
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst overcoating engineering towards high-performance electrocatalysis
Qiang
Liu
 ab,
Marco
Ranocchiari
b and
Jeroen A.
van Bokhoven
*ab
ab,
Marco
Ranocchiari
b and
Jeroen A.
van Bokhoven
*ab
aInstitute for Chemical and Bioengineering, ETH Zurich, Vladimir Prelog Weg 1, 8093 Zurich, Switzerland. E-mail: jeroen.vanbokhoven@chem.ethz.ch
bLaboratory for Catalysis and Sustainable Chemistry, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland
First published on 6th December 2021
Abstract
Clean and sustainable energy needs the development of advanced heterogeneous catalysts as they are of vital importance for electrochemical transformation reactions in renewable energy conversion and storage devices. Advances in nanoscience and material chemistry have afforded great opportunities for the design and optimization of nanostructured electrocatalysts with high efficiency and practical durability. In this review article, we specifically emphasize the synthetic methodologies for the versatile surface overcoating engineering reported to date for optimal electrocatalysts. We discuss the recent progress in the development of surface overcoating-derived electrocatalysts potentially applied in polymer electrolyte fuel cells and water electrolyzers by correlating catalyst intrinsic structures with electrocatalytic properties. Finally, we present the opportunities and perspectives of surface overcoating engineering for the design of advanced (electro)catalysts and their deep exploitation in a broad scope of applications.
 Qiang Liu | Qiang Liu received a BS (2013) in Chemical Engineering and Technology from Shandong University and a PhD degree (2018) in Chemical Engineering from Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, under the supervision of Prof. Xindong Mu. He then moved to Prof. Johannes A. Lercher's group at the Technical University of Munich (TUM) to learn zeolite catalysis from 2018 to 2020. He is currently a Postdoc at the ETH Zurich under the supervision of Prof. Jeroen A. van Bokhoven. His research interest focuses on the design and synthesis of advanced nanocatalysts for more efficient processes related to clean and sustainable energy. |
 Marco Ranocchiari | Marco Ranocchiari received his master's degree in chemistry in 2005 from the Università degli Studi di Milano in Italy. He completed his PhD at the ETH Zurich in 2009. In 2010, he joined the Paul Scherrer Institute in the Laboratory for Catalysis and Sustainable Chemistry and has been a group leader since 2016. His research focuses on energy-related catalysis and metal–organic frameworks for catalytic applications. He is co-founder of the spin-off novoMOF AG (https://novomof.com), a custom MOF manufacturing company. |
 Jeroen A. van Bokhoven | Jeroen A. van Bokhoven completed a degree in chemistry at Utrecht University (Netherlands) in 1995 and went on to obtain a PhD in inorganic chemistry and catalysis from the same university in 2000 (with honours). From 1999 until 2002, he was the head of the XAS (X-ray absorption spectroscopy) users-support group at Utrecht University. In 2002, he moved to the ETH Zurich, where he worked as a postdoctoral researcher in the group of Professor Prins. In 2006 he obtained an SNF assistant professorship in the Department of Chemistry and Applied Biology. He is the 2008 recipient of the Swiss Chemical Society Werner Prize. Since 2010, Jeroen A. van Bokhoven has been a Chair in Heterogeneous Catalysis at the Institute for Chemical and Bioengineering at ETH Zurich and the Head of Laboratory for Catalysis and Sustainable Chemistry at Paul Scherrer Institute. |
1. Introduction
Heterogeneous catalysts, comprising metal nanoparticles (NPs) and solid supports, are widely applied in thermal- and electro-catalysis fields for clean and sustainable energy. Porous carbon, graphene and metal oxides with high specific surface area are the commonly exploited support materials for dispersing metal NPs.1–3 Supported metal based catalysts with diverse particle size, composition, morphology and metal–support interactions can be generated to show distinct catalytic properties.4–6 However, the encountered unstable performance and easy deactivation as a consequence of metal particle agglomeration, coalescence and Ostwald ripening are the remaining obstacles that impair their wide applications.7,8 The issue concerning catalyst deactivation can become more severe under high temperature and/or harsh reaction conditions, i.e. in an oxidizing and corrosive environment, which calls for innovative design and optimization strategies to fulfil the requirement of boosting the catalytic properties of supported metal catalysts.Electrochemical transformation reactions in polymer electrolyte fuel cells (PEFCs) and water electrolyzers (WECs) with high efficiency and long-term durability have remained the hotspot for renewable energy production, conversion and storage.9–11 In PEFCs, the oxygen (O2) reduction reaction (ORR) proceeds via either the direct 4e− pathway to produce H2O or the 2e− route by forming hydrogen peroxide (H2O2) on the cathode. Oxidation of a fuel molecule, i.e. the hydrogen (H2) oxidation reaction (HOR), proceeds at the anode side of fuel cells. On the other hand, WECs that transform electricity into renewable energy involve the H2 and O2 evolution half-reactions (HER and OER), and are operated on the principle opposite to that of PEFCs. At present, platinum (Pt)-group metals (PGMs) are the most active state-of-the-art electrocatalysts, even though 3d transition metal-based nanomaterials are promising alternatives to PGM counterparts. In addition to the high price and scarcity of PGM catalysts, the intensely sluggish kinetics of ORR and OER, along with their intricate reaction pathways, impede the widespread and commercialization of electrochemical devices.12,13 On the other hand, taking the hydrogen-driven PEFCs as an example, the HOR activity is orders of magnitude higher in an acid electrolyte than that in a basic solution (pH-dependent activity),14 which allows an ultra-low loading (≤0.05 mgPt cm−2) of Pt on the anodic catalyst when contacting with acid media. However, the instability and degradation of Pt, which show dependencies on catalyst characteristics (such as particle size, crystal structure, loading) and operating conditions (electrolyte, limiting potential, temperature and so on), still constitute important performance-limiting factors.15,16 In the process of electrocatalyst design, the implicated trade-off relationship between activity enhancement and intrinsic instability is required to be overcome before the practical application of electrode catalysts.
Reducing the usage of PGMs and developing electrochemically highly active and durable materials are the main subjects of intense research on the wide implementation of energy conversion and storage devices.17–19 Of the numerous efforts, alloying Pt with a second transition metal (i.e. Fe, Co, Ni, Cu, Zn) and the fabrication of core–shell nanostructures (where Pt is conformally deposited as an ultra-thin shell on a more abundant metal core) have been proven beneficial for improving the electrochemical performance.20–25 The alterations of the electronic surface structure of the metal by foreign transition metal atoms and the lattice mismatch-derived strains at the interface of metal nanodomains yield the improvements.26,27 Nevertheless, synthetically controllable design of catalysts with high specific surface area and maintaining the nanostructure of metal particles are not easy because of sintering and dealloying issues in the process of thermal treatment and catalytic reactions. The high dissolution tendency of alloyed transition metals or non-PGM cores often calls for elaborate selection of transition metals and precise modulation of Pt “skin”/skeletal as well as morphological integrity and electronic structure.12,28–30 In terms of developing nanomaterials with enhanced catalytic properties, deploying organic/inorganic barriers or overlayers recently appears to be one of the favourable countermeasures, which promises to reconcile high activity with improved stability of metal entities.31,32 Polymeric and metal oxide overlayers are the typical coatings that can protect the carbon material (the commonly used catalyst support) from electrochemical oxidation and corrosion, ultimately making the as-synthesized electrocatalysts durable. Moreover, the multiple functions of surface coatings open a plethora of opportunities for enhancing the electrocatalytic efficiencies of nanocatalysts through the as-generated interfacial effects.33 Thus far, some excellent reviews have discussed the recent development in the design of high-performance metal-based catalysts by using surface engineering strategies and their catalysis related applications.34–37 For instance, Lenne et al.34 reviewed the surface functionalization of metal catalysts with organic ligands and the consequent influence on their surface properties and catalytic activities for electro-oxidation and reduction reactions. Although another review from the research group of Yin mentioned the synthesis of encapsulated metal catalysts, it mainly reported the works related to the field of thermo-catalysis.35 In addition, Pt-based materials designed by means of surface-confinement36 and organic–inorganic hybrid concepts37 for application covering only ORR catalysis were also reviewed. Benefiting from the ongoing innovative development of advanced synthetic nanotechnology and material chemistry, surface overcoating engineering has emerged as a powerful strategy for the development of optimal metal-based catalysts to unlock their application potential in various electrocatalytic processes. Therefore, it is increasingly crucial to offer a critical overview of the potential synthesis methods for surface overcoating of advanced precious and non-precious metal based electrocatalysts for application in diverse important electrocatalytic reactions in PEFCs and WECs.
In the present review, we showcase a comprehensive summary of the versatile surface overcoating strategy for electrocatalyst design with a special focus on their applications in the research of PEFCs and WECs. Specifically, the fabrication of the surface of metal nanoparticles and the modification of support materials (such as carbon and metal oxides) based on a vast variety of overcoating methodologies are surveyed. High-temperature solid oxide fuel cells will not be covered in this review, although there are a number of works on capping Pt electrodes with a metal oxide overlayer for stabilizing the catalyst surface and improving their catalytic performance.38–40 Next, we discuss the progress of surface overcoating-derived efficient electrocatalysts for PEFCs that involve electrochemical reactions such as ORR, HOR, and MOR/EOR (in direct alcohol (i.e. methanol, ethanol) fuel cells, DAFCs) and WECs comprising OER, HER and overall water splitting. The consequent benefits from surface overcoating engineering are considered and the electrocatalysts surveyed include both precious and non-precious metal based electrocatalysts. Finally, we describe the future research opportunities and perspectives associated with the design and deep exploitation of emerging heterogeneous catalysts fabricated by surface overcoating engineering.
2. Surface overcoating methodologies for the nanostructuring of metal based electrocatalysts
In this section, surface overcoating methodologies for the nanostructuring of high-performance electrocatalysts employed in the research of PEFCs and WECs are overviewed. As depicted in Fig. 1, the nanofabrication of metal NPs as well as the modification of catalyst supports (such as carbon materials and metal oxides) through surface overcoating engineering by the utility of different foreign coatings, including organic capping agents/polymers, inorganic carbon nanoshells and metal oxides, is summarized and discussed in the following.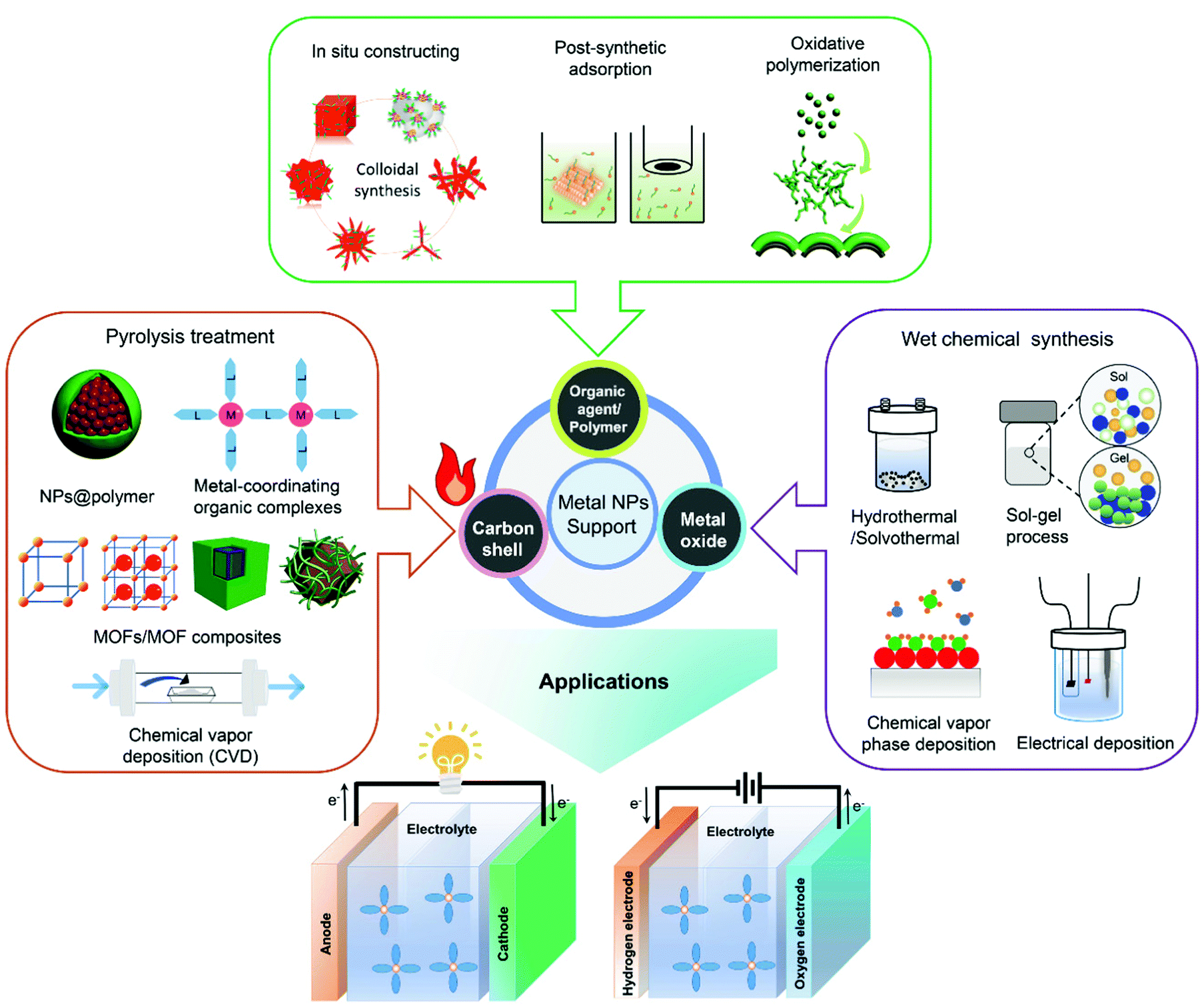 | ||
| Fig. 1 Overview of a variety of overcoating methodologies utilizing organic agents/polymers, inorganic carbon nanoshells and metal oxides as overcoating materials for high-performance electrocatalysis. | ||
2.1 Overcoating of metal nanoparticles with organic capping agents and polymers
Organic capping agents and polymers on metal surfaces are difficult to stabilize and are prone to decompose at high temperature (i.e. >300 °C) under thermal catalysis conditions.41 By contrast, surface modification of metal particles with organic species without the need of chemical- or thermal-removal has proven effective at enhancing electrocatalytic performance.34,36,37,42,43 Creating metal–organic structures is effective in modulating the electronic structure of the metal and alleviating the surface site-access blockage due to the adsorption of poisonous spectator species (i.e. Cl−, Br−, OH−, PO43−, SO42−) and crossover-obtained reactants (i.e. methanol, ethanol).34,37 This section will first start by presenting the synthetic approaches for overcoating metal NPs with organic capping agents and polymers.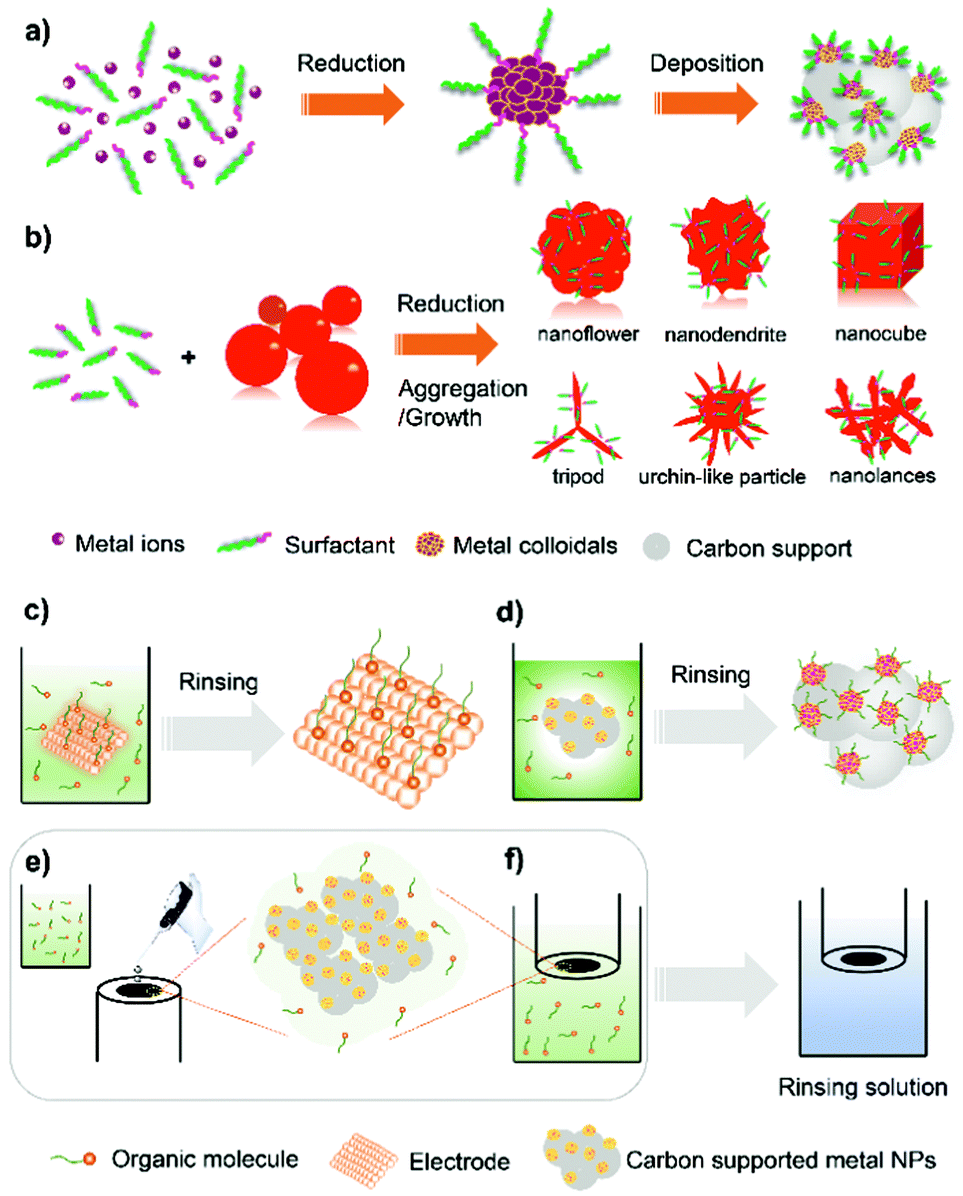 | ||
| Fig. 2 (a) Synthetic procedures for the preparation of carbon-supported metal NPs patterned with organic surfactants via the colloidal reduction method. (b) The synthesis of Pt-based nanostructures with different compositions, shapes and morphologies through chemical reduction routes. (c–f) Schematic illustration of the procedures for the post-synthetic construction of organic architectures on the surface of electrocatalysts. | ||
Surfactants are also useful for engineering metal nanostructures (NSs). It is possible to realize enhanced electrocatalytic properties by utilizing the synergistic effects between the unique metal NSs and the electronic modification by surfactants on the surface.51–57 Recently, intensive works by the research group of Chen and Lee have pointed to the development of various Pt-based NSs through chemical reduction routes using polyallylamine hydrochloride (PAH) as the metal particle stabilizing and shape/morphology controlling agent (Fig. 2b). A number of polyallylamine (PAA)-functionalized Pt-based NSs, including Pt nanocubes,52 Pt–Pd alloy nanoflowers,53 highly branched Pt nanolance assemblies,54 Pd–Pt bimetallic core–shell nanodendrites,55 long-spined sea-urchin-like Pt nanostructures (Pt-LSSUs),56 and Pt tripods,57 have been reported for efficient electrocatalysis. Solution pH (∼0.5–9), temperature, and the amount and type of reduction agents (such as formaldehyde, hydrazine hydrate) are critical parameters for control over the shapes and morphologies of metal NSs. For instance, sea urchin-like Pt-LSSUs characterized by a three-dimensional (3D) interconnected structure and a high branching degree were formed by using hydrazine hydrate (N2H4·H2O).56 In an alkaline aqueous solution, K2PtCl4 as a Pt precursor was mixed with PAA by heating at 80 °C. The formation of Pt-LSSUs was started after adding the N2H4·H2O reductant. As the –NH2 groups of PAA could selectively cap Pt{100} facets, the fast reduction kinetics favours the selective deposition of Pt atoms in the 〈111〉 direction with high growth rates, leading to the formation of Pt branches dominated by {111} facets and surface-functionalized with PAA molecules. The adsorption of PAH on metal NSs through metal–N chemical bonding can be verified by performing catalyst characterization techniques such as energy-dispersive X-ray mapping (EDX-mapping) and X-ray photoelectron spectroscopy (XPS). In most cases, PAH molecules are found loosely packed on the surface of Pt NSs, making it possible for the permeation of the electrolyte without disturbing electrocatalytic processes.
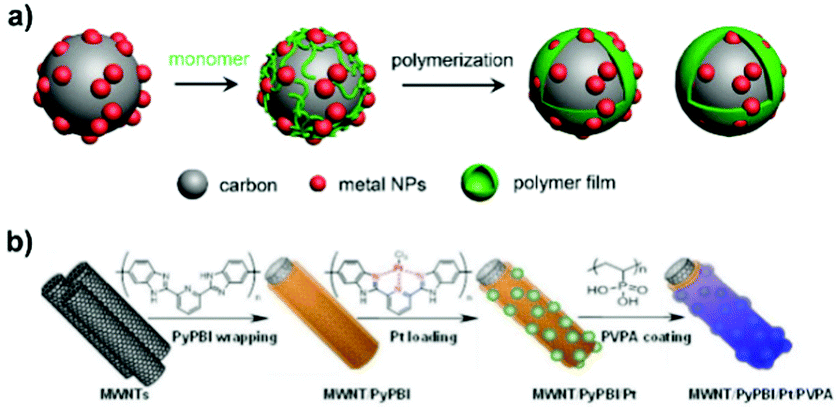 | ||
| Fig. 3 Schematic illustration of the synthesis of polymer-overcoated electrocatalysts. (a) M/C@polymer (M: metal) synthesized by the oxidative polymerization of organic monomers. (b) Polymer double-shelled Pt-based material prepared through sequential overcoating with PyPBI and PVPA. Reproduced from ref. 75 with permission from Springer Nature. Copyright 2015. | ||
Polydopamine (PDA) coating also manifests wide applications because of its robust structure and strong adhesive strength to the surface.69–71 One commonly applied synthetic procedure for PDA overcoating is the oxidative polymerization of dopamine in Tris–HCl buffer solution (Trizma base + hydrochloric acid, pH 8.5) dispersed with the solid catalyst at room temperature, followed by sequential separation, rinsing and drying steps. Controlling the polymerization time allows for tuning the thickness of the PDA overlayer. PDA overcoating is more controllable due to the mild polymerization condition as compared to PANI and Ppy coating.69
From the research works of Yang et al. and Nakashima et al., the wrapped pyridine-containing polybenzimidazole (poly[2,2′-(2,6-pyridine)-5,5′-bibenzimidazole], PyPBI) on a carbon support serves as the glue for highly dispersed Pt NPs.72,73 When multiwalled carbon nanotube-supported Pt NPs are overcoated with a PyPBI layer, their electrocatalytic oxidation performance is favoured.74 Based on these advances, they demonstrated a distinct type of polymer double-shelled electrocatalyst, of which the metal particles and carbon support are separately overcoated with polymer layers.75 As illustrated in Fig. 3b, the steps in the fabrication of a polymer double-shelled electrocatalyst include (i) mixing the carbon material with N,N-dimethylacetamide solution containing PyPBI, (ii) loading Pt NPs onto the PyPBI-wrapped carbon support by a conventional polyol reduction method, and (iii) mixing the above Pt-loaded material with poly(vinylphosphonic acid) (PVPA) aqueous solution. Each step involves the additional separation/rinsing and drying procedure. This strategy is very flexible and can be used to design a large variety of electrocatalysts by elaborate selection of carbon support materials and polymer coatings.76–79 The reinforcement of electrocatalytic performances versus the uncoated catalyst makes it effective in fabricating metal-based nanomaterials with extra modification of polymers.
2.2 Overcoating of metal nanoparticles with inorganic carbon nanoshells
The inorganic carbon nanoshell doped with heteroatoms (such as N, P, S, B, etc.) is another widely exploited overcoating material as in most cases it is capable of improving catalyst's electrical conductivity and providing additional active surface sites for electrocatalysis. In this section, we will discuss a wide assortment of synthetic protocols being developed for overcoating carbon nanoshells on the surface of metal NPs as the emerging efficient electrocatalysts.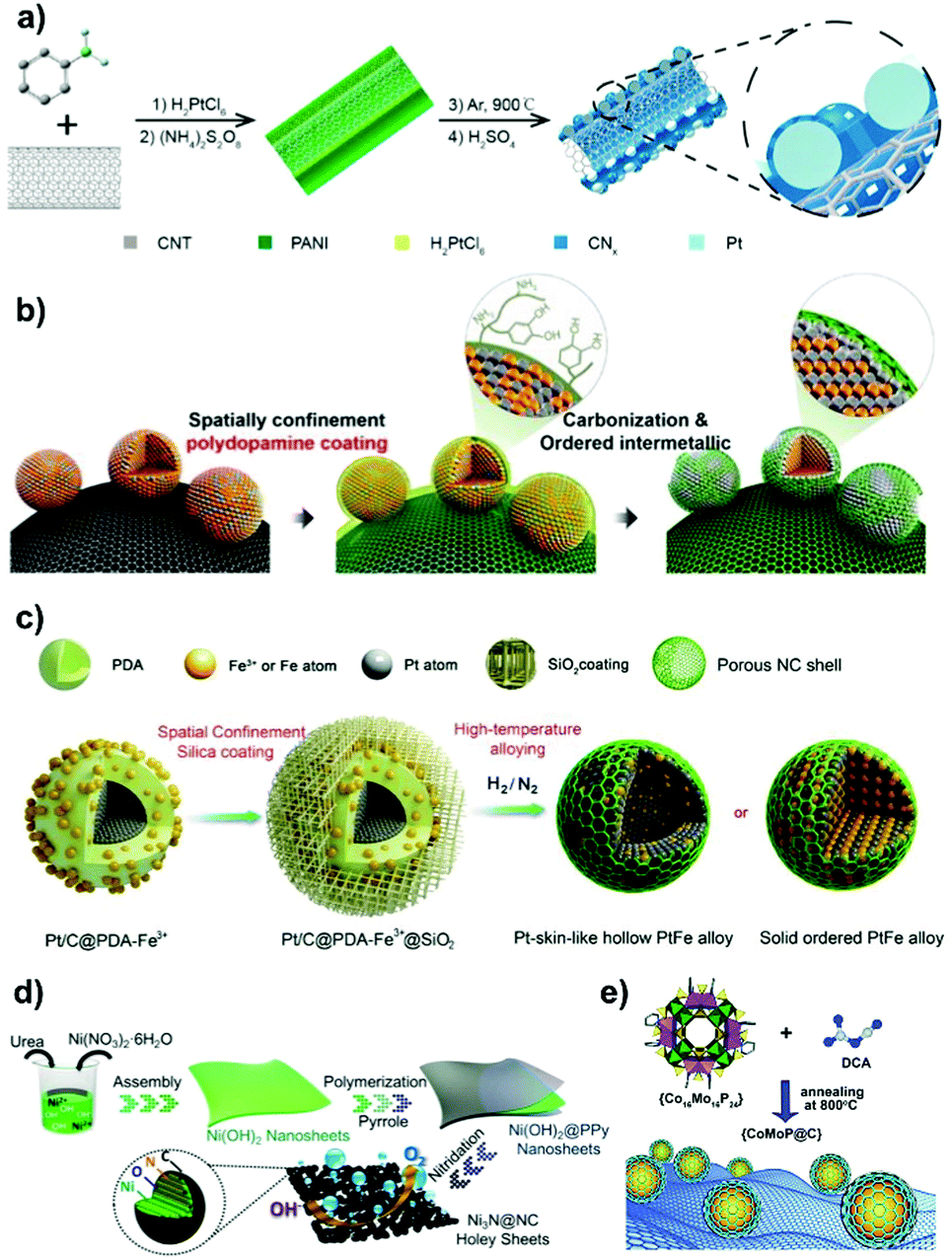 | ||
| Fig. 4 Carbon nanoshell-overcoated electrocatalysts. (a) Pt@CNx/CNT was partially covered with a PANI-derived N-doped carbon shell through the one-pot process. Reproduced from ref. 83 with permission from American Chemical Society, Copyright 2015. (b) Ordered face-centered tetragonal PtFe NPs were obtained by PDA overcoating and carbonization. Reproduced from ref. 89 with permission from American Chemical Society, Copyright 2015. (c) Synthesis of PtFe alloy NPs with an ordered or hollow nanostructure through PDA overcoating and the spatial confinement effect by SiO2 outer coating. Reproduced from ref. 92 with permission from Royal Society of Chemistry, Copyright 2019. (d) Synthesis of N-doped carbon overcoated Ni3N holey sheets through the polymerization of pyrrole on Ni(OH)2 nanosheets and the followed high temperature carbonization–nitridation step. Reproduced from ref. 100 with permission from American Chemical Society, Copyright 2018. (e) Direct pyrolysis of the mixture of dicyandiamide and P-rich polyoxometalate to obtain CoMoP@C. Reproduced from ref. 110 with permission from Royal Society of Chemistry, Copyright 2017. | ||
In terms of overcoating with carbon nanoshells, the optimization of the pyrolysis temperature and carbon nanoshell thickness with the aim of improving electrocatalytic properties is in high demand. A comparison with pyrolysis at lower temperatures, i.e. 600 and 700 °C, indicates that pyrolysis at higher temperature (900 °C) is a good practice to generate carbon nanoshells with a higher degree of graphitization; consequently, the corrosion-resistant carbon nanoshell confers high durability on the resultant catalyst.84 However, care should be taken on the issues related to metal nanoparticle aggregation/sintering and the loss of active surface area at even higher pyrolysis temperatures.
A carbon nanoshell with thickness that is either too thin or too thick will either weaken the protection effect on metal NPs, or kinetically influence reactant transfer from the outer phase into the inner active sites.85 Tong et al.86 precisely tuned the carbon overlayer thickness by controlling the growth of the glucose-containing polymer on Pt/CNTs. It is worth noting that the electrochemical surface area (ECSA) is the accessible area of electrode catalysts to electrolyte components and reactants for catalyzing electrocatalytic reactions. In this work, a thick carbon nanoshell (2 nm) induces 5 times lower ECSA of Pt/CNTs@C compared to uncoated Pt/CNTs. In contrast, the diffusion of oxygen is facilitated on the catalyst overcoated with a 0.8 nm-thin carbon nanoshell for the process of ORR catalysis.
Fabricating ordered Pt-enriched nanoalloys or core–shell nanostructures could fulfill the on-demand requirements of reducing precious Pt loading and improving electrochemical efficiencies.87,88 However, high-temperature annealing treatment on Pt-based nanoalloys usually induces the formation of larger metal NPs with broad size distributions. It results in extremely deteriorated ECSA and inefficient utilization of active surface sites. Impressively, the in situ overcoating of the carbon nanoshell onto metal nanoparticles is able to minimize the aggregation and sintering of metal NPs during annealing treatment. For example, the in situ carbonization of the PDA overlayer on carbon-supported PtFe NPs resulted in a metal particle size close to that before annealing treatment (Fig. 4b).89 By contrast, the catalyst without dopamine coating underwent obvious particle coalescence, forming particles up to tens of nanometers in size. Based on the protective aid of PDA, Pt-rich PtNi alloy particles without a carbon nanoshell were synthesized by pyrolyzing the Ni2+-impregnated Pt/C–PDA composite under a H2-containing atmosphere.90 In addition to improvement in the resistance of metal NPs to sintering, it was shown in this work that PDA also behaved as a sponge to adsorb Ni ions for a precise control over the Pt–Ni ratio in the resulting Pt-alloyed electrocatalyst.
A space-confined pyrolysis strategy was recently developed by Wei's group91,92 to prepare carbon nanoshell-coated PtFe NPs with ordered or Pt-skin-like hollow structures (Fig. 4c). In catalyst synthesis, commercial Pt/C (20 wt% loading of Pt with a diameter of 2–4 nm) was initially overcoated with an oxidatively polymerized PDA overlayer based on the procedure described in Section 2.1.3. After dispersing Pt/C@PDA in an aqueous solution containing Fe3+, the outer-coating of the inorganic SiO2 shell on the catalyst was proceeded by the hydrolysis and polycondensation of tetraethoxysilane (TEOS) catalyzed by HCl in a water/ethanol mixture, followed by the slow evaporation of solvents. Upon annealing treatment, PDA overlayers get transformed into a 2 nm-thin N-doped carbon shell, and the outer silica shell provides a spatially confined space to avoid metal NP aggregation/sintering, while the first formed PtFe alloy intermediates undergo subsequent phase transition, which can be controlled by modulating the content of Fe3+ and annealing temperature during the synthesis process.92
In addition to polymers, heteroatom-containing organic compounds such as thiourea and cyanamide have also become the precursor of choice to yield the heteroatom-doped carbon nanoshell on supported metal nanoparticles. For example, Chen et al.93 prepared a Fe@S,N-DC catalyst via two major steps: (i) pyrolysis of activated carbon in the presence of thiourea (CH4N2S) to obtain the S,N-codoped carbon support (S,N-DC) and (ii) embedding Fe NPs into the carbon matrix by microwave radiation treatment of Fe(OAc)2OH and hexamethylenetetramine, which serve as Fe and N,C sources, respectively. Jin et al.94 synthesized a graphene-supported g-C3N4@CoO core–shell catalyst, consisting of a CoO core and a Co-doped g-C3N4 (graphitic carbon nitride) outer nanoshell, through pyrolysis of the mixture of cyanamide-Co complex and graphene oxide support at 550 °C.
Similar to polymers, organic compounds especially those with heteroatom-containing groups such as –NH2 and –NH– can coordinate with metal ions to form metal-coordinating organic complexes, making them promising precursors for the carbon nanoshell and the support of metal NPs.104–108 For example, a one-step high pressure pyrolysis of ferrocene and cyanamide at 700 °C was reported by Hu et al.104 to prepare Fe3C/C hollow spheres overcoated with 4–9 layers of graphitic carbon. A negligible amount of nitrogen and Fe metal was observed on the surface of Fe3C/C, indicating that it was another mechanism rather than that catalyzed by Fe–Nx moieties that accounted for the high ORR activity. The removal of the carbon nanoshell induced dissolution of Fe3C into the electrolyte accompanied by an obvious ORR activity loss. The authors therefore claimed that Fe3C sites did not have direct contact with the electrolyte but instead made the outer carbon nanoshell active for the ORR catalysis. Analogously, trimetallic FeCoNi alloys overcoated with N-doped carbon nanotubes were generated following the protocol of direct pyrolysis of dicyandiamide–metal nitrate complexes at 800 °C in an inert atmosphere.106
Such a versatile strategy has made it possible to elaborately select platform compounds such as the phosphorus (P) moiety-coordinated counterparts to produce metal phosphides with surface overcoating of carbon nanoshells.108–111 For example, Pu et al.108 prepared a N,P-codoped carbon-encapsulated ruthenium diphosphide catalyst (RuP2@NPC) by pyrolysis of phytic acid (PA) cross-linked Ru complexes and melamine at 900 °C. In the work of Lv et al.,109 cobalt phosphonate hybrid precursors were first fabricated by hydrothermally treating the mixture of cobalt chloride and sodium salt of ethylene diamine tetra(methylene phosphonic acid) (EDTMPS) at 140 °C. Subsequently, the phase-controlled CoxP@NC was obtained by H2 reduction of as-made precursors with different Co/P molar ratios at 700 °C. In addition, Ma et al.110 demonstrated that a carbon nanoshell-coated CoMoP@C catalyst was generated through pyrolysis of the mixture of P-rich polyoxometalate (Co16Mo16P24) and dicyandiamide (DCA) at 800 °C in a N2 atmosphere (Fig. 4e). DCA not only serves as a reducing agent to catalyze the deoxygenation of Co16Mo16P24, but also protects the CoMoP particles from aggregation during the process of carbonization and in situ phosphidation. In the engineered CoMoP@C, the single crystalline CoMoP core with the overcoated 2–4 graphitic carbon layer is favourable for electrocatalytic reactions. We note that, it is also applicable to generate metal phosphide catalysts by carrying out post-phosphorization treatment on as-made metal@C materials using e.g. NaH2PO2 as the P source at moderate-to-high temperature.112
Direct pyrolysis of the MOF nanostructure can feasibly aid in isolating metal sites within the MOF-derived carbon matrix with a large surface area.116–120 Metal nodes in MOFs could serve as the source of metal NPs, while the organic linkers transform into a porous carbon matrix and affect the composition of encapsulated metal entities after high-temperature pyrolysis. Shi et al.118 developed a MOF-directed strategy to synthesize a graphitic carbon nanoshell-coated MoC electrocatalyst (named nanoMoC@GS) with high porosity. As illustrated in Fig. 5a, Mo(CO)6 is first mixed with trimesic acid (H3BTC) in an oxygen-free dimethylformamide (DMF) solvent and then solvothermally treated under argon to form Mo3(BTC)2. Subsequently, carbonization of Mo3(BTC)2 at 700–900 °C under an inert atmosphere yields nanoMoC@GS. Metal carbides are formed by the diffusion of carbon atoms into the interstices of metal atoms, which atomically contact with the organic linkers in MOFs.118,120 This atomic interaction can effectively make metal NPs highly resistive towards aggregation during the carbonization process.
