
Biofunctionality with a twist: the importance of molecular organisation, handedness and configuration in synthetic biomaterial design
Simone I. S.
Hendrikse
 *ab,
Rafael
Contreras-Montoya
c,
Amanda V.
Ellis
a,
Pall
Thordarson
b and
Jonathan W.
Steed
c
*ab,
Rafael
Contreras-Montoya
c,
Amanda V.
Ellis
a,
Pall
Thordarson
b and
Jonathan W.
Steed
c
aDepartment of Chemical Engineering, The University of Melbourne, Melbourne, VIC 3010, Australia. E-mail: shendriksela@unimelb.edu.au
bSchool of Chemistry, University of New South Wales (UNSW), Sydney, NSW 2052, Australia
cDepartment of Chemistry, Durham University, Durham DH1 3LE, UK
First published on 30th November 2021
Abstract
The building blocks of life – nucleotides, amino acids and saccharides – give rise to a large variety of components and make up the hierarchical structures found in Nature. Driven by chirality and non-covalent interactions, helical and highly organised structures are formed and the way in which they fold correlates with specific recognition and hence function. A great amount of effort is being put into mimicking these highly specialised biosystems as biomaterials for biomedical applications, ranging from drug discovery to regenerative medicine. However, as well as lacking the complexity found in Nature, their bio-activity is sometimes low and hierarchical ordering is missing or underdeveloped. Moreover, small differences in folding in natural biomolecules (e.g., caused by mutations) can have a catastrophic effect on the function they perform. In order to develop biomaterials that are more efficient in interacting with biomolecules, such as proteins, DNA and cells, we speculate that incorporating order and handedness into biomaterial design is necessary. In this review, we first focus on order and handedness found in Nature in peptides, nucleotides and saccharides, followed by selected examples of synthetic biomimetic systems based on these components that aim to capture some aspects of these ordered features. Computational simulations are very helpful in predicting atomic orientation and molecular organisation, and can provide invaluable information on how to further improve on biomaterial designs. In the last part of the review, a critical perspective is provided along with considerations that can be implemented in next-generation biomaterial designs.
 Simone I. S. Hendrikse | Simone I. S. Hendrikse received her MSc (2014) and PhD (2018) degrees in Biomedical Engineering at the Eindhoven University of Technology (TU/e), the Netherlands, supervised by Prof. P. Y. W. Dankers and Prof. E. W. Meijer. During her PhD she developed, investigated and tested supramolecular biomaterials on both their fundamental properties as well as their suitability for the culture of organoids. After undertaking short postdoctoral appointments at the TU/e and the University of Melbourne (UoM), Australia, in the groups of Profs. Amanda V. Ellis and Sally L. Gras, she started as a McKenzie Fellow at the UoM in 2019 working on artificial DNA hybrid materials. Her interests include biomimetic synthetic materials, supramolecular hydrogels, hybrid materials, and cell–material interactions. |
 Rafael Contreras-Montoya | Rafael Contreras-Montoya obtained both his MBiotech (2014) and PhD in Chemistry (2019) from the University of Granada (Spain), supervised by Prof. Luis Álvarez de Cienfuegos and Dr Juan J. Díaz-Mochón. He studied the application of peptide-based supramolecular gels to obtain composite materials, specifically composite protein crystals. He continued this research as postdoctoral researcher at Dr José A. Gavira's group at the Laboratory for Crystallographic Studies (Armilla, Granada, Spain). In 2020, he moved to the United Kingdom to take on a postdoctoral research associate position at Durham University with Prof. Jonathan W. Steed. Since 2021, he is working as a Marie Skłodowska-Curie postdoctoral fellow at the Cancer Research UK Edinburg Centre (University of Edinburgh, UK), where he is supervised by Prof. Asier Unciti-Broceta. |
 Amanda V. Ellis | Professor Amanda V. Ellis is the Head of Department of Chemical Engineering at the University of Melbourne, Australia. She graduated from the University of Technology, Sydney in 2003 and has undertaken postdoctoral appointments at Rensselaer Polytechnic Institute, New Mexico State University and Callaghan Innovations, New Zealand. She has been a Professor and Australian Research Future Fellow at Flinders University, South Australia. As an applied chemist/nanotechnologist her work focuses on the surface and interfacial chemistries for supramolecular design and device application. |
 Pall Thordarson | Pall Thordarson (Palli) was born on a farm in Vopnafjörður in the north-east of Iceland. He completed his PhD in Organic Chemistry at the University of Sydney, Australia in 2001. He joined the University of New South Wales School of Chemistry in 2007 where he is now a professor, a Chief Investigator at the ARC Centre of Excellence in Convergent Bio-Nano Science and Technology and a staff member of the Australian Centre for Nanomedicine. Palli's main research activities are within the broad areas of nanomedicine, supramolecular and systems chemistry, with recent focus on peptide and RNA interactions. |
 Jonathan W. Steed | Jonathan W. Steed was born in London, UK in 1969. He obtained his BSc (1990) and PhD (1993) degrees at University College London, working on organometallic chemistry. Between 1993 and 1995 he was a NATO postdoctoral fellow at the University of Alabama and University of Missouri. In 1995 he was appointed as a Lecturer at Kings College London and in 2004 he joined Durham University where he is currently Professor of Inorganic Chemistry. Steed is co-author of the book Supramolecular Chemistry (3rd Ed 2021) and over 350 research papers. He is the recipient of a number of awards including the 2021 Tilden Prize and is a Wolfson Research Merit Award Holder (2018). His interests are in solid state and pharmaceutical materials chemistry, supramolecular gels, crystallization and structural chemistry. |
Key learning points(1) Inter- and intra-molecular order in native biomolecules are essential to life on earth as molecular organisation, handedness and spatial distribution are crucial in biomolecule recognition and interaction.(2) Careful molecular design and preparation strategies are a foundation to creating ordered and helical bio-mimetic nanostructures. (3) Computational simulations provide invaluable insights into nanoscale molecular packing and can be used to screen molecular building blocks to predict desired physicochemical properties a priori, including intermolecular cohesion and secondary structure. (4) Including structural order into biomaterial design is demonstrated to improve cell interactions in both covalent and supramolecular systems and is anticipated to be even more important in achieving improved reciprocal interactions with living systems and hence better performance in biomedical applications. (5) In addition to molecular order, higher complexity and other design parameters, including mechanical properties, need to be matched according to the cell type and application in mind, paying attention to the dynamic nature and traction forces exerted by cells. |
Introduction
After the discovery of handedness and chirality in natural molecules in the 19th century, by Louis Pasteur, it became clear that molecular handedness is an important aspect in life on earth.1 Molecules with the same chemical composition but that are non-superimposable mirror images of each other, i.e., enantiomers, have the same physical and chemical properties except when interacting with polarised light or other chiral molecules. This phenomenon came to prominence in the 1960s, when the drug thalidomide (sold as brand names that include Softenon and Contergan), was prescribed to pregnant women to reduce morning sickness, and resulted in the birth of thousands of children with severe birth defects. The drug contained a mixture of both (R)- and (S)-thalidomide (i.e., racemic), of which the (R)-enantiomer is effective, whereas the (S)-isomer is highly toxic. Since this tragedy, federal guidelines around drugs have tightened and the significance of chirality in drug design and development has become increasingly important.2Why is Nature so selective towards chiral molecules? The building blocks of life – amino acids, nucleotides and saccharides – are synthesised as homochiral molecules with mostly one handedness.3 The origin of this phenomenon is still under debate, and can have an evolutionary and/or physical origin.4 Nevertheless, homochirality in biomolecules is necessary to translate the chirality at the molecular level into three-dimensional functional folds, as a racemic mixture will give disordered and hence inactive structures. However, the opposite enantiomers can be present as well, although are typically correlated with diseases. Normally, peptides and proteins are synthesised with L-amino acids, however, upon aging, an inversion of configuration from L- to D-amino acids sometimes arises.5 Especially in long-living or permanent proteins, D-aspartic acid accumulates in many tissues, including dentine, bone, brain, heart and liver. This amino acid inversion can cause conformational changes, a loss of protein functioning, or trigger protein aggregation. Not surprisingly, protein isomerisation is enhanced in neurodegenerative diseases, such as Alzheimer's disease.6
From the amino acid sequence (i.e., primary structure) that encodes a particular protein, the formation of secondary structures in solution is triggered by non-covalent interactions, mainly driven by hydrophobic interactions and hydrogen bonds. Regions adopt a right-handed α-helical structure, displaying approximately 3.6 amino acids per turn, in which a single intermolecular hydrogen bond per amino acid pair connects a carbonyl oxygen with an amide hydrogen four amino acids downstream, or extended beta-sheets where two hydrogen bonds per pair run between adjacent strands’ carbonyl oxygens and amide hydrogens in a parallel or anti-parallel direction (Fig. 1).7 Although single amino acids have intrinsic propensities to favour α-helical or β-sheet structures, the sequence order, solvation and environment dictate the overall fold.8 According to a simple model neglecting hydrogen bonding, the fold is a result of maximising entropy, in which the α-helix has overlapping excluded volume from adjacent turns (with an optimal pitch to radius ratio c* = 2.5122), while an increasing helix radius locally unwinds the helix forming β-sheets.9 The next hierarchical layer is the formation of so-called tertiary and quaternary structures that include double- or triple-helices, and the formation of fibrils of laterally aligned helices, or alternatively, globular structures composed of multiple subunits, respectively. These layers of hierarchical ordering are a result of chiral amplification of the monomeric units through a three-dimensional (3D) structure fold by non-covalent interactions that is favoured by a reduction in free energy. Although partially or wrongly folded structures (i.e., kinetically trapped) can arise, typically the thermodynamically favoured structure is the one that is functional.
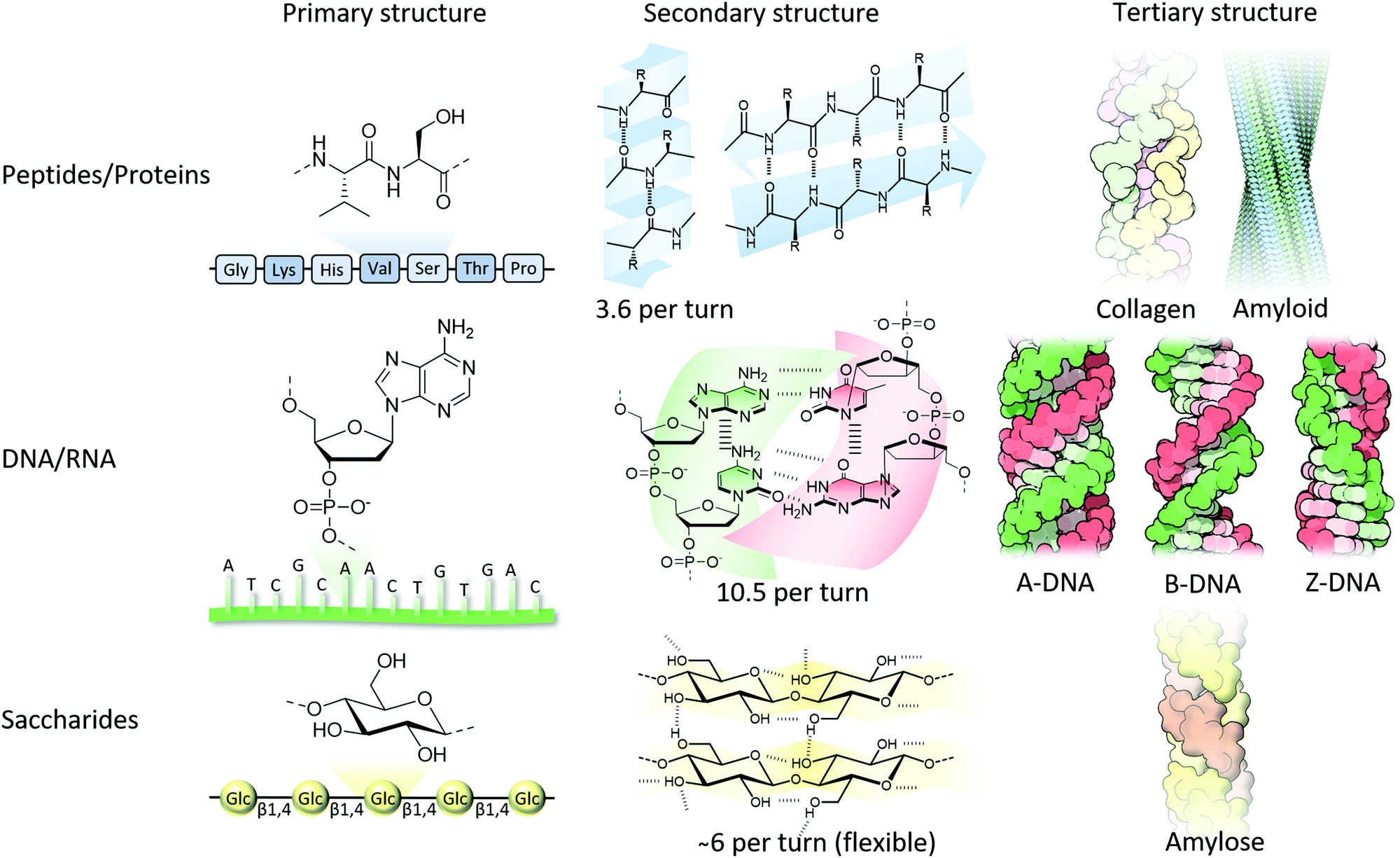 | ||
| Fig. 1 From molecular structure to hierarchical formation of functional structures composed of peptide, nucleotide and saccharide building blocks. Collagen, amyloid and DNA tertiary structures are adapted from ref. 16 with permission from the RCSB PDB, copyright 2015. | ||
As well as right-handed α-helical structures in proteins, DNA also typically adopts a right-handed, double helical structure driven by complementary Watson–Crick nucleobase pairing (adenine (A) binds to thymine (T), and cytosine (C) to guanine (G)) between adjacent single stranded DNA (ssDNA) strands as well as π–π interactions between lateral nucleobases (purines and pyrimidine) in the same strand (Fig. 1). Less commonly observed is Hoogsteen base-pairing (i.e., one nucleobase is in the syn rather than anti configuration). The most common form of DNA is the B-form, having about 10 nucleotides and a pitch of 3.4 nm per turn, whereas in the event of DNA–RNA binding and RNA–RNA duplex formation, a tighter double helical structure (i.e., A-form) is formed composed of about 11 nucleotides and a pitch of 2.8 nm per turn. When DNA is copied, and upon binding of RNA polymerase, DNA flips into a Z-DNA configuration having a left-handed twist instead of the usual right-handed rotation.10 This helix inversion was accidentally discovered in 1972 when DNA was placed in an aqueous solution containing a high salt concentration,11 although the molecular structure was only resolved in 1979.12 Since this time there has been considerable debate on how this helix inversion takes place. Several models have been proposed,13 and with the help of molecular dynamics simulations this includes local stretching and transient Watson–Crick hydrogen-bond disruption followed by an asynchronised base flipping.14 Although Z-DNA has been found in actively transcribed regions of the genome, and has important roles in, amongst others, gene expression, it also has been associated with human diseases, including cancers, immunogenic conditions and neurodegenerative disorders.15 The Z-form is more disordered and stretched compared to the A- and B-form, containing about 12 nucleotides and a pitch of 4.5 nm per turn.
Although proteins are typically synthesised containing L-amino acids, saccharides are synthesised as D-analogues. Polysaccharides comprise of monosaccharide building blocks attached to one another using α- or β-glycosidic linkages and can be a linear or branched polymer. Due to a large array of hydroxyl groups (and sometimes together with negatively charged carboxylates and/or sulfates as found in proteoglycans), many non-covalent interactions between chains and surrounding water are formed, followed by the consequent formation of higher-order hierarchical structures. A well-known example is the formation of thick cellulose fibrils due to lateral alignment of the single cellulose fibres that are immobilised by a multivalent array of hydrogen bonding between the hydroxyl side groups (Fig. 1).16 Although far less investigated due to their flexible nature, many polysaccharides have been shown to form helical structures similar to their protein and nucleotide relatives. The microscopic fibrils that are formed in cellulose have an intrinsic right-handed twist,17 whereas amylose, containing the same glucose repeats, although with α(1–4) rather than β(1–4) glycosidic bonds, forms left-handed single-helical chains.18 Another example can be found in amylopectin, which is a large, highly branched molecule, that has chains intertwined and tightly packed in double-helical chains providing crystalline regions separated by amorphous regions (branching points) consequently giving a highly organised and hierarchical structure in starch granules.19 Interestingly, the macroscopically visible chirality in plant growth (i.e., over 90% of plant vines are right-handed) is dictated by alignment in cellulose in the plant cell walls guided by intracellular microtubules.20
The peculiar development of these well organised structures from the early Earth has resulted in the parallel evolution of proteins and enzymes that recognise and bind these biomolecules by their unique chiral signature. Natural enzymes, like proteinases, are intrinsically asymmetric and therefore stereoselectively bind L-amino acid containing proteins in contrast to their D-enantiomers.21 Although enzymes exist that bind both enantiomers, e.g., racemases, they operate with a chiral preference. As such, in drug discovery, it is crucial to develop enantiomerically pure compounds since their enantiomers can be non-active or even toxic. Learning from this field, in biomaterials science, the presence of a chiral preference in the cellular environment cannot be ignored as the predominant aim is to selectively interact with cellular components for a wide variety of biomedical applications, including for regenerative medicine, combatting antimicrobial resistance and tissue engineering. Therefore, upon the introduction of exogeneous biomaterials to cells, the organisation and hence spatial distribution of functional groups should be considered to allow better reciprocal interactions with living systems.
In this tutorial review we first highlight selected examples of the first steps in synthetic bio-inspired ordered structures that contain peptides, nucleotides and/or saccharides. Design considerations will be highlighted as well as external techniques to induce order. Experimental techniques can elucidate a great deal of information about morphology, order and configuration, although complementary computational simulations can resolve the nanoscopic packing in more detail. They can also be used in the rational design of biomaterials with desired secondary structure. The review concludes with a perspective and considerations that could be employed for the design of ordered biomaterials.
Synthetic bio-inspired ordered structures
Understanding and mimicking the development and evolution of chiral proteins and enzymes in Nature requires a sophisticated approach to the development of biomaterials. Looking at the progress made in drug discovery over the last decades shows a paradigm shift from small molecules towards more complex structures with a distinct ‘secondary’ structure. Although, small molecules have demonstrated important applications as, for example, molecular inhibitors or activators in biomedical applications due to their ability to easy fit in the binding pocket of proteins,22 a large proportion of protein–protein interactions occur via helical elements spreading over a large surface area.23 The inhibition of these protein–protein interactions is considered very important, since many diseases, including pathogen–host interactions, arise from abnormal protein–protein interactions.24 This indicates that the secondary structure, or more specifically functional group orientation, is crucial in biomolecular interactions.25,26 Several strategies have been used to mimic the α-helical structure involved in facilitating protein–protein binding, which includes hydrocarbon stapling, using β-peptides, and lactam bridges among others.25,27 Hence a helical conformation can be vitricated upon installing unfavourable steric interactions or strain in its planar conformation, rendering non-helical and even achiral aggregates into energetically favoured helices (i.e., containing a stereogenic axis rather than stereogenic centre).28 Lessons learned from drug discovery can be translated to biomaterial design, to more effectively interact with extra- and intra-cellular components, both in healthy and diseased cells as well as microorganisms. The sections below highlight seminal contributions that have been made in synthetic bio-inspired pre-organised biomaterial design that include peptides, nucleotides and/or saccharides.Peptide-based assemblies
Large proteins present in the extracellular matrix (ECM) between cells are key in tissue function, as they provide structural integrity, resist compressive forces as well as facilitate cell–cell and cell–matrix communication, collectively influencing cell fate. Understanding cellular behaviour, and to be able to manipulate and control its fate, is the central aim in biomaterials engineering. Common practise is to use full length ECM proteins (e.g., laminin, collagen and vitronectin) as biomaterials (e.g., as coating) for the culture of (stem)cells as they contain all the proteins’ bio-functionalities along with its synergistic binding sites to enable adhesion-dependent cell culture, to regulate cell plasticity and/or to support self-organisation into microtissue (e.g., organoids). However, these extracted, or recombinant, ECM proteins are heterogenous and are difficult to spatially functionalise or might even interfere with other (downstream) signalling pathways. As such, synthetic peptide versions of these ECM proteins have been explored for their ability to mimic active sites of the full protein. These peptide mimics include, amongst others, the amino acid sequences Arg-Gly-Asp (RGD, fibronectin), Ile-Lys-Val-Ala-Val (IKVAV, laminin) and Gly-Phe-Hyp-Gly-Glu-Arg (GFOGER, collagen, Hyp and O denote hydroxyproline).29The RGD sequence is the most widely studied cellular adhesive peptide. It is present in fibronectin, as well as in other ECM proteins.30 The key interaction site for cell adhesion in fibronectin is a highly exposed RGD-containing loop that has a hairpin-like conformation. The glycine residue renders the key part of this loop flexible which is why scrambling this sequence (e.g., RDG or RAD) diminishes cellular adhesion. In synthetic fibronectin mimics it has been shown that displaying the RGD sequence in a loop31 or circular form,32 compared to the linear form, influences integrin binding selectivity. Moreover, the sequence Pro-His-Ser-Arg-Asn (PHSRN) present in the neighbouring (i.e., 9th) domain of full length fibronectin has been shown to play a synergistic effect, of which the distance between the two sequences is crucial, as competitive binding to integrin receptors inhibits cellular adhesion.33 This demonstrates the importance of amino acid orientation into space for specific cellular binding. This discovery represents a huge leap forward in biomaterials design, transforming inert materials into bioactive biomaterials by incorporating RGD sequences.34
Hydrogels are the most used materials to mimic the ECM as they are composed of a 3D network of polymers, chemically crosslinked and/or physically entangled, retaining typically up to 99% water in a similar way to living systems. The polymers generate a mechanically supportive matrix whereas their porous nature allows the diffusion of molecules, such as nutrients and oxygen. Hydrogels based on the non-covalent self-assembly of peptides are particularly interesting for mimicking the ECM as they are highly biocompatible and biodegradable.35 Their self-assembly is typically driven by hydrophobic effects and hydrogen bond formation (and e.g., β-sheet formation), similar as to in amyloidosis in vivo, which is the aggregation of denatured proteins, commonly seen in diseases like Alzheimer's.
In seminal work by the Gazit36 and Ulijn37 groups, 9-fluorenylmethyloxycarbonyl (Fmoc)-functionalised dipeptides (Fmoc-Phe-Phe) were developed that form fibres by π–π interactions between lateral phenylalanine residues, and the formation of antiparallel β-sheets held together by hydrophobic interactions and hydrogen bonding.38 Due to its crystalline character, fibres bundle together to form thick fibrils. The obtained pristine fibrous structures can be enriched with RGD sequences by attaching the RGD sequence to the end of the self-assembled peptide, and by subsequent co-assembly with Fmoc-Phe-Phe, to display RGD sequences on the fibre periphery.39 Hydrogels formed from the self-assembled fibrils have been shown to support anchorage dependent fibroblast growth and spreading in 3D. This work resulted in the foundation of the company BiogelX™ which commercialises hydrogels based on Fmoc derivatives of small peptide sequences found in the ECM as scaffolds for cell culture. Various bio-functionalised peptide amphiphiles with short ECM mimetic sequences have been developed by the group of Stupp.40 Amongst others, peptide amphiphiles containing the neurite-promoting laminin sequence IKVAV were developed and shown to support the differentiation of 3D encapsulated murine neural progenitor cells into neurons. Whereas in covalent polymers the RGD spacing should be tightly regulated to allow strong integrin binding, in supramolecular polymers ECM-mimicking monomers can migrate within the polymer backbone to allow optimal binding. As such, the epitope concentration can be significantly reduced to only 5 mol%. Since the short ECM mimetic peptides, that are installed at the end of supramolecular monomers, might be too close to the polymer backbone, and hence hinder epitope availability and consequently binding affinity, it has been shown that increasing the linker length with 5 glycines improved cell spreading.41 Although binding availability is improved upon increasing linker length, the gained flexibility can negatively influence binding affinity as well.
Not only has ‘bio-functionalisation’ been shown to influence cellular fate of peptide-based assemblies with ECM mimics, the helical direction of the fibres has also been shown to be important.42 Supramolecular polymers based on C2-symmetric L-phenylalanine derivatives self-assemble into left- or right-handed helices depending on the number of methylene units incorporated, also known as the ‘odd–even’ effect (Fig. 2A).43 Investigating the effect of chirality on 2D cellular adhesion and proliferation of mouse embryonic fibroblasts (NIH 3T3) and endothelial cells (Hy926) revealed that both cellular adhesion and proliferation is significantly higher on left-handed films than on right-handed surfaces. Moreover, left-handed helices obtained from L-phenylalanine derivatives displayed improved 3D cell culture and osteogenesis in mesenchymal stem cells (MSC) differentiation, whereas D-phenylalanine derivatives, that form right handed helices, favour adipogenesis in MSC differentiation, higher cytotoxicity towards cancer cells and higher protein absorption.44 Collectively, these results demonstrate (i) the high stereospecificity of cell–ECM interactions which goes beyond discerning molecular asymmetry, and (ii) the relevance of supramolecular chirality in the design of ECM mimicking biomaterials.
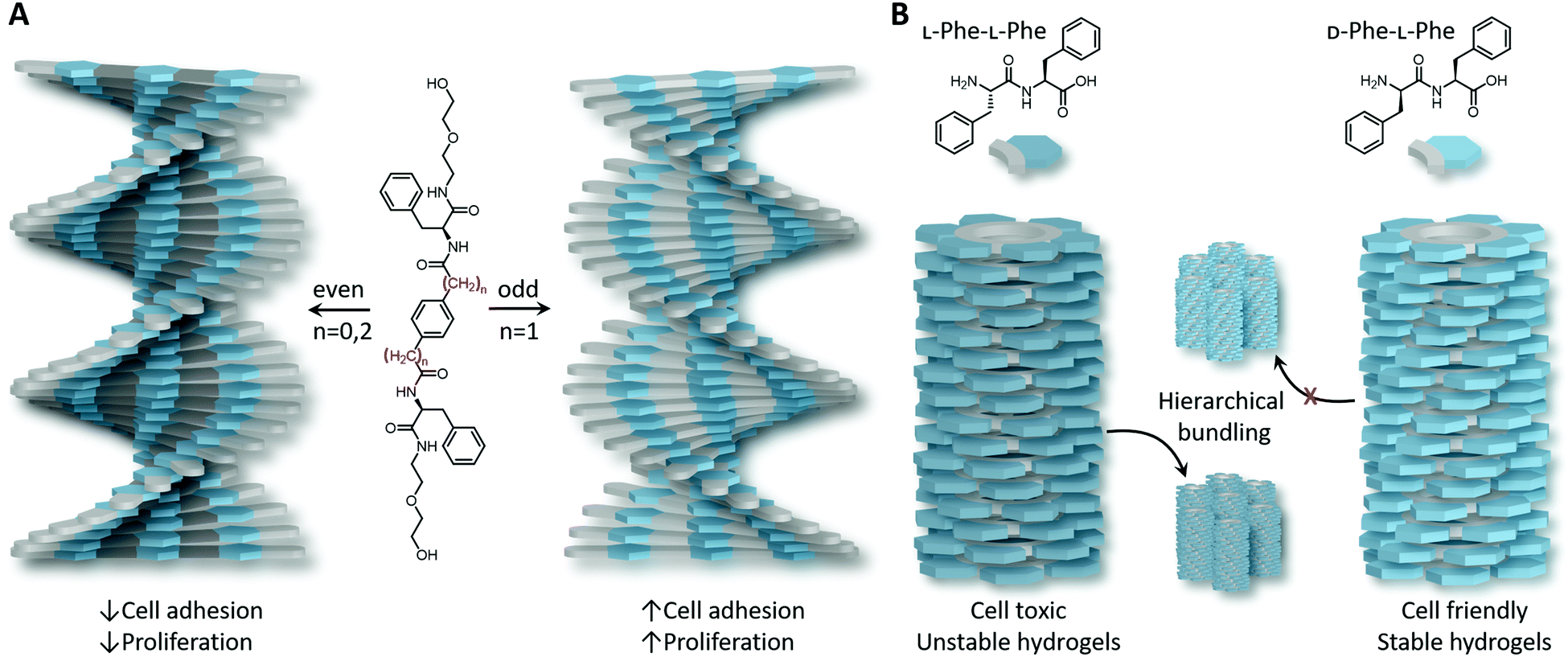 | ||
| Fig. 2 Synthetic self-assembled peptides. (A) Supramolecular polymers based on C2-symmetric L-phenylalanine self-assemble into left- or right-handed helices depending on the number of carbons present between the core and L-phenylalanine (i.e., odd–even effect). (B) L-Phe–L-Phe self-assembles into hierarchical and unstable hydrogels, whereas changing only one stereocentre, i.e., to D-Phe–L-Phe, resulted in self-supporting hydrogels in which cells could be cultured. Slight differences in molecular design and stereochemistry have a profound impact on the helicity of supramolecular polymers and consequently on how cells interact. | ||
Due to the inherent stereoselectivity present in Nature, cells typically favour binding to L-amino acids over their D-isomers. However, exogenous administrated peptides and proteins composed of solely L-amino acids are rapidly cleared in vivo by proteases, which limits their use in a wide variety of biomedical applications. In order to increase their life-time, D-amino acids have been investigated and have already found great potential as they are resistant to degradation, have low immunogenicity and can improve therapeutic potency of peptide-based drugs by five orders of magnitude.45,46 However, care must be taken when considering amino acid isomerisation as it can completely alter side chain orientation and therefore affect secondary structure formation and hence protein binding.47,48 Even given this, the potential of using heterochiral monomers, containing both L- and D-amino acids, has still been explored.
Supramolecular self-assembly is tightly dependent on a well-balanced hydrophobic-to-hydrophilic balance along with minimised steric hindrances, and thus optimal packing, hence order, in the laterally stacked monomers. As a result, atomic spatial arrangement and backbone flexibility are key parameters. It has been shown by Marchesan et al.49 that the handedness of heterochiral peptide-based assemblies is dictated by the nanoscopic packing of the monomers and can be controlled by tuning the molecular design of monomers. Whereas homochiral tripeptides did not form hydrogels, the inclusion of a D-amino acid in the middle of two phenylalanine units was shown to form rigid hydrogels by hierarchical bundling. The presence of linear amino acid side chains was shown to result in efficient packing of the units into hierarchically formed thick fibrils, whereas branched side chains formed thinner, although denser and more interconnected networks rather than bundles. Extending this research to Phe-Phe dipeptides showed that by changing the chirality of just one stereocentre (i.e., from L-Phe–L-Phe to D-Phe–L-Phe), hierarchical bundling was suppressed and transparent, self-supportive and stable hydrogels where cells could be cultured were generated as compared to cell-toxic and unstable hydrogels formed by L-Phe–L-Phe (Fig. 2B).50 The effect of amino acid sequence on nanoscale packing was also highlighted by the group of Stupp on peptide amphiphile self-assembly.40 For example, upon substituting alanine by more hydrophobic valines (Ala6 to Val4Ala2), first helix unwinding was observed followed by a higher degree of twisting than the homoalanine sequence (i.e., shorter pitch).51 Moreover, the same group revealed that a higher lipid membrane interaction was obtained for L-peptide amphiphiles compared to their D-enantiomers, together with a contradictory reduce in cell viability.52 This unexpected reduction in cell viability might be attributed to slow membrane destabilisation and/or peptide amphiphile engulfment within cells due to cell's traction forces able to disrupt the weak cohesion between monomers, both facilitated by electrostatic interactions between negatively charged cellular membranes and positively charged end-functionalised peptide amphiphiles. Consequently, the hydrophobicity, charge, steric hindrance and internal cohesion (and hence internal dynamics) between monomers in supramolecular polymers can affect cell viability and question the biocompatibility of the synthetic system. Altogether, this demonstrates the importance in molecular design of the monomeric building blocks and the subsequent nanoscale packing in the supramolecular polymers they form upon self-assembly.
The significance of introducing internal order in biomaterials has not only been demonstrated for supramolecular polymers, it has also been shown of importance in covalent polymeric systems, in which the backbone is fixated rather than transient. In a study by Hammond and coworkers,53 cell-adhesive peptides, that have both the RGD and PHSRN sequence (i.e. PHSRN-K-RGD), were grafted to 8-arm poly(ethylene glycol) and two polypeptides with different secondary structures crosslinked in a hydrogel network. Poly(γ-propargyl-L-glutamate), that forms a rigid α-helical conformation, was shown to display superior attachment and spreading of 2D cultured induced-pluripotent stem cell-derived endothelial cells as compared to poly(γ-propargyl-D,L-glutamate), containing 50% D- and 50% L-glutamate that forms a random-coil secondary structure, and the 8-arm poly(ethylene glycol) control. Strong attachment and spreading in α-helical containing hydrogels were also observed in the absence of adhesive peptides, also confirming that an ordered secondary structure, with potentially the influence of local nanoscale stiffness, can enhance cell responses.
Taken together, including an ECM mimetic peptide in biomaterial design is a good start to enable cell adhesion, however, having control over conformation, helicity and order can push cell interactions to the next level and can improve favourable reciprocal biomolecule interactions.
Nucleotide-based assemblies
Nucleotide-based biomaterials are promising materials for a wide variety of biomedical applications, including for gene therapy, clinical diagnostics and DNA purification amongst others.54 The sequence specificity of the Watson–Crick base pairing augments these biomaterials with high programmability and spatial control. This unique property has been widely utilised to create DNA materials with controlled shapes and sizes by a technique known as DNA origami.55 Typically, a long ssDNA is scaffolded into a particular design in a computer-aided program (e.g., cadnano and ATHENA56) and neighbouring strands are stapled together by short complementary sequences that cross-over in the scaffolded structure holding the desired 2D or 3D shape together. Key to the formation of these structures are the programmable unique sequences, cross-overs that are designed by taking advantage of the periodic turn of dsDNA (∼10.5 nucleotides per turn), and a slow temperature cooling ramp to allow correct folding.Next to the famous double helix, different secondary and tertiary structures can also form in Nature, that include hairpins, triple helices, i-motifs and G-quadruplexes. Not surprisingly, aptamers, which are short oligonucleotide sequences that can fold into small 3D structures, have shown great affinity and selectivity towards a wide variety of targets, including proteins, peptides, carbohydrates and small molecules, and comprise the ability to distinguish between enantiomers in a similar way to antibodies.57 Despite their great promise, extremely small aptamers that could potentially target small molecules and toxins, are more challenging to obtain, due to a reduced ability to fold in a unique 3-dimensional fold, and hence a decrease in specificity and stability.
Synthetic biomaterials have been designed that include either only nucleobases, nucleosides (also (deoxy)ribose) or full nucleotides, either naturally occurring or modified. Due to the highly negative charge present on the nucleotide backbone, nucleotide conjugates, including block copolymers and inorganic conjugates, typically form spherical micelles as a result of electrostatic repulsion.58,59 Though, 1D fibrous structures can be obtained when the hydrophilic-to-hydrophobic balance is restored or by the help of cosolvents.60,61 In another approach to circumvent the charged backbone, nucleobases (i.e., without (deoxy)riboses and negatively charged phosphates) have been attached to polymers, including peptide backbones. Peptide nucleic acids (PNAs) have been intensely investigated and have shown great promise in a wide range of applications, due to their increased stability and ability to hybridise to natural DNA and RNA by strand invasion.62 This invasion is very effective as hybridisation of PNA–DNA is stronger than DNA–DNA and PNA–PNA hybridisation partly due to its neutral character, reducing repulsive interactions that are present during DNA–DNA duplex formation. PNAs have shown to be able to bind to hairpin, quadruplex and RNA targets as well, of which the binding is typically stabilised by overhanging nucleotides. However, PNAs in their unbound state have the tendency to collapse into globular structures, probably due to a hydrophobic collapse of the nucleobases. Therefore, backbone modifications have been included to pre-organise the polymer by using α- or γ-modifications with respect to the amine conjugated nucleobase unit (Fig. 3A). A right-handed helical twist is obtained upon γ-modifications with L-amino acids, whereas left-handed helices can be obtained using D-amino acids. Upon increasing the number of L-serine γ-modifications resulted in an increase in DNA and RNA affinity, observed as an increase in melting temperature (Fig. 3A).63
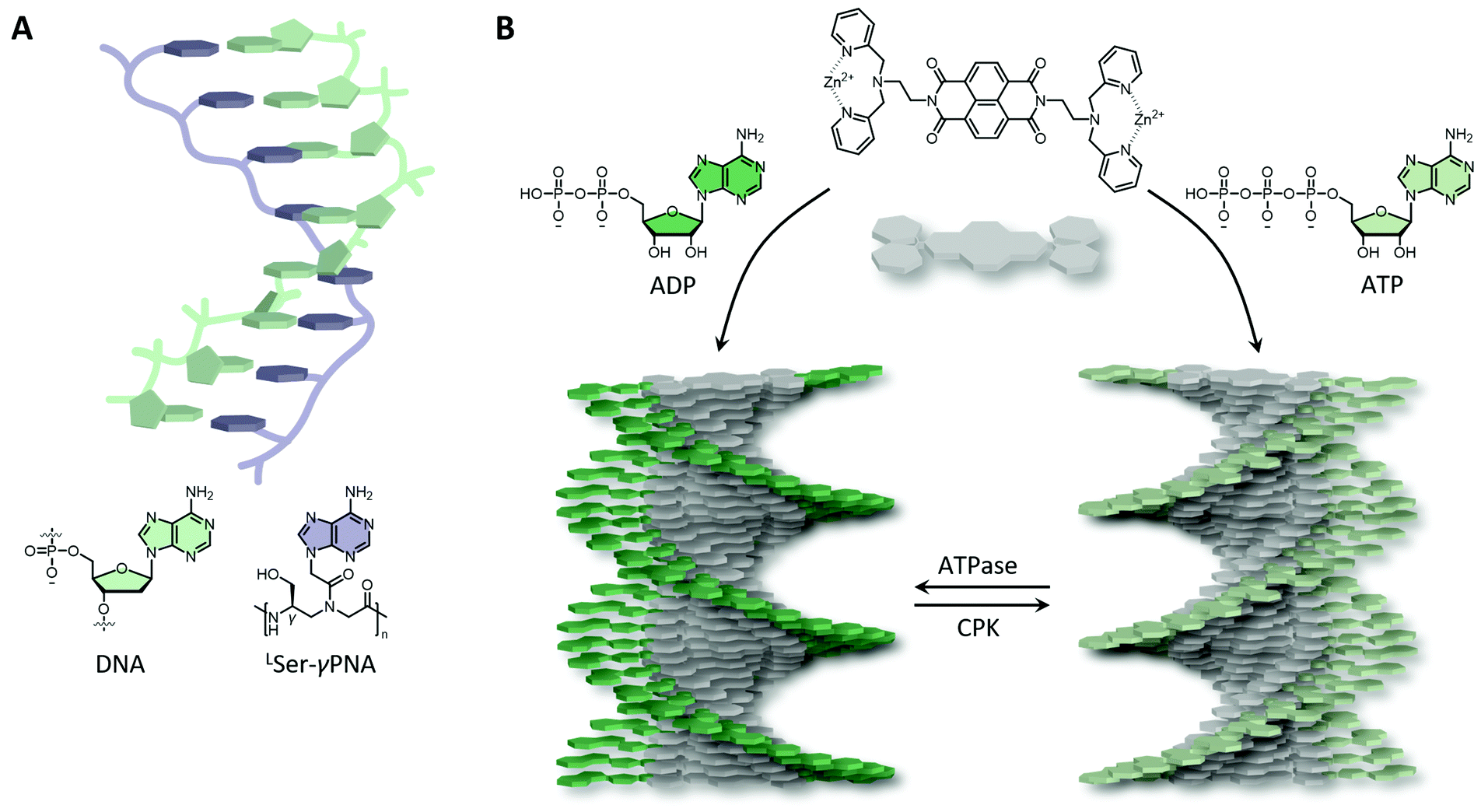 | ||
| Fig. 3 Synthetic DNA mimics. (A) L-Serine γ-backbone modified peptide nucleic acids (LSer-γPNA) have been shown to stabilise DNA and able to bind DNA with high affinity. (B) ATP and ADP initiate the ordered self-assembly of naphthalene diimide with zinc(II) dipicolylethylenediamine receptors into right- or left-handed helical assemblies, respectively. The helicity could be inverted by dephosphorylation (ATPase) or recovery of ATP by creatine phosphokinase (CPK). | ||
Other peptide–nucleotide hybrid materials have been developed that have shown remarkable properties. Shape shifting nanofibers were obtained from DNA block copolymers by Park and coworkers60 when either the polarity of the co-solvent was changed, or upon protease treatment. Nanofibres were initially obtained due to π–π interactions between phenylalanine side chains and disrupted hydrogen bonding, whereas strengthening these hydrogen bonds upon increasing solvent polarity or backbone cleavage resulted in a transition to nanosheets due to lateral hydrogen bonding alignment.
Short ssDNA and RNA have been used as templates to fabricate chiral supramolecular polymers of small organic molecules, including naphthalene, anthracene and porphyrins amongst others, into both left- and right-handed nanofibers driven by non-covalent interactions.64 Control, and the ability to switch between helicities, can be achieved by altering ionic strength, pH, and cooling rate. Likewise, the binding of adenosine–triphosphate (ATP) facilitated by cationic ammonium or guanidinium, and metal coordinated organic groups, such as dipicolyl ethylene diamine, have also been shown to induce supramolecular chirality. Remarkably, the helicity of such a system could be tuned by ATP dephosphorylation, yielding right-handed helices upon ATP binding, and left-handed helices upon adenosine diphosphate (ADP) binding (Fig. 3B). Interestingly, such materials can be augmented with out-of-equilibrium and autonomous behaviour, and hence life-like properties similar to actin filaments present in Nature, upon the addition of enzymes.65 ATP binding initiates supramolecular self-assembly, whereas enzymes (e.g., ATPases) trigger depolymerisation due to ATP hydrolysis. By recovering ATP using e.g., creatine phosphokinase (CPK), the system reassembles resulting in a living and transient supramolecular system.
Saccharide-based assemblies
Many biomaterials developed today contain naturally derived polysaccharides, such as alginate and chitosan, because of their close resemblance to the ECM, high biocompatibility, bioactivity and biodegradation, and high abundancy, allowing readily extraction from renewable resources. Moreover, their gelling properties are well understood, e.g., upon the addition of salts, such as calcium for alginate or phosphates for chitosan, in an aqueous solution, a crosslinked network is obtained of which the mechanical properties can be tuned by playing with the weight percentage and the salt composition. Although significantly less studied, synthetic saccharides might be better alternatives as naturally derived polysaccharides suffer from batch-to-batch variations in composition, have a large polydispersity, and have unpredictable degradation patterns. However, several challenges remain that include (i) a significant long chain of repeating saccharides is necessary to support strong binding (i.e., multivalency effect) as single saccharide units contain only weak attractive interactions for non-covalent interactions; (ii) the synthesis typically requires multiple steps and is relatively low yielding; (iii) the secondary structure is influenced both by the environment (i.e., solvents, salts and metals) as well as functional group composition (e.g., sulphates, phosphates) and configuration (e.g., α- versus β-glycosidic linkage); and (iv) finding the delicate balance between water solubility and self-assembly is challenging, as the saccharides can be either too hydrophilic (e.g., due to sulphates, phosphates as is often seen in proteoglycans) or insoluble due to crystallinity (e.g., cellulose). In Nature, this delicate balance together with its hierarchical structure is anticipated to be under enzymatic control, as the introduction of different saccharide moieties in a regular covalent pattern can create disorder (i.e., a kink and hence termination of conformational order). As such, most synthetic systems rely on simple mono- or di-saccharides and/or make advantage of an amphiphilic design similar to self-assembled peptides (i.e., hydrophobic followed by a hydrophilic block).66Compared to the non-covalent interactions that drive peptide and amphiphile assembly – mainly hydrophobic effects and hydrogen bonding – the intrinsically weak intermolecular carbohydrate–carbohydrate interactions are typically overlooked, despite that these are of utmost importance in key cellular processes, including in cell and pathogen interactions.67 These weak interactions allow carbohydrates to screen (cell) surfaces, and upon recognition, cooperativity is initiated to tighten the binding by the multivalent effect. Multiple saccharides line up and bind to their host to reach a synergistic binding that is significantly stronger than the sum of the individual components, collectively giving the multivalent carbohydrate its biological function. Next to inter- (and also intra-) molecular interactions, water plays an important aspect in carbohydrate ordering as well, yielding remarkable gelling properties that can effectively resist tensile forces (e.g., as found in cartilage). Although investigating chiral amplification and hierarchical order in synthetic saccharide-based systems in aqueous solutions is still in its infancy, seminal contributions have been reached in this field.
As highlighted above, a well-balanced hydrophobic-to-hydrophilic ratio is key in driving the supramolecular assembly of biomaterials. For carbohydrate-functionalised materials, in particular, this is quite challenging due to the increased disorder in the system as a consequence of increased free rotation of glycosidic bonds, the different conformations that carbohydrates can adapt, their non-planar character, and the ability for only weak non-covalent donor and acceptor interactions. Similar to bulk microphase separation,68 which is dependent on the volume fraction of the blocks, linker length (i.e., degree of polymerisation) and degree of block incompatibility (i.e., Flory–Huggins parameter), micelles are formed when the carbohydrate headgroup is too hydrophilic and bulky, whereas vesicles can be formed when the linker length increases, and fibrous structures when all three parameters are in balance, as was illustrated by pioneering work by Lee and coworkers.69
Carbohydrate-functionalised birefringent hydrogels with micron scale fibre alignment were developed by Hudalla et al.70 The incorporation of the monosaccharide N-acetylglucosamine at the end of a self-assembling peptide building block drives the anisotropic fibre alignment by carbohydrate–carbohydrate interactions and solvent depletion in a molecular crowded environment (Fig. 4A). Carefully balanced carbohydrate interactions have been shown to be crucial as diglycosylated disaccharides and unglycosylated hydroxyl analogues were unable to form aligned nanofibers. The densely packed aligned glycosylated peptide nanofibers were shown to be resistant against non-specific proteins, mammalian cells and bacteria binding, yet endowing specific interactions to lectins. This highlights the importance of molecular packing and functional group orientation in supramolecular assembly in facilitating biomolecule interactions. Chiral amplification of monosaccharides was also obtained upon the incorporation of glucose and mannose to achiral supramolecular amphiphiles, a result of a combination of hydrophobic effects of the core and intermolecular carbohydrate–carbohydrate interactions.71 Fibrous structures with opposite helicity were obtained due to α- versus β-glycosylation (Fig. 4B). More hydrophilic and bulky disaccharide-functionalised amphiphiles were unable to form ordered fibres, however, upon the co-assembly with glucose-functionalised monomers, chiral amplification and hence carbohydrate–carbohydrate interactions were restored.
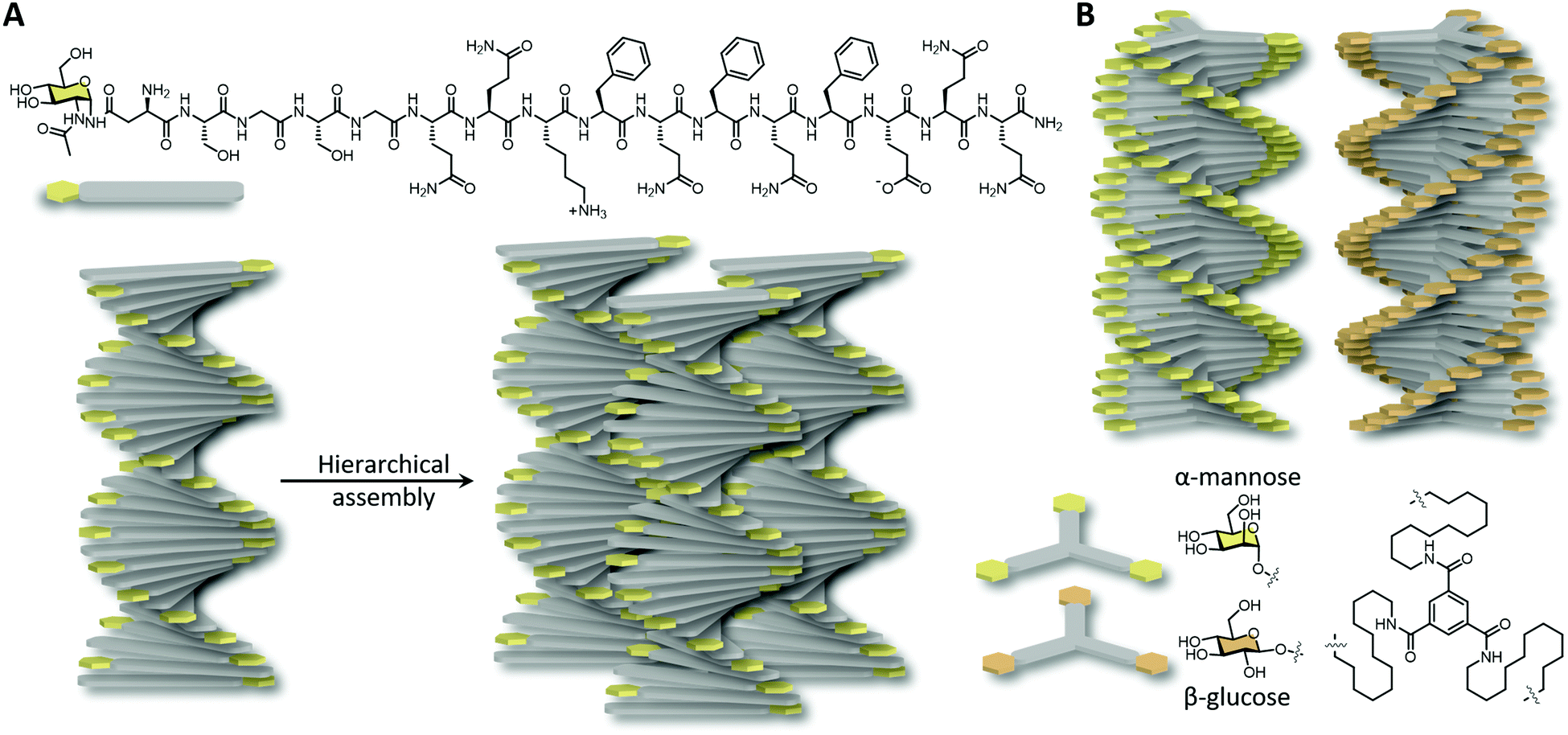 | ||
| Fig. 4 Synthetic saccharide functionalised self-assembled nanofibers. (A) N-Acetylglucosamine functionalised peptides assembled into ordered nanofibers and subsequently into hierarchical bundles by solvent depletion. (B) Mirror image nanofibers obtained by substituting α-mannose with β-glucose at the end of C12 functionalised benzene-1,3,5-tricarboxamide. | ||
Computational simulations
The nanoscopic packing of the atoms within bio-inspired polymers is in direct relation to the macroscopic structures they form, including helical fibres. In order to study the formation of these structures, typically, a combination of several techniques is performed, that include spectroscopic (e.g., UV-vis, circular dichroism, fluorescence), microscopic (atomic force microscopy, cryogenic-transmission electron microscopy) and scattering (e.g. dynamic light scattering, small angle X-ray scattering) techniques, to arrive at a molecular picture of these polymers. Although these techniques provide information concerning polymer morphology and links to (dis)assembly pathways, a more comprehensive understanding of the nanoscopic molecular packing can be obtained when this data is complemented with molecular simulations.In molecular dynamics (MD) simulations, the polymeric system is built in a confined simulation box using empirical force fields, and the molecular motion of the included molecules is followed over time in either a bottom-up or top-down approach.72 In the bottom-up approach, the formation of a self-assembled polymer and hence the formation of non-covalent bonds starting from the monomeric building blocks is probed, whereas in a top-down approach, an already assembled polymer, with an idealised pre-determined structure, is simulated over time to probe different structural configurations. Although multiple packing motifs underpinning these alternative configurations might arise, these simulations can provide the potential energy or order of the aggregate, which determine the overall stability of the aggregate. By simulating the formation of the non-covalent interactions over time, key drivers of the growth process can be determined (e.g., hydrophobic or π–π interactions), and self-assembled pathways formed, including kinetic intermediates. Importantly, a fundamental understanding of the secondary structure can be obtained.
Information concerning the nanoscale molecular packing is usually obtained using classic all-atom MD simulations. This technique is, however, limited by the use of 106 particles and 100 ns simulation time, which does not adequately simulate the formation of bigger and more complex aggregates, and the formation of long-range order. To overcome this limitation, coarse-grain simulations, that simplifies the molecules into adhesive beads (i.e., 4 heavy atoms including their hydrogen atoms are represented as 1 bead), and enhanced sampling techniques, to sample the kinetic pathways involved, including intermediates, in the formation of the final assembled polymer, can be employed. Also, these techniques have their disadvantages, although collectively provide invaluable information into self-assembly and secondary structure formation. By comparing experimental findings with the performed computational simulations a complete fundamental understanding spanning over several length scales can be obtained. In a study by Marrink et al., MD simulations and semiempirical quantum chemical calculations to predict UV-vis, circular dichroism and infrared absorption spectra were performed, next to experimentally acquired electron microscopy and spectroscopy, to elucidate the self-replication mechanism of peptide macrocycles.73 This complementary investigation showed good agreement between calculated and experimentally acquired spectra, and revealed a cooperative self-assembly process driven by alignment between aromatic headgroups and β-sheet forming peptides giving favourable hydrophobic interactions and dipole–dipole interactions, with the inner core being collapsed to expel unfavourable water interactions (Fig. 5A).
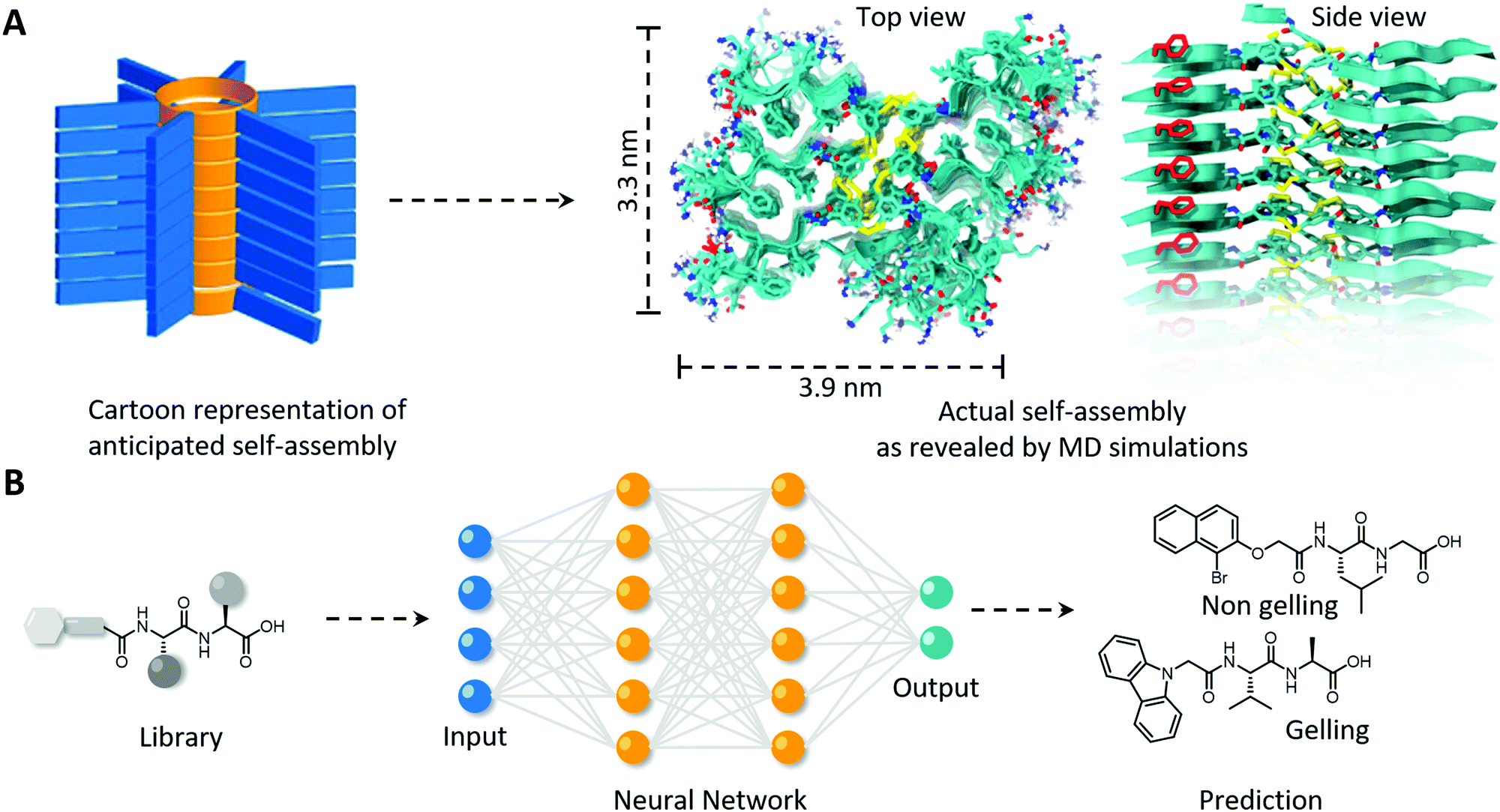 | ||
| Fig. 5 Using computational simulations to (A) elucidate the nanoscale packing in molecular self-assembly of peptide macrocycles and (B) the in silico prediction of the gelation of a virtual library of mono- and di-peptides utilising a neural network. (A) Modified and reproduced with permission from ref. 73. Copyright 2017 American Chemical Society. | ||
Computational simulations not only complement experimental findings to elucidate the full molecular picture of self-assembled polymers, they can also be used to predict supramolecular self-assembly propensity, gel formation and secondary structure formation, and hence allows the in silico selection and development of bio-inspired polymers with desired morphology and order. Several machine learning methods to generate quantitative structure–property relations were explored by Berry and coworkers to predict the gelation of an extensive library of mono- and di-peptides (Fig. 5B).74 They revealed that among the machine learning methods utilised, neural networks performed the best in successfully predicting gelation propensity, followed by support vector machines and random forests, which were experimentally confirmed with test compounds. The models also revealed that the five physicochemical properties; (i) number of rings, (ii) predicted molecular aqueous solubility, (iii) polar surface area, (iv) A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP (i.e., octanol–water partition coefficient) and (v) number of rotatable bonds, dictated the prediction of gel formation. Predicting the secondary structure a priori is more challenging as initial configuration is usually derived from experimentally derived databases, and simulations usually only display small rearrangements. Initial steps in this space have been made, including in revealing unexpecting α-helical intermediates in β-sheet forming peptides, and predicting melting curve trends of short DNA–polymer conjugates and collagen-like peptides, however, needs to be further explored for more complex synthetic biopolymers and non-natural (e.g., amino acid and nucleobase analogues) building blocks.72,75
logP (i.e., octanol–water partition coefficient) and (v) number of rotatable bonds, dictated the prediction of gel formation. Predicting the secondary structure a priori is more challenging as initial configuration is usually derived from experimentally derived databases, and simulations usually only display small rearrangements. Initial steps in this space have been made, including in revealing unexpecting α-helical intermediates in β-sheet forming peptides, and predicting melting curve trends of short DNA–polymer conjugates and collagen-like peptides, however, needs to be further explored for more complex synthetic biopolymers and non-natural (e.g., amino acid and nucleobase analogues) building blocks.72,75
Future directions and opportunities
By including order and helicity in synthetic bioinspired materials the field of biomaterials will be revolutionised, with a subsequent move closer to more real-life and ‘sophisticated’ materials and applications, all of which are anticipated to be able to interact with living systems more accurately, and in a reciprocal manner, than their static and simple relatives. However, by simply including order in biomaterials, biocompatibility is not guaranteed. In fact, there are more parameters that should be taken into consideration and in order to support reciprocal interactions across several length scales, and hence the ability to support more complex functions, the creation of higher order hierarchical structures is required.Other design parameters to be taken into consideration
Although this review so far has focussed on the importance of chirality and order in biomaterials to achieve improved biomolecule and cell interactions, the mechanical and physical properties of the biomaterial have been shown to dictate the cell viability and differentiation over time as well, as a consequence of cells’ intrinsic response to remodel its environment. Despite that these parameters are highly dependent on the cell type and biomaterial used, some general trends have been observed. Next to the well-known importance of the elastic modulus76 and rate of degradation77 that influences stem cell fate and cell growth, the ability to dissipate cell-induced stress (stress relaxation) and the ability to allow dynamic rearrangements in biomaterial design has shown crucial importance too. In this regard, the Mooney lab78 showed seminal contributions by utilising non-degradable alginate hydrogels in which the stress relaxation was tuned (time at which stress is reduced to half its value, τ1/2 from 1 hour to 1 minute) while keeping the polymer concentration and elastic modulus constant. In fast relaxing hydrogels (τ1/2 of about 1 minute), a significant enhancement in cell spreading, proliferation and osteogenic differentiation of 3D cultured murine mesenchymal stem cells was mediated by integrin adhesion, RGD clustering and cytoskeletal (i.e. actomyosin) contractility, indicating faster ability of cells to mechanically remodel the matrix. Although the interface between engineered matrices and cells has shown to require dynamic remodelling, a study by Burdick and coworkers79 showed that the deposition of human mesenchymal stem cells’ secreted proteins, i.e. nascent proteins, can dictate stem cell fate as well as early as within a few hours after cell encapsulation, while masking RGD-presentation of covalent degradable and dynamic non-degradable hydrogels.Given that cells induce traction forces to remodel the surrounding matrix raises a challenge for supramolecular hydrogels build from solely small molecular building blocks; since these building blocks are held together by weak non-covalent interactions, these monomers can be released from their scaffold and internalised (e.g., by endocytosis) or can even actively penetrate in the cell membrane (typically when the amphiphile contains a positive net charge as highlighted before). As such it is important to investigate and control the nanoscale packing, and hence dynamic property, of these monomeric building blocks within their fibrous support to match the cell's remodelling and traction forces. The influence of the internal dynamics of synthetic supramolecular biomaterials was investigated by Dankers et al.80 while employing ureidopyrimidinone functionalised supramolecular hydrogels. In excessively dynamic hydrogels, presentation of RGD ligands to cells was shown to be ineffective, whereas cell spreading and migration was significantly enhanced upon tuning the dynamic profile and RGD availability of the supramolecular system. As such, cell compatibility and the ability to control cell fate is dependent on several parameters that need to be carefully optimised to match the desired cell type specific response for a given application.
Towards higher order hierarchical self-assembly
Nature comprises of many high-order hierarchical structures that are self-assembled at different length scales. The obtained highly ordered structures give rise to a plethora of functionalities, ranging from structural properties to signal transduction. Discrepancies in their molecular orientation is associated with diseases. However, recreating these hierarchical structures in a laboratory is challenging, due to a lack in ability to control the nanoscale to macroscale organisation and the absence of chaperon proteins.81 Next to self-assembly by careful design considerations already highlighted previously, several external techniques have been successfully applied to induce molecular alignment and nanofibril arrangements. These include, amongst others, artificial spinning, 3D printing, layer-by-layer-assembly, and external field induced crystallisation.81–83 Moreover, multi-component materials can be fabricated by mixing in distinct bio-functionalised polymers that can either co-assemble (i.e., intercalate within the same fibres) or self-sort (i.e., non-woven networks). Although increased complexity can bring in multi-functionality and synergistic effects, care has to be taken considering that the different components can interact or even interfere with the self-assembly properties of the individual components.Towards hybrid ordered materials
Chiral amplification in supramolecular polymers has been widely investigated, first in organic media and more recently in aqueous solutions. Although their inherent dynamic properties augment the obtained biomaterials with adaptive and life-like properties, a lower stability and toughness as compared to covalent polymers might limit certain biomedical applications, and their delicate molecular design to obtain the desired self-assembly still remains challenging for more complex materials. As such, enriching covalent polymers with hydrogen bonding moieties has been shown to give rise to interesting materials with strain-stiffening84 and super tough properties.85,86 More recently, hybrid polymers based on supramolecular peptide amphiphiles crosslinked by methacrylate head-groups, mixed with poly(N-isopropylacrylamide) and augmented with light-switchable spiropyran, were shown to support macroscopic mechanical actuation.87 Both macroscopic unidirectionality and rotational movement was achieved by controlling and localising the light exposure. It is envisioned that more exciting materials in aqueous solutions can be obtained by combining covalent and supramolecular interactions, which can be further enriched with an intrinsic order for improved communication with living systems.Reducing experimental load
Current biomaterial designs are still heavily dependent on trial-and-error approaches to match the desired chemical-, physical- and biological properties. As highlighted in this review, computational simulations bear great potential to predict material properties a priori, including the propensity for gelation. In addition, current rapid progress in machine learning and artificial intelligence would further progress this field to obtain more accurate and more complex predictions, including in intermolecular cohesion and secondary structure. On the other hand, automated systems allow fast high-throughput experimental screening with smaller volumes and hence materials required, although still require a fair amount of iterations to be screened and can still be laborious and time consuming, and the influence of biomaterial order and helicity is typically overlooked. As such, design of experiment (DoE)88 approaches can be implemented to reduce the number of conditions by half, a quarter or even more (i.e., fractional factorial of which the resolution depends on the number of parameters to be explored), still being able to subtract similar information as doing a full factorial screening. Yet, it provides additional information as it allows analysing data in a statistically relevant manner, where key and negligible contributors (free of cofounding interactions) are defined, as well as multi-factor interactions, that collectively help streamlining the optimisation of the experimental design and outcomes. Hence, this approach can be used to screen and optimise several parameters in parallel to arrive at a desired biomaterial design for a selected biomedical application.Conclusions
To conclude, this tutorial review has provided insights into how molecular organisation, spatial atomic configuration and helicity can be used as a tool to develop biomaterials, in addition to the important design parameters already widely explored, such as mechanical properties, degradation and stress relaxation, with more ‘life-like’ properties as compared to materials composed of randomly dispersed polymers. It is envisioned that including order and spatial functional group arrangement improves ligand accessibility and reciprocal interactions with proteins, cells and tissue and aids in achieving better performance in a diverse range of biomedical applications.Realising such great control in self-assembly, and hence the helicity in synthetic materials, is already achieved by (i) molecular design in which the stereochemistry and hydrophilic-to-hydrophobic balance is crucial; (ii) including steric constraints to favour helicity, such as lactam bridges and employing β-peptides; or (iii) utilising external forces and techniques to induce molecular alignment, e.g., 3D printing and layer-by-layer assembly. Next steps towards improved, and hence next-generation, biomaterial designs should consider (i) incorporating several bio-active components, e.g., different ECM-derived peptides that can work synergistically; (ii) increasing complexity using multicomponent single- and multiple polymeric systems; (iii) inducing hierarchical order to achieve reciprocal interactions that span across several length scales (i.e., from nanoscopic interactions at the atom scale to synergistic whole organ level at the micron scale, Fig. 6). In addition to these considerations that aim to build more complex and sophisticated biomaterials with internal order, the amphiphilic nature of the supramolecular polymers, including molecular packing and internal positive charges, on one hand, and the traction forces exerted by cells due to mechanotransduction on the other hand, also need to be regarded as these can induce cell apoptosis, due to e.g. internalisation, membrane distortion or loss of support.
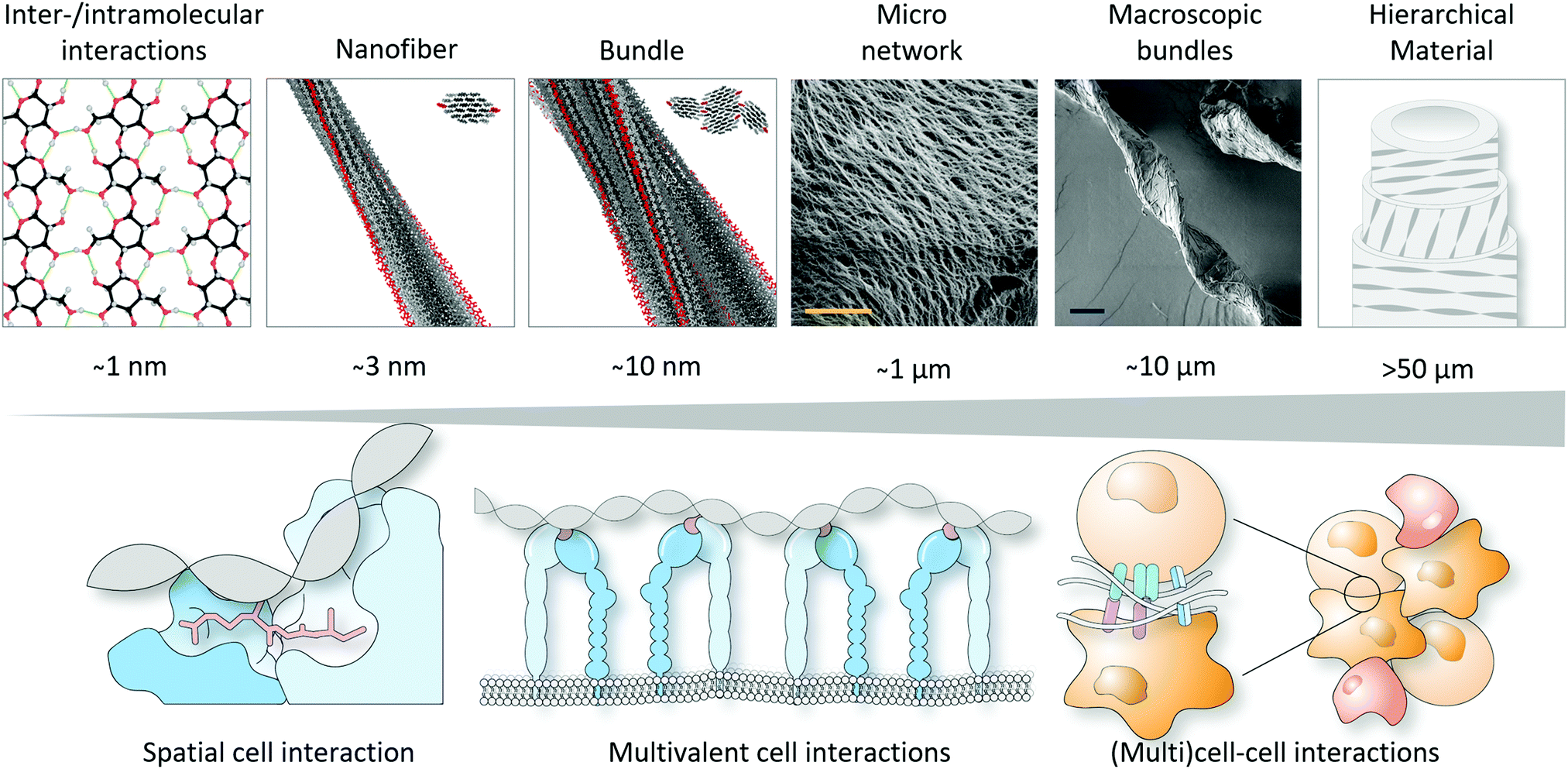 | ||
| Fig. 6 Towards hierarchical materials that can match cell interactions across several length scales. Part of this figure is modified and reproduced from ref. 17. Copyright 2019 Springer Nature. | ||
Since there is no single material that suits all cell types and hence multiple (biomedical) applications, every type of biomaterial typically needs to be optimised according to the desired application by undergoing several iterations to match the desired biological responses. This screening of several parameters is unfortunately still stuck in a laborious trial-and-error process. However, rapid progress in computational simulations and artificial intelligence, potentially in conjunction with implementing statistical DoE approaches, bears great potential to reduce this laborious hurdle in a cost-effective manner and in a multi-disciplinary setting. As such, combining knowledge across several disciplines, from drug discovery to computer science, is key to create more sophisticated biomaterials, and realising improved biomedical applications for the near future.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by funding from the Australian Research Council (ARC), Discovery Projects (DP190100055, SISH and AVE) and (DP190101892, PT), the McKenzie Fellowship from the University of Melbourne (SISH), and the Engineering and Physical Sciences Research Council (EP/S035877/1, RC-M and JWS).Notes and references
- S. F. Mason, Nature, 1984, 311, 19–23 CrossRef CAS PubMed.
- W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760–770 CrossRef CAS PubMed.
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2010, 2, a002147 Search PubMed.
- A. Guijarro and M. Yus, The Origin of Chirality in the Molecules of Life: A Revision from Awareness to the Current Theories and Perspectives of this Unsolved Problem, Royal Society of Chemistry, Cambridge, 2008 Search PubMed.
- S. Ritz-Timme and M. J. Collins, Ageing Res. Rev., 2002, 1, 43–59 CrossRef CAS PubMed.
- G. Li, K. DeLaney and L. Li, Nat. Commun., 2019, 10, 5038 CrossRef PubMed.
- J. M. Berg, L. Stryer and J. L. Tymoczko, Biochemistry, 5th edition, W. H. Freeman, New York, 2002 Search PubMed.
- K. Fujiwara, H. Toda and M. Ikeguchi, BMC Struct. Biol., 2012, 12, 18 CrossRef CAS PubMed.
- Y. Snir and R. D. Kamien, Science, 2005, 307, 1067 CrossRef CAS PubMed.
- G. Wang and K. M. Vasquez, Front. Biosci., 2007, 12, 4424–4438 CrossRef CAS PubMed.
- F. M. Pohl and T. M. Jovin, J. Mol. Biol., 1972, 67, 375–396 CrossRef CAS PubMed.
- A. H. J. Wang, G. J. Quigley, F. J. Kolpak, J. L. Crawford, J. H. van Boom, G. van der Marel and A. Rich, Nature, 1979, 282, 680–686 CrossRef CAS PubMed.
- M. A. Fuertes, V. Cepeda, C. Alonso and J. M. Pérez, Chem. Rev., 2006, 106, 2045–2064 CrossRef CAS PubMed.
- M. A. Kastenholz, T. U. Schwartz and P. H. Hünenberger, Biophys. J., 2006, 91, 2976–2990 CrossRef CAS PubMed.
- S. Ravichandran, V. K. Subramani and K. K. Kim, Biophys. Rev., 2019, 11, 383–387 CrossRef CAS PubMed.
- D. S. Goodsell, S. Dutta, C. Zardecki, M. Voigt, H. M. Berman and S. K. Burley, PLoS Biol., 2015, 13, e1002140 CrossRef PubMed . Molecule of the Month illustrations adapted from DOI: 10.2210/rcsb_pdb/mom_2001_11, DOI: 10.2210/rcsb_pdb/mom_2015_9 and DOI: 10.2210/rcsb_pdb/mom_2000_4.
- A. Paajanen, S. Ceccherini, T. Maloney and J. A. Ketoja, Cellulose, 2019, 26, 5877–5892 CrossRef CAS.
- A. Buléon, P. Colonna, V. Planchot and S. Ball, Int. J. Biol. Macromol., 1998, 23, 85–112 CrossRef.
- G. Eggleston, J. W. Finley and J. M. deMan, in Principles of Food Chemistry, ed. J. M. deMan, J. W. Finley, W. J. Hurst and C. Y. Lee, Springer International Publishing, Cham, 2018 Search PubMed.
- M. Nakamura and T. Hashimoto, Symmetry, 2020, 12, 2056 CrossRef CAS.
- V. S. Lamzin, Z. Dauter and K. S. Wilson, Curr. Opin. Struct. Biol., 1995, 5, 830–836 CrossRef CAS PubMed.
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301–317 CrossRef CAS PubMed.
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS.
- D. P. Ryan and J. M. Matthews, Curr. Opin. Struct. Biol., 2005, 15, 441–446 CrossRef CAS.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- J. Taechalertpaisarn, R.-L. Lyu, M. Arancillo, C.-M. Lin, L. M. Perez, T. R. Ioerger and K. Burgess, Org. Biomol. Chem., 2019, 17, 3267–3274 RSC.
- Z. A. Wang, X. Z. Ding, C.-L. Tian and J.-S. Zheng, RSC Adv., 2016, 6, 61599–61609 RSC.
- M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2016, 45, 1542–1556 RSC.
- I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS.
- M. D. Pierschbacher and E. Ruoslahti, Nature, 1984, 309, 30–33 CrossRef CAS.
- S. E. Ochsenhirt, E. Kokkoli, J. B. McCarthy and M. Tirrell, Biomaterials, 2006, 27, 3863–3874 CrossRef CAS PubMed.
- T. G. Kapp, F. Rechenmacher, S. Neubauer, O. V. Maltsev, E. A. Cavalcanti-Adam, R. Zarka, U. Reuning, J. Notni, H.-J. Wester, C. Mas-Moruno, J. Spatz, B. Geiger and H. Kessler, Sci. Rep., 2017, 7, 39805 CrossRef CAS PubMed.
- Y. Feng and M. Mrksich, Biochemistry, 2004, 43, 15811–15821 CrossRef CAS PubMed.
- S. L. Bellis, Biomaterials, 2011, 32, 4205–4210 CrossRef CAS.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935–3953 RSC.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- S. Sur, F. Tantakitti, J. B. Matson and S. I. Stupp, Biomater. Sci., 2015, 3, 520–532 RSC.
- X. Dou, N. Mehwish, C. Zhao, J. Liu, C. Xing and C. Feng, Acc. Chem. Res., 2020, 53, 852–862 CrossRef CAS PubMed.
- J. Liu, F. Yuan, X. Ma, D.-I. Y. Auphedeous, C. Zhao, C. Liu, C. Shen and C. Feng, Angew. Chem., Int. Ed., 2018, 57, 6475–6479 CrossRef CAS PubMed.
- Y. Wei, S. Jiang, M. Si, X. Zhang, J. Liu, Z. Wang, C. Cao, J. Huang, H. Huang, L. Chen, S. Wang, C. Feng, X. Deng and L. Jiang, Adv. Mater., 2019, 31, 1900582 CrossRef PubMed.
- D. M. Veine, H. Yao, D. R. Stafford, K. S. Fay and D. L. Livant, Clin. Exp. Metastasis, 2014, 31, 379–393 CrossRef CAS PubMed.
- B. D. Welch, A. P. VanDemark, A. Heroux, C. P. Hill and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16828–16833 CrossRef CAS.
- M. Garton, S. Nim, T. A. Stone, K. E. Wang, C. M. Deber and P. M. Kim, Proc. Nat. Acad. Sci. U. S. A., 2018, 115, 1505–1510 CrossRef CAS.
- C. Li, M. Pazgier, J. Li, C. Li, M. Liu, G. Zou, Z. Li, J. Chen, S. G. Tarasov, W.-Y. Lu and W. Lu, J. Biol. Chem., 2010, 285, 19572–19581 CrossRef CAS PubMed.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CAS.
- S. Kralj, O. Bellotto, E. Parisi, A. M. Garcia, D. Iglesias, S. Semeraro, C. Deganutti, P. D’Andrea, A. V. Vargiu, S. Geremia, R. De Zorzi and S. Marchesan, ACS Nano, 2020, 14, 16951–16961 CrossRef CAS PubMed.
- M. H. Sangji, H. Sai, S. M. Chin, S. R. Lee, I. R. Sasselli, L. C. Palmer and S. I. Stupp, Nano Lett., 2021, 21, 6146–6155 CrossRef CAS.
- K. Sato, W. Ji, Z. Álvarez, L. C. Palmer and S. I. Stupp, ACS Biomater. Sci. Eng., 2019, 5, 2786–2792 CrossRef CAS PubMed.
- H. He, M. Sofman, A. J. S. Wang, C. C. Ahrens, W. Wang, L. G. Griffith and P. T. Hammond, Biomacromolecules, 2020, 21, 566–580 CrossRef CAS.
- S. I. S. Hendrikse, S. L. Gras and A. V. Ellis, ACS Nano, 2019, 13, 8512–8516 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- M. Glaser, S. Deb, F. Seier, A. Agrawal, T. Liedl, S. Douglas, M. K. Gupta and D. M. Smith, Molecules, 2021, 26, 2287 CrossRef CAS PubMed.
- A. Ruscito and M. C. DeRosa, Front. Chem., 2016, 4, 14 Search PubMed.
- S. K. Albert, X. Hu and S.-J. Park, Small, 2019, 15, 1900504 CrossRef PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS.
- C.-J. Kim, J.-e. Park, X. Hu, S. K. Albert and S.-J. Park, ACS Nano, 2020, 14, 2276–2284 CrossRef CAS.
- M.-P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS PubMed.
- B. A. Armitage, Drug Discovery Today, 2003, 8, 222–228 CrossRef CAS.
- A. Dragulescu-Andrasi, S. Rapireddy, B. M. Frezza, C. Gayathri, R. R. Gil and D. H. Ly, J. Am. Chem. Soc., 2006, 128, 10258–10267 CrossRef CAS PubMed.
- M. Balaz, S. Tannir and K. Varga, Coord. Chem. Rev., 2017, 349, 66–83 CrossRef CAS.
- A. Mishra, S. Dhiman and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 2740–2756 CrossRef CAS PubMed.
- A. Mittal, Krishna, Aarti, S. Prasad, P. K. Mishra, S. K. Sharma and B. Parshad, Mater. Adv., 2021, 2, 3459–3473 RSC.
- A. Brito, S. Kassem, R. L. Reis, R. V. Ulijn, R. A. Pires and I. Pashkuleva, Chem, 2021, 7, 2943–2964 CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- B.-S. Kim, D.-J. Hong, J. Bae and M. Lee, J. Am. Chem. Soc., 2005, 127, 16333–16337 CrossRef CAS PubMed.
- A. Restuccia, D. T. Seroski, K. L. Kelley, C. S. O’Bryan, J. J. Kurian, K. R. Knox, S. A. Farhadi, T. E. Angelini and G. A. Hudalla, Chem. Commun., 2019, 2, 53 CrossRef.
- S. I. S. Hendrikse, L. Su, T. P. Hogervorst, R. P. M. Lafleur, X. Lou, G. A. van der Marel, J. D. C. Codee and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 13877–13886 CrossRef CAS.
- P. W. J. M. Frederix, I. Patmanidis and S. J. Marrink, Chem. Soc. Rev., 2018, 47, 3470–3489 RSC.
- P. W. J. M. Frederix, J. Idé, Y. Altay, G. Schaeffer, M. Surin, D. Beljonne, A. S. Bondarenko, T. L. C. Jansen, S. Otto and S. J. Marrink, ACS Nano, 2017, 11, 7858–7868 CrossRef CAS PubMed.
- J. K. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016, 7, 4713–4719 RSC.
- A. Jayaraman, A. Kulshreshtha, P. Taylor and A. Prhashanna, Foundations of Molecular Modeling and Simulation: Select Papers from FOMMS 2018, Springer Nature, Singapore, 2021 Search PubMed.
- J. H. Wen, L. G. Vincent, A. Fuhrmann, Y. S. Choi, K. C. Hribar, H. Taylor-Weiner, S. Chen and A. J. Engler, Nat. Mater., 2014, 13, 979–987 CrossRef CAS PubMed.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H.-P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed.
- C. Loebel, R. L. Mauck and J. A. Burdick, Nat. Mater., 2019, 18, 883–891 CrossRef CAS PubMed.
- M. Diba, S. Spaans, S. I. S. Hendrikse, M. M. C. Bastings, M. J. G. Schotman, J. F. van Sprang, D. J. Wu, F. J. M. Hoeben, H. M. Janssen and P. Y. W. Dankers, Adv. Mater., 2021, 33, 2008111 CrossRef CAS.
- S. Ling, D. L. Kaplan and M. J. Buehler, Nat. Rev. Mater., 2018, 3, 18016 CrossRef CAS.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- K. Ariga, X. Jia, J. Song, J. P. Hill, D. T. Leong, Y. Jia and J. Li, Angew. Chem., Int. Ed., 2020, 59, 15424–15446 CrossRef CAS PubMed.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. Van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS PubMed.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- C. Li, A. Iscen, H. Sai, K. Sato, N. A. Sather, S. M. Chin, Z. Álvarez, L. C. Palmer, G. C. Schatz and S. I. Stupp, Nat. Mater., 2020, 19, 900–909 CrossRef CAS PubMed.
- S. A. Weissman and N. G. Anderson, Org. Process Res. Dev., 2015, 19, 1605–1633 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |