
Structural characterisation methods for supramolecular chemistry that go beyond crystallography
Niklas
Geue
 a,
Richard E. P.
Winpenny
b and
Perdita E.
Barran
*a
a,
Richard E. P.
Winpenny
b and
Perdita E.
Barran
*a
aMichael Barber Centre for Collaborative Mass Spectrometry, Manchester Institute of Biotechnology, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: perdita.barran@manchester.ac.uk
bDepartment of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 24th November 2021
Abstract
Supramolecular chemistry has grown rapidly over the past three decades, yet synthetic supramolecular chemists still face several challenges when it comes to characterising their compounds. In this review, we present an introduction to structural characterisation techniques commonly used for non-crystalline supramolecular molecules, e.g. nuclear magnetic and electron paramagnetic resonance spectroscopy (NMR and EPR), mass spectrometry (MS), ion mobility mass spectrometry (IM-MS), small-angle neutron and X-ray scattering (SANS and SAXS) as well as cryogenic transmission electron microscopy (cryo-TEM). We provide an overview of their fundamental concepts based on case studies from different fields of supramolecular chemistry, e.g. interlocked structures, molecular self-assembly and host–guest chemistry, while focussing on particular strengths and weaknesses of the discussed methods. Additionally, three multi-technique case studies are examined in detail to illustrate the benefits of using complementary techniques simultaneously.
 Niklas Geue | Niklas Geue is a second-year PhD student in the groups of Prof. Perdita Barran and Prof. Richard Winpenny at The University of Manchester, UK. He received his Bachelor degree in Chemistry from Leipzig University in 2019, before he worked on different projects in Analytical Supramolecular Chemistry at the University of New South Wales in Sydney and the University of California, Los Angeles. His work in Manchester now focuses on the use of ion-mobility mass spectrometry for the characterisation of supramolecular systems like heterometallic rotaxanes. In his spare time, Niklas is an enthusiastic chess player regularly competing in tournaments on the state and national level. |
 Richard E. P. Winpenny | Richard E. P. Winpenny obtained both his degrees from Imperial College, London; his PhD studies with Prof. David Goodgame involved synthesis of coordination polymers. After a period at Texas A&M University, working as a postdoctoral fellow with Prof. John Fackler, Jr., he moved to a lectureship at the University of Edinburgh. In 2000, after ten years in the frozen wastes of Northern Britain, he was appointed to the Chair of Inorganic Chemistry at the University of Manchester. Since 2018 he has held an EPSRC Established Career Fellowship and an ERC Advanced Grant. He won the Royal Society of Chemistry Tilden Prize in 2011 and Ludwig-Mond Prize in 2016, and led the team that won the RSC 2021 Dalton Division Horizon Prize. He is a Fellow of the Learned Society of Wales and a member of the Academia Europaea. |
 Perdita E. Barran | Perdita E. Barran obtained her first degree from The University of Manchester in Chemistry with Industrial Experience and her PhD from The University of Sussex, under the supervision of Prof. Tony Stace and Prof. Sir Harry Kroto. After working as a postdoctoral researcher at the University of Sussex and UC Santa Barbara with Prof. Mike Bowers, she moved to The University of Edinburgh, where she helped establish a proteomics centre SIRCAMS. She was awarded an EPSRC Advanced Research Fellowship and after a period as a Senior Lecturer and Reader, she moved back to The University of Manchester, to direct the Michael Barber Centre for Collaborative Mass Spectrometry. She has won the Royal Society of Chemistry Joseph Black award (2009), Theophilus Redwood award (2019) The American Chemical Society Measurement Science Lectureship (2020) and led the team that won the RSC Analytical Division Robert Boyle Horizon Award (2021). |
Key learning points(1) The reader will repeat and/or learn basic concepts for a range of structural characterisation methods and get new insights into evaluating the obtained data critically.(2) The reader will come to the conclusion that, although X-ray crystallography is still the method of choice, a range of different techniques exist that can characterise supramolecular compounds in a precise and unambiguous manner. The reader will also gain a practical understanding of the discussed tools including their particular strengths and weaknesses. (3) After reading this tutorial review, the reader will hopefully agree with our evaluation that multi-technique approaches show a great utility for the characterisation of non-crystalline supramolecular molecules. |
1 Introduction
Starting with the discovery of the inclusion complexes in the late 19th century, supramolecular chemistry developed into an independent field of research until the mid-1960s.1 From then on, the discipline received considerably more attention culminating in the 2016 chemistry Nobel Prize for molecular machines. Supramolecular chemists initially were driven by the synthesis of ‘chemically beautiful’ molecules, but more recently there has been a shift in focus to synthesise compounds with potential application in a range of disciplines including medicine,2,3 catalysis4,5 and materials science.6In general, supramolecular compounds comprise different types of molecules, e.g. host–guest-complexes, interlocked molecules or self-assembled structures that are held together via non-covalent bonding. Such interactions may be weak (although not always) and can be tuned to be reversible. The combination of inter- or intramolecular non-covalent interactions, much inspired by naturally occurring biological complexes, provides structural complexity and molecular diversity in the supramolecular regime.7 There are naturally considerable challenges that can occur during the synthesis of supramolecules and, as for other synthesised molecules, accurate and rapid characterisation is critical to hone the synthetic processes.8 For supramolecular compounds, a major limitation arises with molecules that do not form crystals that diffract, rendering X-ray crystallography redundant. Additionally, even if a given supramolecular complex does allow crystallographic data to be obtained, this will not necessarily inform on the different conformations that it can adopt in solution or other phases.
Considering this, a concomitant need exists to find convincing alternatives for characterisation of the structure(s) and stability of supramolecular compounds. For successful translation of these compounds to applied research and bulk manufacture, structural characterisation should encompass high-throughput techniques. To date principal challenges for structural characterisation include: distinguishing closely related species, including isomers; circumventing or preventing aggregation as well as managing purification and stability issues during the analytical process. Depending on the molecule, different structural characterisation techniques or increasingly a combination of techniques may be pertinent.
In the first part of the present review (Sections 2–6), we discuss relevant techniques based on literature examples and consider their strengths and weaknesses for structural characterisation of supramolecular molecules. We consider nuclear magnetic and electron paramagnetic resonance spectroscopy (NMR and EPR), mass spectrometry and ion mobility mass spectrometry (MS and IM-MS), small angle X-ray and neutron scattering approaches (SAXS and SANS) and cryogenic transmission electron microscopy (cryo-TEM, Fig. 1). In the second part (Sections 7–9), we cover examples of use and emphasize how multi-technique approaches can overcome challenges that cannot be solved by a single method.
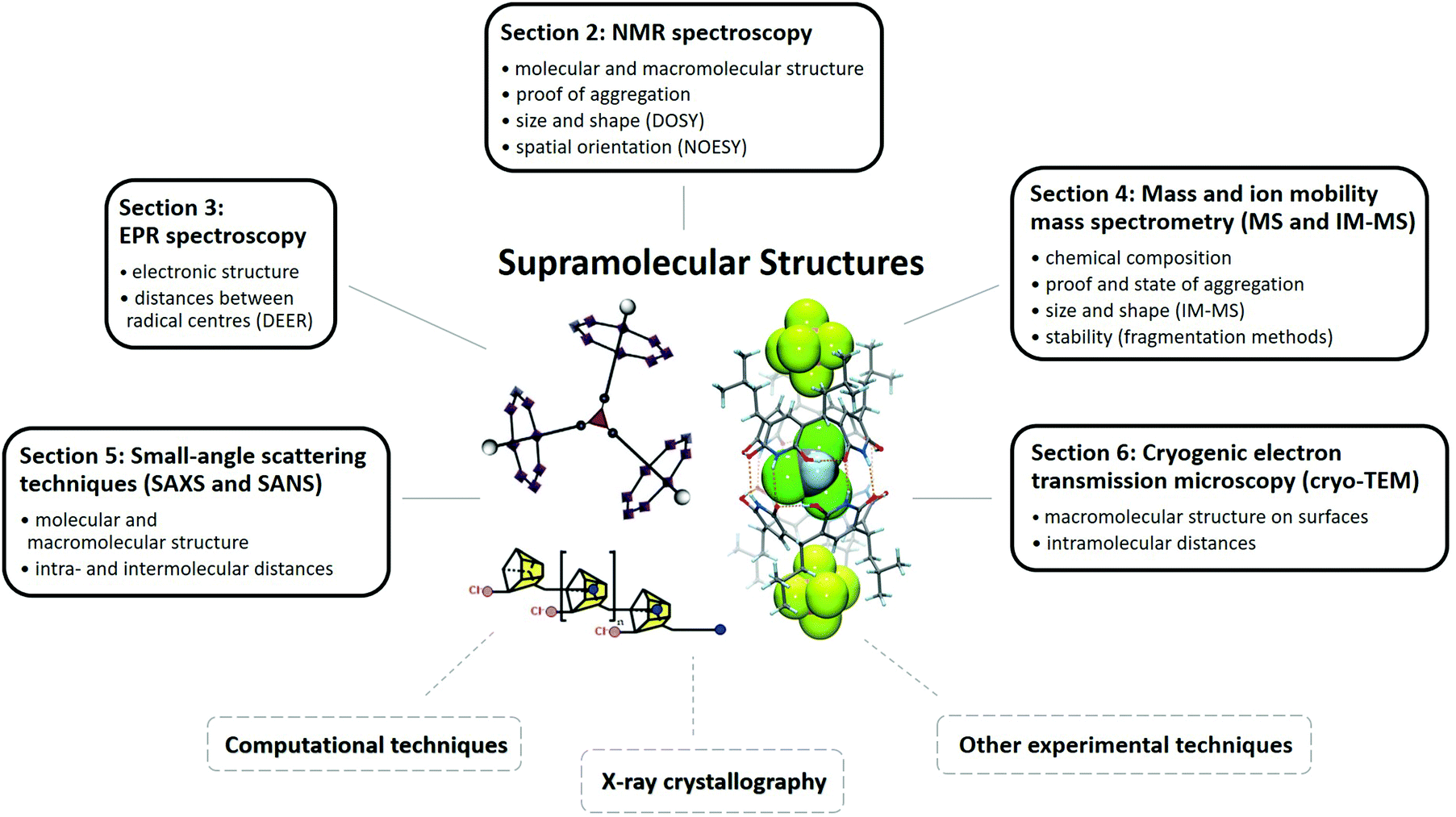 | ||
| Fig. 1 Schematic illustration of techniques used for the structural characterisation of supramolecules. Common findings of the different methods are noted in the corresponding boxes. Reproduced from ref. 19 with permission from the Chinese Chemical Society (CCS), Shanghai Institute of Organic Chemistry (SIOC), and the Royal Society of Chemistry, as well as from ref. 68 with permission from The Author(s), Copyright © 2016, and from ref. 82 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Copyright © 2017. | ||
Due to the limited scope of this review, not every technique can be discussed, but detailed information about the application of other methods for supramolecular structures can be found elsewhere. This applies for dynamic light scattering (DLS),9 X-ray powder diffraction,10,11 scanning tunnelling microscopy (STM),12 magnetic measurements13 and circular dichroism spectroscopy (CD).14
2 Nuclear magnetic resonance spectroscopy
Technique
NMR spectroscopy is amongst the most popular structural characterisation methods in synthetic chemistry research. It combines easy sample preparation and wide applicability to a range of molecules with rich structural detail, in particular regarding through-bond and through-space interactions. Consequently, NMR spectroscopy is also commonly used to characterise supramolecular compounds, e.g. host–guest complexes.15–17Besides its established advantages, the application of NMR spectroscopy also contains drawbacks for the structural characterisation of supramolecular molecules. For example, the sample must be relatively pure and have a high solubility which can be problematic.18 Additionally, when dealing with very large (supramolecular) compounds, 1H- and 13C-NMR spectra become hard to interpret eventually leading to ambiguous data due to overlapping signals. This is also more often the case with highly symmetric molecules due to the similar/analogous chemical environments of relevant atoms.8
2D NMR
Complementary to 1D NMR approaches, several two-dimensional techniques have emerged as powerful tools for the characterisation of supramolecular compounds in solution.15 Perhaps the most relevant is diffusion ordered spectroscopy (DOSY), which seeks to distinguish different molecules on the basis of their diffusion coefficients. This is achieved by a combination of radio-frequency pulses, as used in 1D NMR, and magnetic field gradients, which provide additional spatial information.19 Other 2D approaches, e.g. correlation spectroscopy (COSY) and nuclear Overhauser effect spectroscopy (NOESY), are also applicable.20Solid-state NMR
Another, rapidly growing technique for supramolecular chemistry is solid-state NMR (ssNMR), with which it is possible to characterise solid crystalline and, for our purpose more importantly, amorphous substances.15 The method is influenced by anisotropic nuclear interactions, which are averaged to zero in solution, but strongly affect the behaviour of nuclear spins in the solid phase. ssNMR is a wide and complex field and is beyond the scope of this review. Further information can be found in the literature.21–24Literature
Several reviews have been published on the use of solution-NMR spectroscopy for supramolecular chemistry with most of them having a special focus on DOSY.15,17,19,25–27 In the following paragraphs, two examples are presented highlighting the application of solution-NMR techniques to characterise host–guest complexes and supramolecular oligomers.Study example A
Our first study discusses the host–guest behaviour of resorcin[4]-arenes, a class of bisphenolic ligands consisting of four methylene-bridged resorcinol units20 (structure 1 in Fig. 2A). Although the first molecule of this type was synthesised in 1980,28 the characterisation of their host–guest complexes in solution were initially realised in 2006. Here a combination of 1H-NMR and 2D NMR techniques were used to prove that hexameric resorcin[4]-arene cages, which were structurally characterised in earlier studies,29,30 are able to encapsulate six glutaric acid guests.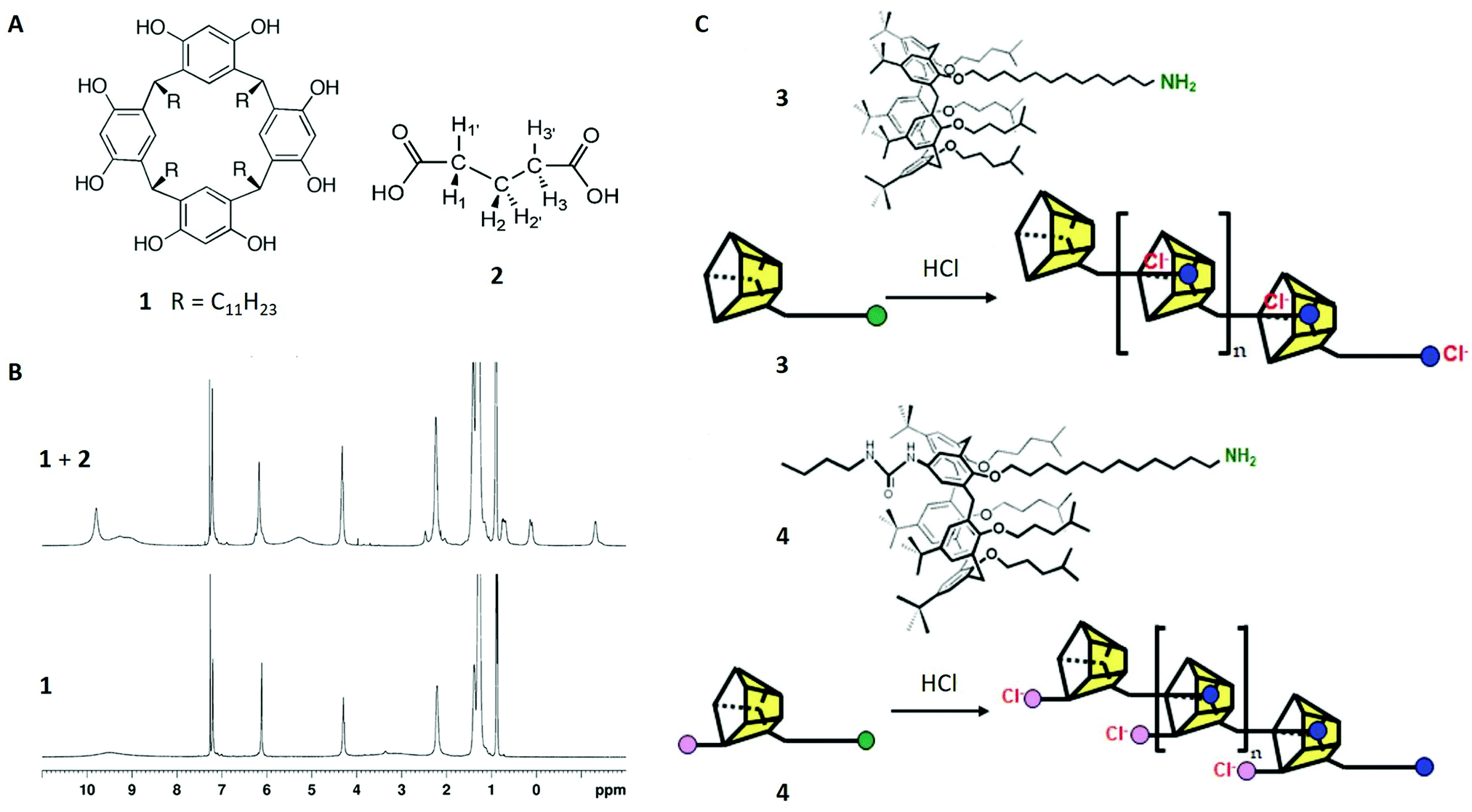 | ||
| Fig. 2 (A) Structures of resorcinarene 1 and glutaric acid 2. (B) 1H-NMR spectra of 1 (30 mM) in CDCl3 with (top) and without (bottom) glutaric acid (90 mM). (C) Structures of calix[5]arenes 3 and 4 and schematic of their oligomerisation processes. Reproduced with permission from ref. 19 with permission from the Chinese Chemical Society (CCS), Shanghai Institute of Organic Chemistry (SIOC), and the Royal Society of Chemistry, and from ref. 20 with permission from National Academy of Sciences, U.S.A., Copyright © 2006. | ||
When comparing the 1H-NMR spectra of resorcin[4]arene 1 with and without glutaric acid 2, different guest signals of 2 are found upfield-shifted. Additionally, integration yielded a host–guest stoichiometry of around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating six enclosed guest molecules. The extend of the encapsulation process was shown to increase with higher concentrations of glutaric acid and longer reaction times, which was readily observable due to the high sensitivity of the host aryl peak at 6.1 ppm. This supported the proposed encapsulation mechanism and additionally indicated a slowly occurring exchange mechanism at the expense of solvent molecules, in this case chloroform.
:1, indicating six enclosed guest molecules. The extend of the encapsulation process was shown to increase with higher concentrations of glutaric acid and longer reaction times, which was readily observable due to the high sensitivity of the host aryl peak at 6.1 ppm. This supported the proposed encapsulation mechanism and additionally indicated a slowly occurring exchange mechanism at the expense of solvent molecules, in this case chloroform.
In order to assign the different upfield-shifted guest peaks unambiguously, 2D techniques (COSY and NOESY) were applied and localised the H1/H3 signals at 0.7 ppm, H1′/H3′ at 0.1 ppm and H2/H2′ at −1.3 ppm (Fig. 2A and B). Although this might be surprising at first view, the broken symmetry (“enantiotopic pairs of geminal hydrogens become diastereotopic upon encapsulation”)20 was consistent with the formation of a chiral capsule, which was already found in the crystalline structure.
Taking this data in combination, the absence of an encapsulation process would have been unlikely. Nevertheless, conclusive proof was found with 2D DOSY spectroscopy which recorded the diffusion coefficients of glutaric acid and of the hexameric cage, isolated and also when combined in solution. As expected, the free glutaric acid diffuses faster (D = 1.05 × 10−5 cm2 s−1) than the hexameric cage (D = 0.24 × 10−5 cm2 s−1) due to its smaller size. By contrast, when measuring the encapsulated guest, the diffusion coefficient is significantly reduced (D = 0.24 × 10−5 cm2 s−1), showing approximately the same value as the cage. Therefore, it was proven that the complex diffuses through solution as one supramolecular entity.
Study example B
This example considers non-covalently bound oligomers, another relevant molecular class in the field of supramolecular chemistry.31 In particular, a calix[5]arene, consisting of four methylene-bridged phenyl-ether units and a single 12-aminododecyl substituent (structure 3 in Fig. 2C), was synthesised and studied with 1H-NMR and DOSY NMR in the absence and the presence of acids. The aim of this study was to investigate possible oligomerisation of the calix[5]arene induced by the protonation of the amino-group and its subsequent inclusion in another calix[5]arene molecule (Fig. 2C top). This was suggested by the 1H-NMR spectrum of 3 with HCl, showing different peak sets of either the unthreaded groups at the ends of the oligomer or the threaded core units.Intramolecular self-inclusion was another possible interpretation of the data, but this could be discounted with the use of DOSY NMR. In case of this hypothetical event, the diffusion coefficients of 3 in acidic solution would have remained similar compared to the monomer, whereas the experimental data shows a strong decrease of this value. This observation could only be explained with oligomers (or polymers).
To investigate the properties of this system further, picric acid was applied as a proton source, to remove the anion from the oligomer after protonation. This was intended to lead to longer chains, which was confirmed by lower diffusion coefficients in the DOSY experiment. Furthermore, the diffusion coefficient of the picrate anion was higher than in free solution, which suggested a loose attachment to the oligomer.
In a subsequent study,32 the authors continued to investigate the mechanistic details of these systems by using another monomeric calix[5]arene 4 – this time with an additional urea moiety being able to bind chloride ions (Fig. 2C bottom). In combination with HCl and under similar conditions, 4 showed a significant lower diffusion coefficient indicating higher order polymers compared to 3 and confirming the advantage of anion removal for oligomerisation.
3 Electron paramagnetic resonance spectroscopy
Technique
Electron paramagnetic resonance spectroscopy (EPR), also known as the “older sibling” of NMR, is one of the oldest spectroscopic techniques. The general concept is similar to NMR, but with the difference that electron spins are excited instead of their atomic nuclei analogues. Thus, only species with unpaired electrons (free radicals) are observable with EPR, which is its biggest weakness.33 However, when compared to the “younger sibling”, it offers two important advantages: higher sensitivity (micromolar concentrations are detectable, which is particularly useful for supramolecular molecules) and the possibility to obtain spatial information about the distances between radical centres via double electron–electron resonance spectroscopy (DEER), a method that will be introduced later.18,34Although supramolecular systems with persistent, free radicals are occasionally desired, with potential applications in quantum technology, they are not common unless a metal site is present. If not inherent to the organic supramolecule under analysis, paramagnetic species have to be introduced to the system either covalently, via “spin-labelling”, or non-covalently, via “spin-probing” to obtain an EPR signal.18 We will discuss case studies for both approaches (study examples C and D) as well as an example of heterometallic rotaxanes from our groups (study example E). More detailed information on the use of EPR spectroscopy in supramolecular chemistry can be found in dedicated reviews.18,33–37
Study example C
In the first two discussed study examples, nitroxides are used as the free radicals in the supramolecular system. These species are commonly applied as spin probes and spin labels due to their high stability, and also because of their sensitive EPR properties with respect to different chemical environments.18 This behaviour is due to the hyperfine interaction between the radical electron and the 14N nucleus (spin = 1), which is commonly referred to as a(N). This value, as well as the spin density on the nitrogen, increases when exposed to a surrounding with a higher polarity, which makes it possible to clearly distinguish between free and complexed species.18A subclass of nitroxides are benzyl tert-butylnitroxides (BTNB, structures 5–7 in Fig. 3A), which offer conformational information in the EPR spectrum due to coupling to the two benzylic protons.38 This interaction, expressed by the constant a(H) (Fig. 3B),38 is particularly relevant for structural characterisation.39 This study investigated, among other things, the inclusion of three differently substituted BTNB radicals in β-cyclodextrin and its influence on the radical conformation. For that purpose, the EPR spectrum of each BTNB was recorded in water (Fig. 3B for the spectrum of 5) and the a(H) values were compared to the corresponding parameters in the spectra, where the macrocycle was present.
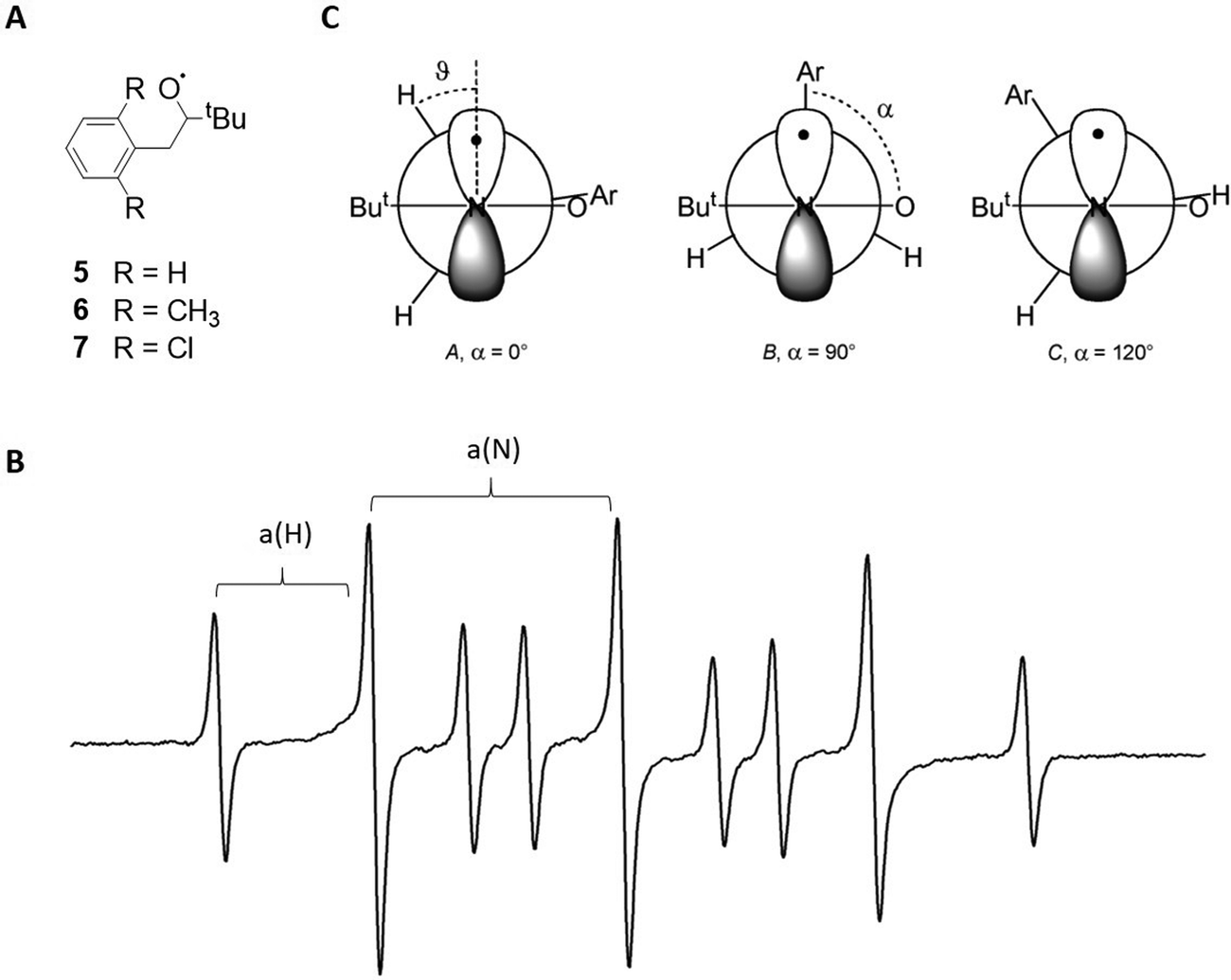 | ||
| Fig. 3 (A) Structures of the benzyl tert-butylnitroxides 5–7. (B) EPR spectrum of 5 in water at room temperature. Hyperfine interactions a(N) and a(H) are labelled. (C) Different possible nitroxide conformations for 5–7. Reproduced from ref. 39 with permission from the Royal Society of Chemistry and from ref. 37 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Copyright © 1999. | ||
The magnitudes of a(H) could be predicted for three possible radical conformations in water (Fig. 3C) and were compared to experimental values. It was demonstrated that the parameters of 5 fit very well to a dihedral angle of 120° between the aryl–C and N–O bonds (referred to as α in the following), which corresponds to structure C, whereas the dimethyl- and dichlorine-substituted species 6 and 7 likely adopt conformation A with α = 0° (Fig. 3C).
When including the radicals in the β-cyclodextrin macrocycle, both the a(H) and the a(N) values decrease due to a less polar environment. For 5, the decline is significantly stronger than expected from theoretical predictions, but fits well to a conformation change from C to B. Conversely, the change of a(H) is as expected for 6 and 7, suggesting the same conformation A in- and outside of the macrocycle (Fig. 3C).
A related technique to EPR is double electron–electron resonance (DEER) also called pulsed electron–electron double resonance (PELDOR). With this method, it is possible to measure the distance between two radical centres in the same molecule and to compare it with results obtained from other methods including theoretical calculations. This is particularly useful for interlocked molecules, where the narrowness of the distance distribution can yield information about the conformational flexibility of the system.18 This was for example demonstrated by Pievo et al. in 2012, where the authors measured the end-to-end distance between two 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) radicals in the axle of a [3]-rotaxane.40 Based on the distance distribution, the results showed that the inclusion in two cucurbit[6]uril macrocycles significantly reduces the flexibility of the diradicalic thread. Further theoretical details about DEER are beyond the scope of this review and can be found elsewhere.41
Study example D
Another practical example by Franchi et al., introduced the paramagnetic moieties (in this case TEMPO radicals) via covalent bonds (spin-labelling).42 This approach requires more synthetic effort, but offers the benefit of measuring interactions between non-covalently bound molecules if both possess paramagnetic groups.Here an axle with a 4,4′-bipyridinium- and a dialkylammonium recognition site was labelled with a TEMPO radical and reacted with a crown ether that was also TEMPO-labelled forming the rotaxane 8 (Fig. 4). Following a well-known mechanism, the binding position of the macrocycle changes with the pH-value. This shuttling process, which for example starts at the dialkylammonium group under acidic conditions and ends at the bipyridinium site after adding a base, was investigated with DEER spectroscopy. The analysis of the spectra in both states surprisingly showed that the distance between the radicals remains similar after the transfer to the bipyridinium group. Additionally, the distance distribution narrowed significantly down during this process, which suggests a smaller conformational flexibility. In accordance with crystallographic data and stochastic dynamic calculations, a strong geometric change of the crown ether was demonstrated during the shuttling process (Fig. 4).18
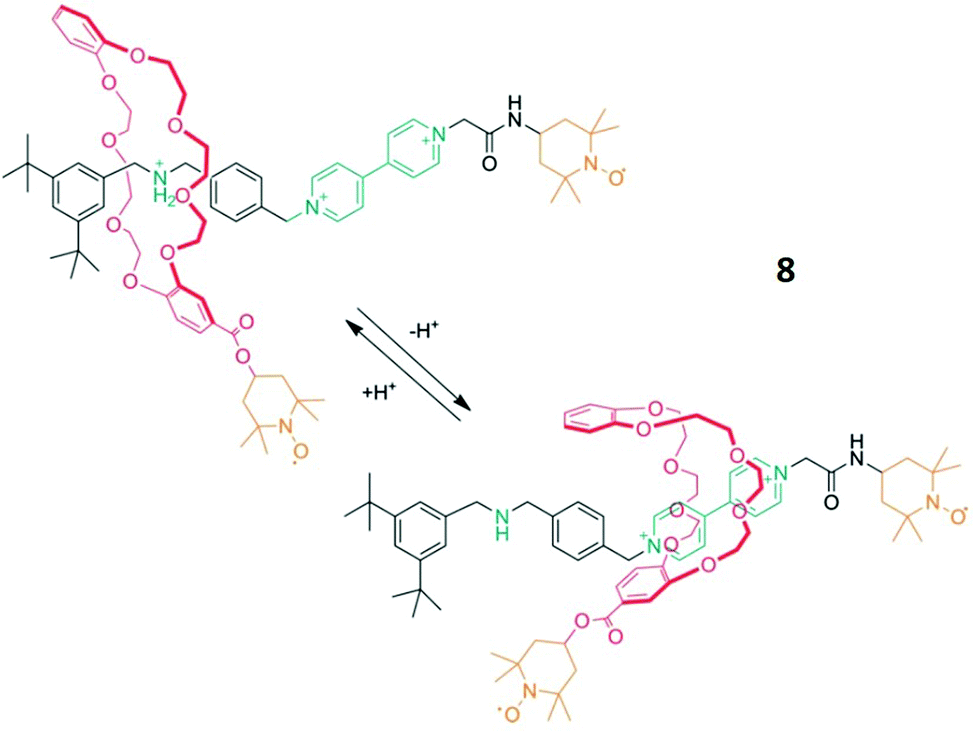 | ||
| Fig. 4 Schematic of the molecular shuttling process of rotaxane 8. Reproduced from ref. 18 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Copyright © 2020. | ||
Study example E
A more complex problem for DEER spectroscopy is presented by polymetallic cage complexes where the electron spin is not localized on individual free radicals but is spread around multiple metal sites. An example of this is the study of [3]- and [4]-rotaxanes reported by Lockyer et al.43 The structures obtained on single crystals contain two polymetallic components, a heterometallic {CrIII7NiII} ring and a central {CrIIINiII2} triangle. Anti-ferromagnetic coupling between the metals within the components make each an effective S = 1/2 centre and therefore good subjects for EPR spectroscopy. Two supramolecular assemblies were crystallised, a [3]-rotaxane where two {CrIII7NiII} rings surround the central {CrIIINiII2} triangle, and a [4]-rotaxane where three rings are present around the triangle.DEER spectroscopy was used at T = 5 K in a frozen solution to measure contacts between the {CrIII7NiII} rings. As the S = 1/2 state is delocalized around the ring, the DEER signal is due to the distribution of metal–metal contacts between the metals in the rings. This gives a complex distribution which fits well with the metal–metal distances that can be calculated from the single crystal distributions. There are however subtle differences, particularly for the [3]-rotaxane. In the frozen solution, as measured by DEER, it appears that the two rings attached at the periphery of the supramolecular assembly, tip away from one another making the longest metal–metal distances greater in solution than in the crystalline phase. For the more symmetric [4]-rotaxane the three rings around a triangle have much less conformational flexibility and therefore show a closer agreement between the metal–metal distances in the crystalline and solution phases.
4 Mass spectrometry and ion mobility spectrometry
Technique
In the early days of supramolecular chemistry, mass spectrometry was not considered to be a suitable characterisation method due to a mistaken belief that ionisation would prevent the successful transfer of intact supramolecular molecules in the mass spectrometer. As it happens, soft ionisation methods, such as electrospray ionisation (ESI) and matrix assisted laser desorption ionisation (MALDI), are well able to preserve large biological macromolecules into the gas phase and such methods can be repurposed for the analysis of supramolecular compounds.1Sample transfer from solution phase to the vacuum of the mass analysers may influence the strength of non-covalent interactions, especially those competing with or depending on solvent or counterions. Electrostatic interactions will be stronger in the absence of any screening effects from solvent which can alter the lowest energy conformation. This argument, for example, also applies to hydrogen bonds, whereas hydrophobic interactions are weakened in the gas phase due to the absence of polar solvent molecules. The transfer from solution to the desolvated state may affect other properties, as described previously,44,45 but for samples stable in hydrophobic solvents, with a low dielectric constant, it is likely that the gas phase structure (where the dielectric constant is 1) will be highly similar to the solvated one.
The use of mass spectrometry can provide several insights beyond the exact mass and isotopic distribution of the compound under investigation. For example, the method is more tolerant of mixtures or unpurified compounds than NMR and EPR and can be used to investigate the stoichiometry of a complex as well as to count the number of stabilising counter ions.45
Tandem mass spectrometry
In addition to classic MS measurements, tandem mass spectrometry (MS/MS, MS2) has emerged as a powerful tool to investigate supramolecular molecules. In MS/MS, an ion of interest is m/z-selected and activated with collisions, photons or electrons resulting in the cleavage of covalent and non-covalent bonds. Tandem MS provides a diagnostic fragmentation spectrum which can be used to examine the stability of the molecule of interest and to provide insight to its structure.46Ion mobility mass spectrometry
Ion mobility mass spectrometry is a useful technique for the structural characterisation of supramolecular molecules. Ion mobility separates ions according to their mobility through a cell filled with drift gas, across which a weak electric field is applied. A typical ion mobility experiment injects a pulse of ions into the drift cell and measures the time it takes them to traverse the cell. The mobility of an ion is defined by the influence of the number of collisions that it has with the drift gas, which is directly related to its size and to the interactions that the atoms within the ion have with the drift gas. A more mobile (smaller) ion will have fewer collisions and take less time to traverse the cell than a less mobile (larger) ion. The experimental drift time can be converted to a drift gas and temperature dependent collisional cross section (CCS). The CCS of any given ion can be used to define its size, and the CCS of a set of candidate structures can be computationally calculated for comparison with experimental data. As well as directly measuring the structure of a given ion, the inclusion of ion mobility to mass spectrometry experiments also allows isobaric (same mass) isomers to be distinguished.47Literature
The use of mass spectrometry for supramolecular chemistry has been reviewed regularly since the late 90's,1,7,8,44 whereas ion mobility spectrometry has more recently been adopted by the community.47–49 Here, we present two examples that illustrate some of the strengths of these methods for structural characterisation.Study example F
Sawada et al. have used ESI-MS in a series of elegant experiments that distinguish inclusion complex enantiomers. Their method involved examination of one host enantiomer with a pseudo-racemic mixture of the guest, in which one enantiomer is isotopically labelled.1,50 Both diastereotopic complexes were formed, but in different ratios depending on the proportion of the guest and the preferred binding. The isotopic labelling of one enantiomer means that the resulting complexes are distinguishable in the mass spectrum. To rule out any possible effects of the isotopic labelling, the experiment is repeated with the other host enantiomer labelled.51 This approach may suffer if the diastereoselectivities in solution and gas phase differ or if the ionised form of the compound is significantly different from that in solution, although it should still provide comparable information.Study example G
A recent study by Kruve et al. was able to determine the topology of interlocked molecules using a combination of MS/MS and IM-MS. The main question addressed was how to distinguish different types of isobaric interlocked macrocycles.52 The authors used collision induced dissociation (CID) coupled with travelling wave ion mobility spectrometry (TWIMS) to distinguish a macrocycle from a [2]catenane (Fig. 5). This was possible because the fragmentation of the macrocycle generated a single linear fragment, whereas the [2]catenane dissociates to produce both a cycle and a linear chain. If these fragments had different masses, the CID experiment would have been sufficient with two different masses that are visible in the fragment spectrum for the catenane, and only one for the normal macrocycle. However, if the fragments are isobaric, then the use of ion mobility enabled unambiguous identification due to the lower mobility of the linear form compared to the cyclic fragment, whereas only one peak at all would have been observed upon fragmenting the macrocycle.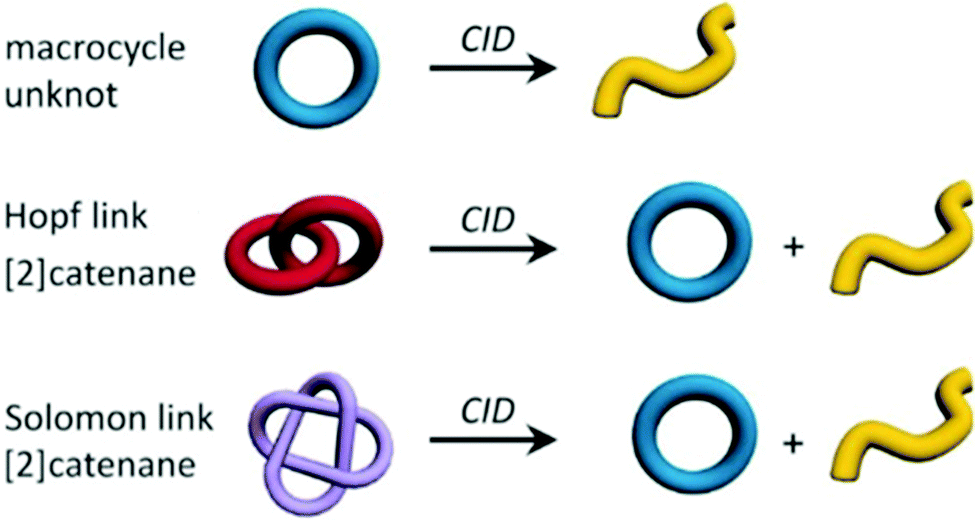 | ||
| Fig. 5 Topologies of the three discussed structures (macrocycle, Hopf link and Solomon link) including the expected fragment topologies after CID experiments. Reproduced from ref. 52 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Copyright © 2019. | ||
A more challenging example of this experimental workflow compared a Hopf link with a Solomon link, which are both [2]catenanes (Fig. 5). Here, the fragments from the Solomon links could be distinguished on the basis of their CCS values, as they are more densely packed than Hopf links. As well as providing structural parameter for given compounds in terms of their CCS, IM-MS can also provide the range of conformations that can be adopted by converting the experimental arrival time distribution to a CCS distribution.53
Instead of enumerating the CCS values, Kruve et al. here introduced the so-called “floppiness parameter” Fl, which is the quotient of the charge-corrected arrival time of the parent ion and the charge- and mass-corrected arrival time of the least entangled (often linear) fragment ion (eqn (1)).52
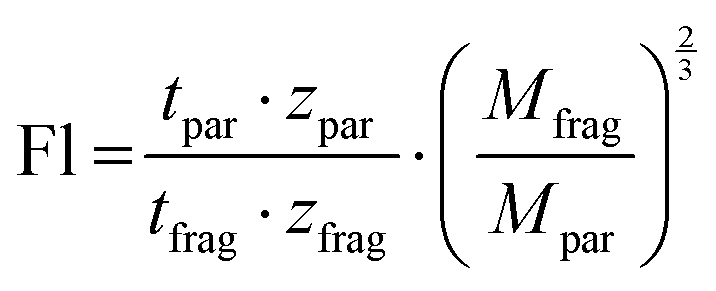 | (1) |
5 Small-angle neutron and X-ray scattering
Techniques
Small-angle scattering techniques are powerful methods to investigate structures of supramolecular assemblies in solution or of soft matter, e.g. gels. Neutron (SANS) and X-ray scattering (SAXS) often require that samples are transported to specialist beam lines, although for example SAXS is increasingly available also as a lab-based technique.The general concept behind their operation is based on measurements of the deflection of neutrons (SANS) or X-rays (SAXS) after their interaction with the target compound that are significantly larger than the radiation wavelength.56,57 The deflected signals can be derived to provide structural information regarding the size and shape of the molecule. If the deflection angle is very low, the technique is commonly referred to as “small-angle”.56,57
Although often employed at the same site, and with beams of the same wavelength, SAXS and SANS measurements each provide different information. Neutrons impart significantly smaller energies to the analyte than X-rays, thus, may be the better choice for non-destructive analysis. Another difference is that neutrons are able to penetrate deeper into the sample than X-rays, which makes them more suitable for in situ analysis.57
Additionally, whilst X-rays interact with the electrons of a species, neutrons scatter based on interactions with atomic nuclei which is mass dependent and leads to an isotopic effect. This is advantageous when it becomes necessary to enhance the contrast between target structure and background (solvent contrast variation), which is commonly applied for SANS.58 Different mixtures of H2O and D2O can for example access a wide range of scattering length densities due to the mass differences of the hydrogen isotopes, which would not be possible in the same way for the non-mass-dependent SAXS. Different examples of solvent contrast variation for SANS of supramolecular structures are known from the literature.59,60
Further information about the use of SAXS and SANS in the supramolecular regime can be found elsewhere.61–65 In the following, we will discuss three case studies highlighting the use of small-angle scattering techniques for the characterisation of supramolecular structures.
Study example H
Kumari et al. describe the manipulation of bis-urea gel networks, both non-fluorinated (structure 9 in Fig. 6A) and fluorinated (structure 10 in Fig. 6B) with a method called thin-film shearing.66 This technique involves the rapid rotation of gels with a particular speed and a certain tilt angle (relative to the horizontal position), which leads to a thinner film.66 To assess the effect of this process, it was essential to analyse and characterise the entanglement of the gel networks before and after.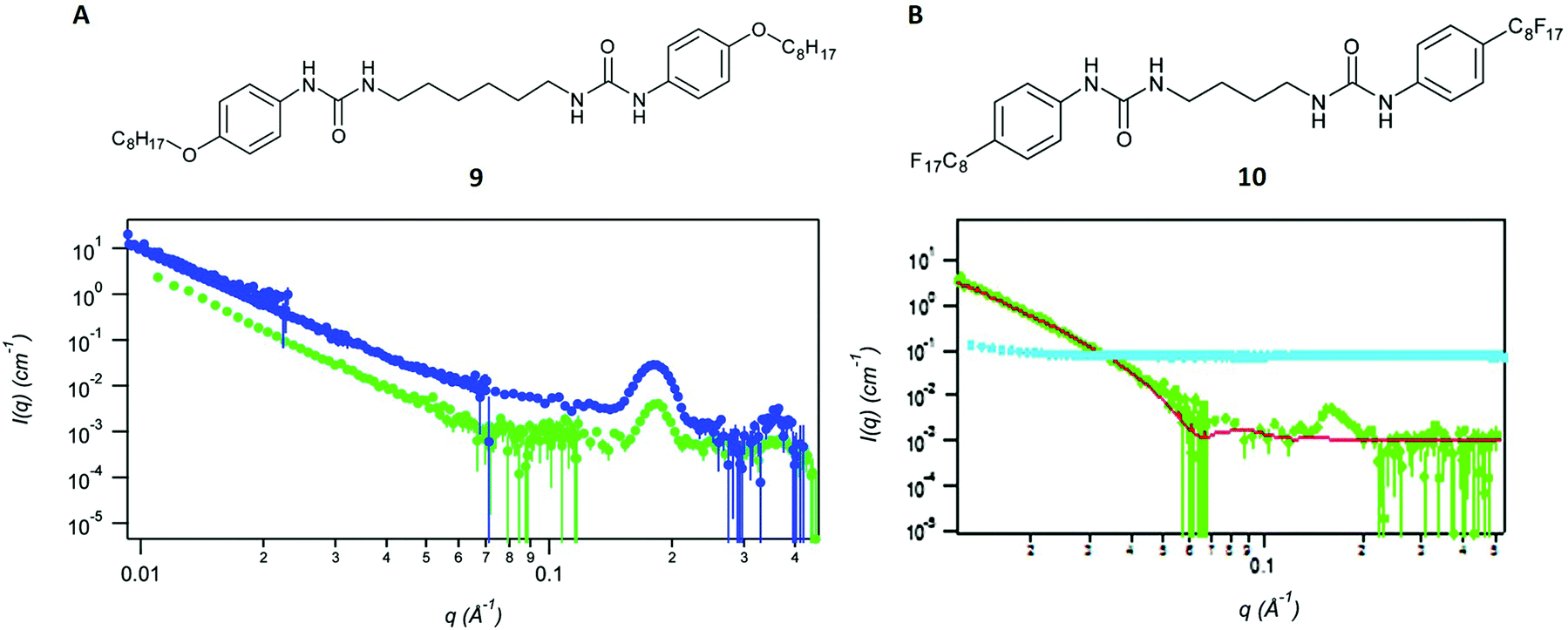 | ||
| Fig. 6 (A) Structure of the non-fluorinated gel 9 and SANS curves without further treatment (blue dots) and after thin-film shearing (green dots). (B) Structure of the fluorinated gel 10 and SANS curves without further treatment (green dots) and after thin-film shearing (cyan dots). Reproduced from ref. 66 with permission from the Royal Society of Chemistry. | ||
The analyses after thin-film shearing were performed with SANS involving neutrons of the wavelength λ = 5 Å. A range of scattering vectors q, which depend on the neutron wavelength λ and the scattering angle θ, were determined with a movable detector. Higher q-values are generally associated with local gel structure at shorter distances, whereas the lower q-region in the spectrum refers to the global structure in the bulk network (Fig. 6). The obtained scattering curves were fitted with a fractal flexible cylinder model61,66,67 and compared between the fluorinated and non-fluorinated bis-urea gels. The results of an earlier study, in which the authors also applied SANS, suggested a lamellar structure for both gels in their native state.67 This conclusion and several other characteristic distances were derived from the model.
Because of the previous results, both materials were also expected to yield similar scattering curves after the thin-film shearing process. However, this was only the case for the non-fluorinated gel 9, which showed the same peak in the high q-region and an analogously decreasing curve part in the lower q-region66 (Fig. 6A). The fluorinated gel 10, by contrast showed a flat scattering intensity. The curve did also not show any new peaks or features, suggesting the complete disruption of the network and the absence of any architecture after thin-film shearing (Fig. 6B).
Study example I
In the second example, taken from prior work from our groups, we synthesised and characterised several [n]-rotaxanes that are potentially relevant as spin qubits in quantum information processing.68 Similar to study example E, these particular molecules were built of amine axles and paramagnetic {CrIII7NiII} rings which coordinated around the amine nitrogen atoms. In combination with other metal centres, a wide range of high-order hybrid organic–inorganic rotaxanes were formed via molecular self-assembly. In this case we were able to apply X-Ray crystallography for most of the synthesised structures, although not for the presumably triangular [7]-rotaxane 11 (Fig. 7A).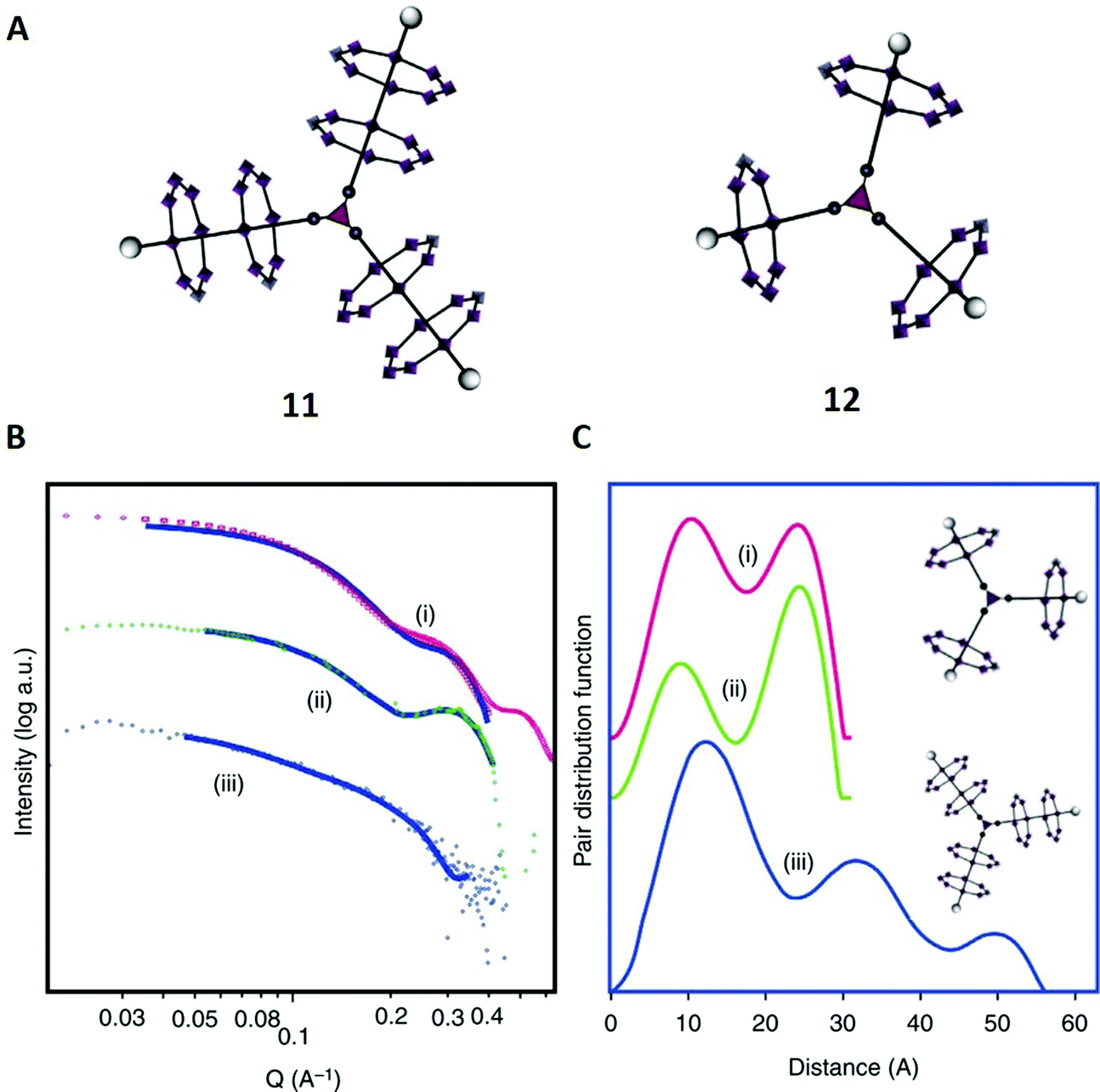 | ||
| Fig. 7 (A) Structures of the [7]-rotaxane 11 and the [4]-rotaxane 12. (B) SAXS data; (i) calculated plot from crystal structure of 12, (ii) experimental data from 12, (iii) experimental data from 11. The solid lines show the fits that were used for the pair distribution function curves shown in (C). (C) Pair distance distribution functions; (i) fit to calculated SAXS data for 12, (ii) fit to experimental data collected for 12 and (iii) fit to experimental data collected for 11. Reproduced from ref. 68 with permission from The Author(s), Copyright © 2016. | ||
To investigate its structure further in solution, SAXS was used for 11 and the [4]-rotaxane 12 (Fig. 7B), which had already been fully characterised with X-ray crystallography. A pair distance distribution function (PDDF) was obtained from each SAXS data set (Fig. 7C). Such X-ray scattering data provides a probability for the distance between electrons in the investigated molecule.69
For the PDDF of compound 12, two peaks were observed at distances of 12 Å and 22 Å (curve ii) that perfectly matched with the reference function calculated from crystallographic data (curve i). Then, we moved on to the characterisation of 11 (curve iii), where we obtained three maxima at 12 Å, 32 Å and 50 Å. The first peak was very similar in 11 and 12, and suggestive of intra-annular contacts that are similar in both structures. With this information, the assignment of the second peak of 12 to ring–ring interactions was possible.
Coming back to 11, it was suggested that the higher maxima in 11 at 32 Å and 50 Å were also associated with contacts between different rings, and, because of their high values, probably between an inner ring and an outer ring of different branches (32 Å) and between two outer rings (50 Å). Thus, the SAXS data was consistent with the model and in accordance with the proposed structure 11. Notably, atomistic molecular dynamics simulations (AMDS, comparable to study example J) later confirmed our predictions for similar structures.43
Study example J
As indicated above, SAXS is even more powerful when supported by AMDS to predict the distributions of distances that can be seen in solution. AMDS is a molecular mechanics method for calculating solution structures of large molecules, and allows to account for interactions with solvent molecules. An example of this is a study of a [13]-rotaxane, that contains twelve heterometallic {CrIII7NiII} rings assembled around a {Pd6} octahedron.70 Notably, the [13]-rotaxane crystallises with six further anionic {CrIII7NiII} rings encapsulated within the [13]-rotaxane. The structure was not predicted and proving that it existed in solution phase prior to crystallization could only be achieved by SAXS supported by AMDS.The SAXS and respective PDDF data are rich and extend out to 65 nm, with four maxima at around 8, 25, 38 and 52 nm (Fig. 8A and B).70 It suggests that a very large supramolecular assembly is present, but only the combination with AMDS allows the structural complexity to be unpicked. In general, once the simulation of the solution structure is completed, it can then be modified in silico and predict SAXS data for different geometries (Fig. 8C–G) that can be compared with the experimental spectra. The results show firstly that the entire structure is maintained, including the six anionic {CrIII7NiII} rings present (Fig. 8B, blue and red trace). The green traces shown in Fig. 8 are the calculated SAXS curves if the anionic rings were lost (Fig. 8D) – the overall size is identical to that for the entire molecule and hence only by simulating the SAXS using AMDS it can be demonstrated that the entire molecule is intact.
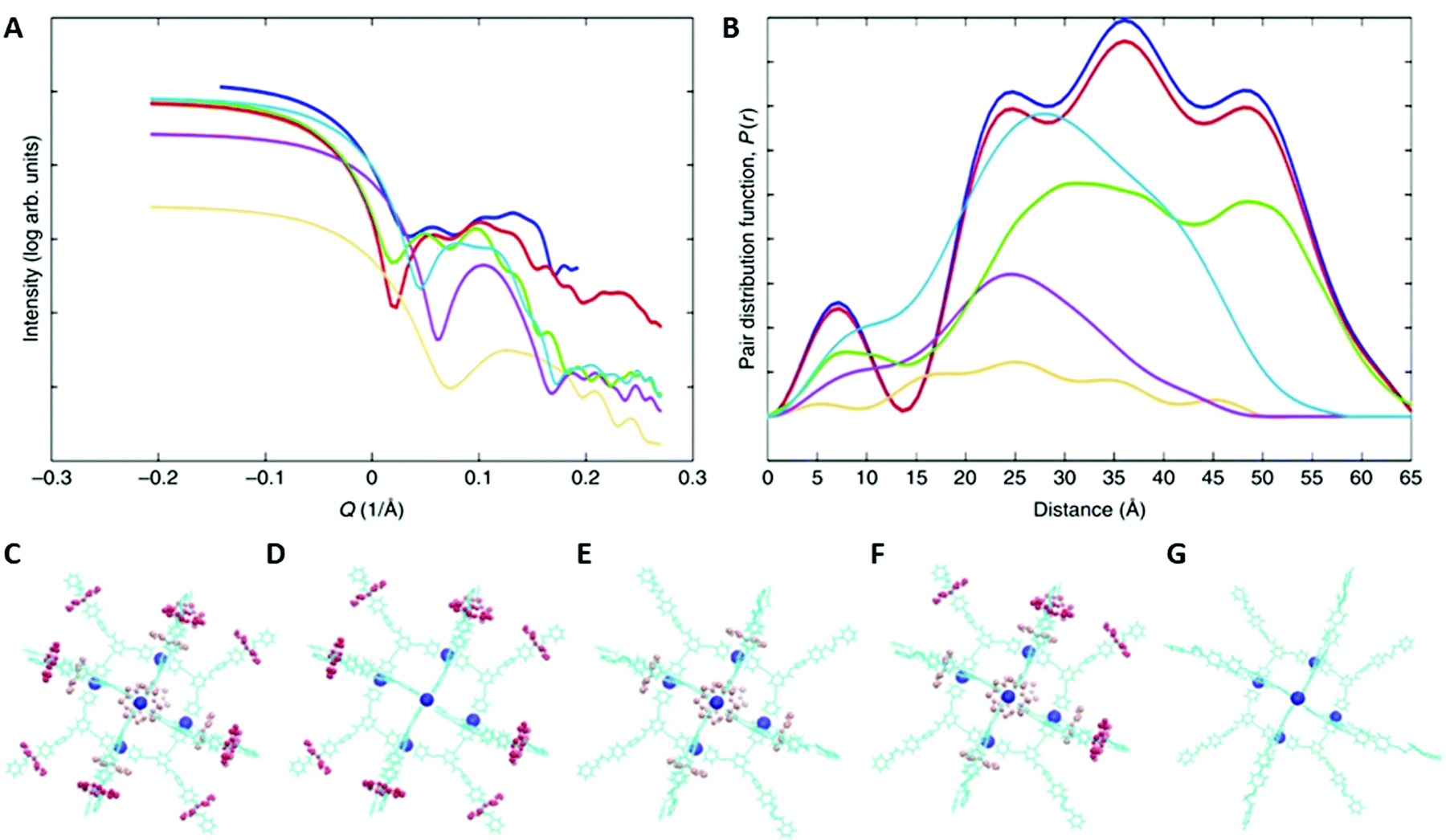 | ||
| Fig. 8 SAXS data for a [13]-rotaxane and modelling of this data. (A) SAXS data recorded as a 25 mM solution of the [13]-rotaxane in THF at T = 298 K. (B) shows the pair distance distribution functions. In (A) and (B) the experimental result is shown in blue. The calculated traces are: the whole structure (red, shown in C); the structure lacking the six ring anions (green, shown in D); the structure lacking the rings on the thread of the [2]-rotaxanes (purple, shown in E); the structure lacking half the rings from the rotaxanes (cyan blue shown in F); the {Pd6} core and organic threads only (orange, shown in G). Reproduced from ref. 70 with permission from The Author(s), Copyright © 2019. | ||
6 Cryogenic transmission electron microscopy
Techniques
Microscopic techniques provide useful complementary tools with which to investigate structures of supramolecular assemblies. In contrast to the previously discussed methods, microscopy enables the direct imaging of macromolecular structures without having the need to interpret complex spectra. A wide range of microscopic techniques were successfully applied to supramolecular molecules with all of them showing different strengths and weaknesses.12,71 However, cryogenic transmission electron microscopy (cryo-TEM) has arguably emerged as the most promising tool and will therefore be discussed further in this section.Cryo-TEM have had a major impact on the structural characterisation of biological compounds, however its use for supramolecular molecules has not received as much attention.71 Compared to traditional TEM, cryo-TEM facilitates the resolution of structures without staining, which can be necessary to enhance the contrast between sample and substrate. Staining is preferentially avoided as it can cause structural changes, but even without it, for TEM each sample has to be dried onto a substrate, which can lead to artefact inclusion.72 Avoiding both staining and drying is therefore a useful improvement for an unambiguous characterisation of supramolecular molecules, which can readily be achieved by cryo-TEM.71,72
Before the collection of the image, the sample is frozen and therefore fixed, which allows a static and in-depth characterisation in the samples’ solvated state. This advantage comes at the cost of complex and expensive sample preparation and measurement. Other strengths include the possibility to use cryo-TEM for very small or inhomogeneous samples, overcoming the limits of other microscopic techniques.71,72
Cryo-electron tomography
Usually, cryo-TEM provides only two-dimensional images, although 3D shots can also be obtained. This is most convincingly achieved by cryo-electron tomography (cryo-ET), where several 2D-images are recorded from different angles. The series of images is combined to yield a 3D representation of the sample that is called tomogram.71,73Literature
In the following, we will show how cryo-TEM and cryo-ET can be applied to characterise supramolecular assemblies. cryo-TEM has recently been reviewed as a tool for the characterisation of soft matter72 and also in comparison to conventional TEM approaches.71 Further details about the use of cryo-ET in supramolecular chemistry can also be found elsewhere.73Study example K
This case study concerns cyanine dyes, a class of organic compounds that have become famous for their self-assembly behaviour in solution. Due to strong dipole–dipole interactions, those molecules tend to align in particular polymeric configurations, leading to the formation of aggregates with altered properties. The most established ones are J-aggregates, which show red-shifted absorption bands and often improved fluorescence properties, making them interesting prospective tools for biomedical imaging, self-driving cars or photovoltaics.74–78 In a recent study, the well-known cyanine dye C8O3 was functionalised with aminopropanediol yielding four isomers with slightly different head groups (Fig. 9A).79 The authors main goal was to investigate how head group stereochemistry and conformation affect the self-assembly properties of these dyes. For that purpose, their J-aggregates were formed in water and structurally characterised by cryo-TEM.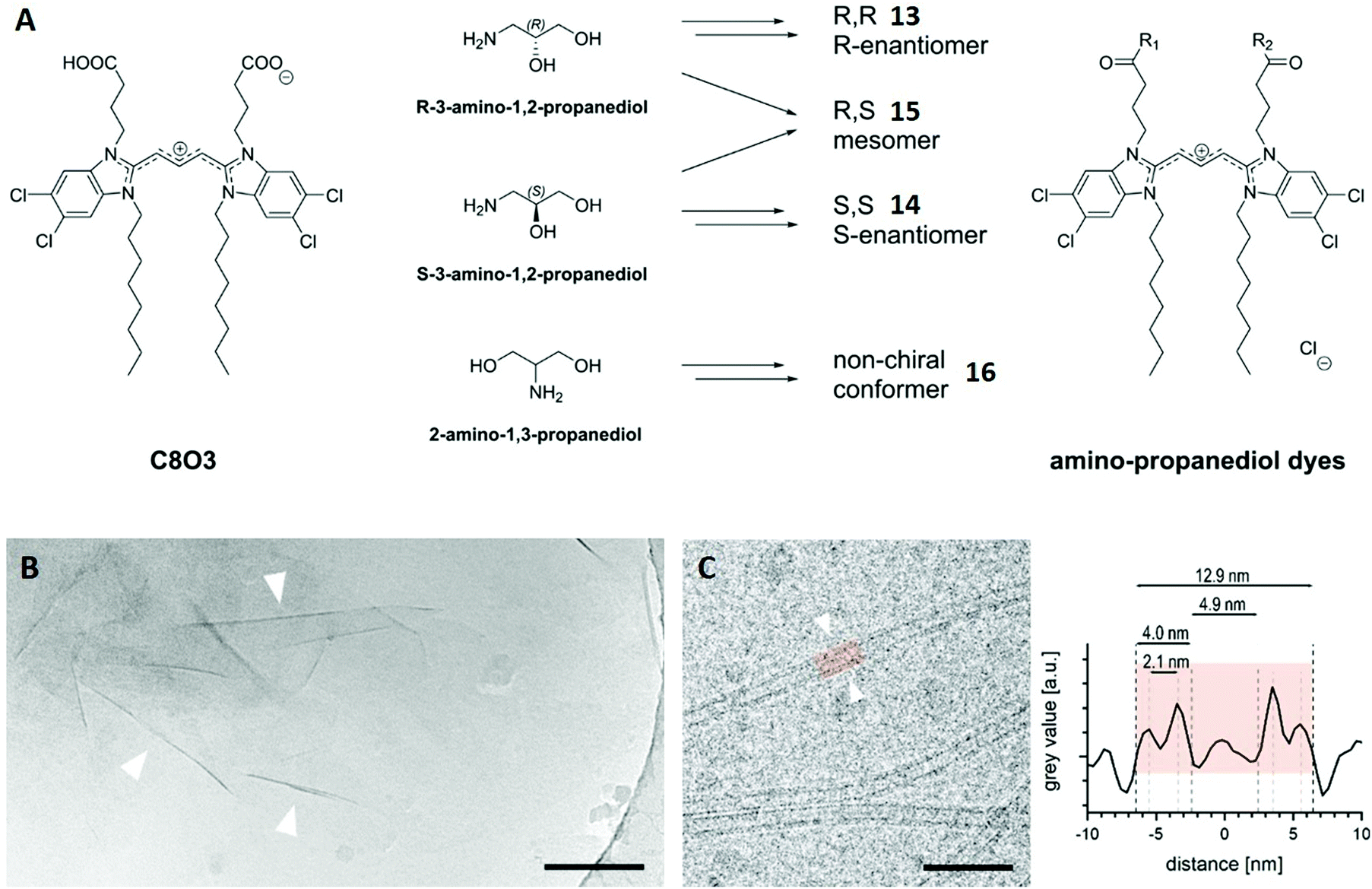 | ||
| Fig. 9 (A) Chemical structure of C8O3 and schematic synthesis of the four cyanine dye isomers 13–16. (B) Cryo-TEM image of a 0.1 mM solution of the enantiomer 13. Wrinkles of the observed sheets are marked with arrowheads. Scale bar: 200 nm. (C) Cryo-TEM image of a 1 mM solution of mesomer 15 showing individual tubes (left). Line plot across the highlighted tube-section (right) allows the characterisation of the tube and its different distances. Scale bar: 50 nm. Reproduced from ref. 79 with permission from The Authors, Copyright © 2020. Published by Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Cryo-TEM revealed sheet-like assemblies for both enantiomers 13 and 14 with no tendency to form stacked sheets. The thickness of a selected sheet was roughly estimated to 6 nm using the wrinkles in the cryo-TEM image (arrowheads in Fig. 9B). Conversely, the mesomeric isomer 15 did not exhibit sheet-like aggregates, but instead nanotubes. The grey value of the cryo-TEM image, which represents the spatially located electron density, was averaged and plotted along its central axis for a selected tube section (Fig. 9C). This revealed a double-layer architecture for the tubular assembly and also provided distances for the outer (12.9 nm) and inner diameter (4.9 nm) as well as the wall thickness (4.0 nm) of this particular tube. The outer diameter was averaged for hundreds of tubes after different time intervals, showing an almost constant value (12.5–12.7 nm) with a small standard deviation.
Several twisted tube bundles were also observed in cryo-TEM images of 15 (Fig. 10A). In order to investigate their structure, a tomogram (cryo-ET) was reconstructed from 65 images at tilt angles between −65° and 63° for a selected tube bundle. The slice along the boxed bundle showed a hexagonal arrangement of five tubes around a sixth tube in the centre (Fig. 10B), which could be followed along the long axis in the tomogram. Complete 360° rotations were used to reconstruct the entire bundle volume (Fig. 10C). Interestingly, the wall thickness was the same for the inner (between the tubes) and outer walls. Thus, the double-layered wall was not present inside the bundle, suggesting a formation mechanism during which one of the layers was lost.
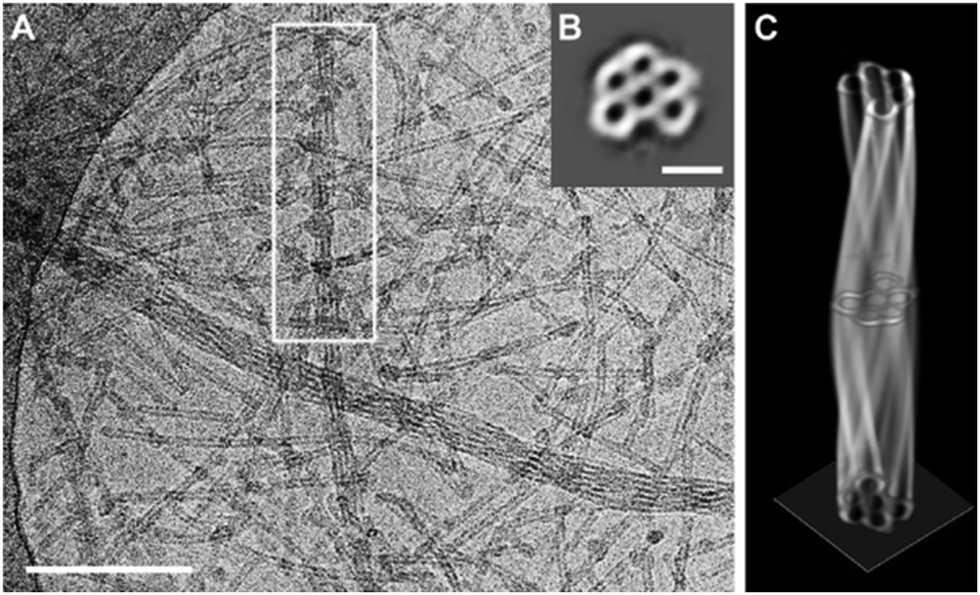 | ||
| Fig. 10 (A) Cryo-TEM image of a 1 mM solution of the mesomer 15 showing twisted tube bundles. Scale bar: 200 nm. (B) Cryo-ET image of 15 perpendicular to the long axis of the marked bundle. Summed up over 65 images from different tilt angles. Scalebar: 50 nm. (C) Constructed 3D model of the marked bundle of 15. Reproduced from ref. 79 with permission from The Authors, Copyright © 2020. Published by Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Finally, conformer 16 exhibited needle-shaped crystals in cryo-TEM, but tubular bundles and isolated tubes were also found in very small amounts. This indicated the dominance of crystallisation over supramolecular aggregation for dye 16, which was in accordance with its observed fast precipitation.
After discussing the most relevant techniques for the structural characterisation of supramolecular molecules, we move to present three examples where supramolecular systems were successfully characterised by multi-technique approaches. In particular, we highlight the powerful interplay of the already discussed techniques while also mentioning the impact of other methods.
7 Host–guest complex formation of pyridine[4]arenes
Molecular recognition describes the specific binding between two molecules via non-covalent interactions, for example between a host and a guest molecule.80,81 This concept represents a major challenge in supramolecular chemistry because it is particularly important for catalytic processes and molecular sensors. In this context, binding preferences of macrocycles towards cationic, neutral or anionic guests are a critical aspect of fundamental supramolecular research.In the first case study, the complexation behaviour of tetraisobutyloctahydroxypyridine[4]arene towards hexafluoro-phosphate anions (PF6−) and solvent molecules was investigated.82 Octahydroxypyridine[4]arenes are very similar to the already discussed resorcin[4]arenes (Fig. 2A), with the difference that these molecules are based on pyridine instead of benzene scaffolds (structure 17 in Fig. 11A). Prior to this study, contradicting reports existed proposing different binding sites for octahydroxypyridine[4]arenes and resorcinarenes, especially regarding anionic guests, promoting the authors to investigate this topic further with a complementary set of different methods.82
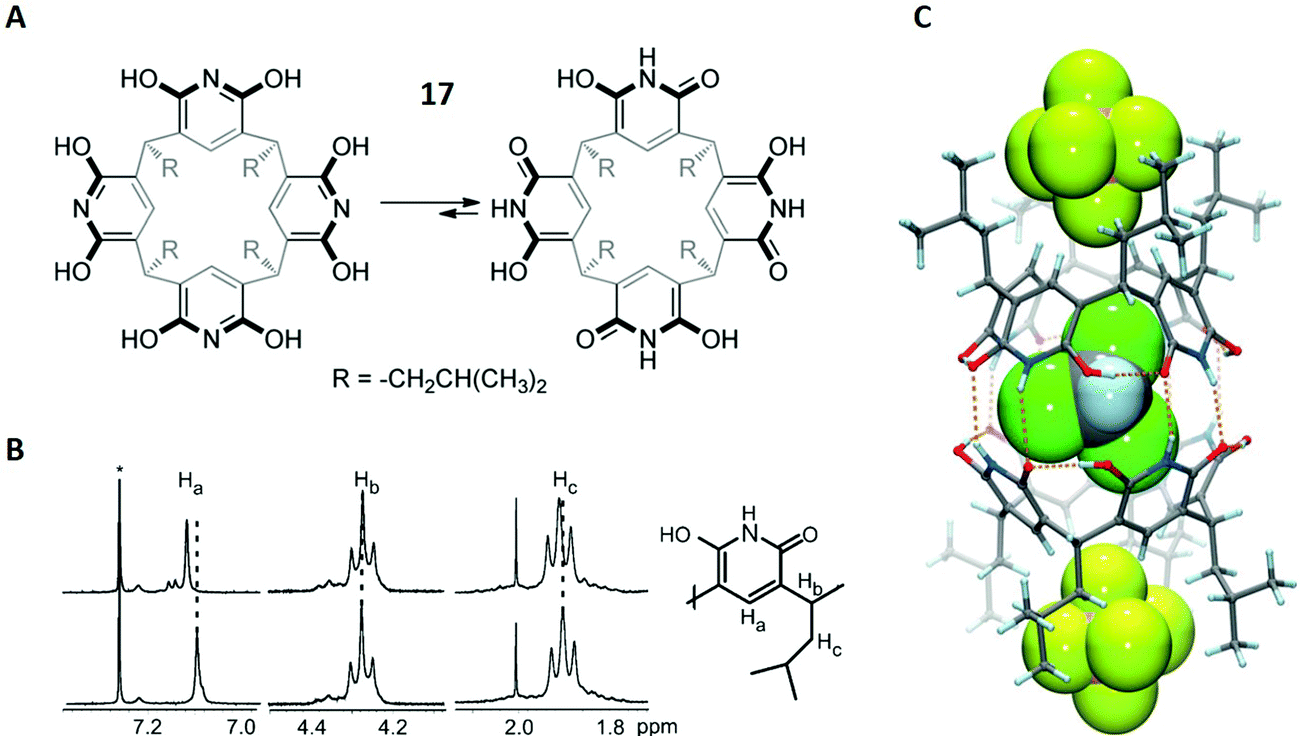 | ||
| Fig. 11 (A) Structures of both tautomeric forms of 17. (B) Selected region of the 1H-NMR spectra of 17 (bottom) and 17 + PF6− (top). CDCl3 is labelled with an asterix. (C) Crystal structure of [172 + 2 PF6(exo) + CHCl3(endo)]2−. PF6− and CHCl3 are shown in spacefilling mode. Dashed orange lines represent hydrogen bonds. Reproduced from ref. 82 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Copyright © 2017. | ||
Kiesilä et al. started with the characterisation of 17 and PF6− in chloroform solution and examined the resulting complex with different NMR methods. Previously, an equilibrium between hexameric and dimeric aggregates was described for a similar octahydroxypyridine[4]arene, in which the authors assigned a diffusion coefficient of D = 0.37 × 10−5 cm2 s−1 to the dimer.83 This value matched well with D = 0.411 × 10−5 cm2 s−1, which was obtained by DOSY NMR on three different resonances in the 1H-NMR spectrum.
After the authors had proved that 17 occurs as a dimer in solution (172), the 1H-NMR spectrum was used to obtain information on the binding site of the PF6− anion. Notably, the spectrum showed downfield shifts for the hydrogen atoms Ha and Hc, suggesting interactions of the CH-units with the PF6− ion (Fig. 11B). This result was in favour of exo binding (PF6− outside the cavity). Additionally, a comparison between the 19F-NMR spectra of PF6− and 17 + PF6− also showed a downfield shift upon complexation. This strongly indicated exo complexation, since an upfield shift would have been expected for endo binding (PF6− inside the cavity). Thus, with all the gathered information, it became clear that PF6− is non-covalently bound outside the cage in chloroform solution.
Although this is not the scope of this review, it should be noted that the authors were able to obtain a crystal structure of 17 + PF6− (Fig. 11C). The structure showed that PF6−, like in solution, is attached to rather than encapsulated in the dimeric cavity 172, although it is located in the pockets of the hydrophobic isobutyl groups. However, a chloroform molecule was found in the inside of the cage, representing the first solid-state structure in which endo complexation of a solvent simultaneously occurred to anionic exo binding.
Furthermore, the complexation behaviour of 17 in combination with PF6− was investigated in the gas phase. ESI-MS showed peaks for the deprotonated dimer [172 − H]−, its solvent adducts [172 − H + S]− and different ternary complexes [172 + PF6 + S]−, which occurred in different solvents S (Fig. 12A for S = acetone). It should also be noted that the authors did not find any signal corresponding to a host–guest complex with the monomeric 17, clearly suggesting the dimer as the driving molecule for inclusion complex formation.
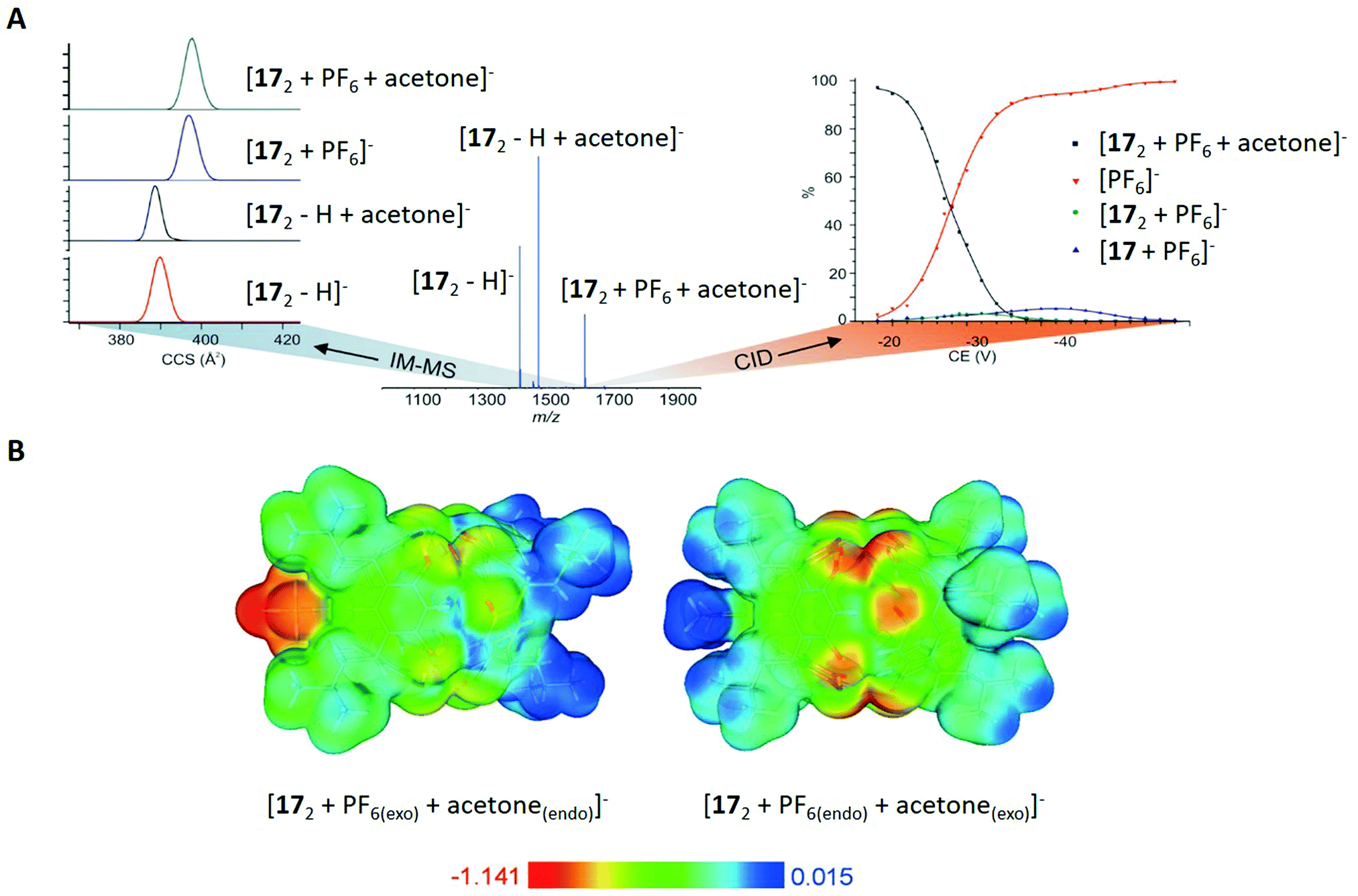 | ||
| Fig. 12 (A) Left: Ion mobility spectra for selected ions of 172, centre: ESI-MS spectrum of 17 with an equimolar amount of NH4PF6 in acetone, right: collision induced dissociation (CID) curves for the ternary complex [172 + PF6 + acetone]−. (B) Calculated electrostatic potentials for the ternary complexes [172 + PF6(exo) + acetone(endo)]− and [172 + PF6(endo) + acetone(exo)]− mapped on the isosurface of the total electron density (0.003 a.u.). Reproduced from ref. 82 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Copyright © 2017. | ||
For the determination of the binding sites of S and PF6−, different ternary ions [172 + PF6 + S]− were m/z-selected and activated in a tandem mass spectrometry experiment, showing the dominant release of the PF6− anion as the first fragmentation step (Fig. 12A on the right side). This was clearly in favour of the already observed structure in solution and the solid state, because the exo bounded species is expected to leave the complex before the endo molecule due to differences in binding energy.
Additional proof came from ion mobility mass spectrometry, which showed smaller CCS values for the species [172 − H]− and [172 + S − H]− compared to [172 + PF6]− and [172 + PF6 + S]− (with S = acetone, ΔCCS = 8 Å, Fig. 12A on the left side). This again underlines the exo complexation of PF6−, since the biggest change in CCS values should be caused by the species that is attached to the cavity rather than encapsulated in its interior. Moreover, the authors assumed a spherical shape for the molecules and calculated their diameter based on CCS values. For the dimeric species in general, the comparison of those values (between 2.2 and 2.3 nm) with the hydrodynamic radius obtained from DOSY NMR (2.0 nm) and with the X-ray crystal structure (1.9 nm) proved similar structures of the host–guest complex in solution, gas phase and in the solid state.
Complementary to the discussed experiments, the authors applied density functional calculations (DFT) for the inclusion complexes in vacuum. In agreement with the gas phase experiments, the complex [172 + PF6(exo) + S(endo)]− was energetically favoured compared to [172 + PF6(endo) + S(exo)]− (with S = acetone). Calculated electrostatic potential surfaces were used to illustrate the cause for the observed behaviour, showing that the polarised and electron-rich interior is barely suitable for anion binding (Fig. 12B). However, the electron-poor exterior (isobutyl chains) was revealed to have a better affinity towards anions.
In summary, Kiesilä et al. demonstrate the benefit of applying several analytical approaches to shed light on the complexation behaviour of an octahydroxypyridine[4]-arene in solution, gas phase and the solid state. Based on the obtained results from different techniques and contrary to prior studies, they revealed that exo binding is the main path of complexation for PF6− anions.82 Additionally, they observed a similar behaviour for other anions and solvents, suggesting a general trend of this interesting phenomenon.
8 Self-assembly of supramolecular polymers via hydrophobic interactions
Hydrophobic interactions show a significant lack of directionality compared to other non-covalent interactions, which represents a major problem for the rational design of supramolecular polymers, particularly in aqueous solution. A decade ago the design of polymers based on pairwise hydrophobic interactions, which are comparable to hydrogen bonds in terms of directionality, was only possible with protected π-surfaces.84 This changed with the work of Ustinov et al. who used benzene scaffolds and substituted them with perylene diimide groups (PDIs, Fig. 13A).84 The PDIs additionally carried polyethylene glycol units and were either located above (1,3,5 substituents) or below (2,4,6 substituents) the phenyl plane, favouring the construction of stacked polymers (Fig. 13B). The authors synthesised two compounds 18 and 19 and studied their self-assembly behaviour in solution with several techniques.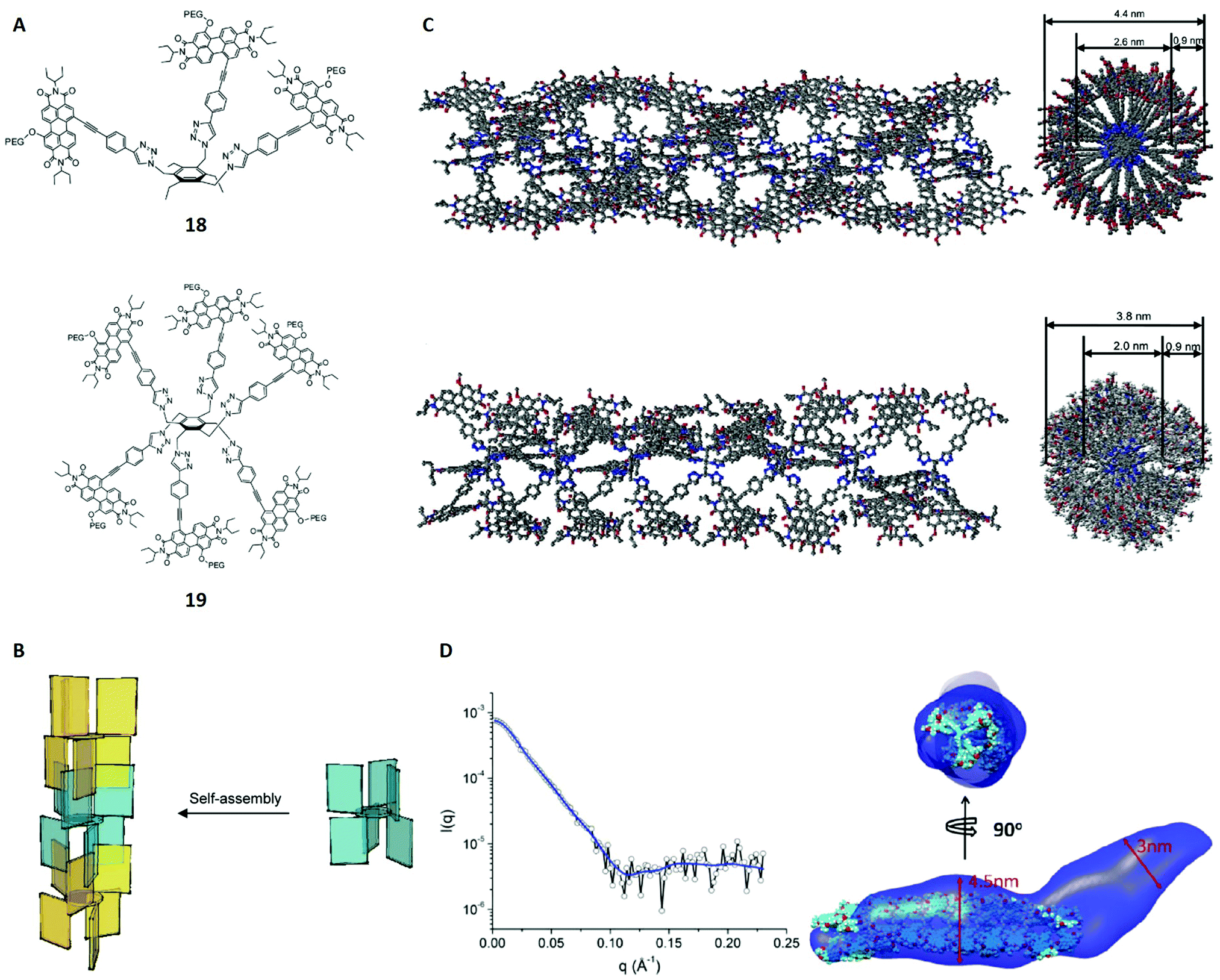 | ||
| Fig. 13 (A) Structures of the monomers 18 and 19. (B) Schematic self-assembly pattern for structure 19. Different colours were used for alternate molecules. (C) Modelled polymers for 18 (top, 22 molecules used) and 19 (bottom, 7 molecules used) in side (left) and top view (right). Polyethene glycol units and hydrogen atoms were omitted for clarity. (D) Left: SAXS spectrum of 1 × 10−5 M 19 in water/THF (70:30, v/v) including the envelope fit (blue line). Right: Two views of the overlay of the SAXS envelope of 19 (blue) and the molecular model from (C). Reproduced from ref. 84 with permission from American Chemical Society, Copyright © 2011. | ||
Initially, UV-vis and fluorescence spectra suggested the occurrence of aggregation in water/THF mixtures based on a hypsochromic shift of the absorption band and other peak patterns that are characteristic for cofacial aggregation of PDIs. In order to structurally characterise the assumed polymers, cryo-TEM was applied and revealed fiber-like aggregates for 18 and 19 in a water/THF mixture (7:3, v/v). External (4.4 ± 0.3 nm and 3.8 ± 0.4 nm) and internal diameters (2.6 ± 0.2 nm and 2.0 ± 0.2 nm), wall thicknesses (0.9 ± 0.2 nm for both) and tube lengths (166 ± 81 nm and 183 ± 96 nm) could be derived for the aggregates of 18 and 19 from the cryo-TEM images. Additionally, dynamic light scattering (DLS) was applied to determine the particle size distributions.85 The hydrodynamic radii obtained from DLS (219 ± 39 nm and 162 ± 62 nm) agreed with the tube length values from cryo-TEM within the margin of error. Moreover, molecular modelling calculations were performed, showing that pairwise interactions through the PDI surfaces fit the cryo-TEM imaged fibres very well for both aggregates (Fig. 13C). The models also suggested that the self-assembly of 19 yields fibres with cavities of around 1 nm3, making it an interesting candidate for the encapsulation of hydrophobic molecules.
Further structural proof came from EPR spectroscopy, which is known to give a signal for partially reduced radical anions of the PDI groups.86 Notably, its peak width depends on the number of PDI groups associated with the electron hopping process, making it possible to confirm or contradict the proposed model of pairwise interactions. After reduction with sodium dithionite, both aggregates showed an EPR signal with a width of 3.01 G in accordance with electron hopping between two PDI groups (pairwise interactions).86
As the last complementary technique, SAXS was applied on the assemblies of 18 and 19 in order to confirm the results of the theoretical model. For that purpose, structural envelopes were reconstructed according to the literature,87 yielding a cylinder-like shape for both aggregates (Fig. 13D). The cross sectional diameters obtained from SAXS (≈5.0 nm and 3.0–4.5 nm) agree roughly with the model parameters. Further comparison of the derived envelopes and the modelled structures is illustrated and also show a good agreement. Thus, the SAXS studies are consistent with the modelled and imaged structures.
Another interesting analysis, arguably also related to the structural characterisation of 18 and 19 in their aggregated forms, is the determination of the average number of monomers in a polymer (polymerisation degree). The authors showed that this is possible with fluorescence spectroscopy, because they were able to assign one peak/shoulder (≈620 nm) to the monomers and a second band (≈700 nm) to the assemblies. Due to broad overlapping signals, reliable deconvolution was not possible for the assembly of 18. However, the authors obtained average polymerisation degrees between 10 and 140 for 19 in solution, with the value depending on the concentration and the temperature of the system. These studies shed light on thermodynamic properties of polymeric 19, but further details are beyond the scope of this review.
In conclusion, Ustinov et al. present the first example of supramolecular polymers that are based on pairwise hydrophobic interactions between unprotected π-surfaces. With this study, the authors implemented the starting point for a new direction in the design process of hydrophobic polymers. Determining the polymeric structures of 18 and 19 in solution was achieved by a powerful combination of UV-vis and fluorescence spectroscopy, cryo-TEM, DLS, molecular modelling, EPR as well as SAXS. The discussed in-depth characterisation of both assemblies would hardly have been possible with fewer methods, again emphasising the relevance of multi-technique approaches for supramolecular structures.
9 Type III-B rotaxane dendrimers
The word dendrimer, which is derived from the greek word dendron (engl.: tree), describes a class of hyperbranched molecules with repetitive units (similar to fractals). These molecules are known as established compounds and potential candidates for a wide range of applications, among others drug delivery, catalysis and chemical sensing.88 Combining these compounds with rotaxanes leads to a new class of supramolecular molecules called rotaxane dendrimers, which can be divided into three different types (I, II and III) and subtypes (A, B and C).89–91In this case study, Kwan et al. report the synthesis of type III-B rotaxane dendrimers of three (G1–G3) and dendrons† of four generations (G1–G4, Fig. 14A).91 They used a HPF6 dibenzyl-ammonium (DBA) thread featuring two acetylene groups and a functionalised crown ether macrocycle as building blocks. Dendrons (G1–G4) and dendrimers (G1–G3) were formed via a copper catalysed azide–alkyne cycloaddition (CuAAC) applied before and after modifying functional groups at the branching points, in detail depending on the particular step in the process.
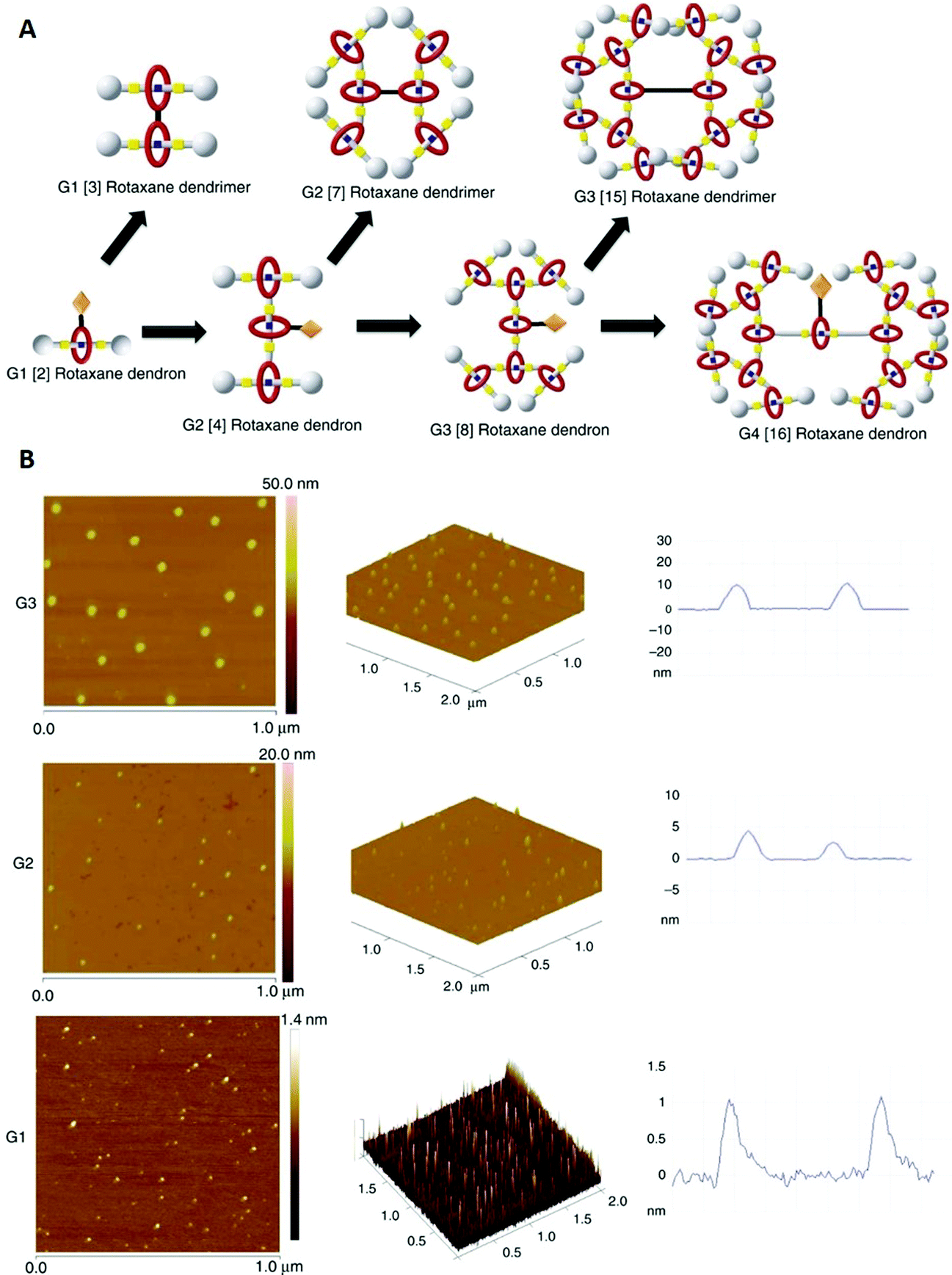 | ||
| Fig. 14 (A) Schematic of the different type III-B rotaxane dendrimers and dendrons. (B) Left and centre: AFM pictures of G1, G2 and G3 dendrimers on a mica surface. Right: Height profiles of the corresponding AFM images. Reproduced from ref. 91 with permission from The Author(s), Copyright © 2018. | ||
In order to characterise the desired compounds, 1H- and 13C-NMR spectroscopy were applied for both dendrimers and dendrons. For example, when comparing the isolated thread with the G1 dendron, the proton signal in the amine group showed a significant downfield shift and a split to a triplet due to the formed hydrogen bond with the macrocycle. Aromatic proton signals of both thread and macrocycle shifted upfield caused by shielding and stabilisation effects. Furthermore, when comparing the three dendrimer NMR spectra, most signals become broader and less resolved for higher generations. This is due to the higher number of repeating units, clearly indicating the synthesis of more complex species. Further details on the 1D-NMR spectra assignments can be found in the original manuscript and the corresponding ESI.
Proof of purity came from DOSY NMR, which showed a single and pure component for each dendrimer (G1–G3). Moreover, two-dimensional NOESY NMR was performed to check the correct position of the macrocycles within the rotaxane dendrimers. The authors found clear cross peaks between the aromatic protons of the thread and aliphatic protons of the macrocycle in each spectrum, which confirmed the expected location of the crown ether.
Although the discussed 1D and 2D NMR spectra were in favour of the desired products, they were hardly sufficient for an unambiguous characterisation. As mentioned in Section 2, highly symmetric and big molecules (like the described rotaxane dendrimers and dendrons) are difficult to distinguish with NMR since the chemical environments of the signals are often similar and can overlap.8 However in this case, ultimate proof was obtained from ESI-MS. Different charge state peaks of the formula [M − z PF6]z+ (z = 2, 3, 6, 13, depending on the molecule) were found for all dendrimers and dendrons confirming the correct synthetic products.
The authors further tested the molecules’ shuttling behaviour caused by the pH-sensitivity of the rotaxane moieties. After deprotonation, strong signal shifts were observed in the 1H-NMR spectra compared to the protonated species. The amine group protons significantly shifted upfield, suggesting the loss of the hydrogen bond to the macrocycle. Contrary, the protons located at the triazole units, which were formed via CuAAC, shifted downfield indicating the new location of the macrocycle. This was also confirmed by NOESY, which showed cross peaks through-space between the triazole unit and the crown ether macrocycle, and a density-functional based tight binding calculation (DFTB+), revealing the preference of the crown ether for the triazole unit in the deprotonated species. Furthermore, the PF6− counterions should have been completely removed upon deprotonation. This was shown by 31P-NMR, which did not yield any signal. Additionally, MALDI-MS confirmed the synthesis of the deprotonated G1 and G2 dendrimers, whereas the authors could not obtain the high molecular-weight peak for G3.
In order to proof the size of the rotaxane dendrimers, dynamic light scattering (DLS, Section 8) was applied and yielded hydrodynamic radii for the dendrimers of all three generations (G1 = 1.80 ± 0.14 nm, G2 = 3.50 ± 0.21 nm and G3 = 4.57 ± 0.81 nm). As expected, the higher generation dendrimers were bigger and this trend was also confirmed by hydrodynamic radii and diameters obtained from DOSY. DOSY was also used to investigate the size of the deprotonated molecules, showing slightly larger species compared to the original cationic species. This suggested an unfolding process caused by molecular shuttling, which was also indicated by the DFTB+ study.
Additionally, atomic force microscopy (AFM) was used to probe the size/height of the synthesised dendrimers on a mica surface (Fig. 14A and B). AFM is a well-established high-resolution microscopic technique, which is mainly used for topographic imaging of surfaces.92 Here Kwan et al. reported nearly spherical morphologies for all dendrimers, but unsurprisingly different averaged molecule heights/sizes (G1 ≈ 1.15 nm, G2 ≈ 3.94 nm and G3 ≈ 10.96 nm, Fig. 14B right). For the G1 and G2 dendrimers, all three techniques (DLS, DOSY and AFM) showed similar sizes, whereas the size of the G3 dendrimer is significantly higher in AFM images. The authors suggested a decrease in adsorption forces for the higher generation dendrimers as a possible explanation, which could reduce flattening on the mica surface and therefore yield a higher height than measured by DLS and DOSY in solution. In accordance with the hydrodynamic radii obtained from DLS and DOSY, AFM also revealed a significant height increase for the deprotonated species.
In summary, Leung and co-workers synthesised novel Type III-B rotaxane dendrimers through copper catalysed azide–alkyne cycloadditions and were able, among other things, to isolate a G3 Type III-B rotaxane dendrimer for the first time. This was a major advance in the difficult design process of these complex, but also beautiful molecules. Dendrimer and dendron were characterised by a combination of NMR and MS methods, whereas DLS, DOSY NMR and AFM were additionally applied to investigate the dendrimer sizes. Overall, this case study is another impressive example of synthetic supramolecular chemistry and corresponding structural characterisation, which was once again achieved by different complementary analytical techniques.
10 Conclusions and outlook
This review has provided an overview of the most relevant techniques for the structural characterisation of supramolecular molecules with a focus on techniques that do not require the material to crystallise. Fundamental concepts to introduce NMR, EPR, MS, IM-MS, SANS, SAXS and cryo-TEM have been presented and illustrated with examples from different areas of supramolecular chemistry. We also presented three in-depth case studies in which the authors used multiple techniques to address research questions related to host–guest complexes, self-assembled polymers and interlocked structures, while focussing on the powerful and dynamic interplay of the already discussed methods. Further analytical techniques and theoretical calculations were touched in order to provide a rounded introduction to the interesting and important topic of analytical supramolecular chemistry.When examining the particular strengths and weaknesses of the highlighted methods, we found in particular NMR and MS/IM-MS to be the most convincing alternatives to X-ray crystallography. Most supramolecular structural characterisation problems can be addressed with one or both of these techniques, yielding reliable results in solution and gas phase. However, different shortcomings described in this article, frequently lead to the need of using complementary techniques like EPR, SANS, SAXS and cryo-TEM, among others. Although these methods are less common and not always applicable, they also provide valuable results for a range of systems (Fig. 1).
Additionally to the experimental techniques previously discussed, theoretical calculations have emerged as game changers in supramolecular chemistry. The rapid progress made in the last decades (e.g. in DFT calculations and empirical methods) is inevitably connected to the massive advance of the field. Because this development will likely continue in the future, it deserves even more attention in the supramolecular regime.
Generally the future of supramolecular chemistry, which is still in its early days, can hardly be overestimated. Supramolecular chemists already managed to find solutions on urging challenges of our time and laid the foundation for even more promising applications in the nearer future. Structurally characterisation will remain a decisive aspect of this development, in particular when high-throughput methods are needed for moving to an industrial scale. From all the discussed methods, we believe that mass spectrometry, coupled with ion mobility spectrometry, is perhaps the most suitable for that purpose.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. G. is grateful for funding through the President's Doctoral Scholar Award by The University of Manchester. The authors would like to thank Sandra Piel, RWTH Aachen, and Dr. Lennart Ramakers, Wageningen University & Research, for helpful discussions and comments as well as Prof. Marco Lucarini, University of Bologna, for providing relevant EPR spectra. This work was supported by an EPSRC Established Career Fellowship (EP/R011079/1) and a European Research Council Advanced Grant (ERC-2017-ADG-786734) to R. E. P. W.References
- B. Baytekin, H. T. Baytekin and C. A. Schalley, Org. Biomol. Chem., 2006, 4, 2825–2841 RSC
.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13–26 CrossRef PubMed
- H. Cui and B. Xu, Chem. Soc. Rev., 2017, 46, 6430–6432 RSC
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC
- J. L. Casas-Hinestroza, M. Bueno, E. Ibáñez and A. Cifuentes, Anal. Chim. Acta, 2019, 1081, 32–50 CrossRef CAS PubMed
-
J. Ujma, F. Benoit and P. E. Barran, Comprehensive Supramolecular Chemistry II, Elsevier, 2nd edn, 2017, vol. 6, pp. 389–403 Search PubMed
- A. Wishard and B. C. Gibb, Supramol. Chem., 2019, 31, 608–615 CrossRef CAS PubMed
-
C. Tedesco, Comprehensive Supramolecular Chemistry II, Elsevier, 2nd edn, 2017, vol. 2, pp. 45–73 Search PubMed
-
G. D. Enright, S. Takeya and J. A. Ripmeester, Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons, Ltd, 2012 Search PubMed
-
A. G. Slater, P. H. Beton and N. R. Champness, Comprehensive Supramolecular Chemistry II, Elsevier, 2nd edn, 2011, vol. 2, pp. 1440–1448 Search PubMed
-
E. L. Gavey and J. M. Rawson, Comprehensive Supramolecular Chemistry II, Elsevier, 2nd edn, 2017, vol. 2, pp. 151–180 Search PubMed
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211–5233 RSC
- M. R. Hansen, R. Graf and H. W. Spiess, Chem. Rev., 2016, 116, 1272–1308 CrossRef CAS PubMed
- J. J. Alcázar, N. Geue, V. Valladares, A. Cañete, E. G. Pérez, L. García-Río, J. G. Santos and M. E. Aliaga, ACS Omega, 2021, 6, 10333–10342 CrossRef PubMed
- Y. Cohen, S. Slovak and L. Avram, Chem. Commun., 2021, 57, 8856–8884 RSC
- M. Lucarini, Eur. J. Org. Chem., 2020, 2995–3008 CrossRef CAS
- Y. Cohen and S. Slovak, Org. Chem. Front., 2019, 6, 1705–1718 RSC
- T. Evan-Salem, I. Baruch, L. Avram, Y. Cohen, L. C. Palmer and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12296–12300 CrossRef CAS PubMed
- M. R. Chierotti and R. Gobetto, Chem. Commun., 2008, 1621–1634 RSC
-
M. R. Chierotti and R. Gobetto, Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons, Ltd, 2012 Search PubMed
- H. W. Spiess, Isr. J. Chem., 2014, 54, 16–24 CrossRef CAS
-
A. Comotti, S. Bracco and P. Sozzani, Comprehensive Supramolecular Chemistry II, Elsevier, 2nd edn, 2017, vol. 2, pp. 75–99 Search PubMed
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS PubMed
-
Y. Cohen, L. Avram, T. Evan-Salem, S. Slovak, N. Shemesh and L. Frish, Analytical Methods in Supramolecular Chemistry, Wiley, 2nd edn, 2012, pp. 197–285 Search PubMed
- L. Avram and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586–602 RSC
- A. G. S. Hogberg, J. Org. Chem., 1980, 45, 4498–4500 CrossRef
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2002, 124, 15148–15149 CrossRef CAS PubMed
- L. Avram and Y. Cohen, Org. Lett., 2003, 5, 3329–3332 CrossRef CAS PubMed
- S. Pappalardo, V. Villari, S. Slovak, Y. Cohen, G. Gattuso, A. Notti, A. Pappalardo, I. Pisagatti and M. F. Parisi, Chem. – Eur. J., 2007, 13, 8164–8173 CrossRef CAS PubMed
- C. Capici, Y. Cohen, A. D’Urso, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi, R. Purrello, S. Slovak and V. Villari, Angew. Chem., Int. Ed., 2011, 50, 11956–11961 CrossRef CAS PubMed
- M. M. Roessler and E. Salvadori, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC
- E. Mezzina, R. Manoni, F. Romano and M. Lucarini, Asian J. Org. Chem., 2015, 4, 296–310 CrossRef CAS
- E. G. Bagryanskaya and S. R. A. Marque, Electron Paramagn. Reson., 2017, 25, 180–235 Search PubMed
- M. Lucarini and E. Mezzina, Electron Paramagn. Reson., 2011, 22, 41–70 CAS
- P. Franchi, M. Lucarini and G. Pedulli, Curr. Org. Chem., 2005, 8, 1831–1849 CrossRef
- M. Lucarini, B. Luppi, G. F. Pedulli and B. P. Roberts, Chem. – Eur. J., 1999, 5, 2048–2054 CrossRef CAS
- P. Franchi, C. Casati, E. Mezzina and M. Lucarini, Org. Biomol. Chem., 2011, 9, 6396–6401 RSC
- R. Pievo, C. Casati, P. Franchi, E. Mezzina, M. Bennati and M. Lucarini, ChemPhysChem, 2012, 13, 2659–2661 CrossRef CAS PubMed
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed
- P. Franchi, V. Bleve, E. Mezzina, C. Schäfer, G. Ragazzon, M. Albertini, D. Carbonera, A. Credi, M. Di Valentin and M. Lucarini, Chem. – Eur. J., 2016, 22, 8745–8750 CrossRef CAS PubMed
- S. J. Lockyer, S. Nawaz, A. Brook, A. J. Fielding, I. J. Vitorica-yrezabal, G. A. Timco, N. A. Burton, A. M. Bowen, R. E. P. Winpenny and E. J. L. Mcinnes, J. Am. Chem. Soc., 2020, 142, 15941–15949 CrossRef CAS PubMed
- C. A. Schalley, Mass Spectrom. Rev., 2001, 20, 253–309 CrossRef CAS PubMed
- L. Cera and C. A. Schalley, Chem. Soc. Rev., 2014, 43, 1800–1812 RSC
- P. Bayat, D. Lesage and R. B. Cole, Mass Spectrom. Rev., 2020, 1–23 Search PubMed
- E. Kalenius, M. Groessl and K. Rissanen, Nat. Rev. Chem., 2019, 3, 4–14 CrossRef CAS
- L. Polewski, A. Springer, K. Pagel and C. A. Schalley, Acc. Chem. Res., 2021, 54, 2445–2456 CrossRef CAS PubMed
- O. H. L. Williams and N. J. Rijs, Front. Chem., 2021, 9, 682743 CrossRef CAS PubMed
- M. Sawada, Y. Takai, T. Kaneda, R. Arakawa, M. Okamoto, H. Doe, T. Matsuo, K. Naemura, K. Hirose and Y. Tobe, Chem. Commun., 1996, 1735–1736 RSC
- G. Hornung, C. A. Schalley, M. Dieterle, D. Schroder and S. Derek, Chem. – Eur. J., 1997, 3, 1866–1883 CrossRef CAS
- A. Kruve, K. Caprice, R. Lavendomme, J. M. Wollschläger, S. Schoder, H. V. Schröder, J. R. Nitschke, F. B. L. Cougnon and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 11324–11328 CrossRef CAS PubMed
- A. P. France, L. G. Migas, E. Sinclair, B. Bellina and P. E. Barran, Anal. Chem., 2020, 92, 4340–4348 CrossRef CAS PubMed
- P. A. Faull, K. E. Korkeila, J. M. Kalapothakis, A. Gray, B. J. McCullough and P. E. Barran, Int. J. Mass Spectrom., 2009, 283, 140–148 CrossRef CAS
- J. R. N. Haler, D. Morsa, P. Lecomte, C. Jérôme, J. Far and E. De Pauw, Methods, 2018, 144, 125–133 CrossRef CAS PubMed
- D. A. Jacques and J. Trewhella, Protein Sci., 2010, 19, 642–657 CrossRef CAS PubMed
-
L. A. Feigin and D. I. Svergun, Structure Analysis by Small-Angle X-Ray and Neutron Scattering, Springer Science, 1987 Search PubMed
- O. Dunne, M. Weidenhaupt, P. Callow, A. Martel, M. Moulin, S. J. Perkins, M. Haertlein and V. T. Forsyth, Eur. Biophys. J., 2017, 46, 425–432 CrossRef CAS PubMed
- H. Endo, K. Mayumi, N. Osaka, K. Ito and M. Shibayama, Polym. J., 2011, 43, 155–163 CrossRef CAS
- A. Dawn, M. Mirzamani, C. D. Jones, D. S. Yufit, S. Qian, J. W. Steed and H. Kumari, Soft Matter, 2018, 14, 9489–9497 RSC
-
H. Kumari, Comprehensive Supramolecular Chemistry II, Elsevier, 2nd edn, 2017, vol. 2, pp. 289–302 Search PubMed
-
A. C. Toma and T. Pfohl, Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons, Ltd, 2012 Search PubMed
-
C. D. Putnam, M. Hammel, G. L. Hura and J. A. Tainer, Q. Rev. Biophys., 2007, 40, 191–285 Search PubMed
- P. Lindner and G. Wignall, MRS Bull., 1999, 24, 34–39 CrossRef CAS
- S. H. Chen, E. Y. Sheu, J. Kalus and H. Hoffman, J. Appl. Crystallogr., 1988, 21, 751–769 CrossRef CAS
- H. Kumari, S. R. Kline, S. R. Kennedy, C. Garvey, C. L. Raston, J. L. Atwood and J. W. Steed, Chem. Commun., 2016, 52, 4513–4516 RSC
- H. Kumari, S. E. Armitage, S. R. Kline, K. K. Damodaran, S. R. Kennedy, J. L. Atwood and J. W. Steed, Soft Matter, 2015, 11, 8471–8478 RSC
- A. Fernandez, J. Ferrando-Soria, E. M. Pineda, F. Tuna, I. J. Vitorica-Yrezabal, C. Knappke, J. Ujma, C. A. Muryn, G. A. Timco, P. E. Barran, A. Ardavan and R. E. P. Winpenny, Nat. Commun., 2016, 7, 1–6 Search PubMed
- H. Liu and P. H. Zwart, J. Struct. Biol., 2012, 180, 226–234 CrossRef CAS PubMed
- J. Ferrando-Soria, A. Fernandez, D. Asthana, S. Nawaz, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, C. A. Muryn, F. Tuna, G. A. Timco, N. D. Burton and R. E. P. Winpenny, Nat. Commun., 2019, 10, 1–7 CrossRef CAS PubMed
- L. E. Franken, E. J. Boekema and M. C. A. Stuart, Adv. Sci., 2017, 4, 1–9 Search PubMed
- J. Watt, D. L. Huber and P. L. Stewart, MRS Bull., 2019, 44, 942–948 CrossRef
- J. Harapin, M. Eibauer and O. Medalia, Structure, 2013, 21, 1522–1530 CrossRef CAS PubMed
- J. L. Bricks, Y. L. Slominskii, I. D. Panas and A. P. Demchenko, Methods Appl. Fluoresc., 2018, 6, 012001 CrossRef PubMed
- A. P. Deshmukh, D. Koppel, C. Chuang, D. M. Cadena, J. Cao and J. R. Caram, J. Phys. Chem. C, 2019, 123, 18702–18710 CrossRef CAS
- A. P. Deshmukh, A. D. Bailey, L. S. Forte, X. Shen, N. Geue, E. M. Sletten and J. R. Caram, J. Phys. Chem. Lett., 2020, 11, 8026–8033 CrossRef CAS PubMed
- A. Bailey, A. Deshmukh, T. Atallah, U. Barotov, M. Pengshung, E. Sletten and J. Caram, ChemRxiv, 2021 DOI:10.33774/chemrxiv-2021-rjmvz
- A. Deshmukh, N. Geue, N. Bradbury, T. Atallah, C. Chuang, M. Pengshung, J. Cao, E. Sletten, D. Neuhauser and J. Caram, ChemRxiv, 2021 DOI:10.33774/chemrxiv-2021-ql3b7-v2
- B. Schade, A. K. Singh, V. Wycisk, J. L. Cuellar-Camacho, H. von Berlepsch, R. Haag and C. Böttcher, Chem. – Eur. J., 2020, 26, 6919–6934 CrossRef CAS PubMed
- E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS PubMed
- K. Ariga, H. Ito, J. P. Hill and H. Tsukube, Chem. Soc. Rev., 2012, 41, 5800–5835 RSC
- A. Kiesilä, L. Kivijärvi, N. K. Beyeh, J. O. Moilanen, M. Groessl, T. Rothe, S. Götz, F. Topić, K. Rissanen, A. Lützen and E. Kalenius, Angew. Chem., Int. Ed., 2017, 56, 10942–10946 CrossRef PubMed
- T. Evan-Salem and Y. Cohen, Chem. – Eur. J., 2007, 13, 7659–7663 CrossRef CAS PubMed
- A. Ustinov, H. Weissman, E. Shirman, I. Pinkas, X. Zuo and B. Rybtchinski, J. Am. Chem. Soc., 2011, 133, 16201–16211 CrossRef CAS PubMed
-
J. Jeevanandam and M. K. Danquah, Research Advances in Dynamic Light Scattering, Nova Science Pub Inc, 2020 Search PubMed
- T. M. Wilson, T. A. Zeidan, M. Hariharan, F. D. Lewis and M. R. Wasielewski, Angew. Chem., Int. Ed., 2010, 49, 2385–2388 CrossRef CAS PubMed
- D. I. Svergun and M. H. J. Koch, Rep. Prog. Phys., 2003, 66, 1735–1782 CrossRef CAS
- E. Abbasi, S. F. Aval, A. Akbarzadeh, M. Milani, H. T. Nasrabadi, S. W. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Nanoscale Res. Lett., 2014, 9, 1–10 CrossRef CAS PubMed
- J. W. Lee and K. Kim, Top. Curr. Chem., 2003, 1, 111–140 CrossRef PubMed
-
C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, 2016 Search PubMed
- C. S. Kwan, R. Zhao, M. A. Van Hove, Z. Cai and K. C. F. Leung, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed
- F. J. Giessibl, Rev. Mod. Phys., 2003, 75, 949–983 CrossRef CAS
Footnote |
| † Dendrimer-like structure with a chemically addressable group, often precursor of dendrimers. |
| This journal is © The Royal Society of Chemistry 2022 |