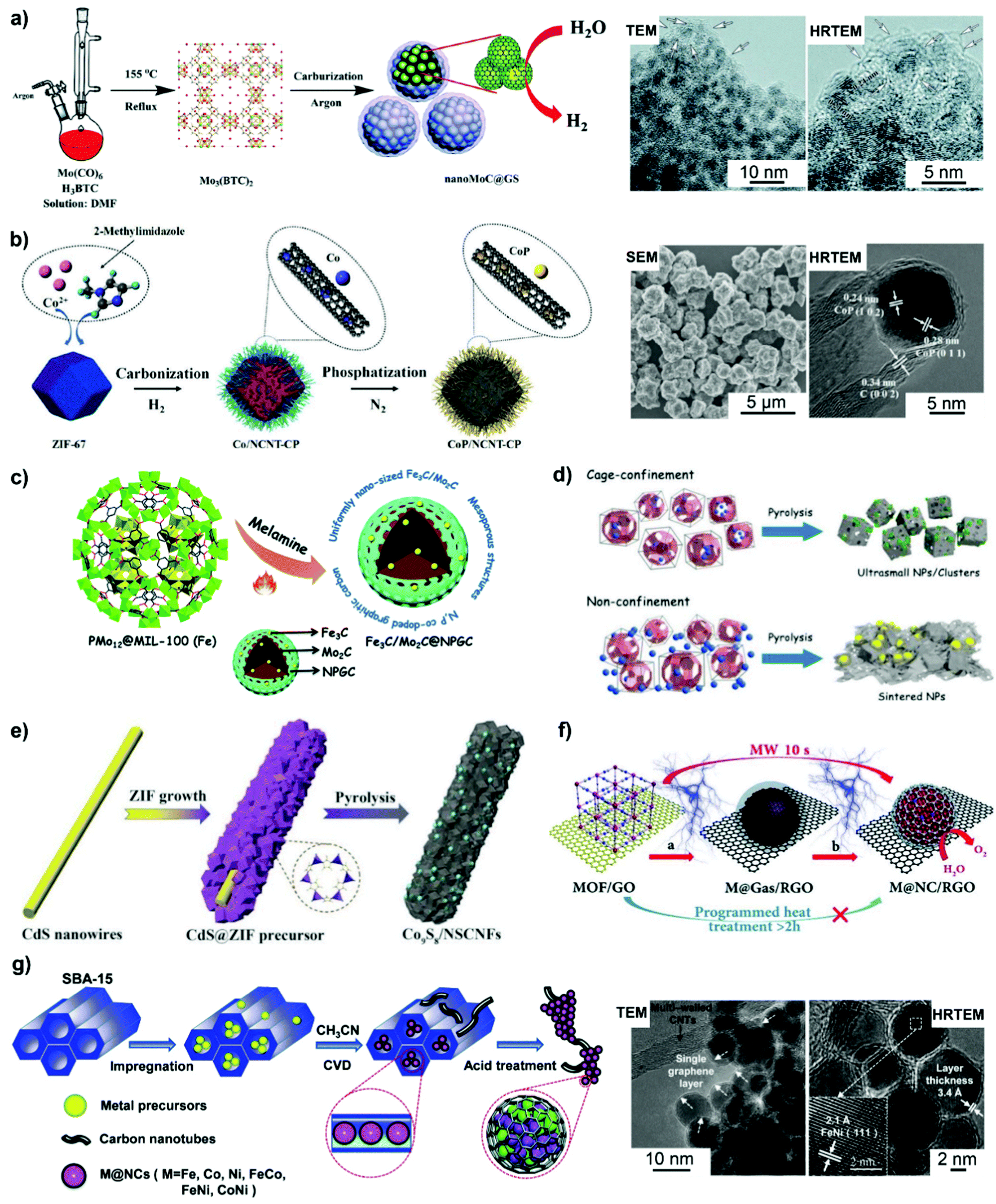 | ||
| Fig. 5 Carbon nanoshell-overcoated electrocatalysts obtained from MOFs or MOF nanocomposites. (a) Mo3(BTC)3 MOF synthesis and its carbonization to nanoMoC@GS under 700 °C. Reproduced from ref. 118 with permission from Royal Society of Chemistry, Copyright 2016. (b) CoP/NCNT-CP was prepared by the carbonization of ZIF-67 under H2 and subsequent phosphatization treatment in N2. Reproduced from ref. 119 with permission from American Chemical Society, Copyright 2019. (c) N,P-Codoped graphitic carbon-embedded Fe3C/Mo2C was obtained through pyrolysis of PMo12@MIL-100(Fe). Reproduced from ref. 127 with permission from Royal Society of Chemistry, Copyright 2016. (d) Ultra-small WC NPs were produced on the basis of cage-confinement of MOFs. Reproduced from ref. 129 with permission from American Chemical Society, Copyright 2017. (e) A synthetic procedure for N,S-codoped carbon fiber embedded Co9S8. Reproduced from ref. 132 with permission from Wiley-VCH, Copyright 2018. (f) Ultrafast microwave-assisted CVD-like synthesis of M@NC/RGO. Reproduced from ref. 135 with permission from Royal Society of Chemistry, Copyright 2018. Chemical vapor phase deposition (CVD) also favours the generation of carbon nanoshell-overcoated electrocatalysts. (g) Graphene layer-overcoated transition metal or metal alloy NPs obtained by CVD technology. Reproduced from ref. 136 with permission from Royal Society of Chemistry, Copyright 2016. | ||
In addition, the transition metal moieties such as Co and Fe in MOFs could catalyze the formation of carbon nanotubes (CNTs) under pyrolysis conditions, helping in obtaining CNT-encapsulated metal based catalysts.119,120 For example, Co-containing ZIF-67 polyhedra (ZIF: zeolitic imidazolate framework) were pyrolyzed at 900 °C in a H2 atmosphere, along with low-temperature phosphatization treatment to produce N-doped carbon nanotube (NCNT)-embedded CoP NPs dispersed on carbon polyhedra (500 nm of average diameter) (Fig. 5b).119 In the process of carbonization, Co ions get reduced into metallic Co, which could promote the formation and growth of NCNTs. Subsequently, on phosphatization treatment, metallic Co species react with the PH3 species generated from the decomposition of NaH2PO2, leading to the formation of CoP NPs encased in NCNTs, which is underpinned by high resolution TEM (HRTEM) analysis.
Prussian blue analogues (PBAs) with the chemical formula K3[M(CN)6] or MII3[MIII(CN)6]2 (where M represents the transition metals), as a subclass of MOFs, have been investigated to fabricate carbon nanoshell-overcoated electrocatalysts with promising catalytic properties.121–124 One can additionally supplement active sites by introducing metal precursors during the synthesis of PBAs to design well-alloyed or core–shell nanostructures. In an exemplary work reported by Jiang et al.,124 an Ir-doped Prussian-blue analogue (Co3[Co(CN)6]2) was first prepared by mixing the H2IrCl6 precursor with Co(CH3COO)2, K3[Co(CN)6]2 and PVP in an aqueous solution, followed by hydrothermal treatment at 100 °C for 5 h. On direct pyrolysis, IrCo@NC was generated composed of N-doped graphene shell-overcoated IrCo alloy NPs. The CN− groups from the PBA served as the source of carbon and nitrogen during the annealing treatment. As a consequence of the phase transformation of Co moieties at elevated temperatures, regulation of the structure phase of IrCo NPs can be achieved. In this work, the lower temperature-pyrolyzed catalyst, i.e. IrCo@NC-500, was characterized to possess a hexagonal IrCo phase with surface covering of the N-enriched graphene overlayer. The graphene overlayer was able to transfer the adjacent carbon atoms into catalytic active sites and form strong interactions with the encapsulated metal NPs. Similarly, binary transition metal-based FeCo@C125 and NiFe@C126 have been generated following the protocol of annealing a wide assortment of PBA-based nanomaterials.
The unique pore structures and interconnected channels of MOFs render them as promising hosts for introducing catalytic metal moieties. An example of this is the direct pyrolysis of fabricated polyoxometalate (POM)-incorporated MOF materials, which could make the introduction of transition metals into metal carbide electrocatalysts feasible. In this sense, POMs serve as the metal sources, while MOFs get transformed into a carbon matrix with additional metal sites after carbonization. Li et al.127 reported for the first time the synthesis of a Fe3C/Mo2C-containing N,P co-doped carbon catalyst by pyrolyzing POM@MOF-100(Fe) with melamine (Fig. 5c). The POM@MOF-100(Fe) precursor was intentionally synthesized by hydrothermally treating the mixture of FeCl3, H3PMo12O40 and trimethyl 1,3,5-benzenetricarboxylate at 130 °C for 72 h, followed by sequential centrifugation, washing and drying steps. POM@MOF-100(Fe) was then mixed with melamine by ball milling before the pyrolysis treatment. Melamine provides the source of carbon and nitrogen, while the Fe species derived from MOF-100(Fe) catalyzes the formation of graphitized carbon during the process of carbonization. Subsequent HRTEM and EDX-mapping images verified that the confinement effect of MOF-100 benefited the high dispersion of Mo2C NPs, which were overcoated with uniform graphitic carbon layers. The resulted catalyst possessed a characteristic mesoporous structure, facilitating the mass transfer process during catalytic reactions. This work highlights the new opportunities for the design of advanced non-precious metal-based electrocatalysts by the utility of POM-incorporated MOF nanomaterials.
Metal precursors could also be incorporated into the as-synthesized MOFs featured with open channels and large pores by using suitable post-synthesis approaches.128,129 The confinement effect of MOF nanomaterials is beneficial to form more uniform dispersion relative to that of metal NPs supported on open-structured solid materials. As a typical example, in order to rationally avoid WC sintering into larger particles during high-temperature synthesis, Xu et al.129 developed a cage-confinement pyrolysis route where MAF-6 (rho-[Zn(eim)2]) was selected as a host to isolate the guest metal sites-W(CO)6 (Fig. 5d). Compared to the in situ mixing method, a low-temperature vapor adsorption procedure, that is, heating the mixture of MAF-6 and W(CO)6 in a sealed setup at 85 °C for 24 h, was more effective for the confinement and isolation of W(CO)6 within the MAF-6 framework to form W(CO)6@MAF-6. Subsequently, WC@NPC was yielded through the carbonization of the W(CO)6@MAF-6 precursor at 980 °C in N2 for 5 h. Owing to the pivotal role of the cage-confinement effect, WC with much smaller particle size (ca. 2 nm) relative to most reported metal carbides was coated with 1–2 graphitic carbon layers.
Combining MOFs with other structure-oriented materials provides the route for fabricating complex composites with multiple compositions and specific nanostructures.125,130–135 For example, coating MOFs with a polymer overlayer could prevent the aggregation of MOF-derived NPs during pyrolysis.131 PVP-coated NiFe-PBA131 and NiFeMn-PBA134 could be transformed into a unique 3D network composed of N-doped carbon-overcoated heterojunctions after carbonization, compared to that generated without the hybrid PVP polymer. In addition, Wu et al.132 reported a facile method to realize the formation of Co9S8 NPs encapsulated by N,S-doped carbon nanofibers (Fig. 5e). CdS@ZIF hybrids (ZIF polyhedra-wrapped CdS nanowires) were first prepared by quickly pouring the methanolic solution of 2-methylimidazole (2-MIM) and PVP into the mixed solution of zinc nitrate/cobalt nitrate and CdS nanowires. On pyrolyzing the CdS@ZIF precursor in an inert atmosphere, the ZIF shell was transformed into a Co,N-codoped carbon matrix. Concomitantly, CdS got reduced into evaporable Cd and S; the latter can be doped into the carbon matrix and react with Co species for the generation of N,S-doped carbon nanofiber-encapsulated Co9S8 NPs with uniform dispersion. HRTEM measurements validated the overcoating of Co9S8 NPs with a few carbon layers, which have an interplanar spacing of 0.34 nm. The obtained nanocatalyst shows the features of a large surface area and a mesoporous structure, resulting in abundant accessible active sites and a fast mass transfer process for electrocatalytic OER catalysis.
Furthermore, Bu et al.135 demonstrated that graphene oxide (GO) was employed in an ultrafast microwave-assisted route for converting PBA nanomaterials into N-doped graphene shell-coated transition metal NPs (i.e. FeCoNi, CoNi, FeNi, Co) supported on reduced GO (RGO) (Fig. 5f). This strategy induces the decomposition of PBAs into metals and carbon- and nitrogen-containing gases. The in situ formed gases could deposit onto the surface of the metal via a so-called chemical vapor deposition-like process, which is dramatically different from the conventional programmed heating procedure. During this well-controlled process, GO gets reduced into RGO and works as a microwave susceptor by rapidly creating heat to promote the uniform conversion of PBA nanomaterials within a seconds time. RGO could also facilitate the high dispersion of metal NPs derived from PBA nanomaterials. This strategy developed in this work is simple and controllable, which will expand the application in the synthesis of other kinds of metal@NC materials.
2.3 Overcoating of the metal catalyst with inorganic metal oxides
Metal oxides (i.e. SiO2 and TiO2) feature the advantages of good thermal stability, high acid resistance and tunable structural porosity, thus, showing potential as overcoating materials for rationally fabricating high-performance electrocatalysts. Metal oxide overlayer-encapsulated materials are an emerging class of electrocatalysts, and the corresponding viewpoint has already been provided by Esposito.138 The silica overlayer has been reported to improve catalyst durability in the way of stabilizing metal NPs towards out-diffusion and suppressing their migration, agglomeration and sintering under high temperature and/or severe reaction conditions.139–141 Conventional sol–gel chemistry of silica could help in achieving the overcoating of silica onto the surface of metal catalysts. Takenaka et al.140 coated a silica overlayer onto carbon black (CB)-supported Pt–Co via the hydrolysis and polycondensation of TEOS and 3-aminopropyl-triethoxysilane (APTES) in the presence of a base – trimethylamine – followed by a high-temperature calcination step (Fig. 6a). In the overcoating process, APTES with amino groups can strongly interact with the carbon support and the silica precursor derived from APTES provides nucleation sites for the successive hydrolysis of TEOS on the surface of the catalyst.141 A benefit from silica overlayer coating is that a narrower particle size distribution is obtained on SiO2/Pt–Co/CB than that found with Pt–Co/CB, the sample without the SiO2 overlayer.140 Likewise, Pt–Co NPs on SiO2/Pt–Co/CB are inclined to have a higher alloying degree, whereas those on Pt–Co/CB are prone to agglomerate in the absence of silica.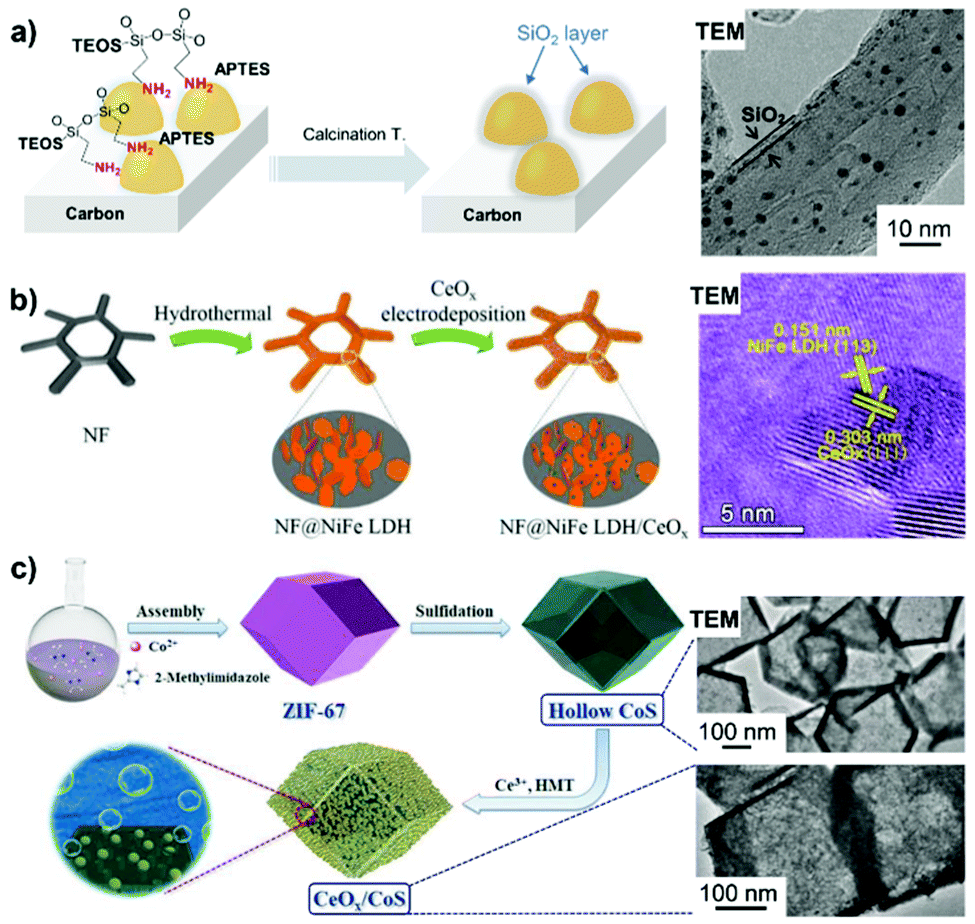 | ||
| Fig. 6 Inorganic metal oxide-overcoated electrocatalysts prepared by different synthetic methods. (a) Overcoating carbon-supported metal particles with a silica oxide layer by the successive hydrolysis of APTES and TEOS, along with calcination treatment. Reproduced from ref. 141 with permission from American Chemical Society, Copyright 2014. (b) The hydrothermal and subsequent electrodeposition process for obtaining NF@NiFe LDH/CeOx. Reproduced from ref. 144 with permission from American Chemical Society, Copyright 2018. (c) Schematic of the synthesis of hollow CoS particles with surface-covered CeOx by thermal treatment in the presence of Ce3+ precursor and HMT. Reproduced from ref. 145 with permission from Wiley-VCH, Copyright 2018. | ||
It is worth noting that the coating of silica would not be simple when unsupported metal NPs are implicated especially those belonging to the smaller particle size regime (<10 nm), owing to the weak interaction and high interfacial energy between metal and silica coating. In this sense, elaborate synthesis approaches are required to fabricate well-controlled core–shell nanostructures in the context of potential application in catalysis, which have been comprehensively reviewed by Gao et al.35
Kang et al.142 prepared porous TiO2 thin overlayer-coated Pt NPs by first wrapping as-formed Pt NPs with a TiO(OH)2 layer by the hydrolysis of titanium(IV) butoxide in ammonia-containing ethanol solution. Subsequently, TiO2-skin on the surface of Pt was attained through annealing treatment at a moderate temperature of 350 °C in an Ar atmosphere, where TiO(OH)2 was allowed to be converted into a TiO2 overlayer. In addition to the aforementioned wet chemical synthesis approaches, atomic layer deposition (ALD) is becoming an efficient technology for overcoating metal oxides on the surface of metal catalysts. In an exemplified report, Kim et al.143 carried out TiO2 ALD overcoating on the surface of carbon-supported Co NPs, producing an active OER electrocatalyst after optimizing the thickness of the TiO2 overlayer by controlling the number of ALD cycles. We will present the brief discussions of the metal oxide ALD process in Section 2.4.3.
Metal oxides also find application in surface overcoating of metal oxide nanosheets,144 porous and hollow metal-based nanostructures145 and metal thin film electrodes.146–149 For example, Wang et al.144 first constructed NiFe layered double hydroxide-coated nickel foam (NF@NiFe LDH) by hydrothermally treating NF in a solution of metal nitrates. CeO2 NPs were then electrodeposited onto NF@NiFe LDH at a current density of 0.25 mA cm−2 in a three-electrode cell to yield the 3D self-supporting NF@NiFe LDH/CeOx electrode catalyst (Fig. 6b). From the HRTEM images of NF@NiFe LDH/CeOx, a visible interface structure was identified between the NiFe LDH(113) plane and the overcoated CeOx(111) plane. Their local electron potential difference introduced abundant oxygen vacancies, which could promote the electron/charge transfer and create more active surface sites for the alkaline HER. In addition, coating an ultra-thin overlayer of a metal oxide (i.e. SiO2, VOx and CeOx) on the metal electrode surface could form a semi-permeable membrane by controlling the transfer of reactants or products to achieve a higher electrical selectivity. As such, electrodeposition of MnOx thin film onto a glassy carbon-supported IrOx electrode endowed the OER catalysis with improved efficiency over the chorine evolution reaction in acid solution for potential water splitting application.146 An example of metal oxide overcoating on porous metal-based nanostructures comes from the report of Xu et al.145 First, an amorphous CoS hollow nanocage was formed by refluxing ZIF-67 particles in an ethanol solution of thioacetamide. Subsequently, a hybrid nanostructure (CeOx/CoS) was constructed by in situ overcoating CeOx NPs (with an average diameter of 5 nm) onto ZIF-67-derived hollow CoS polyhedra via a thermal treatment using Ce(NO3)3 and hexamethylenetetramine at 180 °C for more than 6 h (Fig. 6c). The resultant CeOx/CoS nanostructure, containing CoS polyhedra decorated with CeOx particles, possesses the features of a highly porous structure and a large surface area, which are favourable for electrocatalytic OER activity. It was found that an optimized loading of CeOx was profitable for tailoring the metal electronic structure, forming a large number of defective sites and limiting the oxidation/dissolution of catalytically active Co species.
2.4 Modification of the carbon support by surface overcoating
Carbon black is the most used low-aspect-ratio support material for PGM electrocatalysts, including Vulcan XC72, Ketjen EC 300J, Ketjen EC 600JD and Black pearls 2000. However, carbon corrosion in terms of electrochemical oxidation (C + 2H2O → CO + 4H+ + 4e−) and chemical oxidation (C + H2O → CO + H2 or C + 2H2O → CO2 + 2H2) is a severe issue under shut-down/start-up operating conditions and/or in an oxidizing environment, in addition to the issue of the easy deactivation of supported metal NPs. Moreover, the presence of PGM-based NPs on carbon support materials could accelerate the corrosion of carbon through the oxidation pathways. The literature review highlights that surface overcoating engineering on carbon black and high-aspect-ratio carbon nanotubes can bring in benefits such as mitigation of carbon corrosion, improvement in the dispersion of metal NPs, and formation of strong metal–support interactions. These benefits of surface-overcoating modifications on the carbon support will be discussed in Section 3.5.1. In this section, we showcase the synthetic methodologies being developed for overcoating of carbon support materials with polymers, carbon species, and metal oxides by some representative body of works.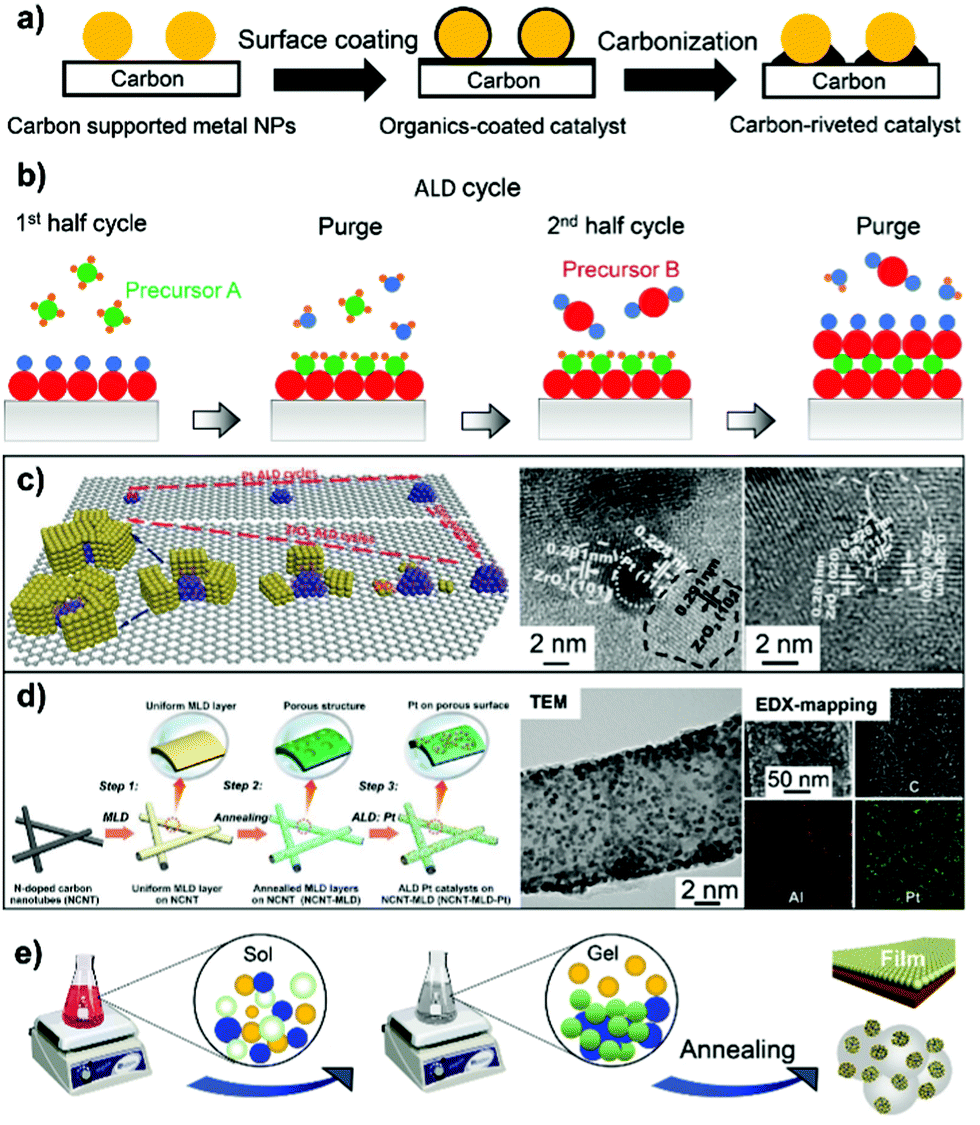 | ||
| Fig. 7 (a) Schematic illustration of carbon rivet formation on a carbon-supported metal catalyst using organic coatings as the carbon precursor. (b) Schematic of the process of ALD cycles. (c) Overcoating ZrO2 onto the N-doped carbon nanotube by ALD cycle number control. Reproduced from ref. 191 with permission from Wiley-VCH, Copyright 2015. (d) MLD of AlOx species onto the carbon nanotube to support the well-dispersed Pt NPs. Reproduced from ref. 193 with permission from Elsevier, Copyright 2019. (e) Schematic of the sol–gel process for the preparation of carbon–metal oxide composites. | ||
In addition, graphitic carbon nitride (g-C3N4) materials feature the advantages of high mechanical stability in an acidic/oxidative environment, and possess abundant Lewis acid and base sites (i.e. –NH groups and N atoms on the triazine/heptazine ring) for high dispersion of metals by forming strong electronic interactions. Introducing g-C3N4 materials onto the surface of carbon has been demonstrated to stabilize carbon-supported PtRu NPs for enhanced MOR electrocatalysis.173–175 Direct one-pot pyrolysis of the mixture of carbon-supported metal material and melamine is an effective method for g-C3N4 overcoating.173 On the other hand, the carbon nitride nanosheet-coated carbon, named C@g-C3N4, was reported to be fabricated through thermal polymerization of the mixture of urea and carbon support, followed by an exfoliation process at decomposition temperature (600 °C) under an inert atmosphere.174,175
Chemical vapor phase deposition. Among the boomingly deployed chemical vapor phase deposition technologies, atomic layer deposition (ALD) and molecular layer deposition (MLD) are, in particular, gaining in popularity for the rational design and fabrication of advanced nanomaterials from an atomic level. The readers are referred to the excellent reviews that highlight the status and prospects of ALD and MLD technologies for sustainable energy-related applications.182–187
The ALD process generally proceeds as sequential, self-limiting surface reactions by exposing the substrate to separate precursors (such as trimethylaluminum (TMA) and water vapor used in the best known Al2O3 ALD process) in near-vacuum conditions. Each deposition cycle tends to form an atomically thick layer on the surface of the substrate (Fig. 7b). ALD can be used to deposit many kinds of coatings such as metal oxides, nitrides, and sulfides, and allows control over the uniformity of surface coatings with high accuracy. ALD overcoating of metal oxide layers with the thickness controlled by deposition cycle numbers could endow the as-fabricated catalysts with distinct catalytic reactivities.188–190 Researchers have specifically developed the area-selective ALD technology in an attempt to precisely control overcoating sites and expose more preferred surface.187 An organic mask molecule, i.e. oleylamine, is used to prevent the deposition of metal oxide coatings on the surface of metal nanoparticles. Oleylamine molecules can be removed by a high-temperature annealing treatment afterwards. As such, typical examples of area-selective ALD coating of ZrO2 nanocages onto Pt-supported N-doped carbon nanotubes (NCNTs)191 and TaOx ALD overcoating onto Pt-supported carbon black192 have been reported by Sun's group. As illustrated in Fig. 7c, upon increasing ZrO2 ALD cycles from 20, 30 to 50, an open or holy nanocage is formed around the Pt nanoparticle without deposition on its surface. Thermal stability evaluation by heating the as-synthesized catalysts to 600 °C indicated that the particle size of Pt on ALD30ZrO2–Pt/NCNT (with 30 ALD cycles) increased from 2.3 to 2.7 nm, while that of ALD50ZrO2–Pt/NCNT600 °C (with 50 ALD cycles) showed a nearly constant particle size distribution. ALD50ZrO2–Pt/NCNT600 °C also displayed 9 and 10 times higher electrochemical stability relative to the ALDPt/NCNT without ZrO2 overcoating and the reference Pt/C catalyst, respectively, after 4000 potential cycles between 0.6 and 1.2 VRHE.191 Therefore, the encapsulation of Pt NPs within ALD overcoating-derived ZrO2 nanocages could allow the catalytic efficacy of supported metal-based electrocatalysts to be maximized.
MLD is an extended technology after ALD for the preparation of polymer-based organic materials and inorganic–organic hybrid materials.183 Metal-based metalcones and organic ethylene glycol (EG) are the most commonly used precursors for inorganic–organic thin-film coatings. The removal of organic linkers such as EG by thermal or chemical treatment is necessary for the generation of porous metal oxide overlayers. For developing a stable Pt ORR electrocatalyst, Zhang et al.193 for the first time applied MLD technology to stabilize Pt NPs. MLD alumina oxide (AlOx) overlayers on N-doped carbon nanotubes were fabricated by alternatively introducing trimethylaluminium and glycerol precursors into a Gemstar-8 ALD system. MLD AlOx overlayers feature a porous structure with enriched pores after thermal treatment at 400 °C in air (Fig. 7d). As verified by TEM and EDX-mapping images, the enriched pores on AlOx-overcoated carbon nanotubes serve as the anchor sites for interacting with Pt NPs, which accounts for the observed high dispersion of the metal. Consequently, the MLD strategy imparts the Pt-based catalyst with markedly improved ORR performance, overpassing the catalyst in which the Pt NPs are not coated with the MLD-derived metal oxide overlayer.
Wet chemical synthesis. For wet chemical synthesis, the introduction of metal oxides onto the carbon support could be divided into two approaches, including the hydrothermal-induced deposition–precipitation process and the chemical sol–gel process via electrostatic interactions. Limitedly, a CeO2-decorated carbon support was reported to be obtained through the direct carbonization of Ce-MOFs.194 The deposition of metal particles on carbon–metal oxide composites, as a step involved in metal based catalyst preparation, can be performed by (i) one-pot co-precipitation with a metal hydroxide onto the carbon support followed by annealing treatment; or (ii) post-immobilization via the conventional polyol reduction method; or (iii) direct “solid” method involving impregnation, heating and gas reduction steps. Selecting a reliable method requires care to meet the requirements of metal particle size and size-distribution as well as metal chemical states for specific applications in catalysis.
In the deposition–precipitation process, a base such as NaOH and urea facilitates the first conversion of metal salt precursors into metal hydroxides and then their simultaneous deposition on the surface of the carbon support.195,196 Thermal decomposition of as-precipitated metal hydroxides on the surface of carbon helps in the formation of homogeneous and crystalline metal oxide-overcoated carbon composites. On the other hand, in the sol–gel chemical process, metal alkoxide compounds (M(OR)n), including ethyloxide, isopropoxide, and butoxide compounds, are initially hydrolyzed into M(OH)m(OR)n−m species in the presence of water or in an acid solution under moderate hydrothermal conditions.197–199 Concomitantly, the as-formed M(OH)m(OR)n−m species undergo a complex polycondensation to form a well-dispersed metal oxide network on the surface of carbon after annealing treatment (Fig. 7e). Based on this strategy, Akalework et al.200 overcoated multiwall carbon nanotubes (MWCNTs) with an ultra-thin TiO2 film. The ultra-thin TiO2 was a valuable coating for improvement in the dispersion of Pt with smaller particle size (1.6 nm) and a higher ECSA value (285.5 m2 g−1), giving rise to a much higher electrocatalytic efficiency than that found with the uncoated Pt-MWCNT and commercial Pt/C catalyst. Similarly, TiO2-coated carbon nanotubes (TiO2/CNTs) were one-pot synthesized through the non-boiling isothermal hydrolysis process.201 SnO2–C composites with different SnO2 loading were prepared via the sol–gel process.202 Besides metal alkoxide compounds, metal chlorides such as NbCl5,203 SnCl2,204 and TaCl5,205 metal acetylacetonate (i.e. Zr(acac)4),206 and metal acetate (i.e. manganese acetate)207 are also employed to prepare carbon–metal oxide nanocomposites, mostly by the process of thermal hydrolysis-deposition in anhydrous alcohol or an alcohol/water system.
Other synthesis methods. There are other attempts such as the direct air-annealing,208 self-assembly-decomposition,209 and a physical pulse-laser ablation in liquids (PLAL) method210 for preparing carbon–metal oxide composites. In particular, highly dispersed MoOx species were introduced onto carbon nanotubes (CNTs) through the decomposition of phosphomolybdic acid (HPMo, Keggin-type), which were self-assembled on the surface of functionalized CNTs.209 Besides, TiOx or SnOx derived from the PLAL method were found to have abundant oxygen defects, which could reduce GO into rGo and the Pt precursor (PtCl42−) to Pt(0) NPs. This in situ reduction process was crucial for the uniform distribution of small Pt NPs and the formation of strong interactions between Pt and the metal oxide, leading to enhanced mass activity, stability and excellent CO-antipoisoning ability towards the MOR catalysis.210
2.5 Modification of the metal oxide support by surface overcoating
Overcoating with carbon nanoshells has been widely exploited to modify the surface of the metal oxide support with examples pertaining to CeO2,211,212 Mn3O4,213 and TiO2.214–217 Similar to the approach described for metal nanoparticles and the carbon support, direct pyrolysis of the composites containing organic compounds or polymers (such as ionic liquid,212 Ppy,214 xylose,215 furfuryl alcohol216) is adapted for overcoating of the metal oxide support with carbon nanoshells. For example, through direct carbonization of β-cyclodextrin on the mesoporous CeO2 support (MP-CeO2) at 400 °C, Chu et al.211 prepared carbon-CeO2 composites comprising CeO2 that were partly coated with carbon layers. Pt NPs were then deposited on the peripheral interface between MP–CeO2 and carbon layer by means of the microwave-assisted polyol reduction method. Carbon overcoating on the MP–CeO2 support could produce a large number of oxygen-containing species, i.e. –COOR group, which serve as anchoring sites for Pt deposition. An optimized 10.4 wt% loading of carbon species was effective for an acceptable catalytic performance of Pt/C40–CeO2. Upon further increasing the carbon content to 15.9 wt%, the contact between Pt NPs and CeO2 support was obstructed, resulting in poor electrocatalytic property. As another example, the synthetic process for overcoating the carbon layer onto the Mn3O4 support to fabricate the Pt/Mn3O4–carbon triple-junction structure is illustrated in Fig. 8a.213 Starting from Mn3O4 as the catalyst support, dopamine undergoes sequential oxidative polymerization and high-temperature carbonization to result in the formation of MnO@C. With the increase in the concentration of dopamine in the synthetic solution, the carbon nanoshell thickness also shows an increasing trend after carbonization (Fig. 8b–e). Subsequently, the deposition of Pt NPs was proceeded via a galvanic replacement process, whereby Pt precursors were allowed to diffuse through the carbon nanoshell to chelate and grow onto the surface of inner MnO, while MnO could be concurrently oxidized into Mn3O4. The final structure of the catalyst relied on the thickness of the carbon shell, since a thicker (i.e. 7.1 nm) carbon layer was observed to delay the inward diffusion of PtCl42− which induced the deposition of Pt on the outer surface of carbon. These works clearly imply that it is important to control the amount or the shell thickness of carbon on the metal oxide support to benefit the electrocatalytic performance of metal based catalysts.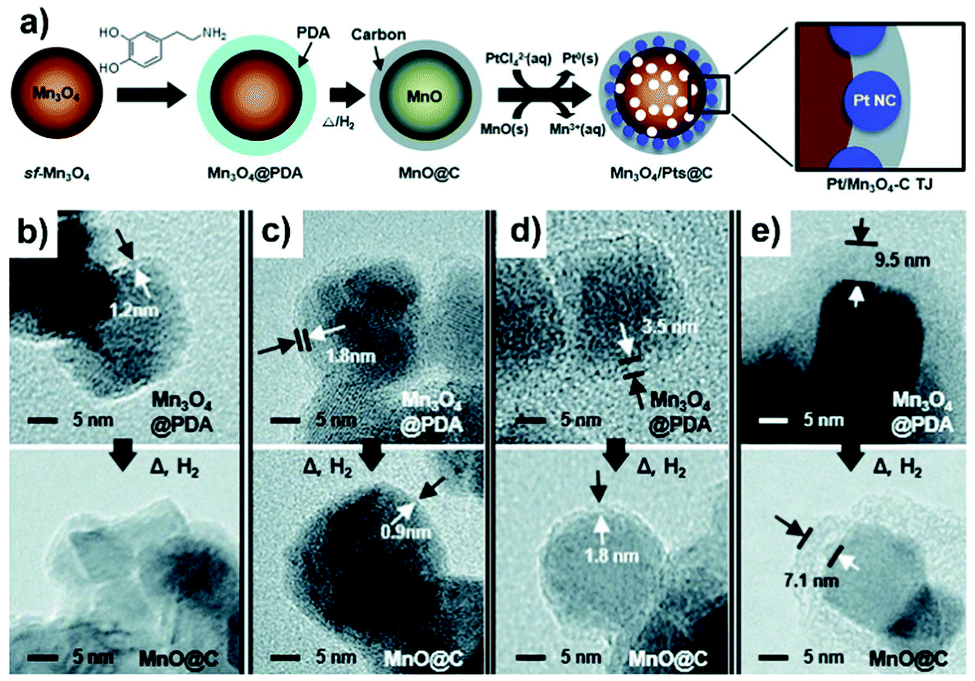 | ||
| Fig. 8 (a) Schematic illustration of the preparation process of the Pt/Mn3O4–carbon triple-junction structure using Mn3O4, dopamine, and PtCl42− as the support, the precursor for the carbon overlayer and the precursor of Pt NPs, respectively. (b–e) TEM images of the overcoated metal oxide support having different outer layer thickness before and after high temperature pyrolysis. Reproduced from ref. 213 with permission from Royal Society of Chemistry, Copyright 2017. | ||
2.6 Comparison of different surface overcoating strategies for the design of high-performance electrocatalysts
A comparison of a diversity of surface overcoating methodologies utilizing organic capping agents/polymers, inorganic carbon nanoshells and metal oxides is presented in Table 1, and the pros and cons of each process are also overviewed. High-performance electrocatalysts can be rationally designed via surface overcoating engineering by a suitable selection of overcoating materials and synthetic methodologies. The advantages of using organic capping agents and polymers as the surface coatings include (i) a large number of organic matters featuring specific electron-withdrawing/donating properties can be selected to manipulate the electron density of the metal surface by constructing unique metal–organic interfacial nanostructures and (ii) the corresponding catalytic benefits vary with the organic matters functionalized on the surface of metals when they are applied for a range of electrochemical reactions. However, it is precisely these advantages that might cause a lot of work for screening optimal organic species in the design and optimization of high-performance electrocatalysts in the laboratory. The stability of organic molecules coordinated with the metal needs to be paid attention when the surface-functionalized catalyst is applied under harsh operating conditions. Carbon nanoshells, as the stable coating material, are generally fabricated on the catalyst surface through pyrolysis and/or deposition of organic precursors at high temperatures. Synthetic conditions in terms of temperature, gas environment and coating amount need to be elaborately optimized since the engineered catalyst structural properties would significantly impact the electron/mass transfer process and further the catalytic efficiencies during catalysis. The introduction of the carbon nanoshell may influence metal particle properties such as the size, morphology and surface composition, in addition to the high-energy input in this strategy. On the basis of the existing literature, wet chemical synthesis and atomic layer deposition are the most used processes for the surface overcoating of metal catalysts with metal oxides. In particular, the ALD technology is able to allow control over the deposition location (that is the selective area atomic layer deposition) and the overlayer thickness from an atomic scale, although this technology along with MLD is not economically viable. In comparison with ALD/MLD, wet chemical synthesis tends to be more affordable for large scale modification of metal catalysts. It is important to perform wet chemical synthesis under the conditions that are suitable for the transformation of metal precursors (i.e. by hydrolysis, polycondensation), the further formation of an inorganic network and the final deposition onto the catalyst substrate in the solid–liquid phase. Metal oxide layers as-fabricated on the surface of catalysts are usually not uniform when using such a complex process, thus leading to limitations in terms of reliability. Nevertheless, note that the integration of surface coatings with metal NPs and/or support materials into a unique nanostructure could effectively endow the designed nanocatalysts with synergistic contributions for clean energy-based electrocatalysis especially in PEFCs and WECs.| Coatings | Processes | Pros | Cons |
|---|---|---|---|
| Organic capping agent/polymer | In situ capping through colloidal synthesis | One-pot method for organic-capped metal nanostructures; abundantly available organic capping agents | Wastage of most of the as-added organic molecules; several synthetic parameters affect particle properties |
| Post-synthetic construction through e.g. the dipping method | Simple and easy method | Specific solvent for dissolving the organic modifier | |
| Oxidative polymerization in liquid phase | Uniform thin film; relatively high loading | Control of film thickness; special oxidant added; impurities generated | |
| Inorganic carbon nanoshell | High-temperature pyrolysis of polymers or metal–organic composites including MOFs | Simple; abundantly available carbon precursors; potential scale-up | Induction of metal particle structural evolution under heating conditions; high energy required; control of carbon shell homogeneity and graphitization degree |
| Chemical vapor phase deposition | Thin-layer carbon shell; high uniformity | High energy required; toxic and explosive gases as carbon precursors | |
| Inorganic metal oxide layer | Wet chemical synthesis | Simple and efficient; commercially viable | Limitation of controlling the layer thickness, morphology and uniformity |
| Atomic layer deposition/molecular layer deposition | Atomic control over layer thickness; ultra-thin coating film; high uniformity | Low coating rate; economic viability | |
| Electrodeposition | Easy control by applying a slight voltage; cost-effective | Pre-coating the catalyst onto the electrode; specific devices equipped with electrode and electrolyte |
3. Overcoated electrocatalysts for polymer electrolyte fuel cells
Proton-exchange membrane fuel cells (PEMFCs) and anion-exchange membrane fuel cells (AEMFCs) are among the most explored fuel cell technologies. PGM-based nanomaterials are the active electrocatalysts so far for energy-related small molecule activation reactions such as the ORR, HOR and MOR in PEFCs. Metal NPs supported on fuel cell electrocatalysts easily suffer from the issues of coalescence, Ostwald ripening and dissolution, attributed to their weak interactions with the support and the highly acidic operating conditions.15,16 Due to the high potential (1.5–2.0 V) of the cathode under shut-down/start-up conditions a severe carbon corrosion could be induced, which, in turn, leads to the decrease of metal–support interactions accompanied by metal NP agglomeration and detachment from the support.15 Moreover, the serious site-blockage and poisoning effects by spectator species, i.e. PO43−, SO42−, CO, and methanol (the crossover species in direct methanol fuel cells (DMFCs)), still remain as problems that hinder the improvement of catalytic efficiencies for low-temperature fuel cells.37 Owing to the above performance degradation mechanisms, in most cases the electrocatalysts cannot meet the fuel cell performance targets. Several limitations, in essence, pose considerable challenges to optimal electrocatalyst design: (i) the trade-off between electrocatalytic activity and long-term durability; (ii) the sacrificing of power performance and durability when decreasing the loading of PGMs (in order to reduce cost).12,16 As summarized in the above section, vast research has been focused on the surface overcoating strategy for the fabrication of metal NPs and the modification of support materials. Herein, recent advances of both precious and non-precious metal-based electrocatalysts fabricated by surface overcoating engineering for efficient application in H2–O2 fuel cells (PEMFCs and AEMFCs involving the ORR and HOR) and direct alcohol fuel cells (anodic reactions such as MOR and EOR) are presented in Sections 3.1–3.3. A comparison of the surface coatings suitable for above-mentioned catalytic processes is summarized in Section 3.4. The benefits of utilizing overcoating-derived support materials, i.e. porous carbon and metal oxides, for metal-supported electrocatalyst design and optimization will be discussed in Section 3.5.3.1 Overcoated electrocatalysts for the ORR
The ORR catalysis on the cathode is at the heart of fuel cells since its sluggish kinetics require a higher loading of Pt, thereby becoming the active area of research. The cathodic ORR is a multielectron process involving the reacting active intermediates such as *OOH, *O and *OH. The ORR in acid and alkaline solution can proceed by the following pathway: O2 + 4H+ + 4e− → 2H2O (acidic condition) and O2 + 2H2O + 4e− → 4OH− (alkaline condition). Depending on the nature of the electrocatalyst, a partial reduction, the two-electron pathway: O2 + 2H+ + 2e− → H2O2 (acidic condition) and O2 + H2O + 2e− → HO2− + OH− (alkaline condition), can take place on the cathode to form H2O2. ORR is oftentimes regarded as a prototype for developing advanced concepts that can be applied to other catalytic systems.218 In this section, the categories related to both precious and non-precious metal based ORR electrocatalysts are described.Organic material-overcoated precious metal based ORR electrocatalysts. Engineering metal based electrocatalysts with high resistance towards the site-blocking by spectator species from the electrolyte is desirable for catalytic improvements in performance for the cathodic ORR in PEFCs.31,42,47 Surface-patterned organic molecules such as cyanide (CN−) groups and amine could hinder the adsorption of non-covalent bulky adsorbates (SO42− and PO43− ions) on the metal, leading to favoured ORR activity in acid solution.44,59 The limitation on the CN−-patterned Pt surface is that the noncovalent interactions formed between the anionic CN− adlayer and the positively hydrated K+(H2O)x cluster would block O2 adsorption and result in activity deactivation in a basic KOH electrolyte.59 Electrostatic interactions were recently utilized by Jung et al.,219 who fabricated self-assembled monolayers of L-cysteine on Pt for efficient ORR catalysis. On the one hand, the –COOH groups of L-cysteine become negatively charged (–COO−) in KOH solution, and can repulse excess OH− and attract the hydrated K+(H2O)x cluster to avoid the blocking of Pt active sites (Fig. 9a). On the other hand, when immersing in acidic H3PO4, the –NH2 groups of L-cysteine are protonated to form positively charged –NH3+ species, which could capture the PO43− ions by forming strong hydrogen bond and electrostatic interactions, and thus inhibiting their interactions with Pt. In these ways, the hindering by various spectator ions is remitted, as indicated by the decreased adsorption charge densities on Pt (Fig. 9b). As a result, the ORR specific activity on L-cysteine-patterned Pt is much higher than that found with the unmodified Pt catalyst in both KOH and H3PO4 solutions (Fig. 9c).
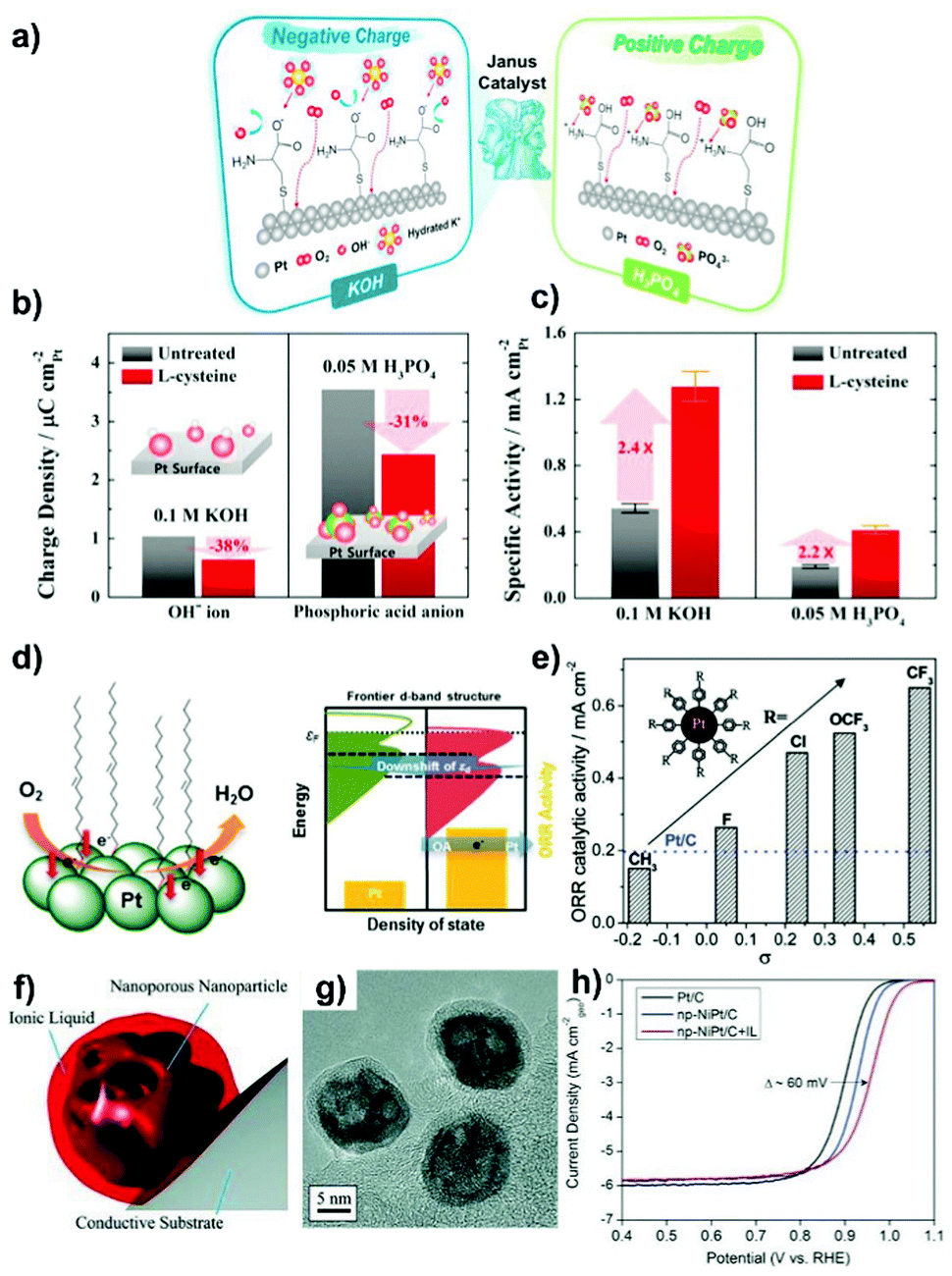 | ||
| Fig. 9 (a) Schematics of the electrostatic interaction between charged L-cysteine molecules on the Pt surface and spectator ions in KOH and H3PO4 solutions. (b) Anion adsorption charge densities obtained from CVs and (c) ORR specific activities (at 0.9 VRHE) on untreated and L-cysteine-decorated Pt polycrystalline structure. Reproduced from ref. 219 with permission from Elsevier, Copyright 2015. (d) Schematic of the Pt d-band structure change after surface-capping of OA molecules. Reproduced from ref. 222 with permission from American Chemical Society, Copyright 2013. (e) Variation of the ORR specific activity (at 0.9 VRHE) with Hammet substituent constant (σ). Reproduced from ref. 224 with permission from American Chemical Society, Copyright 2012. (f) Cartoon illustrating the encapsulation of nanoporous NiPt NPs by an IL. (g) HRTEM image of NiPt NPs encapsulated within the [MTBD] [beti] IL, supported on carbon. (h) ORR curves for Pt/C, np-NiPt/C, and np-NiPt/C + IL in O2-saturated 0.1 M HClO4 at 60 °C with 20 mV s−1 and 1600 rpm. Reproduced from ref. 226 with permission from Wiley-VCH, Copyright 2013. | ||
In DMFCs, the issue associated with methanol crossover from the anode to cathode of the cell remains as a barrier to the development of ORR electrocatalysts. Improving the methanol-tolerance of the cathodic Pt-based electrocatalyst is thus of vital importance for achieving electrical selectivity and high efficiency of ORR. Polymer layers and organic matters such as PAH,51,54,56 PVPA/PyPBI75 and ionic liquid ([MTBD][bmsi])220 have been fabricated onto Pt-based catalysts by using in situ or post-synthesis approaches to improve their anti-poisoning ability towards methanol. The improved methanol-tolerance can be attributed to the steric hindrance effect from the surface coatings. Lu et al.221 reported that a higher pyridine coverage (θS,H > 50%) on the surface of the Pt/C ORR electrocatalyst was necessary for hindering the adsorption of methanol. As a result, ORR specific activity linearly increased with θS,H and a 6 times enhancement was achieved when the Pt surface was saturated with pyridine (θS,H = 80%). In addition to the steric hindrance effect, surface-overcoated organic molecules have profound effects on the electronic surface structure of metal entities.51,52,56 As indicated from CO stripping measurement, the coordination of pyridine molecules made the Pt surface electron-rich, which could facilitate the adsorption of O2 and weaken the adsorption of the *O intermediate.221 ORR mass activity was found to be determined by the competition between the steric and electronic effects from pyridine. Accordingly, the electronic effect dominated the ORR process when θS,H < 50%. The authors also demonstrated the existence of both steric and electronic effects from 4-dimethylaminopyridine and triphenylphosphine ligands functionalized on the Pt surface. Other ligands such as oleylamine (OA) and butylamine exhibited a negligible electronic effect on the ORR in acidic solution. Chung et al.222 observed from synchrotron-based spectroscopy that the d-band center of Pt downshifted after surface-construction with OA (Fig. 9d), a fact ascribed to the increase of electron density in the frontier d states of Pt. The smaller d-band vacancy contributed to the decreased adsorption strength of bulky PO43− ions on the Pt surface, resulting in enhanced ORR kinetics. In this study, the frontier d-band structure of metals represents the suitable descriptor of the ORR activity.
The strong adsorption of OHad on the metal surface causes the sluggish ORR kinetics.64,223 Metal intrinsic structural properties with respect to their electronic structures strongly determine the adsorption of OHad. Electron delocalization on Pt is thus reasonable for changing metal affinity with Oad and OHad species and enabling the regulations on ORR catalytic activity. Zhou et al.224 manipulated the electronic surface structure of Pt using a variety of phenyl-based organic molecules with different electron-withdrawing para-groups (such as CH3, F, Cl, OCF3, CF3). As shown in Fig. 9e, the ORR activity increases in line with the increased electronegativity of para-groups, according to their Hammet substituent constants (σ). The transfer of electrons from Pt to the surface organic molecules could decrease the electron density of Pt and weaken the adsorption of oxygen-containing species (OHad, Oad), which eventually lead to a positive effect on ORR catalysis. In addition, the organic agent-functionalized Pt catalysts show a higher stability than “naked” Pt/C under similar conditions.
The OHad–water network is a stable form of OHad after its incorporation into water hexagonal networks. In addition, the interface water, known as the source of oxygenated species, is regarded as the site-blocker for ORR electrocatalysts. Typically, surface engineering of Pt with a hydrophobic ionic liquid layer has been considered as the effective strategy to efficiently destabilize OHad species and repel water from the surface intermediates and products while maintaining the accessibility of active sites to the reactants, resulting in high resistance of the catalyst to surface oxidation and thus accelerated ORR activities.63,225–227 In the work reported by Snyder et al.,226 markedly improved ORR kinetics in performance was observed on nanoporous NiPt nanoparticles encapsulated by a hydrophobic, protic ionic liquid (IL, [MTBD][beti]) (Fig. 9f and g). This IL coating is not only able to provide a nonaqueous environment to prevent the oxidation of Pt, but also brings in the benefit of high solubility of O2 near the metal particle surface. As shown in Fig. 9h, a nearly 60 mV reduction of overpotential for ORR is a clear reflection of the decrease of the surface coverage of OHad. Such an IL-coated catalyst exhibits an order of magnitude larger half-cell specific activity than the commercial Pt/C catalyst.
Based on the existing literature, it should be stressed that there is an inevitable loss of the active surface area following the organic surface modification.47,49 It is therefore necessary to optimize the amount or the coverage of the organic material on the surface of the metal to maintain the adsorption and desorption of reactants and products with the aim of realizing an acceptable electrocatalytic performance. Meanwhile, the findings from a small number of studies especially on ionic liquid layer coatings of Pt-based catalysts suggest comparable ECSA values to the unmodified counterparts.227,228 In most cases, the coordination stability of organic–metal nanostructures has been ensured at least in the potential range where the electrochemical reactions take place.221 On the other hand, the ionic liquid layer for example was found to accelerate the dissolution of Mo from PtNiMo NPs because of its strong complexing capability, leading to worse durability performance.228 Therefore, comprehensive interface theory and understanding the dynamic properties of the interfacial metal–organic structure during the electrochemical processes are highly desired in further research works.62
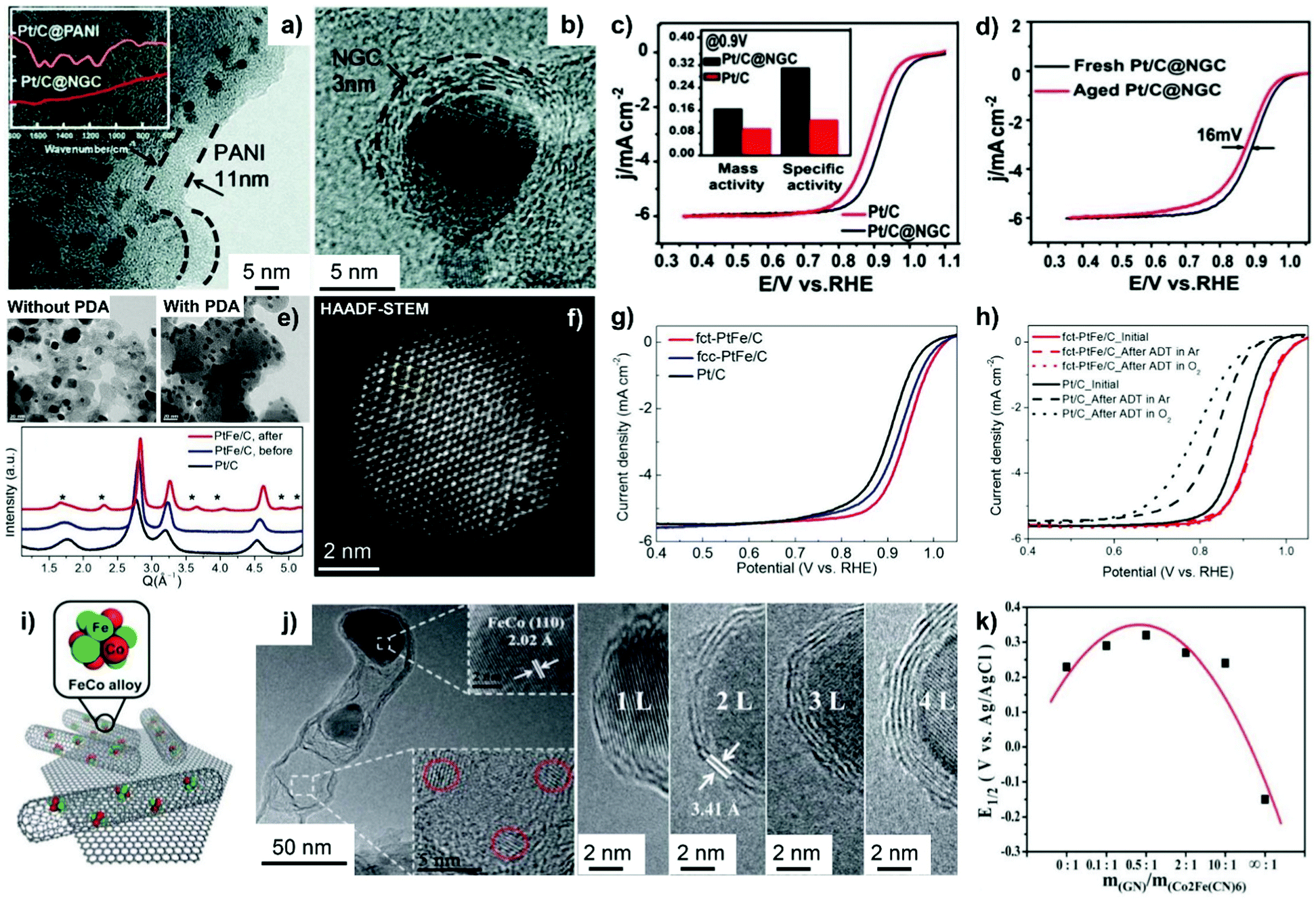 | ||
Fig. 10 HRTEM images of (a) Pt/C@PANI and (b) Pt/C@NGC. (c) Polarization curves of Pt/C and Pt/C@NGC in an O2-saturated 0.1 M HClO4 solution at 60 °C at 10 mV s−1 and 1600 rpm. The inset in (c) represents the mass activities and specific activities at 0.9 VRHE for Pt/C and Pt/C@NGC. (d) Polarization curves of Pt/C@NGC before and after 1500 cycles under a condition similar to that in (c). Reproduced from ref. 230 with permission from Royal Society of Chemistry, Copyright 2014. (e) TEM images (upper) of PtFe NPs after annealing at 700 °C with and without surface coating of PDA; high-resolution powder diffraction patterns (bottom) of PtFe/C before and after annealing in comparison with Pt/C. (f) HAADF-STEM image of annealed fct-PtFe NPs. (g) ORR polarization curves of Pt/C, fcc-PtFe/C, and fct-PtFe/C at the rotating disc electrode (RDE). (h) Polarization curves of fct-PtFe/C and Pt/C before and after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ADT cycles. Reproduced from ref. 89 with permission from American Chemical Society, Copyright 2015. (i) Illustration of Pod(N)-FeCo/GNs with the FeCo (110) crystal plane and smaller NPs confined within pod-like CNTs. (j) HRTEM images show the cross-sections of FeCo NPs covered by 1–4 layers of graphitic carbon wall. (k) Half-wave potential (E1/2) of RDE polarization curves as a function of m(GN)/m(Co2Fe(CN)6). Reproduced from ref. 242 with permission from Royal Society of Chemistry, Copyright 2013. 000 ADT cycles. Reproduced from ref. 89 with permission from American Chemical Society, Copyright 2015. (i) Illustration of Pod(N)-FeCo/GNs with the FeCo (110) crystal plane and smaller NPs confined within pod-like CNTs. (j) HRTEM images show the cross-sections of FeCo NPs covered by 1–4 layers of graphitic carbon wall. (k) Half-wave potential (E1/2) of RDE polarization curves as a function of m(GN)/m(Co2Fe(CN)6). Reproduced from ref. 242 with permission from Royal Society of Chemistry, Copyright 2013. | ||
In order to form ordered face-centered tetragonal (fct)-structured PtFe NPs with small particle size, Chung et al.89 overcoated a N-doped carbon nanoshell onto as-synthesized carbon-supported disordered PtFe NPs by in situ carbonization of the PDA overlayer. High-resolution powder diffraction and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) characterization verified the formation of 6.5 nm fct-PtFe NPs that are supported on the carbon (Fig. 10e and f). The PDA-derived carbon shell with less than 1 nm thickness could efficiently avoid PtFe NP coalescence. The fct-PtFe NPs showed a weak affinity to oxygen-containing species and thereby a higher ORR activity than fcc-PtFe and monometallic Pt/C (Fig. 10g). Extremely high durability was observed on the ordered fct-PtFe catalyst (Fig. 10h), which is because of the higher resistance of Fe atoms towards oxidation and dissolution after electronic coupling with Pt. The strong binding of N dopants with Pt atoms was also speculated to be responsible for stability improvement. In addition, structurally ordered intermetallic PtFe alloy NPs were formed via a thermally driven interfacial diffusion alloying route in the presence of the as-fabricated inner PDA layer and outer silica shell.92 In this case, ordered PtFe NPs exhibited a 600% increase in mass activity and specific activity to the ORR in comparison with Pt/C. A higher stability was additionally observed on the PtFe alloy due to the ordered atomic arrangement and robustness of the N-doped carbon nanoshell. Moreover, a PDA-derived N-doped graphitic carbon layer (NGC, 0.76 nm thickness) was overcoated on the carbon-supported PtNi3 alloy catalyst to overcome the stability issue under acidic conditions, as reported by Sun et al.231 In the preparation process, overcoating as-synthesized PtNi3/C with the PDA layer was first performed, followed by high-temperature pyrolysis to obtain the final catalyst, which was named PtNi3/C@NGC. Structural characterization indicated that the PtNi3 particle holds the merit of a Pt-rich surface and a Ni-rich core, likely attributed to the higher adsorption energy of Pt with N atoms (from PDA coating) than that of Ni. As a result, the utilization efficiency of Pt on the PtNi3/C@NGC sample was improved, showing 1.16 A mgPt−1 of mass activity and 2.9 mA cm−2 of specific activity towards the ORR. During the accelerated durability test between 0.6 and 1.0 VRHE for 10000 cycles under an O2 atmosphere, PtNi3/C@NGC displayed negligible loss of ECSA and half-wave potential, which were much superior than PtNi3/C and commercial JM-Pt/C catalysts.
MOF-derived nanostructures are also corroborated to bring in the benefits of enhancing the electrocatalytic performance of PGM-based catalysts.232–234 Wu et al.233 reported that a ZIF-67-derived ultra-thin 2D carbon layer could stabilize Pt NPs distributed on reduced graphene oxide. It is the synergy of Pt with Co–Nx–C active sites (pyrolyzed from ZIF-67) that improved the ORR activity. Theoretical studies revealed that O*, OOH* and H2O2 were formed only on Co–N4–C sites. The generated H2O2 could spillover to Pt(111) sites to produce H2O when Pt(111) was in the vicinity of Co–N4–C sites. In addition, a carbon layer-coated PtNi ORR electrocatalyst, named PtNi@C, was obtained from the pyrolysis of ZIF-like nanomaterials, which were synthesized by a one-pot solvothermal process from Ni2+, Pt2+ and 2-MIM.234 Several factors contributed to the high ORR efficiencies, including the optimized adsorption energy of oxygen-containing species after alloying Pt with Ni atoms, the modulated electronic structure of PtNi by the N-doped carbon nanoshell, and the physical barrier functionality of carbon coatings towards metal agglomeration and dissolution. Regardless of the type of organic materials used as the carbon precursor, the pyrolysis conditions should be precisely controlled as the thickness and graphitization degree of the carbon nanoshell determine the catalytic efficacy of metal@C materials.84–86
Inorganic metal oxide overlayer-fabricated precious metal based ORR electrocatalysts. Silica oxide is the most investigated coating material among the metal oxides employed for the design of high-performance fuel cell electrocatalysts. Takenaka et al.140,141 have demonstrated that silica overlayer-coated Pt-based electrocatalysts exhibit enhanced ORR efficiencies. The selection of silica precursors usually has an effect on the features of the obtained silica overlayer with regard to porosity and hydrophilic/hydrophobic properties. Catalyzed by a base such as the ammonia water solution, the C2H5O–Si bonds in TEOS could hydrolyze into Si–OH groups, which could further undergo Si–O–Si bond formation with a high polycondensation degree. Unlike C2H5O–Si bonds, the CH3–Si bonds in the methyltriethoxysilane (MTEOS) precursor cannot hydrolyze into Si–OH groups. As a result, the silica overlayer (CH3–SiO2) derived from MTEOS shows larger pore size and hydrophobic surface property. The higher ORR activity of SiO2/Pt/CNT(MTEOS) was mainly attributed to the promoted diffusion of oxygen and water in the larger pores the catalyst featured.141 The micropore size of the TEOS-derived silica overlayer can be enlarged by using organic NH2(CH2)nNH2 as a template to benefit the diffusion of reactants and products on SiO2/Pt/CNT during the ORR process.235 The benefits from coating with silica layers are not limited to the well-known aspects in terms of preventing metal NPs from degradation and acting as the selective membrane for oxygen diffusion onto the active sites. As reported from the work of Park et al.,236 the silica overlayer with hydrophilic Si–OH surface species could help in increasing the proton conductivity especially in low-humidity conditions. According to them, the overcoating of Pt/C with silica layers enabled the preparation of the anodic catalyst layer for PEFCs with a low ratio of ionomer to carbon. As such, SiO2-coated Pt/C showcased a better electrocatalytic performance than the commercially uncoated Pt/C catalyst.
Metal–polymer composites and overcoated metallic catalysts. Polymers can incorporate transition metal sites into their matrix to work as non-precious electrocatalysts in fuel cells.71 To mimic the atomic configuration in cobalt porphyrins, for instance, conductive polypyrrole (PPY) loaded on Vulcan XC 72 carbon was selected as the entrapping matrix to prepare the Co–PPY–C composite.238 After entrapping into the PPY polymer, Co species were observed to strongly interact with the N atoms in pyrrole units by forming Co–N bonds. In this case, the as-formed CoNx species were active for the achievement of a power density of ca. 0.14 W cm−2 in H2–air fuel cells at 0.4 V. Likewise, the Co–PPY–C composite was extremely stable, without showing obvious activity drop after 100 h cell operations.
Transition-metal-derived N-doped carbon materials are normally produced from the pyrolysis of metal–heteroatom-containing composites or metal-coordinating complexes.94,239–241 Dong et al.240 obtained N,S-codoped carbon matrix-coated Co NPs through the in situ reduction of Co9S8 with melamine in the process of carbonization. S species could facilitate N element doping into the carbon matrix with a high richness of defects, which served as the additional active sites for the alkaline ORR catalysis. The realized high durability was assigned to the protective aid from carbon overlayers. Similarly, in order to inhibit the leaching of Fe into acid solution during the process of ORR, heteroatom-doped carbon-coated Fe catalysts with core–shell nanostructures have been developed. In the study of Park et al.,241 Fe@NSC20-7001-9001 (NSC: N,S-codoped carbon shell) served as the best performing catalyst, by showing an enhanced half-wave potential (0.794 V) and comparable ORR activity (0.864 mA cm−2) to Pt/C (0.811 V and 1.18 mA cm−2@0.85 V). The half-wave potential negatively shifted by only 41 mV after the stability test with 10000 potential cycles between 0.7 and 1.2 VRHE in O2-saturated 0.5 M H2SO4. Recently, Chen et al.93 studied the impact of S-doping of carbon layers on the ORR catalysis catalyzed by Fe@S,N-DC, which was composed of Fe encapsulated within S,N-codoped defective carbon layers (S,N-DC). In comparison with the S-free counterpart, the S,N-DC surface was characterized by enriched electron density, which could reduce the barrier towards the electron transfer from Fe to the S,N-DC shell, thus boosting the ORR activity.
Overcoated metal alloy and metal oxide catalysts. As demonstrated by Wu et al.,239 a PANI–FeCo–C catalyst was successfully fabricated through the pyrolysis of aniline, carbon black, cobalt(II) nitrate, iron(III) chloride and (NH4)2S2O8 oxidant under an inert atmosphere followed by acid leaching and second heating treatment. Upon pyrolysis at elevated temperatures, PANI got gradually converted into spherical carbon particles and resulted in a more graphitic structure when the temperature reached 900 °C. The resultant PANI–FeCo–C was the best-performing catalyst by exhibiting high activity and remarkable operative stability after 700 h at 0.4 V in H2–air fuel cells. Higher temperature (950 °C, 1000 °C) led to a non-uniform morphology for the carbon particles with a high loss of surface area and, thus an inferior catalytic performance.
In order to study the influence of carbon wall thickness on the ORR, Deng et al.242 pyrolyzed the mixture of graphene nanosheets (GNs) and Co2Fe(CN)6 to form highly dispersed FeCo alloy NPs, which were encapsulated by pod-like carbon nanotubes (Pod(N)) with fewer walls. GNs were effective in controlling the growth of carbon nanotubes. From the catalyst, named Pod(N)–FeCo/GNs, it was found that the thickness of the carbon wall distributes within 3–4 layers over the metal cores (Fig. 10i and j). ORR activity based on the half-wave potential (E1/2) of RDE polarization curves displays a volcano-shaped relationship with the GNs/Co2Fe(CN)6 mass ratio. 0.5/1 is an optimized ratio to obtain a maximum power density (328 mW cm−2) on Pod(N)–FeCo/GNs (Fig. 10k). More than 60% increase of power density was attained with respect to Pod(N)–FeCo (derived from direct-pyrolysis of Co2Fe(CN)6) in H2–O2 fuel cell tests. Theoretical studies indicated that the N dopants on the surface of Pod(N) could promote the adsorption of O2. Such a promotion effect appeared to decrease with an increase in the thickness of the carbon wall. It reasonably explained the observation that the catalyst with a thinner carbon wall displayed much higher activity towards the ORR catalysis. In addition, ORR catalytic efficiencies can be enhanced in most cases by the synergy of the metal alloy with the heteroatom-doped carbon nanoshell.95,125
Metal oxide core–shell nanostructures such as CoO@Co-g-C3N4 were reported to exhibit high ORR activity, long-term durability and good methanol-tolerance ability.94 The outer Co-doped g-C3N4 shell on the CoO@Co-g-C3N4 catalyst was assigned as the active site for ORR. In addition, Gao et al.243 demonstrated the preparation of N-doped carbon-overcoated Fe3O4 through the pyrolysis of in situ-polymerized PANI-coated MIL-101-Fe MOF nanocomposites. The PANI overlayer served as a highly conductive skeleton and the source of nitrogen dopants in the catalyst obtained. The authors found that the PANI-derived N-doped carbon nanoshell was crucial for facilitating O2 adsorption to enhance the ORR activity. They also investigated the effects of pyrolysis temperature and PANI coating amount on the ORR. From the optimal catalyst that was pyrolyzed at 900 °C with the initial mass ratio of aniline to MOFs at 1.5, a positive ORR onset potential was observed, close to that of the benchmark Pt/C catalyst in alkaline solution.
Overcoated other kinds of metal-based compounds. Carbon nanoshell-overcoated Fe3C-based electrocatalysts have been developed by pyrolyzing organic compounds for the ORR.120,244 In the catalyst overcoated with N-doped carbon shells, the coordination nests consisting of Fe3C and N atoms are active for the alkaline ORR catalysis.245 Additionally introducing heteroatoms such as boron (B) into the N-doped carbon matrix could enhance the ORR activity over the Fe–B, N–C active sites.246 In addition, Karuppannan et al.102 recently reported a nitrogen- and fluorine-enriched carbon-overcoated Fe/Fe3C ORR electrocatalyst (NFC@Fe/Fe3C). They examined the effect of pyrolysis temperature on the structural and electrocatalytic properties of NFC@Fe/Fe3C. On increasing the temperature above 900 °C, there was a decreasing trend of the contents of nitrogen and fluorine in the carbon matrix, and the Fe3C phase was converted into the FeC phase through carbon diffusion into face-centered cubic iron crystals. NFC@Fe/Fe3C-9, a sample that was pyrolyzed at 900 °C, exhibited the best ORR performance with a half-wave potential of 0.87 V, which was 16 mV higher than that of commercial Pt/C in 0.1 M KOH. It also showed exemplary durability, where 26 mV degradation after 50
000 cycles (0.6–1.0 VRHE) in alkaline solution and 27 mV decay after 30000 cycles (0.6–1.0 VRHE) in acid media were attained. The electrical interactions between the outer carbon shell and the inner metal core have been similarly observed on carbon-coated C@Co-P/C,247 N-rich carbon shell-coated CoNP@NC/NG248 and B,N-codoped graphitic carbon shell-encased Co2P,249 representing the efficient ORR electrocatalysts.
To sum up, surface overcoating-derived ORR electrocatalysts can be fabricated in a rational way, which requires a suitable choice of coatings including organic matters, inorganic carbon nanoshells and metal oxide layers. By integrating the benefits such as the modulated electronic structure of metal species, the sterically hindered process of metal coalescence/sintering and dissolution as well as the regulated adsorption behaviour of spectator species, the as-made precious metal based electrocatalysts could expectantly exhibit strengthened catalytic properties for the cathodic ORR in PEFCs. With the aid of the protection of transition metal NPs from contacting with the harsh electrolyte environment, along with the synergistic effects between heteroatom-doped carbon coatings and metal cores, remarkably reinforced electrocatalytic activity and durability can also be achieved on non-precious metal based electrocatalysts for the ORR catalysis.
3.2 Overcoated electrocatalysts for the HOR
As the anode reaction of a H2–O2 fuel cell, the HOR in acid and alkaline solution proceeds via the following: H2 → 2H+ + 2e− (acidic condition) and H2 + 2OH− → 2H2O + 2e− (alkaline condition). In an acidic solution, the only intermediate on the metal surface is the adsorbed hydrogen (Had); thus, the hydrogen binding energy (HBE) is widely accepted as the activity descriptor parameter for the acidic HOR.250 When switching the acidic operating environment to a basic one, the HOR shows markedly slow kinetics even when catalyzed by Pt-based catalysts. We note, in passing, that recently Ru-alloyed electrocatalysts such as PtRu,251,252 RuIr,253 and IrPdRu254 have been innovatively developed as the most active materials for the alkaline HOR. Massive efforts have also been devoted for an in-depth understanding of the mechanism of the alkaline HOR with the aim of designing active and stable electrocatalysts.255,256 HBE257 and hydroxyl binding energy (OHBE, also known as oxophilicity)251 are among the proposed theories for the alkaline HOR. In the elementary HOR process in an alkaline solution, the adsorbed molecular H2 dissociates into surface adsorbed Hadvia a Tafel step (H2 + 2M* → 2M*–Had) or the Heyrovsky step (H2 + OH− + M* → M*–Had + H2O + e−), followed by a Volmer step (M*–Had + OH− →M* + H2O + e−) from where H2O and one electron are produced. The anodic HOR process mostly follows the Tafel–Volmer pathway and the Heyrovsky–Volmer pathway on catalytic active sites. Notably, as the attractive alternative to PEMFCs, AEMFCs open up the possibilities of applying earth-abundant and low-cost catalysts for the alkaline HOR, since non-PGM based electrocatalysts have been developed with Pt-like performance for alkaline ORR.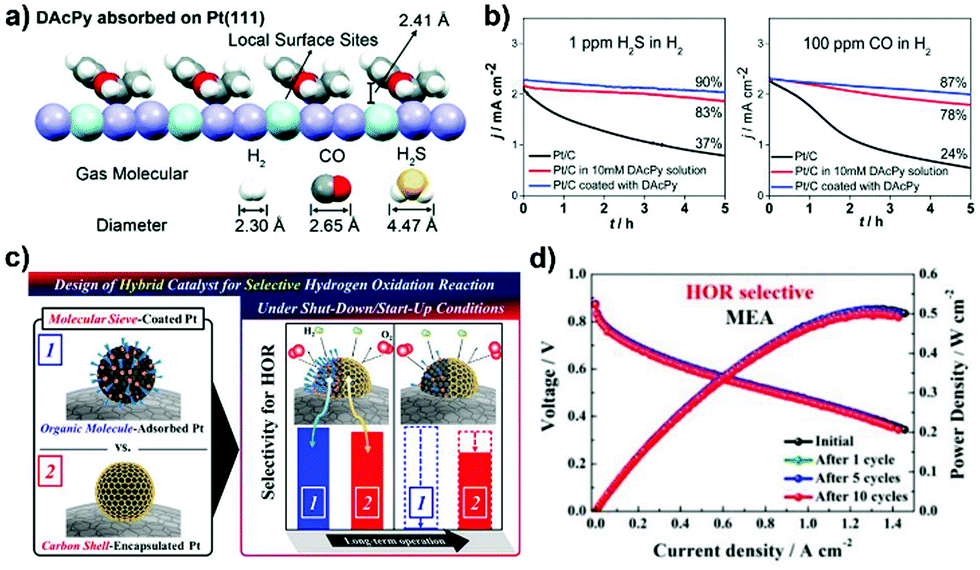 | ||
| Fig. 11 (a) The side view of the proposed model for reduced-form DAcPy adsorbed on Pt(111). (b) H2S and CO tolerance tests in a 0.5 M H2SO4 solution saturated with 1 ppm H2S and 100 ppm CO balanced H2 gas at 0.1 VRHE and 1600 rpm. Reproduced from ref. 258 with permission from Royal Society of Chemistry, Copyright 2018. (c) Schematic illustration (left) of molecular sieve-coated Pt catalysts using organic materials and carbon shells; the changes in HOR selectivity (right) of the catalysts during the long-term operations in PEMFCs. (d) Changes in the single-cell performance of the HOR-selective MEA after shut-down/start-up cycles. Reproduced from ref. 261 with permission from American Chemical Society, Copyright 2019. | ||
Under shut-down/start-up conditions, unexpected ORR on the anode side because of the crossover of O2 through the membrane could cause a high cathode potential, which could lead to oxidative corrosion of carbon supports and performance degradation of electrocatalysts. Surface engineering of Pt NPs with coatings acts as a kind of promising strategy to overcome this issue. As earlier reported by Marković's group, patterning Pt with a self-assembled monolayer of calix[4]arene molecules featuring a wide rim structure endowed the catalyst with a high selectivity for the HOR.63,64 High occupation of Pt by the calix layer could not change HOR activity since it has fast kinetics in an acidic electrolyte. In contrast, the ORR was inhibited because of the insufficient sites for O2 adsorption and O–O bond cleavage on the Pt surface following the organic patterning. Similarly, an enhanced HOR selectivity was attained by using fine-tuned dodecanethiol/Pt ensemble sites after optimizing the concentration of surface organic molecules.260 In this study, high durability was achieved from the cell test, although part of thiol groups would be detached from the surface. The instability of organic molecules coated on the Pt/C catalyst was also revealed from the work of Jang et al.261 Oleylamine-coated Pt NPs supported on carbon material were first prepared, denoted as Pt@C/C ASP. Following annealing at 600 °C and 900 °C in an Ar atmosphere, Pt@C/C ASP got converted into a nanoporous carbon molecular sieve-encapsulated Pt catalyst, named Pt@C/C 600 and Pt@C/C 900, respectively. The HOR selectivity and electrochemical stability of the attained catalysts were evaluated by collecting ORR and HOR polarization curves after every 1000 cycles between 0.05 and 1.05 VRHE in Ar-saturated 0.1 M HClO4. The results indicated that the ORR activity of Pt@C/C ASP was totally recovered after 3000 cycles due to the detachment of organic molecules, whereas that of Pt@C/C 900 was slightly changed and showed an excellent long-term durability combined with a high selectivity to the anodic HOR (Fig. 11c). According to the authors, a higher pyrolysis temperature promised the formation of nanoporous carbon shells featuring a higher carbon density and smaller pore size, which were responsible for the selective permeation of H2. The potential application of Pt@C/C 900 as the anode catalyst in PEMFCs was evidenced by transforming it into a membrane electrode assembly (MEA). In comparison with the conventional MEA with severe degradation after the first cycle, the HOR-selective MEA showed well maintained single-cell performance after running for 10 shut-down/start-up cycles (Fig. 11d).
Coating with a metal oxide overlayer can also boost the HOR efficiencies of PGM-based electrocatalysts. Owing to the oxygen-deficient and high oxophilic character, CeOx has been applied in the development of Pd–CeOx nanocomposites with intimate contact between Pd and CeOx for the alkaline HOR.262 Speck et al.263 fabricated a quasi-2D Pd/CeOx buried interface by ALD technology to investigate the HOR activity and stability in an alkaline electrolyte. In this work, the accelerated alkaline HOR activity was ascribed to the bifunctional mechanism where the adsorption/dissociation of H2 takes place on Pd sites and the CeOx film provides OH− to facilitate the desorption of strongly adsorbed H on Pd via the Volmer step. Benefiting from the semipermeable property of the CeOx film, much high stability of Pd was achieved since it could not only prevent the metal from oxidation but also impair the outward diffusion of metal ions to the bulk electrolyte. In addition to CeOx, TiOx encapsulation of Pt NPs by forming strong metal support interactions (SMSIs) is effective at tuning the HOR selectivity. Stühmeier et al.264 reported that partial-encapsulation of Pt NPs with TiOx overlayers through reductive heating treatment could hinder the process of Pt oxidation and oxygen reduction, while allowing superior HOR selectivity and activity at high potentials. The influence of SMSIs on the electrochemical behavior of Pt was investigated by Geppert et al.265 Coating with a TiOx thin-film could reduce the ability of Pt to catalyze the ORR and OER, while no influence was observed on the HOR even above 1.2 VRHE. These findings were attributed to the selective permeability of the TiOx film, allowing proton and hydrogen diffusion but impairing the transport of oxygenated species.
:Ni2+ ratio on the Ni interfacial surface oftentimes determines the HOR performance. For instance, a volcano-type dependence of HOR specific activity was revealed on Ni/NiOx electrodes, in which the catalyst with 30% coverage of NiOx on its surface showed a 14-fold increase.285 In addition, when the molar ratio of Ni(OH)2/Ni was around 1.1/1, maximum HOR activity was obtained on the Ni(OH)2-decorared Ni/C catalyst that was generated by an electrochemical oxidation method.286
Stability is one of the important criteria for pursuing cost-effective HOR electrocatalysts. Under a higher anodic potential (i.e. 0.7 V), metallic Ni-based catalysts can be fully oxidized in the long-term HOR process, causing performance deterioration.287 Apart from alloying with another metal, overcoating of Ni NPs with inorganic nanoshells was proven to be efficient for improving the antioxidation capability of Ni-based catalysts. For example, Gao et al.288 revealed that coating with a few-layer hexagonal boron nitride (h-BN) shell resulted in weakened interaction of O, H and OH species on the Ni surface and thus a better alkaline HOR performance catalyzed by the retained metallic Ni. Later, the group of Zhuang reported carbon shell-overcoated Ni catalysts for the alkaline HOR.289 The Ni catalysts were generated by vacuum pyrolysis of nickel acetylacetonate under different temperatures. In this study, Ni@C-500 °C delivered the highest utilization of Ni active sites and the best HOR activity among the catalysts obtained. Although a higher pyrolysis temperature, i.e. 600 °C and 700 °C, benefited the formation of carbon shells with a higher graphitization degree and a higher percentage of Ni0, the ECSA of Ni@C catalysts showed a markedly decreased trend since the carbon shells would hinder the interaction of the electrolyte with the inner Ni cores. This study demonstrates that carbon shells are able to prevent the passivation of metallic Ni during the electrochemical HOR process. On the other hand, the thickness of the carbon nanoshell needs an elaborate control for achieving high HOR activity, as the intrinsic HOR activity can be more than 100% increased when decreasing the thickness of carbon from three to one layer.290 It is worth noting that, not limited to Ni, the heteroatom-doped carbon layer has also been applied to fabricate transition metal carbides, i.e. W2C@N,PC (N,P-doped carbon encased W2C NPs), as the promising alternatives to Pt/C for the HOR catalysis across a whole pH range.291
In spite of the progress related to the transition metal based materials, the alkaline HOR activities of these catalysts are still inferior to those catalyzed by PGM-based electrocatalysts. According to the cell performance tests in AEMFCs, a power density of around 1000–1200 mW cm−2 can be achieved on PtRu292 and CeOx–Pd/C262 anodic catalysts. On the other hand, the power density obtained from Cr@Ni293 and Ni3Mo/C268 was only 70 and 120 mW cm−2, respectively. In addition, non-precious metal catalysts usually have a thick layer on the anode because of the need of high loading, which would have a pronounced impact on the mass transfer of reactants and water management.289 Accordingly, facile synthetic strategies along with the combination of advanced characterization technologies with theoretical calculations are desired to engineer highly active and stable HOR electrocatalysts for practical application in AEMFCs.
3.3 Overcoated electrocatalysts for direct alcohol fuel cells
The direct alcohol fuel cells, in particular, the direct methanol fuel cells (DMFCs), are promising portable power sources due to the high power density and high efficiency. In DMFCs, the poisoning of PGM-based electrode catalysts caused by the carbonaceous intermediate, i.e. CO, usually impairs the cell performance. Intriguingly, the strategy of catalyst overcoating engineering provides the opportunities of improving the anti-poisoning capability of electrocatalysts towards CO in the process of MOR. The underlying origin of the CO anti-poisoning ability of polymer double-shelled electrocatalysts for highly efficient MOR catalysis was proposed by Yang et al.76,77,79 The PyPBI overlayer could stabilize Pt–OHad by forming Pt–OH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) species through hydrogen bonding. The stabilized Pt–OHad then facilitates the oxidation of CO to CO2 (Pt–(CO)ad + Pt–OHad → CO2 + H+ + e−). On the other hand, the PVPA coatings with multi OH groups could undergo hydration reactions to accelerate the formation of Pt–OHad (Pt + H2O → Pt(OH)ad + H+ + e−), which consume further Pt–(CO)ad species to generate active Pt surface. They demonstrated that the kinetics of MOR can be accelerated because of the fast oxidation of CO to CO2 in the presence of multi OH groups from the polymer overlayers. As mentioned in Section 3.1, the employment of pyridine molecules on the cathodic Pt/C catalyst helped to significantly avoid the adsorption of methanol and ensured a high ORR efficiency.221 However, as a consequence of the cooperation of steric and electronic effects from pyridine, when pyridine-capped Pt/C was used as the anodic catalyst for the MOR, the MOR mass activity linearly decreased with the increased coverage of pyridine on the catalyst surface. This work indicates that the impact of organic ligands on metal-based catalysts needs specific study and understanding for implementation in various catalytic reactions.
species through hydrogen bonding. The stabilized Pt–OHad then facilitates the oxidation of CO to CO2 (Pt–(CO)ad + Pt–OHad → CO2 + H+ + e−). On the other hand, the PVPA coatings with multi OH groups could undergo hydration reactions to accelerate the formation of Pt–OHad (Pt + H2O → Pt(OH)ad + H+ + e−), which consume further Pt–(CO)ad species to generate active Pt surface. They demonstrated that the kinetics of MOR can be accelerated because of the fast oxidation of CO to CO2 in the presence of multi OH groups from the polymer overlayers. As mentioned in Section 3.1, the employment of pyridine molecules on the cathodic Pt/C catalyst helped to significantly avoid the adsorption of methanol and ensured a high ORR efficiency.221 However, as a consequence of the cooperation of steric and electronic effects from pyridine, when pyridine-capped Pt/C was used as the anodic catalyst for the MOR, the MOR mass activity linearly decreased with the increased coverage of pyridine on the catalyst surface. This work indicates that the impact of organic ligands on metal-based catalysts needs specific study and understanding for implementation in various catalytic reactions.
A typical example of a carbon nanoshell-encapsulated catalyst developed with high activity for the MOR is Pt3CoRu/C@NC, as reported by Wang et al.294 On the surface of commercial Pt/C, they built two space-confined interfaces by overcoating of the PDA layer followed by the addition of Ru3+ and Co3+ atoms and the sequential overcoating with the inorganic SiO2 shell. The inner PDA layer not only serves as the precursor of N-doped carbon but also provides chelating sites for Ru3+ and Co3+ atoms. In the process of H2-assisted pyrolysis, the outer SiO2 shell could avoid PDA decomposition loss and prevent as-formed Pt3CoRu NPs from coalescence and sintering. The obtained Pt3CoRu/C@NC in this example, when used as the MOR catalyst, showed 4.2 times higher mass activity (0.97 mA μgPt−1) and 5.3 times higher specific activity (1.6 mA cm−2) compared to that of the commercial Pt/C catalyst. CO stripping voltammograms showed that the onset potential (0.35 V) of COad oxidation on Pt3CoRu/C@NC was more negative compared to that on Pt/C with 0.82 V, pointing to a higher CO-antipoisoning ability during the MOR catalysis. Electrical modification of Pt with Ru and Co is responsible for facilitating the methanol dehydrogenation process and the removal of surface-generated COad species. Pt3CoRu/C@NC also possesses an improved structural and chemical stability because of its ordered structure and the protective aid of the carbon nanoshell.
In addition, membrane-coated electrocatalysts for the MOR were also studied on the basis of the strategy of controlling transport phenomena on the catalyst surface.138 A silicon oxide (SiOx)-buried Pt thin film electrode was fabricated by Robinson et al.295 as a highly active MOR electrocatalyst. The overcoating-derived permeable SiOx overlayer with less than 10 nm thickness could prevent metal Pt from corrosion and maximize the density of active sites at the metal|metal oxide interface. In this work, a distinct increase of CO-tolerance ability was indicated by collecting CO stripping voltammetry curves, from which the CO oxidation peaks were found to shift to negative potentials when increasing the thickness of the SiOx overlayer. In addition, silanol groups on SiOx played a promoting role in the removal of COad species from the surface of Pt (which is known as the bifunctional mechanism) and thereby a 2-fold activity enhancement towards the MOR compared to the bare Pt thin film electrode was achieved.
3.4 Comparison of the surface coatings suitable for the electrochemical reactions in PEFCs
In view of the fact that the organic and inorganic coatings as discussed above are all beneficial for the electrochemical reactions in PEFCs, the surface overcoating strategy could usher in emerging opportunities for demand-guided design of electrocatalysts to deal with the practical application use. Generally, the benefits from coatings on metal NPs are most frequently reported in terms of stability improvement and reaction kinetics control. The overcoating layer especially the carbon nanoshell and metal oxide could act as a physical barrier toward the metal NP coalescence/sintering and detachment from the surface of the catalyst. The direct contact of the electrode catalyst with the highly oxidative or corrosive electrolyte could also be avoided by the coating overlayers, thereby suppressing the metal dissolution, surface passivation and catalytic deactivation. It is therefore expected that carbon nanoshells and metal oxide overlayers are more suitable when considering the improvement of both structural and chemical stability of metal catalysts for the ORR, HOR and alcohol electrooxidation reactions under different operating conditions in PEFCs.The mechanisms of overcoating in controlling the reaction kinetics of metal catalysts can be mainly classified into the following three categories: (i) size-selective eletrocatalysis, (ii) electronic interactions and modulations, and (iii) overlayer-assisted electrocatalysis (can also be called the bifunctional mechanism). Depending on the molecular size of reactants and/or spectator species, the coating overlayer can be rationally constructed to control their transport on/to the metal surface. As such, extremely high catalytic selectivity toward the HOR vs. ORR (under shut-down/start-up conditions in H2–O2 fuel cells)63,64,260,261,265 and the ORR vs. MOR (the case associated with methanol crossover in DMFCs)51,54,56,75,219–221 has been achieved. ORR and HOR catalytic activity can be additionally enhanced because of the abundant active sites free from blocking by poisoning species.224–227,258,259 Moreover, the organic surfactant/polymer and heteroatom-doped carbon nanoshell can bring in the electronic effects on metals by modulating their affinity to reactants and active surface intermediates (i.e. H*, OH*, OOH*) with appropriate binding states and, thus boosting the ORR and HOR activities. However, the reports on the electronic effects of metal oxides especially the silica overlayer for catalyzing the electrochemical reactions such as ORR and MOR are limited. Furthermore, polymer and metal oxide overlayers are the suitable coating of choice for overlayer-assisted electrocatalysis. In particular, this concept has been validated to be efficient for accelerating alcohol electrooxidation reaction and circumventing the CO-poisoning issue in DMFCs.76,77,79,295 By providing hydroxyl groups on the surface, metal oxides can also facilitate the sorption behaviour of H during the alkaline HOR process.263 Based on these advances, it is important to select a suitable coating overlayer for enhancing the efficiencies of metal based catalysts in diverse electrochemical reactions. It is also necessary to exploit the knowledge of the stability of coatings on the metal surface under a variety of conditions. Any single effect or the co-existence of these catalytic effects as discussed above may influence catalyst's catalytic performance, which also depends on the operating conditions, i.e., testing configurations. Reasonable differentiation and quantitative understanding of these beneficial effects are still in their developing stage, which calls for great experimental efforts to deal with the practical distribution of overcoating-derived electrocatalysts in PEFCs.
3.5 Overcoated supports for the electrocatalysts in PEFCs
Inhibiting the corrosion of the carbon support. Polymers65,79,151 and metal oxides such as SnO2,204 Ta2O5,205 SiO2,296 and ZrO2297 can efficiently protect the carbon support from oxidative corrosion under operating conditions. From XPS quantitative analysis, unobvious carbon loss presumably by the way of carbon oxidation to CO2 or CO was observed on Pt/C@PANI.65 PANI coatings endowed the carbon support with extremely robust functionality, which could reduce the probability of Pt NP degradation. It was verified by the findings from the accelerated stress tests (0–1.2 VRHE) that, only 30% ECSA loss was observed on Pt/C@PANI, lower than that (83%) of Pt/C. In addition, based on the cyclic voltammetry (CV) curves of the polymer double-shelled NanoPC/PyPBI/Pt/PVPA catalyst,79 a comparison with that of commercial CB/Pt showed a smaller hydroquinone–quinone (HQ/Q) redox peak even after 50
000 potential cycles (1.0–1.5 VRHE) in 0.1 M HClO4 at 60 °C, indicating a higher resistance towards corrosion after overcoating the carbon support with polymers. In this work, the origin of the high stability of the carbon material was speculated to be the multipoint acid–base interactions between outer surface-coated PVPA and PyPBI, the latter was overcoated on nanoporous carbon (NanoPC).
The mechanism of inherent stability improvement by overcoating PBI polymer layers on carbon black was studied by Fujigaya et al.158 Based on N2 physical-adsorption analysis, they discerned the conclusion that PBI overlayers would preferentially coat the gate of micropores and interior mesopore sites, rather than the surface of micropores inside the carbon support. Since the inner micropore surface has a high degree of defects, PBI overlayers could efficiently suppress the penetration of the electrolyte into micropores, and thus preventing the carbon matrix from being oxidatively corroded. This protection mechanism is believed to be applicable to other kinds of polymer-overcoated carbon-supported metal catalysts.
Improving the dispersion of metal nanoparticles. Carbon materials with a highly crystalline graphitic structure and good electrical conductivity are ideal supports for electrocatalysts. Insufficient functional groups on the surface of the carbon support usually weaken the stabilization effect on metal NPs. Such limitation on the carbon support could be overcome by overcoating with organic materials such as PyPBI,72,73 PANI,150–152 PDA,156 Ppy,157 and PBI159,160 or by surface-grafting NH2- and SO3H-containing functional groups.298 Aniline molecules adsorbed on CNTs have been reported to serve as the separate coordination sites for Pt4+ and strongly interact with the reduced Pt(0) particles to have a high dispersion.299
Polymer overcoatings on the carbon support could help in the fine control of the size and size-distribution of supported metal NPs through strong coordinations,72,73,152,159 although the electrical conductivity of the catalyst may be affected. We note that, owing to the fact that polymer overlayers preferentially block the gate of micropores in carbon,158 the deposition of metal entities in the micropores could be suppressed;151,160 therefore, the utilization efficiency of metal active sites is expectantly raised. In this respect, PyPBI-coated carbon black allowed less Pt loading with the smallest particle size (2.4 ± 0.2 nm) and the highest ECSA (120.8 ± 12.0 m2 gPt−1) on CB/PyPBI/Pt0.1/PVPA (0.1 is the weight ratio between Pt feed and polymer-coated carbon support).78 A record MOR mass activity (1860 mA mgPt−1) was thus achieved, together with 10 times higher durability than commercial CB/Pt. Also, as higher as 92% utilization efficiency of Pt was realized on CNT/E-HBM/Pt (E-HBM: end-capped hyperbranched sulfonated macromolecules), outperforming that (64%) of CNT/Pt without polymer overcoating on the CNT support.300 Electrochemical tests on CNT/E-HBM/Pt showed 20 and 1.4 times higher durability and ORR mass activity than the reference CB/Pt catalyst, respectively.
As a consequence of the formation of metal–N bonding, when metal NPs are loaded on polymer-overcoated carbon support materials, stabilized metal species with high dispersibility could be achieved. A high electrocatalytic activity is related to the improved utilization efficiency of active sites. It can also be attributed to the favourable electronic structure of metal nanostructures modulated by the coordination sites (or the defects) from the carbon support. An example of this is the immobilization of PtCo alloy NPs on PNIPAM-functionalized carbon (C-PNIPAM).169 In this work, the electronic structure of PtCo was remarkably changed by surface Co–N species, which were formed between Co atoms and PNIPAM. The formation of Co–N bonds not only indirectly enhanced the electron transfer from Co to Pt, but also hindered the oxidation of surface active sites. As a result, PtCo/C-PNIPAM displayed higher ORR activity and degradation resistance, in comparison with PtCo/C without PNIPAM functionalization. However, it was recently demonstrated by Kato et al.167 that the PBI overlayer on the carbon support could not impose an effect on the electronic structure of PtNi nanoframes immobilized but, instead, tended to stabilize Pt–(OH)ad species under the ORR conditions for activity improvement. Indeed, they found the electronic effect from the corresponding N-doped carbon-supported PtNi catalyst. These results indicate the importance of manipulating the microscopic environment of N atoms at the metal/support interface for efficient electrochemical reactions, which is expected to be a general scenario applicable for other heteroatoms.
The Nafion ionomer is widely used as the proton conductor and catalyst binder during the fabrication of the MEA. In most cases, the distribution of Nafion on the MEA is rather inhomogeneous and its content often has a large impact on the accessibility of reactants to metal sites. As from the recent works by Yang et al.301 and Jayawickrama et al.,160 when PyPBI and PBI adhesive overlayer-coated carbon materials were applied as the supports for the loading of Pt NPs, a homogeneous coating of the Nafion ionomer on the MEA was achieved. It was attributed to the existing acid–base interactions between the acid sulfonic group of the Nafion ionomer and the basic imidazole group of polymer coatings.301 The consequence is that, the formation of a long-range proton conduction path is facilitated, and metal utilization efficiency and structural stability are improved as well. The above results indicate the high possibilities of the polymer overcoating strategy for efficient electrocatalyst design, although the intrinsic aspects that are responsible for enhanced catalytic performance need careful studies in each case.
Forming strong metal–support interfacial structures. A wide assortment of metal oxides such as TiO2,190,199 AlOx,193 ZrO2,191 CeO2,194 Ta2O5,205,302 MnOx207 and MoOx209 have been investigated for the surface modification of carbon supports. Semiconducting TiO2 doped with Ta/Nb197 and N303 atoms with improved electrical conductivity is also employed. Apart from mitigating the corrosion of the carbon support and promoting the dispersion and utilization efficiency of the metal,193,200,210,296,297 metal oxide species in the form of particles or thin films could strongly interact with the metal NPs immobilized on carbon support materials.189,192,194,204,205,207 Recently, the studies of surface and interfacial interactions are becoming extremely vital for engineering advanced heterogeneous catalysts.304 Thus far, remarkable progress has been made in the understanding of metal–support interactions on metal-supported electrocatalysts, in particular, in which metal oxide coatings are present on the surface.
A metal–metal oxide–graphene triple junction was formed after the deposition of 2 nm Pt NPs onto indium oxide (ITO) nanocrystal-decorated graphene hybrids.305 This triple junction is distinguishable from TEM images (Fig. 12a and b) and its formation is thermodynamically favourable, as revealed by DFT calculations. Electrochemical tests on Pt–ITO–graphene hybrids show remarkable ORR activity and stability in comparison to Pt–graphene (Fig. 12c and d) and most of the other Pt metal based catalysts. Since then, creating a metal–metal oxide–support triple-phase boundary has paved the way for developing highly efficient ORR electrocatalysts such as Pt–TiO2/C,171 Pt/MnO2/GS (GS: graphene sheet),306 Pt–SnO2–prGO (prGo: protected and reduced graphene oxide)307 and ALD–TaOx–Pt/C.192 Particularly, we note in passing that area-selective ALD was readily applied for the selective nucleation and growth of TaOx particles (with 2.7 ± 0.5 nm diameter) around the carbon-supported Pt NPs.192 The triple junction structure of Pt–TaOx–C was imperative for the formation of strong metal–support interactions and, thereby stabilizing the as-immobilized Pt NPs and minimizing the ECSA loss during the electrocatalytic processes in PEFCs.
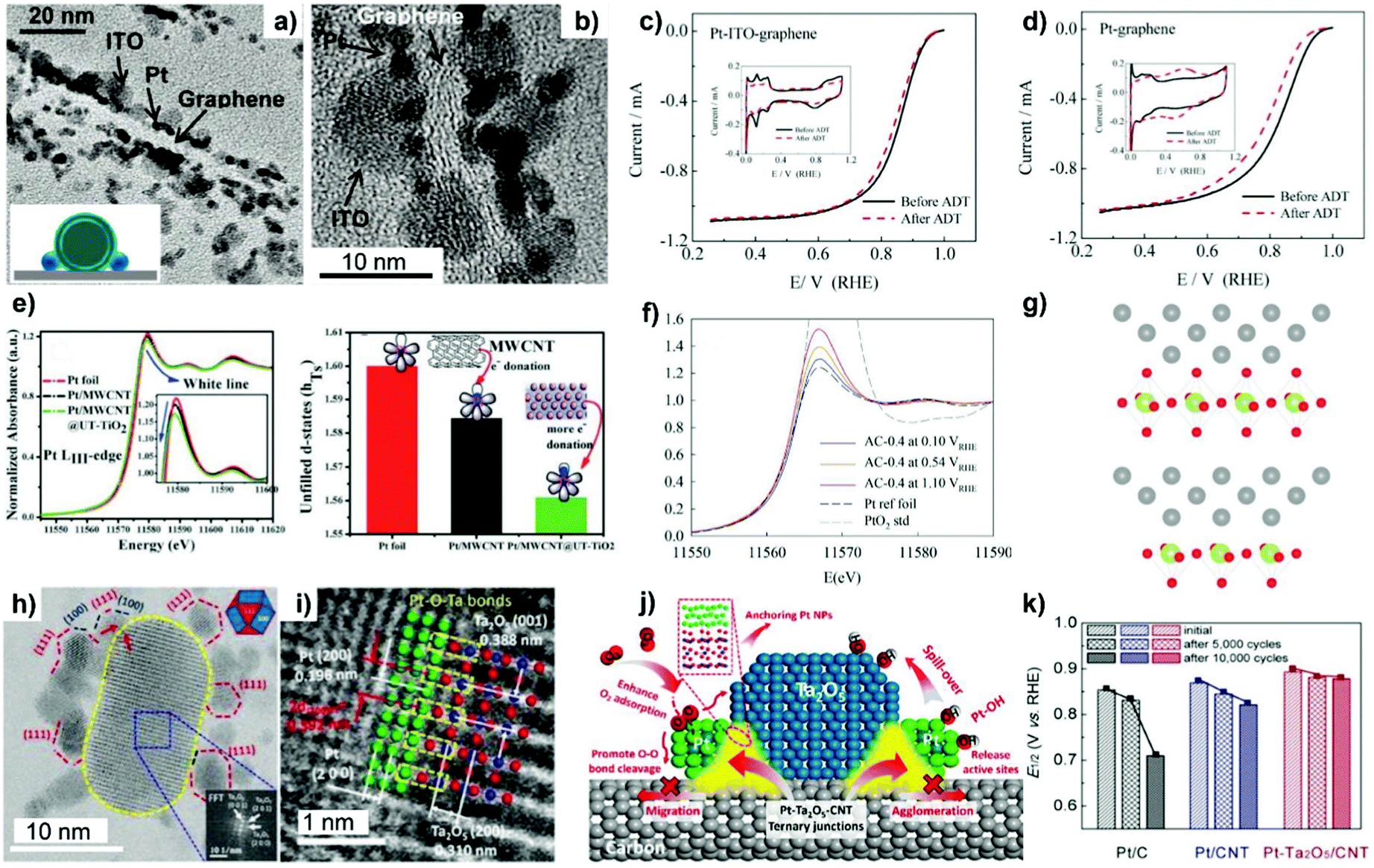 | ||
| Fig. 12 (a and b) Cross-section TEM images of Pt–ITO–graphene hybrids. The inset in (a) is the schematic structure of the Pt–ITO–graphene nanocomposite. LSV ORR polarization curves of Pt–ITO–graphene (c) and Pt–graphene (d) in O2-saturated 0.1 M HClO4 (5 mV s−1, 1600 rpm). Inset: CVs in N2-saturated 0.1 M HClO4 (50 mV s−1). Reproduced from ref. 305 with permission from American Chemical Society, Copyright 2011. (e) Pt L3-edge XANES spectra (the inset shows an enlarged region of peaks), while the right bar graph is the calculated unfilled d-states for Pt foil and other catalysts. Reproduced from ref. 200 with permission from Royal Society of Chemistry, Copyright 2012. (f) XANES spectra of AC-0.4 collected at the Pt L3-edge at 0.1 VRHE, 0.54 VRHE, and 1.1 VRHE in O2-saturated 0.1 M HClO4. (g) Interfaces between Pt and the saturated Nb2O5 (up) and unsaturated NbOx (down); silver, green, and red balls represent Pt, Nb, and O, respectively. Reproduced from ref. 308 with permission from American Chemical Society, Copyright 2017. (h) HAADF-STEM image (the inset shows the FFT pattern in the blue region) and (i) enlarged image of Pt–Ta2O5/CNT. (j) Schematic of the mechanisms responsible for the improved ORR activity and durability on Pt–Ta2O5/CNT. (k) Half-wave potential (E1/2) values after 5000 and 10000 cycles. Reproduced from ref. 309 with permission from American Chemical Society, Copyright 2019. | ||
Both geometric and electrical modifications occur on the metal, metal oxide and carbon support, which, in turn, induce the formation of unique interfacial active sites. Akalework et al.200 performed XPS and X-ray absorption spectroscopy (XAS) characterization to gain an in-depth understanding of metal–support interactions on ORR electrocatalysts. One of the findings of the negative shift of the Pt 4f XPS peak compared to uncoated Pt–MWCNTs is indicative of the interaction of Pt with ultra-thin TiO2-overcoated MWCNTs through forming SMSIs. As illustrated in Fig. 12e, the white line intensity (the d-band vacancy indicator) of the Pt L3-edge X-ray absorption near edge structure (XANES) follows the trend Pt foil > Pt–MWCNT > Pt–MWCNT@UT-TiO2. The formation of SMSIs and the decrease of Pt d-band vacancy provide evidence for the efficient ORR electrocatalytic property of Pt-MWCNT@UT-TiO2.
The nature of metal and metal oxide interactions (MMOIs) was intensively studied by Jia et al.308 in a representative Pt/NbOx/C system, where NbOx was saturated on amorphous carbon (AC) while it was unsaturated on the graphene nanoflake (GF) support. Electron transfer from Pt to NbOx was firmly verified by the collected Pt L3-edge and Nb K-edge XANES spectra. The in situ XANES spectra of the Pt L3-edge for AC-0.4 (Pt/Nb ratio is 0.4), especially that collected at 0.1 VRHE, show higher white line intensity than that of Pt foil, indicating the formation of Pt–O interactions between Pt and O species of NbOx (Fig. 12f). Until the potential reached 1.1 VRHE, the Pt–O peak shows a drastic increase and Pt–Nb interactions still exist on AC-0.4. Nevertheless, Pt on Pt-NbOx/GF was found to mainly interact with Nb during the entire voltage cycling. These findings point out that the composition of the metal–metal oxide (MMO) interface determines the interaction of Pt with either oxygen atoms or metal cations in metal oxides. As illustrated in Fig. 12g, Pt atoms interact with Nb in oxygen-deficient NbOx, whereas Pt–O interactions are first dominant on saturated NbOx followed by the formation of Pt–Nb bonds when O atoms immigrate onto Pt particles. It is worth noting that the as-formed Pt–Nb species are not the active sites for the ORR. In fact, the shorter Pt–Pt bonds resulting from Pt–O interactions impart the ORR process with high activity. Small-sized and low-coordinated Pt NPs are stabilized by NbOx with the formation of MMIO, leading to enhanced ORR activity and durability of the studied MMO system.
Gao et al.309 developed a carbon nanotube (CNT) on which 8.8 nm Ta2O5 NPs were uniformly dispersed to anchor Pt by forming metal–metal oxide–carbon junctions. HAADF-STEM images indicate that the Pt species, adjacent to the interface between highly crystalline Ta2O5 and carbon, tend to expose the {111} and {100} planes and form Pt–O–Ta bonds by lattice fringes overlapping at the Pt–Ta2O5 interface (Fig. 12h and i). Similar to the Pt–NbOx/C described above,308 the electron transfer was directed from Pt to Ta2O5, as revealed by the lower binding energy shift of Ta 4f XPS and the higher Pt L3-edge XANES white line intensity of Pt–Ta2O5/CNT. Pt–Ta2O5/CNT displays higher ORR activity and superior long-term durability than commercial Pt/C and Pt/CNT (Fig. 12k). The improvement in electrocatalytic properties is attributed to the combined features, including the particularly exposed Pt{111} and {100} planes, the electron-deficient Pt (active for O–O bond cleavage), and the spillover effect of Ta2O5 by reducing the formation of OHad on Pt sites (Fig. 12j). In addition, the strong metal–metal oxide interactions formed at the Pt–Ta2O5–carbon interface and the highly hindered Pt–O oxidation are advantageous towards inhibiting Pt NPs from migration/sintering and dissolution.
In short, the benefits from surface overcoating engineering on carbon materials in terms of preventing the carbon support from corrosion, improving the metal dispersion degree and forming strong metal–support interactions have been discussed. Further studies of surface overcoating-derived carbon-supported electrocatalysts should be focused on correlating the structural–performance relationships to provide novel guidelines for progress in the design of high-performance electrocatalyst.
Lee's group reported a Pt NP-deposited Mn3O4 catalyst with lower ORR mass activity than commercial Pt/C, owing to the smaller ECSA and poor conductivity.311 They further developed carbon thin layer-coated Mn3O4 nanocrystals as a suitable support for Pt NP deposition.213 Pt was exclusively deposited at the interfacial area between the Mn3O4 core and the outer carbon nanoshell, resulting in the formation of a Pt/Mn3O4–carbon triple-junction (TJ) structure. The afforded Mn3O4/Pts@C1.8nm (the catalyst overcoated with a 1.8 nm-thin N-doped carbon overlayer) exhibited significantly higher mass activity than Mn3O4/Pts and Pt/C, which was attributed to the synergistic effect of the TJ structure. Control-synthesized catalysts including Mn3O4/Pts@C700°C having larger Pt NPs, Mn3O4@C7.1nm/Pts without the TJ structure and Mn3O4/Pts–CB with physically mixed phase all showed inferior electrocatalytic performance in comparison with Mn3O4/Pts@C1.8nm. Fabricating a triple-junction nanostructure comprising metal particles, metal oxide and carbon support is readily applicable for the design of various electrocatalysts with promising properties.171,192,305–309
Increasing the thickness of the carbon shell on the metal oxide support is beneficial for the improvement of electrical conductivity and the uniform distribution of Pt NPs with high ECSA.217 A thicker carbon shell (i.e. 5.0 nm) on TiO2, however, could hinder the spillover of –OH species from TiO2 to Pt, and thus, resulting in inferior CO-antipoisoning ability during the MOR process. It indicates that providing –OH species from the metal oxide by rationally controlling carbon shell thickness is profitable for kinetic enhancement. Such a OH promotor mechanism also holds true for the case of SnO2-supported Pd NPs for the alcohol electro-oxidation reactions, i.e. MOR and EOR, in alkaline solution.312 The formation of reactive oxygenated species (OHads) and their delivery to Pd NPs in principle would be limited when overcoating the SnO2 support with carbon shells. Interestingly, the overcoating-derived catalyst, named Pd–(SnO2@C)/C, showed remarkable electrocatalytic properties. The proposed rationale for this finding was that the enhancement of electrical interactions between Pd and carbon shell-overcoated SnO2 acted as the dominant contribution to the high activities of alcohol electro-oxidation reactions. The carbon overlayer could electrically modify the SnO2 support and subsequently alter the surface adsorption of reaction intermediates, i.e. aldehyde and COad, to remove them from the catalyst surface and release metal active sites. The electronic interaction mechanism reported in this work could probably provide a guideline for the design of high-performance electrocatalysts in direct alcohol fuel cell applications.
4. Overcoated electrocatalysts for water electrolyzers
Water electrolyzers produce H2 and O2via the water splitting process by the application of electrical energy, representing attractive energy storage devices. Electrochemical water splitting comprises the two-electron cathodic hydrogen evolution reaction (HER) and the four-electron anodic oxygen evolution reaction (OER). Pt-Based materials are the most efficient HER electrocatalysts, while IrOx and RuOx are active for catalyzing the OER.313 Owing to the high price and scarcity of precious metals, numerous research efforts have been devoted to the design of low-cost transition metal-based nanomaterials for overall water splitting in acid and alkaline environments.314 Of particular note, there is often a high demand on long-term durability when electrocatalysts are applied for water splitting in acid solution. To date, rational synthetic strategies are still desirable to improve the utilization efficiency of active sites and enhance the electrocatalytic properties of both precious and non-precious metal catalysts to deal with the efficient application demands in WECs.4.1 Overcoated electrocatalysts for the OER
The OER, a half-reaction including HO*, O* and HOO* intermediate reactive species, takes place in acid and alkaline solution as follows: 2H2O → O2 + 4H+ + 4e− (acidic condition) and 4OH− → O2 + 2H2O + 4e− (alkaline condition). OER catalysis shows sluggish kinetics due to the multistep proton coupled electron transfer involved in O–H bond breaking and O–O bond formation. The performance of OER electrocatalysts is usually compared on the basis of some critical parameters such as the overpotential needed to achieve a current density of 10 mA cm−2, Tafel slope (the relationship between current density and overpotential), faradaic efficiency (the charge utilization efficiency for producing desired products) and long-term durability, i.e. by recording chronoamperometry curves over operation time.Earth-abundant transition metal-based OER electrocatalysts especially those applied in an alkaline environment have recently attracted growing research interest. Carbon materials are still the widely employed supports for OER electrocatalyst design, taking advantage of the features such as high structural and chemical stability, and excellent conductivity. In this section, a large number of surface overcoating-derived precious and non-precious metal based OER electrocatalysts will be discussed by correlating their intrinsic structural properties with the electrochemical performance.
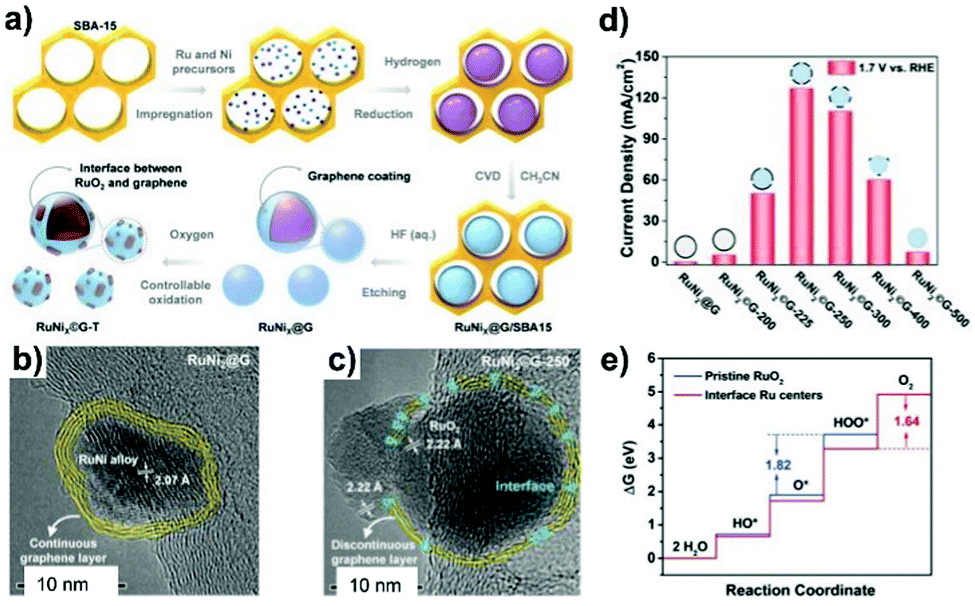 | ||
| Fig. 13 (a) Schematic of the synthesis of RuNi2@G-T with an interface between RuO2 and graphene. HRTEM image of (b) RuNi2@G and (c) RuNi2@G-250; the latter has a unique interfacial structure between RuO2 and graphene. (d) The current densities at 1.7 VRHE of RuNi2@G samples treated under different oxidation temperatures. (e) Free energy profiles for the OER over RuO2 and the interface Ru centers between RuO2 and graphene with an armchair edge at zero potential (U = 0). Reproduced from ref. 137 with permission from Wiley-VCH, Copyright 2020. | ||
Compared with RuO2, IrOx is the most suitable OER electrocatalyst benefiting from its high durability in acidic solution. Fujigaya et al.161 have demonstrated that PBI overcoating of multi-walled CNTs (MWCNTs) was effective in anchoring small IrOx NPs and improving their utilization efficiency for a highly active OER catalysis in 0.05 M H2SO4. Owing to the superior electrochemical stability of MWCNTs, the as-made MWCNT/PBI/Ir catalyst also exhibited an excellent durability. As chloride species often suppress the OER activity and make metal catalysts dissolved, an efficient control of chlorine evolution reaction (CER) is of vital importance for saline water electrolysis. It has been validated that the OER selectivity in the presence of impurities can be well regulated by overcoating a metal oxide on the surface of active sites. An example of this is the electrodeposited MnOx thin film serving as a physical transfer barrier towards the diffusion of chloride ions onto the inner IrOx catalytically active sites, which by itself made no contribution to the OER activity.146 The diffusion of water, protons and O2 species from the electrolyte to the active sites remained unaffected in the presence of the metal oxide overlayer on metal sites, as described in this case.
Metal, metal (hydro)oxide and metal alloy. Co-Based materials containing Co2+ and Co3+ species are active for the OER catalysis. TiO2 ALD overcoating onto a carbon-supported cobalt catalyst, i.e. Co/C, could enhance its OER property.143 In this work, OER activity was found to increase with ALD cycle number. 60 cycles of TiO2 ALD (0.79 mg cm−2 loading on Co/C) showed 2.5 times higher activity than commercial Pt/C in alkaline solution. An in-depth study revealed that the enhanced activity was attributed to the oxidation of Co by the TiO2 overlayer through the formation of Ti–O–Co bonds. Co with a high oxidation state was believed to be the active sites for the OER and, the rate determining step, viz., the reaction of adsorbed OH− with O* to form OOH* species, was accelerated. It is well known that Co-based oxide materials tend to undergo structural self-reconstruction, which leads to a boosted OER activity. In this case, the real active site on graphitic, nanocarbon-coated Co catalyst (named Co@NC-600) for the alkaline OER was identified to be CoOOH/CoO2 with high oxidation states, which was in situ electrochemically formed from metallic Co species.315 Yu et al.316 synthesized a polyfluoroaniline (PFANI)-coated Co(OH)2 catalyst featured with a core–shell structure for efficient OER application. The small overpotential (265 mV) to achieve a current density of 10 mA cm−2 was assigned to several positive aspects such as the optimized electronic structure of Co2+ in Co(OH)2 by the fluorine anion from the PFANI layer, the hydrophilic property of Co(OH)2@PFANI and the accelerated charge transfer process after overcoating with 1.7 nm-thin PFANI. In addition, g-C3N4 nanosheet coatings with 10 nm layer thickness can strongly couple with Co(OH)2 nanowires to form abundant active sites and promote the process of electron transfer.317 As a result, the g-C3N4-overcoated Co(OH)2 nanowires, named Co(OH)2@g-C3N4, manifested a surpassed OER activity in 1.0 M KOH with 320 mV overpotential at a current density of 10 mA cm−2, as well as long-term durability, in contrast to the performance of PGM catalysts (such as IrO2, RuO2, Pt/C) and individual Co- and g-C3N4-based materials.
As already discussed, overcoating metal active sites (i.e. Pt, IrOx) with an ultra-thin metal oxide overlayer is valuable for improving electrical reactivity and chemical stability.146,295 This strategy is applicable for the design of non-precious metal-based OER electrocatalysts as well. For example, Obata et al.318 reported the strategy of overcoating an anodic-deposited CeOx layer on the surface of NiFeOx to enhance its stability for the OER in alkaline solution. The permselective CeOx layer can not only ensure the permeation of OH− and O2 while preventing redox ions from diffusing through, but also inhibit the dissolution of Fe species during catalysis. In addition, it was validated that the interfacial effects resulting from CeOx overcoating on metal (hydro)oxide materials such as Ni(OH)2, bimetallic NiFe-hydroxide and spinel Co3O4 could induce strong electronic interactions on the as-coupled interface and enable tuning the electroadsorption of oxygenated intermediates and thus being responsible for the enhanced OER catalysis in an alkaline electrolyte.319–321
After studying CNT-confined Fe NPs,322 Cui et al.136 introduced single layer graphene onto 3d transition metals (Fe, Co, Ni, and FeNi, FeCo, CoNi alloys) by a CH3CN CVD process for the alkaline OER. FeNi@C showed the best OER activity among the catalysts, as shown by 280 mV overpotential to deliver a current density of 10 mA cm−2 and excellent durability after 10000 cycles, outperforming that of the commercial IrO2 catalyst (Fig. 14a). As determined by DFT calculations, the ΔG(O*) − ΔG(HO*) (the activity descriptor) of FeNi@C was 1.48 eV, which is close to the volcano peak. Electron transfer between metals and N dopants was found to enhance the adsorption of reactive intermediates, as reflected by the optimal ΔG(O*) − ΔG(HO*) in Fig. 14b. In addition, effective control of the carbon coating process and careful identification of structural properties of the catalyst are important to be considered.126,135 During the preparation of N-doped graphitic carbon-coated NiFe NPs by pyrolyzing NiFe-PBA materials, Ni appeared to be more prone to promote the carbon graphitization degree than Fe.126 The carbon shell with spherical (high Ni/Fe ratio) and tubular (low Ni/Fe ratio) morphology can be respectively obtained by tuning the Ni/Fe ratios in PBA precursors. XPS and XAS results revealed the electron donation from metallic Fe to the carbon shell, while there was no electron transfer for the case of Ni. Instead, Ni was found to strain the NiFe lattice, ascribed to the observation that both Fe–Fe and Ni–Ni bonds shifted in comparison with Fe and Ni foil. In addition to the modifications on the local and electrical structure of NiFe NPs, the N dopants in the carbon shell could make the carbon surface positively charged to facilitate the formation of oxygenated OH* and O* species, and ultimately enhancing the OER activity.134 Such a synergistic effect was also observed in single-walled CNT-embedded FeCo NPs323 and FeNi@N-doped graphene.98 Furthermore, the changes in alloy composition (i.e. FeCoNi alloy) by incorporating a third metal into a binary alloy could tune the electron transfer numbers between metal alloy and overcoated graphene layer;324 thus, the adsorption of surface intermediates was manipulated to achieve enhanced OER properties.107 Moreover, it was reported that pyrolytic treatment of PBA nanomaterials325 and ZIF polyhedra coatings133 could induce the metal-doping of the outer N-doped carbon shell. In these cases, both the synergistic effects and electron penetration from metal-doped NC shells to metal cores benefited the surface-dependent OER process.
 | ||
| Fig. 14 (a) Durability test of FeNi@NC in an alkaline electrolyte in comparison to IrO2. (b) The calculated negative overpotential (ηcal) against ΔG(O*) − ΔG(HO*) on different catalysts. Reproduced from ref. 136 with permission from Royal Society of Chemistry, Copyright 2016. (c) Overpotentials of the catalysts required to reach j = 10 mA cm−2. (d) Chronoamperometric responses at 1.50 VRHE (Ej=10 of Ni3N@NC-24). Reproduced from ref. 100 with permission from American Chemical Society, Copyright 2018. (e) Polarization curves of N-CoS2@graphene and other compared samples in 1.0 M KOH at 2 mV s−1. (f) Long-term stability measurements on N-CoS2@graphene in 1.0 M KOH. Reproduced from ref. 333 with permission from Elsevier, Copyright 2020. | ||
Metal phosphides, nitrides and chalcogenides. Metal phosphides and metal nitrides have been vigorously studied as active OER electrocatalysts. Introduction of a porous nanostructure onto active metal sites enables enhanced OER electrocatalytic properties. For example, Ni–Ni PBA direct pyrolysis-derived porous carbon coating on NiP nanoplates could improve catalyst conductivity, facilitate the charge transfer and reactant diffusion process, and promote the exposure of abundant active sites.326 In this work, the in situ-formed Ni oxide species served as the primary active sites for the alkaline OER catalysis. Likewise, Yu et al.327 reported that, in addition to the function of improving electrical conductivity, the carbon nanoshell could cooperate with the 3D Ni8P3 nanosheet for reducing the adsorption energy of intermediate catalytic species and help in protecting the Ni8P3 nanosheet from dissolution. As a result, the carbon-coated Ni8P3 nanosheet, named C@Ni8P3, exhibited better OER activity by offering lower overpotential (267 mV) to deliver a current density of 10 mA cm−2 than that (283 mV@10 mA cm−2) of uncoated Ni8P3. The decreased Tafel slope (51 mV dec−1vs. 78 mV dec−1) indicated facilitated electrokinetics of water oxidation after engineering carbon nanoshell coatings on the Ni8P3 nanosheet. In addition to the monometallic phosphides mentioned above, overcoating-derived bimetallic phosphides such as carbon-incorporated hollow-structured NiCoP nanoboxes,128 nanoporous carbon-coated Co0.7Fe0.3P@C328 and Co3FePx/C329 have been demonstrated as efficient OER electrocatalysts, in which the synergistic effects between the carbon coating and the metal core are suggested.
Overcoating-derived metal nitrides fabricated for catalyzing the process of OER are limited. Here we present the N-doped carbon-coated Ni3N holey sheets (Ni3N@NC) developed by Yuan et al.100 As displayed in Fig. 14c, overcoating a 4 nm-thin carbon shell enables a lower overpotential of 260 mV to obtain a current density of 10 mA cm−2 on Ni3N@NC-24 in 0.1 M KOH, whereas that of IrO2, NiO@NC, NC and Ni3N is determined to be 300, 350, 510, and 360 mV, respectively. Ni3N@NC-24 is also stable in alkaline solution by maintaining 94.4% of the initial activity after 10 h operation, more excellent than that (60.2%) of IrO2 under the same experimental condition (Fig. 14d). Further increasing carbon shell thickness to 8 nm for the case of Ni3N@NC-48 is inferior for the OER activity as explained by the access-blocking of the active Ni3N core.
Metal chalcogenides such as Co-based chalcogenides have drawn increasing research interest because of the features of earth-abundance and high activity for the OER catalysis. There are several reports on the synthesis of Co-based chalcogenides by utilizing the surface overcoating strategy.145,330–333 In particular, on the N,S-codoped carbon nanofiber-encapsulated Co9S8 catalyst for the OER tests in 1.0 M KOH solution, the observed overpotential of 302 mV at a current density of 10 mA cm−2 and the low Tafel slope of 54 mV dec−1 surpassed the performance of commercial RuO2.132 Apart from Co9S8, CoS2 ultrafine NPs overcoated with 1–5 layers of N-doped graphene were prepared by sequential pyrolysis of ZIF-67 and sulfurization with S.333 The resulted N-CoS2@graphene catalyst offers 205 mV overpotential to reach a geometrical current density of 10 mA cm−2 and a low Tafel slope of 65 mV dec−1 in 1.0 M KOH solution (Fig. 14e). In durability tests by keeping at 10 and 100 mA cm−2 current density, the potentials of N-CoS2@graphene are negligibly reduced (Fig. 14f), whereas the CoS2 NPs without overcoating with a graphene layer are found to be degraded rapidly. Fig. 15 is the comparison and summary of some representative surface overcoating-derived non-precious metal based electrocatalysts for efficient OER catalysis in alkaline solution (1.0 M KOH).126,132,136,316,317,323,325,327,329,333
 | ||
| Fig. 15 Comparison of the reported OER electrocatalysts in terms of the required overpotential at a current density of 10 mA cm−2 and Tafel slope in 1.0 M KOH solution. The corresponding electrochemical parameters for some of the uncoated catalysts are also plotted (orange color) for comparison. | ||
In brief, surface overcoating can bring various benefits for both precious and non-precious metal based electrocatalysts applied in the OER catalysis. The few reports of polymer- or metal oxide-coupled non-precious metal catalysts are limited to the applications in the OER in alkaline solution. In view of their high electrical conductivity, chemical stability and electronic interaction effects on the metal core, heteroatom-doped carbon nanoshells are more suitable compared with the polymer and metal oxide overlayer for the design of high-performance OER electrocatalysts. Typically, the improved inherent structural and chemical stability is attributed to the presence of the carbon shell with a few layers. Based on this functionality of the carbon shell, we expect that, electrocatalysts especially those composed of cost-effective 3d transition metals will be developed to be promising substitutes to precious metals for acidic OER applications. Moreover, by rationally controlling the thickness of the carbon shell, the electron transfer process can be facilitated by means of changing the adsorption behaviour of surface reactive intermediates on metal NPs, thus enhancing the OER in both acid and alkaline solutions.
4.2 Overcoated electrocatalysts for the HER
Hydrogen has shown great potential as a promising energy carrier employed in H2–O2 fuel cells for clean and sustainable energy. In addition to production from conventional industrial water–gas shift reaction, hydrogen can be produced by a cleaner and carbon-neutral way, i.e. water splitting process in water electrolyzer devices. One of the half-reactions of the HER, a two-electron-transfer reaction, takes place in acid and alkaline solution as follows: 2H+ + 2e− → H2 (acidic condition) and 2H2O + 2e− → H2 + 2OH− (alkaline condition). In general, there are two reaction steps involved in the HER process regardless of the type of electrolyte. The formation of adsorbed H (Hads) by the discharge of H2O or hydronium ions (H3O+) is the first step, which is known as the Volmer reaction. As the following second step, the production of H2 proceeds by either electrochemical desorption (Heyrovsky reaction) or chemical desorption (Tafel reaction) of Hads. Both the Volmer–Tafel and Volmer–Heyrovsky processes are involved in the HER. HER electrocatalysts based on precious metals and/or low-cost transition metals have been tremendously developed to reduce cost and adapt well under complex operating conditions.115,314 Specifically, the HER shows more sluggish kinetics in alkaline solution compared to that in an acidic system, mainly owing to the energy barrier for the generation of H+ from water dissociation.334 DFT calculations and recent experimental results have demonstrated that the HBE and oxophilicity can be considered as the dominant thermodynamic descriptors for HER activity,13,335 as similarly described for the HOR. Based on the Sabatier rule, the binding of catalysts with reactant molecules or intermediates should neither be too strong nor too weak. Hereto, optimizing the HBE on various surface sites is a reasonable solution for engineering optimal HER electrocatalysts for application in WECs. Similar to OER, some critical indexes such as overpotential, Tafel slope, exchange current density, and faradaic efficiency are used to elucidate the HER activity of designed electrocatalysts. In this section, surface overcoating-derived electrocatalysts favourable for the HER catalysis will be discussed.N-Doped hollow porous carbon polyhedron-overcoated Pt (Pt@NHPCP) was demonstrated as an efficient HER electrocatalyst through the pyrolysis of the Pt-encapsulated ZIF-8 nanomaterial.337 In addition, an ultra-thin 2D carbon layer carbonized from ZIF-67 was able to stabilize Pt NPs on the RGO support.233 The synergistic effect between Pt and Co–Nx–C active sites (ZIF-67 derivatives) contributed to the improved HER activity in both acid and alkaline solutions. DFT studies revealed that the benefited HER activity in acid solution was originated from the facilitated sorption behaviour of H species (with the lowest HBE of 0.23 V), while in basic media it was mainly due to the accelerated water dissociation (OH–H bond cleavage) to form Had, which remains the rate-limiting step in the alkaline HER process.
Overcoating an ultra-thin overlayer of metal oxide on the Pt electrode could aid in the achievement of high HER selectivity. Labrador et al.338 prepared a buried Pt/SiOx interface by overcoating the SiOx film with different thickness on a smooth Pt thin film. CV curves indicated that Pt was totally located beneath the SiOx overlayer and H+ was selectively accessible to Pt sites via diffusion through the SiOx membrane-like coating layer, while the diffusion of Cu2+ was blocked. As a result, the HER occurring at the Pt/SiOx interface exhibited high selectivity in the presence of Cu2+, a model poison for Pt which suppresses the HER activity. Indeed, a platinum oxide (PtOx) interlayer was identified at the Pt/SiOx interface and its properties can be regulated by the thickness of the SiOx or Pt film to result in strengthened electrocatalytic performance.339
Other precious metals such as Ru340 and Ag341 have been investigated by overcoating or encapsulating with carbon nanoshells for efficient HER catalysis. In particular, a recent work by Li et al.340 reported the preparation of a thin layer of N-doped carbon-coated Ru catalyst by thermally annealing polydopamine-coated Ru NPs (Ru@PDA). To investigate the effect of the Ru crystalline structure on the HER activity, the authors annealed the Ru@PDA composite at elevated temperatures. Ordered hexagonal-close-packed (hcp, (100) (002) surface) and fully crystallized Ru NPs with a diameter of 4 nm were formed when the pyrolysis temperature reached 700 °C. Higher temperature for example 800 °C resulted in the overgrowth of Ru NPs with a lower ECSA. Ru@NC-700 worked as the best HER catalyst in 0.5 M H2SO4 among the prepared catalysts, as shown by a small overpotential (27.5 mV) to reach a current density of 10 mA cm−2 and Tafel slope (37 mV dec−1) which was nearly close to that of Pt/C. Furthermore, the stability of hcp-Ru@NC-700 was at least 10 times better than that of Pt/C because of the protective aid of the carbon nanoshell.
As alloying with 3d transition metals can lower the loading of PGMs, ultra-thin carbon shell-coated NiRu,342 RuCo,343 and IrCo124 have been prepared through the direct pyrolysis of Ru- or Ir-incorporated MOF nanomaterials. For example, after pyrolyzing Ir-doped Co-PBA at 500 °C, the resultant IrCo@NC-500 catalyst exhibited even lower overpotential (45 mV) to deliver a current density of 10 mA cm−2 in N2-saturated 1.0 M KOH solution, which is 20% above the activity of 20 wt% Pt/C (Fig. 16a and b).124 A rapid electron transfer process occurred on IrCo@NC-500 as indicated by the smaller Tafel slope (80 mV dec−1) than that (101 mV dec−1) of 5% Pt/C (Fig. 16c). Durability evaluation in 0.5 M H2SO4 showcased only a slight cathodic current delay over the initial 5000 cycles due to the leaching of outer-exposed Co species; then, a stable current was reached after 10000 cycles because of the protection of the inner core by the graphene shell from leaching into the acidic solution. Theoretical studies demonstrated that the charge transfer from the metal core to the outer graphene surface facilitated the adsorption of H* on the graphene overlayer, eventually resulting in enhanced HER activity.
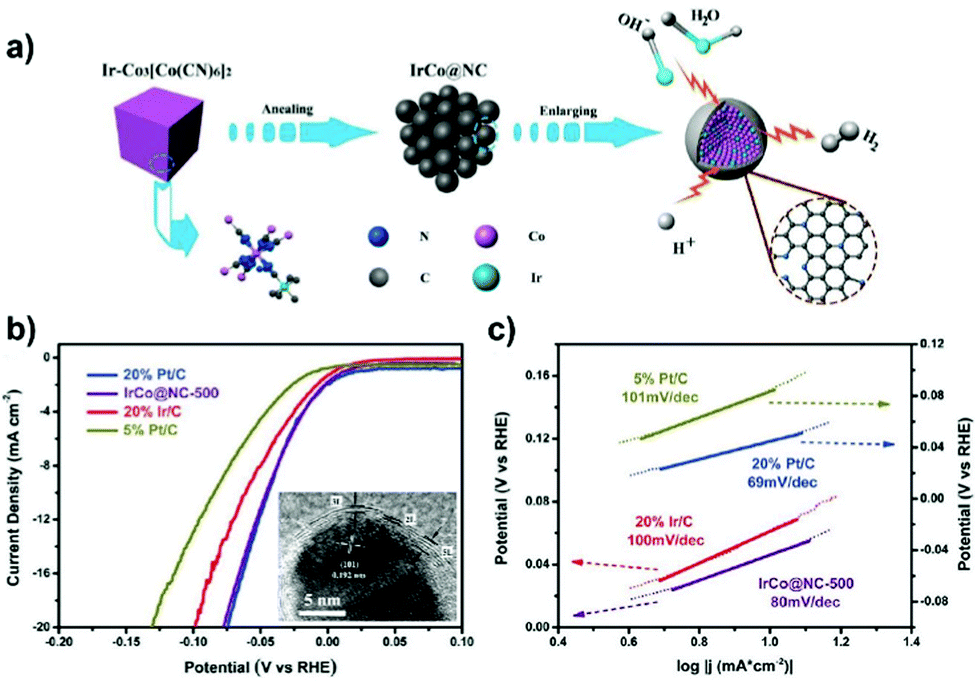 | ||
| Fig. 16 (a) Schematic illustration of the synthesis of IrCo@NC by direct pyrolysis of Ir–Co–PBA. (b) LSV polarization curves and (c) Tafel slopes of IrCo@NC-500, Ir/C, and Pt/C in N2-saturated 1.0 M KOH solution. The inset in (b) is the HRTEM image of IrCo@NC-500. Reproduced from ref. 124 with permission from Wiley-VCH, Copyright 2018. | ||
Precious metal-based phosphides hold promise for enhancing the HER activity as well, as a consequence of the regulations on the electronic structure of the metal by incorporated non-metallic P.108,250 It is known that electrocatalysts with Gibbs free energy of hydrogen adsorption (ΔGH*) close to 0 eV are active for catalyzing the HER. However RuP2 has a negative ΔGH*, meaning that hydrogen atoms are strongly bound to active sites, which results in a hindered process of proton and electron transfer and thereby a delayed HER activity. With this consideration, Pu et al.108 introduced a N,P-codoped carbon shell to improve the HER performance of Ru2P NPs. DFT calculations revealed a lower absolute ΔGH* value of RuP2@NPC than RuP2, NPC and C. As a result, RuP2@NPC exhibited an overpotential of 38 mV at a current density of 10 mA cm−2 in 0.5 M H2SO4, smaller than that of control prepared catalysts. RuP2@NPC also worked well in neutral (overpotential of 57 mV in 1.0 M PBS (phosphate buffered saline)) and alkaline media (overpotential of 52 mV in 1.0 M KOH) with comparable activity but excellent durability than the commercial Pt/C catalyst. In addition, HER performance of metal phosphides can be optimized through control over P content during the phosphating process.344 A suitable amount of the P precursor (NaH2PO2) in the IrCl4/NaH2PO2-blended polymer (which was pyrolyzed under high temperature) was responsible for modulating Ir electronic configuration and tuning the thickness of the N,P-codoped carbon shell to allow an efficient electron transfer process for the HER in a wide pH range.
Transition metal, metal (hydro)oxide, metal alloys. Non-precious 3d transition metals (i.e. Fe, Co, Ni, Cu) could catalyze the HER process due to the presence of plenty of unpaired d-orbital electrons for facilitating hydrogen chemisorption. Carbon nanoshell-coated transition metal based catalysts have been specifically reported to exhibit reinforced HER performance.345–347 Tavakkoli et al.348 prepared single-shell carbon-coated Fe NPs (abbreviated as SCEINs) decorated on single-walled carbon nanotubes (SWNTs) by the ethylene CVD process. Metallic Fe NPs with a diameter of 2.6 nm were overcoated with an outer single-layer carbon shell, as evidenced by the HRTEM images in Fig. 17a and b. SCEINs/SWNTs displayed high HER activity with about 0 VRHE onset potential and 40 mV dec−1 Tafel slope, with the latter being close to that (37 mV dec−1) of Pt/C, when measured in N2-saturated 0.5 M H2SO4 (Fig. 17c and d). There was no observable activity degradation after 1000 potential cycles in the range of −0.15 to 0.15 VRHE, pointing to an excellent durability as a result of the protection of single-shell carbon. In addition, Co species encapsulated within carbon materials have been developed for efficient HER.103,349–352 An ultra-thin N,B-codoped carbon cage-encapsulated Co catalyst, named Co@BCN, was produced from the pyrolysis of ZIF-67 materials mixed with H3BO3.352 ZIF-67 comprising Co metal nodes and organic linkers serves as the metal precursor and self-sacrificing template. When applied for hydrogen production via the HER, the BCN shell on Co@BCN can protect the Co metal from being corroded and oxidized, which results in high stability in extremely corrosive conditions. Dopants such as N, O, and B in the carbon nanoshell are beneficial for synergistically tuning ΔGH* near to the zero value, thus enhancing the HER activity.103,352
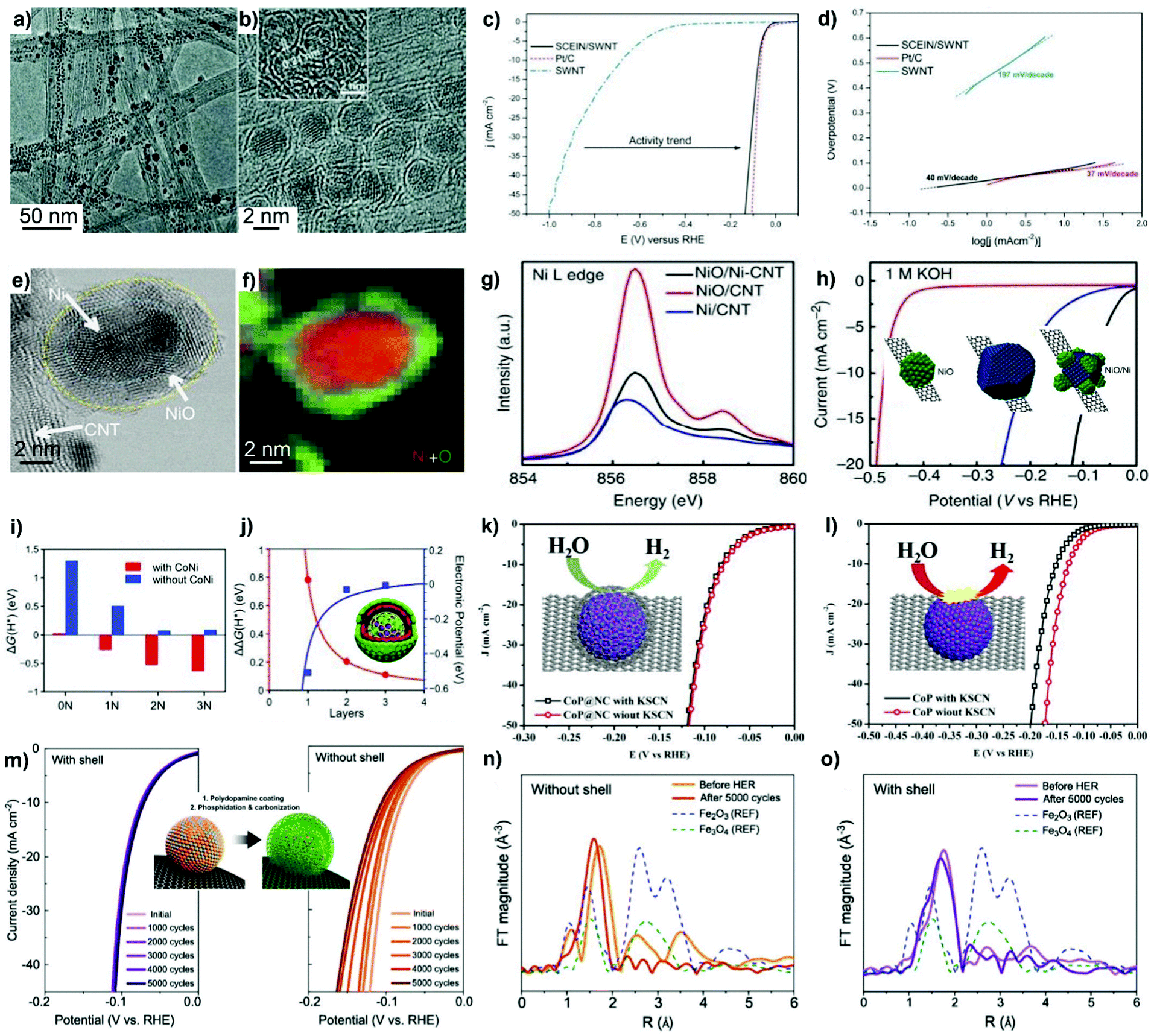 | ||
| Fig. 17 (a) TEM and (b) HRTEM images of SCEIN/SWNT; the inset in (b) represents the (110) lattice plane of Fe particles in SCEINs. (c) Polarization curves at 50 mVs−1 in 0.5M H2SO4 and (d) Tafel slopes of SWNT, SCEIN/SWNT, and Pt/C. Reproduced from ref. 348 with permission from Wiley-VCH, Copyright 2015. (e) Bright-field STEM image and (f) chemical map for Ni and O distribution of NiO/Ni-CNT. (g) Ni L edge XANES spectra and the HER linear sweep voltammetry (h) of three hybrid materials in 1.0 M KOH. Reproduced from ref. 356 with permission from Springer Nature, Copyright 2014. (i) The free energy of H adsorption (ΔG(H*)) on pure and N-doped (one, two, or three N atoms per shell) graphene shells with and without an enclosed CoNi cluster. (j) ΔΔG(H*) (red line) and electronic potential (blue line) as a function of graphene layer numbers, where ΔΔG = ΔG (without metal) − ΔG (with metal); inset: schematic illustration of a CoNi particle coated with three-layer graphene. Reproduced from ref. 105 with permission from Wiley-VCH, Copyright 2015. HER polarization curves of CoP@NC (k) and CoP (l) with and without the addition of KSCN in 0.5 M H2SO4. Reproduced from ref. 369 with permission from American Chemical Society, Copyright 2017. (m) Polarization curves of FeP NPs with (left) and without (right) carbon shell after different cycle tests. EXAFS spectra of FeP NPs without (n) and with (o) carbon shell. Reproduced from ref. 371 with permission from American Chemical Society, Copyright 2017. | ||
In view of the fact that non-precious metal based electrocatalysts exhibit excellent OER properties in alkaline conditions compared to those implemented in acid systems, there are many cost-effective HER electrocatalysts being developed with high performance for alkaline water electrolysis. In particular, there is considerable interest in engineering Ni-based alloys and heterostructured composites as the promising electrocatalysts for the HER in alkaline electrolytes.353 It has been well documented by the group of Markovic that capping the Pt surface with 3d transition metal hydr(oxy)oxide nanoclusters was effective at boosting the alkaline HER activity.354,355 They successfully applied such a hetero-junction concept to modify the Ni surface with Ni(OH)2 nanoclusters. The obtained Ni(OH)2/Ni catalyst showed 4 times higher alkaline HER activity than the bare Ni surface, mainly attributed to the promoted dissociation of water.334 Gong et al.356 demonstrated that pyrolysis-induced partial reduction of Ni(OH)2 could afford NiO/Ni nano-hybrids distributed on the carbon nanotube. The oxidized carbon nanotube played an important role in the formation of the unique Ni@NiO core–shell nanostructure (Fig. 17e and f) by impending the reduction of Ni oxides into Ni particles and preventing them from undergoing further sintering. Compared with the control samples, i.e. Ni/CNT and NiO/CNT, NiO/Ni-CNT showed <100 mV overpotential to deliver a current density of 10 mA cm−2 at a loading of 0.28 mg cm−2 in 1.0 M KOH (Fig. 17g and h). The highly effective alkaline HER activity stemmed from the synergistic effect on the NiO/Ni interface in terms of providing the sorption sites for OH− on NiO and facilitating H adsorption on Ni0. It is known that Ni0 at the NiO/Ni interface is much more active than the bulk Ni0. Notably, the NiO/Ni heterostructure was unstable under the long-term HER condition, which was caused by the oxidation of Ni0. The authors accordingly prepared the nanoscale Ni core being coated with the Cr2O3-blended NiO overlayer to display a superior stability in basic solution.357 The addition of Cr2O3 could prevent the oxidation of the Ni core, thus maintaining the abundant NiO/Ni nanointerfaces for the efficient HER catalysis. In addition to metal oxides, carbon nanoshells have been deployed to enhance the long-term durability of the Ni surface for passivation or oxidation, as reported by Doan et al.358 The coating of the Ni surface with a graphitic carbon layer was done by pyrolyzing the mixture of Ni2+-cupferron complex in the presence of carbon support at 700 °C. Surface analysis showed the amount of Ni0 on the carbon-coated catalyst as high as 46%, whereas no Ni0 (but 76% of Ni(OH)2) was observable on the catalyst without carbon shell embedding. The protection of the Ni0 state from oxidation by carbon coating was evidenced from in situ XAS measurements.
Carbon coatings were also able to deactivate the formation of hydride on the Ni surface. Consequently, the carbon-embedded Ni catalyst featuring an optimal ratio of Ni0:NiOx exhibited a superior HER performance. These advances demonstrate the importance of a favourable ratio between Ni0 and NiOx on Ni-based electrocatalysts for alkaline HER catalysis.
Layered double hydroxides (LDHs) and their derivatives are promising electrocatalysts for alkaline water splitting due to their unique electron distribution. However, the poor conductivity and insufficient active sites impede their application in alkaline HER. Recently, Wang et al.144 performed surface overcoating engineering on the NiFe LDH with CeOx by using a electrodeposition method. The enhanced alkaline HER activity in this work was credited to the facilitated H2O adsorption on the oxygen vacancies created at the NiFe LDH/CeOx interface. Of note, the development of carbon shell-overcoated metal oxides as electrocatalysts toward the HER catalysis is rarely investigated. The carbon shell encapsulation strategy was applied to optimize the acidic HER performance of a WOx-based catalyst (WOx@C/C) by means of improving electrical conductivity, increasing charge transfer rate, and optimizing hydrogen adsorption energy.359 There are also few reports on the utility of polyoxometalate (POM)-incorporated MOF nanomaterials to fabricate metal oxide based HER electrocatalysts.360,361 For example, Tang et al.361 prepared MoO2@PC-RGO consisting of MoO2, P-doped nanoporous carbon (PC) and reduced graphene oxide (RGO) through the pyrolysis of the one-pot formed POM-MOFs/GO nanocomposite. For the HER in acidic solution, MoO2@PC-RGO exhibited an onset potential of 0 mV, a Tafel slope of 41 mV dec−1, and longer durability. It was described that the well-dispersed MoO2 particles protected by the carbon shell, the P dopants derived from POM and the porous structure of MoO2@PC-RGO synergistically led to improvement in HER performance.
Metal alloys consisting of metal atoms mixed at the nano scale can tune the HBE on active sites and result in synergistic effects which enhance their HER performance. The overcoating strategy has been applied for engineering metal alloys such as CoNi,105 FeCo,362 NiCu363 and NiFe364 with carbon nanoshells as HER electrocatalysts. By means of pyrolyzing the lamellar complex EDTA-CoNi (EDTA: ethylenediaminetetraacetate), Deng et al.105 prepared uniform CoNi nanoalloys which are encapsulated within ultra-thin graphene shells with only 1–3 layers. DFT calculation results in Fig. 17i show that combining CoNi with the N-graphene shell with a varied amount of N dopants could positively tune H adsorption energy. The promotion effect on electron transfer on CoNi declines with the increasing layers of the graphene shell, as illustrated by the H adsorption difference [ΔΔG(H*)] of the graphene layer with or without CoNi (Fig. 17j). The effect of the thickness of the carbon shell on the HER activity was observed on the NiCu@C catalyst as well.363 More importantly, in this case, an acceptable HER performance was achieved by a delicate control over the carbon shell thickness even without the incorporation of any dopants such as N, P, B, O and so on. These results indicate that controlling the thickness of the carbon nanoshell is an important strategy to optimize the electronic structure of the catalyst with superior HER activity.
Transition metal phosphides, carbides, chalcogenides. The application of transition metal phosphides, carbides, and chalcogenides in HER catalysis has been intensively exploited in the past decade.327,365,366 Benefiting from the suitable hydrogen adsorption free energy and good conductivity after P incorporation, there is a body of research works that focus on developing efficient metal phosphides for the HER.97,109,110,326 In particular, carbon nanoshell-coated CoP catalysts were demonstrated through high-temperature carbonization and phosphidation treatment on Co-based zeolitic imidazolate frameworks such as ZIF-67119,367,368 and ZIF-9.369 The incorporation of P in the above works is normally carried out by post-synthetic phosphidation treatment using e.g. NaH2PO2 at 200–500 °C. Impressively, Wang et al.370 developed a N,P-codoped carbon shell-coated Cu3P-based highly active HER electrocatalyst from single Cu-MOF with N- and P-containing linkers in its framework. As for the HER activity, Yang et al.369 reported that CoP@NC overcoated with an ultra-thin N-doped carbon shell (2–4 layers) was better than CoP@C and CoP in both 0.5 M H2SO4 and 1.0 M KOH solutions. Meanwhile, only 2 mV degradation after 1000 potential cycles from 0.05 to −0.2 VRHE and negligible current density loss (at 0.1 V overpotential) were detected after 24 h of continuous operation. The authors further conducted poisoning experiments using thiocyanate ions (SCN−) to identify the active sites on CoP@NC. As shown in Fig. 17k and l, CoP has 22 mV increase of overpotential after adding SCN−, while that of CoP@NC with a N-doped carbon nanoshell is only 5 mV, indicating that carbon atoms that are adjacent to the N dopants in the carbon shell are the active sites for HER catalysis. Similarly, after overcoating of the N,P-codoped carbon shell (derived from ZIF-67) on the CoP nanoarray, the carbon atom-adjacent heteroatoms featuring electron donor–acceptor properties could provide abundant active sites for HER catalysis.368
As discussed in Section 2.2, heteroatom-containing polymers and organic compounds are suitable precursors for fabricating a porous carbon shell on metal particles. In this context, Chung et al.371 prepared polydopamine pyrolysis-derived carbon shell-coated FeP NPs with a diameter of 4 nm. The afforded FeP/C with a N-doped carbon shell exhibits better HER activity (71 mV overpotential at 10 mA cm−2, 52 mV dec−1 Tafel slope) and excellent durability compared to FeP without carbon coating, a single carbon shell without Fe as well as oxidized FeOx/C (Fig. 17m). By comparing the Fe K-edge X-ray absorption fine structure (EXAFS) before and after the durability tests, and referring to the standard data of Fe2O3 and Fe3O4 (Fig. 17n and o), the authors discovered that the first coordination shell signal of the uncoated sample slightly shifted to the oxidized Fe state after 5000 cycles, whereas that of carbon-coated FeP/C remained in the stable valence state. The above findings indicated that the carbon nanoshell could protect FeP NPs from oxidation, which was further evidenced by performing theoretical studies.
There are also reports on HER electrocatalysts based on different kinds of metal phosphides such as MoP, FeP and Cu3P, which are derived from carbon overcoating by the pyrolysis of organic matters, along with in situ or post-synthetic phosphidation treatment.107,121,372,373 Li et al.107 demonstrated that embedding MoP NPs in the Mo- and P-doped carbon layer realized the formation of Mo–C and P–C bonds. The electron transport process on the prepared MoP@C was favoured to result in extremely lower overpotential and Tafel slope for the HER catalysis in acid and alkaline solutions in comparison with that of the uncoated MoP counterpart. Likewise, Zhao et al.373 recently synthesized MoP NPs encapsulated within hollow-spherical-structured N-doped carbon thin layers (MoP@NCHSs) to study the synergistic effect between MoP NPs and N dopants for enhancing HER performance. In this work, the N content and configurations were detailed from K-edge XANES and N 1s XPS spectra. The higher HER activity in alkaline solution was proposed to be originated from the large pyridinic-N ratio on MoP@NCHSs-900 which was pyrolyzed at 900 °C. Pyridinic-N–Mo was the active site for HER, as revealed by performing pyridinic N poisoning experiments and DFT calculations. In other words, pyridinic-N was effective in inhibiting the strong adsorption of OH*, and the pyridinic-N–Mo interactions can weaken the adsorption of H on Mo by electron transfer.
Carbon matrix-embedded transition metal carbides such as Mo2C/G-NCS,374 MoCx@C,375 β-Mo2C@C,376 WC@C,129 W2C@WC1−x@C,377 and W/W2C@NPC378 are also under extensive investigations for HER applications, owing to the features of good conductivity and excellent thermal and electrical stability against sintering. Similar to metal phosphides noted above, heteroatoms such as N and P elements doped in carbon shells can activate the adjacent carbon atoms to be the active sites on metal carbides for the catalytic generation of H2.111,379–382 Moreover, the incorporation of transition metal moieties (such as Fe, Co, Ni) at the atomic scale into metal carbide based HER electrocatalysts has been demonstrated to be a rewarding strategy for achieving the manifestation of synergistic effects.116,127,383,384 In addition to the capability of promoting the formation of a carbon nanoshell and the high dispersion of metal carbide particles during carbonization, the incorporated transition metals could modulate the surface electronic structure of the catalyst to affect H binding on the active sites and thus are responsible for the favourable HER performance.
Nanomaterials (especially those derived from MOFs) featuring hollow structures could maximize the exposure of active sites and facilitate the mass transfer kinetics for efficient catalysis. Nevertheless, it is challenging in most cases to fabricate such catalysts because of the easy collapse or destruction of the structure of applied precursors during the high-temperature pyrolysis. Impressively, surface overcoating engineering could help in architecturing hollow-structured and robust molybdenum based carbides for HER catalysis, as demonstrated by Xu et al.385 Prior to pyrolytic treatment, they overcoated the POM-MOF (Ni) precursor with a thin layer of carbon nitride polymer by the self-polymerization reaction of hexamethylenetetramine in ethanol solution dispersed with hollow-structured POM-MOF (Ni) at 170 °C. A totally destroyed morphology with severe metal particle agglomeration was observed after the direct annealing of the POM-MOF (Ni) precursor at 800 °C. On the other hand, overcoating with the carbon nitride polymer could efficiently avoid the collapse of hollow POM-MOF precursors and prevent metal particles from coalescence or detachment during the process of carbonization. As a result, the carbon layer-coated hollow-structured molybdenum carbide together with the Ni-MOFs-derived Ni nanoparticle made the catalyst exhibit remarkable HER activity and long-term durability in alkaline solution.
Transition metal chalcogenides are also garnering research attention for the HER.386,387 It was reported that overcoating the carbon shell onto MoS2 was profitable for conductivity improvement, mass transfer dynamics and protection of the MoS2 nanosphere from collapse and stacking.388 Similar to the pyridinic-N–Mo sites formed on MoP@NCHSs-900,373 pyridinic-N sites on N-doped carbon coated NiSe2 nano-octahedra (NiSe2@NC) could determine the corresponding HER activity which showed a linear correlation with the content of pyridinic-N.389 For the preparation of NiSe2@NC, mixed-linker Ni-MOFs consisting of O-coordinating anionic carboxylate linkers and N-coordinating neutral ligands appeared to be good candidates for selective selenization. After a deliberate control over the content and configuration of N in the carbon nanoshell by using different N-ligands, NiSe2@NC-PZ (PZ: pyrazine), in particular, exhibited a lower overpotential of 162 mV at 10 mA cm−2 and Tafel slope (88 mV dec−1) in alkaline media than other NiSe2@NC hybrids.
To briefly conclude, facile catalyst engineering via the introduction of foreign overcoatings on metal based catalysts is valuable for tackling the performance degradation. Non-precious metal based electrocatalysts have been fabricated as the favourable alternatives to precious metal counterparts for the HER catalysis. To our delight, carbon coating engineering is most widely exploited for enhancing the intrinsic activity of metal catalysts toward the HER, as discussed above based on the existing literature. The heteroatom dopants in the carbon nanoshell can positively modify H adsorption free energy with a nearly zero value to potentially optimize the electronic structure of the metal and the process of H formation/binding and thus boosting the HER efficiencies in both acid and alkaline environment. For comparison, representative surface overcoating-derived electrocatalysts based on precious and non-precious metals for efficient HER catalysis in both acid107,233,340,342,348,361,363,369,378,381 and alkaline solutions107,108,124,342,343,374,375,378,389 are summarized in Fig. 18.
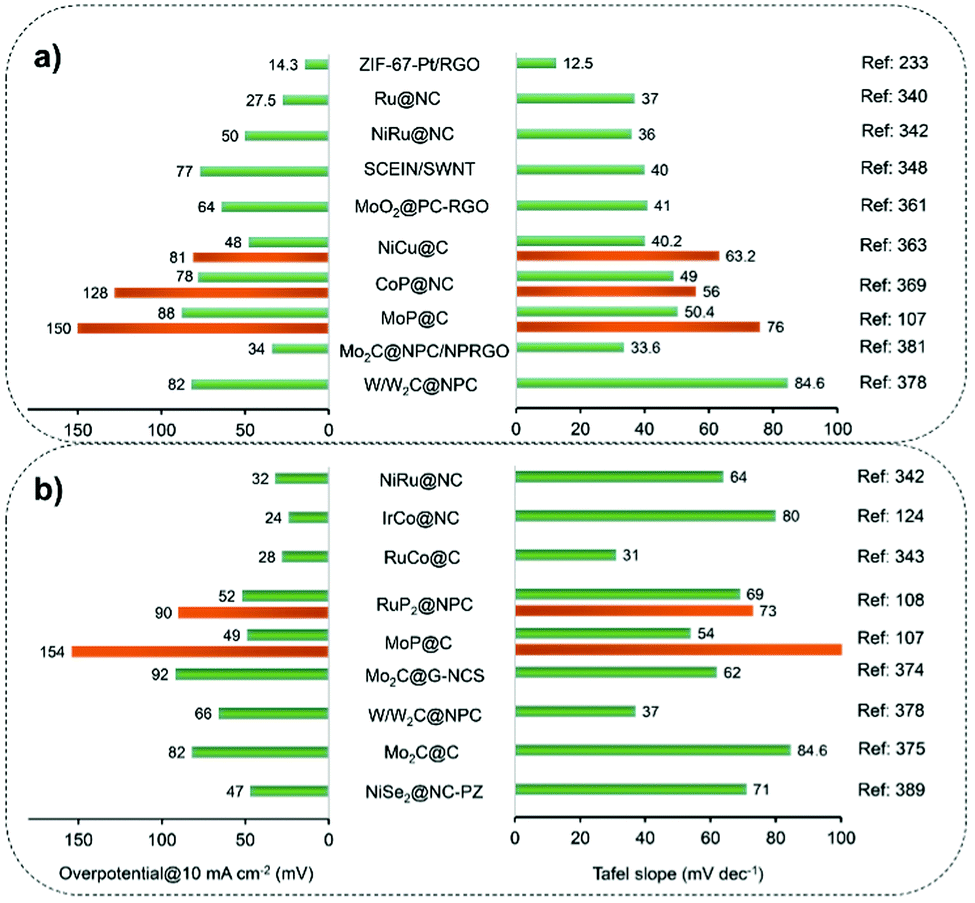 | ||
| Fig. 18 Comparison of the reported HER electrocatalysts in terms of the required overpotential to reach a current density of 10 mA cm−2 and Tafel slope in (a) 0.5 M H2SO4 and (b) 1.0 M KOH electrolytes. The electrochemical parameters for some of the corresponding uncoated catalysts are also plotted (orange color). | ||
4.3 Overcoated bifunctional electrocatalysts for overall water splitting
Electrochemical overall splitting of water powered by renewable energy appears promising for pure H2 and O2 production via the HER and OER. Owing to the sluggish kinetics of OER and the high demand on catalyst stability, developing high-efficiency bifunctional overall water splitting electrocatalysts in all pH range is practically meaningful and imperative. Pt/C and RuOx or IrOx are the benchmark catalysts for HER and OER, respectively. Recently, cost-effective water splitting electrocatalysts with satisfactory performance are becoming desirable. Some specially designed catalysts have already displayed similar or even better performance than that found with Pt/C‖RuO2 and/or Pt/C‖IrO2 counterparts.The overpotential (the difference value between the required potential and the thermodynamic potential (1.23 V, 25 °C)) for OER/HER and the voltage needed to reach a certain current density (10 mA cm−2) are the parameters that are generally used to evaluate the overall water splitting performance of bifunctional OER/HER electrocatalysts.
In the development of precious metal-based water splitting electrocatalysts, alloying treatment and/or constructing core–shell nanostructures with abundant 3d transition metals has been demonstrated to result in a positively perturbed electronic structure of the metal for efficient electrocatalysis. There are several works that focus on carbon nanoshell-encapsulated precious metal electrocatalysts for overall water splitting.390–392 For example, Li et al.391 prepared Co@Ir core–shell NPs encapsulated in N-doped porous carbon through the galvanic replacement of IrCl3 with the ZIF-67-derived Co/NC nanomaterial in an aqueous solution for 24 h, followed by centrifugation and drying step. The formation of a core–shell structure with a shell of thin layer of Ir on Co@Ir could promote the exposed proportion of Ir and further improve its utilization efficiency. Electrical measurements on Co@Ir/NC-10% (10% is the initial mass amount of Ir) displayed the best performance in terms of superior OER activity than IrO2 and close HER activity to Pt/C, along with excellent long-term durability. The authors evaluated further the practical overall water splitting performance of Co@Ir/NC-10% in basic solution by assembling the catalyst onto carbon fiber paper, and showed potential application under the applied 1.7 VRHE. Following a similar galvanic replacement strategy, Ni@Ru core–shell NPs overcoated with a 2–5 nm-thin graphitic carbon nanoshell displayed a potential of 1.612 V at 10 mA cm−2 for overall water splitting in alkaline solution, outperforming the reference Pt/C and RuO2 catalysts.392
Combining the merits of the carbon matrix with non-precious metal species by using various overcoating methodologies, along with the phosphorization or sulfurization treatment, promises cost-effective electrocatalysts based on abundant metals for efficient overall water splitting reactions. Recently, boosting research activities have been dedicated to the development of surface overcoating-derived bifunctional overall water splitting electrocatalysts, including metallic transition metals and alloys,117,324,345,393–398 transition metal (hydro)oxides,144,399–402 metal phosphides,130,131,327,403–406 metal carbides407–409 and metal chalcogenides.101,410,411 Some typical examples of these works are presented below.
Li et al.399 developed N-doped carbon-coated Co–Mn oxide (CoMnO@CN) by utilizing a solvent evaporation-induced self-assembly method (Fig. 19a). After the slow evaporation of solvent and the calcination step, 5 nm-sized CoMnO particles were assembled into a highly ordered superlattice structure with the overcoating of a 2 nm-thin amorphous carbon layer. The obtained CoMnO@CN exhibited the highest OER current density (308 mA cm−2) at 1.65 VRHE in alkaline solution compared with structure disordered CoMnO@CN and the catalyst without N doping (Fig. 19b and c).The HER performance of CoMnO@CN superlattices also surpassed that of most earth-abundant metal based HER catalysts (Fig. 19d). According to the authors, CoMnO NPs in the CoMnO@CN catalyst favour the OER, while the outer carbon shells not only provide additional active sites for HER, but could also synergistically protect the inner CoMnO NPs from aggregation or dissolution during the electrocatalytic processes. When working as both the electrodes in an alkaline water electrolyzer, CoMnO@CN superlattices realized 54 and 108 mA cm−2 current density with an applied potential of 1.7 V and 1.8 V, respectively. High stability was also achieved after 10 h operation with an applied potential of 1.5 V (Fig. 19e).
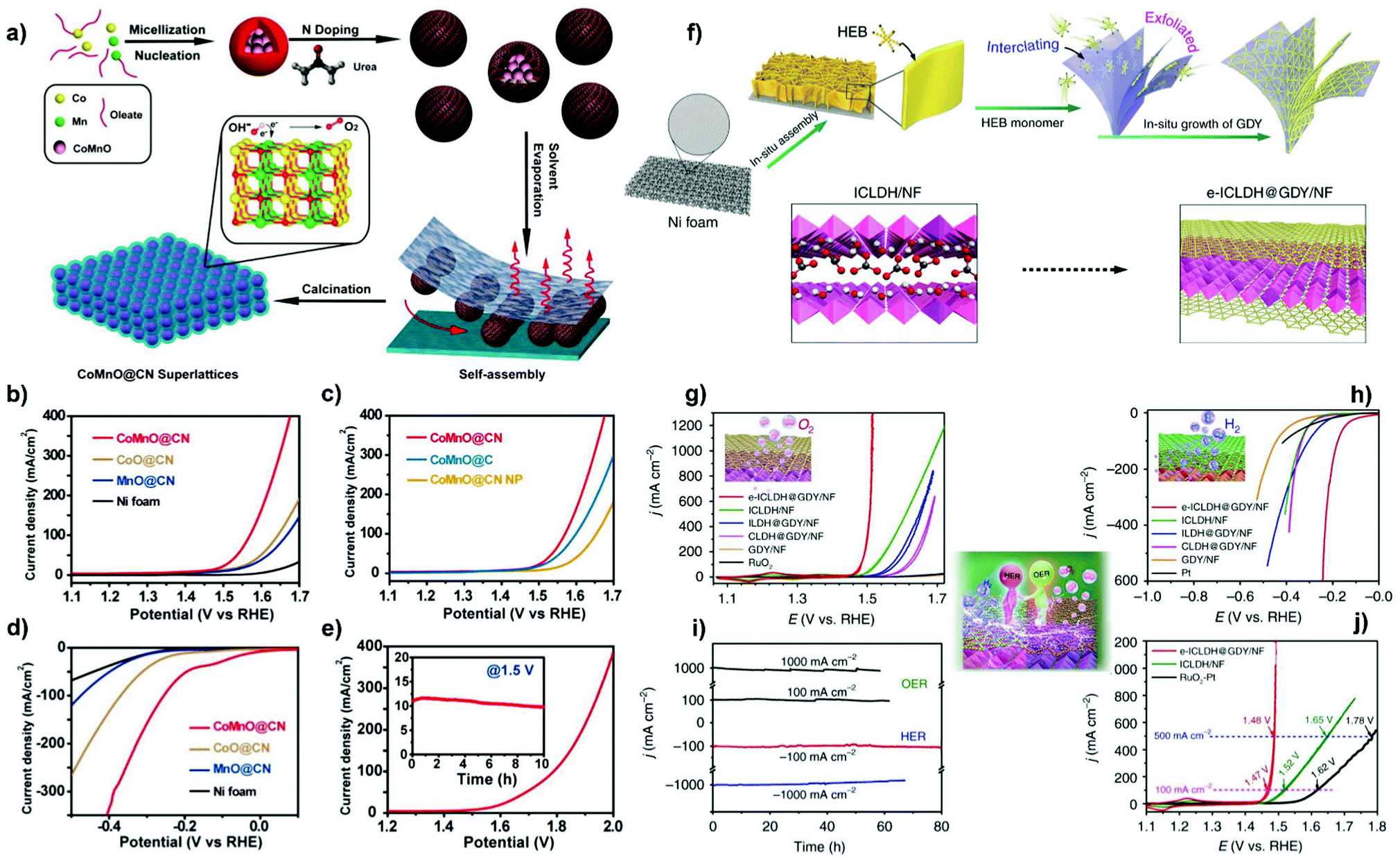 | ||
| Fig. 19 (a) Synthetic procedures for the CoMnO@CN superlattice structure. (b and c) OER and (d) HER LSV curves of CoMnO@CN compared with other control samples in a 1.0 M KOH electrolyte in a 3-electrode configuration. (e) Overall water splitting measurements of CoMnO@CN in a two-electrode configuration. The inset represents the chronoamperometric curve at an applied potential of 1.5 V across the electrodes. Reproduced from ref. 399 with permission from American Chemical Society, Copyright 2015. (f) Schematic of the synthesis of e-ICLDH@GDY/NF structures. (g) OER and (h) HER polarization curves of as-synthesized different samples. (i) Chronopotentiometric curves of e-ICLDH@GDY/NF for OER and HER at 100 and 1000 mA cm−2. (j) The activities of pristine ICLDH/NF and e-ICLDH@GDY/NF towards the overall water splitting reaction with iR-correction. Reproduced from ref. 421 with permission from Springer Nature, Copyright 2018. | ||
Note that, in addition to the surface overcoating strategy, there are other strategies being developed for improving the performance of metal based catalysts toward the electrochemical water splitting, such as doping, defect engineering and designing unique nanoarchitectonics and so on.412,413 A number of 3d transition metal-based nanomaterials including metal oxides, nitrides, and sulfides have been and are being rationally designed as good candidates for overall water splitting reactions. For example, Zhang et al.414 designed NiMoO4−x/MoO2 by combining oxygen vacancies and nanointerfaces between NiMoO4−x and MoO2 as the efficient water splitting catalyst. In this case, a voltage of 1.56 V was required for a current density of 10 mA cm−2. Porous MoO2 nanosheets on nickel foam were developed by the group of Cui as the active and stable bifunctional overall water splitting catalyst, showing a cell voltage of 1.53 V for a current density of 10 mA cm−2 together with at least 24 h of durability.415 Li et al.416 reported that 3D self-supported Fe-doped Ni2P nanosheet arrays showed promising overall water splitting performance with 1.49 V of cell voltage at 10 mA cm−2 and excellent stability at 1.7 V. Recently, Yu et al.417 reported 3D core–shell NiMoN@NiFeN supported on porous Ni foam for the alkaline seawater splitting. When working as the bifunctional electrode catalyst at 60 °C, NiMoN@NiFeN achieved a current density of 500 and 1000 mA cm−2 at a record voltage of 1.608 and 1.709 V, respectively. In addition, a cell voltage as low as 1.43 V at 10 mA cm−2 and excellent long-term durability (100 h@100 mA cm−2) were achieved on the NiCoMo–OH catalyst, which was prepared by Hao et al.418 through a chloride corrosion method. Moreover, a NiMoOx/NiMoS heterostructure was engineered in the work of Zhai et al.419 to accelerate water splitting kinetics, which required a voltage of 1.60 and 1.66 V respectively to reach a current density of 500 and 1000 mA cm−2 as well demonstrated excellent long-term durability.
The overall water splitting cell voltage and durability of overcoated materials developed as bifunctional water splitting electrocatalysts in 1.0 M KOH solution are summarized in Table 2. It is worth stressing that surface overcoating engineering can be regarded as one of the promising strategies for improving catalyst's overall water splitting performance. Typically, coupling carbon with ultra-thin Co-doped NiO (Ni0.82Co0.18O) on Ni foam enabled to feature unique 3D configuration and abundant exposed active sites, ensuring an ultralow voltage of 1.42 V at a current density of 10 mA cm−2 when catalyzing the overall water splitting.400 Recently, Qian et al.420 reported Ni NPs encapsulated with ultra-thin N-doped graphene coupled with MoO2 nanosheets for catalyzing the water splitting at large current density. In detail, the prepared Ni@C-MoO2/NF electrode exhibited a voltage of 1.54, 1.61 and 1.67 V at 10, 50 and 100 mA cm−2, respectively. Impressively, it can be held for over 196 h at 1000 mA cm−2 in 1.0 M KOH. These superior performances were ascribed to the following factors: (i) the electron transfer between Ni NPs and N atoms from the carbon shell optimized the surface electronic structure, thus enhancing the OER/HER activity; (ii) the Ni@C structure was prone to have a more efficient electron transfer and faster faradaic process on the electrode–electrolyte interface; and (iii) the self-supported MoO2 nanosheets on Ni foam improved the utilization efficiency of active sites and facilitated the mass transfer process during the reaction. Moreover, when employed as both anode and cathode catalysts in a water electrolyzer, C@Ni8P3 (the Ni8P3 nanosheets surface-coated with carbon shell) can deliver a current density of 10 mA cm−2 at a lower cell voltage (1.65 V) than Ni8P3 without carbon coating with a voltage of 1.79 V in 1.0 M KOH.327 C@Ni8P3 also exhibited an excellent long-term durability performance, whereas Ni8P3 was found to be considerably degraded.
| Electrocatalyst | Cell voltage@10 mA cm−2 [V] | Durability [h] | Substrate | Ref. |
|---|---|---|---|---|
| Fe1.0Co1.1Ni1.4-NC | 1.52 | 20 (@10 mA cm−2) | Carbon cloth | 117 |
| Co@N-CS/N-HCP | 1.545 | 24 (@30 mA cm−2) | Carbon cloth | 397 |
| Ni@NC | 1.60 | 50 (@1.62 V) | Ni foam | 345 |
| Ni@C-MoO2/NF | 1.54 | 196 (@1000 mA cm−2) | Ni foam | 420 |
| Cu@C | 1.645 | 24 (@10 mA cm−2) | Ni foam | 398 |
| NiFe LDH/CeOx | 1.51 | 10 (@10 mA cm−2) | Ni foam | 144 |
| e-ICLDH@GDY/NF | 1.43 | 60 (@100 mA cm−2) | Ni foam | 421 |
| FeCH@GDY/NF | 1.49 | 25 (@10 mA cm−2) | Ni foam | 422 |
| CoMnO@CN | 1.50 | 10 (@10 mA cm−2) | Ni foam | 399 |
| Ni0.82Co0.18O@C | 1.42 | 20 (@10 mA cm−2) | Ni foam | 400 |
| Ni-NiFe2O4@C | 1.57 | 20 (@20 mA cm−2) | Carbon cloth | 401 |
| FeNiP/PG | 1.58 | 10 (@10 mA cm−2) | Carbon cloth | 130 |
| NC-NiFeOx@NiFe-P | 1.59 | 20 (@10 mA cm−2) | Ni foam | 131 |
| NiFeOx@NiFe-P | 1.81 | — | Ni foam | 131 |
| C@Ni8P3 | 1.65 | 25 (@10 mA cm−2) | Ni foam | 327 |
| Ni8P3 | 1.79 | — | Ni foam | 327 |
| CoP@C@rGo | 1.50 | 24 (@10 mA cm−2) | Carbon paper | 406 |
| Co9S8@NOSC-900 | 1.60 | 10 (@50 mA cm−2) | Ni foam | 101 |
| NCT-NiCo2S4 | 1.60 | 15 (@10 mA cm−2) | Ni foam | 411 |
In another study by Hui et al.,421 the synthesis of graphdiyne-wrapped iron-cobalt LDH nanosheets (CoFe-LDH) with sandwiched structure grown on Ni foam and their application as a bifunctional overall water splitting electrode were reported. In this case, graphdiyne (GDY), a 2D monolayer of sp/sp2-hybridized carbon material, was introduced into the interlayer of CoFe LDH by using hexaethynylbenzene monomers through in situ polymerization (Fig. 19f). The uniform growth of the GDY film on the CoFe LDH surface with intimate contact induced the complete exfoliation of bulk LDH into ultrathin LDH nanosheets being wrapped with GDY on both sides of surfaces. Notably, due to its high electronic conductivity and the richness in charged carbon atoms, the GDY layer was found to play a critical role in enhancing the catalytic performance of CoFe LDH for the overall water splitting. The OER activity test on the as-fabricated catalyst indicated a low overpotential of 216, 249, and 278 mV to deliver 10, 100, and 1000 mA cm−2 current densities (Fig. 19g). To achieve such current densities, an overpotential of 43, 215 and 256 mV was respectively required for the HER catalysis (Fig. 19h). The excellent OER performance was mainly ascribed to Fe doping and GDY incorporation which induced enhanced charge transfer from LDH to GDY. On the other hand, the origin of the enhancement of HER activity was enabled by the facilitated electron transport and the abundant active sites after GDY layer wrapping. The superior durability (Fig. 19i) of e-LDH@GDY/NF to the OER and HER was attributed to the protective aid from the GDY overlayer. The performance of GDY-wrapped CoFe LDH for overall water splitting was evaluated in 1.0 M KOH in a two electrode configuration. As shown in Fig. 19j, 1.43, 1.46 and 1.49 V was respectively measured to achieve 10, 100 and 1000 mA cm−2 current densities. The electrolyzer displayed an enhanced long-term stability (over 60 h) at the current density of 100 mA cm−2 under a constant voltage of 1.56 V. A faradaic efficiency of 97.40% for the O2 evolution was achieved. The authors also revealed the decisive role of the GDY overlayer in improving the electrocatalytic efficiencies of ultrathin GDY-wrapped iron carbonate hydroxide nanosheets for the overall water splitting in WECs.422
The above achieved progress indicates that the encapsulated metal catalysts constructed based on surface overcoating engineering are a kind of advanced architecture for catalyzing high-performance electrochemical reactions. Accordingly, both precious and non-precious metal based electrocatalysts designed and optimized by surface overcoating engineering have been validated to be efficient for application in WECs. By controlling the synthetic aspects such as the elaborate selection of overcoating materials, pyrolysis conditions and heteroatom dopants, the chemical composition, spatial structural properties of electrocatalysts as well as the nature of active sites can be rationally optimized to result in outstanding overall water splitting performance.
5. Summary and outlook
This review summarizes a variety of surface overcoating methodologies implemented for engineering high-performance electrocatalysts, which are potentially applied for sustainable energy-related transformations. Substantial advances in nanoscience and material chemistry have benefited the fabrication of metal nanoparticles and the modification of catalyst supports by the utility of overcoating materials, including organic matters, inorganic carbon nanoshells and metal oxides. Surface-engineered nanostructures featuring the promoted synergistic effect of metal sites and foreign coatings can remarkably enhance the process of electrocatalysis. Despite the research progress, effectiveness and structure–catalytic function of surface overcoating-derived electrocatalysts as discussed in this review, the development of electrochemically highly active and durable nanocatalysts is still ongoing to achieve their large-scale distributions in renewable energy conversion and storage devices. Therefore, the identified opportunities and perspectives associated with the surface overcoating engineering for designing optimal heterogeneous catalysts should be considered and are detailed below.(1) Approximate selection of overcoating materials for precise design and optimization of more advanced electrocatalysts. In addition to the manifestation of steric hindrance effects, surface overcoatings enable one to dramatically modulate the electronic surface structure of metal entities and tune the adsorption behaviour of reactants and key intermediate catalytic species; consequently, electrocatalytic performance varies with the overcoating materials introduced. It is worth noting that catalyst electrocatalytic activity, which can be descripted by the binding energy states of surface intermediates (i.e. H*, OH*, OOH*), follows the intrinsic catalytic behaviour of metals, the pH of electrolytes, and the operating environment (such as the cell performance tests where electrocatalysts are fabricated into MEAs). Therefore, the specific selection of surface overcoating materials should be compatible with specifically investigated applications in the areas related to electrochemical energy storage and conversion. Currently, organic capping agents/polymers, inorganic carbon nanoshells and metal oxides have been successfully employed as the favourable surface overcoating materials. With the blooming synthetic development of metal nanostructures as well as support materials, the exploitation of innovative overcoating materials such as MOFs and/or MOF-derivatives becomes increasingly crucial for the fabrication of multifunctional nanocomposites with superior efficacy when applied in PEFCs, WECs and beyond. We refer the readers to a recent review, which describes the synergistic advantages of MOF coatings in broad applications.423
(2) In-depth understanding of the interfacial mechanism at different scales. Electrical coupling, regulating the structure and mobility of metal NPs, and controlling surface adsorption kinetics are among the most rewarding benefits from catalyst overcoating engineering. As noted, the functional groups of N-containing organic compounds serve as the coordination sites for controllable synthesis of metal entities with desired phase compositions and morphological integrities. Heteroatom-doped carbon nanoshells derived from the carbonization of organic composites could afford a plethora of functionalities, including the modulation of the binding energy states of reactants and intermediate reactive species and the limitation posed on the probability of metal nanoparticle coalescence and sintering. In addition, in many cases the strong metal–metal oxide interaction is a critical factor as well for enhancing electrocatalytic properties via specific modification pathways. Despite the widely accepted role of the carbon nanoshell in stabilizing metal NPs, there is still a dearth of in-depth studies on the carbonization of organic matters and the concurrent overcoating process on the surface of metal entities during the process of high-temperature pyrolysis. Moreover, an improved mechanistic understanding for enhancing electrocatalytic properties and the synergistic effect between metal particles and overcoatings is still imperative. Intensive in situ and operando characterization technologies such as XRD, X-ray absorption (XPS, XANES, EXAFS), TEM/STEM and infrared spectroscopy (IR) in conjunction with theoretical calculations could enable detailed structural and fundamental observations on the genuine active sites and their evolution processes under the real-time synthetic and operating conditions.
(3) Broadening the application of the surface overcoating strategy. Besides the emerging applications in PEFCs (involving HOR, ORR, alcohol oxidation reactions) and WECs (comprising OER, HER, overall water splitting) as featured in this review article, the surface overcoating strategy also shows increasing potential for fabricating advanced nanomaterials, which are already applied in thermal-catalytic reactions,188,424–428 redox flow batteries,429,430 metal–air batteries,431,432 solid oxide fuel cells,433–435 lithium-ion batteries or capacitors,69,436,437 ammonia electrosynthesis,438,439 CO2 electrocatalytic reduction,440–442 and biotechnology.443,444 One could obtain highly dispersed fine metal nanoparticles with the aid of organic or inorganic overlayers after annealing treatment. As appealing in the thermal catalysis field, the carbon nanoshell derived from organic capping agents or polymers has been employed as a sacrificial protector to inhibit metal particles from migration/coalescence and sintering. A simple removal of carbon species by post-oxidation treatment enables the formation of surface clean and ultra-stable Co, Mo, Au, Pd, and Pt catalysts.445–448 One can also achieve the control of catalytic selectivity in gas-phase thermal reactions by the utility of the reactant sieve property of the carbon overlayer.449 As for manipulating catalytic activity, after overcoating reducible metal oxides onto Pt/SiO2, CO oxidation activity followed the trend TiO2 > CeO2 > Pt/SiO2 > Ta2O5 ≈ Nb2O5.450 It is assigned to the formation of a metal–metal oxide interface, which is also beneficial for Pt with high resistance towards sintering under high temperature. In another report, the Cu–ZrO2 interface formed on ZrO2-overcoated Cu/SiO2 was efficient for methanol synthesis from CO2 hydrogenation.451 Moreover, dopamine polymerization-derived N-doped carbon shell-coated SnO2,452 SnFe2O4,453 and silicon454 have already exhibited excellent rate capability and long-term stability in lithium-ion batteries. Intriguingly, the implementation of the surface overcoating strategy is holding great potential for engineering of optimal heterogeneous catalysts with a broad scope of applications.
Suitably engineered electrocatalysts play crucial roles in the practical applications of electrocatalysis-based energy conversion and storage devices. Great progress in the rational design of nanomaterials based on the surface overcoating strategy has been made for the assurance of electrochemical transformations with high efficiencies. We hope that this review can be a useful reference and serves to provide instructive inspirations for catalysis research communities. Surface overcoating engineering will continue being one of the crucially developed strategies for the design, fabrication and optimization of advanced heterogeneous catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 875524. This work does not reflect the official opinion of the European Union and responsibility for the information lies entirely with the authors.References
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245–248 CrossRef CAS PubMed.
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- H. You, S. Yang, B. Ding and H. Yang, Chem. Soc. Rev., 2013, 42, 2880–2904 RSC.
- Y. Dai, P. Lu, Z. Cao, C. T. Campbell and Y. Xia, Chem. Soc. Rev., 2018, 47, 4314–4331 RSC.
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- E. D. Goodman, J. A. Schwalbe and M. Cargnello, ACS Catal., 2017, 7, 7156–7173 CrossRef CAS.
- T. W. Hansen, A. T. DeLaRiva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 1808066 CrossRef PubMed.
- Y. Cong, B. Yi and Y. Song, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- K. Ehelebe, J. Knöppel, M. Bierling, B. Mayerhöfer, T. Böhm, N. Kulyk, S. Thiele, K. J. J. Mayrhofer and S. Cherevko, Angew. Chem., Int. Ed., 2021, 60, 8882–8888 CrossRef CAS PubMed.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- K. Singh, E. B. Tetteh, H.-Y. Lee, T.-H. Kang and J.-S. Yu, ACS Catal., 2019, 9, 8622–8645 CrossRef CAS.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- S. Mukerjee, S. Srinivasan, M. P. Soriaga and J. McBreen, J. Electrochem. Soc., 1995, 142, 1409–1422 CrossRef CAS.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 RSC.
- Y. Kang, J. B. Pyo, X. Ye, T. R. Gordon and C. B. Murray, ACS Nano, 2012, 6, 5642–5647 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- S. T. Hunt, M. Milina, A. C. Alba-Rubio, C. H. Hendon, J. A. Dumesic and Y. Román-Leshkov, Science, 2016, 352, 974–978 CrossRef CAS PubMed.
- J. Li, Z. Xi, Y.-T. Pan, J. S. Spendelow, P. N. Duchesne, D. Su, Q. Li, C. Yu, Z. Yin, B. Shen, Y. S. Kim, P. Zhang and S. Sun, J. Am. Chem. Soc., 2018, 140, 2926–2932 CrossRef CAS PubMed.
- Z. Xia and S. Guo, Chem. Soc. Rev., 2019, 48, 3265–3278 RSC.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- A. Garg, M. Milina, M. Ball, D. Zanchet, S. T. Hunt, J. A. Dumesic and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2017, 56, 8828–8833 CrossRef CAS PubMed.
- D. Göhl, A. Garg, P. Paciok, K. J. J. Mayrhofer, M. Heggen, Y. Shao-Horn, R. E. Dunin-Borkowski, Y. Román-Leshkov and M. Ledendecker, Nat. Mater., 2020, 19, 287–291 CrossRef PubMed.
- Y. J. Sa, D.-J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T.-Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- Z.-Z. Jiang, Z.-B. Wang, D.-M. Gu and E. S. Smotkin, Chem. Commun., 2010, 46, 6998–7000 RSC.
- H. Li, C. Chen, D. Yan, Y. Wang, R. Chen, Y. Zou and S. Wang, J. Mater. Chem. A, 2019, 7, 23432–23450 RSC.
- Q. Lenne, Y. R. Leroux and C. Lagrost, ChemElectroChem, 2020, 7, 2345–2363 CrossRef CAS.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- Y. Nie and Z. Wei, Prog. Nat. Sci., 2020, 30, 796–806 CrossRef CAS.
- M. Sharma, N. Jung and S. J. Yoo, Chem. Mater., 2018, 30, 2–24 CrossRef CAS.
- I. Chang, S. Ji, J. Park, M. H. Lee and S. W. Cha, Adv. Energy Mater., 2015, 5, 1402251 CrossRef.
- S. Hong, S. Oh, H. J. Kim, Y. Lim, J. An and Y.-B. Kim, J. Electrochem. Soc., 2017, 164, F1301–F1306 CrossRef CAS.
- J. W. Shin, S. Oh, S. Lee, J.-G. Yu, J. Park, D. Go, B. C. Yang, H. J. Kim and J. An, ACS Appl. Mater. Interfaces, 2019, 11, 46651–46657 CrossRef CAS PubMed.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- Y. Y. J. Tong, Chem. Soc. Rev., 2012, 41, 8195–8209 RSC.
- K. Miyabayashi, H. Nishihara and M. Miyake, Langmuir, 2014, 30, 2936–2942 CrossRef CAS PubMed.
- Y.-H. Chung, S. J. Kim, D. Y. Chung, H. Y. Park, Y.-E. Sung, S. J. Yoo and J. H. Jang, Chem. Commun., 2015, 51, 2968–2971 RSC.
- F. Mirkhalaf, J. Paprotny and D. J. Schiffrin, J. Am. Chem. Soc., 2006, 128, 7400–7401 CrossRef CAS PubMed.
- Z.-Y. Zhou, X. Kang, Y. Song and S. Chen, Chem. Commun., 2011, 47, 6075–6077 RSC.
- Z.-Y. Zhou, J. Ren, X. Kang, Y. Song, S.-G. Sun and S. Chen, Phys. Chem. Chem. Phys., 2012, 14, 1412–1417 RSC.
- A. M. Hofstead-Duffy, D.-J. Chen and Y. Y. J. Tong, Electrochim. Acta, 2012, 82, 543–549 CrossRef CAS.
- W. Liu and H. Wang, Surf. Sci., 2016, 648, 120–125 CrossRef CAS.
- J. E. Newton, J. A. Preece, N. V. Rees and S. L. Horswell, Phys. Chem. Chem. Phys., 2014, 16, 11435–11446 RSC.
- Q. Xue, J. Bai, C. Han, P. Chen, J.-X. Jiang and Y. Chen, ACS Catal., 2018, 8, 11287–11295 CrossRef CAS.
- G. Fu, K. Wu, X. Jiang, L. Tao, Y. Chen, J. Lin, Y. Zhou, S. Wei, Y. Tang, T. Lu and X. Xia, Phys. Chem. Chem. Phys., 2013, 15, 3793–3802 RSC.
- G. Fu, K. Wu, J. Lin, Y. Tang, Y. Chen, Y. Zhou and T. Lu, J. Phys. Chem. C, 2013, 117, 9826–9834 CrossRef CAS.
- G. Fu, X. Jiang, M. Gong, Y. Chen, Y. Tang, J. Lin and T. Lu, Nanoscale, 2014, 6, 8226–8234 RSC.
- F.-M. Li, X.-Q. Gao, S.-N. Li, Y. Chen and J.-M. Lee, NPG Asia Mater., 2015, 7, e219 CrossRef CAS.
- G.-R. Xu, B. Wang, J.-Y. Zhu, F.-Y. Liu, Y. Chen, J.-H. Zeng, J.-X. Jiang, Z.-H. Liu, Y.-W. Tang and J.-M. Lee, ACS Catal., 2016, 6, 5260–5267 CrossRef CAS.
- G.-R. Xu, J. Bai, L. Yao, Q. Xue, J.-X. Jiang, J.-H. Zeng, Y. Chen and J.-M. Lee, ACS Catal., 2017, 7, 452–458 CrossRef CAS.
- Y. Ding, B.-Q. Miao, Y.-C. Jiang, H.-C. Yao, X.-F. Li and Y. Chen, J. Mater. Chem. A, 2019, 7, 13770–13776 RSC.
- D. Strmcnik, M. Escudero-Escribano, K. Kodama, V. R. Stamenkovic, A. Cuesta and N. M. Marković, Nat. Chem., 2010, 2, 880–885 CrossRef CAS PubMed.
- B. Genorio, D. Strmcnik, R. Subbaraman, D. Tripkovic, G. Karapetrov, V. R. Stamenkovic, S. Pejovnik and N. M. Marković, Nat. Mater., 2010, 9, 998–1003 CrossRef CAS PubMed.
- B. Genorio, R. Subbaraman, D. Strmcnik, D. Tripkovic and V. R. Stamenkovic, Angew. Chem., Int. Ed., 2011, 50, 5468–5472 CrossRef CAS PubMed.
- J. Bai, N. Jia, P. Jin, P. Chen, J.-X. Jiang, J.-H. Zeng and Y. Chen, J. Energy Chem., 2020, 51, 105–112 CrossRef.
- S.-i. Yamazaki, M. Asahi and T. Ioroi, Electrochim. Acta, 2019, 297, 725–734 CrossRef CAS.
- S.-i. Yamazaki, M. Asahi, N. Taguchi, T. Ioroi, Y. Kishimoto, H. Daimon, M. Inaba, K. Koga, Y. Kurose and H. Inoue, ACS Catal., 2020, 10, 14567–14580 CrossRef CAS.
- S. Chen, Z. Wei, X. Q. Qi, L. Dong, Y.-G. Guo, L. Wan, Z. Shao and L. Li, J. Am. Chem. Soc., 2012, 134, 13252–13255 CrossRef CAS PubMed.
- X. Sun, N. Zhang and X. Huang, ChemCatChem, 2016, 8, 3436–3440 CrossRef CAS.
- A.-L. Wang, H. Xu, J.-X. Feng, L.-X. Ding, Y.-X. Tong and G.-R. Li, J. Am. Chem. Soc., 2013, 135, 10703–10709 CrossRef CAS PubMed.
- K. S. Kim, Y. Hong, H. C. Kim, S.-I. Choi and J. W. Hong, Chem. – Eur. J., 2019, 25, 7185–7190 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- L. Shen, H. Lv, S. Chen, P. Kopold, P. A. van Aken, X. Wu, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1700142 CrossRef PubMed.
- C. M. Parnell, B. Chhetri, A. Brandt, F. Watanabe, Z. A. Nima, T. K. Mudalige, A. S. Biris and A. Ghosh, Sci. Rep., 2016, 6, 31415 CrossRef CAS PubMed.
- T. Fujigaya, M. Okamoto and N. Nakashima, Carbon, 2009, 47, 3227–3232 CrossRef CAS.
- K. Matsumoto, T. Fujigaya, K. Sasakic and N. Nakashima, J. Mater. Chem., 2011, 21, 1187–1190 RSC.
- T. Fujigaya, M. Okamoto, K. Matsumoto, K. Kaneko and N. Nakashima, ChemCatChem, 2013, 5, 1701–1704 CrossRef CAS.
- Z. Yang and N. Nakashima, Sci. Rep., 2015, 5, 12236 CrossRef CAS PubMed.
- Z. Yang, M. R. Berber and N. Nakashima, J. Mater. Chem. A, 2014, 2, 18875–18880 RSC.
- Z. Yang, C. Kim, S. Hirata, T. Fujigaya and N. Nakashima, ACS Appl. Mater. Interfaces, 2015, 7, 15885–15891 CrossRef CAS PubMed.
- Z. Yang, I. H. Hafez, M. R. Berber and N. Nakashima, ChemCatChem, 2015, 7, 808–813 CrossRef CAS.
- Z. Yang, I. Moriguchi and N. Nakashima, ACS Appl. Mater. Interfaces, 2016, 8, 9030–9036 CrossRef CAS PubMed.
- K. Cheng, Z. Kou, J. Zhang, M. Jiang, H. Wu, L. Hu, X. Yang, M. Pan and S. Mu, J. Mater. Chem. A, 2015, 3, 14007–14014 RSC.
- J. Liu, W. Li, R. Cheng, Q. Wu, J. Zhao, D. He and S. Mu, Langmuir, 2019, 35, 2580–2586 CrossRef CAS PubMed.
- K. Ham, S. Chung and J. Lee, J. Power Sources, 2020, 450, 227650 CrossRef CAS.
- L. Guo, W.-J. Jiang, Y. Zhang, J.-S. Hu, Z.-D. Wei and L.-J. Wan, ACS Catal., 2015, 5, 2903–2909 CrossRef CAS.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J.-H. Jang, Y.-H. Cho, T. Lim, Y.-E. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- H. Yamada, H. Kato and K. Kodama, J. Electrochem. Soc., 2020, 167, 084508 CrossRef CAS.
- X. Tong, J. Zhang, G. Zhang, Q. Wei, R. Chenitz, J. P. Claverie and S. Sun, Chem. Mater., 2017, 29, 9579–9587 CrossRef CAS.
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- D. Y. Chung, S. W. Jun, G. Yoon, S. G. Kwon, D. Y. Shin, P. Seo, J. M. Yoo, H. Shin, Y.-H. Chung, H. Kim, B. S. Mun, K.-S. Lee, N.-S. Lee, S. J. Yoo, D.-H. Lim, K. Kang, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 15478–15485 CrossRef CAS PubMed.
- Y. Cho, W. H. Lee and H. Kim, J. Electrochem. Soc., 2016, 164, F65–F70 CrossRef.
- Q. Wang, S. Chen, F. Shi, K. Chen, Y. Nie, Y. Wang, R. Wu, J. Li, Y. Zhang, W. Ding, Y. Li, L. Li and Z. Wei, Adv. Mater., 2016, 28, 10673–10678 CrossRef CAS PubMed.
- X. Zou, S. Chen, Q. Wang, X. Gao, J. Li, J. Li, L. Li, W. Ding and Z. Wei, Nanoscale, 2019, 11, 20115–20122 RSC.
- S. Chen, Y. Yan, P. Hao, M. Li, J. Liang, J. Guo, Y. Zhang, S. Chen, W. Ding and X. Guo, ACS Appl. Mater. Interfaces, 2020, 12, 12686–12695 CrossRef CAS PubMed.
- J. Jin, X. Fu, Q. Liu and J. Zhang, J. Mater. Chem. A, 2013, 1, 10538–10545 RSC.
- X.-W. Gao, J. Yang, K. Song, W.-B. Luo, S.-X. Dou and Y.-M. Kang, J. Mater. Chem. A, 2018, 6, 23445–23456 RSC.
- Z. Wang, B. Li, X. Ge, F. W. Thomas Goh, X. Zhang, G. Du, D. Wuu, Z. Liu, T. S. Andy Hor, H. Zhang and Y. Zong, Small, 2016, 12, 2580–2587 CrossRef CAS PubMed.
- S. Yang, L. Chen, W. Wei, X. Lv and J. Xie, Appl. Surf. Sci., 2019, 476, 749–756 CrossRef CAS.
- S. A. Shah, Z. Ji, X. Shen, X. Yue, G. Zhu, K. Xu, A. Yuan, N. Ullah, J. Zhu, P. Song and X. Li, ACS Appl. Energy Mater., 2019, 2, 4075–4083 CrossRef CAS.
- S. Li, R. Wang, X. Yang, J. Wu, H. Meng, H. Xu and Z. Ren, ACS Sustainable Chem. Eng., 2019, 7, 11872–11884 CrossRef CAS.
- Y. Yuan, Y. Zhou, H. Shen, S. A. Rasaki, T. Thomas, J. Wang, C. Wang, J. Wang and M. Yang, ACS Appl. Energy Mater., 2018, 1, 6774–6780 CrossRef CAS.
- S. Huang, Y. Meng, S. He, A. Goswami, Q. Wu, J. Li, S. Tong, T. Asefa and M. Wu, Adv. Funct. Mater., 2017, 27, 1606585 CrossRef.
- M. Karuppannan, J. E. Park, H. E. Bae, Y.-H. Cho and O. J. Kwon, Nanoscale, 2020, 12, 2542–2554 RSC.
- H. Wang, B. Hou, Y. Yang, Q. Chen, M. Zhu, A. Thomas and Y. Liao, Small, 2018, 14, 1803232 CrossRef PubMed.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- Z. Li, L. Cai, M. Song, Y. Shen, X. Wang, J. Li, J. Wang, P. Wang and L. Tian, Electrochim. Acta, 2020, 339, 135886 CrossRef CAS.
- G. Li, Y. Sun, J. Rao, J. Wu, A. Kumar, Q. N. Xu, C. Fu, E. Liu, G. R. Blake, P. Werner, B. Shao, K. Liu, S. Parkin, X. Liu, M. Fahlman, S.-C. Liou, G. Auffermann, J. Zhang, C. Felser and X. Feng, Adv. Energy Mater., 2018, 8, 1801258 CrossRef.
- Z. Pu, I. S. Amiinu, Z. Kou, W. Li and S. Mu, Angew. Chem., Int. Ed., 2017, 56, 11559–11564 CrossRef CAS PubMed.
- X. Lv, J. Ren, Y. Wang, Y. Liu and Z.-Y. Yuan, ACS Sustainable Chem. Eng., 2019, 7, 8993–9001 CrossRef CAS.
- Y.-Y. Ma, C.-X. Wu, X.-J. Feng, H.-Q. Tan, L.-K. Yan, Y. Liu, Z.-H. Kang, E.-B. Wang and Y.-G. Li, Energy Environ. Sci., 2017, 10, 788–798 RSC.
- G. Yan, C. Wu, H. Tan, X. Feng, L. Yan, H. Zang and Y. Li, J. Mater. Chem. A, 2017, 5, 765–772 RSC.
- C. Pi, C. Huang, Y. Yang, H. Song, X. Zhang, Y. Zheng, B. Gao, J. Fu, P. K. Chu and K. Huo, Appl. Catal., B, 2020, 263, 118358 CrossRef CAS.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- Z. Chen, H. Qing, K. Zhou, D. Sun and R. Wu, Prog. Mater. Sci., 2020, 108, 100618 CrossRef CAS.
- Q. Liang, J. Chen, F. Wang and Y. Li, Coord. Chem. Rev., 2020, 424, 213488 CrossRef CAS.
- X. Li, L. Yang, T. Su, X. Wang, C. Sun and Z. Su, J. Mater. Chem. A, 2017, 5, 5000–5006 RSC.
- M. Khalid, A. M. B. Honorato, G. T. Filho and H. Varela, J. Mater. Chem. A, 2020, 8, 9021–9031 RSC.
- Z. Shi, Y. Wang, H. Lin, H. Zhang, M. Shen, S. Xie, Y. Zhang, Q. Gao and Y. Tang, J. Mater. Chem. A, 2016, 4, 6006–6013 RSC.
- L. Wang, J. Cao, X. Cheng, C. Lei, Q. Dai, B. Yang, Z. Li, M. A. Younis, L. Lei, Y. Hou and K. Ostrikov, ACS Sustainable Chem. Eng., 2019, 7, 10044–10051 CrossRef CAS.
- H. Peng, M. Zhang, K. Sun, X. Xie, H. Lei and G. Ma, Appl. Surf. Sci., 2020, 529, 147174 CrossRef CAS.
- X. Zhu, M. Liu, Y. Liu, R. Chen, Z. Nie, J. Li and S. Yao, J. Mater. Chem. A, 2016, 4, 8974–8977 RSC.
- J. Su, G. Xia, R. Li, Y. Yang, J. Chen, R. Shi, P. Jiang and Q. Chen, J. Mater. Chem. A, 2016, 4, 9204–9212 RSC.
- B. Singh and A. Indra, Mater. Today Energy, 2020, 16, 100404 CrossRef.
- P. Jiang, J. Chen, C. Wang, K. Yang, S. Gong, S. Liu, Z. Lin, M. Li, G. Xia, Y. Yang, J. Su and Q. Chen, Adv. Mater., 2018, 30, 1705324 CrossRef PubMed.
- J. Xi, Y. Xia, Y. Xu, J. Xiao and S. Wang, Chem. Commun., 2015, 51, 10479–10482 RSC.
- L. Du, L. Luo, Z. Feng, M. Engelhard, X. Xie, B. Han, J. Sun, J. Zhang, G. Yin, C. Wang, Y. Wang and Y. Shao, Nano Energy, 2017, 39, 245–252 CrossRef CAS.
- J.-S. Li, Y.-J. Tang, C.-H. Liu, S.-L. Li, R.-H. Li, L.-Z. Dong, Z.-H. Dai, J.-C. Bao and Y.-Q. Lan, J. Mater. Chem. A, 2016, 4, 1202–1207 RSC.
- P. He, X.-Y. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2017, 56, 3897–3900 CrossRef CAS PubMed.
- Y.-T. Xu, X. Xiao, Z.-M. Ye, S. Zhao, R. Shen, C.-T. He, J.-P. Zhang, Y. Li and X.-M. Chen, J. Am. Chem. Soc., 2017, 139, 5285–5288 CrossRef CAS PubMed.
- F. Bu, W. Chen, M. F. Aly Aboud, I. Shakir, J. Gu and Y. Xu, J. Mater. Chem. A, 2019, 7, 14526–14535 RSC.
- Q. Hu, X. Liu, C. Tang, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, Sustainable Energy Fuels, 2018, 2, 1085–1092 RSC.
- L.-L. Wu, Q.-S. Wang, J. Li, Y. Long, Y. Liu, S.-Y. Song and H.-J. Zhang, Small, 2018, 14, 1704035 CrossRef PubMed.
- B. Xu, X. Wang, X. Yang, Z. Chen, Y. Sun, Q. Liu and C. Li, Electrochim. Acta, 2019, 296, 738–745 CrossRef CAS.
- Q. Hu, X. Liu, C. Tang, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, Electrochim. Acta, 2018, 265, 620–628 CrossRef CAS.
- F. Bu, W. Chen, J. Gu, P. O. Agboola, N. F. Al-Khalli, I. Shakir and Y. Xu, Chem. Sci., 2018, 9, 7009–7016 RSC.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC.
- X. Cui, P. Ren, C. Ma, J. Zhao, R. Chen, S. Chen, N. P. Rajan, H. Li, L. Yu, Z. Tian and D. Deng, Adv. Mater., 2020, 32, 1908126 CrossRef CAS PubMed.
- D. V. Esposito, ACS Catal., 2018, 8, 457–465 CrossRef CAS.
- N. Aoki, H. Inoue, H. Kawasaki, H. Daimon, T. Doi and M. Inaba, J. Electrochem. Soc., 2018, 165, F737 CrossRef CAS.
- S. Takenaka, A. Hirata, E. Tanabe, H. Matsune and M. Kishida, J. Catal., 2010, 274, 228–238 CrossRef CAS.
- S. Takenaka, H. Miyamoto, Y. Utsunomiya, H. Matsune and M. Kishida, J. Phys. Chem. C, 2014, 118, 774–783 CrossRef CAS.
- S. Kang, F. Xia, Z. Hu, W. Hu, Y. She, L. Wang, X. Fu and W. Lu, Electrochim. Acta, 2020, 343, 136119 CrossRef CAS.
- H. J. Kim, D. H. K. Jackson, J. Lee, Y. Guan, T. F. Kuech and G. W. Huber, ACS Catal., 2015, 5, 3463–3469 CrossRef CAS.
- X. Wang, Y. Yang, L. Diao, Y. Tang, F. He, E. Liu, C. He, C. Shi, J. Li, J. Sha, S. Ji, P. Zhang, L. Ma and N. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 35145–35153 CrossRef CAS PubMed.
- H. Xu, J. Cao, C. Shan, B. Wang, P. Xi, W. Liu and Y. Tang, Angew. Chem., Int. Ed., 2018, 57, 8654–8658 CrossRef CAS PubMed.
- J. G. Vos, T. A. Wezendonk, A. W. Jeremiasse and M. T. M. Koper, J. Am. Chem. Soc., 2018, 140, 10270–10281 CrossRef CAS PubMed.
- W. J. Jo, G. Katsoukis and H. Frei, Adv. Funct. Mater., 2020, 30, 1909262 CrossRef CAS.
- B. Endrődi, V. Smulders, N. Simic, M. Wildlock, G. Mul, B. Mei and A. Cornell, Appl. Catal., B, 2019, 244, 233–239 CrossRef.
- B. Endrődi, O. Diaz-Morales, U. Mattinen, M. Cuartero, A. K. Padinjarethil, N. Simic, M. Wildlock, G. A. Crespo and A. Cornell, Electrochim. Acta, 2020, 341, 136022 CrossRef.
- D. He, C. Zeng, C. Xu, N. Cheng, H. Li, S. Mu and M. Pan, Langmuir, 2011, 27, 5582–5588 CrossRef CAS PubMed.
- B. Ye, K. Cheng, W. Li, J. Liu, J. Zhang and S. Mu, Langmuir, 2017, 33, 5353–5361 CrossRef CAS PubMed.
- Y. Wang, Z. Li, S. Xu, Y. Xie and S. Lin, Mater. Res. Bull., 2018, 102, 172–179 CrossRef CAS.
- B. Wu, D. Hu, Y. Kuang, B. Liu, X. Zhang and J. Chen, Angew. Chem., Int. Ed., 2009, 48, 4751–4754 CrossRef CAS PubMed.
- Q.-L. Zhang, T.-Q. Xu, J. Wei, J.-R. Chen, A.-J. Wang and J.-J. Feng, Electrochim. Acta, 2013, 112, 127–132 CrossRef CAS.
- X. Yu, H. Wang, L. Guo and L. Wang, Chem. – Asian J., 2014, 9, 3221–3227 CrossRef CAS PubMed.
- W. Tamakloe, D. A. Agyeman, M. Park, J. Yang and Y.-M. Kang, J. Mater. Chem. A, 2019, 7, 7396–7405 RSC.
- R. Escudero Cid, J. L. Gómez de la Fuente, S. Rojas, J. L. García Fierro and P. Ocón, ChemCatChem, 2013, 5, 3680–3689 CrossRef CAS.
- T. Fujigaya, S. Hirata, M. R. Berber and N. Nakashima, ACS Appl. Mater. Interfaces, 2016, 8, 14494–14502 CrossRef CAS PubMed.
- M. Okamoto, T. Fujigaya and N. Nakashima, Small, 2009, 5, 735–740 CrossRef CAS PubMed.
- S. M. Jayawickrama, Z. Han, S. Kido, N. Nakashima and T. Fujigaya, Electrochim. Acta, 2019, 312, 349–357 CrossRef CAS.
- T. Fujigaya, Y. Shi, J. Yang, H. Li, K. Ito and N. Nakashima, J. Mater. Chem. A, 2017, 5, 10584–10590 RSC.
- Z. Yang and N. Nakashima, J. Mater. Chem. A, 2015, 3, 23316–23322 RSC.
- F. Morales-Lara, V. K. Abdelkader-Fernández, M. Melguizo, A. Turco, E. Mazzotta, M. Domingo-García, F. J. López-Garzón and M. Pérez-Mendoza, J. Mater. Chem. A, 2019, 7, 24502–24514 RSC.
- X.-C. Liu, G.-C. Wang, R.-P. Liang, L. Shi and J.-D. Qiu, J. Mater. Chem. A, 2013, 1, 3945–3953 RSC.
- H. Yang, H. Zou, M. Chen, S. Li, J. Jin and J. Ma, Inorg. Chem. Front., 2017, 4, 1881–1887 RSC.
- M. R. Berber, I. H. Hafez, T. Fujigaya and N. Nakashima, J. Mater. Chem. A, 2014, 2, 19053–19059 RSC.
- M. Kato, R. Nakahoshiba, K. Ogura, S. Tokuda, S. Yasuda, K. Higashi, T. Uruga, Y. Uemura and I. Yagi, ACS Appl. Energy Mater., 2020, 3, 6768–6774 CrossRef CAS.
- T. Fujigaya and N. Nakashima, Adv. Mater., 2013, 25, 1666–1681 CrossRef CAS PubMed.
- N. Jung, S. Bhattacharjee, S. Gautam, H.-Y. Park, J. Ryu, Y.-H. Chung, S.-Y. Lee, I. Jang, J. H. Jang, S. H. Park, D. Y. Chung, Y.-E. Sung, K.-H. Chae, U. V. Waghmare, S.-C. Lee and S. J. Yoo, NPG Asia Mater., 2016, 8, e237 CrossRef CAS.
- Z.-Z. Jiang, Z.-B. Wang, Y.-Y. Chu, D.-M. Gu and G.-P. Yin, Energy Environ. Sci., 2011, 4, 2558–2566 RSC.
- Z.-Z. Jiang, Z.-B. Wang, Y.-Y. Chu, D.-M. Gu and G.-P. Yin, Energy Environ. Sci., 2011, 4, 728–735 RSC.
- Z.-Z. Jiang, Z.-B. Wang, W.-L. Qu, H. Rivera, D.-M. Gu and G.-P. Yin, Nanoscale, 2012, 4, 7411–7418 RSC.
- Q. Zhang, Z. Yang, J. Yang, X. Yu, Y. Ling, Y. Zhang, W. Cai and H. Cheng, Chem. Commun., 2018, 54, 9282–9285 RSC.
- C.-Z. Li, Z.-B. Wang, X.-L. Sui, L.-M. Zhang, D.-M. Gu and S. Gu, J. Mater. Chem. A, 2014, 2, 20139–20146 RSC.
- C.-Z. Li, Z.-B. Wang, X.-L. Sui, L.-M. Zhang and D.-M. Gu, Carbon, 2015, 93, 105–115 CrossRef CAS.
- S.-Y. Huang, P. Ganesan, S. Park and B. N. Popov, J. Am. Chem. Soc., 2009, 131, 13898–13899 CrossRef CAS PubMed.
- Y. Liu, S. Shrestha and W. E. Mustain, ACS Catal., 2012, 2, 456–463 CrossRef CAS.
- V. T. T. Ho, C.-J. Pan, J. Rick, W.-N. Su and B.-J. Hwang, J. Am. Chem. Soc., 2011, 133, 11716–11724 CrossRef CAS PubMed.
- T.-T. Nguyen, V. T. ThanhHo, C.-J. Pan, J.-Y. Liu, H.-L. Chou, J. Rick, W.-N. Su and B.-J. Hwang, Appl. Catal., B, 2014, 154, 183–189 CrossRef.
- A. Kumar and V. Ramani, ACS Catal., 2014, 4, 1516–1525 CrossRef CAS.
- Y. Liu and W. E. Mustain, J. Am. Chem. Soc., 2013, 135, 530–533 CrossRef CAS PubMed.
- A. S. Asundi, J. A. Raiford and S. F. Bent, ACS Energy Lett., 2019, 4, 908–925 CrossRef CAS.
- Y. Zhao and X. Sun, ACS Energy Lett., 2018, 3, 899–914 CrossRef CAS.
- Z. Gao and Y. Qin, Acc. Chem. Res., 2017, 50, 2309–2316 CrossRef CAS PubMed.
- X. Meng, X. Wang, D. Geng, C. Ozgit-Akgun, N. Schneider and J. W. Elam, Mater. Horiz., 2017, 4, 133–154 RSC.
- B. Zhang and Y. Qin, ACS Catal., 2018, 8, 10064–10081 CrossRef CAS.
- K. Cao, J. Cai and R. Chen, Chem. Mater., 2020, 32, 2195–2207 CrossRef CAS.
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS PubMed.
- Y. Chen, J. Wang, X. Meng, Y. Zhong, R. Li, X. Sun, S. Ye and S. Knights, J. Power Sources, 2013, 238, 144–149 CrossRef CAS.
- S. Hussain, H. Erikson, N. Kongi, A. Tarre, P. Ritslaid, M. Kook, M. Rähn, M. Merisalu, V. Sammelselg and K. Tammeveski, J. Electrochem. Soc., 2019, 166, F1284–F1291 CrossRef CAS.
- N. Cheng, M. N. Banis, J. Liu, A. Riese, X. Li, R. Li, S. Ye, S. Knights and X. Sun, Adv. Mater., 2015, 27, 277–281 CrossRef CAS PubMed.
- Z. Song, B. Wang, N. Cheng, L. Yang, D. Banham, R. Li, S. Ye and X. Sun, J. Mater. Chem. A, 2017, 5, 9760–9767 RSC.
- L. Zhang, Y. Zhao, M. N. Banis, K. Adair, Z. Song, L. Yang, M. Markiewicz, J. Li, S. Wang, R. Li, S. Ye and X. Sun, Nano Energy, 2019, 60, 111–118 CrossRef CAS.
- C. Du, X. Gao, C. Cheng, Z. Zhuang, X. Li and W. Chen, Electrochim. Acta, 2018, 266, 348–356 CrossRef CAS.
- F. Xu, D. Wang, B. Sa, Y. Yu and S. Mu, Int. J. Hydrogen Energy, 2017, 42, 13011–13019 CrossRef CAS.
- Y. Li, X. Zhang, S. Wang and G. Sun, ChemElectroChem, 2018, 5, 2442–2448 CrossRef CAS.
- Y.-J. Wang, D. P. Wilkinson, V. Neburchilov, C. Song, A. Guest and J. Zhang, J. Mater. Chem. A, 2014, 2, 12681–12685 RSC.
- Y. Zhuang, W. Ding, Y. Liu, Z. Mou, J. Sun and M. Guan, J. Mater. Sci., 2015, 50, 3875–3882 CrossRef CAS.
- J. Wang, M. Xu, J. Zhao, H. Fang, Q. Huang, W. Xiao, T. Li and D. Wang, Appl. Catal., B, 2018, 237, 228–236 CrossRef CAS.
- N. G. Akalework, C.-J. Pan, W.-N. Su, J. Rick, M.-C. Tsai, J.-F. Lee, J.-M. Lin, L.-D. Tsai and B.-J. Hwang, J. Mater. Chem., 2012, 22, 20977–20985 RSC.
- J. Huang, J. Zang, Y. Zhao, L. Dong and Y. Wang, Mater. Lett., 2014, 137, 335–338 CrossRef CAS.
- S. Hussain, N. Kongi, H. Erikson, M. Rähn, M. Merisalu, L. Matisen, P. Paiste, J. Aruväli, V. Sammelselg, L. A. Estudillo-Wong, K. Tammeveski and N. Alonso-Vante, Electrochim. Acta, 2019, 316, 162–172 CrossRef CAS.
- Z. Ma, S. Li, L. Wu, L. Song, G. Jiang, Z. Liang, D. Su, Y. Zhu, R. R. Adzic, J. X. Wang and Z. Chen, Nano Energy, 2020, 69, 104455 CrossRef CAS.
- Y. Qu, Y. Gao, L. Wang, J. Rao and G. Yin, Chem. – Eur. J., 2016, 22, 193–198 CrossRef CAS PubMed.
- W. Gao, T. Liu, Z. Zhang, M. Dou and F. Wang, J. Mater. Chem. A, 2020, 8, 5525–5534 RSC.
- A. Meng, L. Lin, X. Yuan, T. Shen, Z. Li and Q. Li, ChemCatChem, 2019, 11, 2900–2908 CrossRef CAS.
- Y.-P. Xiao, W.-J. Jiang, S. Wan, X. Zhang, J.-S. Hu, Z.-D. Wei and L.-J. Wan, J. Mater. Chem. A, 2013, 1, 7463–7468 RSC.
- L. Wang, W. Wu, Z. Lei, T. Zeng, Y. Tan, N. Cheng and X. Sun, J. Mater. Chem. A, 2020, 8, 592–598 RSC.
- Z. M. Cui, S. P. Jiang and C. M. Li, Chem. Commun., 2011, 47, 8418–8420 RSC.
- S. Wu, J. Liu, Y. Ye, Z. Tian, X. Zhu and C. Liang, ACS Appl. Energy Mater., 2019, 2, 5577–5583 CrossRef CAS.
- Y.-Y. Chu, Z.-B. Wang, Z.-Z. Jiang, D.-M. Gu and G.-P. Yin, Adv. Mater., 2011, 23, 3100–3104 CrossRef CAS PubMed.
- Y. Y. Chu, J. Cao, Z. Dai and X. Y. Tan, J. Mater. Chem. A, 2014, 2, 4038–4044 RSC.
- D.-G. Lee, H. Jeong, K.-W. Jeon, L. Zhang, K. Park, S. Ryu, J. Kim and I. S. Lee, J. Mater. Chem. A, 2017, 5, 22341–22351 RSC.
- J. Zhu, M. Xiao, X. Zhao, C. Liu and W. Xing, J. Power Sources, 2015, 292, 78–86 CrossRef CAS.
- W. Li, Y. Bai, F. Li, C. Liu, K.-Y. Chan, X. Feng and X. Lu, J. Mater. Chem., 2012, 22, 4025–4031 RSC.
- X. Wu, W. Zhuang, L. Lu, L. Li, J. Zhu, L. Mu, W. Li, Y. Zhu and X. Lu, Appl. Surf. Sci., 2017, 426, 890–896 CrossRef CAS.
- Y. Chang, C. Yuan, Y. Li, C. Liu, T. Wu, B. Zeng, Y. Xu and L. Dai, J. Mater. Chem. A, 2017, 5, 1672–1678 RSC.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- N. Jung, H. Shin, M. Kim, I. Jang, H.-J. Kim, J. H. Jang, H. Kim and S. J. Yoo, Nano Energy, 2015, 17, 152–159 CrossRef CAS.
- Y. Tan, C. Xu, G. Chen, N. Zheng and Q. Xie, Energy Environ. Sci., 2012, 5, 6923–6927 RSC.
- L. Lu, Z. Wang, S. Zou, Y. Zhou, W. Hong, R. Li, L. Xiao, J. Liu, X.-Q. Gong and J. Fan, J. Mater. Chem. A, 2018, 6, 18884–18890 RSC.
- Y.-H. Chung, D. Y. Chung, N. Jung and Y.-E. Sung, J. Phys. Chem. Lett., 2013, 4, 1304–1309 CrossRef CAS PubMed.
- H. S. Casalongue, S. Kaya, V. Viswanathan, D. J. Miller, D. Friebel, H. A. Hansen, J. K. Nørskov, A. Nilsson and H. Ogasawara, Nat. Commun., 2013, 4, 2817 CrossRef.
- Z.-Y. Zhou, X. Kang, Y. Song and S. Chen, J. Phys. Chem. C, 2012, 116, 10592–10598 CrossRef CAS.
- M. Asahi, S.-i. Yamazaki, N. Taguchi and T. Ioroi, J. Electrochem. Soc., 2019, 166, F498–F505 CrossRef.
- J. Snyder, K. Livi and J. Erlebacher, Adv. Funct. Mater., 2013, 23, 5494–5501 CrossRef CAS.
- G.-R. Zhang, M. Munoz and B. J. M. Etzold, Angew. Chem., Int. Ed., 2016, 55, 2257–2261 CrossRef CAS PubMed.
- M. George, G.-R. Zhang, N. Schmitt, K. Brunnengräber, D. J. S. Sandbeck, K. J. J. Mayrhofer, S. Cherevko and B. J. M. Etzold, ACS Catal., 2019, 9, 8682–8692 CrossRef CAS PubMed.
- W. Song, Z. Chen, C. Yang, Z. Yang, J. Tai, Y. Nan and H. Lu, J. Mater. Chem. A, 2015, 3, 1049–1057 RSC.
- Y. Nie, S. Chen, W. Ding, X. Xie, Y. Zhang and Z. Wei, Chem. Commun., 2014, 50, 15431–15434 RSC.
- K. Sun, J. Li, F. Wang, W. He, M. Fei, Z. Lu, H. Zhang, J. Liu and Z. Zou, Chem. Commun., 2019, 55, 5693–5696 RSC.
- G. Chen, H. Shan, Y. Li, H. Bao, T. Hu, L. Zhang, S. Liu and F. Ma, J. Mater. Chem. A, 2020, 8, 10337–10345 RSC.
- W. Wu, Z. Zhang, Z. Lei, X. Wang, Y. Tan, N. Cheng and X. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 10359–10368 CrossRef CAS PubMed.
- W. Li and S. Zou, ACS Appl. Energy Mater., 2019, 2, 2769–2778 CrossRef CAS.
- S. Takenaka, T. Miyazaki, H. Matsune and M. Kishida, Catal. Sci. Technol., 2015, 5, 1133–1142 RSC.
- K. Park, T. Ohnishi, M. Goto, M. So, S. Takenaka, Y. Tsuge and G. Inoue, Int. J. Hydrogen Energy, 2020, 45, 1867–1877 CrossRef CAS.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- Z. Dong, M. Li, W. Zhang, Y. Liu, Y. Wang., C. Qin, L. Yu, J. Yang, X. Zhang and X. Dai, ChemCatChem, 2019, 11, 6039–6050 CrossRef CAS.
- H.-S. Park, S.-B. Han, D.-H. Kwak, J.-H. Han and K.-W. Park, J. Catal., 2019, 370, 130–137 CrossRef CAS.
- J. Deng, L. Yu, D. Deng, X. Chen, F. Yang and X. Bao, J. Mater. Chem. A, 2013, 1, 14868–14873 RSC.
- S. Gao, B. Fan, R. Feng, C. Ye, X. Wei, J. Liu and X. Bu, Nano Energy, 2017, 40, 462–470 CrossRef CAS.
- Y. Hou, T. Huang, Z. Wen, S. Mao, S. Cui and J. Chen, Adv. Energy Mater., 2014, 4, 1400337 CrossRef.
- K. Vezzù, A. B. Delpeuch, E. Negro, S. Polizzi, G. Nawn, F. Bertasi, G. Pagot, K. Artyushkova, P. Atanassov and V. Di Noto, Electrochim. Acta, 2016, 222, 1778–1791 CrossRef.
- K. Yuan, S. Sfaelou, M. Qiu, D. Lützenkirchen-Hecht, X. Zhuang, Y. Chen, C. Yuan, X. Feng and U. Scherf, ACS Energy Lett., 2018, 3, 252–260 CrossRef CAS.
- J. Ryu, N. Jung, D.-H. Lim, D. Y. Shin, S. H. Park, H. C. Ham, J. H. Jang, H.-J. Kim and S. J. Yoo, Chem. Commun., 2014, 50, 15940–15943 RSC.
- X. Zhong, Y. Jiang, X. Chen, L. Wang, G. Zhuang, X. Li and J.-G. Wang, J. Mater. Chem. A, 2016, 4, 10575–10584 RSC.
- C. Han, X. Bo, Y. Zhang, M. Li, A. Wang and L. Guo, Chem. Commun., 2015, 51, 15015–15018 RSC.
- Z. Pu, R. Cheng, J. Zhao, Z. Hu, C. Li, W. Li, P. Wang, I. S. Amiinu, Z. Wang, M. Wang, D. Chen and S. Mu, iScience, 2020, 23, 101793 CrossRef CAS PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- K. Elbert, J. Hu, Z. Ma, Y. Zhang, G. Chen, W. An, P. Liu, H. S. Isaacs, R. R. Adzic and J. X. Wang, ACS Catal., 2015, 5, 6764–6772 CrossRef CAS.
- J. Ohyama, D. Kumada and A. Satsuma, J. Mater. Chem. A, 2016, 4, 15980–15985 RSC.
- H. Wang and H. D. Abruña, J. Am. Chem. Soc., 2017, 139, 6807–6810 CrossRef CAS PubMed.
- Y. Cong, B. Yi and Y. Song, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- L. Su, D. Gong, Y. Jin, D. Wu and W. Luo, J. Energy Chem., 2022, 66, 107–122 CrossRef.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef PubMed.
- T. Wang, Z.-X. Chen, S. Yu, T. Sheng, H.-B. Ma, L.-N. Chen, M. Rauf, H.-P. Xia, Z.-Y. Zhou and S.-G. Sun, Energy Environ. Sci., 2018, 11, 166–171 RSC.
- M. Sun, Y. Lv, Y. Song, H. Wu, G. Wang, H. Zhang, M. Chen, Q. Fu and X. Bao, Appl. Surf. Sci., 2018, 450, 244–250 CrossRef CAS.
- S.-W. Yun, S.-A. Park, T.-J. Kim, J.-H. Kim, G.-W. Pak and Y.-T. Kim, ChemSusChem, 2017, 10, 489–493 CrossRef CAS PubMed.
- J. Jang, M. Sharma, D. Choi, Y. S. Kang, Y. Kim, J. Min, H. Sung, N. Jung and S. J. Yoo, ACS Appl. Mater. Interfaces, 2019, 11, 27735–27742 CrossRef CAS PubMed.
- R. K. Singh, E. S. Davydova, J. Douglin, A. O. Godoy, H. Tan, M. Bellini, B. J. Allen, J. Jankovic, H. A. Miller, A. C. Alba-Rubio and D. R. Dekel, Adv. Funct. Mater., 2020, 30, 2002087 CrossRef CAS.
- F. D. Speck, F. S. M. Ali, M. T. Y. Paul, R. K. Singh, T. Böhm, A. Hofer, O. Kasian, S. Thiele, J. Bachmann, D. R. Dekel, T. Kallio and S. Cherevko, Chem. Mater., 2020, 32, 7716–7724 CrossRef CAS.
- B. M. Stühmeier, S. Selve, M. U. M. Patel, T. N. Geppert, H. A. Gasteiger and H. A. El-Sayed, ACS Appl. Energy Mater., 2019, 2, 5534–5539 CrossRef.
- T. N. Geppert, M. Bosund, M. Putkonen, B. M. Stühmeier, A. T. Pasanen, P. Heikkilä, H. A. Gasteiger and H. A. El-Sayed, J. Electrochem. Soc., 2020, 167, 084517 CrossRef CAS.
- G. Zhao, J. Chen, W. Sun and H. Pan, Adv. Funct. Mater., 2021, 31, 2010633 CrossRef CAS.
- A. G. Oshchepkov, G. Braesch, A. Bonnefont, E. R. Savinova and M. Chatenet, ACS Catal., 2020, 10, 7043–7068 CrossRef CAS.
- S. Kabir, K. Lemire, K. Artyushkova, A. Roy, M. Odgaard, D. Schlueter, A. Oshchepkov, A. Bonnefont, E. Savinova, D. C. Sabarirajan, P. Mandal, E. J. Crumlin, I. V. Zenyuk, P. Atanassov and A. Serov, J. Mater. Chem. A, 2017, 5, 24433–24443 RSC.
- M. Wang, H. Yang, J. Shi, Y. Chen, Y. Zhou, L. Wang, S. Di, X. Zhao, J. Zhong, T. Cheng, W. Zhou and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 5771–5777 CrossRef CAS PubMed.
- A. Roy, M. R. Talarposhti, S. J. Normile, I. V. Zenyuk, V. D. Andrade, K. Artyushkova, A. Serov and P. Atanassov, Sustainable Energy Fuels, 2018, 2, 2268–2275 RSC.
- W. Sheng, A. P. Bivens, M. Myint, Z. Zhuang, R. V. Forest, Q. Fang, J. G. Chen and Y. Yan, Energy Environ. Sci., 2014, 7, 1719–1724 RSC.
- S. Qin, Y. Duan, X.-L. Zhang, L.-R. Zheng, F.-Y. Gao, P.-P. Yang, Z.-Z. Niu, R. Liu, Y. Yang, X.-S. Zheng, J.-F. Zhu and M.-R. Gao, Nat. Commun., 2021, 12, 2686 CrossRef CAS PubMed.
- T. Wang, M. Wang, H. Yang, M. Xu, C. Zuo, K. Feng, M. Xie, J. Deng, J. Zhong, W. Zhou, T. Cheng and Y. Li, Energy Environ. Sci., 2019, 12, 3522–3529 RSC.
- F. Yang, P. Han, N. Yao, G. Cheng, S. Chen and W. Luo, Chem. Sci., 2020, 11, 12118–12123 RSC.
- W. Ni, T. Wang, P. A. Schouwink, Y.-C. Chuang, H. M. Chen and X. Hu, Angew. Chem., Int. Ed., 2020, 59, 10797–10801 CrossRef CAS PubMed.
- F. Song, W. Li, J. Yang, G. Han, P. Liao and Y. Sun, Nat. Commun., 2018, 9, 4531 CrossRef PubMed.
- Z. Zhuang, S. A. Giles, J. Zheng, G. R. Jenness, S. Caratzoulas, D. G. Vlachos and Y. Yan, Nat. Commun., 2016, 7, 10141 CrossRef CAS PubMed.
- F. Yang, X. Bao, Y. Zhao, X. Wang, G. Cheng and W. Luo, J. Mater. Chem. A, 2019, 7, 10936–10941 RSC.
- Y. Zhou, Z. Xie, J. Jiang, J. Wang, X. Song, Q. He, W. Ding and Z. Wei, Nat. Catal., 2020, 3, 454–462 CrossRef CAS.
- A. G. Oshchepkov, A. Bonnefont and E. R. Savinova, Curr. Opin. Electrochem., 2021, 26, 100667 CrossRef CAS.
- F. Yang, X. Bao, P. Li, X. Wang, G. Cheng, S. Chen and W. Luo, Angew. Chem., Int. Ed., 2019, 58, 14179–14183 CrossRef CAS PubMed.
- Y. Yang, X. Sun, G. Han, X. Liu, X. Zhang, Y. Sun, M. Zhang, Z. Cao and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 10644–10649 CrossRef CAS PubMed.
- A. G. Oshchepkov, A. Bonnefont, V. A. Saveleva, V. Papaefthimiou, S. Zafeiratos, S. N. Pronkin, V. N. Parmon and E. R. Savinova, Top. Catal., 2016, 59, 1319–1331 CrossRef CAS.
- A. G. Oshchepkov, A. Bonnefont, V. N. Parmon and E. R. Savinova, Electrochim. Acta, 2018, 269, 111–118 CrossRef CAS.
- A. G. Oshchepkov, A. Bonnefont and E. R. Savinova, Electrocatalysis, 2020, 11, 133–142 CrossRef CAS.
- Y. Pan, G. Hu, J. Lu, L. Xiao and L. Zhuang, J. Energy Chem., 2019, 29, 111–115 CrossRef.
- E. S. Davydova, F. D. Speck, M. T. Y. Paul, D. R. Dekel and S. Cherevko, ACS Catal., 2019, 9, 6837–6845 CrossRef CAS.
- L. Gao, Y. Wang, H. Li, Q. Li, N. Ta, L. Zhuang, Q. Fu and X. Bao, Chem. Sci., 2017, 8, 5728–5734 RSC.
- Y. Gao, H. Peng, Y. Wang, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2020, 12, 31575–31581 CrossRef CAS PubMed.
- J. Wang, X. Dong, J. Liu, W. Li, L. T. Roling, J. Xiao and L. Jiang, ACS Catal., 2021, 11, 7422–7428 CrossRef CAS.
- B. Xiong, W. Zhao, L. Chen and J. Shi, Adv. Funct. Mater., 2019, 29, 1902505 CrossRef.
- L. Wang, E. Magliocca, E. L. Cunningham, W. E. Mustain, S. D. Poynton, R. Escudero-Cid, M. M. Nasef, J. Ponce-González, R. Bance-Souahli, R. C. T. Slade, D. K. Whelligan and J. R. Varcoe, Green Chem., 2017, 19, 831–843 RSC.
- S. Lu, J. Pan, A. Huang, L. Zhuang and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611–20614 CrossRef CAS.
- Q. Wang, S. Chen, H. Lan, P. Li, X. Ping, S. Ibraheem, D. Long, Y. Duan and Z. Wei, J. Mater. Chem. A, 2019, 7, 18143–18149 RSC.
- J. E. Robinson, N. Y. Labrador, H. Chen, B. E. Sartor and D. V. Esposito, ACS Catal., 2018, 8, 11423–11434 CrossRef CAS.
- P. Dhanasekaran, A. Shukla, S. V. Selvaganesh, S. Mohan and S. D. Bhat, J. Power Sources, 2019, 438, 226999 CrossRef CAS.
- H. Lv, N. Cheng, T. Peng, M. Pan and S. Mu, J. Mater. Chem., 2012, 22, 1135–1141 RSC.
- L. Xin, F. Yang, S. Rasouli, Y. Qiu, Z.-F. Li, A. Uzunoglu, C.-J. Sun, Y. Liu, P. Ferreira, W. Li, Y. Ren, L. A. Stanciu and J. Xie, ACS Catal., 2016, 6, 2642–2653 CrossRef CAS.
- C.-H. Hsu, H.-Y. Liao and P.-L. Kuo, J. Phys. Chem. C, 2010, 114, 7933–7939 CrossRef CAS.
- Z. Yang, J. Li, Y. Ling, Q. Zhang, X. Yu and W. Cai, ChemCatChem, 2017, 9, 3307–3313 CrossRef CAS.
- Z. Yang, T. Fujigaya and N. Nakashima, J. Power Sources, 2015, 300, 175–181 CrossRef.
- Z. Song, M. N. Banis, L. Zhang, B. Wang, L. Yang, D. Banham, Y. Zhao, J. Liang, M. Zheng, R. Li, S. Ye and X. Sun, Nano Energy, 2018, 53, 716–725 CrossRef CAS.
- W. Yuan, J. Li, L. Wang, P. Chen, A. Xie and Y. Shen, ACS Appl. Mater. Interfaces, 2014, 6, 21978–21985 CrossRef CAS PubMed.
- C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Chem. Rev., 2020, 120, 1184–1249 CrossRef CAS PubMed.
- R. Kou, Y. Shao, D. Mei, Z. Nie, D. Wang, C. Wang, V. V. Viswanathan, S. Park, I. A. Aksay, Y. Lin, Y. Wang and J. Liu, J. Am. Chem. Soc., 2011, 133, 2541–2547 CrossRef CAS PubMed.
- H. Huang, Q. Chen, M. He, X. Sun and X. Wang, J. Power Sources, 2013, 239, 189–195 CrossRef CAS.
- H. Huang, Y. Liu, Q. Gao, W. Ruan, X. Lin and X. Li, ACS Appl. Mater. Interfaces, 2014, 6, 10258–10264 CrossRef CAS PubMed.
- Q. Jia, S. Ghoshal, J. Li, W. Liang, G. Meng, H. Che, S. Zhang, Z.-F. Ma and S. Mukerjee, J. Am. Chem. Soc., 2017, 139, 7893–7903 CrossRef CAS PubMed.
- W. Gao, Z. Zhang, M. Dou and F. Wang, ACS Catal., 2019, 9, 3278–3288 CrossRef CAS.
- Z. Zhang, J. Liu, J. Gu, L. Su and L. Cheng, Energy Environ. Sci., 2014, 7, 2535–2558 RSC.
- K. W. Kim, S. M. Kim, S. Choi, J. Kim and I. S. Lee, ACS Nano, 2012, 6, 5122–5129 CrossRef CAS PubMed.
- T. Qu, Q. Tan, Y. Chen, L. Liu, S. Guo, Y. Chen, Y. Liu, S. Li and Y. Liu, ACS Appl. Energy Mater., 2019, 2, 8449–8458 CrossRef CAS.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- H. Xu, H. Shang, C. Wang and Y. Du, Coord. Chem. Rev., 2020, 418, 213374 CrossRef CAS.
- A. Sivanantham, P. Ganesan, L. Estevez, B. P. McGrail, R. K. Motkuri and S. Shanmugam, Adv. Energy Mater., 2018, 8, 1702838 CrossRef.
- J. Yu, X. Hou, C. Zhai, J. Li, A. Sartorel, M. Carraro, Z. Xin and Y. Huang, ACS Sustainable Chem. Eng., 2020, 8, 6127–6133 CrossRef CAS.
- M. Tahir, N. Mahmood, L. Pan, Z.-F. Huang, Z. Lv, J. Zhang, F. K. Butt, G. Shen, X. Zhang, S. X. Dou and J.-J. Zou, J. Mater. Chem. A, 2016, 4, 12940–12946 RSC.
- K. Obata and K. Takanabe, Angew. Chem., Int. Ed., 2018, 57, 1616–1620 CrossRef CAS PubMed.
- D. Zhao, Y. Pi, Q. Shao, Y. Feng, Y. Zhang and X. Huang, ACS Nano, 2018, 12, 6245–6251 CrossRef CAS PubMed.
- R. Bose, K. Karuppasamy, H. Rajan, D. B. Velusamy, H.-S. Kim and A. Alfantazi, ACS Sustainable Chem. Eng., 2019, 7, 16392–16400 CrossRef CAS.
- B. Qiu, C. Wang, N. Zhang, L. Cai, Y. Xiong and Y. Chai, ACS Catal., 2019, 9, 6484–6490 CrossRef CAS.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- D. Xie, Y. Chen, D. Yu, S. Han, J. Song, Y. Xie, F. Hu, L. Li and S. Peng, Chem. Commun., 2020, 56, 6842–6845 RSC.
- Y. Yang, Z. Lin, S. Gao, J. Su, Z. Lun, G. Xia, J. Chen, R. Zhang and Q. Chen, ACS Catal., 2017, 7, 469–479 CrossRef CAS.
- T. Najam, S. S. A. Shah, W. Ding, Z. Ling, L. Li and Z. Wei, Electrochim. Acta, 2019, 327, 134939 CrossRef CAS.
- X.-Y. Yu, Y. Feng, B. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 RSC.
- J. Yu, Q. Li, N. Chen, C.-Y. Xu, L. Zhen, J. Wu and V. P. Dravid, ACS Appl. Mater. Interfaces, 2016, 8, 27850–27858 CrossRef CAS PubMed.
- Y. Liu, Q. Wang, L. Wu, Y. Long, J. Li, S. Song and H. Zhang, Cryst. Growth Des., 2018, 18, 3404–3410 CrossRef CAS.
- J. Han, G. Chen, X. Liu, N. Zhang, S. Liang, R. Ma and G. Qiu, Chem. Commun., 2019, 55, 9212–9215 RSC.
- J.-Y. Zhao, R. Wang, S. Wang, Y.-R. Lv, H. Xu and S.-Q. Zang, J. Mater. Chem. A, 2019, 7, 7389–7395 RSC.
- Z. Chen, R. Wu, M. Liu, Y. Liu, S. Xu, Y. Ha, Y. Guo, X. Yu, D. Sun and F. Fang, J. Mater. Chem. A, 2018, 6, 10304–10312 RSC.
- Q. Yang, L. Liu, L. Xiao, L. Zhang, M. J. Wang, J. Li and Z. Wei, J. Mater. Chem. A, 2018, 6, 14752–14760 RSC.
- W. Zhang, W. Chen, Q. Xiao, L. Yu, C. Huang, G. Lu, A. W. Morawski and Y. Yu, Appl. Catal., B, 2020, 268, 118449 CrossRef CAS.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K.-C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 5848 CrossRef CAS PubMed.
- G.-R. Xu, J. Bai, J.-X. Jiang, J.-M. Lee and Y. Chen, Chem. Sci., 2017, 8, 8411–8418 RSC.
- J. Ying, G. Jiang, Z. P. Cano, L. Han, X.-Y. Yang and Z. Chen, Nano Energy, 2017, 40, 88–94 CrossRef CAS.
- N. Y. Labrador, E. L. Songcuan, C. D. Silva, H. Chen, S. J. Kurdziel, R. K. Ramachandran, C. Detavernier and D. V. Esposito, ACS Catal., 2018, 8, 1767–1778 CrossRef CAS.
- M. E. S. Beatty, H. Chen, N. Y. Labrador, B. J. Lee and D. V. Esposito, J. Mater. Chem. A, 2018, 6, 22287–22300 RSC.
- Y. Li, L. A. Zhang, Y. Qin, F. Chu, Y. Kong, Y. Tao, Y. Li, Y. Bu, D. Ding and M. Liu, ACS Catal., 2018, 8, 5714–5720 CrossRef CAS.
- X. Ji, B. Liu, X. Ren, X. Shi, A. M. Asiri and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 4499–4503 CrossRef CAS.
- Y. Xu, S. Yin, C. Li, K. Deng, H. Xue, X. Li, H. Wang and L. Wang, J. Mater. Chem. A, 2018, 6, 1376–1381 RSC.
- J. Su, Y. Yang, G. Xia, J. Chen, P. Jiang and Q. Chen, Nat. Commun., 2017, 8, 14969 CrossRef PubMed.
- W.-L. Yu, J.-Q. Chi and B. Dong, J. Mater. Chem. A, 2021, 9, 2195–2204 RSC.
- Y. Xu, W. Tu, B. Zhang, S. Yin, Y. Huang, M. Kraft and R. Xu, Adv. Mater., 2017, 29, 1605957 CrossRef PubMed.
- J. Deng, P. Ren, D. Deng, L. Yu, F. Yang and X. Bao, Energy Environ. Sci., 2014, 7, 1919–1923 RSC.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., Int. Ed., 2014, 53, 4372–4376 CrossRef CAS PubMed.
- M. Tavakkoli, T. Kallio, O. Reynaud, A. G. Nasibulin, C. Johans, J. Sainio, H. Jiang, E. I. Kauppinen and K. Laasonen, Angew. Chem., Int. Ed., 2015, 54, 4535–4538 CrossRef CAS PubMed.
- W. Zhou, Y. Zhou, L. Yang, J. Huang, Y. Ke, K. Zhou, L. Li and S. Chen, J. Mater. Chem. A, 2015, 3, 1915–1919 RSC.
- X. Dai, Z. Li, Y. Ma, M. Liu, K. Du, H. Su, H. Zhuo, L. Yu, H. Sun and X. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 6439–6448 CrossRef CAS PubMed.
- L. Zhang, S. Zhu, S. Dong, N. J. Woo, Z. Xu, J. Huang, J.-K. Kim and M. Shao, J. Electrochem. Soc., 2018, 165, J3271–J3275 CrossRef CAS.
- H. Zhang, Z. Ma, J. Duan, H. Liu, G. Liu, T. Wang, K. Chang, M. Li, L. Shi, X. Meng, K. Wu and J. Ye, ACS Nano, 2016, 10, 684–694 CrossRef CAS PubMed.
- M. Gong, D.-Y. Wang, C.-C. Chen, B.-J. Hwang and H. Dai, Nano Res., 2016, 9, 28–46 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- M. Gong, W. Zhou, M.-C. Tsai, J. Zhou, M. Guan, M.-C. Lin, B. Zhang, Y. Hu, D.-Y. Wang, J. Yang, S. J. Pennycook, B.-J. Hwang and H. Dai, Nat. Commun., 2014, 5, 4695 CrossRef CAS PubMed.
- M. Gong, W. Zhou, M. J. Kenney, R. Kapusta, S. Cowley, Y. Wu, B. Lu, M.-C. Lin, D.-Y. Wang, J. Yang, B.-J. Hwang and H. Dai, Angew. Chem., Int. Ed., 2015, 54, 11989–11993 CrossRef CAS PubMed.
- H. Doan, I. Kendrick, R. Blanchard, Q. Jia, E. Knecht, A. Freeman, T. Jankins, M. K. Bates and S. Mukerjee, J. Electrochem. Soc., 2021, 168, 084501 CrossRef CAS.
- S. Jing, J. Lu, G. Yu, S. Yin, L. Luo, Z. Zhang, Y. Ma, W. Chen and P. K. Shen, Adv. Mater., 2018, 30, 1705979 CrossRef PubMed.
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- Y.-J. Tang, M.-R. Gao, C.-H. Liu, S.-L. Li, H.-L. Jiang, Y.-Q. Lan, M. Han and S.-H. Yu, Angew. Chem., Int. Ed., 2015, 54, 12928–12932 CrossRef CAS PubMed.
- Y. Yang, Z. Lun, G. Xia, F. Zheng, M. He and Q. Chen, Energy Environ. Sci., 2015, 8, 3563–3571 RSC.
- Y. Shen, Y. Zhou, D. Wang, X. Wu, J. Li and J. Xi, Adv. Energy Mater., 2018, 8, 1701759 CrossRef.
- S. Wu, X. Shen, G. Zhu, H. Zhou, Z. Ji, L. Ma, K. Xu, J. Yang and A. Yuan, Carbon, 2017, 116, 68–76 CrossRef CAS.
- X. Yin, L. Yang and Q. Gao, Nanoscale, 2020, 12, 15944–15969 RSC.
- S. Xu, H. Zhao, T. Li, J. Liang, S. Lu, G. Chen, S. Gao, A. M. Asiri, Q. Wu and X. Sun, J. Mater. Chem. A, 2020, 8, 19729–19745 RSC.
- X. Luo, Q. Zhou, S. Du, J. Li, L. Zhang, K. Lin, H. Li, B. Chen, T. Wu, D. Chen, M. Chang and Y. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 42335–42347 CrossRef CAS PubMed.
- X. Huang, X. Xu, C. Li, D. Wu, D. Cheng and D. Cao, Adv. Energy Mater., 2019, 9, 1803970 CrossRef.
- F. Yang, Y. Chen, G. Cheng, S. Chen and W. Luo, ACS Catal., 2017, 7, 3824–3831 CrossRef CAS.
- R. Wang, X.-Y. Dong, J. Du, J.-Y. Zhao and S.-Q. Zang, Adv. Mater., 2017, 30, 1703711 CrossRef PubMed.
- D. Y. Chung, S. W. Jun, G. Yoon, H. Kim, J. M. Yoo, K.-S. Lee, T. Kim, H. Shin, A. K. Sinha, S. G. Kwon, K. Kang, T. Hyeon and Y.-E. Sung, J. Am. Chem. Soc., 2017, 139, 6669–6674 CrossRef CAS PubMed.
- M. Pi, T. Yang, S. Wang and S. Chen, J. Mater. Res., 2018, 33, 546–555 CrossRef CAS.
- D. Zhao, K. Sun, W.-C. Cheong, L. Zheng, C. Zhang, S. Liu, X. Cao, K. Wu, Y. Pan, Z. Zhuang, B. Hu, D. Wang, Q. Peng, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 8982–8990 CrossRef CAS PubMed.
- H. Wei, Q. Xi, X. Chen, D. Guo, F. Ding, Z. Yang, S. Wang, J. Li and S. Huang, Adv. Sci., 2018, 5, 1700733 CrossRef PubMed.
- X. Yang, X. Feng, H. Tan, H. Zang, X. Wang, Y. Wang, E. Wang and Y. Li, J. Mater. Chem. A, 2016, 4, 3947–3954 RSC.
- R. A. Mir and O. P. Pandey, Chem. Eng. J., 2018, 348, 1037–1048 CrossRef CAS.
- I. Kim, S.-W. Park and D.-W. Kim, Nanoscale, 2018, 10, 21123–21131 RSC.
- Q. Zhang, F. Luo, H. Hu, R. Xu, K. Qu, Z. Yang, J. Xu and W. Cai, Chem. Commun., 2019, 55, 9665–9668 RSC.
- Y.-Y. Chen, Y. Zhang, W.-J. Jiang, X. Zhang, Z. Dai, L.-J. Wan and J.-S. Hu, ACS Nano, 2016, 10, 8851–8860 CrossRef CAS PubMed.
- L. Zhang, H. Yang, D. K. J. A. Wanigarathna and B. Liu, Small Methods, 2018, 2, 1700353 CrossRef.
- J.-S. Li, Y. Wang, C.-H. Liu, S.-L. Li, Y.-G. Wang, L.-Z. Dong, Z.-H. Dai, Y.-F. Li and Y.-Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS PubMed.
- Y. Liu, G. Yu, G.-D. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS PubMed.
- L. Peng, J. Shen, L. Zhang, Y. Wang, R. Xiang, J. Li, L. Li and Z. Wei, J. Mater. Chem. A, 2017, 5, 23028–23034 RSC.
- F. Yu, Y. Gao, Z. Lang, Y. Ma, L. Yin, J. Du, H. Tan, Y. Wang and Y. Li, Nanoscale, 2018, 10, 6080–6087 RSC.
- X. Xu, F. Nosheen and X. Wang, Chem. Mater., 2016, 28, 6313–6320 CrossRef CAS.
- Q. Yun, Q. Lu, X. Zhang, C. Tan and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 626–646 CrossRef CAS PubMed.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134 CrossRef PubMed.
- J. Ren, Z. Wang, Y. Feng, B. Guo, K. Yu and Z. Zhu, New J. Chem., 2019, 43, 17502–17510 RSC.
- Z. Huang, S. Yuan, T. Zhang, B. Cai, B. Xu, X. Lu, L. Fan, F. Dai and D. Sun, Appl. Catal., B, 2020, 272, 118976 CrossRef CAS.
- X. Sun, F. Liu, X. Chen, C. Li, J. Yu and M. Pan, Electrochim. Acta, 2019, 307, 206–213 CrossRef CAS.
- D. Li, Z. Zong, Z. Tang, Z. Liu, S. Chen, Y. Tian and X. Wang, ACS Sustainable Chem. Eng., 2018, 6, 5105–5114 CrossRef CAS.
- W. Wu, Y. Wu, D. Zheng, K. Wang and Z. Tang, Electrochim. Acta, 2019, 320, 134568 CrossRef CAS.
- J. Wang, D. Gao, G. Wang, S. Miao, H. Wu, J. Li and X. Bao, J. Mater. Chem. A, 2014, 2, 20067–20074 RSC.
- Y. Hou, Z. Wen, S. Cui, S. Ci, S. Mao and J. Chen, Adv. Funct. Mater., 2015, 25, 872–882 CrossRef CAS.
- M. Li, T. Liu, X. Bo, M. Zhou and L. Guo, J. Mater. Chem. A, 2017, 5, 5413–5425 RSC.
- Q. Mo, N. Chen, M. Deng, L. Yang and Q. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 37721–37730 CrossRef CAS PubMed.
- Z. Chen, Y. Ha, H. Jia, X. Yan, M. Chen, M. Liu and R. Wu, Adv. Energy Mater., 2019, 9, 1803918 CrossRef.
- T. Zhang, R. Cheng, B. Li, C. Wang, Y. Guo, J. Liu and L. Wang, Chem. Commun., 2021, 57, 769–772 RSC.
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun, J. Tang, L. Zhang, A. M. Al-Enizi, Z. Yang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 14305–14312 CrossRef CAS PubMed.
- H. Yang, Z. Chen, W. Hao, H. Xu, Y. Guo and R. Wu, Appl. Catal., B, 2019, 252, 214–221 CrossRef CAS.
- J. Zhang, Y. Jiang, Y. Wang, C. Yu, J. Cui, J. Wu, X. Shu, Y. Qin, J. Sun, J. Yan, H. Zheng, Y. Zhang and Y. Wu, Electrochim. Acta, 2019, 321, 134652 CrossRef CAS.
- Y. Ha, L. Shi, Z. Chen and R. Wu, Adv. Sci., 2019, 6, 1900272 CrossRef PubMed.
- J.-T. Ren, G.-G. Yuan, C.-C. Weng and Z.-Y. Yuan, Electrochim. Acta, 2018, 261, 454–463 CrossRef CAS.
- Z. Peng, Y. Yu, D. Jiang, Y. Wu, B. Y. Xia and Z. Dong, Carbon, 2019, 144, 464–471 CrossRef CAS.
- L. Yang and L. Zhang, Appl. Catal., B, 2019, 259, 118053 CrossRef CAS.
- H. Xu, H. Jia, B. Fei, Y. Ha, H. Li, Y. Guo, M. Liu and R. Wu, Appl. Catal., B, 2020, 268, 118404 CrossRef CAS.
- Z.-Y. Yu, Y. Duan, M.-R. Gao, C.-C. Lang, Y.-R. Zheng and S.-H. Yu, Chem. Sci., 2017, 8, 968–973 RSC.
- Q. Feng, Y. Xiong, L. Xie, Z. Zhang, X. Lu, Y. Wang, X.-Z. Yuan, J. Fan, H. Li and H. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 25123–25132 CrossRef CAS PubMed.
- M. Li, Y. Zhu, H. Wang, C. Wang, N. Pinna and X. Lu, Adv. Energy Mater., 2019, 9, 1803185 CrossRef.
- Q. Hao, S. Li, H. Liu, J. Mao, Y. Li, C. Liu, J. Zhang and C. Tang, Catal. Sci. Technol., 2019, 9, 3099–3108 RSC.
- F. Li, R. Xu, Y. Li, F. Liang, D. Zhang, W.-F. Fu and X.-J. Lv, Carbon, 2019, 145, 521–528 CrossRef CAS.
- H. Sun, X. Xu, Y. Song, W. Zhou and Z. Shao, Adv. Mater., 2021, 31, 2009779 CAS.
- L. Sun, Q. Luo, Z. Dai and F. Ma, Coord. Chem. Rev., 2021, 444, 214049 CrossRef CAS.
- Z. Zhang, X. Ma and J. Tang, J. Mater. Chem. A, 2018, 6, 12361–12369 RSC.
- Y. Jin, H. Wang, J. Li, X. Yue, Y. Han, P. K. Shen and Y. Cui, Adv. Mater., 2016, 28, 3785–3790 CrossRef CAS PubMed.
- Y. Li, H. Zhang, M. Jiang, Q. Zhang, P. He and X. Sun, Adv. Funct. Mater., 2017, 27, 1702513 CrossRef.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- S. Hao, L. Chen, C. Yu, B. Yang, Z. Li, Y. Hou, L. Lei and X. Zhang, ACS Energy Lett., 2019, 4, 952–959 CrossRef CAS.
- P. Zhai, Y. Zhang, Y. Wu, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, L. Sun and J. Hou, Nat. Commun., 2020, 11, 5462 CrossRef CAS PubMed.
- G. Qian, G. Yu, J. Lu, L. Luo, T. Wang, C. Zhang, R. Ku, S. Yin, W. Chen and S. Mu, J. Mater. Chem. A, 2020, 8, 14545–14554 RSC.
- L. Hui, Y. Xue, B. Huang, H. Yu, C. Zhang, D. Zhang, D. Jia, Y. Zhao, Y. Li, H. Liu and Y. Li, Nat. Commun., 2018, 9, 5309 CrossRef CAS PubMed.
- L. Hui, D. Jia, H. Yu, Y. Xue and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 2618–2625 CrossRef CAS PubMed.
- J. Meng, X. Liu, C. Niu, Q. Pang, J. Li, F. Liu, Z. Liu and L. Mai, Chem. Soc. Rev., 2020, 49, 3142–3186 RSC.
- L. Liu, P. Concepción and A. Corma, J. Catal., 2016, 340, 1–9 CrossRef CAS.
- B. P. Le Monnier, F. Wells, F. Talebkeikhah and J. S. Luterbacher, Adv. Mater., 2019, 31, 1904276 CrossRef CAS PubMed.
- M. Makosch, W.-I. Lin, V. Bumbálek, J. Sá, J. W. Medlin, K. Hungerbühler and J. A. van Bokhoven, ACS Catal., 2012, 2, 2079–2081 CrossRef CAS.
- A. C. Chien and J. A. van Bokhoven, Catal. Sci. Technol., 2015, 5, 3518–3524 RSC.
- A. Beck, X. Huang, L. Artiglia, M. Zabilskiy, X. Wang, P. Rzepka, D. Palagin, M.-G. Willinger and J. A. van Bokhoven, Nat. Commun., 2020, 11, 3220 CrossRef CAS PubMed.
- G. Gershinsky, P. Nanikashvili, R. Elazari and D. Zitoun, ACS Appl. Energy Mater., 2018, 1, 4678–4685 CrossRef CAS.
- K. Saadi, P. Nanikashvili, Z. Tatus-Portnoy, S. Hardisty, V. Shokhen, M. Zysler and D. Zitoun, J. Power Sources, 2019, 422, 84–91 CrossRef CAS.
- M. P. Clark, M. Xiong, K. Cadien and D. G. Ivey, ACS Appl. Energy Mater., 2020, 3, 603–613 CrossRef CAS.
- T. Tang, W.-J. Jiang, X.-Z. Liu, J. Deng, S. Niu, B. Wang, S.-F. Jin, Q. Zhang, L. Gu, J.-S. Hu and L.-J. Wan, J. Am. Chem. Soc., 2020, 142, 7116–7127 CrossRef CAS PubMed.
- I. Chang, S. Ji, J. Park, M. H. Lee and S. W. Cha, Adv. Energy Mater., 2015, 5, 1402251 CrossRef.
- J. W. Shin, S. Oh, S. Lee, J.-G. Yu, J. Park, D. Go, B. C. Yang, H. J. Kim and J. An, ACS Appl. Mater. Interfaces, 2019, 11, 46651–46657 CrossRef CAS PubMed.
- J. Seo, N. Tsvetkov, S. J. Jeong, Y. Yoo, S. Ji, J. H. Kim, J. K. Kang and W. Jung, ACS Appl. Mater. Interfaces, 2020, 12, 4405–4413 CrossRef CAS PubMed.
- Z. Wang, Y. Zou, Y. Li and Y. Cheng, Small, 2020, 16, 1907042 CrossRef CAS PubMed.
- U. Nisar, N. Muralidharan, R. Essehli, R. Amin and I. Belharouak, Energy Storage Mater., 2021, 38, 309–328 CrossRef.
- X. Qu, L. Shen, Y. Mao, J. Lin, Y. Li, G. Li, Y. Zhang, Y. Jiang and S. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 31869–31877 CrossRef CAS PubMed.
- W. Li, C. Zhang, M. Han, Y. Ye, S. Zhang, Y. Liu, G. Wang, C. Liang and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 44186–44195 CrossRef CAS PubMed.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- Z. Cai, Y. Zhang, Y. Zhao, Y. Wu, W. Xu, X. Wen, Y. Zhong, Y. Zhang, W. Liu, H. Wang, Y. Kuang and X. Sun, Nano Res., 2019, 12, 345–349 CrossRef CAS.
- P. An, L. Wei, H. Li, B. Yang, K. Liu, J. Fu, H. Li, H. Liu, J. Hu, Y.-R. Lu, H. Pan, T.-S. Chan, N. Zhang and M. Liu, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- R. F. Mendes, F. Figueira, J. P. Leite, L. Gales and F. A. Almeida Paz, Chem. Soc. Rev., 2020, 49, 9121–9153 RSC.
- X. Wang, L. Li, T. Zhang, B. Lin, J. Ni, C.-T. Au and L. Jiang, Chem. Commun., 2019, 55, 474–477 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- W. Zhan, Q. He, X. Liu, Y. Guo, Y. Wang, L. Wang, Y. Guo, A. Y. Borisevich, J. Zhang, G. Lu and S. Dai, J. Am. Chem. Soc., 2016, 138, 16130–16139 CrossRef CAS PubMed.
- W. Zhan, Y. Shu, Y. Sheng, H. Zhu, Y. Guo, L. Wang, Y. Guo, J. Zhang, G. Lu and S. Dai, Angew. Chem., Int. Ed., 2017, 56, 4494–4498 CrossRef CAS PubMed.
- S. Yook, H. Shin, H. Kim and M. Choi, ChemCatChem, 2014, 6, 2836–2842 CrossRef CAS.
- S. Y. Moon, B. Naik, C.-H. Jung, K. Qadir and J. Y. Park, Catal. Today, 2016, 265, 245–253 CrossRef CAS.
- I. Ro, Y. Liu, M. R. Ball, D. H. K. Jackson, J. P. Chada, C. Sener, T. F. Kuech, R. J. Madon, G. W. Huber and J. A. Dumesic, ACS Catal., 2016, 6, 7040–7050 CrossRef CAS.
- B. Jiang, Y. He, B. Li, S. Zhao, S. Wang, Y.-B. He and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 1869–1872 CrossRef CAS PubMed.
- D.-C. Guan, S. Tian, Y.-H. Sun, F. Deng, J.-M. Nan, G.-Z. Ma and Y.-P. Cai, Electrochim. Acta, 2019, 322, 134722 CrossRef CAS.
- D. Gueona and J. H. Moon, Chem. Commun., 2019, 55, 361–364 RSC.
| This journal is © The Royal Society of Chemistry 2022 |