituents did not obviously affect the opto-electronic properties of the compounds, they significantly affected the aggregation and charge transporting properties of the molecules. The axial substituents could act as a steric hindrance to prevent stacking of molecular backbones. As the chlorine atom in M33 was smaller than the phenoxy groups in M40–M42, M33 showed more tight molecular stacking than others. This could be verified by the electron mobilities in blend films. Compared with M40, M41 or M42 with the phenoxy substituent, M33 with a small chlorine substituent exhibited higher electron mobility in the PTB7-Th:M33 blend film. Benefiting from the more balanced charge transport, the solution-processed OSC device based on the PTB7-Th:M33 active layer exhibited a higher PCE of 4.0%.
 | ||
| Fig. 16 Chemical structures of SubPc derivatives as electron acceptors for solution-processed OSCs. | ||
Wang et al. introduced three perylene diimide (PDI) units to the periphery of SubPc to develop star-shaped molecules, M43 and M44 (Fig. 16).202 The PDIs as wing groups could not only broaden the absorption spectrum but also lower the LUMO/HOMO energy levels of SubPcs. Compared with the acetylene bridge linked M43, the single bond linked M44 showed a more twisted molecular geometry, which reduced the intermolecular aggregation interaction, leading to appropriate phase separation and more balanced charge transport in the active layers of solution-processed OSCs. As a result, the device based on M44 exhibited remarkably higher VOC, JSC and FF than those of the M43-based device. The authors also designed two star-shaped SubPc-based electron acceptors, M45 and M46, in which 2-(3-oxo-2,3-dihydroinden-1-ylidene)malononitrile (IC) and fluorinated IC were introduced with thiophene bridges, respectively (Fig. 16).203 Due to the strong intramolecular charge transfer (ICT) between the SubPc core and the peripheral IC groups as well as the extended conjugation, M45 and M46 exhibited wide absorption spectra with the full width at half maximum (FWHM) of over 260 nm together with narrow bandgaps of 1.68 eV and 1.66 eV, respectively. Compared with M46 with fluorinated IC, M45 with IC showed a higher LUMO energy level of −3.90 eV, which could lead to a higher VOC value in OSCs. The solution-processed OSCs with M45 or M46 as the acceptor and PBDB-T as the donor showed a PCE of 4.69% with a VOC of 0.87 V or 3.80% with a VOC of 0.77 V, respectively. These devices exhibited a wide photoresponse in the range of 300–750 nm.
An alternative approach to design solution-processable SubPc-based electron acceptors is to fuse electron-withdrawing peripheral aromatic rings, e.g. imide204 and PDI,205 with SubPc cores. Fig. 16 shows two examples of these compounds, M47 and M48. In M48, a SubPc core is fused with three PDI units. The molecule had a cone-shaped and fully conjugated molecular geometry. The dihedral angle between PDI wings and the cone base was ∼25°. The introduction of the PDI wings significantly broadened the absorption spectrum with a red-shifted absorption peak at 680 nm and a high extinction coefficient of 1.25 × 105 M−1 cm−1. The π-conjugated and fully fused connection between the SubPc core and the electron-withdrawing PDI units obviously reduced the LUMO energy level (−3.81 eV) of M48, which was close to that of the PDI unit itself. The low-lying energy level of M48 made it to match with common electron donors in OSCs. Solution-processed OSCs with M48 as the electron acceptor and PM6 as the electron donor exhibited a PCE of 7.53% with a JSC of 13.97 mA cm−2, a VOC of 0.97 V, and an FF of 0.55. This was the best photovoltaic performance for solution-processed OSCs based on SubPc compounds.
 | ||
| Fig. 17 Chemical structures of the electron donor/acceptor materials discussed in this manuscript. | ||
In summary, SubPc derivatives have been intensively used in vacuum-deposited and solution-processed OSCs. PCEs of 7–8% have been demonstrated. A bottleneck limiting the photovoltaic performance of SubPc derivatives is their low charge carrier mobility, which may stem from their cone-shaped configurations. A rational molecular design of SubPc derivatives needs to be adapted to improve their charge transporting properties and their microstructure in thin film towards enhanced OSC device performance. SubPc derivatives have tunable LUMO/HOMO energy levels and they can be used as either electron donors and electron acceptors in OSCs. Noteworthily, in cyclic voltammetry (CV), SubPc derivatives often show irreversible oxidation waves but sometimes show reversible reduction waves. This means that SubPc derivatives are more stable as radical anions than as radical cations and that SubPc derivatives may be more suitable as n-type materials than as p-type materials. Therefore, in OSC applications, we think that SubPc derivatives are more suitable as electron acceptors than as electron donors.
3.5 BNBP-based small molecular electron acceptors
Double B ← N bridged bipyridine (BNBP) is an electron-deficient building block with planar configuration and it was firstly reported by Liu et al. in 2016. BNBP is always used to design n-type polymer semiconductors for application in OSCs, OFETs and organic thermoelectrics.76,206–208 High-efficiency small molecular electron acceptors always have an A–D–A structure, in which an electron-donating core unit is endcapped by two electron-accepting terminals. In 2018, Liu et al. used the electron-deficient BNBP as the core unit to develop small molecular electron acceptors with wide absorption spectra.209 As shown in Fig. 18a, the BNBP-based small molecular electron acceptor, M49, consisted of BNBP as the core unit, two thiophenes as the bridging units and two strong electron-withdrawing IC as the endcapping groups.209 Four bulky phenyl substituents were attached to the BNBP core unit. They acted as a steric hindrance, preventing over-aggregation of the molecules and large-size phase separation in the active layers of OSCs. Typical A–D–A type small molecular acceptors exhibited only one strong absorption band in the long-wavelength region. In comparison, the BNBP-based small molecular acceptor M49 showed two strong absorption bands in the long-wavelength region (λ = ca. 700 nm) and the short wavelength region (λ = ca. 500 nm). The wide absorption spectrum suggested strong sunlight harvesting capability, which was very desirable for photovoltaic applications.210 This wide absorption spectrum was due to the delocalized LUMO and localized HOMO (Fig. 18b), which partially suppressed the HOMO → LUMO electronic transition. M49 exhibited LUMO/HOMO energy levels of −3.93 eV/−5.34 eV, which matched well with a commercially available polymer electron donor, PTB7-Th. The OSC device based on the PTB7-Th:M49 active layer exhibited a PCE of 7.06% and showed a wide photoresponse wavelength extending to 900 nm (Fig. 18d and e) Table 3.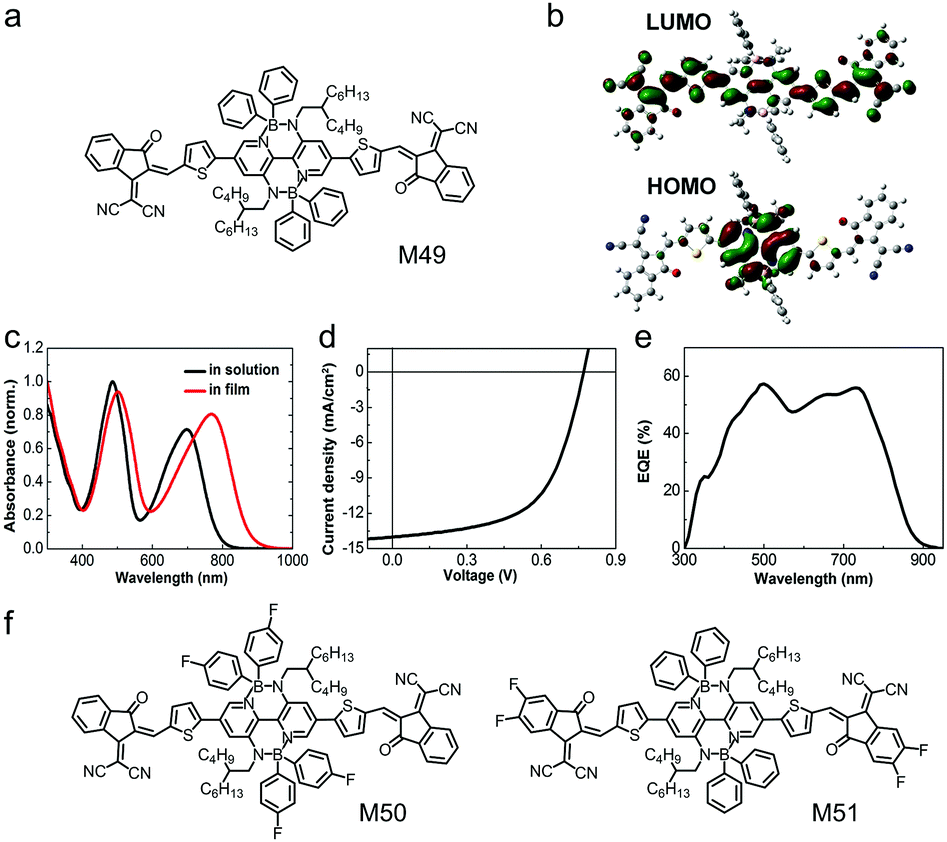 | ||
| Fig. 18 (a) Chemical structure of M49. (b) Kohn–Sham LUMO and HOMO based on density functional theory (DFT) calculations for the model molecule of M49. (c) UV-vis absorption spectra of M49 in solution and in thin film. (d) J–V curve and (e) EQE curve of the device based on the active layer of PTB7-Th:M49. (f) Chemical structures of M50 and M51. Panels (b–e) were reproduced from ref. 209 with permission from the Royal Society of Chemistry, copyright 2017. | ||
| Small molecules | HOMO/LUMO (eV/eV) | E optg (eV) | Device structure | V OC (V) | J SC (mA cm−2) | FF | PCEa (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The maximum PCE value of the corresponding device. | ||||||||
| M49 | −5.34/−3.93 | 1.40 | ITO/PEDOT:PSS/PTB7-Th:M49/Ca/Al | 0.78 | 14.62 | 0.62 | 7.06 | 209 |
| M50 | −5.39/−3.94 | 1.47 | ITO/PEDOT:PSS/PTB7-Th:M50/Ca/Al | 0.73 | 11.40 | 0.60 | 4.96 | 211 |
| M51 | −5.35/−4.02 | 1.33 | ITO/PEDOT:PSS/PTB7-Th:M51/Ca/Al | 0.64 | 8.80 | 0.44 | 2.49 | 211 |
| M52 | −5.73/−3.61 | 2.16 | ITO/PEDOT:PSS/D-OEG:M52/Ca/Al | 1.08 | 2.83 | 0.34 | 1.03 | 216 |
| M53 | −5.92/−3.97 | 1.91 | ITO/ZnO/PTB7-Th:M53/MoO3/Al | 0.80 | 8.97 | 0.44 | 3.12 | 217 |
| M54 | −5.87/−3.91 | 1.86 | ITO/ZnO/PTB7-Th:M54/MoO3/Al | 0.87 | 10.30 | 0.42 | 3.75 | 217 |
| M55 | −5.63/−3.97 | 1.71 | ITO/ZnO/PTB7-Th:M55/MoO3/Al | 0.84 | 10.27 | 0.47 | 4.07 | 217 |
| M56 | −5.82/−3.62 | 2.13 | ITO/ZnO/TTFQX-T1:M56/MoOx/Ag | 1.04 | 9.51 | 0.37 | 3.64 | 218 |
| M57 | — | — | ITO/ZnO/M57:PC71BM/MoO3/Ag | 0.66 | 4.60 | 0.42 | 1.27 | 219 |
| M58 | −4.89/−3.68 | 1.21 | ITO/ZnO/M58:PC71BM/MoOx/Ag | 0.67 | 11.7 | 0.50 | 3.88 | 220 |
| M59 | −5.57/−3.65 | 1.82 | ITO/ZnO/J61:M59/MoO3/Al | 0.77 | 4.10 | 0.47 | 1.49 | 221 |
| M60 | −5.47/−3.72 | 1.59 | ITO/ZnO/J52:M60/MoO3/Al | 0.78 | 10.58 | 0.52 | 4.31 | 221 |
| M61 | −6.24/−3.88 | 2.27 | ITO/ZnO/PTB7-Th:M61/MoO3/Ag | 0.87 | 5.4 | 0.40 | 1.9 | 222 |
| M63 | —/−4.08 | — | ITO/ZnO/PTB7-Th:M63/MoO3/Al | 0.87 | 8.44 | 0.40 | 2.9 | 223 |
In order to tune the LUMO/HOMO energy levels of M49, Liu et al. introduced four electron-withdrawing fluorine substituents to either the organoboron core unit or the endcapping groups and developed M50 or M51 (Fig. 18f).211 Compared to M49, M50 with fluorine substituents at the organoboron core unit exhibited a downshifted HOMO energy level and a blueshifted absorption spectrum. In comparison, M51 with fluorine substituents at the endcapping groups showed a downshifted LUMO energy level and a redshifted absorption spectrum. The difference came from the unique electronic structures of BNBP-based small molecular electron acceptors, i.e. delocalized LUMO over the whole π-conjugated backbone and localized HOMO on the core units. The OSC devices based on M50 or M51 as electron acceptors exhibited a moderate PCE of 2% to 5% probably due to the non-optimal phase separation morphology of the active layers. The two absorption bands of M49 can be tuned by rational molecular design towards panchromatic property and superior sunlight harvesting capability. Liu et al. further developed a series of BNBP derivatives by changing the chemical structures of the core unit, the π-bridging units or the end-capping groups of M49.210 The modification of their chemical structures led to finely tuned energy levels and absorption spectra. A panchromatic absorption spectrum with a FWHM of 280 nm was realized.
A bottleneck limiting the OSC device performance of BNBP-based small molecular electron acceptors is their localized HOMO on the core unit and the large steric hindrance on the core unit. This is detrimental for the electronic interactions between the electron donor and the electron acceptor. Consequently, this would inhibit the photoinduced hole transfer from the BNBP-based small molecular electron acceptors to electron donors. Although the localized HOMO of BNBP-based small molecular electron acceptors leads to wide absorption spectra and excellent sunlight harvesting capability, it may also limit OSC device efficiency.
3.6 Triarylborane-based small molecular electron acceptors
As tri-coordinated boron compounds, triarylboranes possess moderate electron-deficient property because of the p–π* conjugation between the vacant 2p orbital of the boron atom and the π* orbital of the aryl units.67,212–214 Many triarylborane compounds are not adequately stable under ambient conditions because the vacant 2p orbital of the boron atom may be attacked by moisture. The stability issue prevents triarylborane compounds from being used in OSC applications. In 2014, Marder and Jäkle et al. reported two air-stable triarylborane compounds,215 di(thiophen-2-yl)(2,4,6-tri-tert-butylphenyl)borane (BDT) and di(thiophen-2-yl)(2,4,6-tris(trifluoromethyl)phenyl)borane (FBDT), in which the vacant 2p orbital of the boron atom was protected by bulky 2,4,6-tri-tert-butylphenyl (Mes*) and 2,4,6-tris(trifluoromethyl)-phenyl (FMes) groups for excellent air-stability. Moreover, the bulky Mes* or FMes group leads to non-planar configurations of the molecules and inhibits over-crystallization of the molecules in blend films. Therefore, triarylboranes can be used as the core units to design small molecular electron acceptors. Their OSC devices exhibit moderate PCEs of 3–4% with relatively low FF. This may be due to the low electron mobilities and non-optimized phase separation morphology of the triarylborane-based small molecular electron acceptors. However, a great advantage of triarylborane-based small molecular electron acceptors is their excellent solubility, which enables eco-friendly processing of OSCs.The active layers of OSCs are always processed with halogenated or aromatic solvents, which are harmful for the environment and human health. In the mass production of OSCs, the active layers must be processed with eco-friendly solvents, such as alcohol. However, typical electron donor and electron acceptor materials are insoluble in alcohol solvents. Jäkle and Liu et al.216 reported an alcohol-soluble small molecular electron acceptor based on triarylborane in the absence of polar side chains. Fig. 19a shows the chemical structure of the compound, M52. Mes*-substituted dithienylborane was used as the core unit, thiophene was used as the bridging unit and 2-(1,1-dicyanomethylene)rhodanine was used as the endcapping group. M52 exhibited low-lying HOMO/LUMO energy levels of −3.61 eV/−5.73 eV and a good electron mobility of 1.37 × 10−5 cm2 V−1 s−1. The backbone of M52 possessed a large dipole moment (Fig. 19a) and enhanced flexibility (Fig. 19b) because of the embedding of the boron atom, leading to the solubility of M52 in alcohol. OSC devices with M52 as the electron acceptor were fabricated using 1-hexanol as the processing solvent and a PCE of 1.03% was demonstrated. Although this PCE was moderate, this work provided the first alcohol-processed non-fullerene electron acceptor, which was in strong demand for environment-friendly processing of OSCs.
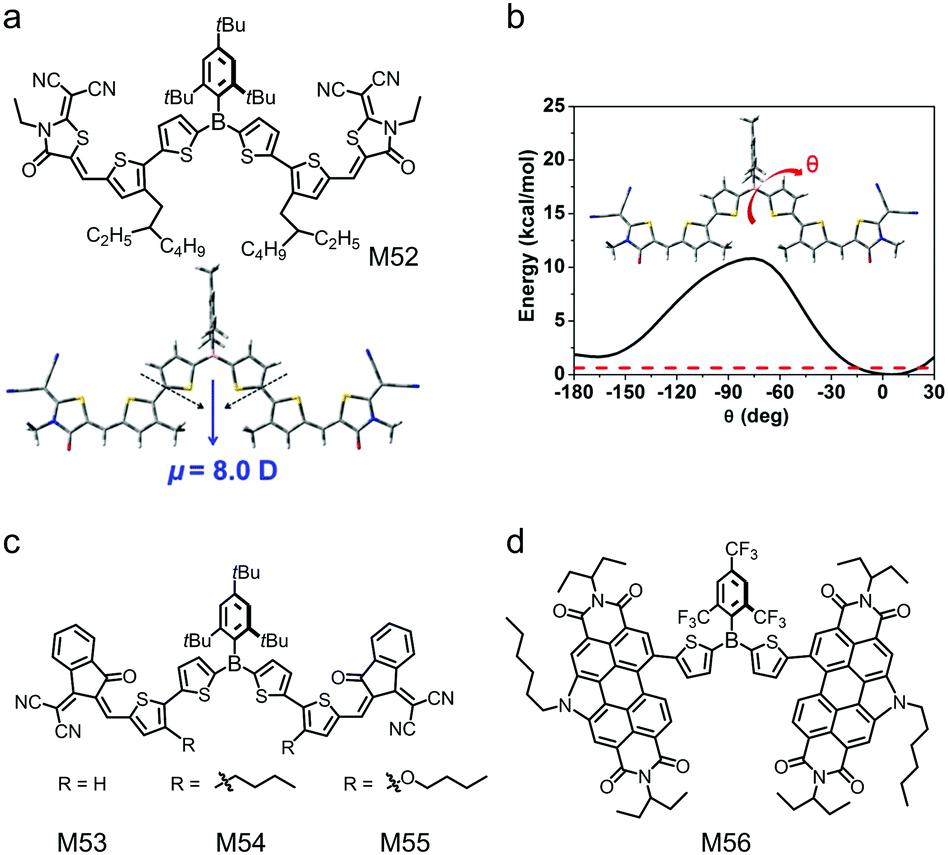 | ||
| Fig. 19 (a) Chemical structure of small molecular acceptor M52, and the optimized configuration and natural dipole moment based on DFT calculations for the model molecule of M52. (b) Potential energy surface scan of the model molecule M52 for dihedral angles (θ) between the two thienyls of the triarylborane unit ranging from −180° to 30°; the red dashed line denotes kBT ≈ 0.6 kcal mol−1 at 298 K. Chemical structures of small molecular acceptors (c) M53, M54, and M55 and (d) M56. Panels (a) and (b) were reproduced from ref. 216 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Liu's group also developed a series of small molecules with triaryborane as the core unit, thiophene as the bridging unit and IC as the endcapping groups (M53, M54 and M55, see Fig. 19c).217 Butyl or butoxy groups were introduced to the thiophene bridging units to tune the optoelectronic properties of the compounds. Compared with M53 and M54, M55 containing butoxy groups showed almost an identical LUMO energy level but an obviously upshifted HOMO energy level, which was ascribed to the electron-donating effect of the alkoxy lateral chains in thiophene units. Consequently, the bandgap of M55 was greatly reduced and the absorption spectrum was redshifted. The introduction of butoxy groups in M55 could form intramolecular S⋯O interactions and improve backbone planarity, which favored molecular stacking in the film state. The ordered stacking in thin film was beneficial for charge transport in OSCs. OSC devices based on these triaryborane-based small molecular electron acceptors showed encouraging PCEs of 3% to 4%. Welch and Jäkle et al. designed M56 by combining the FMes-substituted dithienylborane core with highly electron-deficient PDI units (Fig. 19d).218 Compared to the control compound with unfunctionalized bithiophene as the core unit, M56 with boron functionalized bithiophene had a more twisted molecular geometry, which was expected to prevent the aggregation of the molecules for small-size phase separation morphology. Owing to the p–π* conjugation, M56 exhibited HOMO/LUMO energy levels of −5.82 eV/−3.62 eV, which were lower by 0.20 eV/0.03 eV than those of the control compound, respectively. The OSC device using M56 as the electron acceptor gave a PCE of 3.64%.
3.7 Other organoboron compounds as electron donors or electron acceptors
Besides organoboron small molecules based on BODIPY, SubPc, BNBP and triarylborane, there are some other organoboron small molecules to be used as electron donors or electron acceptors in OSCs. Shimizu et al. synthesized a series of pyrrolopyrrole aza-BODIPY (PPAB) derivatives via titanium tetrachloride-mediated Schiff-base formation reaction of DPP.219 These compounds had planar molecular frameworks and exhibited strong absorption in the Vis/NIR region. Some of them had been tested as electron donors in OSC devices. Fig. 20 shows the chemical structures of two examples of monomeric PPAB (M57) and dimeric PPAB (M58). M58 was an A–D–A type small molecular electron donor with two electron-accepting PPAB terminals connected by an electron-donating cyclopentadithiophene (CPDT) unit. The CPDT unit bearing alkyl side chains not only enhanced the solubility but also improved the interchromophore interactions. Compared to the control molecule with DPP as the terminals, M58 exhibited greatly downshifted LUMO energy levels and much smaller bandgap. As a result, M58 exhibited a maximum absorption wavelength of 845 nm in thin film and an optical bandgap of only 1.21 eV.220 The OSC device with M58 as the electron donor and PC71BM as the electron acceptor showed a PCE of 3.88% with a JSC of 11.7 mA cm−2, a VOC of 0.67 V, and an FF of 0.50.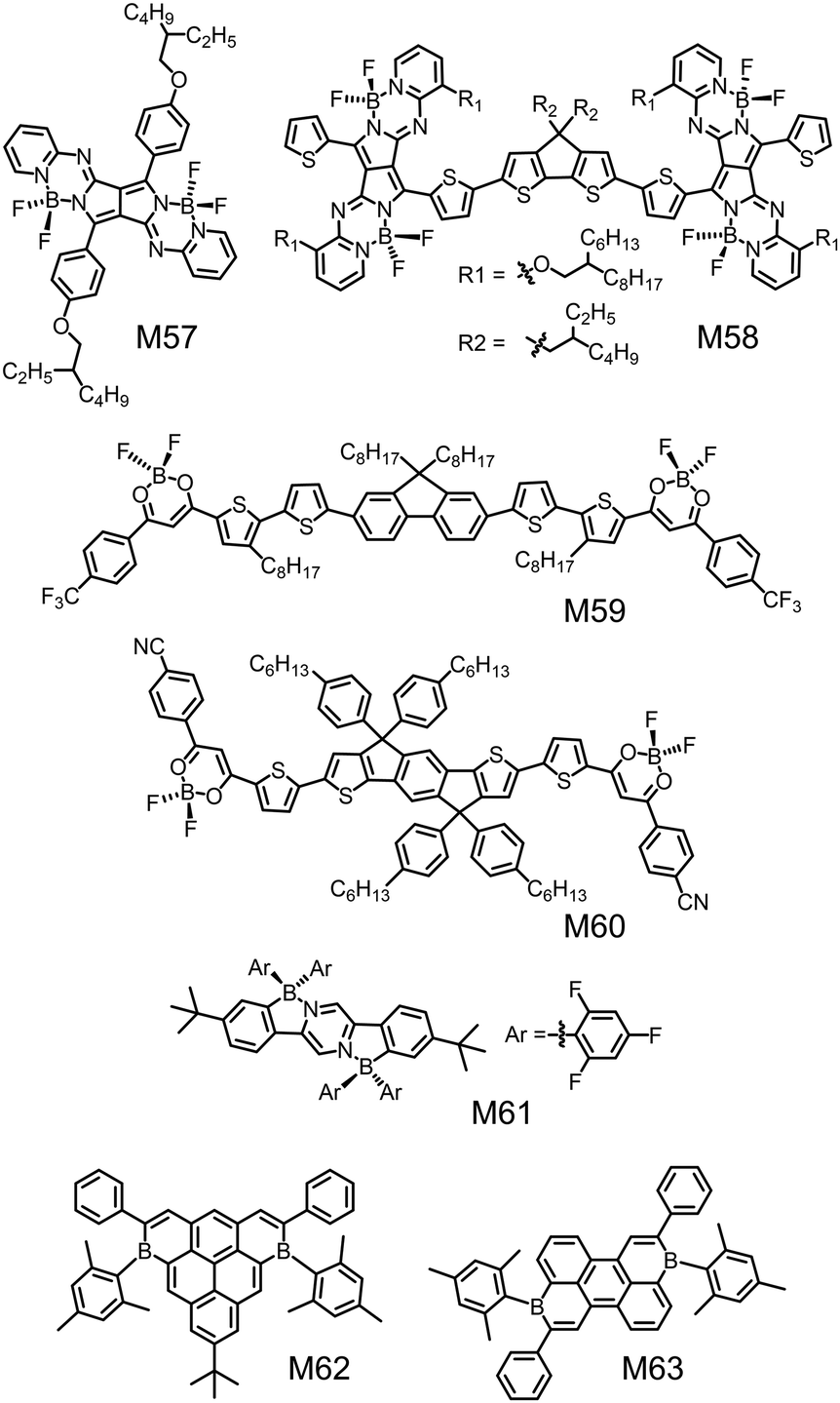 | ||
| Fig. 20 Chemical structures of small molecular donors (M57, M58) and small molecular acceptors (M59–M63). | ||
Yam et al. reported a series of A–D–A type small molecular acceptors, M59 and M60, using the strong electron-deficient difluoroboron-substituted β-diketonate (BF2bdk) moieties as the A units (Fig. 20).221 The single crystal structure of M59 suggested a highly coplanar backbone with small torsion angles (<6°) between the dioxaborine ring and the adjacent thiophene rings. This planar configuration was favorable for molecular packing and charge transport. M59 and M60 exhibited low-lying LUMO energy levels of −3.65 eV and −3.72 eV, respectively, and absorption peak wavelengths of 553 nm and 695 nm, respectively. OSC devices with these BF2bdk-based electron acceptors exhibited a PCE of 4.31%. These results indicate that BF2bdk is an alternative electron-deficient unit to design small molecular electron acceptors.
Piers and Welch et al. reported a family of air and water stable B ← N-doped dihydroindeno[1,2-b]fluorenes through transmetallation via diarylzinc reagents.222 By modifying the substituents on either the boron atom or the flanking aryl rings, the absorption spectra of these molecules could be tuned from the visible region to NIR region. Among these molecules, M61 with 2,4,6-trifluorophenyl substituents on the boron atoms and tert-butyl groups on the flanking aryl rings exhibited low-lying HOMO/LUMO energy levels of −6.24 eV/−3.88 eV. The OSC device with M61 as the electron acceptor and PTB7-Th as the electron donor exhibited a PCE of 1.9%.
Würthner et al. synthesized ambient-stable core- and periphery-tuned boron-doped polycyclic aromatic hydrocarbons (PAHs) by a facile one-pot hydroboration electrophilic borylation cascade/dehygrogenation approach.223Fig. 20 shows the chemical structures of two examples (M62 and M63). The mesityl substitution at the boron center ensured air-stability of the compounds. By modifying the chemical structures of the boron-doped PAHs, their LUMO energy levels could be finely tuned in a wide range from −3.17 eV to −4.08 eV and their absorption spectra could be tuned within the whole visible region. Among them, M63 exhibited a low LUMO energy level of −4.08 eV and a high electron mobility of 3 × 10−3 cm2 V−1 s−1 in OFETs. The OSC device with M63 as the electron acceptor exhibited a PCE of 2.9%.
4. Organoboron polymers for OSCs
Efficient polymer electron donors and polymer electron acceptors are always D–A type conjugated polymers, in which electron-donating units (D) and electron-accepting units (A) are alternatively copolymerized. LUMO and HOMO energy levels of D–A type conjugated polymers can be rationally tuned by modifying the chemical structures of A units and D units, respectively. Moreover, carefully designed D–A type polymers can also have narrow bandgaps, wide absorption spectra, and high hole/electron mobilities. Thereby, D–A type conjugated polymers can fulfill the multiple requirements of polymer electron donors or polymer electron acceptors, and consequently can be used to obtain high-efficiency OSCs. Most of tri-coordinated and tetra-coordinated organoboron building blocks are electron-accepting. Therefore, organoboron building blocks are always used as A building blocks to design D–A type conjugated polymers for OSC applications. The widely used organoboron building blocks include BODIPY derivatives, B ← N bridged thienylthiazole (BNTT), BNBP, BNIDT, BNT, FBDT (triarylborane) (Fig. 21), etc. Noteworthily, excellent OSC device performance has been demonstrated with organoboron polymers. Organoboron polymer electron donors with a PCE of 16% and organoboron polymer electron acceptors with a PCE of 14% have been reported by Duan et al. and Liu et al., respectively.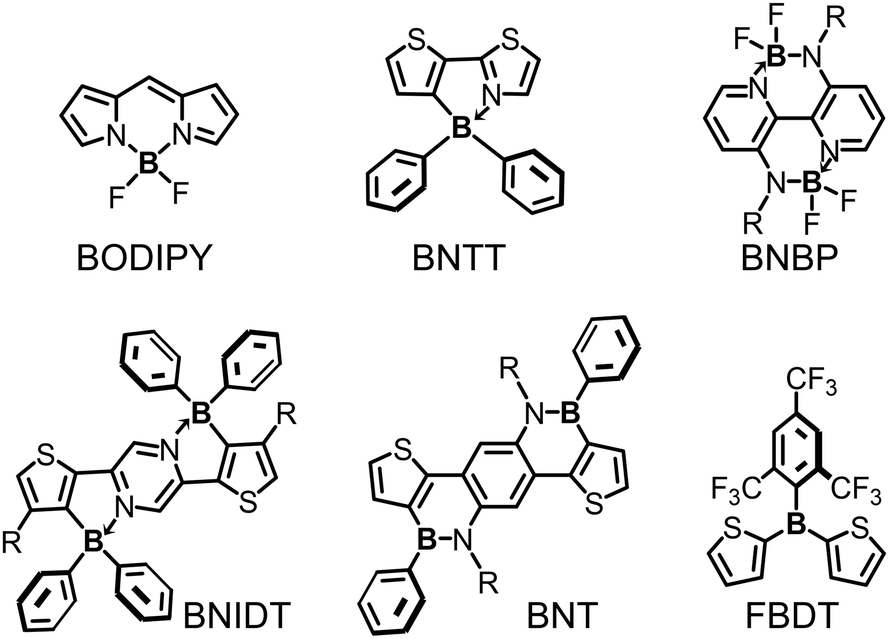 | ||
| Fig. 21 Chemical structures of the building blocks for organoboron polymers in OSCs. | ||
4.1 BODIPY-based polymer donors
With electron-accepting property and rigid planar configuration, BODIPY derivatives have been used as A building blocks to design D–A type polymer electron donors. Most of high-efficiency polymer electron donors show medium bandgaps of 1.7–1.9 eV with absorption spectra in the visible range. In comparison, BODIPY-based polymer donors always possess small bandgaps of around 1.5 eV and show strong light absorption in the NIR region.224–229 The strong sunlight absorbing capability suggests that BODIPY-based polymer donors are an interesting and important class of organic photovoltaic materials.The first BODIPY-based polymer donor was reported by Fréchet et al. in 2010.230 The chemical structures of the two polymer donors, P1 and P2, are shown in Fig. 22. Both polymers exhibited low bandgaps of ca. 1.6 eV and optimal LUMO/HOMO energy level alignment for charge separation when paired with PC61BM. Using PC61BM as the electron acceptor, OSC devices based on P1 and P2 exhibited PCEs of 1.3% and 2.0%, respectively Table 4. Although the OSC device efficiencies were moderate, this work triggered a lot of research on BODIPY-based polymer donors. For example, Qin et al. developed a polymer donor by copolymerizing BODIPY with an organometallic Pt-based building block.231 Skabara et al. reported a polymer donor based on alternating BODIPY unit and bis(3,4-ethylenedioxythiophene) unit.232 Employing α,β-unsubstituted BODIPY as a building block, Chochos et al. designed and synthesized a polymer donor with strong NIR absorption and an onset absorption wavelength of ca. 1100 nm.233 Ramamurthy et al. studied the effect of linking positions of BODIPY building blocks (via α-position or β-position) on the opto-electronic properties of BODIPY-based polymer donors.234 Gros et al. prepared a polymer donor consisting of alternating BODIPY and diethynylthiophene as the backbone and Zn(II)-porphyrin as the pendent group (P3, see Fig. 23a).235 Its absorption spectrum was the sum of absorption spectrum of pendant porphyrin groups and the polymer backbone (Fig. 23b). P3 exhibited an optical bandgap of 1.59 eV, a HOMO energy level of −5.32 eV and a LUMO energy level of −3.73 eV. The optimized OSC device based on the P3:PC71BM active layer showed a PCE of 8.79% with a JSC of 14.48 mA cm−2, a VOC of 0.92 V and an FF of 0.66. The photovoltaic performance of P3 was better than that of the control polymer donor (P(BdP-DEHT)) without pendant porphyrin groups (Fig. 23c). This was related to the broader absorption spectrum and better charge transport of P3 compared to the control polymer donor. The excellent photovoltaic performance of P3 in fullerene-based OSC devices demonstrated the great potential of the BODIPY unit in the molecular design of polymer donors.
 | ||
| Fig. 22 Chemical structures of BODIPY-based polymers P1 and P2. | ||
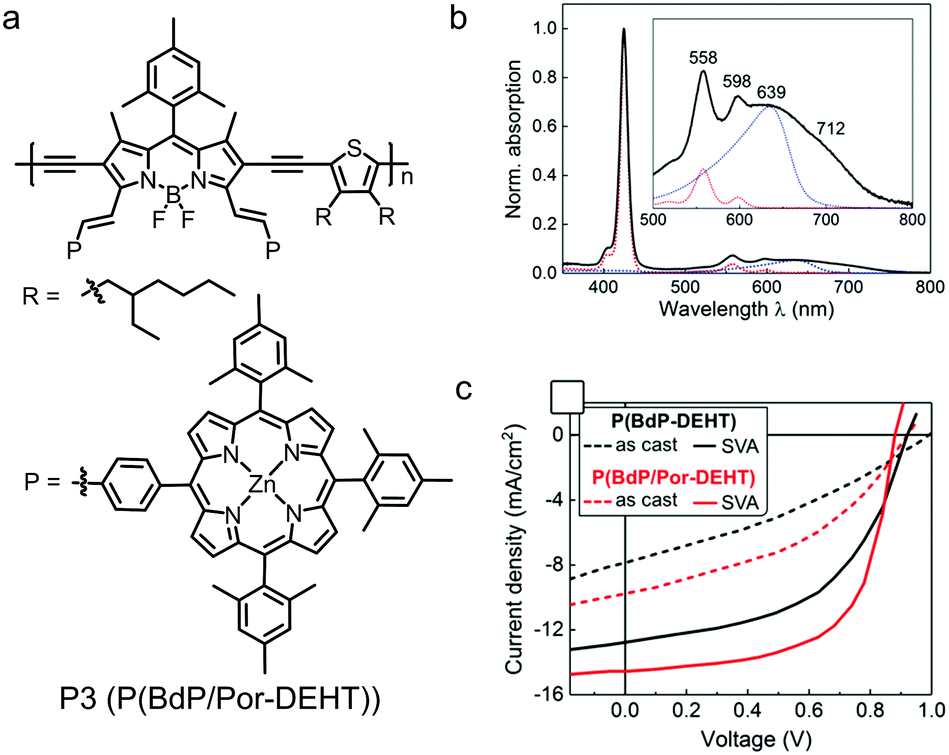 | ||
| Fig. 23 (a) Chemical structure of BODIPY dye P3 (P(BdP/Por-DEHT)). (b) Absorption spectra of P3 (black trace) in solution. (c) J–V curves of the as-cast and solvent vapor annealing (SVA)-treated devices based on the active layers of P3:PC71BM and P(BdP-DEHT):PC71BM. Panels (b) and (c) were reproduced from ref. 235 with permission from the American Chemical Society, copyright 2017. | ||
| Polymers | HOMO/LUMO (eV/eV) | E optg (eV) | Device structure | V OC (V) | J SC (mA cm−2) | FF | PCEa (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The maximum PCE value of the corresponding device. | ||||||||
| P1 | −5.58/−3.73 | 1.61 | ITO/PEDOT:PSS/P1:PC61BM/Al | 0.76 | 4.00 | 0.43 | 1.3 | 230 |
| P2 | −5.45/−3.71 | 1.65 | ITO/PEDOT:PSS/P2:PC61BM/Al | 0.80 | 4.82 | 0.51 | 2.0 | 230 |
| P3 | −5.32/−3.73 | 1.59 | ITO/PEDOT:PSS/P3:PC71BM/PFN/Al | 0.92 | 14.48 | 0.66 | 8.79 | 235 |
| P4 | −5.45/−3.69 | 1.73 | ITO/PEDOT:PSS/P4:PC71BM/PFN/Al | 0.91 | 12.16 | 0.67 | 7.41 | 236 |
| ITO/PEDOT:PSS/P4:SMDPP/PFN/Al | 1.06 | 13.48 | 0.65 | 9.29 | 236 | |||
| P5 | −5.36/−4.05 | 1.31 | ITO/PEDOT:PSS/P5:N2200/PDINO/Al | 0.69 | 1.52 | 0.31 | 0.32 | 237 |
| P5-M | −5.31/−3.90 | 1.41 | ITO/PEDOT:PSS/P5-M:N2200/PDINO/Al | 0.83 | 13.90 | 0.50 | 5.80 | 237 |
| ITO/ZnO/P5-M:BTP-2Cl/MoO3/Ag | 0.77 | 21.44 | 0.60 | 9.86 | 229 | |||
| P6-CC | −5.37/−3.11 | 1.69 | ITO/PEDOT:PSS/P6-CC:P6/LiF/Al | 1.08 | 0.35 | 0.23 | 0.085 | 240 |
| P6 | −5.90/−3.76 | 1.88 | ITO/PEDOT:PSS/P3HT:P6/LiF/Al | 0.78 | 0.47 | 0.38 | 0.14 | 240 |
| P7 | −5.84/−3.80 | 1.63 | ITO/PEDOT:PSS/PTB7-Th:P7/Ca/Al | 0.92 | 11.37 | 0.48 | 5.04 | 243 |
| P8 | −5.81/−3.71 | 1.72 | ITO/PEDOT:PSS/PTB7-Th:P8/Ca/Al | 0.89 | 14.24 | 0.52 | 6.60 | 244 |
| P9 | −5.77/−3.50 | 1.92 | ITO/PEDOT:PSS/PTB7:P9/LiF/Al | 1.09 | 7.09 | 0.44 | 3.38 | 206 |
| ITO/PEDOT:PSS/PCDTBT:P9/Ca/Al | 1.30 | 5.41 | 0.46 | 3.20 | 258 | |||
| P10 | −5.74/−3.59 | 1.93 | ITO/PEDOT:PSS/J61:P10/Ca/Al | 1.01 | 6.42 | 0.47 | 3.04 | 253 |
| P11 | −5.45/−3.87 | 1.56 | ITO/PEDOT:PSS/PTB7:P11/LiF/Al | 0.88 | 7.54 | 0.41 | 2.69 | 254 |
| P12 | −5.84/−3.66 | 1.87 | ITO/PEDOT:PSS/PTB7-Th:P12/Ca/Al | 1.03 | 10.02 | 0.42 | 4.26 | 255 |
| P13-OMe | −5.37/−3.39 | 1.72 | ITO/PEDOT:PSS/P13-OMe:PC71BM/Ca/Al | 0.70 | 2.02 | 0.42 | 0.59 | 256 |
| P13-Me | −5.81/−3.32 | 1.83 | — | — | — | — | — | 256 |
| P13-H | −5.92/−3.43 | 1.91 | — | — | — | — | — | 256 |
| P13-F | −6.04/−3.62 | 1.90 | ITO/PEDOT:PSS/PTB7-Th:P13-F/Ca/Al | 1.09 | 10.13 | 0.47 | 5.16 | 256 |
| P14-OMe | −5.46/−3.48 | 1.66 | ITO/PEDOT:PSS/P14-OMe:PC71BM/Ca/Al | 0.76 | 7.59 | 0.51 | 2.92 | 256 |
| P14-Me | −5.84/−3.42 | 1.81 | — | — | — | — | — | 256 |
| P14-H | −5.77/−3.51 | 1.85 | — | — | — | — | — | 256 |
| P14-F | −5.85/−3.71 | 1.84 | ITO/PEDOT:PSS/PTB7-Th:P14-F/Ca/Al | 1.12 | 7.33 | 0.45 | 3.70 | 256 |
| P15-1T | −5.86/−3.75 | 1.94 | ITO/PEDOT:PSS/J61:P15-1T/Ca/Al | 0.89 | 4.57 | 0.30 | 1.23 | 257 |
| P15-2T | −5.93/−3.54 | 1.90 | ITO/PEDOT:PSS/J61:P15-2T/Ca/Al | 1.06 | 7.26 | 0.46 | 3.57 | 257 |
| P15-3T | −5.75/−3.48 | 1.89 | ITO/PEDOT:PSS/J61:P15-3T/Ca/Al | 1.22 | 11.15 | 0.48 | 6.52 | 257 |
| P15-4T | −5.55/−3.33 | 1.83 | ITO/PEDOT:PSS/P15-4T:PC71BM/Ca/Al | 0.77 | 2.07 | 0.48 | 0.77 | 257 |
| P15-5T | −5.45/−3.11 | 1.74 | ITO/PEDOT:PSS/P15-5T:PC71BM/Ca/Al | 0.81 | 11.62 | 0.62 | 5.79 | 257 |
| P16 | −5.87/−3.62 | 1.86 | ITO/PEDOT:PSS/PTB7-Th:P16/LiF/Al | 1.07 | 12.69 | 0.47 | 6.26 | 259 |
| P17 | −5.74/−3.72 | 1.97 | ITO/PEDOT:PSS/PTB7-Th:P17/Ca/Al | 1.11 | 7.58 | 0.45 | 3.77 | 260 |
| P18 | −5.52/−3.45 | 1.78 | ITO/PEDOT:PSS/CD1:P18/LiF/Al | 1.17 | 13.39 | 0.64 | 10.07 | 261 |
| P18-C28 | −5.58/−3.53 | 1.77 | ITO/ZnO/BD3T:P18-C28/MoO3/Al | 1.12 | 7.16 | 0.63 | 5.06 | 270 |
| P19 | −5.81/−3.42 | 1.95 | ITO/PEDOT:PSS/J61:P19/Ca/Al | 1.24 | 10.12 | 0.57 | 7.09 | 262 |
| ITO/PEDOT:PSS/CD1:P19/Ca/Al | 1.29 | 10.10 | 0.61 | 7.93 | 264 | |||
| P20 | −5.96/−3.46 | 1.93 | ITO/PEDOT:PSS/DR3TBDTC:P20/Ca/Al | 1.11 | 11.18 | 0.65 | 8.01 | 268 |
| P21 | −5.78/−3.65 | 1.82 | ITO/ZnO/BD3T:P21/MoO3/Al | 1.04 | 13.82 | 0.66 | 9.51 | 270 |
| P22 | −5.43/−3.85 | 1.51 | ITO/PEDOT:PSS/PBDB-T:P22/PNDIT-F3N/Al | 0.93 | 8.02 | 0.49 | 4.03 | 273 |
| P23 | −5.71/−3.77 | 1.61 | ITO/PEDOT:PSS/PBDB-T:P23/PNDIT-F3N/Al | 0.93 | 13.01 | 0.70 | 8.78 | 273 |
| P24 | −5.46/−3.79 | 1.57 | ITO/PEDOT:PSS/PBDB-T:P24/PDINO/Al | 0.98 | 4.15 | 0.38 | 1.60 | 275 |
| P25 | −5.71/−3.77 | 1.60 | ITO/PEDOT:PSS/PBDB-T:P25/PDINO/Al | 0.96 | 8.75 | 0.44 | 3.71 | 275 |
| P26 | −5.74/−3.76 | 1.62 | ITO/PEDOT:PSS/PBDB-T:P26/PDINO/Al | 0.96 | 9.20 | 0.47 | 4.23 | 275 |
| P27 | −5.46/−3.63 | 1.38 | ITO/PEDOT:PSS/PTB1:P27/PDINO/Al | 0.80 | 16.90 | 0.54 | 7.25 | 276 |
| P28 | −5.52/−3.45 | 2.07 | ITO/PEDOT:PSS/P28:PC71BM/Ca/Al | 1.00 | 8.31 | 0.45 | 3.82 | 278 |
| P29 | −5.52/−3.46 | 1.86 | ITO/PEDOT:PSS/P29:Y6-BO/PFNBr/Ag | 0.88 | 25.4 | 0.72 | 16.1 | 279 |
| P30 | −5.88/−3.72 | 1.80 | ITO/PEDOT:PSS/PTB7-Th:P30/Ca/Al | 1.00 | 6.51 | 0.43 | 2.83 | 280 |
| P31 | −5.76/−3.58 | 1.83 | ITO/PEDOT:PSS/PTB7-Th:P31/Ca/Al | 1.08 | 4.47 | 0.32 | 1.56 | 280 |
| P32 | —/−3.75 | 2.16 | ITO/PFN-P1/PTB7:P32/MoO3/Ag | 0.40 | 0.94 | 0.31 | 0.12 | 281 |
| P33 | —/−4.35 | 2.07 | ITO/PFN-P1/PTB7:P33/MoO3/Ag | 0.40 | 1.50 | 0.36 | 0.23 | 281 |
BODIPY-based polymer donors also match well with non-fullerene small molecular acceptors to give excellent OSC device performance. Gros et al. reported a BODIPY-based polymer donor P4 (Fig. 24a).236 While the device based on P4:PC71BM showed a PCE of 7.41%, the OSC device with P4 and the small molecular acceptor SMDPP exhibited a PCE of 9.29% together with the photon energy loss (Eloss) of only 0.48 eV. This Eloss value was among the lowest values for OSCs. The improved PCE of the P4:SMDPP device was attributed to enhanced JSC and VOC, which was caused by improved light absorption efficiency of the P4:SMDPP active layer and the higher LUMO energy level of SMDPP, respectively. Alkylthienyl-benzodithiophene (BDT) is a well-known electron-donating building block to design polymer donors. Li et al. copolymerized this BDT unit and BODIPY units to get two D–A type polymer donors, P5 and P5-M (Fig. 24a).237 The methyl substituents on the α-positions of BODIPY in P5-M significantly affected the optoelectronic properties of the polymer donors. Compared with the control polymer P5 without methyl substituents, P5-M with methyl substituents showed higher LUMO/HOMO energy levels, an increased absorption coefficient, higher crystallinity, and dramatically enhanced hole mobility (Fig. 24b). The authors selected a widely used polymer acceptor N2200 to blend with P5 or P5-M to construct all-polymer solar cells (all-PSCs). While the all-PSC device of P5 showed a PCE of only 0.32%, the device of P5-M showed a significantly enhanced PCE of 5.8% with a broad photoresponse from 300 to 900 nm (Fig. 24c). All-PSCs based on polymer acceptor N2200 suffered from low photoresponse in the NIR region. The utilization of polymer donor P5-M with strong NIR absorption in all-PSCs was expected to solve this problem and give high PCE. Xu and Li et al. further blended polymer donor P5-M with small molecular acceptor Y6-2Cl to fabricate OSC devices.229 The devices showed a PCE of 9.86%, which was the highest value reported so far for BODIPY-based polymer donors (Fig. 24d).
 | ||
| Fig. 24 (a) Chemical structures of BODIPY-based polymer donors P4, P5 and P5-M. (b) 2D-GIWAXS patterns of the donor polymers P5 (PBBDT) and P5-M (PMBBDT). (c) J–V curves of the devices based on P5:N2200 and P5-M:N2200 in the dark (dashed line) and under AM1.5G illumination (solid line). (d) J–V curves of the devices based on P5-M:ITIC-2Cl and P5-M:Y6-2Cl in the dark (dashed line) and under AM1.5G illumination (solid line). Panels (b) and (c) were reproduced from ref. 237 with permission from the American Chemical Society, copyright 2019. Panel (d) was reproduced from ref. 229 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Liu et al. designed an annulated BODIPY as a new building block to construct polymer donors with NIR absorption.238 In the chemical structure of the building block, a BODIPY core was fused with two benzene rings for a reduced bandgap and was attached with two alkylthienyl groups as conjugated side chains. The resulting polymers exhibited LUMO/HOMO energy levels of ca. −5.0 eV/−3.5 eV, a hole mobility of ca. 3 × 10−3 cm2 V−1 s−1, and an optical bandgap of ca. 1.3 eV with the maximum absorption wavelength in film exceeding 800 nm. OSC devices of these polymer donors showed a PCE of 2.6%. The unsatisfactory OSC device performance was probably due to the large-size phase separation of the active layers.
At present, the highest PCE for BODIPY-based polymer donors is 9.8%, suggesting that BODIPY derivatives are useful building blocks to design polymer donors. Indeed, the opto-electronic properties of the reported BODIPY-based polymer donors, including LUMO/HOMO energy levels, absorption spectra and hole mobilities, can fulfill the multiple requirements of high-efficiency polymer donors. For further PCE enhancement, great attention should be paid to phase separation morphology of BODIPY-based polymer donors in OSCs. Noteworthily, BODIPY-based polymer donors always exhibit absorption mainly in the far-red or near-infrared region. This property is beneficial for semi-transparent OSCs. However, the application of BODIPY-based polymer donors in semi-transparent OSCs has not been reported until now.
4.2 BNTT-based polymer acceptors
Conjugated polymers containing B ← N units are an important class of polymer electron acceptors for all polymer solar cells (all-PSCs). All-PSCs, which use blends of polymer electron donors and polymer electron acceptors as active layers, have the great advantages of excellent morphology stability and superior mechanical properties. In the past decade, all-PSCs have received great attention and their PCEs have been greatly enhanced together with excellent stability. Since there are far less polymer acceptors than polymer donors, the lack of polymer acceptors limits the development of all-PSCs. The key requirement of polymer acceptors is low-lying LUMO and HOMO energy levels. As B ← N is able to greatly downshift LUMO/HOMO energy levels of π-conjugated systems, polymer acceptors can be designed with B ← N.B ← N bridged thienylthiazole (BNTT, containing one B ← N unit) derivatives were initially reported by Yamaguchi et al. in 2006239 and were adopted to design polymer acceptors by Liu et al. in 2015.240,241 As the BNTT unit is asymmetrical, BNTT-based polymer acceptors are always region-random. Moreover, the large phenyl substituents on the BNTT unit prevent close stacking of polymer backbones. Therefore, BNTT-based polymer acceptors are always amorphous or poorly crystalline. This property is different from typical polymer acceptors, which are semi-crystalline. PCEs of 6–7% have been demonstrated with all-PSCs of BNTT-based polymer acceptors.242,243
B ← N is iso-electronic to the carbon–carbon single bond (C–C). As shown in Fig. 25a, P6-CC is a typical polymer donor with high-lying LUMO and HOMO energy levels. After replacing a C–C unit of P6-CC with a B ← N unit in P6, the LUMO and HOMO energy levels of the resulting polymer were lowered by 0.65 eV and 0.53 eV, respectively (Fig. 25b).240 The LUMO and HOMO energy levels of P6 were −3.76 eV and −5.90 eV, respectively, which were close to those of typical electron acceptors, e.g. PC61BM. This indicated that the replacement of the C–C unit with the B ← N unit converted the polymer from electron donor to electron acceptor. An all-PSC device was fabricated using P6 as the polymer acceptor and P3HT as the polymer donor. This device exhibited a PCE of 0.14%. Although the PCE was low, this preliminary all-PSC device performance experimentally proved that the B ← N unit could be used to design polymer acceptors.
 | ||
| Fig. 25 (a) Chemical structures of the polymer P6-CC containing a C–C unit and the polymer P6 containing a B ← N unit. (b) HOMO/LUMO energy levels of P6-CC and P6. | ||
The poor all-PSC device performance of P6 was preliminarily due to its low electron mobility (μe = 3.4 × 10−7 cm2 V−1 s−1). This was attributed to the large steric hindrance of the phenyl groups on the boron atoms, which inhibited the tight packing of polymer backbones in thin film and hindered the transport of charge carriers between the polymer backbones. To improve the electron mobility of BNTT-based polymer acceptors, Liu et al. extended the length of the repeating units to alleviate the steric hindrance effect of bulky pendant substituents.243 As shown in Fig. 26a, the polymer acceptor P7 was developed by replacing the thieno[3,4-c]pyrrole-4,6-dione-1,3-diyl (TPD) unit in P6 with the long planar isoindigo (IID) unit. According to the optimized configuration obtained by DFT calculations, the length of the repeating unit of P7 was 1.95 nm, which was much longer than that (1.24 nm) of P6. As a result, the π–π stacking distance was reduced from 0.46 nm for P6 to 0.38 nm for P7, and the electron mobility was significantly enhanced by nearly two orders of magnitude from 3.4 × 10−7 cm2 V−1 s−1 for P6 to 2.8 × 10−5 cm2 V−1 s−1 for P7. The PCE of the resulting all-PSC device was improved from 0.12% for P6 to 5.04% for P7 (Fig. 26b).
 | ||
| Fig. 26 (a) Chemical structures and optimized molecular configurations of the repeating units of P6 and P7. (b) J–V curves of the devices based on the active layers of PTB7-Th:P6 and PTB7-Th:P7. (c) Chemical structure of P8. (d) 2D-grazing incidence wide-angle X-ray scattering (2D-GIWAXS) pattern of the P8 film. (e) AFM height images of P8-based blend films with different polymer donors (PTB7-Th, J71 and PffBT4T-2OD). Panels (a) and (b) were reproduced from ref. 243 with permission from the American Chemical Society, copyright 2020. Panels (d) and (e) were reproduced from ref. 242 with permission from the American Chemical Society, copyright 2019. | ||
Liu et al. further developed the polymer acceptor, P8, which was an alternating copolymer of BNTT unit and pyridine-flanked diketopyrrolopyrrole (PyDPP) unit (Fig. 26c).242P8 showed low-lying HOMO/LUMO energy levels of −5.81 eV/−3.71 eV and a high electron mobility of 2.2 × 10−4 cm2 V−1 s−1. Due to its regiorandom structure and large steric hindrance, P8 was amorphous in solid state (Fig. 26d). P8 could match with various polymer donors to give all-PSC devices with high PCEs of 5.2–6.6%.244 When the amorphous P8 was blended with these various polymer donors, the crystallization of polymer donors governed the film-forming process and dominated the phase separation morphology (Fig. 26e). All the polymer donor/P8 blends showed good phase separation morphology and consequently exhibited high PCEs. Recently, Liu et al. developed a new polymer acceptor by copolymerizing a Y-series small molecular acceptor with a BNTT unit.245 Compared with the control polymer acceptor without B ← N, the incorporation of B ← N in the polymer downshifted the LUMO/HOMO energy levels of the polymer by ca. 0.1 eV, and thus promoted efficient photo-induced hole transfer from the polymer acceptor to polymer donor. The OSC device based on this polymer containing the BNTT unit exhibited a PCE of 14.36% with a VOC of 0.89 V, a JSC of 22.62 mA cm−2 and an FF of 0.72. The high device performance demonstrated that BNTT as a building block had potential to design high-performance polymer acceptors.
4.3 BNBP-based polymer acceptors
BNBP,206,246–252 an electron-withdrawing building block based on B ← N, has been intensively studied in the molecular design of polymer acceptors for OSC applications because of four reasons.207 Firstly, owing to the two B ← N units, BNBP exhibits low LUMO/HOMO energy levels. BNBP-based conjugated polymers consequently show low LUMO/HOMO energy levels. Secondly, the two B ← N units lead to the redshifted absorption spectrum of BNBP. This improves the sunlight harvesting capability of BNBP-based conjugated polymers. Thirdly, the two B ← N units fix the planar configuration of BNBP. Therefore, BNBP-based conjugated polymers may show close π-stacking in thin film and possess high electron mobility. Finally, LUMO energy levels of BNBP-based conjugated polymers can be readily tuned, leading to high VOC of the resulting OSC devices. BNBP-based polymer acceptors have become an important class of high-efficiency electron acceptors for OSCs. Their all-polymer photovoltaic devices can afford a PCE of 10% under simulated solar light irradiation and a PCE of 27% under indoor artificial light illumination.The first BNBP-based polymer acceptor, P9 (see Fig. 27a), was an alternating copolymer of BNBP and thiophene.206P9 showed the absorption peak at 622 nm in film, low-lying HOMO/LUMO energy levels of −5.77 eV/−3.50 eV and an electron mobility of 6.9 × 10−5 cm2 V−1 s−1. The all-PSC device based on P9 as the polymer acceptor and PTB7 as the polymer donor exhibited a PCE of 3.38% (Fig. 27b). A homopolymer of BNBP, P10, was synthesized by Yamamoto polycondensation of the di-bromo monomer of BNBP (Fig. 27a).253P10 exhibited an ultra-narrow absorption spectrum in thin film with the FWHM of only 13 nm. The HOMO/LUMO energy levels of P10 were −5.74 eV and −3.59 eV, respectively. P10 could match various polymer donors in all-PSC devices and gave PCEs of 2.44–3.04%. These early works demonstrated the feasibility of designing polymer acceptors using BNBP.
 | ||
| Fig. 27 (a) Chemical structures of BNBP-based polymer acceptors, P9 and P10. (b) J–V curve and EQE spectrum of the device based on the PTB7:P9 blend. Panel (b) was reproduced from ref. 206 with permission from John Wiley and Sons, copyright 2016. | ||
To improve the all-PSC device performance of BNBP-based polymer acceptors, Liu et al. modified their chemical structures to systematically tune their absorption spectra, energy levels and electron mobilities. For example, a polymer acceptor with narrow bandgap and wide absorption spectrum (P11, see Fig. 28a) was developed by copolymerizing BNBP with Th-DPP.254 While P9 showed an optical bandgap of 1.92 eV and an onset absorption wavelength of 646 nm, P11 exhibited an optical bandgap of 1.56 eV and an onset absorption wavelength of 796 nm, indicating much improved sunlight harvesting capability (Fig. 28b). As a result, all-PSC devices based on P11 exhibited broad EQE spectra covering the range of 350–800 nm (Fig. 28c).
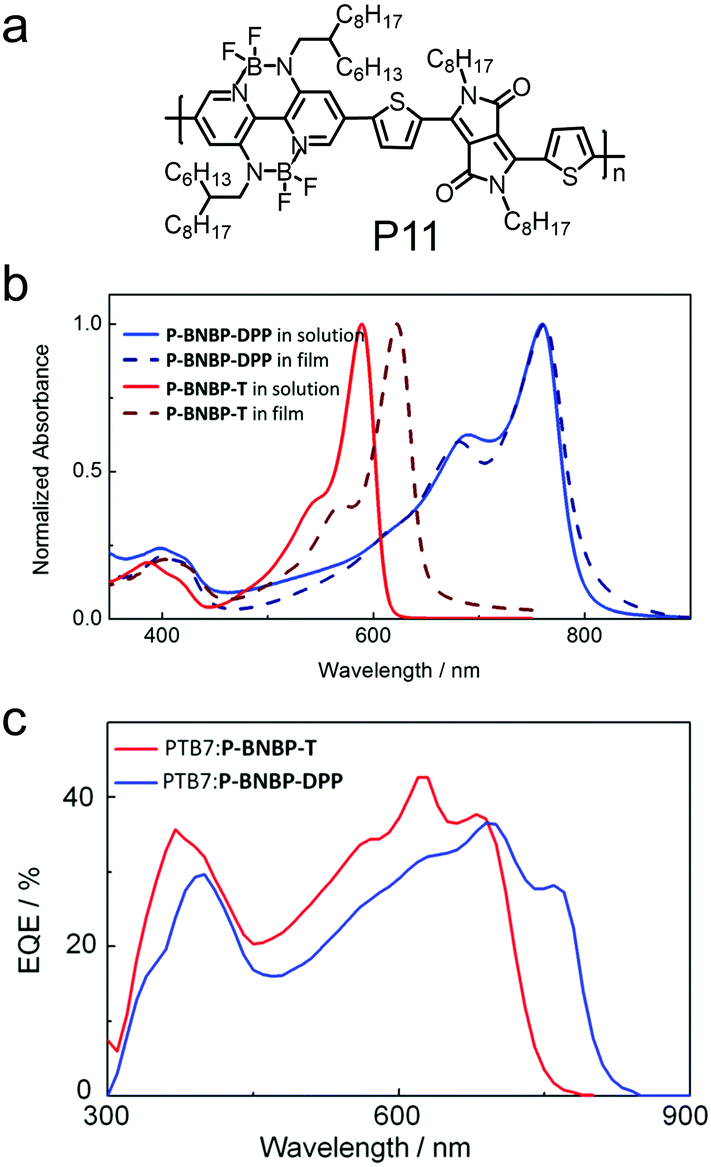 | ||
| Fig. 28 (a) Chemical structure of BNBP-based polymer acceptor P11. (b) The absorption spectra of P9 (P-BNBP-T) and P11 (P-BNBP-DPP) in solution and in film. (c) EQE spectra of the devices based on PTB7:P9 and PTB7:P11 blends. Panels (b) and (c) were reproduced from ref. 254 with permission from the Royal Society of Chemistry, copyright 2016. | ||
LUMO energy levels of BNBP-based based polymer acceptors greatly affect their all-PSC device performance. For example, P12 was designed by replacing the thiophene copolymerizing unit of P9 by selenophene (Fig. 29a).255 This replacement resulted in downshifting of LUMO energy levels from −3.50 eV for P9 to −3.66 eV for P12. Because of the low-lying LUMO energy level, P12 matched well with the benchmark polymer donor, PTB7-Th. The all-PSC device based on PTB7-Th:P12 showed a PCE of 4.26%, which was much higher than that of the PTB7-Th:P9 device (PCE = 2.27%) (Fig. 29b). The downshifted LUMO energy level of P12 also led to slightly decreased VOC of the all-PSC device.
 | ||
| Fig. 29 (a) Chemical structures of P9 and P12, and Kohn–Sham LUMOs of the model compounds of P9 and P12. (b) HOMO/LUMO energy levels of P9 and P12, and J–V curves of the devices based on the PTB7-Th:P9 and PTB7-Th:P12 blends. (c) Chemical structures of the P13 series and P14 series and LUMO energy level alignment for these polymers. (d) Chemical structures of the P15 series and HOMO/LUMO energy level alignment for these polymers. Panel (b) was reproduced from ref. 76 with permission from the American Chemical Society, copyright 2020. | ||
LUMO energy levels of polymer acceptors should be carefully optimized to ensure photo-induced charge separation and to maximize VOC of all-PSC devices. An important feature of BNBP-based conjugated polymers is their tunable LUMO energy levels. Their LUMOs are delocalized over the whole polymer backbones.255 Therefore, their LUMO energy levels can be tuned by modifying the chemical structures of either the BNBP unit or the copolymerizing units. Indeed, LUMO energy levels both as high as −3.2 eV and as low as −4.4 eV have been realized for BNBP-based conjugated polymers. Optimal LUMO energy levels of about −3.6 eV for polymer acceptors can also be readily achieved by molecular design of BNBP-based conjugated polymers.
Liu et al. had finely tuned the LUMO energy levels of BNBP-based polymers. As shown in Fig. 29c, they synthesized a series of polymers with the same backbone of alternating BNBP unit and bithiophene unit.256 By changing the substituents on the BNBP unit or the bithiophene unit, the LUMO energy levels of these polymers could be finely tuned in the range from −3.3 eV to −3.7 eV. Among these polymers, P13-F and P14-F with fluoro substituents on the bithiophene unit showed low LUMO energy levels of below −3.6 eV and could be used as polymer acceptors in all-PSCs. P13-OMe and P14-OMe with methyloxy substituents on the bithiophene unit showed relatively high LUMO/HOMO energy levels and could be used as polymer donors in OSCs. Liu et al. also studied the five polymers (P15 series, see Fig. 29d) consisting of alternating BNBP unit and oligothiophene units of various length.257 With the increasing length of the electron-donating oligothiophene units, the LUMO energy levels of the polymers gradually upshifted from −3.75 eV to −3.11 eV. Among these polymers, P15-3T, which was the alternating polymer of BNBP unit and tri-thiophene unit, showed the most optimal LUMO energy level for polymer acceptor application. The all-PSC device with P15-3T as the electron acceptor exhibited a high PCE of 6.52%. P15-5T, the alternating polymer of BNBP unit and penta-thiophene unit, showed the highest LUMO energy level of −3.11 eV and could work as an electron donor in OSCs. OSC devices with P15-5T as the polymer donor and PC71BM as the electron acceptor showed a PCE of 5.79%.
The VOC of OSCs is strongly related to the LUMO energy levels of electron acceptor materials. The tunable LUMO energy levels of BNBP-based conjugated polymers indicate an opportunity to develop OSCs with remarkably high VOC. For example, an all-PSC device was fabricated by using P9 with a high-lying LUMO energy level as the polymer acceptor and PCDTBT with a low-lying HOMO energy level as the polymer donor.258 An extraordinarily high VOC of 1.3 V was obtained. This VOC was higher by ca. 0.3–0.4 V than the VOC of the corresponding devices based on traditional electron acceptors, such as PC61BM or N2200. The photon energy loss (Eloss) of OSCs, which is the difference between the lowest optical bandgap of donor/acceptor materials and eVOC of the device, is a key factor limiting the PCE of OSCs. OSC devices always have a large Eloss of 0.7–1.0 eV. Due to the tunable LUMO energy levels of the BNBP-based polymer acceptors and the high VOC of the corresponding OSC devices, these devices exhibit a small Eloss of about 0.5–0.6 eV. For example, based on the blend of P16 (Fig. 30a) as the polymer acceptor and PTB7-Th as the polymer donor, an all-PSC device exhibited a small Eloss of 0.51 eV with a PCE of 6.26%.259 The large VOC and the small Eloss of BNBP-based polymer acceptors suggested that there was large room for PCE enhancement.
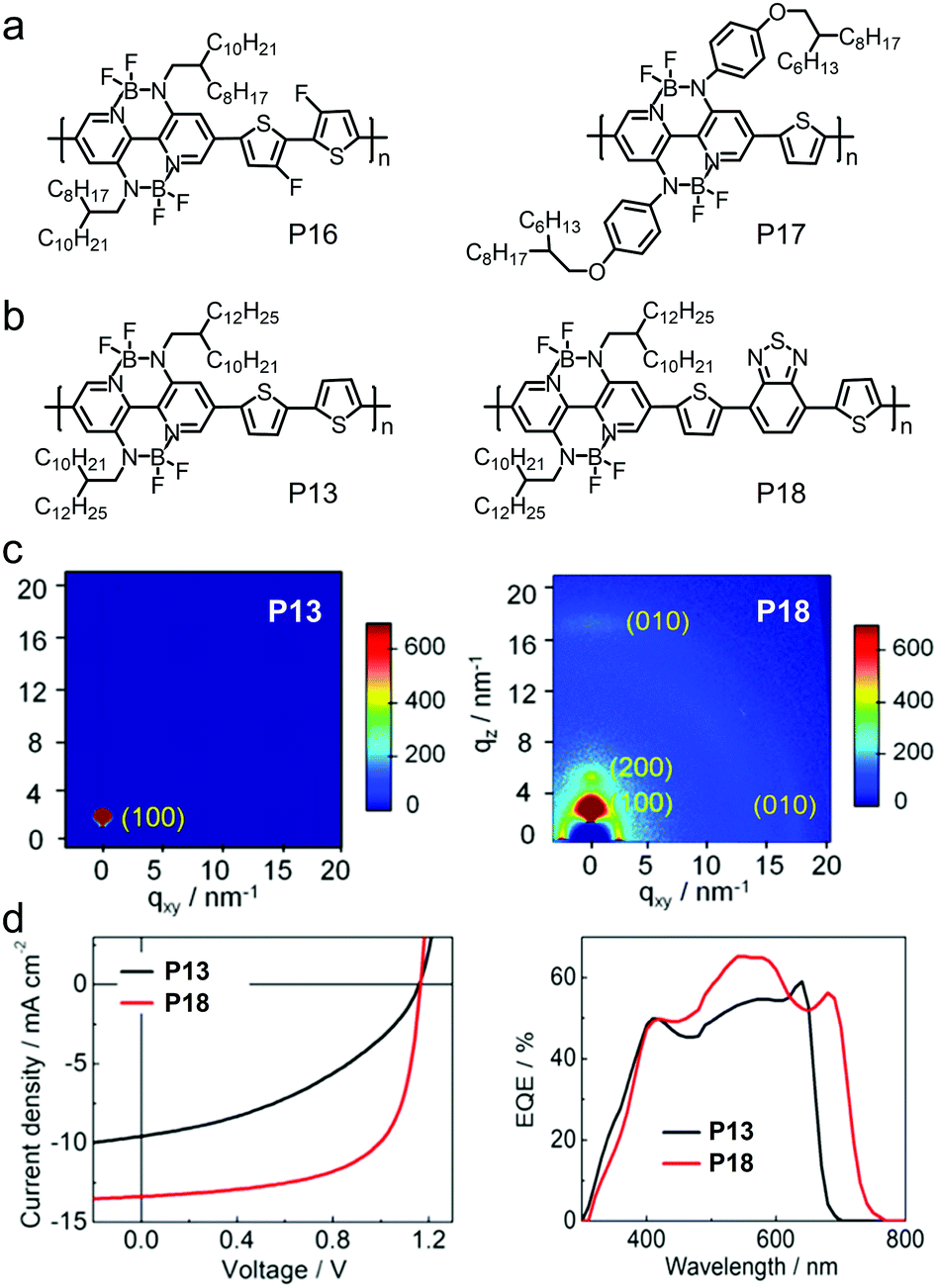 | ||
| Fig. 30 Chemical structures of (a) P16 and P17 and (b) P13 and P18. (c) 2D-GIWAXS patterns of P13 and P18 films. (d) J–V curves and EQE spectra of the devices based on the CD1:P13 and CD1:P18 blends. Panels (c) and (d) were reproduced from ref. 261 with permission from the American Chemical Society, copyright 2020. | ||
BNBP-based conjugated polymers possess excellent electron transporting property, which is proved by their electron mobility of ca. 0.2 cm2 V−1 in OFETs.246 Liu et al. modified the chemical structures of BNBP-based polymer acceptors for enhanced electron mobility towards all-PSC applications. The “conjugated side chains” strategy is widely used to enhance the hole mobility of polymer donors. This strategy was also adopted to improve the electron mobility of BNBP-based polymer acceptors.260 As shown in Fig. 30a, P17 had the same polymer backbone as P9 but with the conjugated alkyloxyphenyl side chains on the BNBP unit. P17 with conjugated alkyloxyphenyl side chains exhibited a smaller π–π stacking distance (dπ–π = 0.36 nm) than that of P9 with alkyl side chains (dπ–π = 0.38 nm). Moreover, the conjugated side chains in P17 could also promote intermolecular π-orbital overlap, which is beneficial for charge transport between polymer chains. As a result, the electron mobility was enhanced from 7.16 × 10−5 cm2 V−1 s−1 for P9 to 3.24 × 10−4 cm2 V−1 s−1 for P17. The PCE of all-PSC devices was also increased from 2.3% for P9 to 3.8% for P17.
A noteworthy BNBP-based polymer acceptor was P18, which was an alternating copolymer of BNBP unit and 4,7-di(thiophen-2-yl)benzo[c][1,2,5]-thiadiazole unit (Fig. 30b).261P18 showed appropriate LUMO/HOMO energy levels of −3.45 eV/−5.52 eV and a high electron mobility of 1.68 × 10−3 cm2 V−1 s−1. In the 2D-GIWAXS pattern of P18 (Fig. 30c), strong (100) and (200) lamellar peaks in the out-of-plane direction and extra (010) π–π stacking peaks in both in-plane and out-of-plane directions could be observed, suggesting ordered packing and mixed face-on/edge-on orientation of the polymer chains. The all-PSC device with P18 as the electron acceptor and CD1 as the electron donor exhibited a PCE of 10.07% with a VOC of 1.17 V, a JSC of 13.39 mA cm−2 and an FF of 0.64 (Fig. 30d). This is among the highest photovoltaic efficiencies reported so far for organoboron polymer acceptors. The incorporation of benzo[c][1,2,5]-thiadiazole into the polymer backbone of P18 greatly affected its opto-electronic properties and thin film morphology, and played an important role in the excellent all-PSC device performance.
Liu et al. have also optimized the phase separation morphology of BNBP-based polymer acceptors in all-PSCs. In the film-forming process of polymer/polymer blends, aggregation of polymer chains in solution can be partially preserved in the resulting film because polymer chains diffuse very slowly in solution. Therefore, the key to optimize the phase separation morphology of polymer/polymer blends in all-PSCs is to control the aggregation of polymer chains in solution. This can be realized by changing the solution preparation method for device fabrication. The traditional method to prepare the solution is to dissolve the polymer donor and polymer acceptor together to get a well-intermixed blend solution (DT method). With this method, the aggregated tendency of polymer chains in solution may be partially disturbed, affecting the polymer stacking in the thin film after spin-coating. To improve polymer ordering and enhance domain purity in thin film, Liu et al. developed a method by dissolving the polymer donor and polymer acceptor individually and then blending them immediately before spin-coating (DI method).262 With J61 as the polymer donor and P19 (Fig. 31a) as the polymer acceptor, while the all-PSC device prepared with the DT method showed a PCE of 5.36%, the device prepared with the DI method exhibited an enhanced PCE of 7.09%. Another approach to control aggregation of polymer chains in solution is to optimize the solution temperature for spin-coating.263 For the all-PSC device based on the CD1:P20 blend, as the solution temperature increased from 30 °C to 90 °C, the polymer chains became more dis-aggregated in solution and were able to diffuse more quickly in the film-forming process. As a result, compared to that with the solution temperature of 30 °C, the all-PSC device prepared with the solution temperature of 90 °C exhibited smaller phase-separation size and a more optimal crystallization degree of the polymers, which led to an improved PCE from 7.7% to 10.0%.
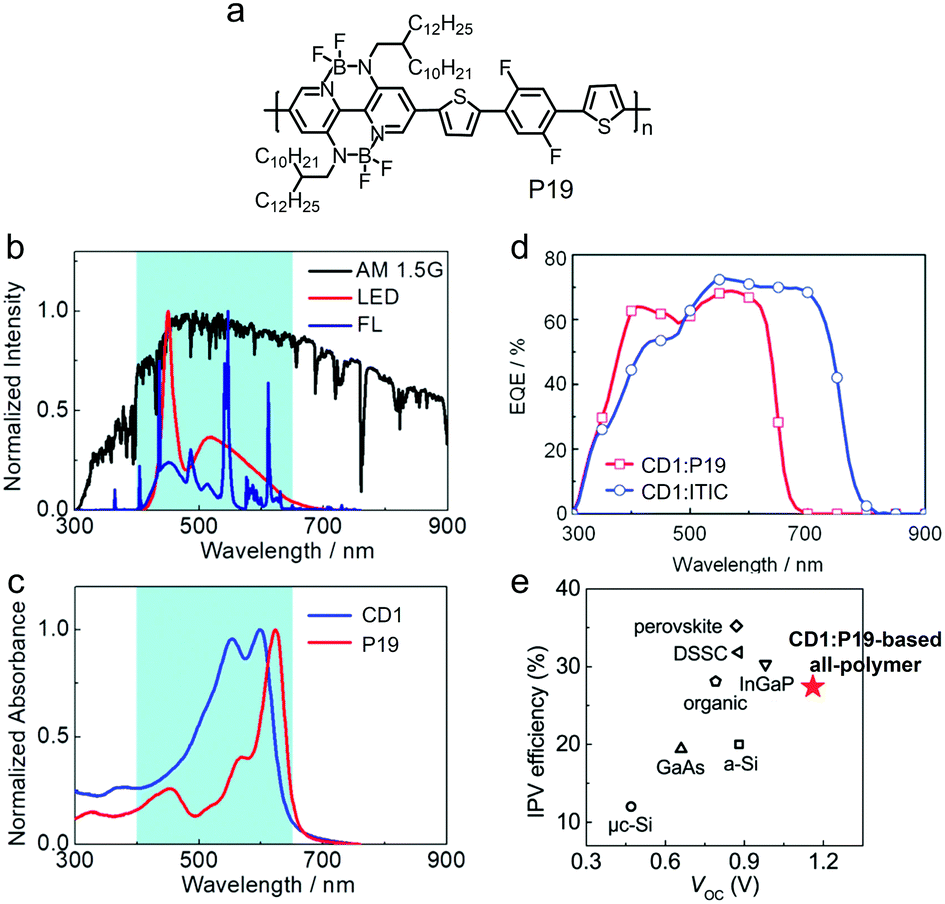 | ||
| Fig. 31 (a) Chemical structure of P19. (b) The AM 1.5G solar spectrum and emission spectra of a white LED and FL. (c) Absorption spectra of CD1 and P19. (d) EQE curves of the photovoltaic cells based on CD1:P19 and CD1:ITIC. (e) The IPV efficiency and VOC distributions of different types of photovoltaic cells. Panels (b–e) were reproduced from ref. 264 with permission from the Royal Society of Chemistry, copyright 2019. | ||
BNBP-based polymer acceptors with bandgaps of ca. 1.9–2.0 eV are suitable for application in indoor photovoltaics (IPVs). The rapid development of Internet of Things (IoT) needs numerous wireless sensors to collect and communicate data. These wireless sensors have power consumption on the microwatt to milliwatt scale. IPVs, which harvests light energy from the indoor environment and generates electricity, is an ideal option to power these sensor nodes for practical applications. BNBP-based polymer acceptors, e.g.P19, have the absorption band mainly in the visible range from 450 nm to 650 nm, which overlaps well with the emission spectra of indoor artificial lighting sources, such as a fluorescent lamp (FL) and light emitting diodes (LEDs). Liu et al. used a blend of CD1 and P19 as the active layer to fabricate an all-polymer photovoltaic device.264 The photo-response of the device was in the wavelength range of 390–630 nm, which lies wholly in the visible region (Fig. 31b–d). Under simulated solar light irradiation, the device showed a PCE of 7.9% with a VOC of 1.29 V and an FF of 0.61. Under fluorescent lamp illumination at the brightness of 2000 lux, the device exhibited a PCE of 27.4% with a VOC of 1.16 V and an FF of 0.67. This was the first report of all-polymer indoor photovoltaics. This PCE was fairly comparable to those of other indoor photovoltaic technologies based on organic materials or inorganic materials (Fig. 31e).
High-efficiency OSCs always use blends of polymer donors and small molecular acceptors as active layers. Small molecular donor/polymer acceptor type (MD/PA-type) OSCs have the merit of excellent thermal stability but have not received widespread attention. This is due to their low PCEs, which come from the poor phase separation morphology of MD/PA blends. BNBP-based polymer acceptors have been used in MD/PA-type OSCs with optimized phase separation morphology and enhanced PCE together with excellent thermal stability.265,266
Liu et al. initially used a commercially available small molecular donor, (p-DTS(FBTTh2)2), to blend with P9 as the polymer acceptor to fabricate MD/PA-type OSC devices.267 The device with the as-cast p-DTS(FBTTh2)2:P9 active layer exhibited a low PCE of 1.00% because of the large-size phase separation morphology. After using a solvent additive and thermal annealing, the PCE of the MD/PA-type OSC device was enhanced to 3.50%. The problem of large size phase separation in MD/PA-type OSC devices can be solved by using small molecular donors with suppressed π-stacking or polymer acceptors with stronger aggregation tendency in solution. As shown in Fig. 32a, DR3TBDTC was a small molecular donor bearing bulky carbazolyl substituents, which acted as steric hindrance groups and suppressed π–π stacking between molecular backbones. A BNBP-based polymer acceptor, P20, was used to pair with DR3TBDTC to fabricate MD/PA-type OSC devices.268 In the as-cast film of DR3TBDTC:P20, P20 formed ordered aggregates and DR3TBDTC was amorphous. No crystallization of DR3TBDTC was observed in the blend film, indicating that the aggregation of P20 governed the film-forming process and dominated the film morphology. After thermal annealing, DR3TBDTC crystallized into nano-sized domains and formed interconnected networks in the active layer (Fig. 32b). As a result, the MD/PA-type OSC device exhibited optimal phase separation morphology and showed a much enhanced PCE of 8.01% (Fig. 32d). The bulky carbazolyl substituents on the small molecular donor played an important role in the excellent active layer morphology of the MD/PA-type OSC devices (Fig. 32c). In addition, the device of DR3TBDTC:P20 showed excellent thermal stability, i.e. maintaining 89% of its initial efficiency after thermal annealing the active layer at 180 °C for 7 days. In contrast, the polymer donor/small molecular acceptor type device of PTB7-Th:EH-IDTBR exhibited a large PCE decrease and retained only 45% of its initial efficiency after 7 days. The high thermal stability of the DR3TBDTC:P20 blend indicated prominent morphological stability, which was mainly attributed to the excellent phase stability of the two materials themselves and the high crystallinity of the blend film. This result further demonstrated the superior intrinsic stability of BNBP-based polymer acceptor materials.
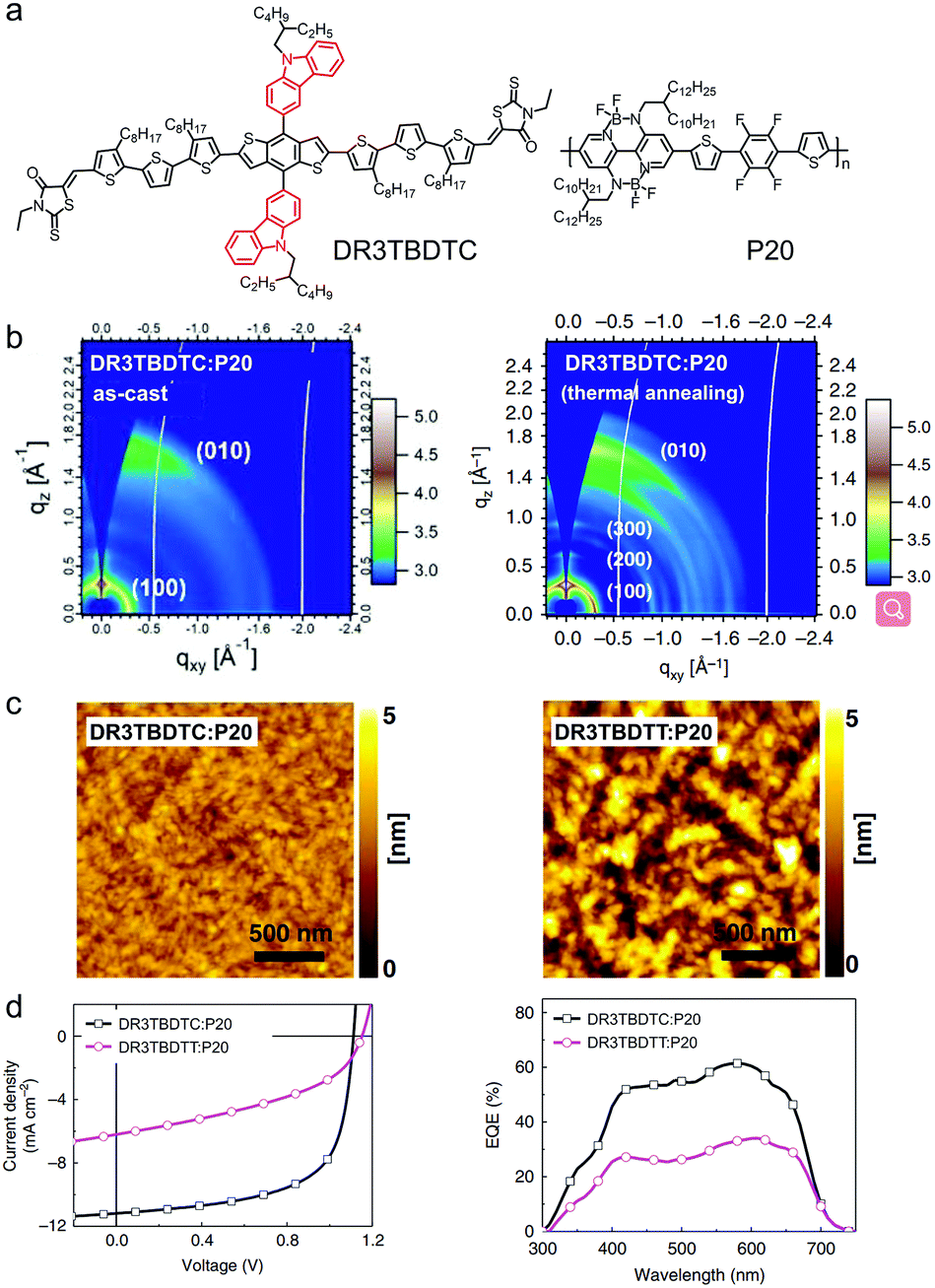 | ||
| Fig. 32 (a) Chemical structures of small molecular donor DR3TBDTC and polymer acceptor P20. (b) 2D-GIWAXS patterns of the as-cast and thermally annealed DR3TBDTC:P20 blend film. (c) AFM height images of DR3TBDTC:P20 and DR3TBDTT:P20 blend films. (d) J–V curves and EQE spectra of the devices based on the DR3TBDTC:P20 and DR3TBDTT:P20 blends. Panels (b–d) were reproduced from ref. 268 with permission from Springer Nature, copyright 2019. | ||
Molecular weights of polymer acceptors affect the aggregation tendency of polymer chains in solution, and thus affect the phase separation morphology of MD/PA blends. For OSC devices based on the DR3TBDTC:P16 blend, by increasing the molecular weight of polymer acceptor P16, the aggregation and crystallization of small molecular donor DR3TBDTC could be suppressed, and small-size phase separated domains of the blends could be obtained.269 The OSC device based on P16 with a number average molecular weight of 117.3 kg mol−1 exhibited an optimized active layer morphology and showed a PCE of 6.4%. The aggregation tendency of polymer acceptors in solution can also be tuned by modifying the chemical structures of the polymers. Fig. 33a shows the chemical structures of two polymer acceptors, P18-C28 and P21.270 The introduction of two extra F substituents on the 2,1,3-benzothiadiazole (BT) unit increased the aggregation tendency of P21 in solution. For P18-C28 with weak aggregation, the active layer of the BD3T:P18-C28 blend exhibited over-crystallization of BD3T, which led to large-size phase separation. In contrast, in the BD3T:P21 blend, the aggregation of P21 was dominant and the crystallization of BD3T was restricted, leading to small-size phase separation in the active layer. While the device of BD3T:P18-C28 exhibited a PCE of 5.06%, the device of BD3T:P21 showed a much enhanced PCE of 9.51% (Fig. 33b). This was the most efficient MD/PA-type OSC device reported so far, indicating the versatility of BNBP-based polymer acceptors. Noteworthily, the MD/PA-type OSC device based on BD3T:P21 exhibited excellent thermal stability. After thermal treatment at 150 °C for 72 hours, the device could retain 84% of its initial PCE value (Fig. 33c and d). This superior device stability at high temperature was attributed to the high phase transition temperatures of the two materials and the excellent thermal stability of morphology in the MD/PA-type active layer.
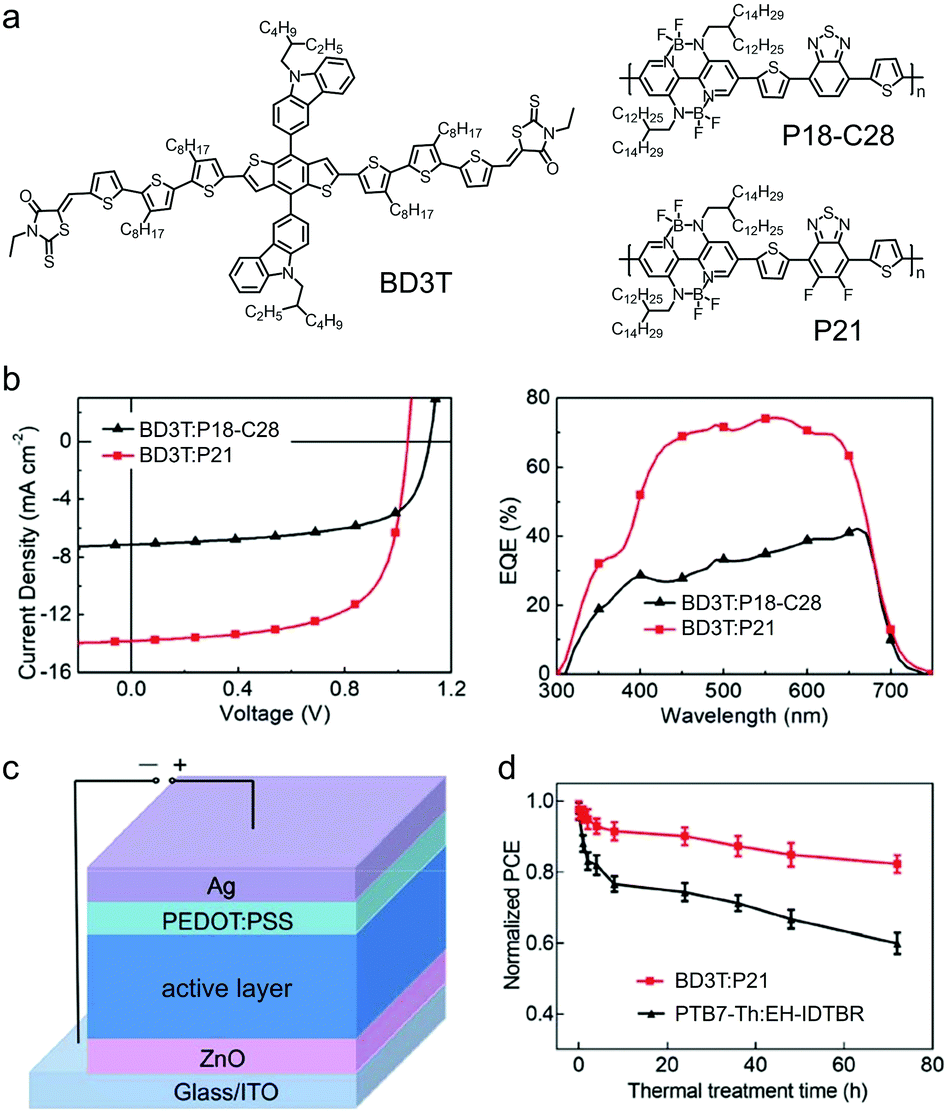 | ||
| Fig. 33 (a) Chemical structures of small molecular donor BD3T and polymer acceptors P18-C28 and P21. (b) J–V curves and EQE spectra of the devices based on the BD3T:P18-C28 and BD3T:P21 blends. (c) The inverted device structure of the OSC operating at 150 °C. (d) The normalized PCE for the OSC devices based on BD3T:P21 and PTB7-Th:EH-IDTBR blends with thermal treatment of the devices at 150 °C for different time periods. Panels (b–d) were reproduced from ref. 270 with permission from the Royal Society of Chemistry, copyright 2020. | ||
In summary, aiming at excellent OSC device performance, the opto-electronic properties of BNBP-based polymer acceptors have been systematically tuned by molecular design. Active layer phase separation morphologies of BNBP-based polymer acceptors have also been optimized by various approaches. In OSC devices, BNBP-based polymer acceptors can give a PCE of 10% when blended with polymer donors or a PCE of 9.5% when blended with small molecular donors. Owing to the medium optical bandgap and the high VOC of most of BNBP-based polymer acceptors, these polymers are very suitable for indoor photovoltaic application and a remarkable PCE of 27% has been demonstrated. In the future, research on BNBP-based polymer acceptors should focus on phase separation morphology optimization and indoor photovoltaic application.
4.4 BNIDT-based polymer acceptors
B ← N brigded indacenodithiophene, BNIDT, was used by Huang et al. to design polymer acceptors with excellent all-PSC device performance. In 2016, Fang et al. initially reported the BNIDT skeleton with four bromo substituents on the two boron atoms.271 The compound was not stable under ambient conditions. Later, Huang et al. added four bulky phenyl groups to the two boron atoms of BNIDT and greatly improved its stability.272 Besides, the synthesis of the BNIDT unit was simple and convenient, only involving a one-pot reaction from 2,5-di(thiophen-2-yl)pyrazine (Pz-T). From X-ray diffraction crystallography, the BNIDT unit exhibited a large layer-to-layer distance of 7.59 Å, which was attributed to the steric effect of the vertical phenyl substituents. Although the BNIDT unit had a large layer-to-layer distance, it had a small backbone distance of 3.58 Å due to the presence of the tilted molecular layer, which could facilitate charge transport between the backbones. The excellent stability, facile synthesis and favorable packing structure of the BNIDT unit made it to be used as a new electron-accepting building block to develop functional materials. An important feature of BNIDT-based polymer acceptors is their low crystallinity with excellent electron transporting property. The four phenyl groups on the two boron atoms act as a steric hindrance, inhibit aggregation of polymer chains in solution and prevent close π-stacking of polymer chains in thin film. Despite their low crystallinity, BNIDT-based polymer acceptors exhibit excellent electron transporting property. Their electron and hole mobilities in OFETs are on the order of 10−3–10−2 cm2 V−1 s−1 and their electron mobilities in OSCs are on the order of 10−5 cm2 V−1 s−1. All-PSC devices of BNIDT-based polymer acceptors show an FF higher than 0.7 and a PCE of 8.78%.273 Recently, a ternary OSC containing BNIDT-based polymer acceptors gave a PCE of 16%.274In the first report of BNIDT-based polymer acceptors, Huang et al. copolymerized BNIDT with thiophene or 3,4-difluorothiophene to synthesize two polymers, P22 and P23 (Fig. 34a).273 In spite of the weak crystallinity, both P22 and P23 exhibited high charge mobilities of 10−3–10−2 cm2 V−1 s−1, which benefited from the close backbone stacking of the BNIDT unit and the coplanar backbone conformation of the polymers. P23 exhibited low-lying HOMO/LUMO energy levels of −5.71 eV/−3.77 eV and a strong sunlight absorption ability with an onset absorption wavelength of 770 nm (Fig. 34b and c). With PBDB-T as the polymer donor, all-PSCs based on P22 and P23 as the polymer acceptors afforded a moderate PCE of 4.03% and a high PCE of 8.78%, respectively (Fig. 34d and e). The enhanced performance in the latter case was ascribed to the favorable film morphology, the high and balanced hole/electron mobilities, depressed bimolecular recombination, efficient exciton dissociation, and charge collection.
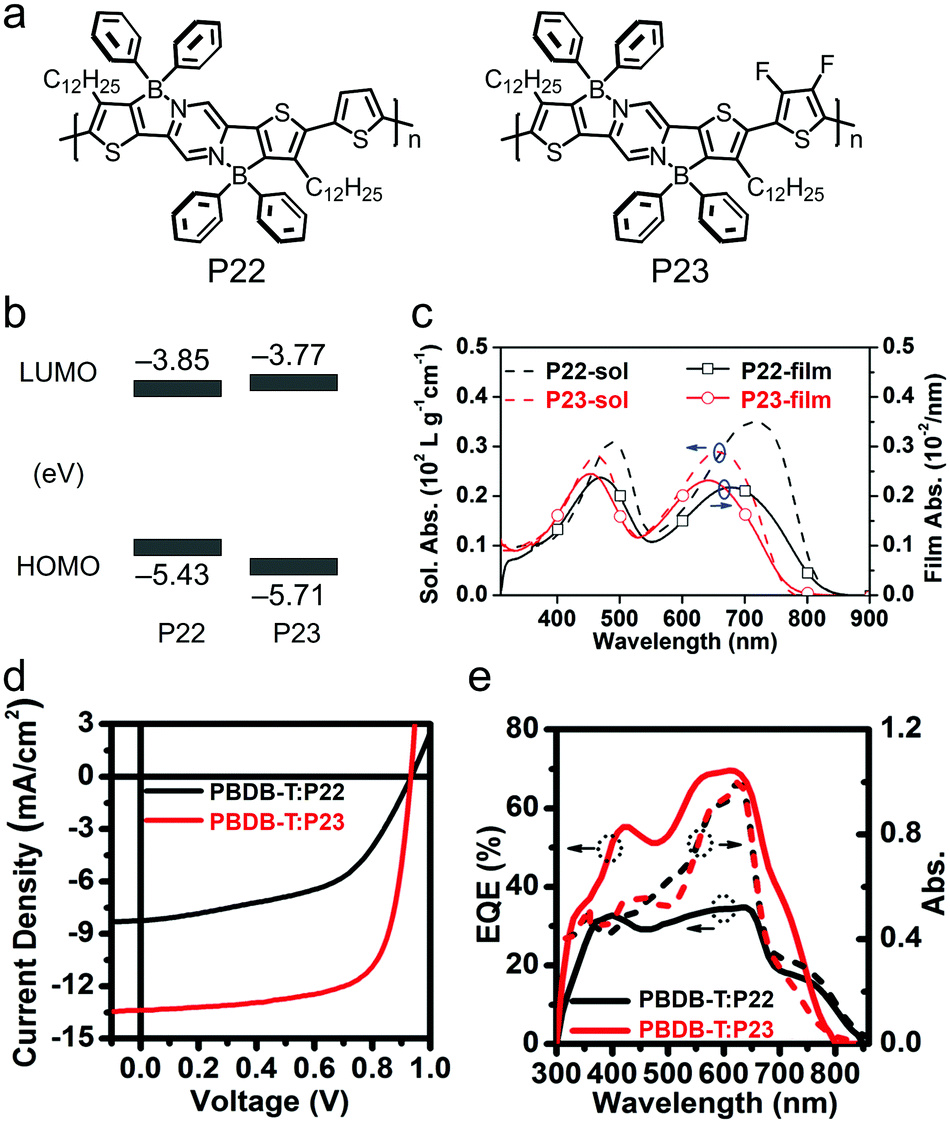 | ||
| Fig. 34 (a) Chemical structures of P22 and P23 containing the BNIDT unit. (b) HOMO/LUMO energy levels of P22 and P23. (c) The absorption spectra of P22 and P23 in solution and in film. (d) J–V curves and (e) EQE spectra of the devices based on the PBDB-T:P22 and PBDB-T:P23 blends. Panels (c–e) were reproduced from ref. 273 with permission from John Wiley and Sons, copyright 2019. | ||
Huang et al. systematically studied the effect of halogenation on the all-PSC device performance of BNIDT-based polymer acceptors.275 They prepared three polymer acceptors (P24–P26) based on alternating BNIDT and BDT units as the backbones but with or without fluorine or chlorine substituents on the BDT units (Fig. 35). The fluorine or chlorine substituents did not affect the LUMO energy levels but decreased the HOMO energy levels of the polymer acceptors. In all-PSC devices, halogenation on the polymer acceptors led to enhanced hole-transfer driving forces and better donor/acceptor miscibility, which further gave rise to higher and more balanced hole/electron mobilities and efficient physical processes.
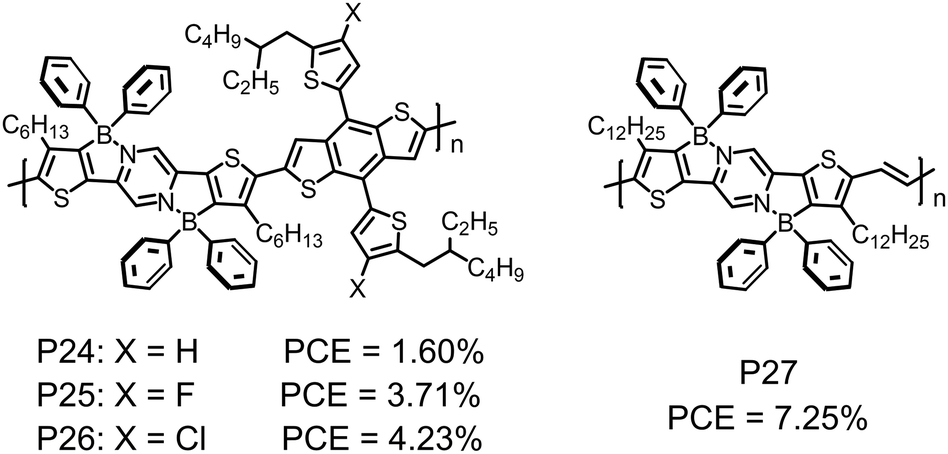 | ||
| Fig. 35 Chemical structures of polymer acceptors P24–P27 containing the BNIDT unit. | ||
To improve the light absorption capacity of B ← N-containing polymer acceptors and increase the photocurrent of the corresponding devices, Huang et al. developed a polymer acceptor P27 by copolymerizing BNIDT and vinyl units (Fig. 35).276P27 exhibited a strong absorption in the 600–900 nm region with the absorption onset wavelength at 900 nm, corresponding to the optical bandgap of 1.38 eV. An all-PSC device was fabricated using PTB1 as the donor and P27 as the acceptor. The EQE spectrum of this device showed wide responses from 300 to 900 nm, which corresponds to the high JSC of 16.90 mA cm−2. The JSC value is the highest value among the JSC's of reported B ← N-containing polymer acceptors.
4.5 Polymer donors containing the B–N unit
The boron–nitrogen covalent bond (B–N) is isoelectronic to the carbon–carbon double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C).277 While the CC bond has a dipole of zero, the B–N bond has a dipole of ca. 1.7 Debye. Therefore, B–N can be used to replace CC in π-conjugated organic molecules with interesting opto-electronic properties. New building blocks based on B–N have been designed to construct polymer electron donors for OSC applications.
C).277 While the CC bond has a dipole of zero, the B–N bond has a dipole of ca. 1.7 Debye. Therefore, B–N can be used to replace CC in π-conjugated organic molecules with interesting opto-electronic properties. New building blocks based on B–N have been designed to construct polymer electron donors for OSC applications.
Yu et al. developed a wide bandgap polymer donor, P28, by copolymerizing pendant-borylated carbazole with BDT (Fig. 36a).278 The B–N bond endowed P28 with increased electron affinity. P28 showed a deep HOMO energy level of −5.52 eV and a wide bandgap of 2.07 eV. The OSC device with the P28:PC71BM active layer exhibited a PCE of 3.82% with a high VOC of 1.0 V.
 | ||
| Fig. 36 Chemical structures of organoboron polymer donors (a) P28 and (b) P29 with boron–nitrogen bonds. (c) The principle of the opposite resonance effect caused by boron and nitrogen atoms. (d) The Jablonski diagram of the electronic states in P29 (ground states (S0), singlet states (S1), triplet states (T1), singlet–triplet energy gap (ΔEST), charge transfer states (CT) and VOC of the OSCs). (e) J–V curves and EQE spectra of the devices based on the active layers of P29 (PBNT-BDD):Y6-BO and PM6:Y6-BO. Panels (c–e) were reproduced from ref. 279 with permission from John Wiley and Sons, copyright 2021. | ||
Recently, Duan et al. reported a new building block BNT featuring B–N bonds and developed a polymer donor, P29.279 An advantage of BNT was the facile synthesis, which was important for low cost OSCs. The di-bromo monomer of BNT could be synthesized by four steps with high overall yields. Compared to the widespread halogenated BDT unit, the easy synthesis of the BNT unit results in lower synthesis cost of the corresponding polymer. According to the single crystal structure of BNT, the boron and the nitrogen atoms formed p–π conjugation with the conjugated backbone. The π–π distance of the adjacent BNT units is 3.59 Å, indicating a weak intermolecular interaction between the units. The polymer donor, P29, was an alternating copolymer of BNT unit and thiophene-flanked benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione (BDD) unit (Fig. 36b). In the test of differential scanning calorimetry (DSC), no melting or crystalline peak was observed in P29. According to 2D-GIWAXS, P29 did not exhibit clear (100) lamellar diffraction signals. These results indicated that P29 has weak crystallinity, which may be due to the weak packing of the BNT unit. Due to the opposite resonance effect of the boron atom and the nitrogen atom in the B–N covalent bond, P29 exhibited a small singlet–triplet gap (ΔEST) between the singlet state (S1) and the triple state (T1) (Fig. 36c). This was beneficial to suppress the recombination loss from charge transfer (CT) state to T1 (Fig. 36d). The OSC device with P29 as the polymer donor and Y6-BO as the small molecular acceptor showed a PCE of 16.1%, which was comparable to that of the device of the well-known polymer donor PM6 with the same acceptor (Fig. 36e). More importantly, owing to the high T1 energy level and the small ΔEST of P29, the OSC device exhibited a non-radiative energy loss of 0.19 eV, which was among the lowest values reported to date for OSCs. The facile synthesis, interesting opto-electronic properties and excellent OSC device performance of P29 proved the great potential of organoboron polymer donors.
4.6 Other organoboron polymers as electron acceptors
Most of conjugated polymers rely on π-conjugation, in which the carbon–carbon single bond and double bond alternate in the main chains. The p–π* conjugation provides a new opportunity to tune the electronic structures of conjugated polymers and substantially expand the scope of conjugated polymers. Liu, Jäkle and coworkers reported two p–π* conjugated polymer acceptors, P30 and P31, by copolymerizing a triarylborane unit (FBDT) with IID or pyridine-flanked DPP units, respectively (Fig. 37a).280 Because of the highly electron-deficient character of the triarylborane unit and IID/PyDPP units, P30 and P31 exhibited low-lying LUMO energy levels of −3.72 eV and −3.58 eV, respectively (Fig. 37b). The electron mobilities of the two polymers were estimated to be ca. 1 × 10−5 cm2 V−1 s−1. The low-lying LUMO energy levels and high electron mobilities of the two triarylborane-based polymers indicated that they could be used as electron acceptors. The all-PSC device with P30 as the polymer acceptor exhibited a PCE of 2.83% (Fig. 37c), which was higher than that of the devices (0.02%) using the control polymers without the boron atom. These results ambiguously proved the photovoltaic applications of p–π* conjugated polymers based on triarylborane.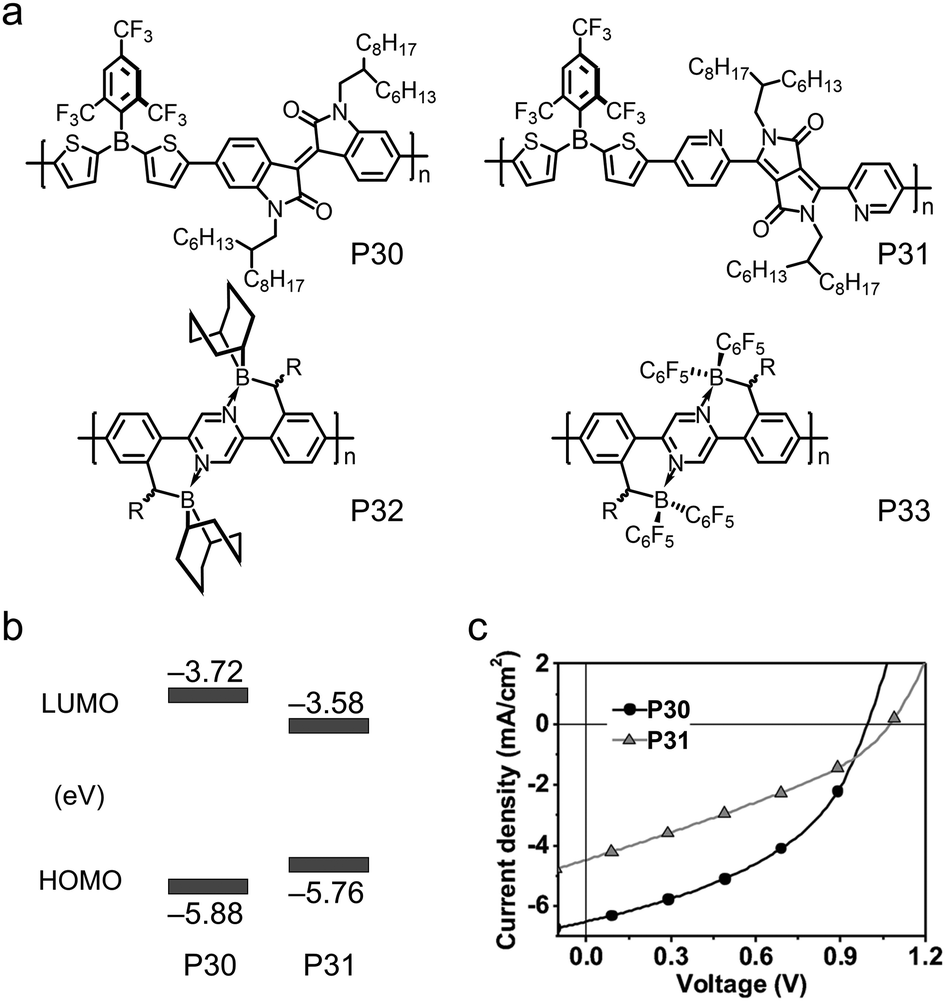 | ||
| Fig. 37 (a) Chemical structures of polymer acceptors, P30–P33. (b) HOMO/LUMO energy levels of P30 and P31. (c) J–V curves of the devices based on the PTB7-Th:P30 and PTB7-Th:P31 blends. Panel (c) was reproduced from ref. 280 with permission from John Wiley and Sons, copyright 2018. | ||
In addition to preparing copolymers by Stille- or Suzuki-type cross-coupling polycondensation, postfunctionalization of polymers is also a way to obtain conjugated polymers to adjust their electronic and structural properties. Pammer, Hauff and coworkers reported a series of ladder polymers featuring intramolecular B ← N bonds via postfunctionalization by hydroboration with different hydroboranes.281Fig. 37a shows two examples of the ladder polymers, P32 and P33, in which alkenyl-functionalized poly(biphenylene-pyrazinylene) (PPz) was postmodified by R2B-H (9H-BBN) and (C6F5)2B-H (BPF-H), respectively. Compared with the substrate polymer PPz (−2.80 eV), postmodification by hydroboration could greatly reduce the LUMO energy levels of the polymers (−3.75 eV for P32 and −4.35 eV for P33). With the decrease of the LUMO energy levels, the bandgaps of P32 and P33 were significantly reduced to 2.16 eV and 2.07 eV, respectively, and the absorption spectra had significant red shift. These polymers exhibited ambipolar charge transport with hole and electron mobilities in the order of 2 × 10−5 cm2 V−1 s−1. The low-lying LUMO energy levels of P32 and P33 enabled them to be used as acceptors in all-PSCs. This result demonstrated the potential of postfunctionalization of polymers to effectively adjust the opto-electronic properties of polymers.
5. Summary and outlook
Organoboron chemistry offers a powerful toolbox to design small molecules or polymers with tunable and excellent opto-electronic properties for OSC applications. In the past decade, a large family of organoboron small molecular donors/acceptors and organoboron polymer donors/acceptors have been developed. They have become an important class of organic photovoltaic materials. At the early stage, scientists have modified the chemical structures of classic organoboron compounds, such as BODIPY and SubPc, towards OSC applications. Later, novel organoboron building blocks, e.g. BNBP, BNIDT and BNT, have been designed and synthesized to develop organic photovoltaic materials. Based on these novel organoboron building blocks, a high PCE and excellent stability have been realized with the resulting OSC devices. At present, PCEs of 16% and 14% have been demonstrated with organoboron electron donors and organoboron electron acceptors, respectively.Despite the great research progress, the OSC device performance of organoboron small molecules and polymers still lags behind that of the best organic photovoltaic materials. To pave the practical application of organoboron photovoltaic materials, great attention should be paid to the following aspects.
(1) New organoboron building blocks. Although plenty of organoboron compounds have been reported, only very few of them have been used to design electron donor or electron acceptor materials in OSCs. In the past several years, the development of organoboron photovoltaic materials has strongly relied on the utilization of some new organoboron building blocks, for example, BNBP, BNIDT, BNT, etc. Indeed, the resulting organoboron photovoltaic materials exhibit some unique properties and show great potential. Great attention should be paid to the molecular design of unknown organoboron building blocks and the utilization of known organoboron compounds.
(2) PCE enhancement. OSC devices based on organoboron materials suffer from low JSC and low FF. EQE higher than 0.70 and FF greater than 0.70 are rarely revealed with organoboron electron donors or acceptors. To enhance the PCE of the organoboron photovoltaic materials, both their opto-electronic properties and their active layer morphology need to be optimized. This is expected to lead to synergistically enhanced JSC and FF without the sacrifice of VOC. So far, only opto-electronic properties of BNBP-based polymer acceptors have been intensively studied. The phase separation morphology optimization of organoboron photovoltaic materials is just in infancy. Provided the opto-electronic property optimization and phase separation morphology optimization are achieved, we believe that there is large room for PCE enhancement of organoboron small molecule and polymer donors/acceptors.
(3) Stability issue. The excellent thermal stability of OSC devices based on organoboron polymer acceptors has already been demonstrated by Liu et al. However, great attention should be paid to the chemical stability of the organoboron photovoltaic materials. Owing to the Lewis acidity or the vacant 2p orbital of the boron atom, organoboron compounds should be volatile to moisture. Indeed, only carefully designed organoboron compounds can possess chemical stability under ambient conditions. The long-term stability of organoboron photovoltaic materials under ambient conditions, e.g. moisture air, need to be evaluated. In-depth studies on the stability issue of organoboron photovoltaic materials should be carried out.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful for the financial support by the National Key Research and Development Program of China (No. 2019YFA0705900) funded by MOST and the National Natural Science Foundation of China (No. 21625403, 21875244).Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Hosenuzzaman, N. A. Rahim, J. Selvaraj, M. Hasanuzzaman, A. B. M. A. Malek and A. Nahar, Renewable Sustainable Energy Rev., 2015, 41, 284–297 CrossRef.
- E. Kabir, P. Kumar, S. Kumar, A. A. Adelodun and K.-H. Kim, Renewable Sustainable Energy Rev., 2018, 82, 894–900 CrossRef.
- F. Creutzig, P. Agoston, J. C. Goldschmidt, G. Luderer, G. Nemet and R. C. Pietzcker, Nat. Energy, 2017, 2, 17140 CrossRef.
- Y. Li, G. Xu, C. Cui and Y. Li, Adv. Energy Mater., 2018, 8, 1701791 CrossRef.
- V. V. Brus, J. Lee, B. R. Luginbuhl, S.-J. Ko, G. C. Bazan and T.-Q. Nguyen, Adv. Mater., 2019, 31, 1900904 CrossRef PubMed.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- Q. Wang, M. Li, Z. Peng, N. Kirby, Y. Deng, L. Ye and Y. Geng, Sci. China: Chem., 2021, 64, 478–487 CrossRef CAS.
- Y. Liu, Z. Zhang, S. Feng, M. Li, L. Wu, R. Hou, X. Xu, X. Chen and Z. Bo, J. Am. Chem. Soc., 2017, 139, 3356–3359 CrossRef CAS PubMed.
- W. Song, R. Peng, L. Huang, C. Liu, B. Fanady, T. Lei, L. Hong, J. Ge, A. Facchetti and Z. Ge, iScience, 2020, 23, 100981 CrossRef CAS PubMed.
- R. Sun, Q. Wu, J. Guo, T. Wang, Y. Wu, B. Qiu, Z. Luo, W. Yang, Z. Hu, J. Guo, M. Shi, C. Yang, F. Huang, Y. Li and J. Min, Joule, 2020, 4, 407–419 CrossRef CAS.
- M. Ren, G. Zhang, Z. Chen, J. Xiao, X. Jiao, Y. Zou, H.-L. Yip and Y. Cao, ACS Appl. Mater. Interfaces, 2020, 12, 13077–13086 CrossRef CAS PubMed.
- W. Liu, J. Zhang, Z. Zhou, D. Zhang, Y. Zhang, S. Xu and X. Zhu, Adv. Mater., 2018, 30, 1800403 CrossRef PubMed.
- F.-C. Chen, Adv. Opt. Mater., 2019, 7, 1800662 CrossRef.
- S. A. Hashemi, S. Ramakrishna and A. G. Aberle, Energy Environ. Sci., 2020, 13, 685–743 RSC.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- Y. Qian, X. Zhang, L. Xie, D. Qi, B. K. Chandran, X. Chen and W. Huang, Adv. Mater., 2016, 28, 9243–9265 CrossRef CAS PubMed.
- M. R. Lee, R. D. Eckert, K. Forberich, G. Dennler, C. J. Brabec and R. A. Gaudiana, Science, 2009, 324, 232–235 CrossRef CAS PubMed.
- K. Forberich, F. Guo, C. Bronnbauer and C. J. Brabec, Energy Technol., 2015, 3, 1051–1058 CrossRef CAS.
- M. Li, F. Igbari, Z.-K. Wang and L.-S. Liao, Adv. Energy Mater., 2020, 10, 2000641 CrossRef CAS.
- K. Feng, J. Huang, X. Zhang, Z. Wu, S. Shi, L. Thomsen, Y. Tian, H. Y. Woo, C. R. McNeill and X. Guo, Adv. Mater., 2020, 32, 2001476 CrossRef CAS PubMed.
- K. Li, Y. Wu, X. Li, H. Fu and C. Zhan, Sci. China: Chem., 2020, 63, 490–496 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- J. Qin, L. Zhang, C. Zuo, Z. Xiao, Y. Yuan, S. Yang, F. Hao, M. Cheng, K. Sun, Q. Bao, Z. Bin, Z. Jin and L. Ding, J. Semicond., 2021, 42, 010501 CrossRef CAS.
- M. Zhang, L. Zhu, G. Zhou, T. Hao, C. Qiu, Z. Zhao, Q. Hu, B. W. Larson, H. Zhu, Z. Ma, Z. Tang, W. Feng, Y. Zhang, T. P. Russell and F. Liu, Nat. Commun., 2021, 12, 309 CrossRef CAS PubMed.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CrossRef CAS.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- Y. He, H.-Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- T. S. van der Poll, J. A. Love, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646–3649 CrossRef CAS PubMed.
- K. Sun, Z. Xiao, S. Lu, W. Zajaczkowski, W. Pisula, E. Hanssen, J. M. White, R. M. Williamson, J. Subbiah, J. Ouyang, A. B. Holmes, W. W. H. Wong and D. J. Jones, Nat. Commun., 2015, 6, 6013 CrossRef CAS PubMed.
- J. Zhou, Y. Zuo, X. Wan, G. Long, Q. Zhang, W. Ni, Y. Liu, Z. Li, G. He, C. Li, B. Kan, M. Li and Y. Chen, J. Am. Chem. Soc., 2013, 135, 8484–8487 CrossRef CAS PubMed.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C.-S. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS.
- H. Hu, P. C. Y. Chow, G. Zhang, T. Ma, J. Liu, G. Yang and H. Yan, Acc. Chem. Res., 2017, 50, 2519–2528 CrossRef CAS.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Nat. Commun., 2016, 7, 13740 CrossRef CAS PubMed.
- J. Wan, X. Xu, G. Zhang, Y. Li, K. Feng and Q. Peng, Energy Environ. Sci., 2017, 10, 1739–1745 RSC.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Q. Yue, W. Liu and X. Zhu, J. Am. Chem. Soc., 2020, 142, 11613–11628 CrossRef CAS PubMed.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- Q. Wei, W. Liu, M. Leclerc, J. Yuan, H. Chen and Y. Zou, Sci. China: Chem., 2020, 63, 1352–1366 CrossRef CAS.
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- Z. Fei, F. D. Eisner, X. Jiao, M. Azzouzi, J. A. Röhr, Y. Han, M. Shahid, A. S. R. Chesman, C. D. Easton, C. R. McNeill, T. D. Anthopoulos, J. Nelson and M. Heeney, Adv. Mater., 2018, 30, 1705209 CrossRef PubMed.
- C. Huang, X. Liao, K. Gao, L. Zuo, F. Lin, X. Shi, C.-Z. Li, H. Liu, X. Li, F. Liu, Y. Chen, H. Chen and A. K. Y. Jen, Chem. Mater., 2018, 30, 5429–5434 CrossRef CAS.
- J. Sun, X. Ma, Z. Zhang, J. Yu, J. Zhou, X. Yin, L. Yang, R. Geng, R. Zhu, F. Zhang and W. Tang, Adv. Mater., 2018, 30, 1707150 CrossRef.
- Z. Zheng, Q. Hu, S. Zhang, D. Zhang, J. Wang, S. Xie, R. Wang, Y. Qin, W. Li, L. Hong, N. Liang, F. Liu, Y. Zhang, Z. Wei, Z. Tang, T. P. Russell, J. Hou and H. Zhou, Adv. Mater., 2018, 30, 1801801 CrossRef.
- T. Jia, J. Zhang, W. Zhong, Y. Liang, K. Zhang, S. Dong, L. Ying, F. Liu, X. Wang, F. Huang and Y. Cao, Nano Energy, 2020, 72, 104718 CrossRef CAS.
- D. Hu, Q. Yang, H. Chen, F. Wobben, V. M. Le Corre, R. Singh, T. Liu, R. Ma, H. Tang, L. J. A. Koster, T. Duan, H. Yan, Z. Kan, Z. Xiao and S. Lu, Energy Environ. Sci., 2020, 13, 2134–2141 RSC.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y.-J. Hwang, H. Li, B. A. E. Courtright, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2016, 28, 124–131 CrossRef CAS.
- X. Meng, L. Zhang, Y. Xie, X. Hu, Z. Xing, Z. Huang, C. Liu, L. Tan, W. Zhou, Y. Sun, W. Ma and Y. Chen, Adv. Mater., 2019, 31, 1903649 CrossRef CAS PubMed.
- Z. Wang, H. Jiang, X. Liu, J. Liang, L. Zhang, L. Qing, Q. Wang, W. Zhang, Y. Cao and J. Chen, J. Mater. Chem. A, 2020, 8, 7765–7774 RSC.
- B. Xiao, A. Tang, J. Zhang, A. Mahmood, Z. Wei and E. Zhou, Adv. Energy Mater., 2017, 7, 1602269 CrossRef.
- Y. Ma, D. Cai, S. Wan, P. Wang, J. Wang and Q. Zheng, Angew. Chem., Int. Ed., 2020, 59, 21627–21633 CrossRef CAS.
- J. Song, C. Li, L. Zhu, J. Guo, J. Xu, X. Zhang, K. Weng, K. Zhang, J. Min, X. Hao, Y. Zhang, F. Liu and Y. Sun, Adv. Mater., 2019, 31, 1905645 CrossRef CAS PubMed.
- Y. Wu, S. Schneider, C. Walter, A. H. Chowdhury, B. Bahrami, H.-C. Wu, Q. Qiao, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2020, 142, 392–406 CrossRef CAS PubMed.
- Y. Tong, Z. Xiao, X. Du, C. Zuo, Y. Li, M. Lv, Y. Yuan, C. Yi, F. Hao, Y. Hua, T. Lei, Q. Lin, K. Sun, D. Zhao, C. Duan, X. Shao, W. Li, H.-L. Yip, Z. Xiao, B. Zhang, Q. Bian, Y. Cheng, S. Liu, M. Cheng, Z. Jin, S. Yang and L. Ding, Sci. China: Chem., 2020, 63, 758–765 CrossRef CAS.
- A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef CAS.
- S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC.
- D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS.
- L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC.
- S.-Y. Li, Z.-B. Sun and C.-H. Zhao, Inorg. Chem., 2017, 56, 8705–8717 CrossRef CAS.
- E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef.
- S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS.
- N. Ikeda, S. Oda, R. Matsumoto, M. Yoshioka, D. Fukushima, K. Yoshiura, N. Yasuda and T. Hatakeyama, Adv. Mater., 2020, 32, 2004072 CrossRef CAS PubMed.
- X. Yin, J. Liu and F. Jäkle, Chem. – Eur. J., 2021, 27, 2973–2986 CrossRef CAS PubMed.
- H. Lu, T. Nakamuro, K. Yamashita, H. Yanagisawa, O. Nureki, M. Kikkawa, H. Gao, J. Tian, R. Shang and E. Nakamura, J. Am. Chem. Soc., 2020, 142, 18990–18996 CrossRef CAS PubMed.
- J. Huang and Y. Li, Front. Chem., 2018, 6, 341 CrossRef PubMed.
- R. Zhao, J. Liu and L. Wang, Acc. Chem. Res., 2020, 53, 1557–1567 CrossRef CAS PubMed.
- G. de la Torre, G. Bottari and T. Torres, Adv. Energy Mater., 2017, 7, 1601700 CrossRef.
- D. Ho, R. Ozdemir, H. Kim, T. Earmme, H. Usta and C. Kim, ChemPlusChem, 2019, 84, 18–37 CAS.
- Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263–12274 CrossRef CAS PubMed.
- J. Huang, X. Wang, Y. Xiang, L. Guo and G. Chen, Adv. Energy Sustainability Res., 2021, 2100016 CrossRef.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- C. Dou, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 9346–9349 CrossRef CAS PubMed.
- X. Y. Wang, J. Y. Wang and J. Pei, Chem. – Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed.
- Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat'ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800–8816 CrossRef CAS PubMed.
- J. B. Gilroy and E. Otten, Chem. Soc. Rev., 2020, 49, 85–113 RSC.
- H. Helten, Chem. – Eur. J., 2016, 22, 12972–12982 CrossRef CAS.
- A. N. Bismillah and I. Aprahamian, Chem. Soc. Rev., 2021, 50, 5631–5649 RSC.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- Y. Min, C. Dou, D. Liu, H. Dong and J. Liu, J. Am. Chem. Soc., 2019, 141, 17015–17021 CrossRef CAS PubMed.
- Y. Min, C. Dou, H. Tian, Y. Geng, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 2000–2004 CrossRef CAS PubMed.
- X. Shao, C. Dou, J. Liu and L. Wang, Sci. China: Chem., 2019, 62, 1387–1392 CrossRef CAS.
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- M. Poddar and R. Misra, Coord. Chem. Rev., 2020, 421, 213462 CrossRef CAS.
- S. Cherumukkil, B. Vedhanarayanan, G. Das, V. K. Praveen and A. Ajayaghosh, Bull. Chem. Soc. Jpn., 2017, 91, 100–120 CrossRef.
- R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC.
- H. Lu, J. Mack, T. Nyokong, N. Kobayashi and Z. Shen, Coord. Chem. Rev., 2016, 318, 1–15 CrossRef CAS.
- E. V. Antina and N. A. Bumagina, Chem. Heterocycl. Compd., 2017, 53, 39–41 CrossRef CAS.
- D. Chen, Z. Zhong, Q. Ma, J. Shao, W. Huang and X. Dong, ACS Appl. Mater. Interfaces, 2020, 12, 26914–26925 CrossRef CAS PubMed.
- Z. Shi, X. Han, W. Hu, H. Bai, B. Peng, L. Ji, Q. Fan, L. Li and W. Huang, Chem. Soc. Rev., 2020, 49, 7533–7567 RSC.
- X. Jiang, S. Li, J. Guan, T. Fang, X. Liu and L. Xiao, Curr. Org. Chem., 2016, 20, 1736–1744 CrossRef CAS.
- Q. Wu, Z. Kang, Q. Gong, X. Guo, H. Wang, D. Wang, L. Jiao and E. Hao, Org. Lett., 2020, 22, 7513–7517 CrossRef CAS PubMed.
- A. Treibs and F.-H. Kreuzer, Justus Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS.
- S. Kolemen and E. U. Akkaya, Coord. Chem. Rev., 2018, 354, 121–134 CrossRef CAS.
- L. Bucher, N. Desbois, P. D. Harvey, G. D. Sharma and C. P. Gros, Sol. RRL, 2017, 1, 1700127 CrossRef.
- B. M. Squeo, V. G. Gregoriou, A. Avgeropoulos, S. Baysec, S. Allard, U. Scherf and C. L. Chochos, Prog. Polym. Sci., 2017, 71, 26–52 CrossRef CAS.
- B. M. Squeo and M. Pasini, Supramol. Chem., 2020, 32, 56–70 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- H. Klfout, A. Stewart, M. Elkhalifa and H. He, ACS Appl. Mater. Interfaces, 2017, 9, 39873–39889 CrossRef CAS PubMed.
- T. Rousseau, A. Cravino, T. Bura, G. Ulrich, R. Ziessel and J. Roncali, Chem. Commun., 2009, 1673–1675 RSC.
- S. Suzuki, M. Kozaki, K. Nozaki and K. Okada, J. Photochem. Photobiol., C, 2011, 12, 269–292 CrossRef CAS.
- N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS.
- E. Bodio and C. Goze, Dyes Pigm., 2019, 160, 700–710 CrossRef CAS.
- L. Jean-Gérard, W. Vasseur, F. Scherninski and B. Andrioletti, Chem. Commun., 2018, 54, 12914–12929 RSC.
- J. Wang, N. Boens, L. Jiao and E. Hao, Org. Biomol. Chem., 2020, 18, 4135–4156 RSC.
- S. Kolemen, Y. Cakmak, T. Ozdemir, S. Erten-Ela, M. Buyuktemiz, Y. Dede and E. U. Akkaya, Tetrahedron, 2014, 70, 6229–6234 CrossRef CAS.
- J. Marques dos Santos, L. K. Jagadamma, N. M. Latif, A. Ruseckas, I. D. W. Samuel and G. Cooke, RSC Adv., 2019, 9, 15410–15423 RSC.
- T. Rousseau, A. Cravino, E. Ripaud, P. Leriche, S. Rihn, A. De Nicola, R. Ziessel and J. Roncali, Chem. Commun., 2010, 46, 5082–5084 RSC.
- J. Liao, Y. Xu, H. Zhao, Y. Wang, W. Zhang, F. Peng, S. Xie and X. Yang, RSC Adv., 2015, 5, 86453–86462 RSC.
- J. Yang, C. H. Devillers, P. Fleurat-Lessard, H. Jiang, S. Wang, C. P. Gros, G. Gupta, G. D. Sharma and H. Xu, Dalton Trans., 2020, 49, 5606–5617 RSC.
- A. Mirloup, N. Leclerc, S. Rihn, T. Bura, R. Bechara, A. Hébraud, P. Lévêque, T. Heiser and R. Ziessel, New J. Chem., 2014, 38, 3644–3653 RSC.
- A. Aguiar, J. Farinhas, W. da Silva, M. E. Ghica, C. M. A. Brett, J. Morgado and A. J. F. N. Sobral, Dyes Pigm., 2019, 168, 103–110 CrossRef CAS.
- A. Sutter, P. Retailleau, W.-C. Huang, H.-W. Lin and R. Ziessel, New J. Chem., 2014, 38, 1701–1710 RSC.
- J. Liao, H. Zhao, Y. Xu, Z. Cai, Z. Peng, W. Zhang, W. Zhou, B. Li, Q. Zong and X. Yang, Dyes Pigm., 2016, 128, 131–140 CrossRef CAS.
- H.-Y. Lin, W.-C. Huang, Y.-C. Chen, H.-H. Chou, C.-Y. Hsu, J. T. Lin and H.-W. Lin, Chem. Commun., 2012, 48, 8913–8915 RSC.
- X. Zhang, Y. Zhang, L. Chen and Y. Xiao, RSC Adv., 2015, 5, 32283–32289 RSC.
- A. Aguiar, J. Farinhas, W. da Silva, M. Susano, M. R. Silva, L. Alcácer, S. Kumar, C. M. A. Brett, J. Morgado and A. J. F. N. Sobral, Dyes Pigm., 2020, 172, 107842 CrossRef CAS.
- T. Jadhav, R. Misra, S. Biswas and G. D. Sharma, Phys. Chem. Chem. Phys., 2015, 17, 26580–26588 RSC.
- T. Rousseau, A. Cravino, T. Bura, G. Ulrich, R. Ziessel and J. Roncali, J. Mater. Chem., 2009, 19, 2298–2300 RSC.
- T. Bura, N. Leclerc, S. Fall, P. Lévêque, T. Heiser, P. Retailleau, S. Rihn, A. Mirloup and R. Ziessel, J. Am. Chem. Soc., 2012, 134, 17404–17407 CrossRef CAS.
- C.-C. Chi, Y.-J. Huang and C.-T. Chen, J. Chin. Chem. Soc., 2012, 59, 305–316 CrossRef CAS.
- W. Liu, J. Yao and C. Zhan, RSC Adv., 2015, 5, 74238–74241 RSC.
- L. Zou, S. Guan, L. Li and L. Zhao, Chem. Res. Chin. Univ., 2015, 31, 801–808 CrossRef CAS.
- R. Mishra, B. Basumatary, R. Singhal, G. D. Sharma and J. Sankar, ACS Appl. Mater. Interfaces, 2018, 10, 31462–31471 CrossRef CAS PubMed.
- G. Thumuganti, V. Gupta and S. P. Singh, New J. Chem., 2019, 43, 8735–8740 RSC.
- İ. Ömeroğlu, A. Şenocak, H. Yetkin, H. Y. Güney, E. Demirbaş and M. Durmuş, J. Porphyrins Phthalocyanines, 2019, 23, 1132–1143 CrossRef.
- P. Erwin, S. M. Conron, J. H. Golden, K. Allen and M. E. Thompson, Chem. Mater., 2015, 27, 5386–5392 CrossRef CAS.
- J. Liao, Y. Xu, H. Zhao, Q. Zong and Y. Fang, Org. Electron., 2017, 49, 321–333 CrossRef CAS.
- W. Liu, J. Yao and C. Zhan, Chin. Chem. Lett., 2017, 28, 875–880 CrossRef CAS.
- G. Tarafdar, J. C. Johnson, B. W. Larson and P. C. Ramamurthy, Dyes Pigm., 2020, 177, 108289 CrossRef CAS.
- W. Liu, A. Tang, J. Chen, Y. Wu, C. Zhan and J. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 22496–22505 CrossRef CAS PubMed.
- L. Xiao, H. Wang, K. Gao, L. Li, C. Liu, X. Peng, W.-Y. Wong, W.-K. Wong and X. Zhu, Chem. – Asian J., 2015, 10, 1513–1518 CrossRef CAS PubMed.
- J. Liao, H. Zhao, Z. Cai, Y. Xu, F. G. F. Qin, Q. Zong, F. Peng and Y. Fang, Org. Electron., 2018, 61, 215–222 CrossRef CAS.
- R. Srinivasa Rao, A. Bagui, G. Hanumantha Rao, V. Gupta and S. P. Singh, Chem. Commun., 2017, 53, 6953–6956 RSC.
- L. Bucher, N. Desbois, E. N. Koukaras, C. H. Devillers, S. Biswas, G. D. Sharma and C. P. Gros, J. Mater. Chem. A, 2018, 6, 8449–8461 RSC.
- G. D. Sharma, S. A. Siddiqui, A. Nikiforou, G. E. Zervaki, I. Georgakaki, K. Ladomenou and A. G. Coutsolelos, J. Mater. Chem. C, 2015, 3, 6209–6217 RSC.
- J. J. Chen, S. M. Conron, P. Erwin, M. Dimitriou, K. McAlahney and M. E. Thompson, ACS Appl. Mater. Interfaces, 2015, 7, 662–669 CrossRef CAS PubMed.
- K. Umezawa, Y. Nakamura, H. Makino, D. Citterio and K. Suzuki, J. Am. Chem. Soc., 2008, 130, 1550–1551 CrossRef CAS PubMed.
- Z.-W. Zhao, Q.-Q. Pan, Y.-C. Duan, Y. Wu, Y. Geng, S.-X. Wu and Z.-M. Su, J. Phys. Chem. C, 2019, 123, 6407–6415 CrossRef CAS.
- T.-y. Li, J. Benduhn, Z. Qiao, Y. Liu, Y. Li, R. Shivhare, F. Jaiser, P. Wang, J. Ma, O. Zeika, D. Neher, S. C. B. Mannsfeld, Z. Ma, K. Vandewal and K. Leo, J. Phys. Chem. Lett., 2019, 10, 2684–2691 CrossRef CAS.
- T.-y. Li, J. Benduhn, Y. Li, F. Jaiser, D. Spoltore, O. Zeika, Z. Ma, D. Neher, K. Vandewal and K. Leo, J. Mater. Chem. A, 2018, 6, 18583–18591 RSC.
- A. Wakamiya, T. Murakami and S. Yamaguchi, Chem. Sci., 2013, 4, 1002–1007 RSC.
- Y. Kubo, K. Watanabe, R. Nishiyabu, R. Hata, A. Murakami, T. Shoda and H. Ota, Org. Lett., 2011, 13, 4574–4577 CrossRef CAS PubMed.
- T.-y. Li, T. Meyer, Z. Ma, J. Benduhn, C. Körner, O. Zeika, K. Vandewal and K. Leo, J. Am. Chem. Soc., 2017, 139, 13636–13639 CrossRef CAS PubMed.
- J. Killoran, L. Allen, J. F. Gallagher, W. M. Gallagher and D. F. O′Shea, Chem. Commun., 2002, 1862–1863 RSC.
- X. Zhang, H. Yu and Y. Xiao, J. Org. Chem., 2012, 77, 669–673 CrossRef CAS PubMed.
- T.-y. Li, T. Meyer, R. Meerheim, M. Höppner, C. Körner, K. Vandewal, O. Zeika and K. Leo, J. Mater. Chem. A, 2017, 5, 10696–10703 RSC.
- T. Mueller, R. Gresser, K. Leo and M. Riede, Sol. Energy Mater. Sol. Cells, 2012, 99, 176–181 CrossRef CAS.
- S. Kraner, J. Widmer, J. Benduhn, E. Hieckmann, T. Jägeler-Hoheisel, S. Ullbrich, D. Schütze, K. Sebastian Radke, G. Cuniberti, F. Ortmann, M. Lorenz-Rothe, R. Meerheim, D. Spoltore, K. Vandewal, C. Koerner and K. Leo, Phys. Status Solidi A, 2015, 212, 2747–2753 CrossRef CAS.
- J. Min, T. Ameri, R. Gresser, M. Lorenz-Rothe, D. Baran, A. Troeger, V. Sgobba, K. Leo, M. Riede, D. M. Guldi and C. J. Brabec, ACS Appl. Mater. Interfaces, 2013, 5, 5609–5616 CrossRef CAS PubMed.
- J. Meiss, F. Holzmueller, R. Gresser, K. Leo and M. Riede, Appl. Phys. Lett., 2011, 99, 193307 CrossRef.
- R. Meerheim, C. Körner, B. Oesen and K. Leo, Appl. Phys. Lett., 2016, 108, 103302 CrossRef.
- M. Lorenz-Rothe, K. S. Schellhammer, T. Jägeler-Hoheisel, R. Meerheim, S. Kraner, M. P. Hein, C. Schünemann, M. L. Tietze, M. Hummert, F. Ortmann, G. Cuniberti, C. Körner and K. Leo, Adv. Electron. Mater., 2016, 2, 1600152 CrossRef.
- A. M. Poe, A. M. Della Pelle, A. V. Subrahmanyam, W. White, G. Wantz and S. Thayumanavan, Chem. Commun., 2014, 50, 2913–2915 RSC.
- W. Liu, J. Yao and C. Zhan, Chin. J. Chem., 2017, 35, 1813–1823 CrossRef CAS.
- G. E. Morse and T. P. Bender, ACS Appl. Mater. Interfaces, 2012, 4, 5055–5068 CrossRef CAS PubMed.
- C. G. Claessens, D. González-Rodríguez and T. Torres, Chem. Rev., 2002, 102, 835–854 CrossRef CAS PubMed.
- C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed.
- K. L. Mutolo, E. I. Mayo, B. P. Rand, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2006, 128, 8108–8109 CrossRef CAS PubMed.
- C.-T. Chou, W.-L. Tang, Y. Tai, C.-H. Lin, C.-H. J. Liu, L.-C. Chen and K.-H. Chen, Thin Solid Films, 2012, 520, 2289–2292 CrossRef CAS.
- N. Beaumont, I. Hancox, P. Sullivan, R. A. Hatton and T. S. Jones, Energy Environ. Sci., 2011, 4, 1708–1711 RSC.
- C.-F. Lin, V. M. Nichols, Y.-C. Cheng, C. J. Bardeen, M.-K. Wei, S.-W. Liu, C.-C. Lee, W.-C. Su, T.-L. Chiu, H.-C. Han, L.-C. Chen, C.-T. Chen and J.-H. Lee, Sol. Energy Mater. Sol. Cells, 2014, 122, 264–270 CrossRef CAS.
- R. Pandey, Y. Zou and R. J. Holmes, Appl. Phys. Lett., 2012, 101, 033308 CrossRef.
- F. Jin, B. Chu, W. Li, Z. Su, X. Yan, J. Wang, R. Li, B. Zhao, T. Zhang, Y. Gao, C. S. Lee, H. Wu, F. Hou, T. Lin and Q. Song, Org. Electron., 2014, 15, 3756–3760 CrossRef CAS.
- H. Gommans, D. Cheyns, T. Aernouts, C. Girotto, J. Poortmans and P. Heremans, Adv. Funct. Mater., 2007, 17, 2653–2658 CrossRef CAS.
- R. Pandey, A. A. Gunawan, K. A. Mkhoyan and R. J. Holmes, Adv. Funct. Mater., 2012, 22, 617–624 CrossRef CAS.
- B. E. Lassiter, J. D. Zimmerman, A. Panda, X. Xiao and S. R. Forrest, Appl. Phys. Lett., 2012, 101, 063303 CrossRef.
- I. J. Curtin and R. J. Holmes, Adv. Energy Mater., 2018, 8, 1702339 CrossRef.
- S. M. Menke, W. A. Luhman and R. J. Holmes, Nat. Mater., 2013, 12, 152–157 CrossRef CAS PubMed.
- B. Verreet, S. Schols, D. Cheyns, B. P. Rand, H. Gommans, T. Aernouts, P. Heremans and J. Genoe, J. Mater. Chem., 2009, 19, 5295–5297 RSC.
- J. D. Dang, D. S. Josey, A. J. Lough, Y. Li, A. Sifate, Z.-H. Lu and T. P. Bender, J. Mater. Chem. A, 2016, 4, 9566–9577 RSC.
- D. Cheyns, B. P. Rand and P. Heremans, Appl. Phys. Lett., 2010, 97, 033301 CrossRef.
- B. Ma, C. H. Woo, Y. Miyamoto and J. M. J. Fréchet, Chem. Mater., 2009, 21, 1413–1417 CrossRef CAS.
- G. Chen, H. Sasabe, T. Sano, X.-F. Wang, Z. Hong, J. Kido and Y. Yang, Nanotechnology, 2013, 24, 484007 CrossRef PubMed.
- C. E. Mauldin, C. Piliego, D. Poulsen, D. A. Unruh, C. Woo, B. Ma, J. L. Mynar and J. M. J. Fréchet, ACS Appl. Mater. Interfaces, 2010, 2, 2833–2838 CrossRef CAS.
- J. Endres, I. Pelczer, B. P. Rand and A. Kahn, Chem. Mater., 2016, 28, 794–801 CrossRef CAS.
- B. Verreet, K. Cnops, D. Cheyns, P. Heremans, A. Stesmans, G. Zango, C. G. Claessens, T. Torres and B. P. Rand, Adv. Energy Mater., 2014, 4, 1301413 CrossRef.
- H. Gommans, T. Aernouts, B. Verreet, P. Heremans, A. Medina, C. G. Claessens and T. Torres, Adv. Funct. Mater., 2009, 19, 3435–3439 CrossRef CAS.
- N. Beaumont, J. S. Castrucci, P. Sullivan, G. E. Morse, A. S. Paton, Z.-H. Lu, T. P. Bender and T. S. Jones, J. Phys. Chem. C, 2014, 118, 14813–14823 CrossRef CAS.
- B. Verreet, B. P. Rand, D. Cheyns, A. Hadipour, T. Aernouts, P. Heremans, A. Medina, C. G. Claessens and T. Torres, Adv. Energy Mater., 2011, 1, 565–568 CrossRef CAS.
- K. L. Sampson, G. E. Morse and T. P. Bender, ACS Appl. Energy Mater., 2018, 1, 2490–2501 CrossRef CAS.
- N. Beaumont, S. W. Cho, P. Sullivan, D. Newby, K. E. Smith and T. S. Jones, Adv. Funct. Mater., 2012, 22, 561–566 CrossRef CAS.
- P. Sullivan, A. Duraud, L. Hancox, N. Beaumont, G. Mirri, J. H. R. Tucker, R. A. Hatton, M. Shipman and T. S. Jones, Adv. Energy Mater., 2011, 1, 352–355 CrossRef CAS.
- K. Cnops, G. Zango, J. Genoe, P. Heremans, M. V. Martinez-Diaz, T. Torres and D. Cheyns, J. Am. Chem. Soc., 2015, 137, 8991–8997 CrossRef CAS PubMed.
- P. Sullivan, S. Schumann, R. Da Campo, T. Howells, A. Duraud, M. Shipman, R. A. Hatton and T. S. Jones, Adv. Energy Mater., 2013, 3, 239–244 CrossRef CAS.
- G. E. Morse, J. L. Gantz, K. X. Steirer, N. R. Armstrong and T. P. Bender, ACS Appl. Mater. Interfaces, 2014, 6, 1515–1524 CrossRef CAS.
- D. S. Josey, G. L. Ingram, R. K. Garner, J. M. Wang, G. J. Evans, Z.-H. Lu and T. P. Bender, ACS Appl. Energy Mater., 2019, 2, 979–986 CrossRef CAS.
- R. K. Garner, D. S. Josey, S. R. Nyikos, A. Dovijarski, J. M. Wang, G. J. Evans and T. P. Bender, Sol. Energy Mater. Sol. Cells, 2018, 176, 331–335 CrossRef CAS.
- D. S. Josey, S. R. Nyikos, R. K. Garner, A. Dovijarski, J. S. Castrucci, J. M. Wang, G. J. Evans and T. P. Bender, ACS Energy Lett., 2017, 2, 726–732 CrossRef CAS.
- K. Cnops, B. P. Rand, D. Cheyns, B. Verreet, M. A. Empl and P. Heremans, Nat. Commun., 2014, 5, 3406 CrossRef PubMed.
- B. Ebenhoch, N. B. A. Prasetya, V. M. Rotello, G. Cooke and I. D. W. Samuel, J. Mater. Chem. A, 2015, 3, 7345–7352 RSC.
- C. Duan, G. Zango, M. García Iglesias, F. J. M. Colberts, M. M. Wienk, M. V. Martínez-Díaz, R. A. J. Janssen and T. Torres, Angew. Chem., Int. Ed., 2017, 56, 148–152 CrossRef CAS PubMed.
- H. Hang, Z. Zhang, X. Wu, Y. Chen, H. Li, W. Wang, H. Tong and L. Wang, J. Mater. Chem. C, 2018, 6, 7141–7148 RSC.
- H. Hang, X. Wu, Q. Xu, Y. Chen, H. Li, W. Wang, H. Tong and L. Wang, Dyes Pigm., 2019, 160, 243–251 CrossRef CAS.
- X. Huang, M. Hu, X. Zhao, C. Li, Z. Yuan, X. Liu, C. Cai, Y. Zhang, Y. Hu and Y. Chen, Org. Lett., 2019, 21, 3382–3386 CrossRef CAS PubMed.
- T. Huang, H. Chen, J. Feng, A. Zhang, W. Jiang, F. He and Z. Wang, ACS Mater. Lett., 2019, 1, 404–409 CrossRef CAS.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS PubMed.
- C. Dou, J. Liu and L. Wang, Sci. China: Chem., 2017, 60, 450–459 CrossRef CAS.
- F. Liu, J. Liu and L. Wang, Acta Phys. – Chim. Sin., 2019, 35, 251–256 CAS.
- F. Liu, Z. Ding, J. Liu and L. Wang, Chem. Commun., 2017, 53, 12213–12216 RSC.
- F. Liu, J. Liu and L. Wang, Chem. – Asian J., 2020, 15, 3314–3320 CrossRef CAS PubMed.
- F. Liu, J. Liu and L. Wang, Org. Chem. Front., 2019, 6, 1996–2003 RSC.
- N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 5112–5113 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- N. Baser-Kirazli, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2020, 59, 8689–8697 CrossRef CAS PubMed.
- X. Yin, J. Chen, R. A. Lalancette, T. B. Marder and F. Jäkle, Angew. Chem., Int. Ed., 2014, 53, 9761–9765 CrossRef CAS PubMed.
- Y. Yu, C. Dong, A. F. Alahmadi, B. Meng, J. Liu, F. Jäkle and L. Wang, J. Mater. Chem. C, 2019, 7, 7427–7432 RSC.
- Y. Yu, B. Meng, F. Jäkle, J. Liu and L. Wang, Chem. – Eur. J., 2020, 26, 873–880 CrossRef CAS PubMed.
- T. A. Welsh, A. Laventure, A. F. Alahmadi, G. Zhang, T. Baumgartner, Y. Zou, F. Jäkle and G. C. Welch, ACS Appl. Energy Mater., 2019, 2, 1229–1240 CrossRef CAS.
- S. Shimizu, T. Iino, A. Saeki, S. Seki and N. Kobayashi, Chem. – Eur. J., 2015, 21, 2893–2904 CrossRef CAS PubMed.
- R. Feng, N. Sato, T. Yasuda, H. Furuta and S. Shimizu, Chem. Commun., 2020, 56, 2975–2978 RSC.
- P. Li, Q. Liang, E. Y.-H. Hong, C.-Y. Chan, Y.-H. Cheng, M.-Y. Leung, M.-Y. Chan, K.-H. Low, H. Wu and V. W.-W. Yam, Chem. Sci., 2020, 11, 11601–11612 RSC.
- M. M. Morgan, M. Nazari, T. Pickl, J. M. Rautiainen, H. M. Tuononen, W. E. Piers, G. C. Welch and B. S. Gelfand, Chem. Commun., 2019, 55, 11095–11098 RSC.
- J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS PubMed.
- A. He, Y. Qin, W. Dai and X. Luo, Dyes Pigm., 2019, 162, 671–679 CrossRef CAS.
- S. P. Economopoulos, C. L. Chochos, H. A. Ioannidou, M. Neophytou, C. Charilaou, G. A. Zissimou, J. M. Frost, T. Sachetan, M. Shahid, J. Nelson, M. Heeney, D. D. C. Bradley, G. Itskos, P. A. Koutentis and S. A. Choulis, RSC Adv., 2013, 3, 10221–10229 RSC.
- D. Baran, S. Tuladhar, S. P. Economopoulos, M. Neophytou, A. Savva, G. Itskos, A. Othonos, D. D. C. Bradley, C. J. Brabec, J. Nelson and S. A. Choulis, Synth. Met., 2017, 226, 25–30 CrossRef CAS.
- B. C. Popere, A. M. Della Pelle and S. Thayumanavan, Macromolecules, 2011, 44, 4767–4776 CrossRef CAS.
- M. Ozdemir, S. W. Kim, H. Kim, M.-G. Kim, B. J. Kim, C. Kim and H. Usta, Adv. Electron. Mater., 2018, 4, 1700354 CrossRef.
- B. Liu, Z. Ma, Y. Xu, Y. Guo, F. Yang, D. Xia, C. Li, Z. Tang and W. Li, J. Mater. Chem. C, 2020, 8, 2232–2237 RSC.
- B. Kim, B. Ma, V. R. Donuru, H. Liu and J. M. J. Fréchet, Chem. Commun., 2010, 46, 4148–4150 RSC.
- W. He, Y. Jiang and Y. Qin, Polym. Chem., 2014, 5, 1298–1304 RSC.
- D. Cortizo-Lacalle, C. T. Howells, S. Gambino, F. Vilela, Z. Vobecka, N. J. Findlay, A. R. Inigo, S. A. J. Thomson, P. J. Skabara and I. D. W. Samuel, J. Mater. Chem., 2012, 22, 14119–14126 RSC.
- B. M. Squeo, N. Gasparini, T. Ameri, A. Palma-Cando, S. Allard, V. G. Gregoriou, C. J. Brabec, U. Scherf and C. L. Chochos, J. Mater. Chem. A, 2015, 3, 16279–16286 RSC.
- G. Tarafdar, U. K. Pandey, S. Sengupta and P. C. Ramamurthy, Sol. Energy, 2019, 186, 215–224 CrossRef CAS.
- L. Bucher, N. Desbois, P. D. Harvey, C. P. Gros and G. D. Sharma, ACS Appl. Mater. Interfaces, 2018, 10, 992–1004 CrossRef CAS PubMed.
- L. Bucher, N. Desbois, P. D. Harvey, C. P. Gros, R. Misra and G. D. Sharma, ACS Appl. Energy Mater., 2018, 1, 3359–3368 CrossRef CAS.
- Y. Guo, D. Xia, B. Liu, H. Wu, C. Li, Z. Tang, C. Xiao and W. Li, Macromolecules, 2019, 52, 8367–8373 CrossRef CAS.
- Y. Wang, J. Miao, C. Dou, J. Liu and L. Wang, Polym. Chem., 2020, 11, 5750–5756 RSC.
- A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170–3173 CrossRef CAS PubMed.
- C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648–3652 CrossRef CAS PubMed.
- Z. Zhang, Z. Ding, C. Dou, J. Liu and L. Wang, Polym. Chem., 2015, 6, 8029–8035 RSC.
- R. Zhao, B. Lin, J. Feng, C. Dou, Z. Ding, W. Ma, J. Liu and L. Wang, Macromolecules, 2019, 52, 7081–7088 CrossRef CAS.
- R. Zhao, C. Dou, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 5313–5317 CrossRef CAS PubMed.
- N. Wang, S. Zhang, R. Zhao, J. Feng, Z. Ding, W. Ma, J. Hu and J. Liu, ACS Appl. Electron. Mater., 2020, 2, 2274–2281 CrossRef CAS.
- Y. Wang, N. Wang, Q. Yang, J. Zhang, J. Liu and L. Wang, J. Mater. Chem. A, 2021, 9, 21071–21077 RSC.
- R. Zhao, Y. Min, C. Dou, B. Lin, W. Ma, J. Liu and L. Wang, ACS Appl. Polym. Mater., 2020, 2, 19–25 CrossRef CAS.
- C. Dong, S. Deng, B. Meng, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 16184–16190 CrossRef CAS PubMed.
- R. Zhao, C. Dou, J. Liu and L. Wang, Chin. J. Polym. Sci., 2017, 35, 198–206 CrossRef CAS.
- R. Zhao, Y. Min, C. Dou, J. Liu and L. Wang, Chem. – Eur. J., 2017, 23, 9486–9490 CrossRef CAS PubMed.
- T. Wang, C. Dou, J. Liu and L. Wang, Chem. – Eur. J., 2018, 24, 13043–13048 CrossRef CAS PubMed.
- X. Long, J. Yao, F. Cheng, C. Dou and Y. Xia, Mater. Chem. Front., 2019, 3, 70–77 RSC.
- X. Long, D. Li, B. Wang, Z. Jiang, W. Xu, B. Wang, D. Yang and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 11369–11373 CrossRef CAS PubMed.
- X. Long, C. Dou, J. Liu and L. Wang, Chin. Chem. Lett., 2018, 29, 1343–1346 CrossRef CAS.
- X. Long, N. Wang, Z. Ding, C. Dou, J. Liu and L. Wang, J. Mater. Chem. C, 2016, 4, 9961–9967 RSC.
- Z. Ding, X. Long, C. Dou, J. Liu and L. Wang, Chem. Sci., 2016, 7, 6197–6202 RSC.
- X. Long, C. Dou, J. Liu and L. Wang, Macromolecules, 2017, 50, 8521–8528 CrossRef CAS.
- J. Miao, H. Li, T. Wang, Y. Han, J. Liu and L. Wang, J. Mater. Chem. A, 2020, 8, 20998–21006 RSC.
- Z. Ding, X. Long, B. Meng, K. Bai, C. Dou, J. Liu and L. Wang, Nano Energy, 2017, 32, 216–224 CrossRef CAS.
- X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504–6508 CrossRef CAS PubMed.
- R. Zhao, Z. Bi, C. Dou, W. Ma, Y. Han, J. Liu and L. Wang, Macromolecules, 2017, 50, 3171–3178 CrossRef CAS.
- R. Zhao, N. Wang, Y. Yu and J. Liu, Chem. Mater., 2020, 32, 1308–1314 CrossRef CAS.
- N. Wang, X. Long, Z. Ding, J. Feng, B. Lin, W. Ma, C. Dou, J. Liu and L. Wang, Macromolecules, 2019, 52, 2402–2410 CrossRef CAS.
- N. Wang, Y. Yu, R. Zhao, Z. Ding, J. Liu and L. Wang, Macromolecules, 2020, 53, 3325–3331 CrossRef CAS.
- Z. Ding, R. Zhao, Y. Yu and J. Liu, J. Mater. Chem. A, 2019, 7, 26533–26539 RSC.
- Z. Zhang, Z. Ding, D. J. Jones, W. W. H. Wong, B. Kan, Z. Bi, X. Wan, W. Ma, Y. Chen, X. Long, C. Dou, J. Liu and L. Wang, Sci. China: Chem., 2018, 61, 1025–1033 CrossRef CAS.
- Z. Zhang, Z. Ding, J. Miao, J. Xin, W. Ma, C. Dou, J. Liu and L. Wang, J. Mater. Chem. C, 2019, 7, 10521–10529 RSC.
- Z. Zhang, Z. Ding, X. Long, C. Dou, J. Liu and L. Wang, J. Mater. Chem. C, 2017, 5, 6812–6819 RSC.
- Z. Zhang, J. Miao, Z. Ding, B. Kan, B. Lin, X. Wan, W. Ma, Y. Chen, X. Long, C. Dou, J. Zhang, J. Liu and L. Wang, Nat. Commun., 2019, 10, 3271 CrossRef PubMed.
- Z. Zhang, T. Wang, Z. Ding, J. Miao, J. Wang, C. Dou, B. Meng, J. Liu and L. Wang, Macromolecules, 2019, 52, 8682–8689 CrossRef CAS.
- J. Miao, B. Meng, Z. Ding, J. Liu and L. Wang, J. Mater. Chem. A, 2020, 8, 10983–10988 RSC.
- C. Zhu, Z.-H. Guo, A. U. Mu, Y. Liu, S. E. Wheeler and L. Fang, J. Org. Chem., 2016, 81, 4347–4352 CrossRef CAS PubMed.
- Y. Li, B. Pang, H. Meng, Y. Xiang, Y. Li and J. Huang, Tetrahedron Lett., 2019, 60, 151286 CrossRef CAS.
- Y. Li, H. Meng, T. Liu, Y. Xiao, Z. Tang, B. Pang, Y. Li, Y. Xiang, G. Zhang, X. Lu, G. Yu, H. Yan, C. Zhan, J. Huang and J. Yao, Adv. Mater., 2019, 31, 1904585 CrossRef CAS PubMed.
- T. Liu, T. Yang, R. Ma, L. Zhan, Z. Luo, G. Zhang, Y. Li, K. Gao, Y. Xiao, J. Yu, X. Zou, H. Sun, M. Zhang, T. A. Dela Peña, Z. Xing, H. Liu, X. Li, G. Li, J. Huang, C. Duan, K. S. Wong, X. Lu, X. Guo, F. Gao, H. Chen, F. Huang, Y. Li, Y. Li, Y. Cao, B. Tang and H. Yan, Joule, 2021, 5, 914–930 CrossRef CAS.
- H. Meng, Y. Li, B. Pang, Y. Li, Y. Xiang, L. Guo, X. Li, C. Zhan and J. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 2733–2742 CrossRef CAS PubMed.
- Y. Xiang, H. Meng, Q. Yao, Y. Chang, H. Yu, L. Guo, Q. Xue, C. Zhan, J. Huang and G. Chen, Macromolecules, 2020, 53, 9529–9538 CrossRef CAS.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- R. G. Brandt, S. G. Sveegaard, M. Xiao, W. Yue, Z. Du, M. Qiu, D. Angmo, T. R. Andersen, F. C. Krebs, R. Yang and D. Yu, J. Mater. Chem. C, 2016, 4, 4393–4401 RSC.
- S. Pang, Z. Wang, X. Yuan, L. Pan, W. Deng, H. Tang, H. Wu, S. Chen, C. Duan, F. Huang and Y. Cao, Angew. Chem., Int. Ed., 2021, 60, 8813–8817 CrossRef CAS PubMed.
- B. Meng, Y. Ren, J. Liu, F. Jäkle and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 2183–2187 CrossRef CAS PubMed.
- M. Grandl, J. Schepper, S. Maity, A. Peukert, E. von Hauff and F. Pammer, Macromolecules, 2019, 52, 1013–1024 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |