
Beyond nylon 6: polyamides via ring opening polymerization of designer lactam monomers for biomedical applications
Maria
Varghese
 and
Mark W.
Grinstaff
*
and
Mark W.
Grinstaff
*
Departments of Chemistry and Biomedical Engineering, Boston University, Boston, MA 02215, USA. E-mail: mgrin@bu.edu
First published on 1st September 2022
Abstract
Ring opening polymerization (ROP) of lactams is a highly efficient and versatile method to synthesize polyamides. Within the last ten years, significant advances in polymerization methodology and monomer diversity are ushering in a new era of polyamide chemistry. We begin with a discussion of polymerization techniques including the most widely used anionic ring opening polymerization (AROP), and less prevalent cationic ROP and enzyme-catalyzed ROP. Next, we describe new monomers being explored for ROP with increased functionality and stereochemistry. We emphasize the relationships between composition, structure, and properties, and how chemists can control composition and structure to dictate a desired property or performance. Finally, we discuss biomedical applications of the synthesized polyamides, specifically as biomaterials and pharmaceuticals, with examples to include as antimicrobial agents, cell adhesion substrates, and drug delivery scaffolds.
 Maria Varghese | Maria Varghese received her combined BS-MS degree (Chemistry major, Biology minor) from Indian Institute of Science Education and Research (IISER), Thiruvananthapuram, India, where she performed undergraduate research on organic synthesis under the mentorship of Professor Kana M. Sureshan. She earned her Ph.D. in Chemistry at Boston University under the mentorship of Professor Mark W. Grinstaff, focusing on the development of sulfated poly-amido-saccharide as anticoagulants and block poly-amido-saccharides for drug delivery applications. She is currently working as a research assistant in the Grinstaff laboratory. |
 Mark W. Grinstaff | Mark W. Grinstaff is the William Fairfield Warren Distinguished Professor, the Distinguished Professor of Translational Research, and a Professor of Biomedical Engineering, Chemistry, Materials Science and Engineering, and Medicine at Boston University. He is also the Director of BU's Nanotechnology Innovation Center and the NIH T32 Biomaterials Program. Mark received his A.B. from Occidental College, earned his Ph.D. under the mentorship of Kenneth S. Suslick at the University of Illinois, and completed his postdoctoral training with Harry B. Gray at the California Institute of Technology. Additional information about his research interest is found at https://www.grinstaff.org. |
Introduction
Nylon polymers are synthesized on the >3 metric ton scale each year, are key constituents of consumer products throughout the industry sectors, and have a rich history from their beginnings in the early 1900's.1 Dr. Wallace H. Carothers, a DuPont scientist in 1935, prepared the first nylon polymer, nylon 66 (Fig. 1A and 2), by the thermally-induced poly-condensation of adipic acid and hexamethylene diamine.2 Nylon 66 was the first truly synthetic useful fiber possessing properties such as low thermal and electrical conductivity, light-weight, and corrosion resistance. It revolutionized the polymer and plastic manufacturing industries with early products from hair-combs to ship propellers to stockings.3 Structurally, nylon 6 (Fig. 1B), first prepared by Paul Schlack in 1938, is similar to nylon 66 with a 5-methylene spacer between the amide linkages.4 However, it is easier to prepare, via ring opening polymerization of ε-caprolactam (Fig. 1C, 4), than nylon 66. Initially, industry used water catalyzed ring-opening polymerization at high temperatures (240 °C to 280 °C) because of the lack of polymerization reproducibility when using a strong base catalyst under anhydrous conditions.5 However, by the 1960s improvements in anionic ring opening polymerization (AROP), to include the use of acylated caprolactam or acylating agents as activators,6 afforded higher rates of polymerization and low residual monomer content.7 Compared to other thermoplastics available at the time, nylon 66 and nylon 6 possessed greater strength, toughness, abrasion resistance, and thermal resistance. Today, the nylon 6 product segment accounts for more than 50% of the global revenue for nylon polymers, estimated at approximately 30 billion US dollars.8,9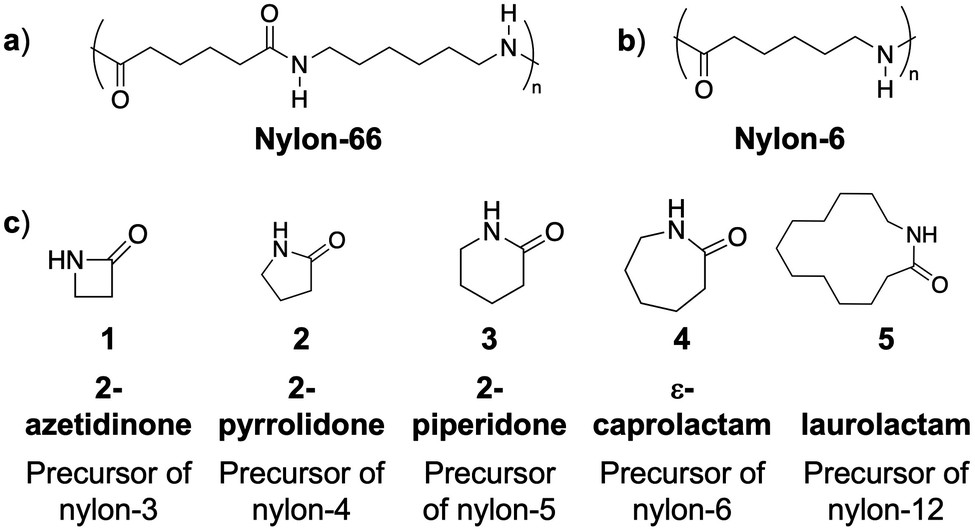 | ||
| Fig. 1 (A) Nylon-66 polymer structure; (B) nylon-6 polymer structure; (C) precursors of common nylon polymers. | ||
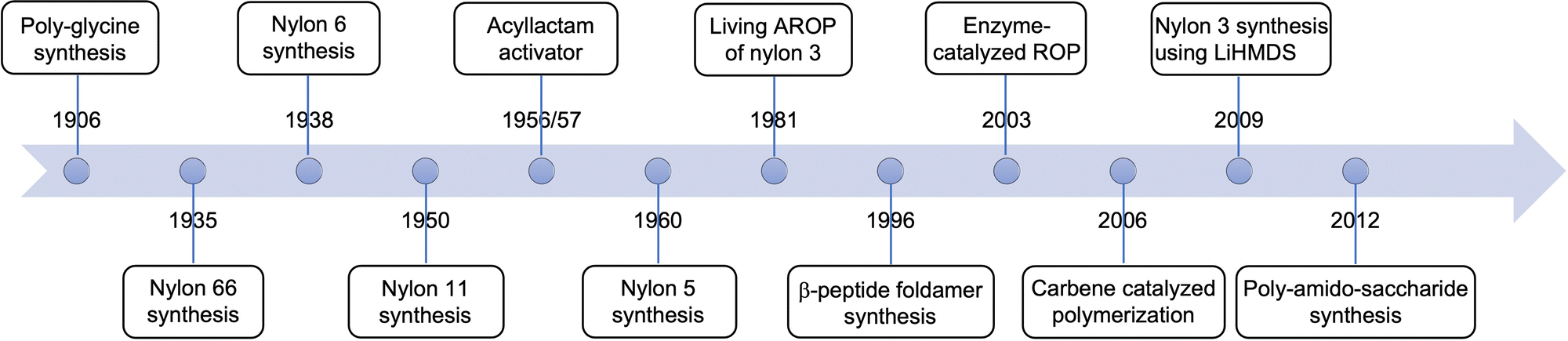 | ||
| Fig. 2 Advancements in nylon synthesis via ring opening polymerization of lactams. | ||
Since these seminal discoveries, additional nylons have been commercialized and ones with increasing chemical complexity are reported. For example, nylon 11 and nylon 12 are commercially prepared by self poly-condensation of 11-aminoundecanoic acid and ring opening polymerization of dodecalactam (Fig. 1C, 5), respectively.10 These higher-carbon number polyamides possess low amide group density, resulting in decreased hydrogen-bonding between polymers and to external agents. This characteristic leads to less crystallinity, resulting in lower melting point and mechanical properties as well as lower absorption of moisture, resulting in higher chemical resistance. Due to these properties, nylons 11 and 12 are used in automotive fuel lines, insulation of wire and cable, as well as medical and food contact applications.10 Other nylon polymers (e.g., nylons 69, 610, 612, and 8) are also reported, but are not widely used.
Nylon polymers with a low number (3, 4 and 5) of carbon atoms in the repeating unit called “low order nylons” are prepared by the anionic ring opening polymerization of 2-azetidinone (Fig. 1, 1), 2-pyrrolidone (Fig. 1, 2) and 2-piperidone (Fig. 1, 3), respectively. These polymers are more hydrophilic due to the abundance of amide bonds in the polymer chain and are more crystalline than nylon 6, nylon 11, or nylon 12. Nylon 5 demonstrates superior ferro-electric property and thermal stability, however, ROP of 2-piperidone to prepare nylon 5 is challenging due to the low ring strain of the 6-membered cyclic-monomer as well as the low solubility of the resulting polymer (due to H-bonding and very high polarity).11 Nylon 4 polymers are biodegradable12,13 and show tenacity, elongation, elastic recovery, and moisture regain similar to cotton and other commercially available polyamide fibers. However, nylon 4 polymers are not commercially synthesized due to their low thermal stability, with melting point just above degradation temperature.14 Nylon 3 polymers or β-peptides, first synthesized by Sebenda et al. in 1981,15 on the other hand are well characterized and studied, especially due to their biomedical applications.
Nylon-2 polymers or polyglycines, first prepared by the ring opening polymerization of α-amino N-carboxyanhydrides (α-NCA) in 190616 are widely studied due to their established syntheses, potential for commercialization, and their protein-mimicking nature due to a greater ratio of amide groups per total atoms in the repeat unit compared to other nylon polymers.17 Polyglycines are susceptible to enzymatic degradation due to their structural similarity to naturally occurring polypeptides. Incorporating higher order nylons, such as nylon-3 in the polymeric structure, slows the degradation rate.18 Polyglycines are finding uses as antibacterial agents, drug and nucleic acid carriers, and building blocks for tissue engineering, which are summarized in excellent reviews by Mazo et al.,17 Hadjichristidis et al.,19 Lu et al.,20 and T. Deming,21,22 and therefore will not be discussed in this review. The timeline of advances in nylon polyamide synthesis via ring opening polymerization of lactams over the past century is summarized in Fig. 2.
Herein, we discuss the various polymerization techniques used to prepare nylon 3 and nylon 6 polymers, including the most widely used anionic ring opening polymerization and less prevalent cationic ROP and enzyme-catalyzed ROP. We then describe new monomers being explored for ROP with increased functionality and stereochemistry. Finally, we provide a detailed account of the biomedical application of polyamides, giving the background necessary to understand current developments as well as to envision future applications. We focus on the advances reported in the past 2 decades. We direct the readers to excellent reviews by Hashimoto for a discussion of ROP reported prior to 2000,23 as well as by Karger-Kocsis et al. for composites produced by AROP of lactams and their manufacturing techniques,24 as these topics will not be discussed. The purpose of this review is to stimulate discussions, highlight recent approaches and success, and provide further motivation for the design, synthesis, and development of new nylon polymers and their evaluation in biomedical applications.
Ring opening polymerization of lactams
1. Anionic ring opening polymerization
Anionic ring opening polymerization (AROP) is the most widely used method for polymerization of lactams. AROP requires a strong, but non-nucleophilic base catalyst, and often uses a lactam-derived imide co-initiator (Fig. 3). The co-initiator can also be produced in situ using acylating agents such as benzoyl chloride, acetyl chloride, or acetic anhydride.25–27 The initiation step encompasses the generation of a lactamate anion by the abstraction of proton by the base, followed by nucleophilic attack on the imide carbonyl group on the co-initiator (if present) (Fig. 3B) or amide carbonyl group on another lactam if not using a co-initiator. The lactamate anion is then regenerated by the proton transfer from an unreacted lactam to the amidate anion formed in the growing polymer backbone. The propagation step involves nucleophilic attack of the lactamate anion on the carbonyl group of the imide in the growing polymer chain.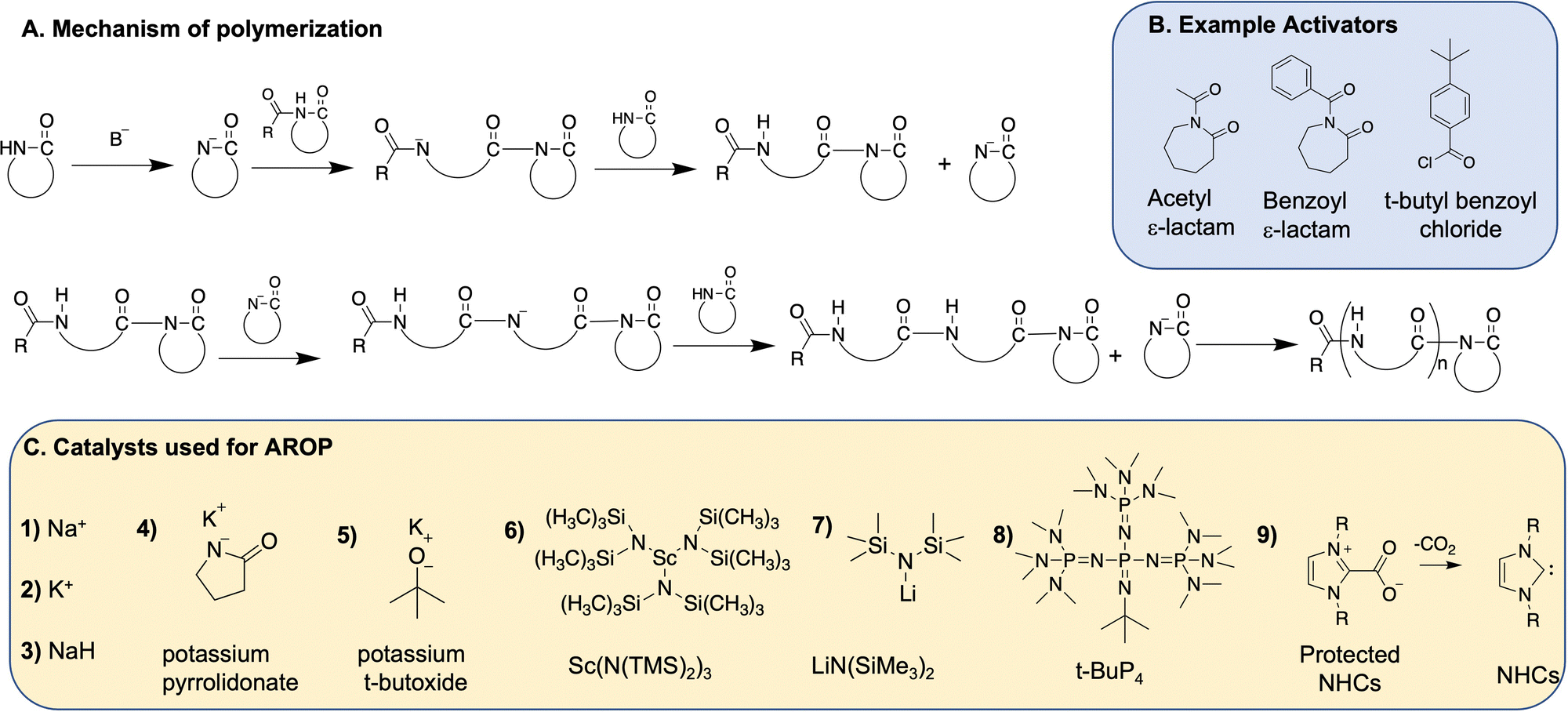 | ||
| Fig. 3 (A) Mechanism of AROP; (B) example activators; and (C) the catalysts used for AROP. | ||
In general, ε-lactam, like caprolactams polymerize similarly to the first report of caprolactam. An extrusion method is often used, wherein the monomer and the catalyst are mixed together, melted, and then polymerized at high temperatures (>180 °C). 2-Pyrrolidone and 2-piperidone undergo a similar polymerization, albeit at lower temperatures (40–50 °C). β-Lactam monomers on the other hand polymerize in solvents such as tetrahydrofuran (THF), dimethyl acetamide (DMAC) with a Lewis acid (e.g., DMAC + LiCl), dimethylsulfoxide (DMSO) or dimethyl formamide (DMF). Addition of the Lewis acid improves monomer solubility.28 The ε-lactam polymerization proceeds with an induction period, which depends on temperature, type and amount of the catalyst/activator formulation,24 while polymerization of other lactams does not require induction periods. Various strong bases initiate the anionic ring opening polymerization of lactams as described below.
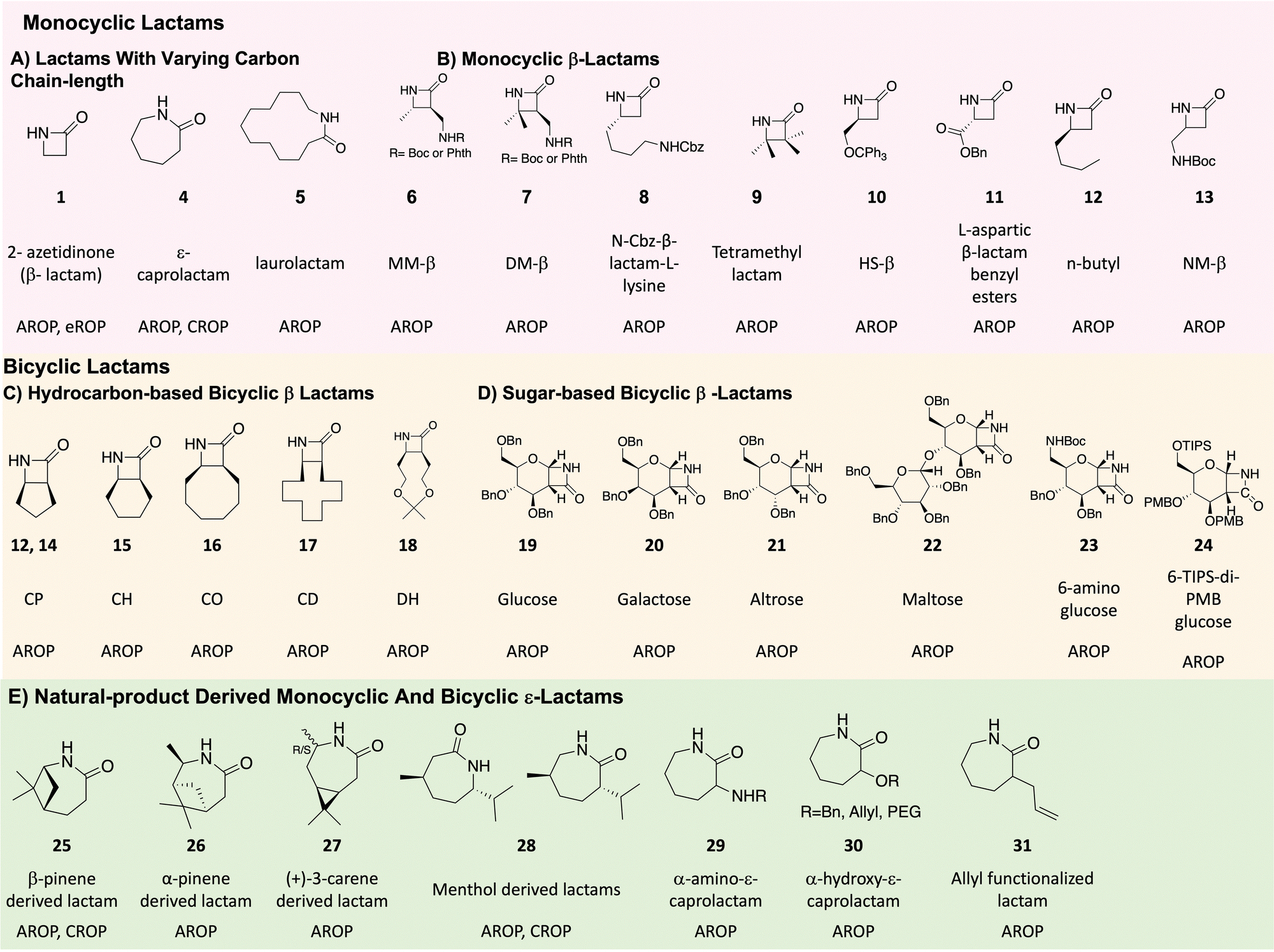 | ||
| Fig. 4 Structure of lactams discussed, their common names, and polymerization method used. | ||
Potassium t-butoxide catalyzes the polymerization of 2-pyrrolidone,12,14,34 while 2-piperidone polymerization is catalyzed by tetramethyl-ammonium 2-oxopiperidin-1-ide11 as well as alkali metals to produce nylon 4 and nylon 5, respectively. Other alkali metal and alkaline earth metal-based catalysts used for AROP of lactams include potassium pyrrolidonate,28,35 and Grignard reagent.36
However Sc(N(TMS)2)3, as reported by Deming et al., is not an effective initiator for highly substituted β-lactams and requires a strong and non-nucleophilic base such as LiN(SiMe3)2 (or LiHMDS, Fig. 3C, structure 7) to initiate the polymerization of monomers 6, 7, 15–17 (Fig. 4), and afford nylon 3 polymers with controlled molecular weights and low dispersities. They also report the use of acylating agents such as 4-tert-butylbenzoyl chloride as co-initiators (Fig. 3B) instead of acylated lactam co-initiator for β-lactam polymerization. Here, the acylated β-lactam co-initiator, which contains the active imide group, forms in situ during polymerization. This polymerization method affords narrow molecular weight distributions with molecular weights comparable to theoretical molecular weights, revealing “quasi-living” polymerization. Using various acid chlorides, a large number of N-terminal end-functionalized polymers are prepared (e.g., thiol terminated nylon 3 polymers by Liu et al.42 and palmitamide terminated amphiphilic nylon 3 polymers by Grinstaff et al.43), thus opening avenues for biomolecule-, polymeric- or surface-conjugations. Moreover, the presence of a C-terminal, β-lactam-derived imide group on the polymer chains, (confirmed by 13C NMR) enables C-terminal functionalisation.44
Additionally, Stahl et al. use the C-terminal imide functionality, along with the “quasi-living” characteristic of the polymerization to prepare di-block nylon 3 polymers.27 The first monomer polymerizes followed by the addition of the second monomer as well as additional LiN(SiMe3)2 resulting in di-block polymers. GPC analysis confirms chain extension by the complete disappearance of the peak corresponding to the first block of the polymer and the shift of the peak to shorter retention time, indicating extension of the polymers. Another one-pot strategy for block-polymer synthesis using β-lactams, as reported by Chan-Park and co-workers,45 involves adding both monomers (Fig. 4, structures 8 and 19) simultaneously and relying on the different polymerization kinetics of the monomers. The monomer with faster polymerization kinetics polymerizes first, forming the first block of the di-block polymer, while the monomer with the slower polymerization kinetics forms the second block. If the two monomers have similar reaction kinetics, the one-pot polymerization yields random copolymers.27
Since alkali metal-based catalysts require high reaction temperatures and Li-metal based reagents are not sustainable due to limited availability of lithium,46 alternative organo-catalyzed AROPs are being explored. Two examples of organo-catalysts used for ROPs are phosphazene bases and N-heterocyclic carbenes, as next described.
t-BuP4 also successfully polymerizes unsubstituted β-lactam (Fig. 4, structure 1) to give unsubstituted nylon 3 polymers, which are inaccessible by metal-based polymerization techniques.28 The polymerization occurs in DMAc solvent with 10 weight% of LiCl (to improve solubility of monomers) at low temperatures (ranging from 25 °C to 80 °C) similar to metal-based polymerization of other complex β-lactams. Varying catalyst to monomer ratio controls molecular weight, with a molecular weight >105 kDa achieved at the catalyst to monomer ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000 (mol mol−1).
:2000 (mol mol−1).
Carbene-catalyzed lactam polymerization, as first discovered by DuPont in 2006, occurs during extrusion of ε-caprolactam in the presence of imidazolium and imidazolium based carbenes, resulting in 84% yield.56 Buchmeiser et al. expanded this technique by building a library of tetrahydropyrimidinium-derived NHCs with varying ring size (5, 6 or 7 membered rings), substituent groups (aliphatic linear, aliphatic cyclic and aromatic) and thermally labile protecting groups (CO2−, MgCl2, and ZnCl2).57 Initiation occurs by first heating the NHC to 90 °C, during which time the carbene deprotects (induction time), followed by deprotonation of the lactam monomer, resulting in “activated” monomers. Next, polymerization transpires upon heating the reaction mixture to above 100 °C. Only the carboxylate-protected NHCs produce polyamides, while the NHCs protected by metals are ineffective in activating the monomer. The yield and efficiency of the polymerization of ε-caprolactam is directly proportional to the basicity of the carbene generated, which in turn is influenced by (i) ring size (6,7-membered > 5-membered) and (ii) electron withdrawing effect of the substituent groups (aliphatic > aromatic).
As reported by Buchmeiser et al., the NHC catalyst also polymerizes laurolactam to give nylon 12 as well laurolactam and ε-caprolactam to afford random copolymers of nylon 6 and nylon 12.58 Unlike the other two studies, an activator is not used. Increasing monomer conversion results in a decrease of molecular weight. The authors attribute this effect to “chain multiplication”, wherein a lactam anion attacks the amide bonds of the polymer, affording shorter polymers. The random co-polymers possess a non-random “blocky” characteristic due to the higher incorporation of ε-caprolactam in the beginning of polymerization versus laurolactam. This result is consistent with the higher ring strain of ε-caprolactam compared to laurolactam, and, hence, greater reactivity for the polymerization.
Use of MgCl2 and LiCl in conjunction with the carboxylate protected NHC (“dual-catalysis”) affords polymers in higher yield. The reaction proceeds even in a non-inert atmosphere (albeit with longer reaction times), resulting in a promising transition metal-free strategy for industrial production of nylon 6 polymers.59 The higher yield obtained in the presence of Lewis acid is rationalized by the formation of an organometallic complex between the metal salts and the lactam monomer, rendering the monomer more electrophilic and thus more susceptible for ring opening polymerization.
While ROP of ε-lactams using NHCs is not a “living” polymerization, molecular weight increases with a decrease in catalyst to monomer ratio, thus providing some control over molecular weight. However, regardless of the use of a Lewis acid one obtains polymers with higher-than-expected molecular weights, indicating the incomplete initiation of the monomer by the carbene.
In short, while NHCs are promising as latent pre-catalysts, since they are inactive until heated to 90 °C and are not as sensitive to air and moisture as metal-based catalysts, further optimizations are required for control in molecular weights and to avoid side reactions.
While AROP is the most widely used polymerization technique for lactams, the anhydrous reaction conditions is a major concern. New advances in nylon 3 polymer synthesis which include water-insensitive ring-opening polymerization of β-amino acid N-thiocarboxyanhydrides (β-NTA) result in polymers with controlled molecular weights and helical secondary conformation.60,61 Moreover, co-polymerization of β-NTA and α-NCA in different solvents affords random-like or block-like polymers, yielding sequence-controlled poly α/β peptides with tuneable proteolysis.18
2. Enzyme-catalyzed ring-opening
While anionic ring-opening polymerization is efficient and highly versatile in terms of monomer structure and control in molecular weight, the technique requires organic reagents and often leads to toxic side-products. Enzyme-catalyzed ring opening polymerization (eROP) is an environmental-friendly alternative for AROP. Novozym 435 (N435) is a commercially available polymer resin with a surface immobilized Lipase B from Candida antarctica (CAL-B).62 Apart from its biological function of hydrolyzing triglycerides, Lipase B in its N435 form is used in biodiesel production, racemic mixture resolution, regioselective reactions, ε-caprolactone polymerization, polycondensation of dimethyl 2,5-furandicarboxylate and aliphatic diamines, and transesterification reactions.62 N435 also catalyzes the ring opening polymerization of lactams. The proposed mechanism of polymerization involves 2 steps: (i) ring opening of the β-lactam by serine-105 of the lipase, resulting in an acyl-enzyme intermediate and a free amine; (ii) attack on the acyl-enzyme by a growing oligomer chain leading to nylon 3; and, (iii) attack on the acyl-enzyme intermediate by a water molecule resulting in free carboxylic acid and chain-termination (Fig. 5).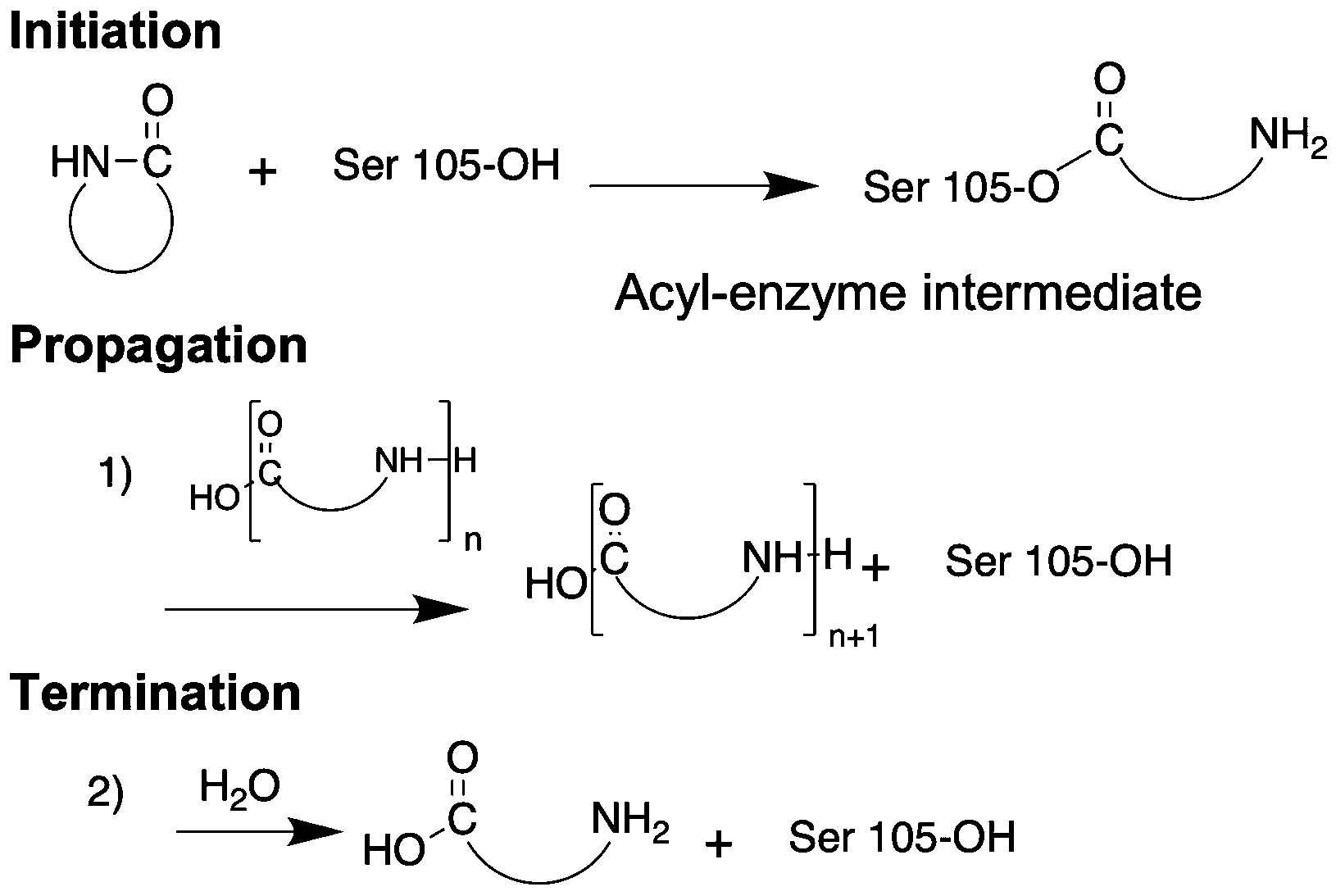 | ||
| Fig. 5 Mechanism of enzyme catalysed ring opening polymerization. | ||
As first reported by Kazlauskas et al., N435 catalyzes the ring opening of 2-azetidinone at temperatures ranging from 55 °C to 80 °C.63 Heating a mixture of N435 and 2-azetidinone at 90 °C for 96 hours in toluene, as described by Loos et al.,64 affords nylon 3 polymers in greater yield (73%). Neither water nor β-alanine initiates the polymerization, confirming enzyme-catalyzed ring opening reaction. Further the enzyme loses only ∼9% activity after 72 hours of heating, thus documentating its stability at high temperature. However, low solubility of the nylon 3 polymers in toluene as well as competition between chain-elongation and chain-termination results in low molecular weight oligomers with an average molecular weight of 586 g mol−1. QM/MM calculations, to study the mechanism of action of CAL-B catalyzed polyamide synthesis confirm the competition between chain-elongation and chain-termination of the catalytic water molecule present in the activation site of the enzyme.65
 | ||
| Fig. 6 Mechanism of cationic ring opening polymerization. | ||
As reported by the Loos group, copolymerization of ε-caprolactone and β-lactam by N435 yields copolymers of poly-ε-caprolactone and poly β-lactam.66 While ε-caprolactone conversion remains constant at different feed ratios, the β-lactam conversion decreases with increasing feed ratio, which the authors attribute to the low solubility of the nylon 3 polymers in toluene.
To summarize, while enzyme-catalyzed reactions possess number of advantages including mild polymerization conditions, non-toxic reagents, and minimal side-products as well as high regio-, enantio-, and chemo-selectivity, enzyme-catalyzed ROP of lactams requires rigorous optimization before widespread application in polyamide synthesis.
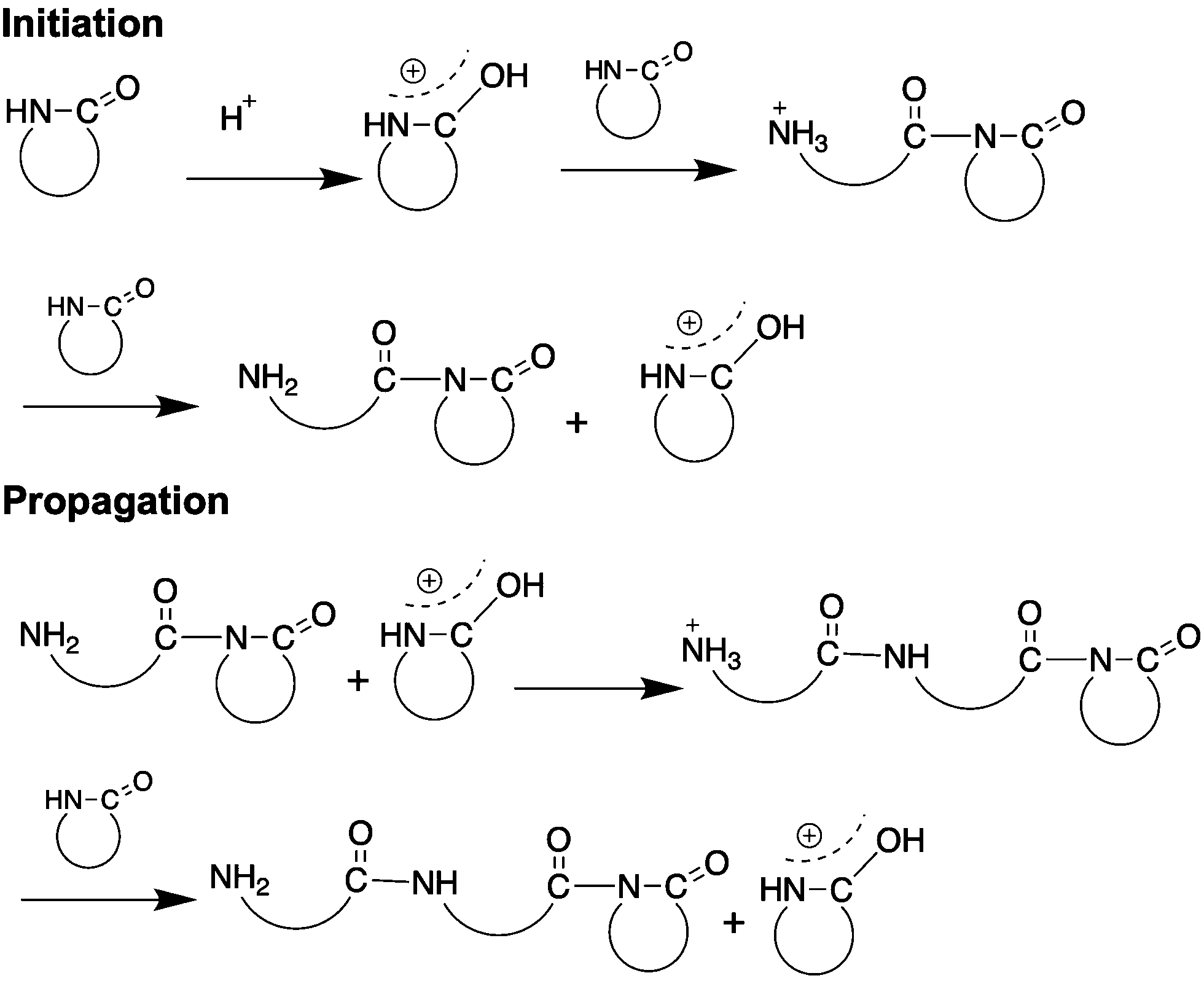
3. Cationic ring opening polymerization
Proton donors such as organic,67 inorganic68 or “solid”69 acids catalyze the cationic ring opening polymerization of lactams (CROP). While cationic ring-opening polymerization is not as widely used as anionic polymerization, CROP affords otherwise inaccessible N-substituted lactam polymers.70 The reaction mechanism depends both on the initiating system and the lactam substitution.70 Briefly, the polymerization begins by protonation of the lactam, resulting in an electrophilic center. The neutral lactam then attacks the protonated lactam yielding the aminoacyl lactam cation as an ammonium salt. The ammonium cation then protonates another lactam monomer, thus regenerating the cationic protonated lactam as well as generating a neutral molecule with an amine end group. The lactam cation then acylates the neutral amine, as it is the strongest nucleophile, resulting in an ammonium salt, thus propagating the polymerization (Fig. 6).70Examples for CROP in recent literature include ROP of terpene β-pinene derived lactams by employing H3PO4 and HCl,68 ROP of ε-caprolactam using sulfonated acids67 and ROP of ε-caprolactam using montmorillonite clay catalyst.69 The reactions proceed without solvent (neat). At temperature >150 °C, shorter polymers (Mn < 8 kDa) form with greater dispersities (Đ between 1.6 and 3.7) compared to AROP.
Lactams used for ring opening polymerization
Due to the advances in chemical methodologies over the past two decades, a wide variety of lactams are available for ROP. Below, we describe various lactams and their polymerization methods as representative examples (Fig. 4).1. Unsubstituted β-lactams
An unsubstituted β-lactam (2-azetidinone, Fig. 4, monomer 1), prepared by chlorosulfonyl isocyanate (CSI) cycloaddition on vinyl acetate followed by KBH4 reduction,64 undergoes anionic ROP in presence of t-BuP428 or lipase B.63 The resulting polymer possess properties such as high capacity of moisture uptake, high glass transition temperature, and high crystallinity.71 Substituted β-lactams are more interesting from a structure, processing, and functionality perspective, and include monocyclic and bicyclic derivatives as described below.2. Substituted β-lactams
Sugar-based β-lactams belong to a new class of bicyclic, substituted β-lactams. As reported by Grinstaff et al., CSI or TCAI cycloaddition with glucal (Fig. 4, 19),78 galactal (Fig. 4, 20),79 altral (Fig. 4, 21)80 and maltal (Fig. 4, 22)81 affords the corresponding sugar β-lactams. LiHMDS initiated polymerization of these monomers yields stereochemically-defined, enantiopure polyamides with degrees of polymerization from 12 to 200. These polymers, termed poly-amido-saccharides, possess many of the favourable properties of polysaccharides to include water-solubility, a helical secondary structure, and multiple hydrogen bonding acceptors and donors.
3. ε-Lactam
ε-Lactams are generally produced via Beckmann rearrangement of oximes, obtained from cyclohexanone derivatives. This method was first reported by O. Wallach in 1900.1 Other natural product-derived lactams are also prepared using a similar method. For example, menthone, a cyclic ketone is directly converted to oxime and then to ε-lactam (Fig. 4, 28) by the Beckmann rearrangement.26 (+)-3-Carene, possessing an endocyclic alkene is either first converted to alcohol by hydroboration25 or to epoxide by CAL-B enzyme,82 and then oxidized to form a ketone. α-Pinene is converted to an alcohol first and then to a ketone,25 while β-pinene, with an exocyclic alkene, is directly oxidized to the ketone.68 All three ketones are then converted to the oxime and then the lactam monomers (Fig. 4, monomers 27, 26 and 25, respectively). In contrast, α-amino-ε-caprolactam (Fig. 4, monomer 29), used to prepare poly-ε-lysine, is prepared by the esterification of commercially available L-lysine monohydrochloride with methanol in thionyl chloride, followed by cyclization in the presence of sodium hydroxide.32Biomedical applications
Nylon 6, nylon 66, and nylon 12 polymers are common components in wound dressings, sutures, catheters, and dental implants and these biomedical applications are reviewed.83–85 Applications for nylon 4 and nylon 5 polymers on the other hand, are limited due to the lack of a reproducible polymerization technique. In contrast, the high water-solubility and degree of functionalizability, availability of robust polymerization techniques, and accessibility to monomers for nylon 3 polymers are advantageous characteristics, and are catalyzing the investigation of these polymers for biomedical applications. Below, we describe a variety of biomedical applications which utilize nylon 3 and nylon 6 polymers.1. Antimicrobial effect
Nylon 3 polymers are finding ever increasing uses as antimicrobial agents. Below are selected examples highlighting the application of nylon 3 polymer as antimicrobial and antifungal agents. The reader is referred to a detailed review on the structural design and antimicrobial properties of these β-polypeptides.86The unique structure and biological activity of host-defense peptides (HDPs) are the inspiration for nylon-based antimicrobial agents. Unlike typical small molecule antibiotics, naturally occurring host-defense peptides, first isolated from silkworm in the 1980s and composed of cationic and hydrophobic amino acids, do not illicit drug resistance.87 The cationic peptides in HDPs interact with the anionic groups in the microbial cell-membrane, while the hydrophobic peptides interact with the lipid components in microbial membrane, resulting in a strong antimicrobial effect via membrane destabilization. However, the low yield, high cost of production, and lack of stability in vivo of natural host-defense peptides precludes their widespread adoption and clinical utility.
Host-defense peptides like cecropins, magainins, and cathelicidins as well as mimicking peptides are sequence specific and adopt a globally amphiphilic alpha-helical conformation. The conformation arises with patches of lipophilic and hydrophilic side chains projected from opposite sides of the helix. In contrast, random nylon 3 co-polymers, prepared from cationic (e.g., Fig. 4, 7) and hydrophobic β-lactams (e.g., Fig. 4, 12), attain non-alpha helical, irregular conformations due to their flexibility but become globally amphiphilic upon interaction with a bacterial membrane.73 This globally amphiphilic conformation disrupts the bacterial cell membrane (Fig. 7A). The ease in preparation of nylon 3 polymers, compared to host-defense peptides, as well as the resistance to protease degradation are noteworthy attributes for these host-defense peptides mimetics.
Specifically, Gellman et al. describe the antibacterial activity of various nylon 3 polymers comprising different cationic and hydrophobic substituents as well as varying ratios of the cationic vs hydrophobic subunits.88 Polymers possessing the moderately hydrophobic cyclohexyl substituent (Fig. 4, 15) and cationic monomethyl amine (Fig. 4, 6) subunit exhibit the most potent antibacterial effect.88 Antibacterial activity is not strongly affected by degree of polymerization, with minimum inhibitory concentration (MIC) decreasing slightly with increasing chain-length for Gram negative E. coli, and MIC increasing slightly with increasing chain-length for Gram-positive S. aureus, B. subtilis, and E. faecium.88 Further, incorporation of β-homoserine and β-homoglycine reduces hemolysis.89 These polymers also exhibit antifungal effects.90–92 Additionally, nylon 3 polymer possessing n-butyl side-groups (monomer 12, Fig. 4),93,94 reported by Liu et al., show superior antimicrobial performance and activity against 45 drug-resistant pathogens. And the polymers are effective in vivo in a Pseudomonas aeruginosa infected rat full-thickness wound model.94 Liu et al. also report antibacterial surface coatings resistant to E. coli and methicillin resistant S. aureus (MRSA) using nylon 3 polymers with monomers 7 and 15 (Fig. 7B).42
Chan-Park et al. describe novel carbohydrate based cationic nylon 3 block co-polymers which display bactericidal activity against both community acquired and hospital-associated MRSA strains (Fig. 7C and D), and exhibit in vivo antibacterial activity superior to vancomycin.45 Unlike the other reports, these co-block polymers prepared from monomers 19 and 8 are non-amphiphilic. Mechanistic studies reveal that the cationic block binds with the anionic wall techoic acid in the cell envelope of MRSA, while the glucose poly-amide block forms a non-fouling corona around the bacteria.95 Moreover, block polymers prepared from monomers 19 and 7 (Fig. 4) reverse antibiotic resistance in carbapenem resistant Gram-negative bacteria by compromising the integrity of the bacterial outer membrane and by deactivation of efflux pump systems.96
Finally, Grinstaff et al. report an amphiphilic carbohydrate-based class of non-cationic antibacterial nylon 3 polymers. Nylon 3 polymers prepared from monomers 19 and 20 (Fig. 4), possessing one or two hydrophobic palmitamide chains at the N-terminal promote or inhibit biofilm formation of Pseudomonas aeruginosa depending on the monomer composition and N-terminal functionalization.43 Random co-polymers of monomers 19 and 20, with one palmitamide chain at the N-terminal lowers surface tension, thus disrupting cell–surface and cell–cell interactions in P. aeruginosa, leading to inhibition of biofilm formation. On the other hand, homopolymers prepared from monomer 20, with a second palmitamide group at the N-terminal do not lower surface tension. Instead, these polymers self-assemble to form rod-like extended structures, which are utilized by the fibrillar adherence proteins of P. aeruginosa, leading to promotion of biofilm formation.
2. Lectin binding
Carbohydrate-binding protein receptors, called lectins, are involved in extracellular recognition, adhesion, cell growth regulation, cancer cell metastasis, and inflammation.97 Example lectins include the asialoglycoprotein receptor displayed on the hepatocyte cell surface that interacts with galactose/N-acetyl-β-galactosamine containing carbohydrate ligand98 and concanavalin A, a lectin present on cell surfaces that binds to nonreducing terminal α-D-mannopyranose and α-D-glucopyranose units.99 Concanavalin A is involved in agglutination of cells, and glycopolymers possessing concanavalin A binding motifs are explored as agglutination inhibitors.100 Polyamides prepared by AROP of glucose based β-lactam monomers78 (Fig. 4, 19) and maltose-based β-lactam monomers81 (Fig. 4, 22) bind to concanavalin A. The glucose-based polyamides having a degree of polymerization ∼50 and maltose polymers with degree of polymerization ∼23 and ∼32 display the highest binding of all polymers. This later result is consistent with the lengths of polymers (∼7 nm, 7.7 nm and 10.8 nm respectively) and the distance between 2 binding sites of Concanavalin A (6.5 nm).3. Cell adhesion
Apart from its well-established application as antimicrobial host-defence peptides, nylon 3 polymers are also being explored as cell adhesive substrates or coatings for tissue culture applications. Nylon 3 polymers are an ideal candidate for preparation of cell adhesive surfaces as they provide a protein-mimetic polyamide structure and are easy to synthesize with highly tuneable side groups.74 Masters et al. report polymers of varying chain-lengths prepared by AROP of monomers with hydrophobic cyclic side groups (15 and 16, Fig. 4), cationic amine side groups (6 and 7, Fig. 4) and uncharged hydrophilic side groups (18, Fig. 4) as coatings for glass slides (Fig. 7E). The polymers are chemically conjugated via the polymeric amine groups on glass slides.74 Copolymers composed of highly hydrophobic subunits (Fig. 4, 6 + 15, 6 + 16) display better NIH-3T3 cell adhesion and growth, as well as uniform spreading based on LIVE/DEAD assay and microscopic images, respectively. The nylon 3 polymer coated surfaces show superior cell adhesion, cell spreading, and morphology compared to other common surfaces used for cell culture such as untreated polystyrene, tissue culture-treated polystyrene, collagen-coated glass, and amine modified glass (Fig. 7F).74The positive controls (tissue culture-treated polystyrene and collagen-coated glass surfaces) display superior cell adhesion in the presence of serum, while adhesion diminished in the absence of serum. Nylon 3 coated glass surfaces on the other hand support substantially greater cell adhesion in the presence and absence of serum. Cell adhesion to synthetic substrates is mediated by an intermediate layer of adsorbed serum proteins, obtained from cell culture media. Adsorption to nylon 3 polymer coated surfaces also follow a similar mechanism, with larger quantities of total protein adsorbed from serum resulting in greater cell adhesion.101 Total serum protein adsorbed on nylon 3 functionalized glass slides (and therefore extent of cell adhesion) directly correlates to (1) number of methyl groups in the cationic subunit, (2) number of carbons in the hydrophobic subunit, and (3) the ratio of number of cationic subunits to total subunits. Polymer composed of monomers 6 and 16 (60:40) and of 7 and 15 (60:40) display the best cell–adhesion. In serum-free conditions, nylon 3 polymer surfaces interact with NIH-3T3 cell–surface proteins–collagen, vitronectin and fibronectin-leading to immobilization. The role of these surface proteins is confirmed by treating NIH-3T3 fibroblasts with plasmin (which degrades cell–surface fibronectin and vitronectin) or collagenase (which degrades collagen) prior to cell adhesion studies. Plasmin treatment led to declined adhesion, indicating greater effect of fibronectin/and or vitronectin in cell adhesion.
Cell adhesion improves with chain-length, with DP > 100 displaying greater cell adhesion. Moreover, in contrast to previous reports, polymers with less hydrophobic cyclohexane and cyclopentane side-groups show higher cell–adhesion at longer chain-lengths.102 Nylon 3 polymer coatings prepared from monomers 6 and 17 (10:90), display high cell adhesion at shorter chain-length (20mer–30mer), and low cell adhesion at longer chain-length (DP > 100), while vice versa is true for polymers prepared from 7 + 15 (60:40). Further, fibroblasts and smooth muscles cells clump on the coated surfaces, while endothelial cells proliferate efficiently and the extent of surface coverage almost doubles from day 1 to day 2 when using coatings of homopolymers composed of monomer 7 (DP = 28, 50, 78 and 90).103 This preferential growth of endothelial cells starting on day 1, directly correlates to the increased ECM deposition by endothelial cells on functionalized surfaces, which promotes higher proliferation rates and lower rates of apoptosis.
Polyethylene glycol-based scaffolds provide a non-adhesive environment to study cell adhesion. Covalent attachment of nylon 3 co-polymers, prepared from monomers 6 and 15 as well as 6 and 16, to PEG hydrogel scaffolds results in increased fibroblast adhesion and spreading compared to PEG-hydrogels bearing arginine-glycine-aspartic acid (RGD) peptides, widely used to promote cell adhesion.74 Moreover, Sieber et al. reports the attachment, growth, and viability of human keratinocytes on nylon 6/PEG blend and nylon 6/PEG copolymer hydrogel coated petri-dishes, the efficacy of which depends on the ratio of nylon 6/PEG and the surface roughness.104
4. Biophysical mimicry of lung surfactant protein
Lung surfactant (LS) is a complex mixture of lipids (∼90% by weight) and proteins (∼10% by weight) present in the alveolar lining of the lungs.105 Lung surfactant proteins include: (1) hydrophilic surfactant protein-A (SP-A) and surfactant protein-D (SP-D) involved in immune response; and (2) hydrophobic surfactant protein-B (SP-B) and surfactant protein-C (SP-C) involved in enhancing the adsorption of lung surfactant to the air–water interface in the lungs. Lack or deficiency of lung surfactant leads to neonatal respiratory distress syndrome106 in pre-term babies and may lead to Acute Respiratory Distress Syndrome (ARDS)/Acute Lung Injury in adults. Surfactant replacement therapy107 treats neonatal respiratory distress syndrome and ARDS in paediatric patients.106 However, the lung surfactants currently employed for surfactant replacement therapy are extracted from mammalian lungs and limited in supply, as well as open to zoonotic infections, thus restricting its clinical use.In contrast to many synthetic lung surfactants which mimic the characteristics of SP-B such as cationic charge, multiple amphiphilic helices, and disulfide bonds, the Gellman group is investigating a hetero-chiral, sequence-random nylon 3 copolymer, possessing global amphiphilicity, but lacks a regular conformation.108 SP-B mimicking polymers (Fig. 7G), composed of lipophilic units (monomers 15 and 16, Fig. 4) mimic the insertion region of SP-B while the cationic units (monomers 6 and 7, Fig. 4) provide the amphiphilic, cationic characteristic of SP-B. A N-terminal octadecanoyl group is included within these polymers to mirror the two hydrophobic palmitoyl groups in SP-C (Fig. 7H).
Surface activity measurements of these lung surfactant mimetics using pulsating bubble surfactometry (static mode and dynamic mode) show that polymers with cationic groups alone are ineffective as a surfactant, while incorporation of hydrophobic groups to the polymer improves the surface tension values. Polymers prepared using monomer 16 exhibit the highest surface activity. Further, surface activity decreases with increasing ratio of cationic subunits while the presence of N-terminal octadecanoyl group improves the surface tension values in dynamic mode. Moreover, these polymers are non-cytotoxic against NIH-3T3 fibroblasts, with IC50 values of polymers comparable to IC50 of other SP-B mimetics.
The mechanism of action involves adsorption of hydrophobic cyclohexane (Fig. 4, 15) and cyclooctane (Fig. 4, 16) groups to the hydrophobic lipid surface with more hydrophobic cyclooctane having stronger interactions, leading to lower surface tension (Fig. 7G). Moreover, cationic side-groups adsorb to the lipid surface by coulombic interactions with the charged lipid head groups, resulting in a further surface tension decrease (Fig. 7H).
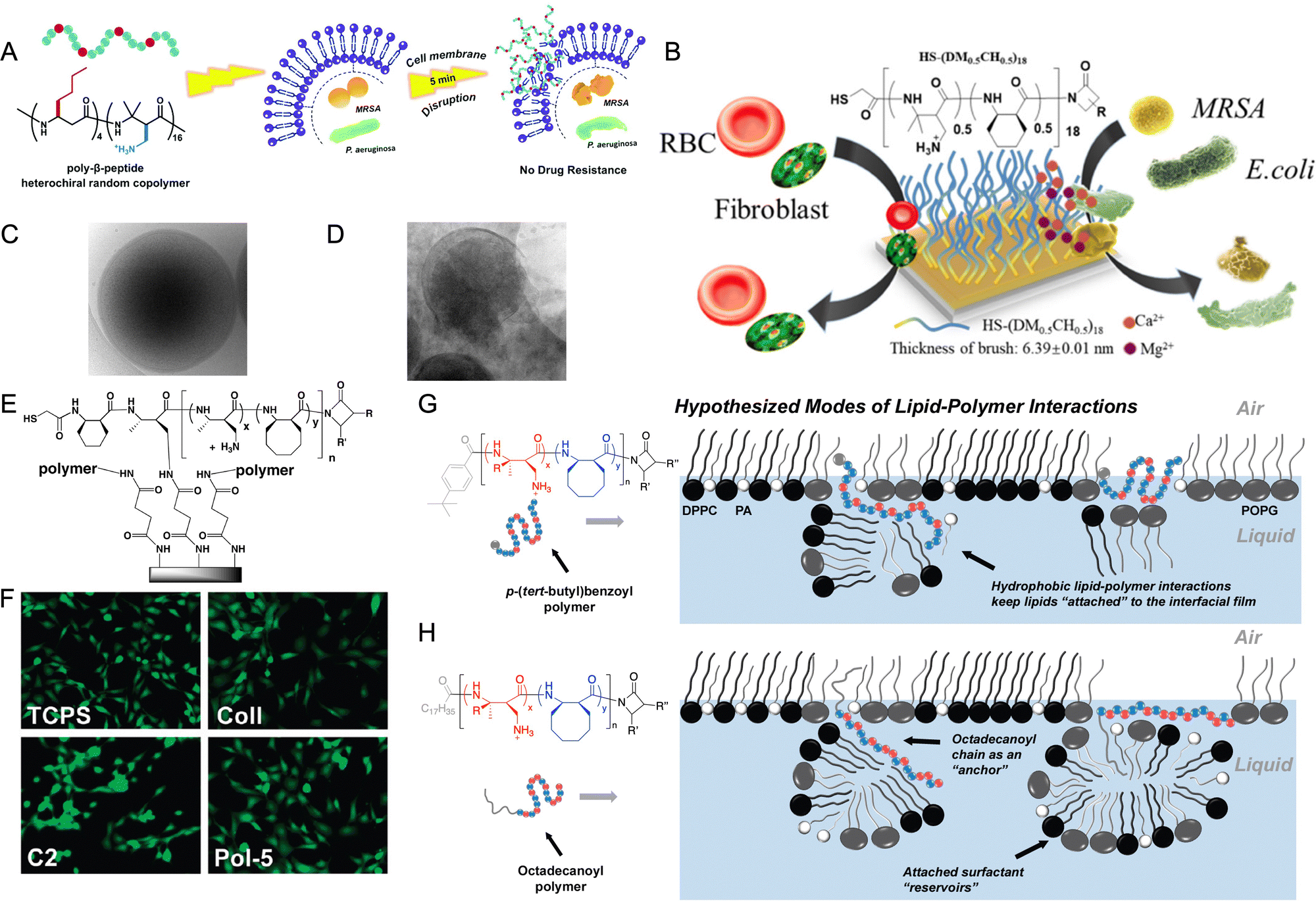 | ||
| Fig. 7 (A) Mechanism of action HDP-mimicking nylon-3 polymers. Adapted with permission from ref. 93. Copyright © Royal Society of Chemistry. (B) Thiol-terminated amphiphilic nylon 3 polymers prepared from monomers 7 and 15 are attached gold surface. The polymer coatings are non-cytotoxic against NIH-3T3 cells and are non-haemolytic but kill Gram-negative E. coli and Gram-positive MRSA bacterial strains. Adapted with permission from ref. 42. Copyright © American Chemical Society; (C and D) cryo-TEM images of untreated control bacteria, with cell envelope intact and cryo-TEM image of polymer-treated (co-block) polymers prepared from monomers 19 and 8 bacteria resulting in cell lysis, respectively. (B and C) Adapted with permission from ref. 45. Copyright © 2019, The Author(s); (E) amphiphilic nylon 3 polymers are immobilized on glass slides using terminal amines. (F) Copolymer prepared from monomers 6 and 16 (Pol-5 in figure) display cell adhesion similar to tissue culture-treated polystyrene (TCPS in figure) and collagen coated glass slides (Coll in figure), and improved cell adhesion than amine modified glass surface (C2 in figure) in the presence of serum. (E and F) Adapted with permission from ref. 74. Copyright © American Chemical Society; (G) mechanism of action of SP-B mimicking polymers for polymers without an octadecanoyl chain. Polymers adopt an amphiphilic conformation and insert into the lipid film, where they are able to retain attached lipids via Coulombic interactions between cationic subunits and charged lipid head groups or hydrophobic interactions between lipophilic subunits. (H) Mechanism of action of SP-C mimicking polymers for polymers with an octadecanoyl chain. Octadecanoylated copolymers act as lipid “anchors” and inserts into lipid acyl chains through hydrophobic hydrocarbon chain–chain interactions, leading to enhanced surface activity. In subfigures G and H, blue spheres represent the lipophilic units while red spheres indicate cationic units. The gray sphere in the “polymer sequence” of A represents the p-(tert-butyl)benzoyl end group, while the gray line extending from the “polymer sequence” in B represents the octadecanoyl chain. (G and H) Adapted with permission from ref. 108. Copyright © American Chemical Society. | ||
5. siRNA delivery
Gene therapy employs nucleic acids such as plasmid DNA, siRNA, or mRNAs to treat diseases by correcting the underlying genetic defect.109 However, delivering naked nucleic acid to a cell is challenging due to its degradation by nucleases, lack of targeting, and limited cellular uptake.109 Traditional nucleic acid delivery agents are immunogenic viral vectors and cytotoxic cationic polymers such as PEI. Amphiphilic nylon 3 polymers possess favourable properties as non-viral vectors such as solubility in water, positive charges for complexation with negatively charged siRNA at physiologic pH, and amphiphilicity. Merkel et al. describe nylon 3 polymers composed of cationic (monomer 7, Fig. 4) and hydrophobic monomers (monomers 14 and 15, Fig. 4) with varying ratios of cationic/hydrophobic monomers and chain-lengths.110 All the polymers are non-cytotoxic up to a concentration of 10 μg mL−1 and condense siRNA at nitrogen-to-phosphate (N/P) ratio of 5 or lower. However, only polymers with cyclopentyl hydrophobic groups (60% hydrophobic, 40% cationic) and molecular weight >30 kDa display >80% uptake of polyplexes in H1299/Luc cell as well as substantial knockdown of a control luciferase gene. The polyplexes are predominantly internalized by cholesterol-dependent clathrin-mediated endocytosis, micropinocytosis, and caveolae-mediated endocytosis, as confirmed by cellular uptake studies in the presence of inhibitors for the different pathways.More recently, Merkel et al. describe random nylon 3 co-polymers, prepared using varying feed ratios of nonmethylated amine β-lactam, di-methylated amine β-lactam (monomer 7, Fig. 4) and cyclopentyl β-lactam (monomer 14, Fig. 4), to condense siRNA and knockdown genes in glioblastoma (Fig. 8A).111 Co-polymers of non-methylated amine β-lactam and cyclopentyl β-lactam form polyplexes of smaller diameter, resulting in better siRNA–polymer packing at an N/P ratio of 3, compared to the other co-polymers. Moreover, polymers with greatest hydrophobic content (i.e., more cyclopentyl β-lactam content), display significantly higher cellular internalization and successful knockdown capabilities. These polyplexes are internalized into cells via clathrin-mediated endocytosis.
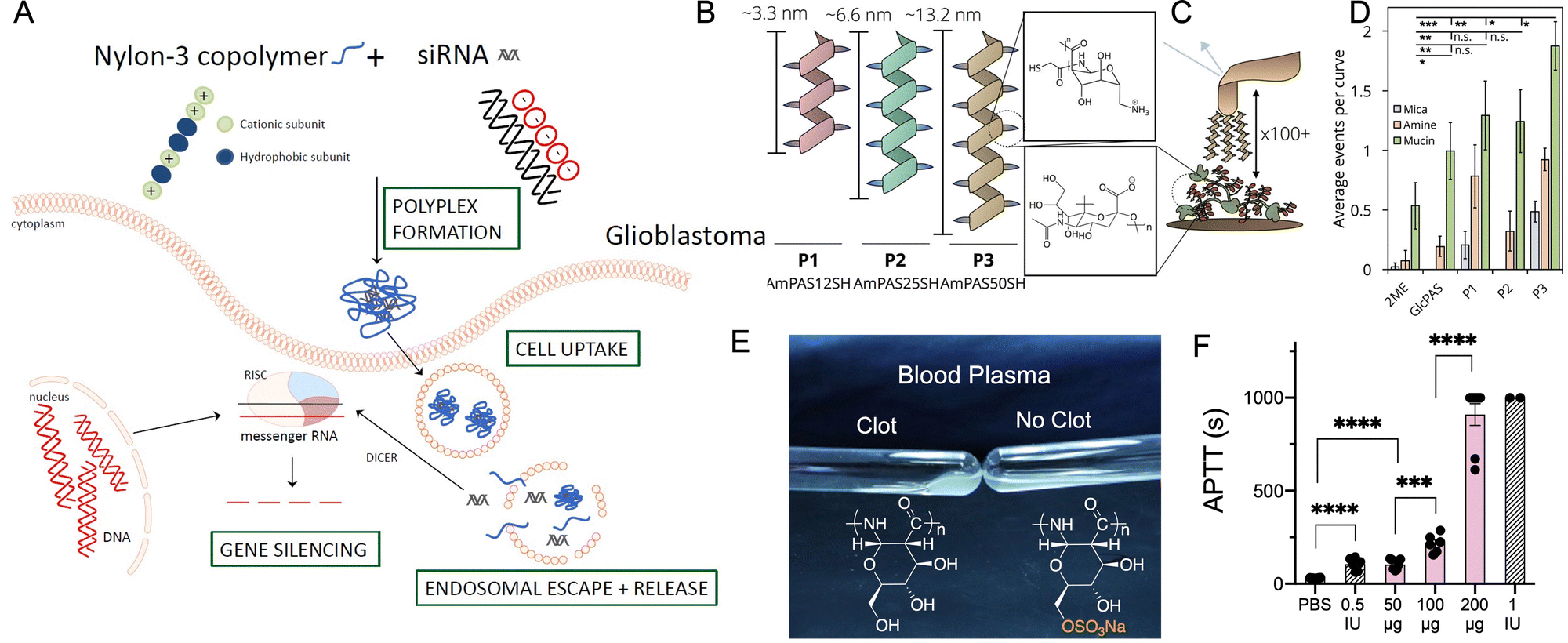 | ||
| Fig. 8 (A) Polyplex formation between nylon 3 polymer and siRNA and mechanism of gene silencing by the polyplexes. Adapted with permission from ref. 111. Copyright © 2019 by the authors; (B) schematic of amine functionalized glucose-based nylon 3 polymers prepared from monomer 23 with degree of polymerization 12, 25 and 50 and their estimated average lengths; magnified: structure of repeating polymer subunit. (C) Schematic of single-molecule force spectroscopy experiment. Magnified: structure of sialic acid in mucin. (D) Average number of adhesion events per curve comparing 2-mercaptoethanol (2ME), non-functionalized glucose-based polyamides and amine functionalized polymers of varying degrees of polymerization. Polymer with degree of polymerization 50 display greatest mucoadhesivity. (B–D) Adapted with permission from ref. 121. Copyright © John Wiley and Sons; (E) representation of blood plasma coagulation in the absence and presence of sulfated glucose-based nylon 3 polymers prepared from monomer 19. (F) In vivo elongation of clotting time (APTT) after intra venous administration of sulfated glucose-based nylon 3 polymers of degree of polymerization 50. (E and F) Adapted with permission from ref. 129. Copyright © Royal Society of Chemistry. | ||
6. Inhibition of ice recrystallization
The inhibition of ice-recrystallization is important in biomedicine, specifically in cell and protein preservation. Since the mid-1900s, the most widely used cryopreservants are DMSO and glycerol,112,113 and these agents display cytotoxic effects at high concentrations. Alternative cryopreservants such as glucose and galactose are less cytotoxic but exhibit lower efficiency.Antifreeze glycoproteins (AFGPs) are natural cryo-preservation agents first discovered in fish serum by DeVries and Wohlschlag in 1969.114 Despite their use in petroleum industry and food industry, the labor-intensive isolation and purification of AFGPs hinders widespread use.115 Polyvinyl alcohol (PVA), with many hydroxyl groups in its structure similar to AFGPS, inhibits ice-recrystallization by adsorbing on the surface of ice crystals.116 Similar to PVA, polyamides prepared by ROP of β-lactams with hydroxyl group functionalities are anti-freeze agents.117 Among the homopolymers and copolymers prepared using β-lactams with one hydroxyl (monomer 10, Fig. 4), 2 hydroxyl (monomer 18, Fig. 4), anionic (monomer 11, Fig. 4), cationic (monomer 7, Fig. 4) and/or hydrophobic side-groups, the two homopolymers of monomers 10 and 18 best inhibit ice-recrystallization. Homopolymers of anionic, cationic or hydrophobic monomers as well as their copolymers of monomers 10 and 18 result in lower levels of inhibition, indicating that the side chain hydroxyl groups are crucial for ice recrystallization inhibition activity. The inhibition decreases at concentrations below 1 mg mL−1 as well as with increases in polymer chain-length. Nylon 3 polymers with hydroxyl side-groups also show minimal cytotoxicity with cytotoxicity increasing with an increase in chain-length and the number of hydroxyl groups per monomer.
7. Protein stabilization
While cationic and hydrophobic nylon 3- and nylon-6 polymers are well studies, anionic polymers are less explored. Carboxylated glucuronic acid poly-amido-saccharides prepared from glucose-based β-lactams (Fig. 4, 19) by selective TEMPO-mediated oxidation of the primary hydroxyl group of glucose PASs stabilize proteins during freeze-thaw cycles. It is thought that the PAS retains water molecules near the protein structure in a similar manner as other protective carbohydrate freezing or lyophilization agents such as trehalose.1188. Mucoadhesion
Chitosan is a cationic biocompatible polysaccharide derived from chitin and used in drug delivery systems due to its mucoadhesivity. The mucoadhesivity of chitosan arises via electrostatic interaction of protonated amines in chitosan with anionic mucin, as well as hydrogen bonding and hydrophobic interactions.119Grinstaff et al. report a chitosan mimicking, mucoadhesive polyamide, synthesized by the AROP of regio selectively amine-functionalized β-lactam monomer (Fig. 4, 23).120 Similar to chitosan, the polymer exerts its mucoadhesive property via its cationic charge, hydrogen bonding capability, and chain entanglement with mucins (Fig. 8B–D). Polymers with a degree of polymerization 50 compared to 25 display the greatest mucoadhesivity as determined by single molecule force microscopy (Fig. 8C and D) and ex vivo studies with mucin coated tissues such as the porcine trachea, ovine esophagus, and porcine duodenum.121 Additionally, unlike chitosan, these polymers are homogeneous in structure and degree of branching, and display excellent control in molecular weight and batch-to-batch consistency.
9. Glycosaminoglycan mimics
Glycosaminoglycans (GAGs) are anionic polysaccharides and include heparin, used as anticoagulant to treat deep vein thrombosis and acute coronary syndrome,122 keratin sulfate and chondroitin sulfate involved in hydration and lubrication of cartilage,123 and heparan sulfate involved in angiogenesis, developmental processes and cell homeostasis.124 While naturally occurring GAGs are widely used as anticoagulants125 the biological origin of these polysaccharides pose many issues such as contamination,126 batch-to-batch variation, and structural heterogeneity.127 Grinstaff et al. report glycosaminoglycan-mimicking sulphated polyamides, with regio-selective128 and non-regioselective functionalization.129 Using an orthogonally protected glucose based β-lactam monomer (Fig. 4, 24), followed by selective deprotection and sulphation, enables synthesis of varied PAS (Mn 2.36 to 14.1 kDa) with sulphate groups only present on the 6′-hydroxyl position of the glucose repeat unit.128 Post sulfation of PASs using SO3·NMe3 affords the non-regio selectively sulphated polymers with an average of one or two sulphates per repeat unit (prepared from monomers 19 and 20, respectively).129 The in vitro anticoagulant effect of non-regio selectively sulphated polymers (Fig. 8E) increases with polymer concentration, chain-length, and degree of sulfation. Anticoagulant activity is a consequence of coagulation factors Xa, XIa and IIa inhibition. The polymers exhibit minimal cytotoxicity and haemolytic effects, and the best anticoagulant polymer, prepared from monomer 19 (with 2 sulphate groups per monomer on average and degree of polymerization 50) displays a dose dependent elongation of clotting time in vivo in a mouse model (Fig. 8F), with bleeding risk comparable to heparin, confirming in vivo efficacy.129Conclusions and future perspectives
Nylon polymers are prevalent in day-to-day life activities as essential components in consumer and healthcare products. While new methods to prepare nylon polymers have emerged, anionic ring opening polymerization (AROP) of lactams remains a powerful method to prepare nylon polymers due to control over molecular weight, availability of different monomers, and efficient polymerization. Furthermore, catalysts with varying basic strengths, nucleophilicity, ionic character, and air and moisture-sensitivity are available for AROP of lactams. That said, continuing developments in cationic- and enzyme-catalyzed ROP of lactams will expand their scope of use and provide access to new polymer compositions.Lactam monomers of increasing structural and chemical complexities are currently available for ROP, thus transitioning nylon polymers from yesterday's simple bulk commodity polymers to biocompatible thermoplastics or bioactive agents. Examples of such monomers includes those possessing functional groups such as alcohols, amines, or carboxylic acids as well as those based on carbohydrates. Due to the high levels of functionalization achieved on the resulting polymers, nylon 3 polymers are approaching the required diversity to create new polymers and materials, based on predefined design requirements. Control of polymer composition and structure is essential to the goal of establishing structure–activity relationships to guide future studies, be it new polymer processing techniques or materials for biomedical applications.
The intent of this article is to provide the background necessary to understand current developments as well as to envision future applications. Importantly, significant opportunities remain on the monomer, polymerization, and biomedical application fronts. First, expanding the diversity of functional groups present on the monomer (or resulting polymer) beyond those currently used to include alkenes, alkynes, halides, phenols, thiols, aldehydes, ketones, sulphides, ammoniums, nitriles, phosphates, etc. will pave the way for further applications of this polymer. Bicyclic monomers are primarily restricted to purely aliphatic systems except for the recently reported carbohydrate analogues – but more complex ring structure are lacking. The new polyamides prepared from the above monomers will provide key data on their chemical, physical, and biological properties, and further inform our knowledge base. Such results will begin to yield general design rules to guide future uses, as today, the polyamide compositions are highly dependent on the application, and trends are difficult to discern across the polymers.
On the polymerization front, high molecular weight nylon polymers and nylon polymers of controlled molecular weights are reported by anionic ring opening polymerization of lactams, but methods to prepare polymers of higher degrees of polymerization (DP > 100) with control in molecular weight and dispersity remains elusive. Polymerizations which afford specific diblocks, triblocks, or other interesting architectures (e.g., dendritic) are of interest and in demand. Discovery of new metal and organic catalysts that are air and/or moisture insensitive or that polymerize diverse monomers will also be key. Enzymatic ring opening polymerization of monomers possessing greater structural and compositional complexity are needed to expand our catalyst toolbox as well as to usher scientific progress from a sustainability and green science standpoint.
Moreover, few nylon 4 and nylon 5 polymers are synthesized or studied due to low thermal stability and a lack of reproducible polymerization techniques, respectively. Incorporating C-2 and C-4 functionalizations on the polymer backbone, similar to the recent report by Cywar et al.,130 as well as N-terminal and C-terminal functionlizations will improve thermal stability for nylon 4 polymers.131 Therefore, we encourage the scientific community to explore the synthesis of thermally stable, functionalized nylon 4 polymers. On the other hand, due to the low ring strain of the six-membered 2-piperidone, nylon-5 polymer synthesis by ROP is thermodynamically and kinetically unfavourable.70 Polymerization leads to crystallization of growing chains and consequent physical termination of polymerization. Therefore, other techniques such as polycondensation of 5-amino valeric acid, with lower control in degree of polymerization and polydispersity need to be explored to obtain nylon 5 polymers. These polymers possess hydrophilicities between nylon 3 and nylon 6, and would likely find utility in biomedical applications.
Another area that requires further study is the natural biodegradation of nylon polymers, especially due to the ubiquitous presence and adverse impacts of non-degradable microplastics on aquatic and terrestrial organisms.132–136 While earlier studies on nylon 6 oligomer degradation by bacteria,137–139 nylon 6,6 degradation by white rot fungi140 and recent studies on nylon 6 polymers by wood degrading fungi141 are key, further exploration of biodegradation pathways of these polymers is essential for a sustainable and environmentally safe future. Incorporation of biodegradable nylon 4 and nylon 2 monomers into higher order nylons is another strategy explored to prepare new classes of nylons with tuneable biodegradability.142
Finally, the successful transition of several of these nylon polymers to in vitro and in vivo studies as devices, therapeutic agents, or macromolecular carriers for drug delivery bodes well for their continued investigation. We encourage the scientific community to explore the utilization of amphiphilic nylon polymers for delivery of small molecules as well as large biomacromolecules such as enzymes, chemokines, RNA, or DNA. As demonstrated by the host-defense peptide, lung surfactant, and antifreeze glycoprotein-mimicking polymers, there is an exciting opportunity for biomimicry of other polypeptides by nylon polymers due to the structural similarity, ease of synthesis, and potential for functionalization. Translation of these polymers to large animal models and first-in human studies has not occurred, and represents the next significant milestone – one that as a community we hope to achieve in the next 5–10 years. Acheiving this milestone will also require highly reproducible polymerizations which are performed on the kilogram scale for good manufacturing practice, as required by governmental regulatory agencies for medical products.
Author contributions
M. V. performed the literature search and background analysis. M. V. and M. W. G. wrote the manuscript.Conflicts of interest
M. V. and M. W. G. are co-inventors on a patent application and the application is available for licensing.Acknowledgements
This work was supported in part by BU, DOD USU (HU0001810012), and the NIH (R01HL164650).References
- P. Matthies and W. F. Seydl, in High Performance Polymers: Their Origin and Development, ed. R. B. Seymour and G. S. Kirshenbaum, Springer Netherlands, Dordrecht, 1986, pp. 39–53 Search PubMed.
- W. Carothers, USPTO, US2130947A, 1938 Search PubMed.
- M. I. Kohan, in High Performance Polymers: Their Origin and Development, ed. R. B. Seymour and G. S. Kirshenbaum, Springer Netherlands, Dordrecht, 1986, pp. 19–37 Search PubMed.
- P. Schlack, USPTO, US2241321A, 1941 Search PubMed.
- P. Matthies and W. F. Seydl, in High Performance Polymers: Their Origin and Development, ed. R. B. Seymour and G. S. Kirshenbaum, Springer Netherlands, Dordrecht, 1986, pp. 39–53 Search PubMed.
- S. Hanshelmut, S. Joachim, S. Hermann and I. Otto, US Pat., US3418314A, 1965 Search PubMed.
- R. M. Joyce and D. M. Ritter, USPTO, US2251519A, 1941 Search PubMed.
- Plastics Polymers & Resins Market Research Report/Nylon market, https://www.fortunebusinessinsights.com/nylon-market-102007, (accessed April 23, 2022).
- Nylon Market Size, Share & Trends Analysis Report By Product (Nylon 6, Nylon 66), By Application (Automobile, Electrical & Electronics, Engineering Plastics, Textiles), By Region, And Segment Forecasts, 2021–2028, https://www.grandviewresearch.com/industry-analysis/nylon-6-6-market, (accessed April 23, 2022).
- G. B. Apgar and M. J. Koskoski, in High Performance Polymers: Their Origin and Development, ed. R. B. Seymour and G. S. Kirshenbaum, Springer Netherlands, Dordrecht, 1986, pp. 55–65 Search PubMed.
- P. Tiedemann, S. Anwar, U. Kemmer-Jonas, K. Asadi and H. Frey, Synthesis and Solution Processing of Nylon-5 Ferroelectric Thin Films: The Renaissance of Odd-Nylons?, Macromol. Chem. Phys., 2020, 221, 1900468 CrossRef.
- K. Tachibana, K. Hashimoto, M. Yoshikawa and H. Okawa, Isolation and characterization of microorganisms degrading nylon 4 in the composted soil, Polym. Degrad. Stab., 2010, 95, 912–917 CrossRef CAS.
- A. Nakayama, N. Yamano, N. Kawasaki and Y. Nakayama, Synthesis and biodegradation of poly(2-pyrrolidone-co-ε-caprolactone)s, Polym. Degrad. Stab., 2013, 98, 1882–1888 CrossRef CAS.
- K. Tachibana, K. Hashimoto, N. Tansho and H. Okawa, Chemical modification of chain end in nylon 4 and improvement of its thermal stability, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2495–2503 CrossRef CAS.
- J. Šebenda and J. Hauer, Living polymerization of lactams and synthesis of monodisperse polyamides, Polym. Bull., 1981, 5, 529–534 CrossRef.
- H. Leuchs, Ueber die Glycin-carbonsäure, Ber. Dtsch. Chem. Ges., 1906, 39, 857–861 CrossRef CAS.
- A. Rasines Mazo, S. Allison-Logan, F. Karimi, N. J. A. Chan, W. Qiu, W. Duan, N. M. O’Brien-Simpson and G. G. Qiao, Chem. Soc. Rev., 2020, 49, 4737–4834 RSC.
- M. Zhou, J. Zou, L. Liu, X. Xiao, S. Deng, Y. Wu, J. Xie, Z. Cong, Z. Ji and R. Liu, Synthesis of poly-α/β-peptides with tunable sequence via the copolymerization on N-carboxyanhydride and N-thiocarboxyanhydride, iScience, 2021, 24, 103124 CrossRef CAS PubMed.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Synthesis of Well-Defined Polypeptide-Based Materials via the Ring-Opening Polymerization of α-Amino Acid N-Carboxyanhydrides, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS PubMed.
- H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Recent advances in amino acid N-carboxyanhydrides and synthetic polypeptides: chemistry, self-assembly and biological applications, Chem. Commun., 2014, 50, 139–155 RSC.
- T. J. Deming, Preparation and development of block copolypeptide vesicles and hydrogels for biological and medical applications, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 283–297 CAS.
- T. J. Deming, Synthesis of Side-Chain Modified Polypeptides, Chem. Rev., 2016, 116, 786–808 CrossRef CAS PubMed.
- K. Hashimoto, Ring-opening polymerization of lactams. Living anionic polymerization and its applications, Progress Polym. Sci., 2000, 25, 1411–1462 CrossRef CAS.
- T. Ageyeva, I. Sibikin and J. Karger-Kocsis, Polymers and Related Composites via Anionic Ring-Opening Polymerization of Lactams: Recent Developments and Future Trends, Polymers, 2018, 10, 357 CrossRef PubMed.
- P. N. Stockmann, D. L. Pastoetter, M. Woelbing, C. Falcke, M. Winnacker, H. Strittmatter and V. Sieber, New Bio-Polyamides from Terpenes: α-Pinene and (+)-3-Carene as Valuable Resources for Lactam Production, Macromol. Rapid Commun., 2019, 40, 1800903 CrossRef PubMed.
- M. Winnacker, S. Vagin, V. Auer and B. Rieger, Synthesis of novel sustainable oligoamides via ring-opening polymerization of lactams based on (−)-menthone, Macromol. Chem. Phys., 2014, 215, 1654–1660 CrossRef CAS.
- J. Zhang, D. A. Kissounko, S. E. Lee, S. H. Gellman and S. S. Stahl, Access to poly-β-peptides with functionalized side chains and end groups via controlled ring-opening polymerization of β-lactams, J. Am. Chem. Soc., 2009, 131, 1589–1597 CrossRef CAS PubMed.
- H. Yang, J. Zhao, M. Yan, S. Pispas and G. Zhang, Nylon 3 synthesized by ring opening polymerization with a metal-free catalyst, Polym. Chem., 2011, 2, 2888–2892 RSC.
- E. Šittler and J. Šebenda, Alkaline Polymerization of Caprolactam. XXI. Dissociation of Alkali Metal Salts of 6-Caprolactam in the Polymerization of 6-Caprolactam, J. Polym. Sci., Part C: Polym. Symp., 2007, 16, 67–77 CrossRef.
- F. Faridirad, S. Ahmadi, M. D. Barmar and G. Naderi, Statistical Analysis of the Catalyst and Activator Level on In Situ Polymerization of Laurolactam during Reactive Melt Blending, J. Thermodyn. Catal., 2016, 7, 1000168 Search PubMed.
- J. Budín, J. Roda, J. Brožek and J. Kříř, Anionic Copolymerization of ε-Caprolactam with ω-Laurolactam, Macromol. Symp., 2006, 240, 78–82 CrossRef.
- Y. Tao, X. Chen, F. Jia, S. Wang, C. Xiao, F. Cui, Y. Li, Z. Bian, X. Chen and X. Wang, New chemosynthetic route to linear ε-poly-lysine, Chem. Sci., 2015, 6, 6385–6391 RSC.
- M. Winnacker, A. Tischner, M. Neumeier and B. Rieger, New insights into synthesis and oligomerization of ε-lactams derived from the terpenoid ketone (−)-menthone, RSC Adv., 2015, 5, 77699–77705 RSC.
- N. C. Kim, J.-H. Kim, J. H. Kim, S. W. Nam, B. S. Jeon and Y. J. Kim, Preparation of Nylon 4 microspheres via heterogeneous polymerization of 2-pyrrolidone in a paraffin oil continuous phase, J. Ind. Eng. Chem., 2015, 28, 236–240 CrossRef CAS.
- K. Hashimoto, T. Oi, J. Yasuda, K. Hotta and M. Okada, Molecular Weight Distribution of Polyamides Obtained in Anionic Polymerization of Methyl-Substituted beta-Lactams and Aminolysis of Their N-Benzoyl Lactams, John Wiley & Sons, Inc, 1997, vol. 35 Search PubMed.
- K. Udipi, R. S. Davé, R. L. Kruse and L. R. Stebbins, Polyamides from lactams via anionic ring-opening polymerization: 1. Chemistry and some recent findings, Polymer, 1997, 38, 927–938 CrossRef CAS.
- T. J. Deming, Living polymerization of α-amino acid-N-carboxyanhydrides, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3011–3018 CrossRef CAS.
- P. Salas-Ambrosio, A. Tronnet, M. Since, S. Bourgeade-Delmas, J.-L. Stigliani, A. Vax, S. Lecommandoux, B. Dupuy, P. Verhaeghe and C. Bonduelle, Cyclic Poly(α-peptoid)s by Lithium bis(trimethylsilyl)amide (LiHMDS)-Mediated Ring-Expansion Polymerization: Simple Access to Bioactive Backbones, J. Am. Chem. Soc., 2021, 143, 3697–3702 CrossRef CAS PubMed.
- Y. Wu, D. Zhang, P. Ma, R. Zhou, L. Hua and R. Liu, Lithium hexamethyldisilazide initiated superfast ring opening polymerization of alpha-amino acid N-carboxyanhydrides, Nat. Commun., 2018, 9, 5297 CrossRef CAS PubMed.
- A. Rodriguez-Galan, S. Muñoz-Guerra, J. A. Subirana, B. Chuong and H. Sekiguchi, Makromolekulare Chemie. Macromolecular Symposia, Wiley Online Library, 1986, vol. 6, pp. 277–284 Search PubMed.
- J. Cheng and T. J. Deming, J. Am. Chem. Soc., 2001, 123, 9457–9458 CrossRef CAS PubMed.
- Y. Qian, F. Qi, Q. Chen, Q. Zhang, Z. Qiao, S. Zhang, T. Wei, Q. Yu, S. Yu, Z. Mao, C. Gao, Y. Ding, Y. Cheng, C. Jin, H. Xie and R. Liu, Surface Modified with a Host Defense Peptide-Mimicking β-Peptide Polymer Kills Bacteria on Contact with High Efficacy, ACS Appl. Mater. Interfaces, 2018, 10, 15395–15400 CrossRef CAS PubMed.
- E. L. Dane, A. E. Ballok, G. A. O’Toole and M. W. Grinstaff, Synthesis of bioinspired carbohydrate amphiphiles that promote and inhibit biofilms, Chem. Sci., 2014, 5, 551–557 RSC.
- J. Zhang, M. J. Markiewicz, B. P. Mowery, B. Weisblum, S. S. Stahl and S. H. Gellman, C-terminal functionalization of nylon-3 polymers: effects of C-terminal groups on antibacterial and hemolytic activities, Biomacromolecules, 2012, 13, 323–331 CrossRef CAS PubMed.
- K. Zhang, Y. Du, Z. Si, Y. Liu, M. E. Turvey, C. Raju, D. Keogh, L. Ruan, S. L. Jothy, S. Reghu, K. Marimuthu, P. P. De, O. T. Ng, J. R. Mediavilla, B. N. Kreiswirth, Y. R. Chi, J. Ren, K. C. Tam, X.-W. Liu, H. Duan, Y. Zhu, Y. Mu, P. T. Hammond, G. C. Bazan, K. Pethe and M. B. Chan-Park, Enantiomeric glycosylated cationic block co-beta-peptides eradicate Staphylococcus aureus biofilms and antibiotic-tolerant persisters, Nat. Commun., 2019, 10, 4792 CrossRef PubMed.
- C. Erickson, Lithium shortage causing bottleneck in energy transition – Lilac Solutions CEO, https://www.spglobal.com/marketintelligence/en/news-insights/latest-news-headlines/lithium-shortage-causing-bottleneck-in-energy-transition-8211-lilac-solutions-ceo-66769303, (accessed April 28, 2022).
- B. Esswein and M. Möller, Polymerization of Ethylene Oxide with Alkyllithium Compounds and the Phosphazene Base “tBuP4,”, Angew. Chem., Int. Ed. Engl., 1996, 35, 623–625 CrossRef CAS.
- L. Dentzer, C. Bray, S. Noinville, N. Illy and P. Guégan, Phosphazene-promoted metal-free ring-opening polymerization of 1,2-epoxybutane initiated by secondary amides, Macromolecules, 2015, 48, 7755–7764 CrossRef CAS.
- A. Molenberg and M. Möller, A fast catalyst system for the ring-opening polymerization of cyclosiloxanes, Macromol. Rapid Commun., 1995, 16, 449–453 CrossRef CAS.
- T. Pietzonka and D. Seebach, The P4-Phosphazene Base as Part of a New Metal-Free Initiator System for the Anionic Polymerization of Methyl Methacrylate, Angew. Chem., Int. Ed. Engl., 1993, 32, 716–717 CrossRef.
- W. Memeger, G. C. Campbell and F. Davidson, Poly(aminophosphazene)s and Protophosphatranes Mimic Classical Strong Anionic Base Catalysts in the Anionic Ring-Opening Polymerization of Lactams, Macromolecules, 1996, 29, 6475–6480 CrossRef CAS.
- J. Chen, M. Li, W. He, Y. Tao and X. Wang, Facile Organocatalyzed Synthesis of Poly(ε-lysine) under Mild Conditions, Macromolecules, 2017, 50, 9128–9134 CrossRef CAS.
- W. He, Y. Tao and X. Wang, Functional Polyamides: A Sustainable Access via Lysine Cyclization and Organocatalytic Ring-Opening Polymerization, Macromolecules, 2018, 51, 8248–8257 CrossRef CAS.
- M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, N-Heterocyclic carbenes (NHCs) as organocatalysts and structural components in metal-free polymer synthesis, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, N-Heterocyclic carbenes (NHCs) as organocatalysts and structural components in metal-free polymer synthesis, Chem. Soc. Rev., 2013, 42, 2142 RSC.
- W. Tam and D. Williamson, USPTO, US 2006/0100365, 2006 Search PubMed.
- S. Naumann, S. Epple, C. Bonten and M. R. Buchmeiser, Polymerization of ε-caprolactam by latent precatalysts based on protected n-heterocyclic carbenes, ACS Macro Lett., 2013, 2, 609–612 CrossRef CAS PubMed.
- S. Naumann, F. G. Schmidt, M. Speiser, M. Böhl, S. Epple, C. Bonten and M. R. Buchmeiser, Anionic ring-opening homo- and copolymerization of lactams by latent, protected N-heterocyclic carbenes for the preparation of PA 12 and PA 6/12, Macromolecules, 2013, 46, 8426–8433 CrossRef CAS.
- H. J. Altmann, M. Steinmann, I. Elser, M. J. Benedikter, S. Naumann and M. R. Buchmeiser, Dual catalysis with an N-heterocyclic carbene and a Lewis acid: thermally latent precatalyst for the polymerization of ε-caprolactam, J. Polym. Sci., 2020, 58, 3219–3226 CrossRef CAS.
- M. Zhou, X. Xiao, Z. Cong, Y. Wu, W. Zhang, P. Ma, S. Chen, H. Zhang, D. Zhang, D. Zhang, X. Luan, Y. Mai and R. Liu, Water-Insensitive Synthesis of Poly-β-Peptides with Defined Architecture, Angew. Chem., Int. Ed., 2020, 59, 7240–7244 CrossRef CAS PubMed.
- D. Zhang, C. Shi, Z. Cong, Q. Chen, Y. Bi, J. Zhang, K. Ma, S. Liu, J. Gu, M. Chen, Z. Lu, H. Zhang, J. Xie, X. Xiao, L. Liu, W. Jiang, N. Shao, S. Chen, M. Zhou, X. Shao, Y. Dai, M. Li, L. Zhang and R. Liu, Microbial Metabolite Inspired β-Peptide Polymers Displaying Potent and Selective Antifungal Activity, Adv. Sci., 2022, 9, 2104871 CrossRef CAS PubMed.
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal.: Sci. Technol., 2019, 9, 2380–2420 RSC.
- S. Park, E. Forró, H. Grewal, F. Fülöp and R. J. Kazlauskas, Molecular Basis for the Enantioselective Ring Opening of β-Lactams Catalyzed by Candida antarctica Lipase B, Adv. Synth. Catal., 2003, 345, 986–995 CrossRef CAS.
- L. W. Schwab, R. Kroon, A. J. Schonten and K. Loos, Enzyme-catalyzed ring-opening polymerization of unsubstituted β-lactam, Macromol. Rapid Commun., 2008, 29, 794–797 CrossRef CAS.
- I. Baum, B. Elsässer, L. W. Schwab, K. Loos and G. Fels, Atomistic Model for the Polyamide Formation from β-Lactam Catalyzed by Candida antarctica Lipase B, ACS Catal., 2011, 1, 323–336 CrossRef CAS.
- E. Stavila, G. O. R. Alberda Van Ekenstein, A. J. J. Woortman and K. Loos, Lipase-catalyzed ring-opening copolymerization of ε-caprolactone and β-lactam, Biomacromolecules, 2014, 15, 234–241 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, A. Basterretxea, D. Mantione, A. Etxeberria, C. Elizetxea, A. de la Calle, S. García-Arrieta, H. Sardon and D. Mecerreyes, Organic-acid mediated bulk polymerization of ε-caprolactam and its copolymerization with ε-caprolactone, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2394–2402 CrossRef CAS.
- M. Winnacker, J. Sag, A. Tischner and B. Rieger, Sustainable, Stereoregular, and Optically Active Polyamides via Cationic Polymerization of ε-Lactams Derived from the Terpene β-Pinene, Macromol. Rapid Commun., 2017, 38, 1600787 CrossRef PubMed.
- D. E. Kherroub, M. Belbachir and S. Lamouri, Cationic ring opening polymerization of ε-caprolactam by a montmorillonite clay catalyst, Bull. Chem. React. Eng. Catal., 2014, 9, 74–80 Search PubMed.
- S. Russo and E. Casazza, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, Netherlands, 2012, vol. 4, pp. 331–396 Search PubMed.
- R. Graf, G. Lohaus, K. Börner, E. Schmidt and H. Bestian, β-Lactams, Their Polymerization and Use as Raw Materials for Fibers, Angew. Chem., Int. Ed. Engl., 1962, 1, 481–488 CrossRef.
- R. Graf, G. Lohaus, K. Börner, E. Schmidt and H. Bestian, β-Lactame, Polymerisation und Verwendung als Faserrohstoffe, Angew. Chem., 1962, 74, 523–530 CrossRef CAS.
- B. P. Mowery, S. E. Lee, D. A. Kissounko, R. F. Epand, R. M. Epand, B. Weisblum, S. S. Stahl and S. H. Gellman, Mimicry of antimicrobial host-defense peptides by random copolymers, J. Am. Chem. Soc., 2007, 129, 15474–15476 CrossRef CAS PubMed.
- M. R. Lee, S. S. Stahl, S. H. Gellman and K. S. Masters, Nylon-3 copolymers that generate cell-adhesive surfaces identified by library screening, J. Am. Chem. Soc., 2009, 131, 16779–16789 CrossRef CAS PubMed.
- S. Chakraborty, R. Liu, Z. Hayouka, X. Chen, J. Ehrhardt, Q. Lu, E. Burke, Y. Yang, B. Weisblum, G. C. L. Wong, K. S. Masters and S. H. Gellman, Ternary Nylon-3 Copolymers as Host-Defense Peptide Mimics: Beyond Hydrophobic and Cationic Subunits, J. Am. Chem. Soc., 2014, 136, 14530–14535 CrossRef CAS PubMed.
- J. Zhang, M. J. Markiewicz, B. Weisblum, S. S. Stahl and S. H. Gellman, Functionally Diverse Nylon-3 Copolymers from Readily Accessible β-Lactams, ACS Macro Lett., 2012, 1, 714–717 CrossRef CAS PubMed.
- J. Zhang, S. H. Gellman and S. S. Stahl, Kinetics of Anionic Ring-Opening Polymerization of Variously Substituted β-Lactams: Homopolymerization and Copolymerization, Macromolecules, 2010, 43, 5618–5626 CrossRef CAS.
- E. L. Dane and M. W. Grinstaff, Poly-amido-saccharides: synthesis via anionic polymerization of a beta-lactam sugar monomer, J. Am. Chem. Soc., 2012, 134, 16255–16264 CrossRef CAS PubMed.
- E. L. Dane, S. L. Chin and M. W. Grinstaff, Synthetic Enantiopure Carbohydrate Polymers that are Highly Soluble in Water and Noncytotoxic, ACS Macro Lett., 2013, 2, 887–890 CrossRef CAS PubMed.
- R. Xiao, E. L. Dane, J. Zeng, C. J. McKnight and M. W. Grinstaff, Synthesis of Altrose Poly-amido-saccharides with β-N-(1 → 2)-D-amide Linkages: A Right-Handed Helical Conformation Engineered in at the Monomer Level, J. Am. Chem. Soc., 2017, 139, 14217–14223 CrossRef CAS PubMed.
- R. Xiao, J. Zeng and M. W. Grinstaff, Biologically Active Branched Polysaccharide Mimetics: Synthesis via Ring-Opening Polymerization of a Maltose-Based β-Lactam, ACS Macro Lett., 2018, 7, 772–777 CrossRef CAS PubMed.
- P. N. Stockmann, D. van Opdenbosch, A. Poethig, D. L. Pastoetter, M. Hoehenberger, S. Lessig, J. Raab, M. Woelbing, C. Falcke, M. Winnacker, C. Zollfrank, H. Strittmatter and V. Sieber, Biobased chiral semi-crystalline or amorphous high-performance polyamides and their scalable stereoselective synthesis, Nat. Commun., 2020, 11, 509 CrossRef CAS PubMed.
- R. Shamey and K. Sinha, A review of degradation of nylon 6. 6 as a result of exposure to environmental conditions, Rev. Prog. Color. Relat. Top., 2003, 33, 93–107 CAS.
- M. Shakiba, E. Rezvani Ghomi, F. Khosravi, S. Jouybar, A. Bigham, M. Zare, M. Abdouss, R. Moaref and S. Ramakrishna, Nylon—A material introduction and overview for biomedical applications, Polym. Adv. Technol., 2021, 32, 3368–3383 CrossRef CAS.
- D. J. Barillo, M. Pozza and M. Margaret-Brandt, A literature review of the military uses of silver-nylon dressings with emphasis on wartime operations, Burns, 2014, 40, S24–S29 CrossRef PubMed.
- Y. Wu, G. Xia, W. Zhang, K. Chen, Y. Bi, S. Liu, W. Zhang and R. Liu, Structural design and antimicrobial properties of polypeptides and saccharide-polypeptide conjugates, J. Mater. Chem. B, 2020, 8, 9173–9196 RSC.
- H. G. Boman and D. Hultmark, Cell-Free Immunity in Insects, Annu. Rev. Microbiol., 1987, 41, 103–126 CrossRef CAS PubMed.
- B. P. Mowery, A. H. Lindner, B. Weisblum, S. S. Stahl and S. H. Gellman, Structure–activity relationships among random nylon-3 copolymers that mimic antibacterial host-defense peptides, J. Am. Chem. Soc., 2009, 131, 9735–9745 CrossRef CAS PubMed.
- S. Chakraborty, R. Liu, Z. Hayouka, X. Chen, J. Ehrhardt, Q. Lu, E. Burke, Y. Yang, B. Weisblum, G. C. L. Wong, K. S. Masters and S. H. Gellman, Ternary nylon-3 copolymers as host-defense peptide mimics: Beyond hydrophobic and cationic subunits, J. Am. Chem. Soc., 2014, 136, 14530–14535 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. P. Falk, B. P. Mowery, A. J. Karlsson, B. Weisblum, S. P. Palecek, K. S. Masters and S. H. Gellman, Structure-activity relationships among antifungal nylon-3 polymers: Identification of materials active against drug-resistant strains of Candida albicans, J. Am. Chem. Soc., 2014, 136, 4333–4342 CrossRef CAS PubMed.
- R. Liu, X. Chen, Z. Hayouka, S. Chakraborty, S. P. Falk, B. Weisblum, K. S. Masters and S. H. Gellman, Nylon-3 polymers with selective antifungal activity, J. Am. Chem. Soc., 2013, 135, 5270–5273 CrossRef CAS PubMed.
- L. A. Rank, N. M. Walsh, R. Liu, F. Y. Lim, J. W. Bok, M. Huang, N. P. Keller, S. H. Gellman and C. M. Hull, A Cationic Polymer That Shows High Antifungal Activity against Diverse Human Pathogens, Antimicrob. Agents Chemother., 2017, 61, e00204–e00217 CrossRef CAS PubMed.
- Q. Zhang, P. Ma, J. Xie, S. Zhang, X. Xiao, Z. Qiao, N. Shao, M. Zhou, W. Zhang, C. Dai, Y. Qian, F. Qi and R. Liu, Host defense peptide mimicking poly-β-peptides with fast, potent and broad spectrum antibacterial activities, Biomater. Sci., 2019, 7, 2144–2151 RSC.
- S. Chen, X. Shao, X. Xiao, Y. Dai, Y. Wang, J. Xie, W. Jiang, Y. Sun, Z. Cong, Z. Qiao, H. Zhang, L. Liu, Q. Zhang, W. Zhang, L. Zheng, B. Yu, M. Chen, W. Cui, J. Fei and R. Liu, Host Defense Peptide Mimicking Peptide Polymer Exerting Fast, Broad Spectrum, and Potent Activities toward Clinically Isolated Multidrug-Resistant Bacteria, ACS Infect. Dis., 2020, 6, 479–488 CrossRef CAS PubMed.
- K. Zhang, C. Raju, W. Zhong, K. Pethe, A. Gründling and M. B. Chan-Park, Cationic Glycosylated Block Co-β-peptide Acts on the Cell Wall of Gram-positive Bacteria as Anti-biofilm Agents, ACS Appl. Bio Mater., 2021, 4, 3749–3761 CrossRef CAS PubMed.
- Z. Si, H. W. Lim, M. Y. F. Tay, Y. Du, L. Ruan, H. Qiu, R. Zamudio-Vazquez, S. Reghu, Y. Chen, W. S. Tiong, K. Marimuthu, P. P. De, O. T. Ng, Y. Zhu, Y. Gan, Y. R. Chi, H. Duan, G. C. Bazan, E. P. Greenberg, M. B. Chan-Park and K. Pethe, A Glycosylated Cationic Block Poly(β-peptide) Reverses Intrinsic Antibiotic Resistance in All ESKAPE Gram-negative Bacteria, Angew. Chem., 2020, 132, 6886–6893 CrossRef.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Lectins: tools for the molecular understanding of the glycocode, Org. Biomol. Chem., 2005, 3, 1593 RSC.
- R. J. Stockert, The asialoglycoprotein receptor: relationships between structure, function, and expression, Physiol. Rev., 1995, 75, 591–609 CrossRef CAS PubMed.
- H. Lis and N. Sharon, Lectins: Carbohydrate-Specific Proteins That Mediate Cellular Recognition, Chem. Rev., 1998, 98, 637–674 CrossRef CAS PubMed.
- K. H. Mortell, M. Gingras and L. L. Kiessling, Synthesis of Cell Agglutination Inhibitors by Aqueous Ring-Opening Metathesis Polymerization, J. Am. Chem. Soc., 1994, 116, 12053–12054 CrossRef CAS.
- R. Liu, K. Z. Vang, P. K. Kreeger, S. H. Gellman and K. S. Masters, Experimental and computational analysis of cellular interactions with nylon-3-bearing substrates, J. Biomed. Mater. Res., Part A, 2012, 100 A, 2750–2759 CrossRef PubMed.
- R. Liu, K. S. Masters and S. H. Gellman, Polymer chain length effects on fibroblast attachment on nylon-3-modified surfaces, Biomacromolecules, 2012, 13, 1100–1105 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. H. Gellman and K. S. Masters, Nylon-3 polymers that enable selective culture of endothelial cells, J. Am. Chem. Soc., 2013, 135, 16296–16299 CrossRef CAS PubMed.
- M. Winnacker, A. J. G. Beringer, T. F. Gronauer, H. H. Güngör, L. Reinschlüssel, B. Rieger and S. A. Sieber, Polyamide/PEG Blends as Biocompatible Biomaterials for the Convenient Regulation of Cell Adhesion and Growth, Macromol. Rapid Commun., 2019, 40, 1900091 CrossRef.
- J. Goerke, Pulmonary surfactant: functions and molecular composition, Biochim. Biophys. Acta, Mol. Basis Dis., 1998, 1408, 79–89 CrossRef CAS.
- J. A. Clements and M. E. Avery, Lung Surfactant and Neonatal Respiratory Distress Syndrome, Am. J. Respir. Crit. Care Med., 1998, 157, S59–S66 CrossRef CAS PubMed.
- T. P. Stevens and R. A. Sinkin, Surfactant replacement therapy, Chest, 2007, 131, 1577–1582 CrossRef PubMed.
- M. T. Dohm, B. P. Mowery, A. M. Czyzewski, S. S. Stahl, S. H. Gellman and A. E. Barron, Biophysical mimicry of lung surfactant protein B by random nylon-3 copolymers, J. Am. Chem. Soc., 2010, 132, 7957–7967 CrossRef CAS PubMed.
- L. Zhu and R. I. Mahato, Expert Opin. Drug Delivery, 2010, 7, 1209–1226 CrossRef CAS PubMed.
- V. Nadithe, R. Liu, B. A. Killinger, S. Movassaghian, N. H. Kim, A. B. Moszczynska, K. S. Masters, S. H. Gellman and O. M. Merkel, Screening nylon-3 polymers, a new class of cationic amphiphiles, for siRNA delivery, Mol. Pharmaceutics, 2015, 12, 362–374 CrossRef CAS PubMed.
- N. Hartl, F. Adams, G. Costabile, L. Isert, M. Döblinger, X. Xiao, R. Liu and O. M. Merkel, The Impact of Nylon-3 Copolymer Composition on the Efficiency of siRNA Delivery to Glioblastoma Cells, Nanomaterials, 2019, 9, 986 CrossRef CAS PubMed.
- C. Polge, A. U. Smith and A. S. Parkes, Revival of spermatozoa after vitrification and dehydration at low temperatures, Nature, 1949, 164, 666 CrossRef CAS PubMed.
- J. E. Lovelock and M. W. H. Bishop, Prevention of freezing damage to living cells by dimethyl sulphoxide, Nature, 1959, 183, 1394–1395 CrossRef CAS PubMed.
- A. L. DeVries and D. E. Wohlschlag, Freezing resistance in some Antarctic fishes, Science, 1969, 163, 1073–1075 CrossRef CAS PubMed.
- A. Ampaw, T. A. Charlton, J. G. Briard and R. N. Ben, Designing the next generation of cryoprotectants–From proteins to small molecules, Pept. Sci., 2019, 111, e24086 CrossRef CAS.
- T. Inada and S.-S. Lu, Inhibition of Recrystallization of Ice Grains by Adsorption of Poly(Vinyl Alcohol) onto Ice Surfaces, Cryst. Growth Des., 2003, 3, 747–752 CrossRef CAS.
- M. J. MacDonald, N. R. Cornejo and S. H. Gellman, Inhibition of Ice Recrystallization by Nylon-3 Polymers, ACS Macro Lett., 2017, 6, 695–699 CrossRef CAS PubMed.
- S. E. Stidham, S. L. Chin, E. L. Dane and M. W. Grinstaff, Carboxylated glucuronic poly-amido-saccharides as protein stabilizing agents, J. Am. Chem. Soc., 2014, 136, 9544–9547 CrossRef CAS PubMed.
- I. A. Sogias, A. C. Williams and V. V. Khutoryanskiy, Why is Chitosan Mucoadhesive?, Biomacromolecules, 2008, 9, 1837–1842 CrossRef CAS PubMed.
- A. S. Balijepalli, A. Hamoud and M. W. Grinstaff, Cationic poly-amido-saccharides: stereochemically-defined, enantiopure polymers from anionic ring-opening polymerization of an amino-sugar monomer, Polym. Chem., 2020, 11, 1926–1936 RSC.
- A. S. Balijepalli, R. C. Sabatelle, M. Chen, B. Suki and M. W. Grinstaff, A Synthetic Bioinspired Carbohydrate Polymer with Mucoadhesive Properties, Angew. Chem., Int. Ed., 2020, 59, 704–710 CrossRef CAS PubMed.
- K. Kaushansky, J. T. Prchal, L. J. Burns, M. A. Lichtman, M. Levi and D. C. Linch, Williams Hematology, 10e, McGraw-Hill Education, New York, NY, 2010 Search PubMed.
- C. W. McILWRAITH, Use of synovial fluid and serum biomarkers in equine bone and joint disease: a review, Equine Veterinary J., 2005, 37, 473–482 CrossRef CAS PubMed.
- V. Cagno, E. D. Tseligka, S. T. Jones and C. Tapparel, Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias?, Viruses, 2019, 11, 596 CrossRef CAS PubMed.
- R. L. Bick, E. P. Frenkel, J. Walenga, J. Fareed and D. A. Hoppensteadt, Hematol. Oncol. Clin. North Am., 2005, 19, 1–51 CrossRef PubMed.
- H. Liu, Z. Zhang and R. J. Linhardt, Lessons learned from the contamination of heparin, Nat. Prod. Rep., 2009, 26, 313–321 RSC.
- H. Bussey, J. L. Francis and T. H. Consensus Group, Heparin Overview and Issues, Pharmacotherapy, 2004, 24, 103S–107S CrossRef CAS PubMed.
- M. Varghese, F. Haque, W. Lu and M. W. Grinstaff, Synthesis and Characterization of Regioselectively Functionalized Mono-Sulfated and -Phosphorylated Anionic Poly-Amido-Saccharides, Biomacromolecules, 2022, 23, 2075–2088 CrossRef CAS PubMed.
- M. Varghese, R. S. Rokosh, C. A. Haller, S. L. Chin, J. Chen, E. Dai, R. Xiao, E. L. Chaikof and M. W. Grinstaff, Sulfated poly-amido-saccharides (sulPASs) are anticoagulants: In vitro and in vivo, Chem. Sci., 2021, 12, 12719–12725 RSC.
- R. M. Cywar, N. A. Rorrer, H. B. Mayes, A. K. Maurya, C. J. Tassone, G. T. Beckham and E. Y.-X. Chen, Redesigned Hybrid Nylons with Optical Clarity and Chemical Recyclability, J. Am. Chem. Soc., 2022, 144, 5366–5376 CrossRef CAS PubMed.
- K. Tachibana, K. Hashimoto, N. Tansho and H. Okawa, Chemical modification of chain end in nylon 4 and improvement of its thermal stability, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2495–2503 CrossRef CAS.
- X. Wu, P. Liu, X. Zhao, J. Wang, M. Teng and S. Gao, Critical effect of biodegradation on long-term microplastic weathering in sediment environments: a systematic review, J. Hazard. Mater., 2022, 437, 129287 CrossRef CAS PubMed.
- World Economic Forum and Ellen MacArthur Foundation and McKinsey & Company, The New Plastics Economy: Rethinking the Future of Plastics.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- E. MacArthur, Beyond plastic waste, Science, 2017, 358, 843 CrossRef PubMed.
- M. A. Hillmyer, The promise of plastics from plants, Science, 2017, 358, 868–870 CrossRef CAS PubMed.
- S. Negoro, Biodegradation of nylon oligomers, Appl. Microbiol. Biotechnol., 2000, 54, 461–466 CrossRef CAS PubMed.
- I. D. Prijambada, S. Negoro, T. Yomo and I. Urabe, Emergence of nylon oligomer degradation enzymes in Pseudomonas aeruginosa PAO through experimental evolution, Appl. Environ. Microbiol., 1995, 61, 2020–2022 CrossRef CAS PubMed.
- S. Negoro, K. Kato, K. Fujiyama and H. Okada, The nylon oligomer biodegradation system ofFlavobacterium andPseudomonas, Biodegradation, 1994, 5, 185–194 CrossRef CAS PubMed.
- T. Deguchi, M. Kakezawa and T. Nishida, Nylon biodegradation by lignin-degrading fungi, Appl. Environ. Microbiol., 1997, 63, 329–331 CrossRef CAS PubMed.
- J. Friedrich, P. Zalar, M. Mohorčič, U. Klun and A. Kržan, Ability of fungi to degrade synthetic polymer nylon-6, Chemosphere, 2007, 67, 2089–2095 CrossRef CAS PubMed.
- R. M. Cywar, N. A. Rorrer, H. B. Mayes, A. K. Maurya, C. J. Tassone, G. T. Beckham and E. Y.-X. Chen, Redesigned Hybrid Nylons with Optical Clarity and Chemical Recyclability, J. Am. Chem. Soc., 2022, 144, 5366–5376 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |