
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoparticles for super-resolution microscopy: intracellular delivery and molecular targeting
Sumit Kumar
Pramanik
 *a,
Sreejesh
Sreedharan
b,
Rajeshwari
Tiwari
a,
Sourav
Dutta
c,
Noufal
Kandoth
c,
Surajit
Barman
c,
Stephen O
Aderinto
d,
Samit
Chattopadhyay
*e,
Amitava
Das
*c and
Jim A
Thomas
*d
*a,
Sreejesh
Sreedharan
b,
Rajeshwari
Tiwari
a,
Sourav
Dutta
c,
Noufal
Kandoth
c,
Surajit
Barman
c,
Stephen O
Aderinto
d,
Samit
Chattopadhyay
*e,
Amitava
Das
*c and
Jim A
Thomas
*d
aCSIR - Central Salt and Marine Chemicals Research Institute, Gijubhai Badheka Marg, Bhavnagar, Gujarat 364002, India. E-mail: sumitpramanik@csmcri.res.in
bHuman Science Research Centre, University of Derby, Kedleston road, DE22 1GB, UK
cDepartment of Chemical Sciences and Centre for Advanced Functional Materials, Indian Institute of Science Education and Research, Kolkata, West Bengal, India. E-mail: nf3938@iiserkol.ac.in; amitava@iiserkol.ac.in
dDepartment of Chemistry, University of Sheffield, Western Bank, Sheffield, S3 7HF, UK. E-mail: James.thomas@sheffield.ac.uk
eDepartment of Biological Sciences, BITS-Pilani, K K Birla Goa Campus, NH 17B, Zuarinagar, Goa 403726, India. E-mail: samitc@goa.bits-pilani.ac.in
First published on 24th November 2022
Abstract
Following an overview of the approaches and techniques used to acheive super-resolution microscopy, this review presents the advantages supplied by nanoparticle based probes for these applications. The various clases of nanoparticles that have been developed toward these goals are then critically described and these discussions are illustrated with a variety of examples from the recent literature.
 Sumit Kumar Pramanik | Sumit Kumar Pramanik is a senior scientist at CSIR-CSMCRI, Bhavnagar, India, and assistant professor at AcSIR. He received his BSc in chemistry from Vidyasagar University and a Master's degree in applied chemistry from Bengal Engineering and Science University. He obtained his PhD in Chemistry at CSIR-Indian Institute of Chemical Biology, Kolkata. Before joining CSIR-CSMCRI, he spent three years as a post-doctoral fellow at Hasselt University, Belgium. His research interests include the synthesis, self-assembly, colloidal and interfacial properties, and application of nanostructured materials in bioimaging, drug delivery, sensing, and optoelectronics. |
 Sreejesh Sreedharan | Sreejesh Sreedharan graduated in 2018 with a PhD in Chemical biology from the University of Sheffield under the supervision of Prof. Jim A. Thomas. He then was a Postdoctoral fellow working with Prof. Katherine Vallis and Prof. Borivoj Vojonovic at the Department of Oncology, University of Oxford where he was carrying out research related to designing multi modal therapeutic imaging probes to track down breast cancer metastases in lymphatic system. He is currently working as Lecturer in Biomedical sciences at University of Derby carrying out research related to developing noval theranostic modalities focused on Liver and Breast cancer. |
 Samit Chattopadhyay | Samit Chattopadhyay was educated at Calcutta University and Jadavpur University. Following post-doctoral studies in the USA (UCONN Health Centre, Farmington, CT and MIT), he returned to India at the National Centre for Cell Science, Pune. A former Director of the CSIR-Indian Institute of Chemical Biology, Kolkata and the CSIR-NEIST, Assam, he is currently a Senior Professor & Shri B. K. Birla & Shrimati Sarala Birla Chair Professor at the Department of Biological Sciences at BITS-Pilani, Goa. He is a fellow of Sir J. C. Bose National Fellowship and is elected fellow of all three major science academies in India. His research focuses on understanding the molecular basis of gene regulation and discovery of therapeutics against cancer. |
 Amitava Das | Amitava Das was educated at Jadavpur University (Kolkata)receiving his PhD degree in 1989. Following research at the Universities of Birmingham and Bristol (UK), he returned to India at CSIR-CSMCRI. After a stint as a Chief Scientist at CSIR-National Chemical Laboratory (Pune), he became Director of CSIR-CSMCRI and Distinguished Professor of the AcSIR. In 2020 he took up a Sr. Professorship in Chemistry at IISER, Kolkata. An elected fellow of all the three major science academies of India and recipient of the SERB-J. C. Bose National Fellowship, his research interests include supramolecular chemistry, photoinduced processes, molecular recognition, biomarkers/bioimaging, and functional nanostructures. |
 Jim A Thomas | Jim A. Thomas carried out his undergraduate studies at the University of Reading. After teaching in the UK and rural Kenya (through the UK development agency VSO), he obtained a PhD in coordination chemistry from University of Birmingham in 1993. Following a Royal Society European Fellowship with Jean-Marie Lehn (Strasbourg) he arrived in Sheffield to work with Prof Chris A Hunter, FRS, but stayed when he was awarded a Royal Society University Research Fellowship. His research focusses on metal ion complexes that recognize ions, bio-ions, and biomolecules and has led to the develop on novel cell probes, therapeutics and theranostics. |
Key learning points(1) A brief overview of key super-resolution microscopy techniques is provided.(2) The working principle of these different nanoscopy techniques for bioimaging are described. (3) A critical assessment of the application of conventional luminescent materials and their use in nanoscopy is presented. (4) The biological significance of various luminescent materials designed at the nanoscale and their organelle/tissue-specificities in super-resolution imaging application is discussed. (5) Potential future challenges, perspectives, and directions for material chemistry applied to nanoscopy are presented. |
Introduction
The cell is the structural and functional unit of living organisms. Exploring the panoply of cellular processes active within its organelles is crucial to a better understanding of many diseases. Such knowledge is vital in the discovery and development of new diagnostics and next-generation therapeutics. With the advent of advanced microscopic and imaging techniques, researchers developed methodologies to separately study each individual cell compartment, facilitating the identification of many inter- and intra-organelle biochemical processes.1,2 In more recent years, super-resolution techniques have addressed many of the limitations of conventional optical microscopy, such as substantial point spread function anisotropy, diffraction-limited resolution, depth-dependent degradation in scattering samples and volumetric bleaching.3 These developments, together with the creation of specially engineered/optimized excitation light, new luminescent dyes, highly sensitive detectors, and reconstruction algorithms, have surmounted the classical diffraction limit of optical microscopy.Nanoscopy techniques even offer the chance to visualize individual molecules as they dynamically interact during intracellular processes that drives cell behaviour.4–7 However, many of these modalities are still not established and thus their true capabilities and potentials are yet to be realized.8,9
Why super-resolution?
Since the 17th century, the optical microscope, which utilizes visible light and a system of lenses to magnify images, has played a pivotal role in unravelling vital questions in biology.10 Yet, despite many technological inventions and manufacturing breakthroughs, obtaining well-resolved images with the minimal aberration of nano-structured objects still remains a challenge. Even when computer-aided optical design and automated grinding methodology are utilized to construct lens components, diffraction effects mean that lateral distances less than half the imaging light wavelength are left unresolved.11,12 Effectively, this means that two objects that are held closer than approximately 220 nm will not image as separate structures. Initial moderate enhancements on this spatial resolution along the lateral and axial axes were achieved through confocal and multiphoton techniques.By utilizing laser excitation and a pinhole-restricted detection system, laser scanning confocal microscopy improves spatial resolution by a factor of 1.4. This improvement is achieved by a pair of re-scanning mirrors between the pinhole and detector that allows for de-coupling of the magnification of the object and scanning spot.13 In multiphoton fluorescence microscopy, nonlinear absorption processes are exploited to reduce the effective size of the excitation point-spread function, but this requires long-wavelength excitation light. Thus, the advantage of laser scanning confocal/multiphoton microscopy is limited mostly to the reduction of background signals that originate from emission sources away from the focal plane,14 and resolution remains limited to >100 nm in another plane. In the last few decades, these limitations have been broken by the development of several super-resolution techniques (Fig. 1).15–17
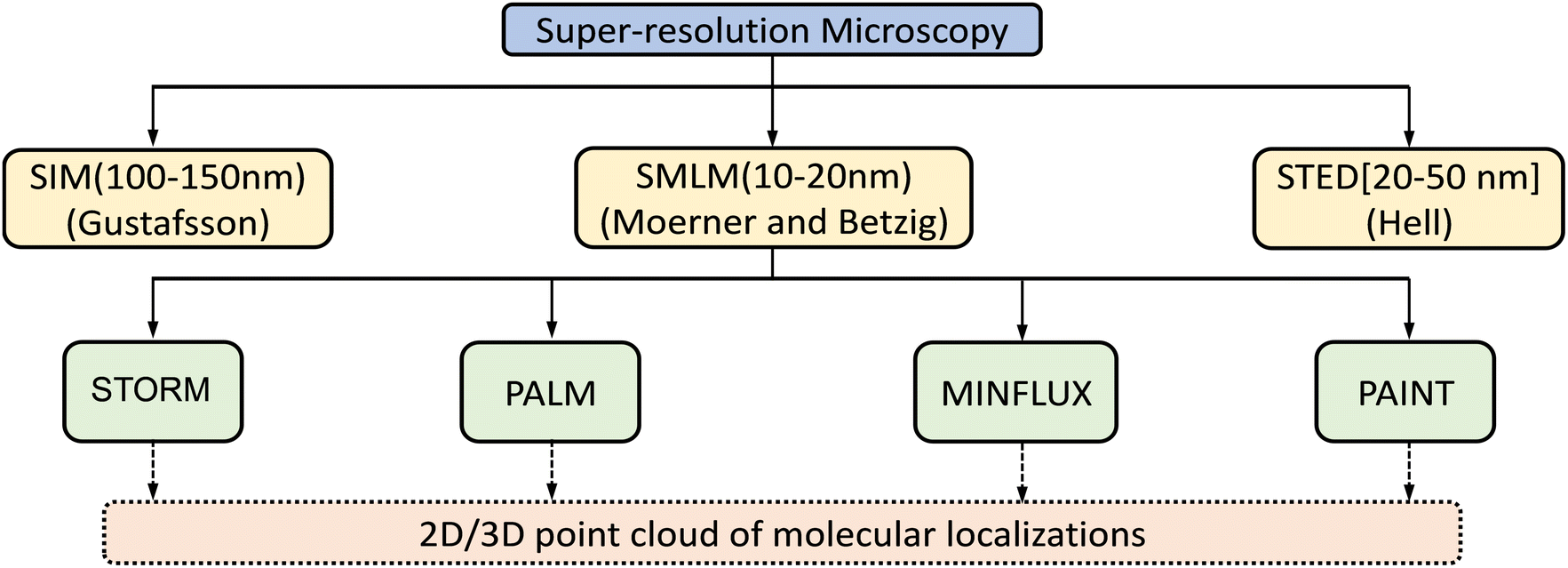 | ||
| Fig. 1 Classification of super-resolution microscopy methods that utilize localization as 2D or 3D point clouds. The abbreviations are: structured illumination microscopy (SIM); single-molecule localization microscopy (SMLM); stimulated emission depletion (STED) microscopy; stochastic optical reconstruction microscopy (STORM); photoactivated localization microscopy (PALM); minimal photon fluxes (MINFLUX); points accumulation for imaging in nanoscale topography (PAINT). | ||
The main super-resolution techniques
The key to the majority of super-resolution methods is to render dye or probe molecules visible for only a short period and/or prevent specific molecules within the specified diffraction region from being detected.12,18 Selective switching between two fluorophore states, usually an ‘Emission ON’ state and ‘Emission OFF’ state, has been utilized to develop sub-diffraction-resolution imaging.19 This methodology, generalized under the acronym RESOLFT—reversible saturable optical fluorescence transitions,20 was first realized by stimulated emission depletion, STED – Fig. 2.18,21–27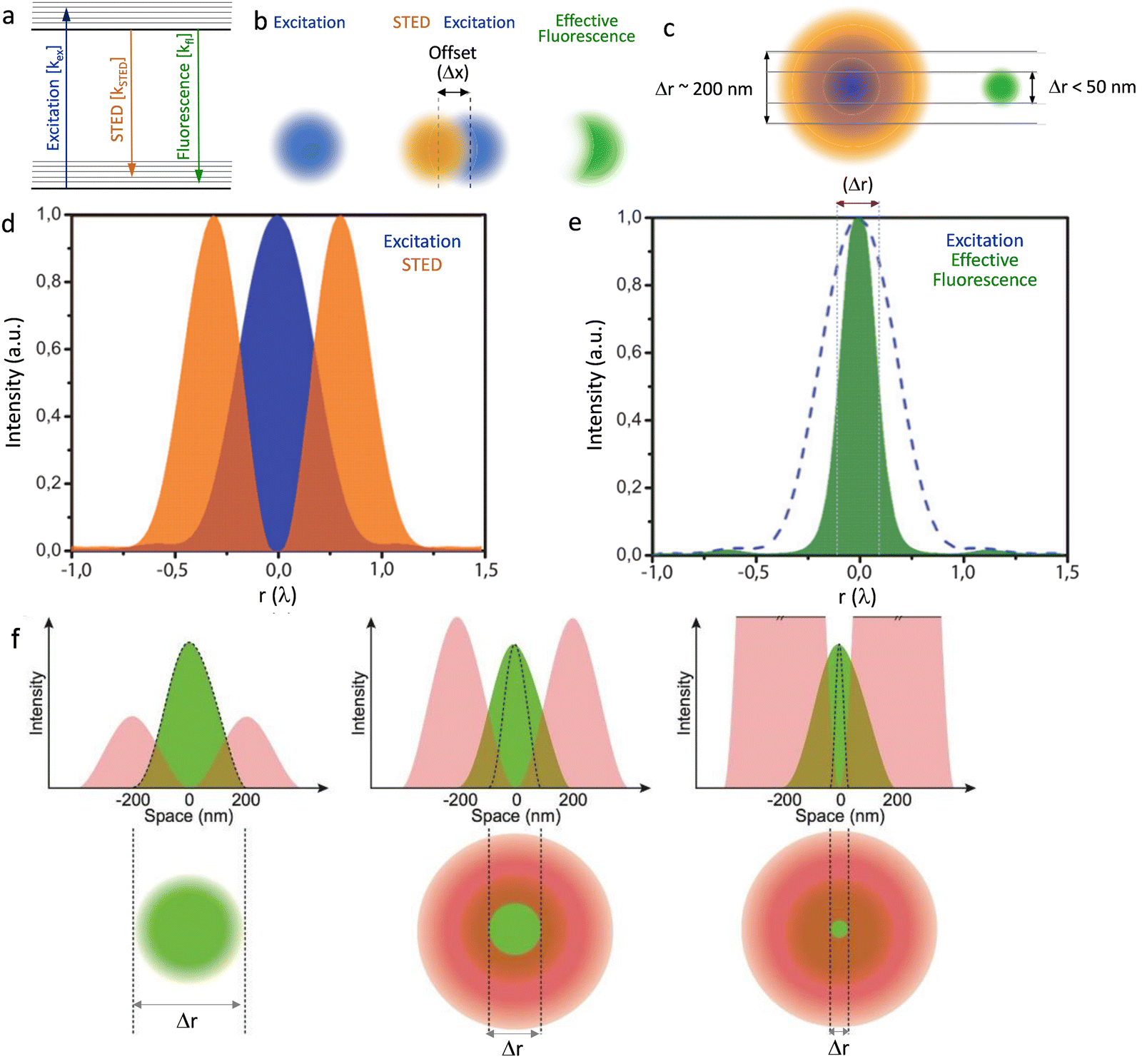 | ||
| Fig. 2 (a) Graphical illustration of the excitation, stimulated emission and fluorescence processes. (b) Two diffraction limited intense STED spots (brown), overlapped with an excitation spot (blue) having an offset (Δx), green emission of the fluorophores in the outer region of the excitation spot are effectively depleted to the ground state by stimulated emission. This attributes to a better resolved fluorescence spot with reduced diffraction limit, in the direction of the offset. (c) A doughnut-shaped STED spot (brown) overlapped with an excitation spot (blue) results a confinement of the area that allows fluorophores to fluoresce. (d) Graphic representation of the overlap of the intensities of the STED donut shaped spot (orange) and excitation spot (blue) in one dimension and (e) a decrease in the width of the effective fluorescence (green) (Δr: FWHM) as a result of stimulated emission depletion. (f) Graphical presentation to reveal the role of intensity of the STED emission in improving resolution (Δr: FWHM of the effective fluorescence). Reproduced with permission from ref. 27, Copyright 2012, Master Thesis, University of Twente, 2012. | ||
Stimulated emission is exploited in STED microscopy to switch a distribution of fluorophores into the required ‘OFF’ state. A sharply focused Laguerre-Gaussian depletion (erase) beam – with either a spiral phase plate or spatial light modulator (SLM) – illuminates a toroidal cross-section of the sample plane being excited by a sharply focused pump beam, forcing luminophores within its volume to de-excite through stimulated emission. The luminophores at the centre of the doughnut-shaped depletion beam are at an intensity minimum and thus are not “turned off”. Any further increase in the intensity of the depletion beam results in an increasingly smaller point spread function (PSF) – see Fig. 2F.
Gated-STED (g-STED) is the most commercially exploited technique that uses ultrashort pulsed excitation and STED lasers with gated detectors to further improve the signal-to-noise ratio and effective spatial resolution of measurements. One drawback of STED is the fact that as high illumination fluxes are required for efficient stimulated emission depletion, luminophores with lower photostability are not suited to this technique.
Another class of super-resolution microscopy involves single-molecule localization microscopy (SMLM) and includes techniques such as stochastic optical reconstruction microscopy (STORM), direct STORM (dSTORM), photoactivated localization microscopy (PALM), and point accumulation for imaging in nanoscale topography (PAINT). For SMLM, conventional wide-field excitation is used and super-resolution is achieved by computationally localizing individual fluorescent molecules through Gaussian peak fitting during the accumulation of thousands of images from sparsely distributed fluorescence molecules (Fig. 3).
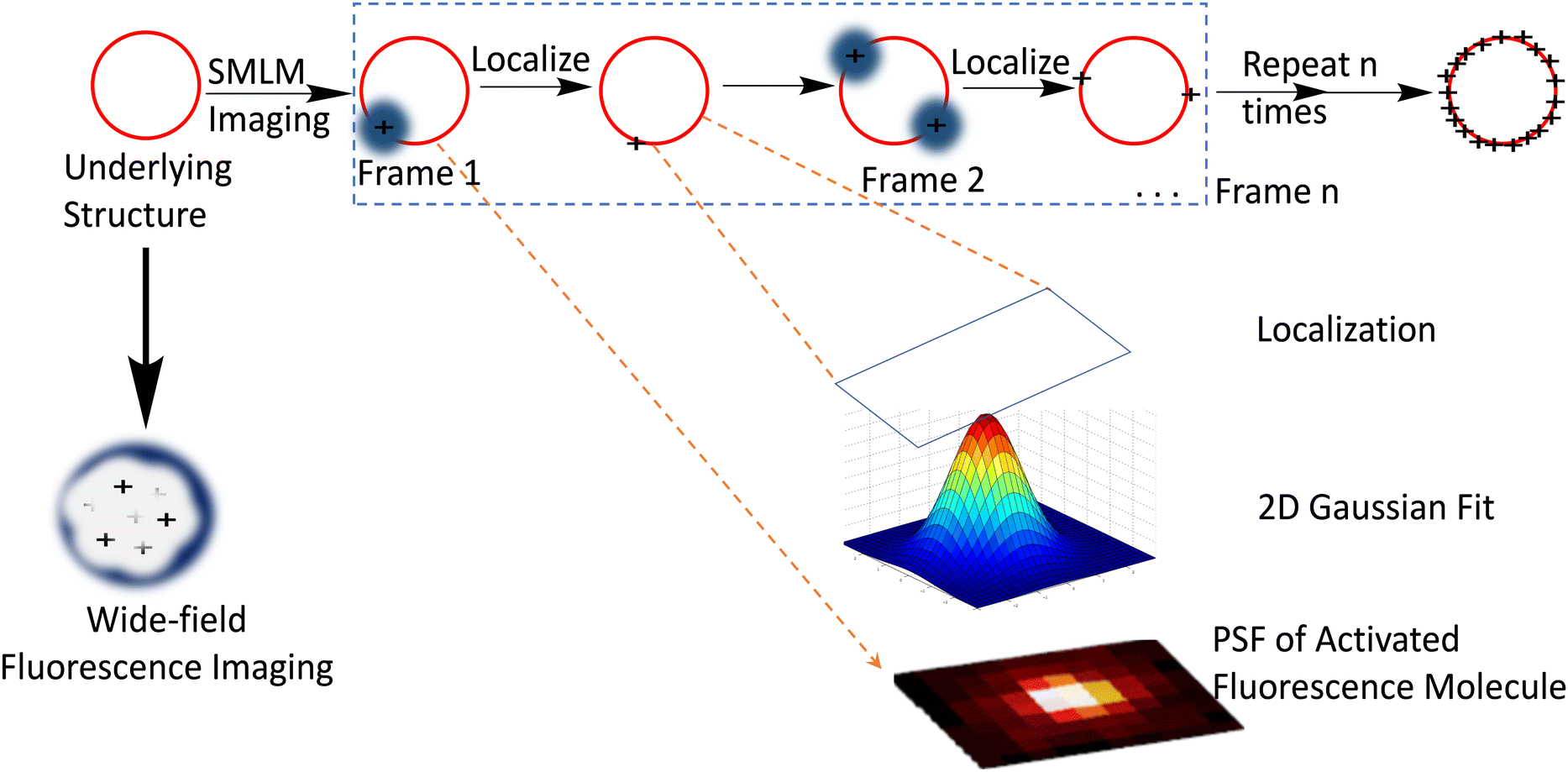 | ||
| Fig. 3 Depiction of SMLM imaging principle: the red circle represents the biological structure below the diffraction limit which is labelled with a suitable dye for visualization with a fluorescence microscope. Compared to the conventional diffraction-limited wide field microscope which produces a rather diffused image, SMLM imaging produces a more resolved image that is constructed from a set of time-separated images, wherein each time frame image contains a small set of excited and imaged labelled object that can be localized using Gaussian point spread functions and accumulated to form the final point-cloud super-resolution image for the structure. Reproduced with permission from ref. 11, copyright 2020, CellPress. | ||
Localized fluorescence is utilized to generate images with high spatial resolution typically ∼20–50 nm, or to define molecular trajectories.23,28–32 In the PAINT technique, an object is imaged continuously by a probe which is only in its fluorescence ON state when bound to an object and is switched OFF when it dissociates from the object or is photobleached. The observed stochastic blinking caused by transient binding depends on the diffusion coefficient and concentration gradient of individual probe molecules.33,34 However, obtaining images with high spatial resolution using a fluorescent marker which is required by the technique to display low to moderate binding to the object adversely influences the data acquisition process.
Generally, issues like fluorescence intensity, photobleaching, optical aberration, and background noise due to intracellular autofluorescence adversely influence the signal-to-noise ratio of both RESOLFT and SMLM and limit their application. Some of these deficiencies are addressed in structured illumination microscopy (SIM), as it utilizes a relatively low illumination light power. In SIM, the object is illuminated by a set of patterned intensities, and an image of the object is reconstructed using a conventional wide-field microscope. This provides an improvement in transverse spatial resolution. Recent advances in high frame rate acquisition, together with low phototoxicity associated with less intense illumination powers, make SIM a workable option for imaging biological dynamics and real-time biochemical processes in living cells at super-resolution.35 The standard Super-Resolution Structured Illumination Microscopy (SR-SIM) technique is suitable for imaging thin samples (<10 μm) with some compromise in temporal resolution, while spot-scanning implementation offers better background rejection and is more suited for thicker specimens (∼10–100 μm).35 Recent advances have made instant SIM (iSIM) the fastest SR-SIM method available, owing to its parallel illumination and analogue processing. This also allows for 10× deeper sample penetration than is obtained with conventional SIM, opening up the possibility of observing previously unvisualized sub-diffraction limited dynamic processes within cells and tissues.
Minimal photon flux (MINFLUX) nanoscopy is a new hybrid strategy that utilizes structured illumination along with single-molecule localization to extract the maximum positional information on a single molecule.36 By targeting single emitters using a doughnut-shaped excitation beam with a central intensity minimum (ideally zero), MINFLUX provides 3D localization of fluorescent labels at a spatial resolution of <5 nm (Fig. 4).37–39 As MINFLUX relies on emission minimization rather than maximization, this data acquisition/processing is typically fast while minimizing photobleaching and signal drift.
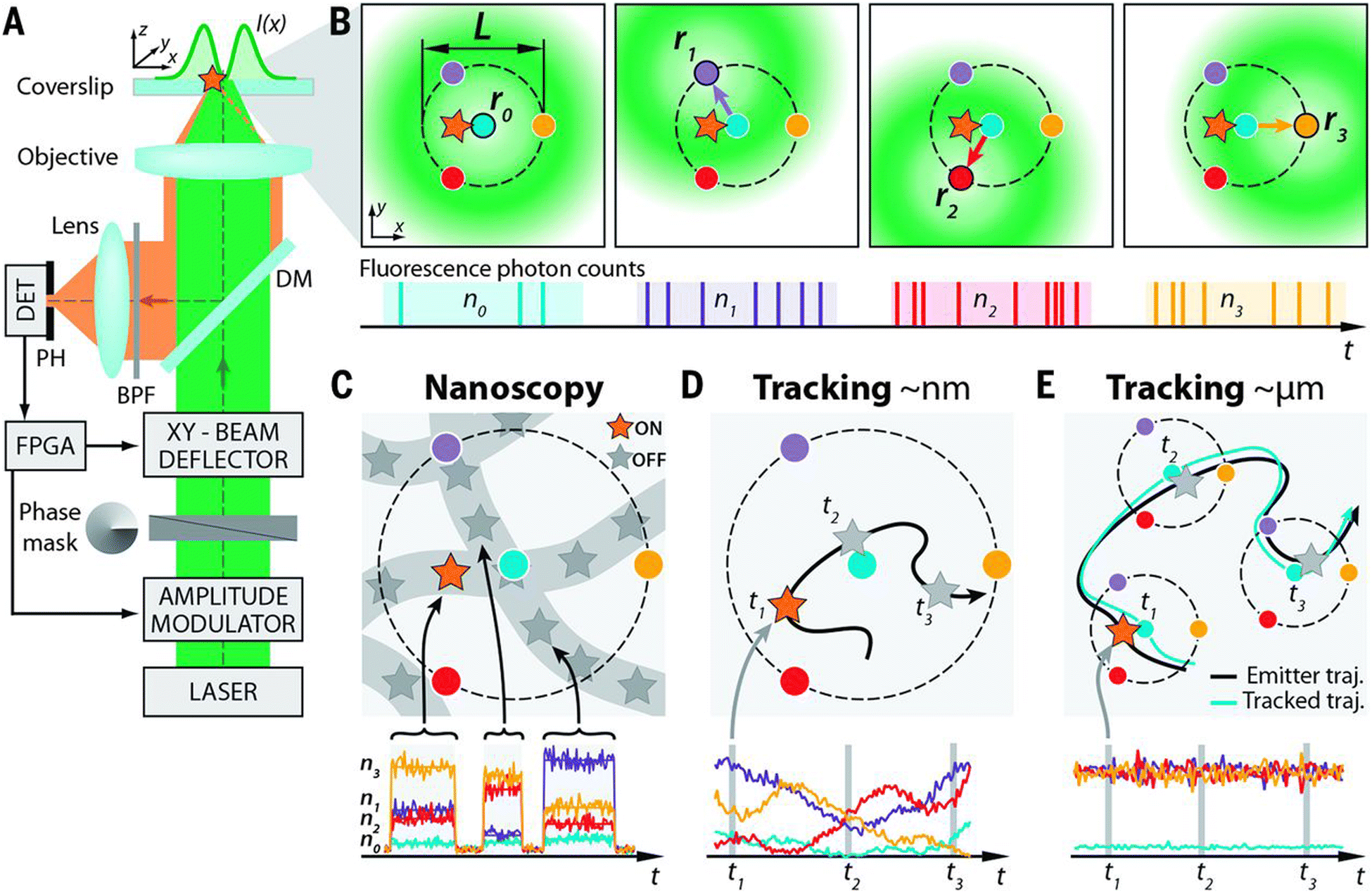 | ||
Fig. 4 Setup, measurement strategy, and various application fields of the 2D MINFLUX implementation: (A and B) (A) simplified setup (details in materials and methods). An excitation laser beam (green) is shaped by a vortex-phase mask forming a doughnut intensity spot in the focal plane of the objective lens. The intensity of the beam is modulated and deflected such that its central zero is sequentially placed at the four focal plane positions ![[r with combining circumflex]](https://www.rsc.org/images/entities/i_char_0072_0302.gif) 0,1,2,3 indicated by blue, violet, red, and yellow dots, respectively. Photons emitted by the fluorescent molecule (star) are collected by the objective lens and directed toward a fluorescence bandpass filter (BPF) and a confocal pinhole (PH), by using a dichroic mirror (DM). The fluorescence photons n0,1,2,3 counted for each doughnut position 0,1,2,3 by the detector (DET) are used to extract the molecular location. Intensity modulation and deflection, as well as the photon counting, are controlled by a field-programmable gate array (FPGA). (B) Diagrams of the positions of the doughnut in the focal plane and resulting fluorescence photon counts. (C to E) Basic application modalities of MINFLUX. (C) Nanoscopy: a nanoscale object features molecules whose fluorescence can be switched on and off, such that only one of the molecules is on within the detection range. They are distinguished by abrupt changes in the ratios between the different n0,1,2,3 or by intermissions in emission. (D) Nanometer-scale (short-range) tracking: the same procedure can be applied to a single emitter that moves within the localization region of size L. As the emitter moves, different fluorescence ratios are observed that allow thelocalization. (E) Micron-scale (long-range) tracking: if the emitter leaves the initial L-sized field of view, the triangular set of positions of the doughnut zeros is (iteratively) displaced to the last estimated position of the molecule. By keeping it around 0 by means of a feedback loop, photon emission is expected to be minimal for n0 and balanced between n1, n2, and n3, as shown. Reproduced with permission from ref. 39, copyright 2016, American Association for the Advancement of Science. 0,1,2,3 indicated by blue, violet, red, and yellow dots, respectively. Photons emitted by the fluorescent molecule (star) are collected by the objective lens and directed toward a fluorescence bandpass filter (BPF) and a confocal pinhole (PH), by using a dichroic mirror (DM). The fluorescence photons n0,1,2,3 counted for each doughnut position 0,1,2,3 by the detector (DET) are used to extract the molecular location. Intensity modulation and deflection, as well as the photon counting, are controlled by a field-programmable gate array (FPGA). (B) Diagrams of the positions of the doughnut in the focal plane and resulting fluorescence photon counts. (C to E) Basic application modalities of MINFLUX. (C) Nanoscopy: a nanoscale object features molecules whose fluorescence can be switched on and off, such that only one of the molecules is on within the detection range. They are distinguished by abrupt changes in the ratios between the different n0,1,2,3 or by intermissions in emission. (D) Nanometer-scale (short-range) tracking: the same procedure can be applied to a single emitter that moves within the localization region of size L. As the emitter moves, different fluorescence ratios are observed that allow thelocalization. (E) Micron-scale (long-range) tracking: if the emitter leaves the initial L-sized field of view, the triangular set of positions of the doughnut zeros is (iteratively) displaced to the last estimated position of the molecule. By keeping it around 0 by means of a feedback loop, photon emission is expected to be minimal for n0 and balanced between n1, n2, and n3, as shown. Reproduced with permission from ref. 39, copyright 2016, American Association for the Advancement of Science. | ||
Theoretical background: how does brightness of a luminophore enhances resolution?
Though biological structures range several orders of magnitude in length scale, the resolution in numerous types of biological light microscopy is very limited.40 The resolution limit of a far-field optical system is quantified by the Rayleigh criterion:41| rR = 0.61 × λ/NA |
To effectively visualize a specific biological sub-structure, most luminescence-based imaging methods depend on observation of a huge number of molecules instantaneously, which is intrinsically limited because the distance between observed molecules is much less than rR. However, super resolution imaging techniques either reduce the size of the observation volume or increase the accessible Fourier space and therefore increase the number of accessible spatial frequencies.
The localization precision σx for point-like objects imaged in two dimensions by fluorescence microscopy is given by;42
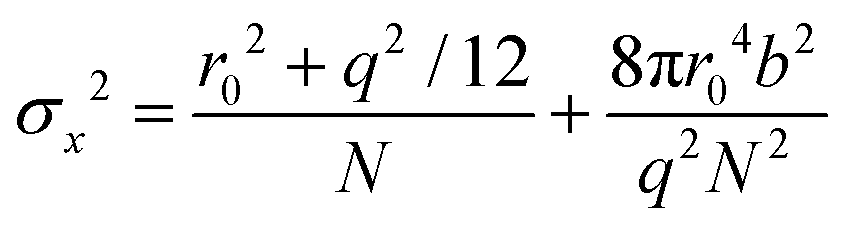
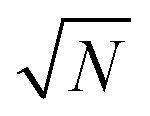 . Therefore, the number of detected photons is crucial for improving localization precision compared to resolution. For a single molecule capable of emitting ∼105 visible photons (λem = 500 nm) before photo-bleaching, positional localization can be as exact as ∼500 nm/(105)0.5, or a few nanometers. Subsequently the finite detection efficiency (typically <5%) of current single-molecule fluorescence microscopes, an estimate of the number of collected photons, is Ncoll = ϕdet/ΦB, where ΦB is the photobleaching quantum yield for the molecule and ϕdet is the detection efficiency, and the two-dimensional localization precision becomes40
. Therefore, the number of detected photons is crucial for improving localization precision compared to resolution. For a single molecule capable of emitting ∼105 visible photons (λem = 500 nm) before photo-bleaching, positional localization can be as exact as ∼500 nm/(105)0.5, or a few nanometers. Subsequently the finite detection efficiency (typically <5%) of current single-molecule fluorescence microscopes, an estimate of the number of collected photons, is Ncoll = ϕdet/ΦB, where ΦB is the photobleaching quantum yield for the molecule and ϕdet is the detection efficiency, and the two-dimensional localization precision becomes40As increased b will increase σx2, it is essential that background noise is minimized, including the background from inactive protein molecules.
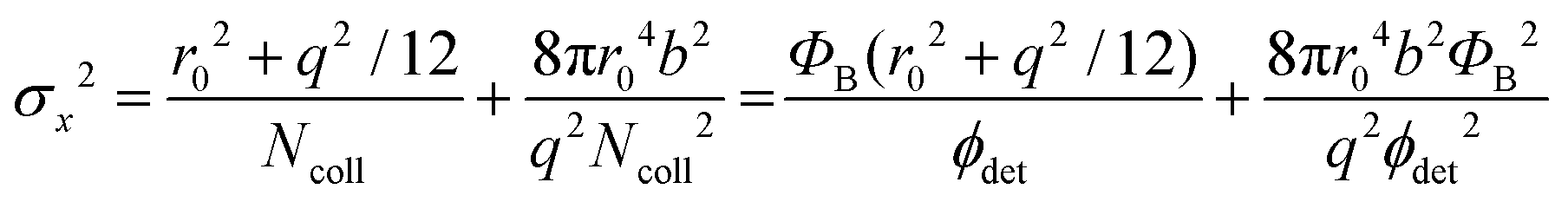
Dyes and probes in super-resolution microscopy
Molecular organic dyes and fluorescent proteins are most commonly used as imaging probes in optical microscopy, as they allow for functionalization to achieve the right balance of lipophilicity and hydrophilicity for efficient internalization and general compatibility with biological objects.43–47 However, for continuous monitoring of single molecules and real-time super-resolution imaging of subcellular organelles, bright photostable probes are demanded, yet many organic dye molecules undergo photobleaching on irradiation.48–51Despite such limitations, appropriately designed chemodosimetric reagents with a fluorescence ON response still provide an distinct advantages in imaging applications.51 Based on mechanistic pathways that allow luminophores to switch between ON and OFF states, SMLM fluorophores belong to five categories; (i) photoswitchable fluorophores that reversibly oscillate between ON and OFF states with an option to ‘blink’ several times (Alexa Fluor 647, ATTO 665, etc.), (ii) photoactivated fluorophores that irreversibly switch between an OFF to ON state, either spontaneously or upon activation by light (PA Janelia Fluor® 549, PA Janelia Fluor® 646, Cy5B, PA-TagRFP, PA-GFP, PAmKate, etc.), (iii) photoconvertible fluorophores that switch irreversibly to another spectral state upon irradiation (Dendra2, mMaple, etc.), (iv) spontaneously blinking dyes that undergo a reversible, pH-dependent chemodosimetric reaction to enable SMLM imaging at a defined media pH (HMSiR39, HEtetTFER40, FRD41, etc.), (v) transiently interacting dyes that have low affinity towards a target. As discussed above, such probes are ideally suited for PAINT as they help to replenish photobleached dyes weakly bound to a target with a fresh supply of transient labels from the bulk medium which counter the loss in signal caused by photobleaching (examples include; nile red, DFHBI-1T, M739, HBR-DOM, etc.). Comparing synthetic dyes and fluorescent proteins, synthetic dyes allow shorter imaging times and better localization precision as synthetic dyes typically have higher photon counts.
In recent years, advances in the materials sciences have provided considerable opportunities to address the shortcomings of extant fluorescent dyes.52 In particular, the design of nanoscopically-sized probes with well-defined optical properties have resulted in a large collection of luminescent nanoparticles such as perovskite quantum dots,53 upconversion nanoparticles (UCNPs),54 semiconductor quantum dots (QDots),55 fluorescent nanodiamond,56 polymer dots and carbon-based graphene nanodots (GQDs).57 Though they are still large relative to dye molecules, they are often comparable in size to fluorescent proteins. And while substantial challenges lie ahead with respect to their complicated surface biochemistry, the many advantages of these nanoparticles are well established, prompting the cell biology and materials science communities to explore their full capabilities in functional subcellular imaging at the nanoscale (Fig. 5).18
 | ||
| Fig. 5 Dissecting eukaryote cells with nanoscopy. (a) Nuclear pores. Left: Averaged direct stochastic optical reconstruction microscopy (dSTORM) image showing the distribution of two nuclear pore (Nup) proteins in the nuclear pore complex (NPC) of Xenopus laevis; right: two possible arrangements for the Nup107–160 complexes based on ground state depletion with individual molecule return (GSDIM) data from a human NPC are traced within the electron density of the cytoplasmic ring of the nuclear pore. Scale bars: 100 nm (left), 25 nm (right). (b) Chromatin domains in Drosophila melanogaster nuclei. (c) Synaptic vesicles in living rat neurons. (d) Cytoplasmic ribosomes in a human cell (e) live nanoscopy of Golgi-derived vesicles. (f) Lysosomes in a living mammalian cell. Scale bar: 1 μm. l | Centrioles in mammalian cells. Scale bar in the right panel: 250 nm. All scale bars are 500 nm unless stated otherwise. (g) Live-cell nanoscopy imaging (left) and 4Pi single-molecule switching nanoscopy (right) of the endoplasmic reticulum in a mammalian cell. Scale bars: 1 μm. (h) Nanoscopy of the cytoskeleton in a mammalian cell. Left, Top: Microtubules (in comparison with a confocal recording); left, bottom: vimentin (in comparison with a confocal recording); right: actin. The scale bar in the right panel is 2 μm. (i) Nanoscopy images of mitochondria in human cells. Left: Outer membrane protein Tom20 (in comparison with a confocal recording); right: F1FOATPase in the inner membrane. (j) | Centrioles in mammalian cells. Scale bar in the right panel: 250 nm. All scale bars are 500 nm unless stated otherwise. (k) Human fibroblasts immunolabelled for the peroxisomal protein PEX5 (red) and the mitochondrial protein Tom20 (green). The arrow shows the colocalization of the two proteins. (l) Focal adhesions in human cells. The colours indicate the vertical (z) coordinate of the labelled actin relative to the substrate. Reproduced with permission from ref. 18, Copyright 2017, Springer Nature. | ||
This review is focused on the recent progress and future potential of such luminescent nanoparticles for use in tracking single molecules and super-resolution imaging of subcellular structures, enabling high-resolution visualization of various biological structures such as mitochondria, lysosome, endoplasmic reticulum, microtubules, chromatin complexes, neurons, clathrin-coated pits, focal adhesion complexes and actin.12,58 We also discuss the challenges associated with their use in biological systems, including intracellular delivery, molecular targeting and quantitative analysis.
In fluorescent quantum dots, QDs
Factors that are crucial for efficient SRM with QDs are; (i) efficient cellular internalization and organelle/tissue-specific localization of the probe molecules, (ii) monovalency of the fluorophore-bioconjugate with an appreciable binding affinity between probe and target, (iii) high emission quantum yields along with appreciable stability towards photobleaching, (iv) stochastic switching of fluorescence states between fluorescence “OFF” and “ON” states.Innumerable research articles have appeared in the literature on the use of QDs for biological imaging since their first demonstration in 1998;59,60 typically, QDs possess excited state lifetimes (>10 ns) that are longer than those of fluorescent dyes and small proteins, and they are stable towards photobleaching on irradiation or chemical degradation under physiological conditions. Due to exciton quantum confinement, they display size/composition-dependent bright fluorescence (450 to 1500 nm), which can be tuned by tailoring their size, shape, and composition.61,62 As they also possess larger two-photon absorption cross-sections (103–104 GM) than typical fluorescent molecular dyes or fluorescent proteins, they have become an attractive choice for applications in SRM.51,63–65 Importantly, surfaces of QD nanocrystals can be post-functionalized with a variety of biomolecules (e.g., antibodies, peptides, proteins, DNA, and vitamins) through well-known conjugation techniques (Fig. 6).66–68
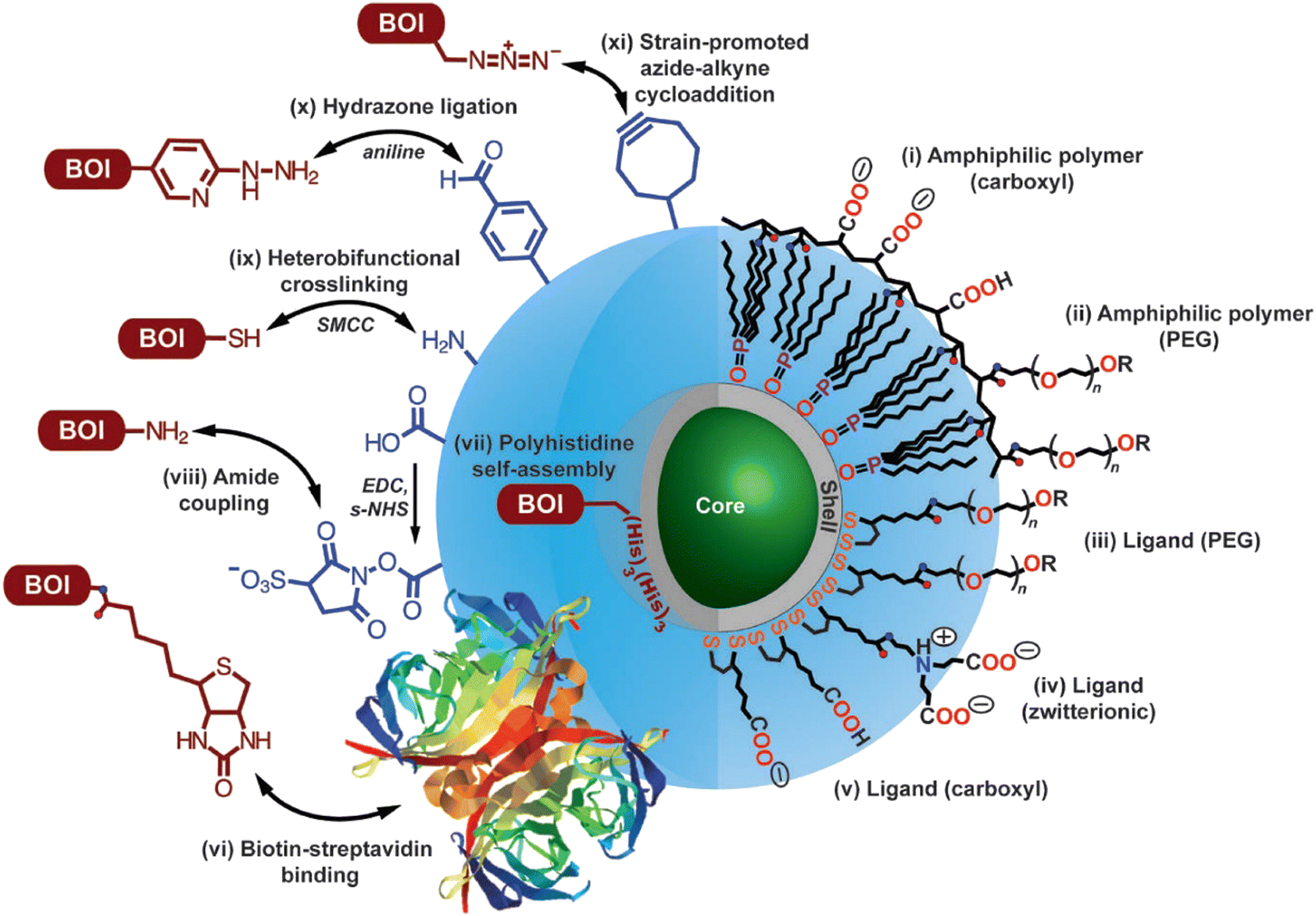 | ||
| Fig. 6 Options for utilizing modified core-shell QDs for target-specific imaging. Surface modification for achieving uniform dispersion involves functionalization with (i) amphiphilic polymer coating with -COOH groups, (ii) PEG oligomers, (iii) dithiol ligand with a distal PEG oligomer, (iv) a zwitterionic functionality, (v) a distal -COOH group. R groups (as shown in this image) include carboxyl, amine, and methoxy; while other functionalities can be introduced (e.g., see vi, x, xi) following appropriate synthetic methodologies. As shown in this image, protocols for conjugating biomolecules of interest are as follows: (vi) biotin–streptavidin binding, (vii) polyhistidine self-assembly on the outher surface of the shell-structure of the QD, (viii) following EDC/s-NHS activation for amide coupling using, (ix) crosslinking reaction utilizing succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (structure not shown), (x) aniline-catalyzed hydrazone ligation, and (xi) strain-promoted azide-alkyne cycloaddition. Double arrow represents conjugation between the functional groups and, in principle, their interchangeability. Reproduced with permission from ref. 68, copyright 2013, Optica. | ||
This also opens up possibilities for multivalent binding via multiple targeting moieties.69,70 Flexibility in emission wavelength, as well as in surface chemistry of bioconjugated QDs, enables their use as nanoprobes for a wide range of applications in nanomedicine and biophotonics, including; near-IR deep-tissue imaging,71 fluorescence imaging in the near-IR (e.g., 1000–1400 nm), single-cell detection, and controlled release of drugs.72
The bright fluorescence and fluorescence intermittency of QD-materials facilitates the spatial and temporal localization of individual QDs, which are more important for SRM techniques like PALM, fPALM, STORM, and dSTORM. However, as QD blinking is stochastic, they are not ideal for localization microscopy and single-molecule tracking experiments. Also, their tendency toward toxicity and nonspecific binding have somewhat restricted their wider application in SRM.73,74
In 1998, Weiss, Alivisatos and their co-workers were the first to use CdSe–CdS core–shell nanocrystals, enclosed in a silica shell for better water dispersibility, for imaging 3T3 mouse fibroblast cells using confocal laser scanning microscopy.60 At the same time, the Nie group used ZnS capped QDs with a CdSe core functionalized with transferrin (an iron transport protein)) for imaging.59 In 2008, Hell and colleagues were first to use Mn2+-doped ZnSe QDs for STED imaging. QD fluorescence was depleted efficiently (90%) and this resulted in a huge number of fluorescent switch ON-OFF cycles before photobleaching was observed, resulting in a factor of ×4.4 improvement in resolution.75 However, the small Stokes’ shifts that are typical of such materials limit the choice of an appropriate depletion beam.76 To address this limitation, the high photostability of commercially available ZnS-coated CdSe QDs and CdTe QDs (Fig. 7a) with an option for repeated STED recordings with large number of frames have been used.
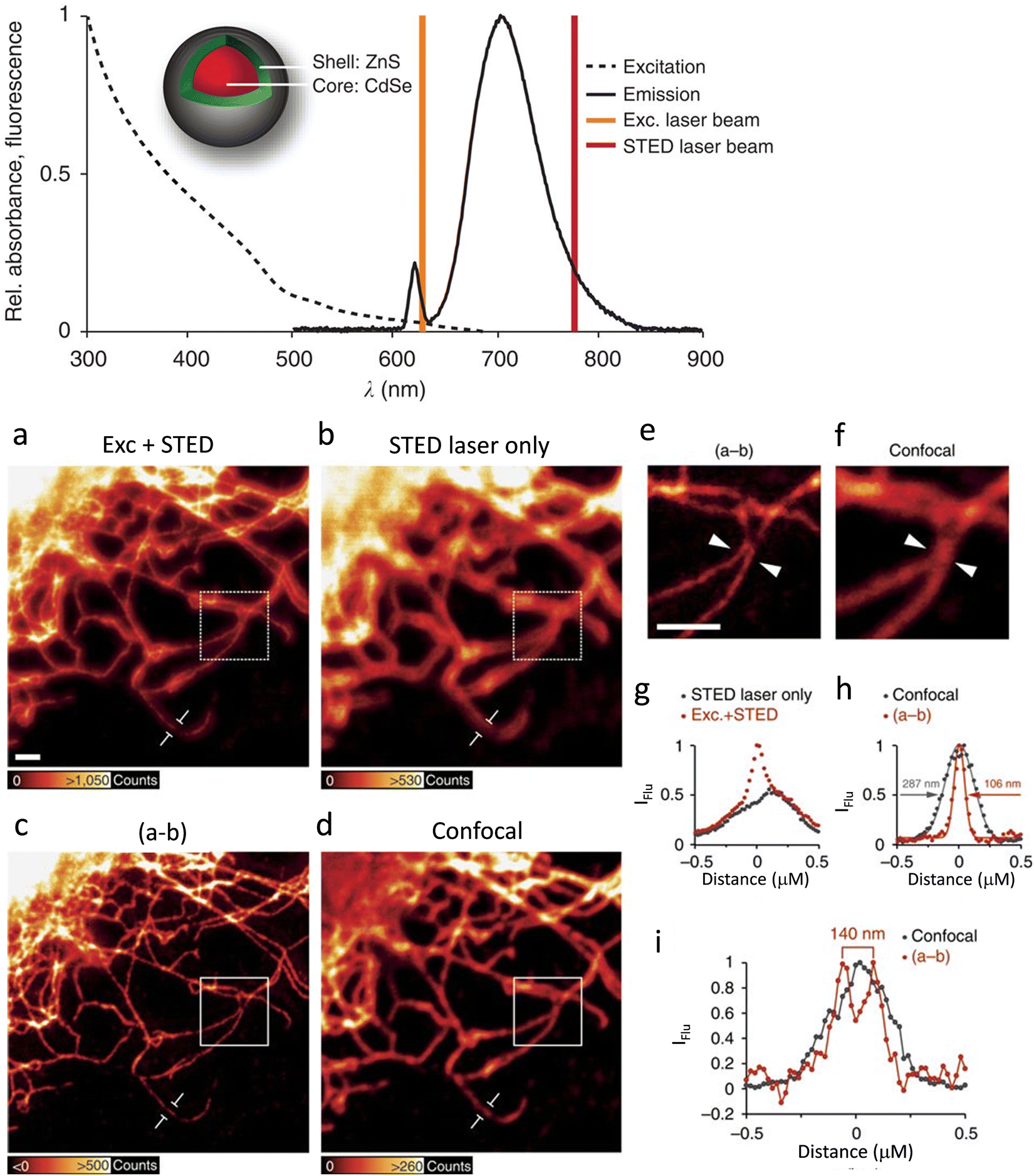 | ||
| Fig. 7 The excitation (- - -) and emission (−) spectra recorded for antibody-coupled ZnS-CdSe QD705 (inset) are shown along with excitation laser line at λexc= 628 nm (brown) and the STED laser line at λSTED = 775 nm (red). Imaging of vimentin fibres with QD705: (a) using λexc = 628 together with λSTED = 775 nm laser beams. Scale bar: 1 mm. (b) Imaged with the λSTED = 775 nm. Both images were obtained with line-wise multiplexing. (c) Deduction of the image (b) from that of (a) using a 3 × 3 median filter). (d) Image of the identical sample region using conventional confocal microscopy with an adjusted lookup tables for better visualization. (e and f) Respectiveimages are inserts from the images shown in c and d (scale bar: 1 μm). (g) Intensity profile for a single vimentin fibre as shown in figures (a) and (b). (h) Intensity profile of the subtracted image (c) and (d) for single vimentin fibre. (i) A plot to show the line profiles (positions indicated with white arrow heads in (e) and (f)) to demonstrate two fibres could be resolved to a distance of 140 nm in the super-resolved image, which was not possible in confocal image. Reproduced with permission from ref. 77, Copyright 2015, Springer Nature. | ||
Results also reveal that the tendency of quantum-dot labels to blink could effectively be suppressed by the combined action of excitation and STED beams. This helped to achieve better resolved and background-free images (Fig. 7b).77 Biofunctionalized QD705 helped to yield a 2.7-fold improvement in resolution (Fig. 7c-i).
Following these reports, Kner et al. reported multiple colour super-resolution imaging with CdSe/ZnS QDs (core/shell) without sacrificing resolution (Fig. 8),78 successfully establishing that blue-shifted QDs could be controlled to maximize the number of imaged localizations. They also used Quenched Stochastic Optical Reconstruction Microscopy (QSTORM) with high photon count QDs to measure microtubule widths and two QDs with widely different excitation wavelengths to separate their emission into discrete channels.
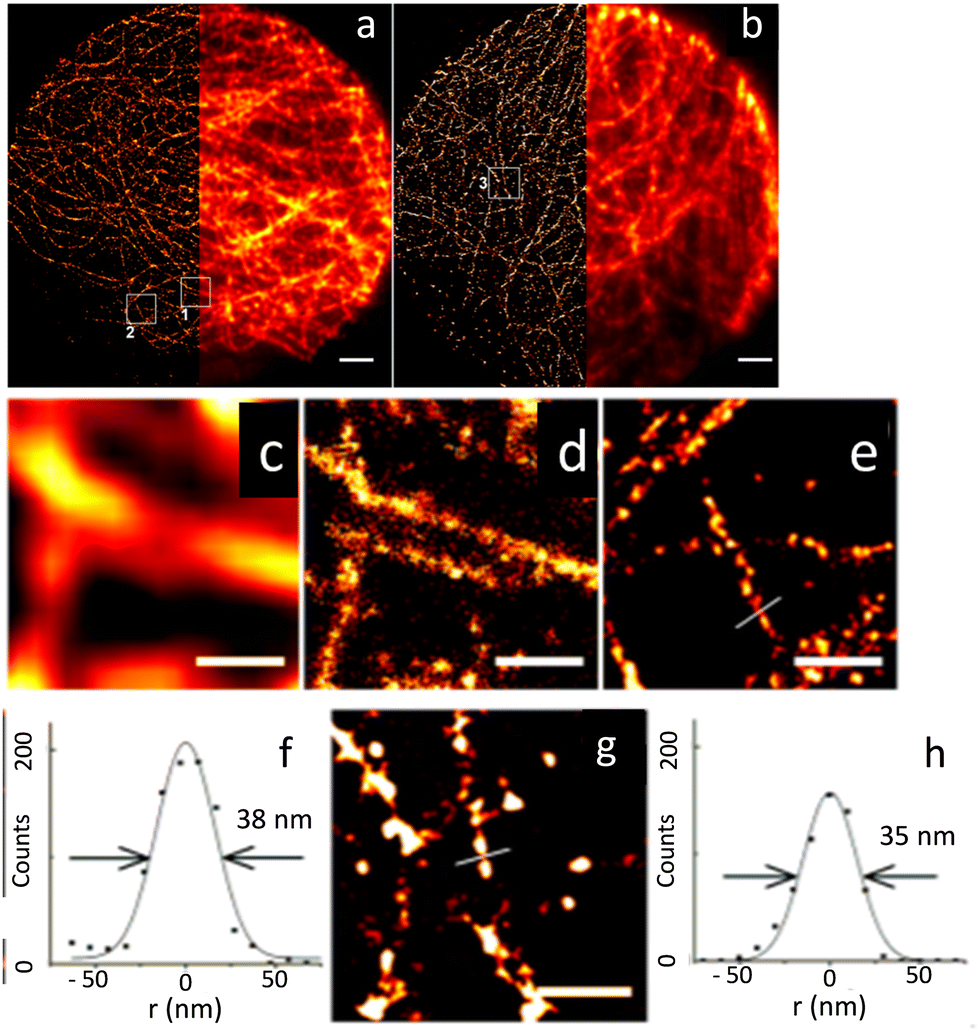 | ||
| Fig. 8 Microscopic images of microtubules in HepG2 cells: (a) STORM using QD565 and (b) wide-field images QD705 (scale bar: 2 μm). Wide-field (c) and STORM (d) images shown in the white square box 1 in (a); scale bar = 500 nm. (e) STORM image of the region shown in box 2 in (a); scale bar = 500 nm. (f) STORM image: cross-section of a microtubule with FWHM of 38 nm and (g) image shown in the white square 3 in (b); scale bar: = 500 nm. A cross-section of a microtubule is shown in (h). Cross-section of a microtubule with FWHM of 35 nm. Reproduced with permission from ref. 78, Copyright 2015, American Chemical Society. | ||
Through STED and SIM super-resolution imaging of microtubule networks in HeLa cells, Xi, et al. have compared the performance of streptavidin-conjugated (SC) cadmium selenium QDs (λem = 775 nm) with that of a conventional super-resolution dye (a cationic dye, ATTO 647N)79 (Fig. 9). STED, SIM and localization-based super-resolution microscopy were also statistically compared, and respective estimated resolutions of ∼60 nm, ∼ 120 nm and ∼ 50 nm were attained.
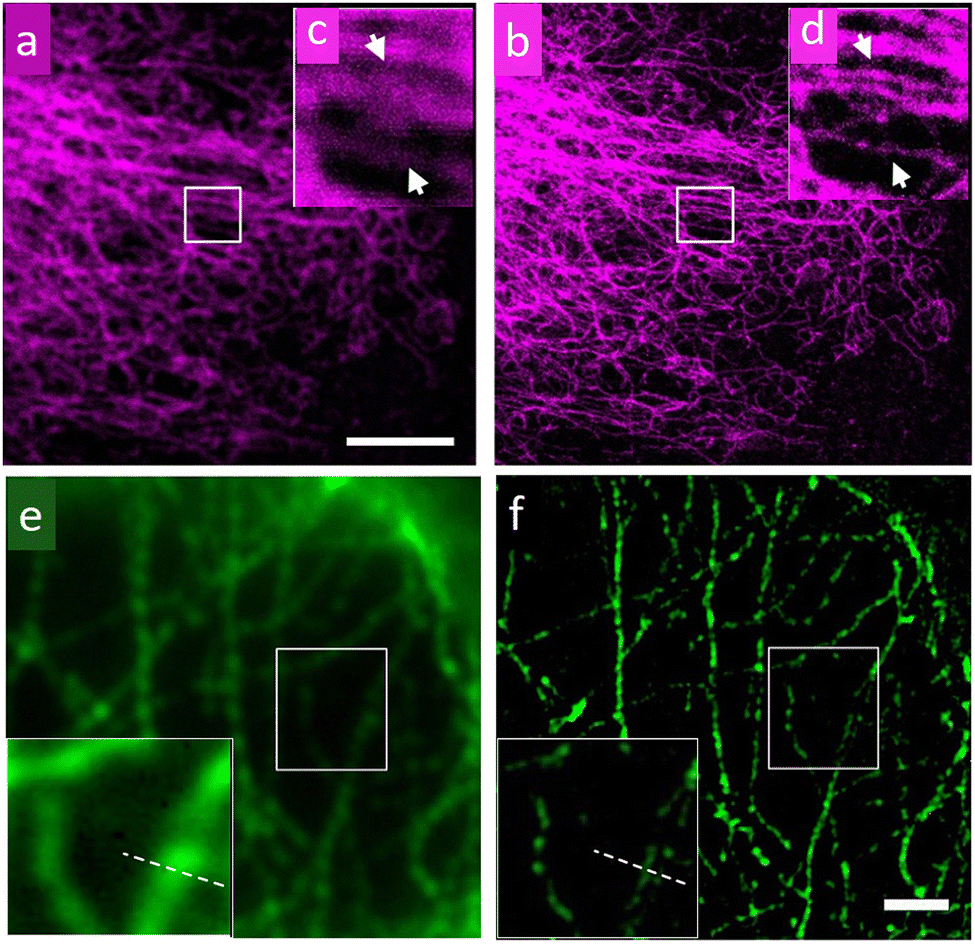 | ||
| Fig. 9 Images of a microtubule network of HeLa cells recorded using confocal and Continuous-Wave (CW) STED microscopy using QDs as a staining agent: (a) confocal, (b) CW STED, (c and d) respective magnified images for selected areas in (a) and (b). (e) Wide-field microscopy image; scale bar: 2 μm. (f) Images of microtubules on HeLa cells by SIM with enlargement as an insert. The wavelength and power density for the STED beam are 775 nm and 200 mW, respectively. The pixel size is 20 nm; scale bar: 5 μm. Reproduced with permission from ref. 79, Copyright 2016, American Chemical Society. | ||
The Ryu group developed a DNA-PAINT technique to further improve the resolution limit of conventional SMLM techniques that utilizes organic dyes.28,80 A spatial resolution of ∼5 nm could be achieved for in in vivo experiments by primarily enhancing the photon number that critically determines spatial resolution.
Specific and monovalent QDs (CdSe:ZnS with λems of 585 nm) were phase transferred from the organic to the aqueous phase and encapsulated with a 50-adenosine phosphorothioate DNA (ptDNA) polymer for the imaging application. Following passivation with ptDNA and tuning of hybridization sequences, monovalent and specific QDs became photo-switchable via QD-PAINT and showed much-improved fluorescence intensity compared to Cy3.80 Due to the narrower full-width-at-half-maximum, FWHM, for surface-modified CdSe:ZnS compared to that of Cy3, a theoretical maximum improvement in spatial resolution by a factor of ∼3 was evaluated for QD-PAINT, this translated into an experimental improvement in resolution of 2.9-fold (Fig. 10) compared to Cy3. As QDs absorb many more photons than Cy3 in identical photon fluxes they also produce much brighter fluorescence, enabling the use of lower laser powers and reducing phototoxic effects on live cells.
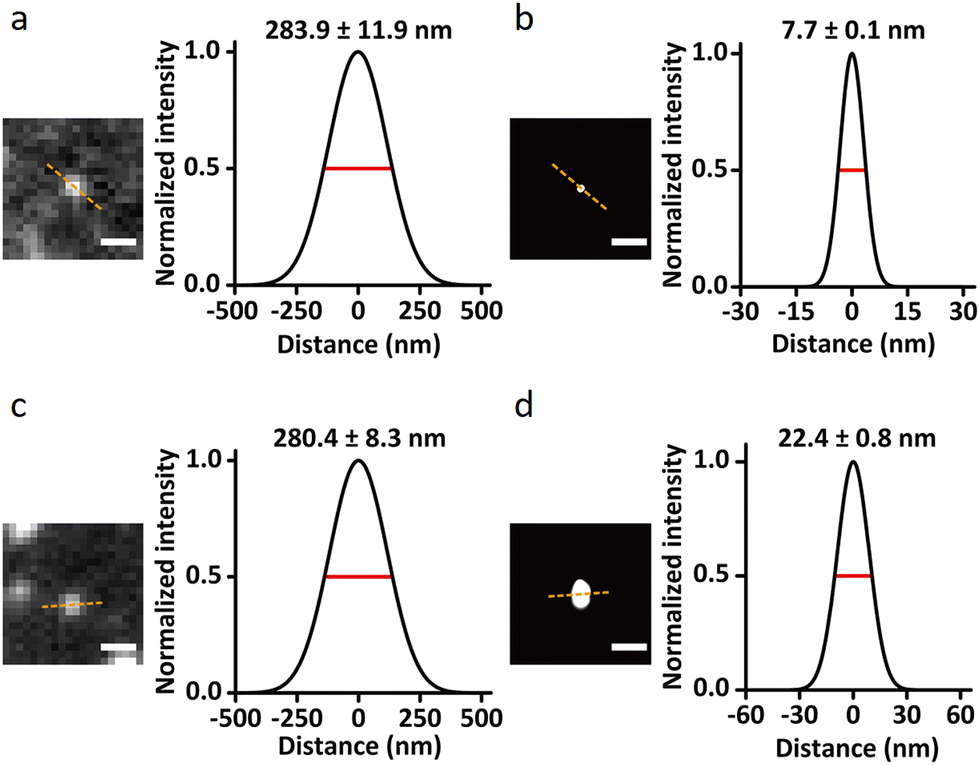 | ||
| Fig. 10 Single-molecule images demonstrating the resolution enhancement through QD-PAINT imaging compared to conventional DNA-PAINT imaging using the cyanine3 (Cy3) fluorophore: images of epidermal growth factor receptor (EGFR) recorded using QD-PAINT (a) before and (b) after reconstruction; while images recorded using DNA-PAINT with Cy3 (c) before and (d) after reconstruction; scale bars: (a and c) 500 nm and (b and d) 100 nm. Cross-sectional histograms show FWHM as the dashed lines for QD-PAINT (a) and (b) after reconstruction; while the analogous images using DNA-PAINT and Cy3 dye (c) and (d) after reconstruction. Reproduced with permission from ref. 80, Copyright 2021, Springer Nature. | ||
Krauss et al. have demonstrated that CdSe/CdS QDs, functionalized with a neuropeptide (BK-QDs; BK: bradykinin) are an improved STORM imaging probe compared to a BK-labelled organic commercial fluorophore (TAMRA-BK).81 STORM images of primary rat hippocampal neuronal cultures using the BK-QD probe showed higher localization accuracy than comparable images obtained using TAMRA-BK (Fig. 11).
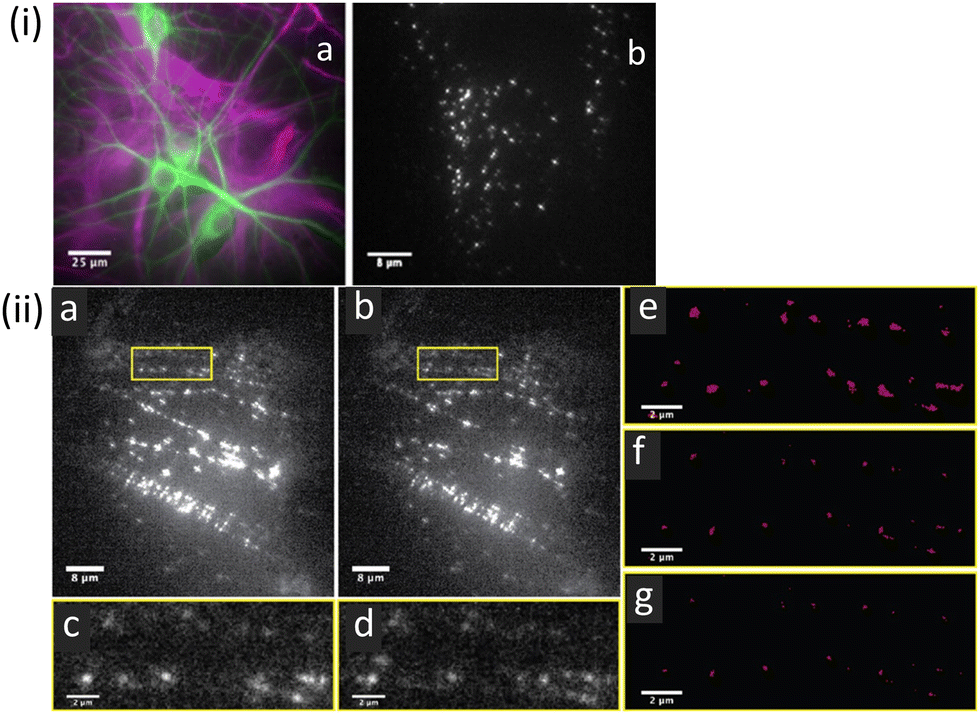 | ||
| Fig. 11 (i) Representative fluorescence images of mixed neuronal glial cultures: (a) hippocampal neuronal glial culture showing astrocytes (purple) and neurons (green) cell types. (b) Image of BKQDs bound to neuronal cultures using a single image frame for the neuronal culture on treatment with BKQDs (30 nM) for 15 min. (ii) STORM images recorded for neuronal cultures stained with BKQDs: (a and b) Frame 1 and frame 1000 out of 1000 frames from a movie; (c and d) enlarged image of the area indicated with the yellow box in a and b, respectively. (e–g) Reconstructed super-resolution images of the area indicated with yellow boxes with localization precisions of (e) 50 nm, (f) 30 nm, and (g) 25 nm to demonstrate the successful localization at all resolution levels. Reproduced with permission from ref. 81, Copyright 2021, American Chemical Society. | ||
This was largely ascribed to much higher fluorescence intensity and a consequentially improved signal-to-noise ratio compared to the organic fluorophore. BK-QDs were also found to “blink” at a faster rate, and -as anticipated- BK-QDs showed higher stability towards photobleaching. These results confirmed that BK-QDs display substantial advantages over organic fluorophores for sub-diffraction limit imaging.
Zhang, et al. prepared anti-mouse IgG (H + L)-conjugated Cd-Se QDs and successfully profiled the distribution and density of integrin αvβ3 in single glioblastoma cells.82 Corresponding SIM images are shown in Fig. 12 and these reveal adherent growth with irregular morphology.
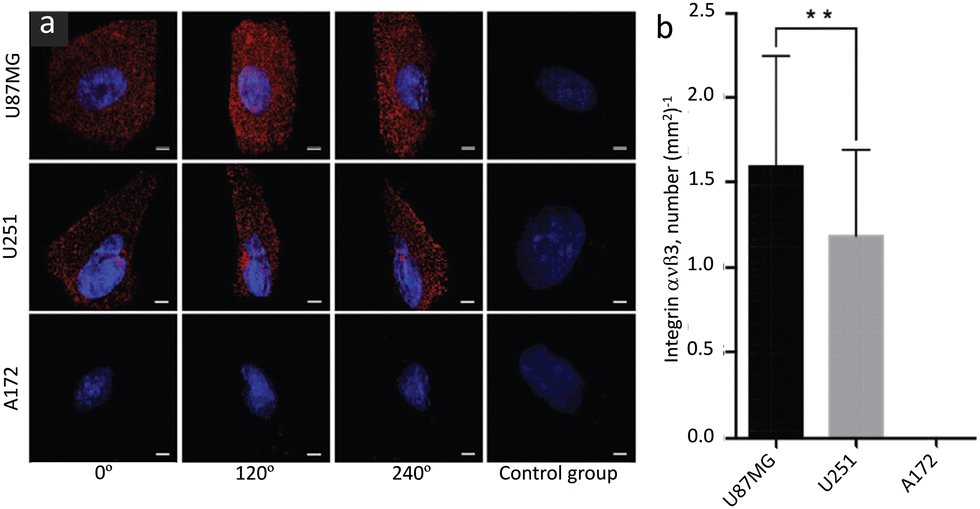 | ||
| Fig. 12 SIM Images of integrin αvβ3 in different glioblastoma cell lines (indicated in the image): (a) images to represent the expression and distribution of αvβ3 in different specified cell lines and viewing angles; images shown in the control panel (right panel) are of a control group without primary antibody. (b) Bar diagram to represent the density of αvβ3 receptors (averaged from 50 individual cells for each cell line). Scale bars: 5 μm. Data are presented as mean ± SD. **p < 0.01. Reproduced with permission from ref. 82, Copyright 2019, The Royal Society of Chemistry. | ||
This study demonstrated the phenotypical heterogeneity of glioblastoma at the single-cell level and revealed how drug vulnerability relates to receptor density on the test cells. It was suggested that this protocol could in principle be exploited for the quantitative profiling of many other surface proteins on cells of interest.
Fluorescent up-conversion nanoscale materials
Unlike d-block transition metal ions, the 4f orbitals of lanthanide ions are hidden under the 6s, 5p, and 5d orbitals, accounting for the narrow emission bands and environmental insensitivity of their spectra.Typically, emission spectra for Ln3+ ions in solid hosts show FWHM of ∼10–20 nm, while this figure varies within a range of ∼25–40 nm, ∼30–50 nm, and ∼100 nm, respectively for Qds, organic dyes and transition metal ions;83 and a narrow FWHM is associated with better image resolutions. Furthermore, as emissions from Ln3+ ions are associated with atomic transitions, they are typically resistant to photobleaching – even on prolonged irradiation. Lower loss in vibrational energy and wider separation between absorption and emission band maxima mean that Ln3+ ions can be excellent luminescence markers, particularly as they frequently display near-infra red, NIR, emission.
Two NIR regions are of particular interest for biological imaging. NIR-I (650 to 950 nm) and NIR-II (1000–1400 nm), define the so-called NIR biological windows that allow imaging with the minimum interference from biological media.84 Importantly, certain Ln3+ ions show NIR excitation through the upconversion phenomenon. These properties make specific La-based reagents/materials ideally suited for exploiting in NIR imaging. Due to these unique emission properties, lanthanide-based upconversion nanoparticles, UCNPs, are being developed as super-resolution imaging probes for diverse applications such as analytical sensing, photodynamic therapy, and clinical diagnostics. Efficient UCNPs are composed of an optically transparent host matrix, a sensitizer, and an activator. To minimize non-radiative losses and maximize radiative emission, an ideal host matrix needs to have a low lattice phonon energy and, as they meet this criterion, NaYF4, NaGdF4, LaF3, and CaF2 have been widely adopted for this role.85–89 Purpose-built syntheses of UCNPs are crucial in developing imaging materials; typically, Yb3+ is used as a sensitizer as it has a high absorption cross-section at ∼980 nm, while Er3+, Tm3+, and Ho3+ are used as activators to favour multiphoton processes (Fig. 13).90 The core–shell structure favours dispersibility in biofluids, improves surface processability, and improves luminescence efficiency by suppressing surface quenching effects.91,92 Various synthetic strategies for surface functionalization of the UCNPs to improve cell membrane permeability and organelle or tissue specificity have been developed.93,94 Simple variations in the composition of the lattice atoms and dopant ions used in the synthesis of the UCNP crystals facilitate their use in multimodal imaging.
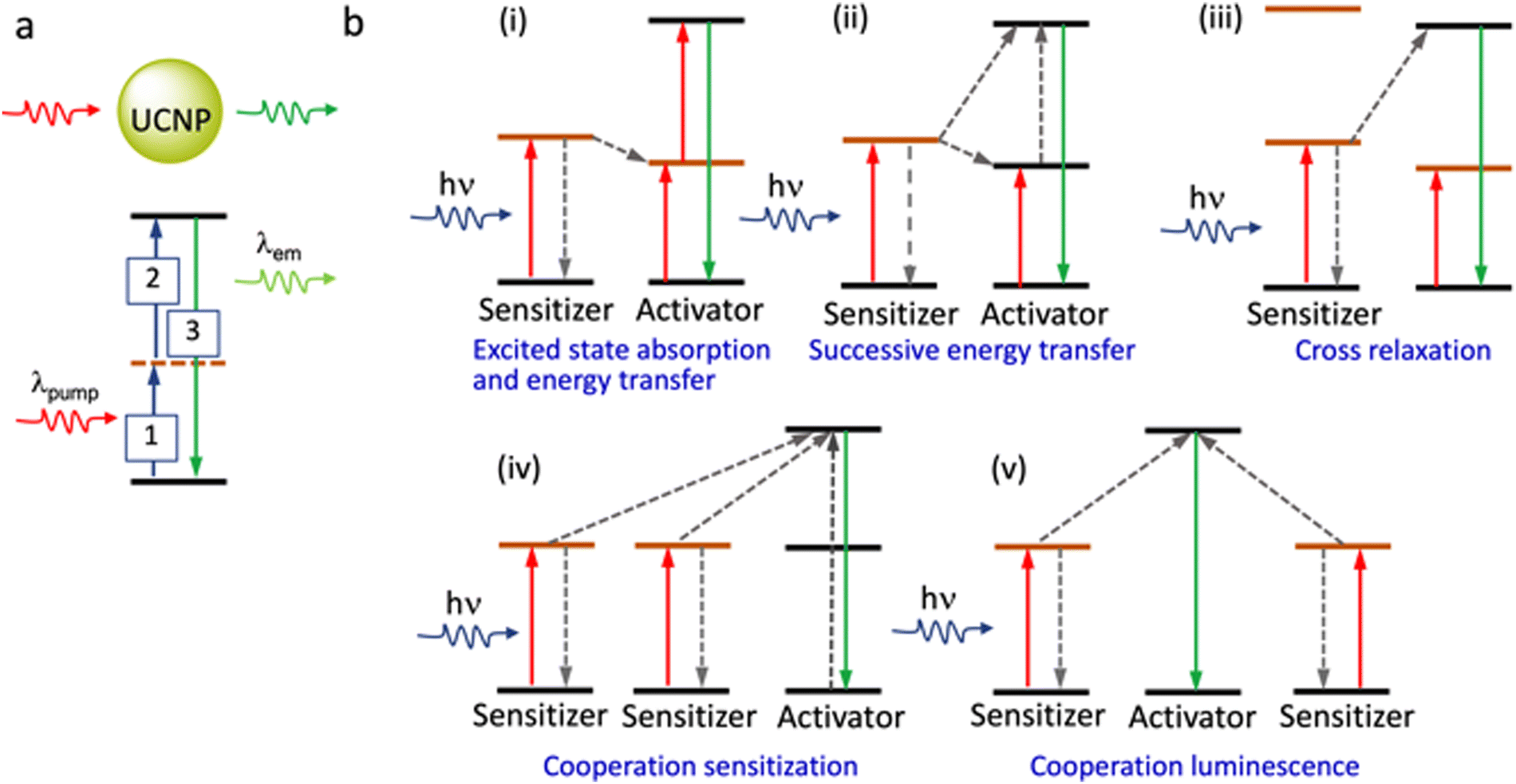 | ||
| Fig. 13 (a) Cartoon presentation of the show the various photo-induced process associated with excitation from the ground state to a metastable excited level (real or virtual), excited state absorption and the eventual relaxation from the higher excited state to the ground state. (b) Different possibilities for energy-transfer from the sensitizer to the activator in the UCNPs. (Energy levels shown are not in scale). | ||
The first report on the use of Y2O2S:Yb3+/Tm3+ particles for tissue imaging appeared in 1999, and related work described the use of Y2O3:Yb3+/Er3+ and Gd2O3:Yb3+/Er3+ for similar applications.95 Since then, a number of literature reports on the use of UCNPs for bioimaging have appeared; however, it is only more lately that their uses in super-resolution imaging have been highlighted.
Typically, to obtain the depletion efficiencies needed in STED imaging the luminescent marker localized to the target object must display a relatively high emission intensity. Toward this aim, Liu, Lu and Yang, and co-workers used UCNPs with high Tm3+ doping for STED microscopy, opting for a 980 nm excitation laser beam and 808 nm depletion beam in their studies.96 Excitation at 980 nm resulted in a population inversion in their intermediate metastable 3H4 state relative to the 3H6 ground level (Fig. 14 and 15). Further, irradiation at 808 nm (matching the upconversion band of the 3H4 → 3H6 transition) initiated amplified stimulated emission to depopulate the 3H4 intermediate level, optically supressing the upconversion pathway to generate blue fluorescence, producing low-power STED imaging. This helped to achieve a ∼28 nm resolution, while the significantly lower saturation intensity resulted in a much better signal-to-noise ratio.
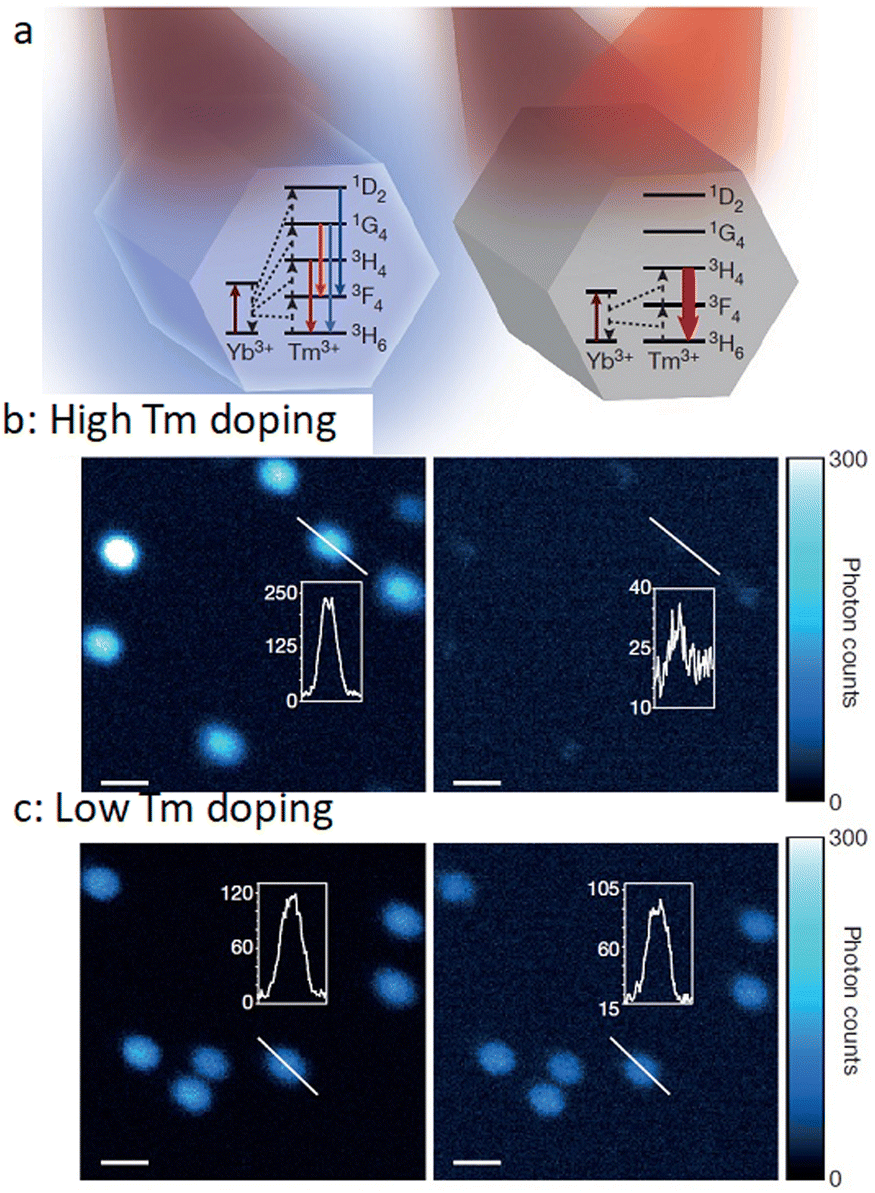 | ||
| Fig. 14 (a) Relative energy levels (not in scale) associated with Yb3+ UCNPs, co-doped with Tm3+, with 980 nm excitation (left panel), and with both 980 nm and 808 nm irradiation (right panel). (b) Confocal images of UCNPs doped with 8% Tm3+ (using 980 nm laser (left panel) and both 980 nm and 808 nm (right panel) dual laser, and (c) analogous results for 1% Tm3+-doping. Reproduced with permission from ref. 96, Copyright 2017, Springer Nature. | ||
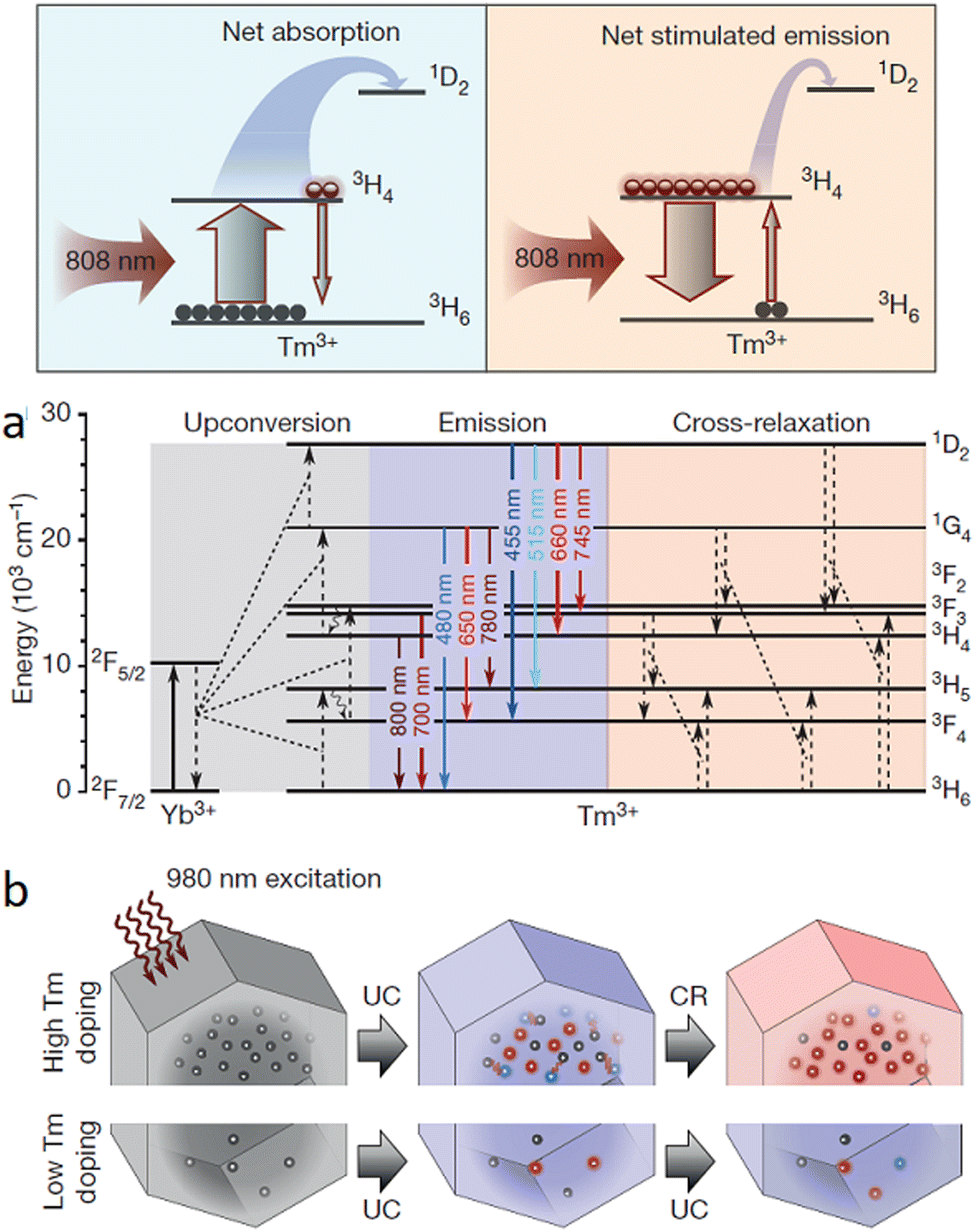 | ||
| Fig. 15 (a) Cartoon energy level diagram to illustrate the net absorption, as well as the stimulated emission between 3H4 and 3H6 levels for Yb/Tm co-doped UCNPs when probed using 980 nm depletion laser and 808 nm excitation laser. (b) Energy level diagram to show possible cross-relaxation pathways for highly doped (with Tm3+) NPs. (c) Cartoon image to show photon-avalanche-like process results from the intense cross-relaxation (CR) between the emitters in a highly doped UCNPs (energy levels for the sensitizers are not shown). Reproduced with permission from ref. 96, Copyright 2017, Springer Nature. | ||
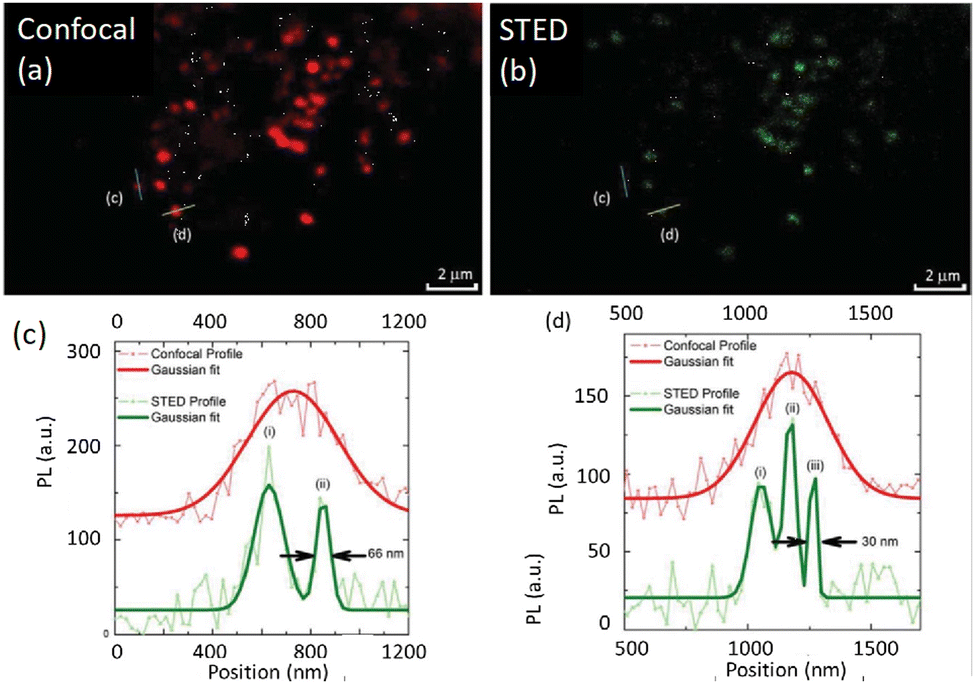
Zhan, He and colleagues developed a UCNP system (NaYF4:18% Yb3+,10% Tm3+) that successfully demonstrated two-colour STED imaging with ∼66 nm resolution using a single pair of excitation/depletion beams.97 Inter-ionic cross-relaxation helped to achieve efficient optical depletion in NaYF4:18% Yb3+, 10% Tm3+ UCNPs with two NIR laser beams centred at 975 nm (excitation) and 810 nm (depletion). The [Tm3+]-dependent depletion efficiency of the 455 nm blue emission band by cross-relaxation processes was also studied.
This process helped to eliminate the need for separate excitation and depletion beams and the associated challenge of aligning multiple laser beams, which constitutes a major obstacle in current multicolour STED protocols. This approach was used to image cytoskeletal desmin protein in HeLa cells by using antibody-conjugated UCNPs (Fig. 16), leading to a lateral imaging resolution of ∼82 nm (<λex 975 nm).
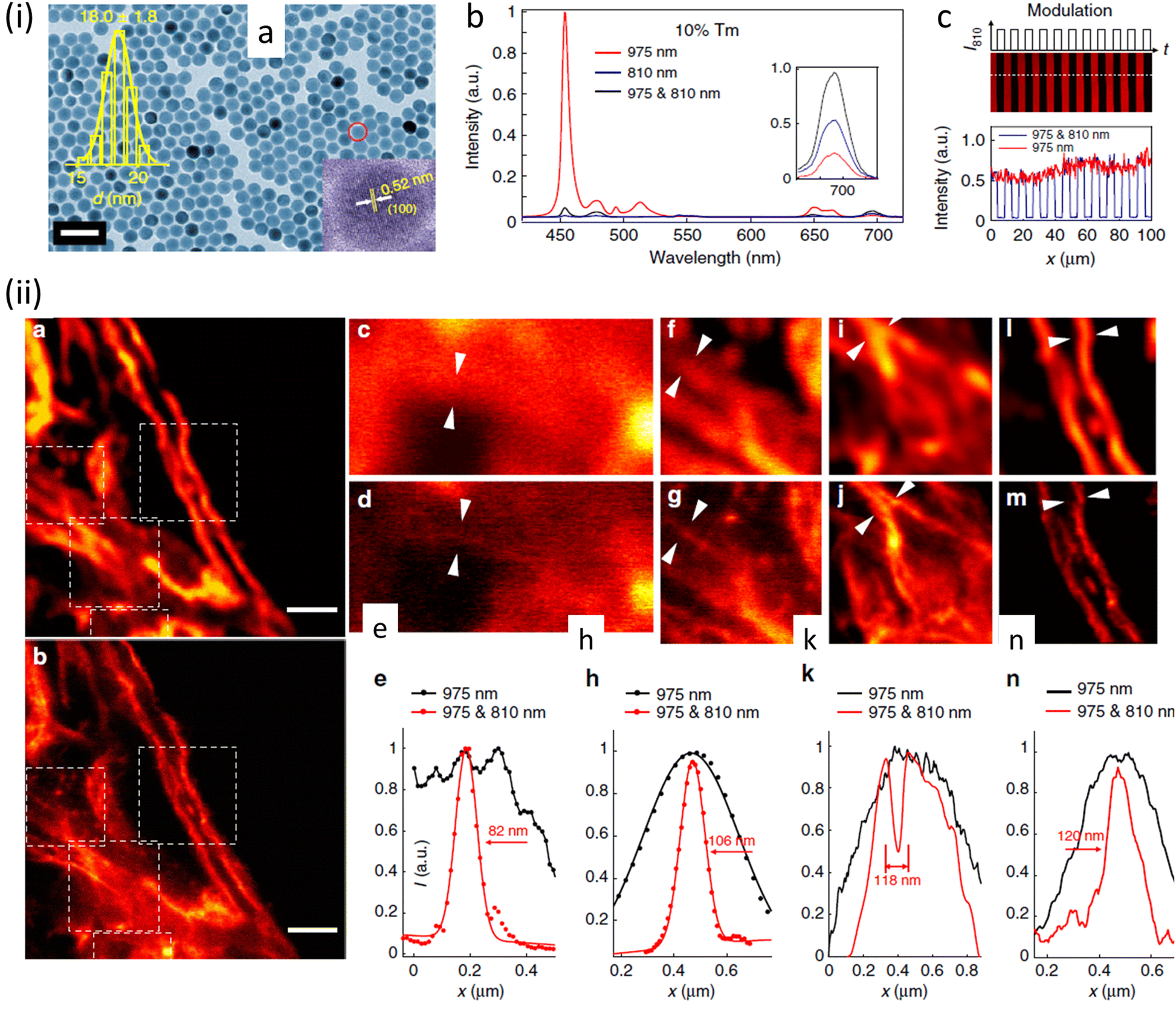 | ||
| Fig. 16 (a) TEM images of (I975455–I975 & 810455)/(I975455) UCNPs. Distribution of diameters of nanoparticle and a HR-TEM image are shown in insets (scale bar: 50 nm). (b) Optical depletion of the 455 nm UC fluorescence of UCNPs with 10% Tm3+ doping: 96% depletion efficiency of the 455 nm emission from UCNPS with 10% Tm3+ doping (λexc = 975 nm (CW), λdepletion = 810 nm (CW)). (c) Images using UCNPs, 10% Tm3+ UCNPs film with modulated 810 nm laser co-irradiation. (ii) SR images of intra-cellular desmin (a cytoskeleton protein) using antibody-conjugated UCNPs: (a) images recorded with 975 nm excitation of cytoskeleton and desmin in HeLa cancer cells incubated with anti-desmin primary antibody using UCNPs bio-conjugated with goat anti-rabbit IgG secondary antibody, (b) identical region imaged using 975 nm excitation and 810 nm STED laser beams; scale bars: 2 μm. (c–n) Enlarged images for selected areas from a and b with corresponding analyses: images in c, f, i, and l are details from a; images in d, g, j, and m are from the white squares in b. (e), (h), (k), (n) are line profiles for areas indicated with arrow heads in c and d, f and g, i and j, l and m. Reproduced with permission from ref. 97, Copyright 2017, Springer Nature. | ||
Jin and co-workers have exploited the facet-selective binding of DNA to NaYF4:Yb,Er nanocrystals to develop imaging technologies.98 The binding energies of various chelating moieties, e.g. oleate anion, oleic acid, phosphodiester bond, and phosphate group on to the two kinds of facets of hexagonal prism-like NaYF4:Yb,Er nanocrystals were also examined to rationalize the observed results. These confirmed that the binding strength of DNA phosphodiester moieties to the surface of NaYF4:Yb,Er nanocrystals was stronger than oleic acid on (001) facets, while it was weaker than oleate anions on (100)/(010) facets. Thus, DNA molecules could effectively replace surfactant molecules on NaYF4:Yb,Er nanocrystals to generate anisotropicity or a hydrophilic surface. The location of DNA molecules was confirmed by analytical methods and was visualized by STORM (Fig. 17).
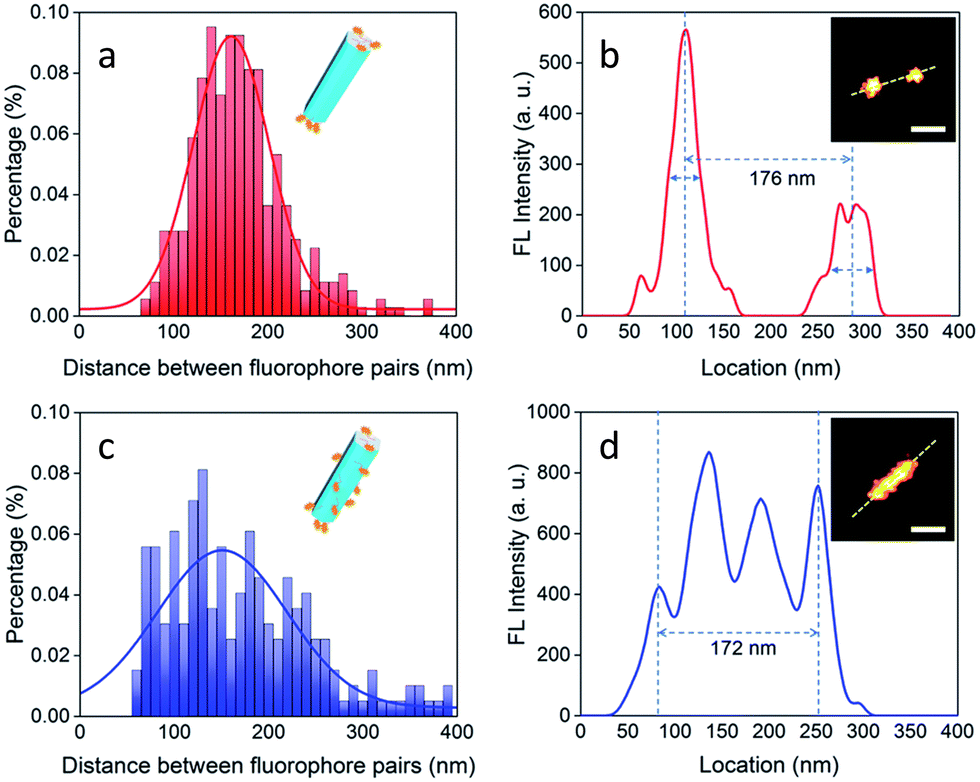 | ||
| Fig. 17 Images demonstrating selective DNA binding to 001 plane of NaYF4:Yb,Er nanorods: (a and b) binding of DNA molecules on end (001) facets: Histogram showing distance between two separated luminescent signals for a population of nanorods of length of ∼170 nm. (Inset (b): Image of a nanorod with end tagged by luminescent ATTO-550). (c and d) UC-nanorods with multiple and random fluorescent clusters. Distribution of separations between two fluorophores in a range of UC-nanorods, evaluated by luminescent signal profile of a representative nanorod (isotropically labelled with ATTO-550). Inset(d): UC-nanorods labelled with ATTO-550. Scale bar: 100 nm. Reproduced with permission from ref. 98, Copyright 2018, The Royal Society of Chemistry. | ||
These studies on facet-selective functionalization of NaYF4:Yb,Er nanocrystals not only provide insights into bio-/nanointerface interactions but have the potential to open up new technology for selective biomolecule functionalization of nanoscale material. Additionally, the controlled self-assembly of UCNPs through tailor-made DNA chemistry suggests that UCNP building blocks could be constructed into more sophisticated functionalized nanostructures.
Monodispersed UCNPs (NaYF4:Yb3+,Tm3+ nanocrystals) were used in studies that revealed the human eye could track a single nanoparticle, emitting over 4200 photons/100 milliseconds, using a simple optical microscope setup (Fig. 18).99 Apart from protein dynamics it was suggested that many sub-cellular functions, such as cytoskeleton rearrangement, and organelle movement and cooperation. involving dynamics at a molecular scale could also be studied using this approach.
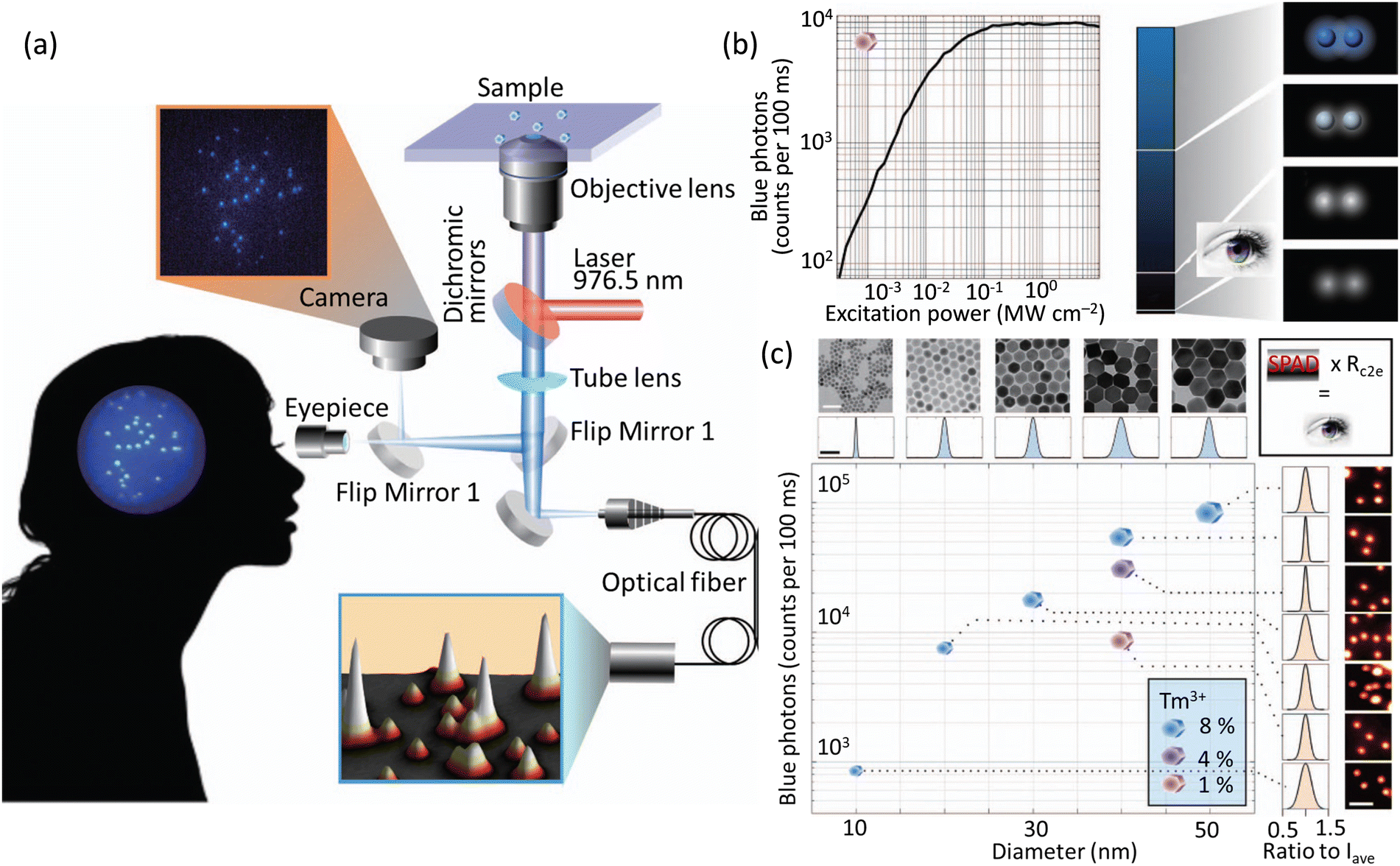 | ||
| Fig. 18 (a) Microscope system to study the sensitivity of a CCD camera and the human eye (interconvertible through flap mirror) in tracking single UCNPs. Fluorescence from the UCNPs is processed by an avalanche photodiode fitted with optical fibre to mimic the duration time for image processing in a human brain. (b) Plot showing emission as a function of laser power using UCNPs with 1 mol% Tm3+-doping (475 ± 25 nm). A region was chosen with 2 UCNPs separated by 3 μm comparable with the central region of the human fovea that have the highest density of cone cells. (c) Plot showing intensity of images related to the blue band of UCNPs vs size of UCNPs (determined by TEM). Intensities for samples with different Tm3+ doping and defined diameter (∼40 nm) is also shown. Standard deviations of the sizes varied from 6.23 to 17.4% from the average intensity (Iave) for 14 patients). Reproduced with permission from ref. 99, Copyright 2018, Springer Nature. | ||
A lanthanide-doped UCNPs (β-NaYF4:Yb,Gd) was developed for tracking lysosomes using SIM microscopy.100 Post-functionalization of NaYF4:Yb,Gd based UCNPs with a lysosome-associated membrane glycoprotein (MAAPGSARRP LLLLLLLLLL) helped to achieve lysosome specificity. Upon excitation at 980 nm, a green emission band at around 525–560 nm was observed due to electronic transitions from the 2H11/2–4I15/2 and 4S3/2–4I15/2 states – Fig. 19.
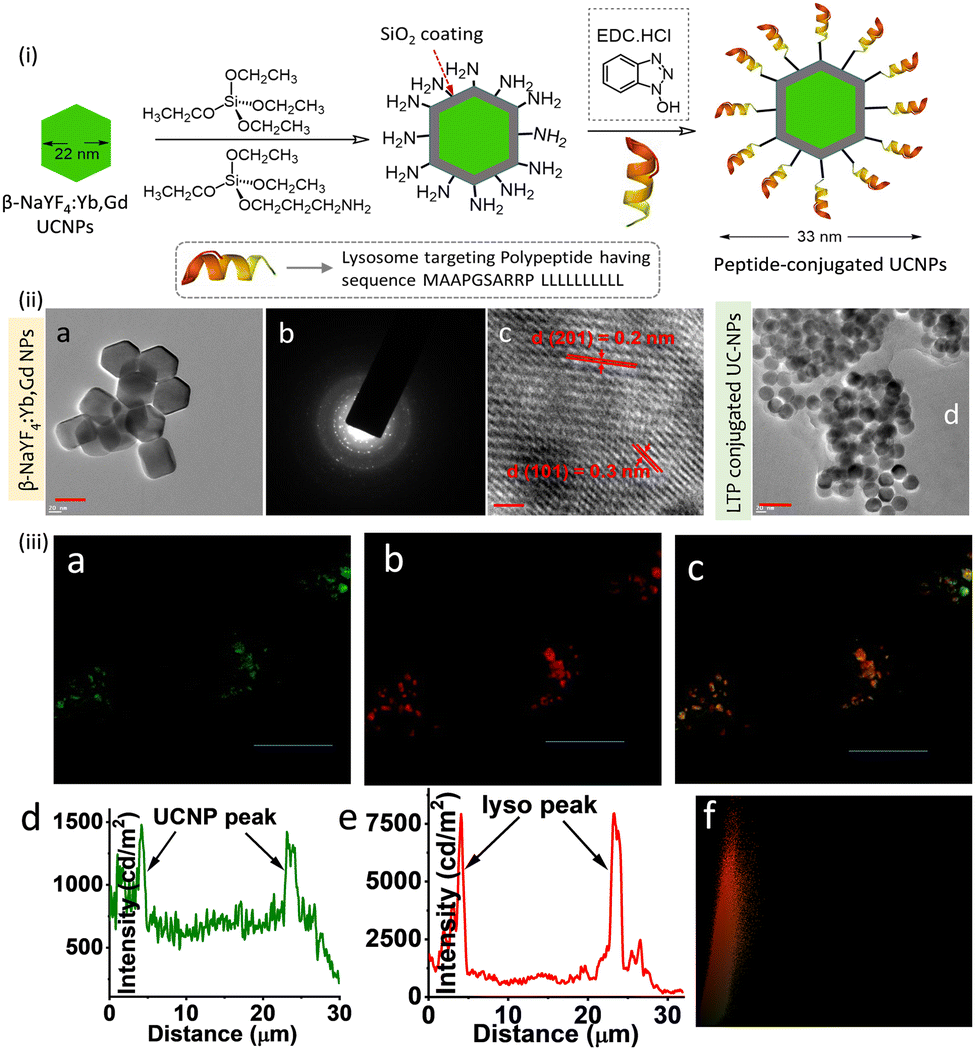 | ||
| Fig. 19 (i) Partial reaction scheme adopted for the synthesis of the lysosomal targeting peptide conjugated UCNPs. (ii) (a–c) TEM images of β-NaYF4:Yb,Gd nanoparticles. (d) TEM images of the UCNPs surface-functionalized with membrane glycoprotein (MAAPGSARRP LLLLLLLLLL). (iii) Co-localization experiments (SIM microscopy) of intracellular localization of UCNPs using LysoTracker probes in RAW cells: (a) in cellular emission of UCNPs with intensity along the traced line shown underneath (panel d). (b) Emission from Lyso-Tracker Deep Red with emission profile (panel e). Merged images and the Pearson co-efficient plot (panel f) confirm the indicating lysosomal localization of the UC-NPs. Excitation at 980 nm was used for all studies. Scale bar for (ii) is 10 nm and for (ii) 10 μm. Reproduced with permission from ref. 100, Copyright 2017, The Royal Society of Chemistry. | ||
The intracellular co-localization experiments using SIM and a commercial lysotracker probe confirmed lysosomal specificity of the UCNPs. It was suggested that these lysosome-targeting UCNPs may be used to study dysfunction in intracellular transport processes associated with lysosomal diseases.
The same research group developed another related (NaYF4:Yb,Gd,Eu)-based UC-nanorod, post-functionalized with triphenyl phosphonium ions, developed as a multifunctional platform for mitochondria imaging (Fig. 20).101 Cationic triphenyl phosphonium ion provided the mitochondrial specificity. Further, the choice of the Gd3+ ion meant that these UC-nanorods were also suitable for magnetic resonance imaging using T1- and T2-weighted MRI phantom images. Thus, the judicious integration of up-conversion fluorescence and Gd3+ provided super-resolution optical imaging and T1–T2 dual-modal magnetic resonance imaging in the same platform.
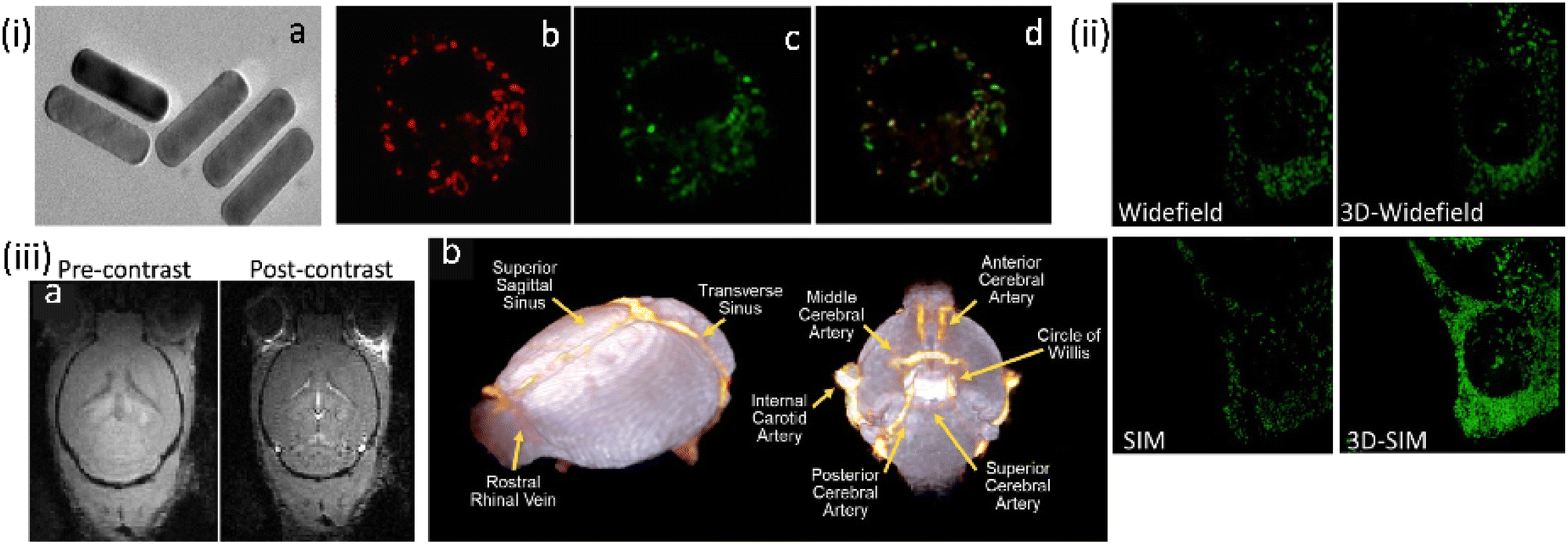 | ||
| Fig. 20 (i) (a) TEM images for β-NaYF4:Yb,Gd,Eu NRs. (b–d) Widefield (WF) microscopy images and the co-localization experiments of UCNPs functionalized with triphenyl phosphonium ion for mitochondrial localization in macrophage RAW 264.7 cells, while (b) being the image for mitotracker green. (ii) Comparison of images for the same area WF vs. 3D-WF vs. SIM vs. 3D-SIM; emissions from UCNRs and MTG fluorescence were recorded in the wavelength ranges of 600–650 nm and 500–550 nm, respectively. (iii) Magnetic Resonance imaging studies: FLASH-3D MR images of (a) mouse brain (axial orientation) captured at 200 mm3 spatial resolution before and after injection of UCNRs, (b) 3D cerebrovascular map of the mouse brain generated using the volume rendering tools in Amira by overlapping the pre-contrast (in white) and post-contrast (in orange) images. Scale bar for (i-a) is 10 nm; for (i-b, c, d and ii) are 10 μm. Reproduced with permission from ref. 101, Copyright 2020, The Royal Society of Chemistry. | ||
Fluorescent carbon dots
Fluorescent carbon dots (CDs) are a relatively new class of nanomaterials that have gained attention for bioimaging owing to their excellent optical properties, which include stability towards photobleaching, high emission quantum yield and size-dependent multicolour and NIR fluorescence. Importantly, their water dispersibility, biocompatibility and intracellular internalization can be tuned through appropriate surface functionalization.57 The high biocompatibility of CDs arises from a combination of their chemical inertness, low toxicity, cell membrane permeation owing to their small sizes (<10 nm) and two-photon activity.102,103 Methodologies for the preparation of the carbon dots are typically categorised as ‘top-down’ or ‘bottom up’ approaches.The top-down method solely uses chemical methods – or a combination of physical and chemical methods – to ‘cleave’ graphene or other carbon allotropes such as C60 fullerene and multi-walled carbon nanotubes.104 However, limitations of this method include wider size distributions, which are associated with changes in spectral properties, and also variations in surface functionalization.104,105
Bottom-up synthetic methods use organic derivatives to produce nanographene with well-defined spectral properties and purpose-built surface functionalization that help to achieve specificity in organelle localisation.104 Importantly, doping with various heteroatoms, such as nitrogen, sulphur, and phosphorus offers the option to tune the emission property of these materials and adds to their versatility as imaging agents.106 These impressive optical properties, along with their surface tunability and their potential for optical blinking have paved the way for carbon dots use in super-resolution imaging.
In 2014, Pompa et al. developed highly photoluminescent multicolour CDs for STED nanoscopy showing a resolution of 30 nm (Fig. 21).107 These CDs were successfully used for imaging MCF 7 cells. Their use in STED demonstrated a >6-fold improvement in spatial resolution compared to conventional confocal microscopy. Importantly, cell viability was not affected by the CDs, and this enabled a complete visualization of cellular events over a relatively long time-window with live cell experiments.
 | ||
| Fig. 21 (a) and (b) Improved spectral resolution of STED compared to confocal imaging using CDs in fixed MCF7 cells (CD concentration: 170 nM, 48 h incubation). (c and d) Representative intracellular emission profiles: raw data shown in light red and best-fit curve in red marked in the respective figure), while raw data for STED is shown in light green and the best fit plot in dark green. Reproduced with permission from ref. 107, Copyright 2014, The Royal Society of Chemistry. | ||
In 2015, Chizhik and co-workers developed CDs that displayed intrinsic dual-colour fluorescence due to two subpopulations of particles of different electric charges (Fig. 22).108 The applicability of CDs as probes for super-resolution optical fluctuation imaging (SOFI) in biological systems has also been studied in two different cell lines; MDCK-II and SAOS-2.
 | ||
| Fig. 22 (a and d) Widefield fluorescence and second-order SOFI images of a Saos-2 cell, respectively. (b, c, e and f) Subareas indicated with white line boxes for the demonstration of a better-resolved image for SOFI. Scale bars 10 μm. Reproduced with permission from ref. 108, Copyright 2016, American Chemical Society. | ||
In SOFI, the influence of probe diffusion was studied by comparing a theoretical model and numerical simulations to establish the impact of sample dynamics on the imaging quality. These studies revealed that over a range of physiological conditions, fluorophore diffusion inflicts a change in the amplitude of the SOFI signal.109 Chizhik and co-workers observed that for both cell types, neutral blue-emissive carbon nanodots were found to penetrate the nuclear membrane and localize to cellular nuclei. Interestingly, the green-emissive CDs were excluded from the nucleus and localized into fibrous, network-like intracellular structures, possibly endosomes or mitochondria. It was proposed that these nanodots are endocytosed and then trafficked along with the lysosomal/endosomal network.
The Huang research group reported on a high-density super-resolution localization imaging method using blinking carbon dots derived from carbon black.110 These carbon dots showed a burst of fluorescence with high photon output (∼8000) and a low duty cycle (∼0.003). The conjugation of carbon dots to goat anti-mouse IgG secondary antibody was established by fluorescence correlation spectroscopy and dynamic light scattering – Fig. 23.
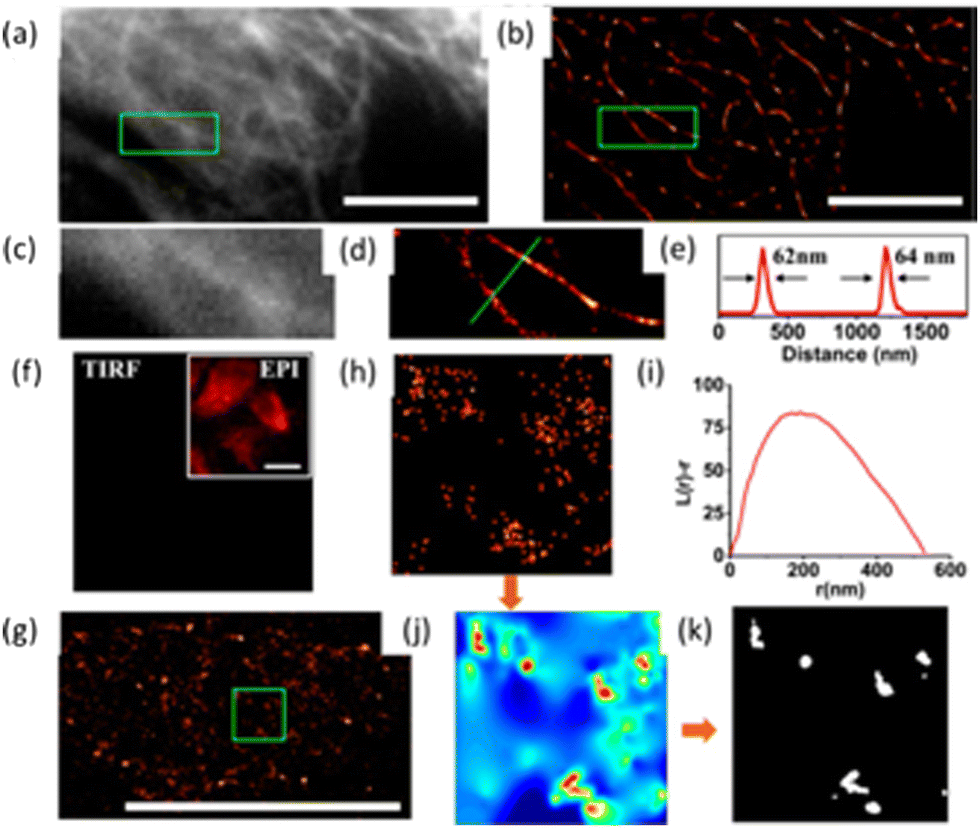 | ||
| Fig. 23 The luminescent property of CDs exploited for plasma membrane imaging (scale bar: 10 μm): (a) Conventional confocal image and (b) super-resolution image of microtubules, while the enlarged section (indicated with a green box shown in (a and b) are presented in (c and d), respectively. (e) Fluorescence intensity profiles for the specific line region, as shown in (d). (f) Image showing the cell membrane stained with CDs in the absence of anti-CCR3 primary antibodies, while the insert shows an autofluorescence image of the same field under epi-illumination. (g) Visualization of the immuno-stained cell membrane with CDs from a representative super-resolution image with an enlargement of a 2 × 2 μm2 region in (g). (i) Ripley's K function analysis of CCR3 clustering in the region. (j) A interpolated cluster map based on Ripley's K function analysis. Colour code: red signifies highly clustered regions, and Blue represents low values for clustering. (k) The corresponding binary cluster image using a threshold. Reproduced with permission from ref. 110, Copyright 2017, American Chemical Society. | ||
Fig. 23 (panels a and b) show the conventional fluorescence image and STORM image of secondary antibodies labelled carbon dot stained microtubules in the same region of the HeLa cell, respectively. The super-resolution microscopy image shows a drastic improvement in the resolution of the microtubule network. This stable blinking feature of the carbon dots allows high-density localization images at a spatial resolution of 25 nm to be acquired.
Haynes and co-workers reported malic acid-derived biocompatible CDs for SRM imaging.111 Interestingly, in the live rainbow trout gill epithelial cell line, the as-synthesized carbon dots showed two distinct intracellular distribution patterns under different excitation wavelengths. Under green-to-yellow laser excitation (488, 514, and 561 nm) (Fig. 24a), filament-like distribution was observed inside the live cells, whereas for blue-to-cyan laser excitation (405 nm) a different distribution of carbon dots (Fig. 24a) was observed. Under green-to-yellow laser excitation, a comparison of live cells treated with carbon dots and MitoTracker Red showed a clear co-localization pattern that suggests the accumulated carbon dots are in, or on, mitochondria (Fig. 24b). Importantly the CDs provided superior imaging in terms of resolution and in tracking mitochondrial movement regions in live cells (Fig. 24c), which reveals the transport of mitochondria-associated CDs along with cytoskeletal elements (e.g., microtubules).112,113
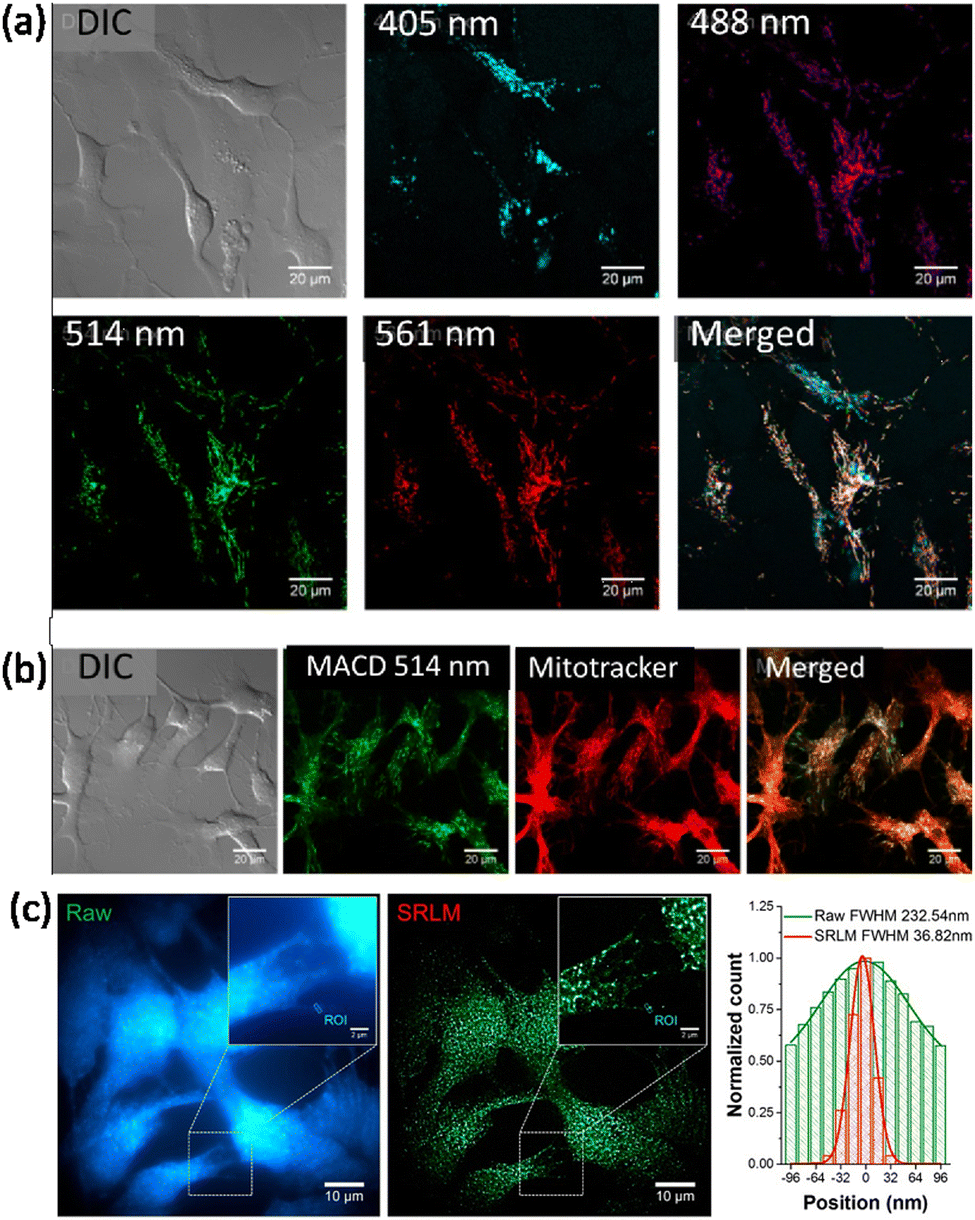 | ||
| Fig. 24 (a) Distribution of malic acid carbon dots (MACDs) in live trout gill epithelial cells. (A) MACDs with different sizes, corresponding to excitation wavelengths 488, 514, and 561 nm, were found to localize at the same intracellular compartments, whereas MACDs that were excited with 405 nm were found to localize in different compartments. (b) Green-to-yellow MACDs (with excitation wavelength ≥ 488 nm) were found mainly in mitochondria, as determined by their co-localization with MitoTracker. (c) A significant improvement of spatial resolution was achieved in SRM imaging monitoring the spontaneous photoblinking of MACDs, illustrated by the fluorescence emission profiles shown in the right panel of the marked rectangle in the images. Reproduced with permission from ref. 111, Copyright 2018, American Chemical Society. | ||
Wu et al. reported nucleolus-targeted red emissive CDs.114 They adopted hydrothermal treatment for the preparation of these CDS using p-phenylene diamine (pP) and various metal ions, which were proposed to act as a catalyst for the generation of the CDs.
This provided a series of red-emitting, metal-free CDs with varying quantum yields. Interestingly, the carbon dots obtained using a nickel catalyst showed a very high affinity towards nucleoli. For STED imaging with live A549 cells, an imaging resolution of 146 nm was achieved (Fig. 25). Additionally, these CDs offered an improvement over the commercial nucleolar dye, SYTO RNASelect, which showed green emission and could only stain nucleoli in fixed/dead cells.
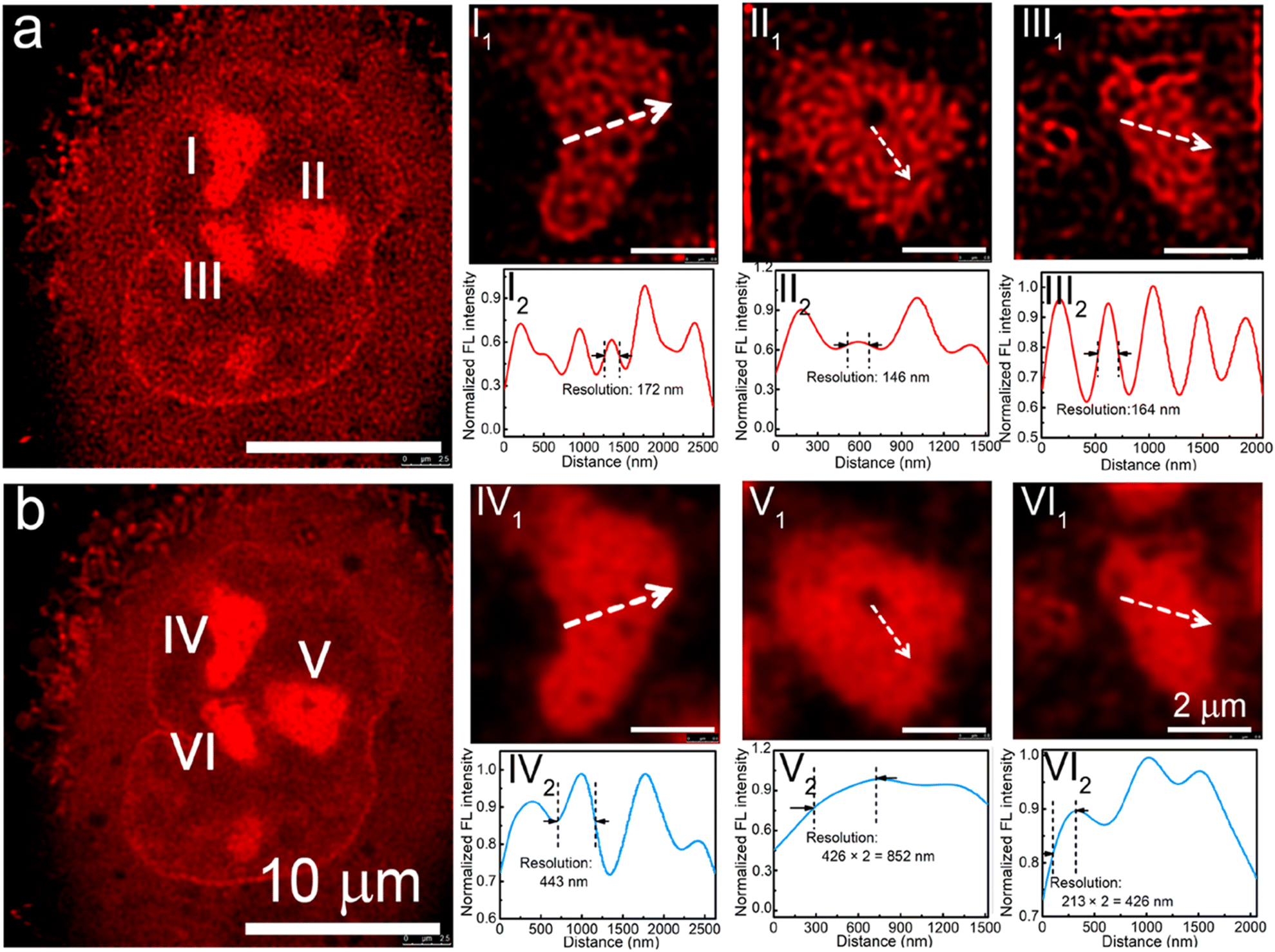 | ||
| Fig. 25 (a) STED and (b) confocal images of a A549 cell using pPCDs synthesized with Ni(II-catalyst. I1, II1, and III1 in (a) are enlarged STED images of the nucleoli of the A549 cell, while I2, II2, and III2 are corresponding fluorescence intensity profiles of the marked lines. IV1, V1, and VI1 are enlarged confocal images of the nucleoli of the A549 cell in b, while IV2, V2, and VI2 are corresponding fluorescence intensity profiles of the marked lines in IV2, V2, and VI2. Reproduced with permission from ref. 114, Copyright 2019, American Chemical Society. | ||
Zhang, et al. reported CDs surface functionalised with the cationic charge could be used for STED imaging of nucleic acid structures in HeLa cells and live C. elegans worms.115 It was found that these CDs interact with double-stranded DNA and single-stranded RNA differently producing spectrally distinguishable signals (Fig. 26). This facilitated real-time monitoring of DNA and RNA motion and localization. Fig. 26 shows images of CD-labelled chromosomes of HeLa cells in prophase and 3D reconstructed models revealing tangled DNA chains in the chromosomes. These studies also confirmed that the CDs could visualize RNA networks in a single nucleolus and are compatible with imaging the ultrastructure of live cells, potentially providing a tool for basic cell biology and clinical diagnosis.
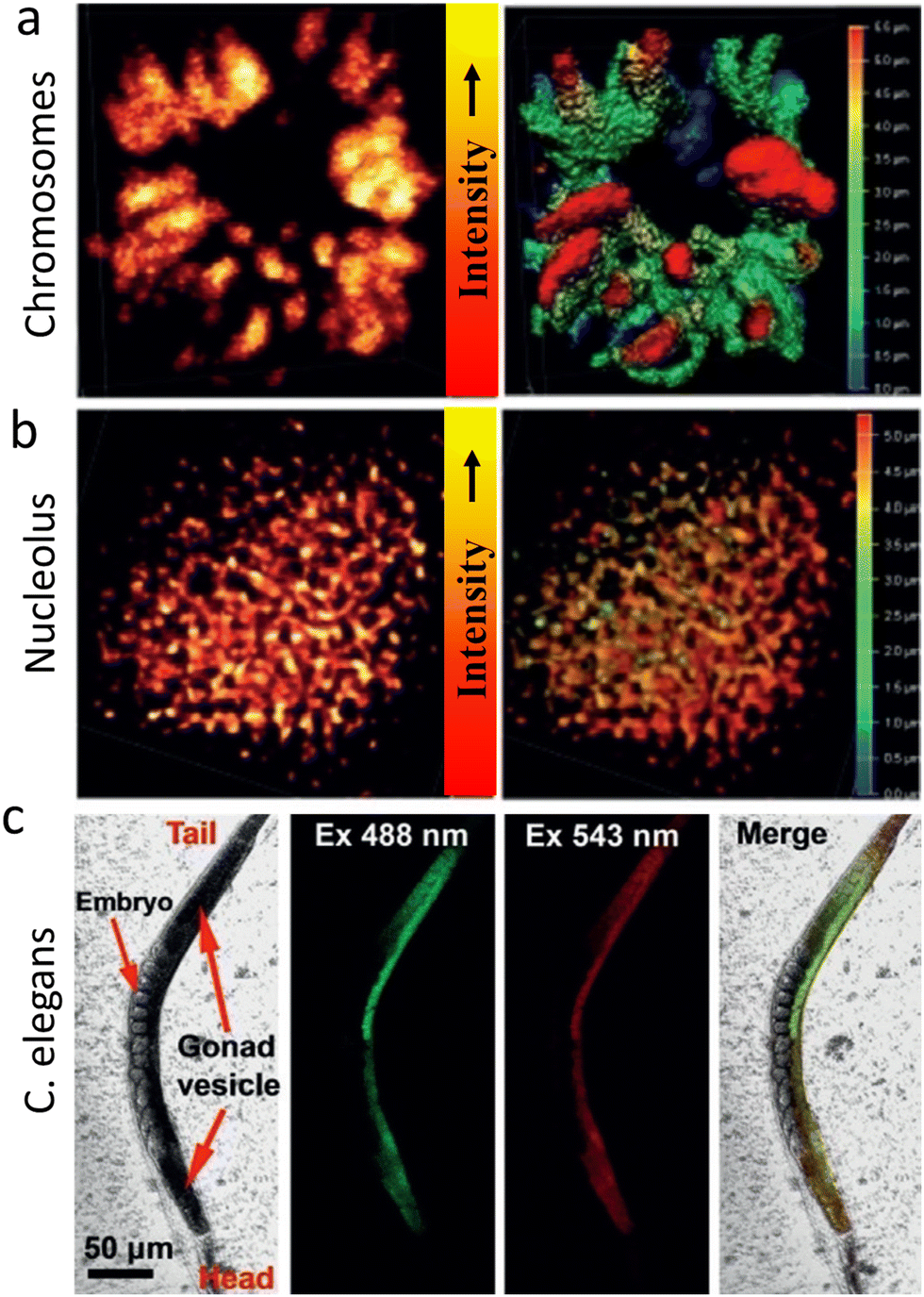 | ||
| Fig. 26 (a) STED microscopy of chromosomes at prophase using λext of 488 nm, λdepletion of 595 nm and a collection window of 500–560 nm. A Z-stack STED image of the chromosome 3D structure (step 0.12 mm) is shown in the right panel. (b) STED image of a nucleolus (λext of 560 nm, λdepletion of 595 nm and a collection window of 570–650 nm). A Z-stack 3D image of a nucleolus structure (step 0.12 mm) is shown in the right panel. (c) Imaging of C. elegans with CDs after incubation for 4 h. The CDs enter the gonad vesicle but not the developing embryos. Reproduced with permission from ref. 115, Copyright 2019, Wiley-VCH. | ||
Das, and his co-workers reported the preparation of graphene-based CDs from neem root extracts as a starting material through a simple and facile hydrothermal method.116 These as-synthesized CDs were utilized for recording SIM images of RAW cells, in which a distinct lysosomal localization was observed. An endocytosis pathway was proposed for the internalization of these CDs, accounting for the translocation from the extracellular environments into intracellular lysosomes (Fig. 27).
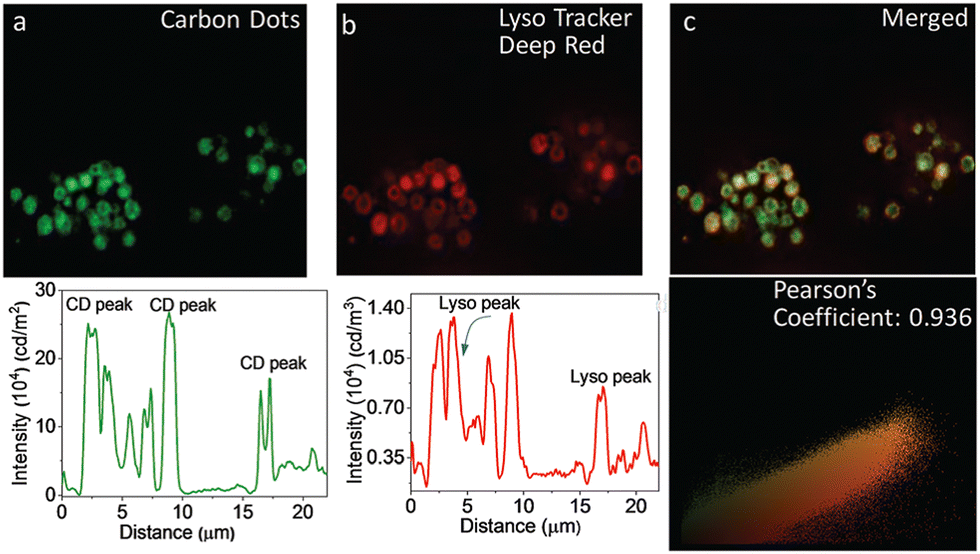 | ||
| Fig. 27 Images to reveal the intracellular localization of CDs and a LysoTracker probe in RAW cells: WF-microscopic images of intracellular emission of CDs (panel a) with intensity profile shown underneath. Emission from commercially available LysoTracker Deep Red (panel b) and the corresponding intensity profile is shown below. (Panel c) shows the overlap of green and red fluorescence, confirming the lysosomal localization of CDs. (Panel d): Pearson coefficient = 0.936. Reproduced with permission from ref. 116, Copyright 2019, The Royal Society of Chemistry. | ||
Imaging studies with zebrafish confirmed that the CDs were preferentially localized in the yolk sac region, suggesting they could be used as luminescent imaging agents for the digestive system.116,117
The nucleolus helps in coordinating cellular stress responses, including during cancer growth and treatment. Consequently, nucleolus-based diagnostics and therapeutics can be used for the precise identification of nuclear stress responses. To address this need, He and co-workers have developed CDs with RNA-binding motifs and fluorescent blinking domains.118 To measure of the nucleolar ultrastructure during the stress responses, they used these CDs to stain various cell types, including mouse (murine breast cancer 4T1, normal CHO, and colorectal cancer CT26 cells) and human cell (glioblastoma U87, human embryonic kidney 293A, and lung cancer A549 cells) lines.
While conventional wide-field microscopy provided too low resolution for detailed studies, imaging by dSTORM, revealed distinct distributions of RNA in the nucleoli of different cells, Fig. 28, demonstrating the potential of these modified CDs for analysing nucleolar ultrastructure and thus distinguishing subtle responses to stressors.
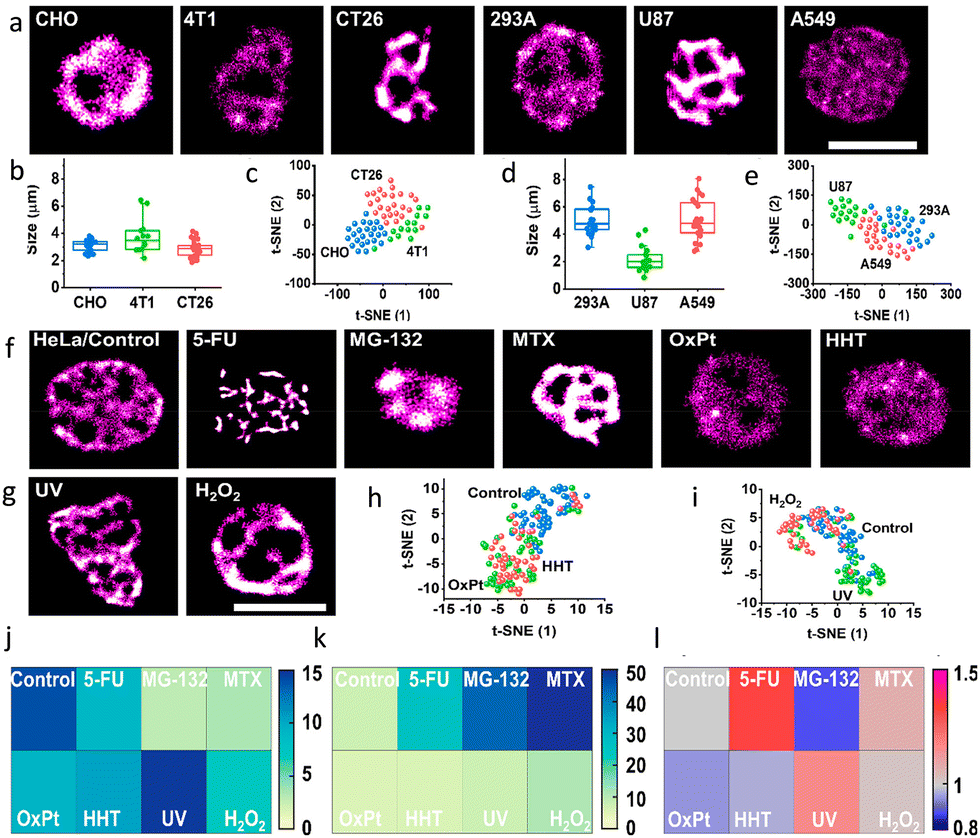 | ||
| Fig. 28 Identifying nucleolar stress response using RBP-CDs: (a) dSTORM images of various cell lines indicated in the respective image. (b and d) Nucleolar size distributions and (c and e) t-SNE plots to reveal the high-dimensional feature data sets of the mouse (CHO, 4T1, CT26) and human (293A, U87, A549) cell lines. dSTORM images of HeLa cells (f) respective images of the HeLa Cells in the absence (control) and presence of different drugs and (g) with different external stimuli as mentioned in individual panels: (j) RNA area, (k) RNA density, and (l) coefficient of variation of nucleoli under various stress conditions. Scale bar, 5 μm. Reproduced with permission from ref. 118, Copyright 2021, American Chemical Society. | ||
Wu, et al. have prepared quaternized CDs (qCDs) that were utilized to simultaneously provide bacterial imaging and antibacterial properties.119 Amine-functionalized carbon dots were post-synthetically grafted with lauryl betaine so they could selectively detect Gram-positive bacteria and display bactericidal properties. A comparison of STED and CLSM images of S. aureus cells (Fig. 29) revealed that, unlike the CLSM images, STED images could be used to confirm that most of the qCDs were localised at the surface of the bacteria and distributed around the membrane in small aggregates.
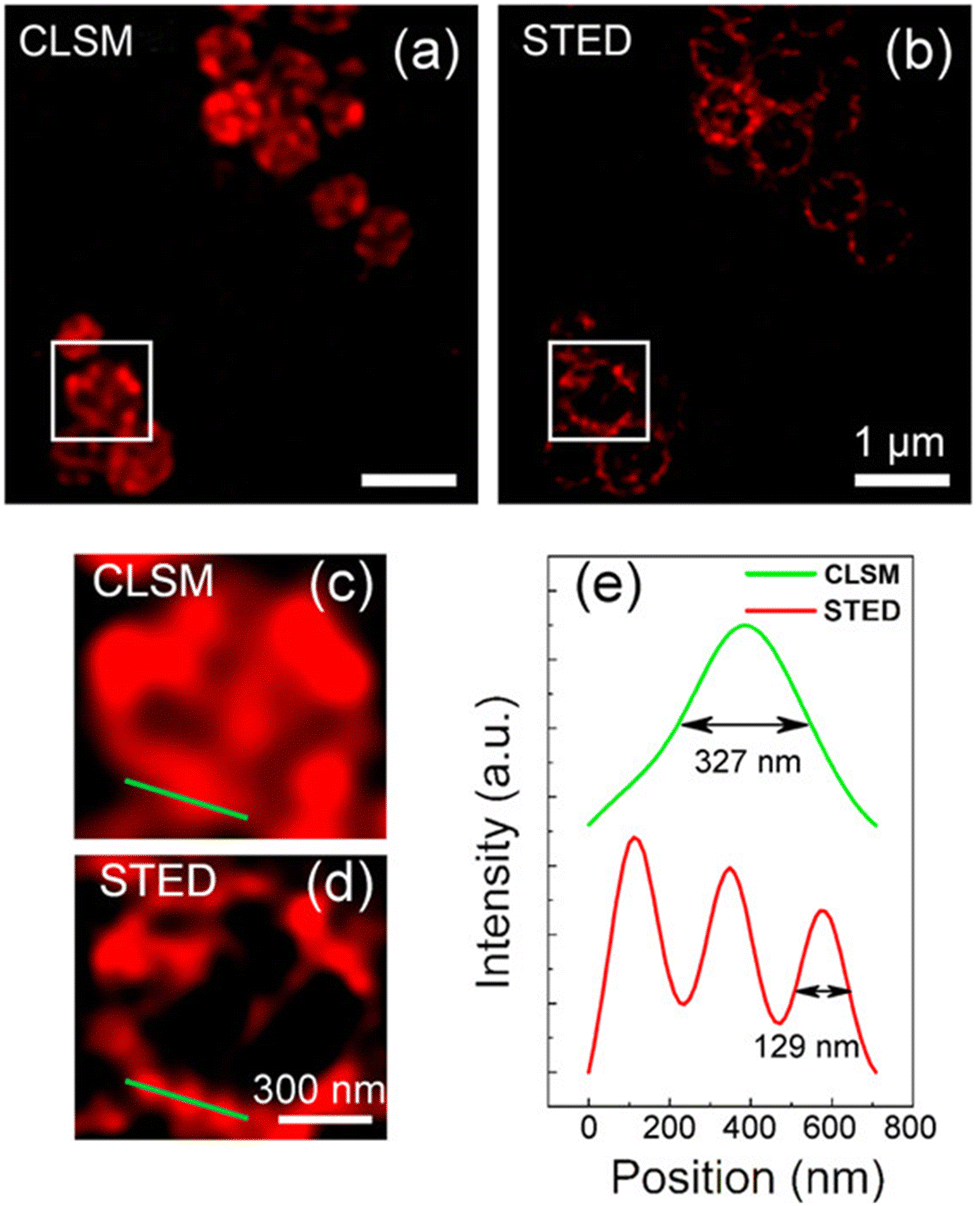 | ||
| Fig. 29 (a and b) Images revealing the superiority of STED over CLSM: images of S. aureus after treatment with qCDs for 1 h. Enlarged section of the boxed areas in (a) and (b) are shown in (c) and (d), respectively. (e) Representative intensity profiles extracted from qCDs-C12 on the surface of S. aureus after Gaussian fitting in CLSM and STED modes. Reproduced with permission from ref. 119, Copyright 2016, American Chemical Society. | ||
Semiconductor polymer dots (semiconductor PDots)
Semiconductor PDots have been exploited in biological imaging owing to their unique photophysical properties and favourable biocompatibility.120,121 Among these materials, photoblinking semiconductor polymer dots are the most widely used for multicolour super-resolution imaging, SOFI nanoscopy and STED imaging. PDots doped with redox-active molecules have been utilized for localization-based super-resolution imaging of bacterial shapes with a localization accuracy of ∼5 nm.122 Besides super resolution imaging, the PDots have also been widely used as biosensors,123,124 photoacoustic imaging,125,126 molecular afterglow imaging,127 and cancer theranostics.Sun and co-workers recently reported two types of photoblinking PDots derived from different semiconducting polymers, namely poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1′,3}-thiadazole)](PFBT) and poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-(1-cyanovinylene-1,4-phenylene)] (CN-PPV). PDots of PFBT and CN-PPV were utilized for SOFI imaging of subcellular structures with minimal or no detectable photo-bleaching during the acquisition time (Fig. 30).128 Images obtained after 2nd-order SOFI with necessary deconvolution analysis showed a much-improved signal to background noise ratio and spatial resolution in comparison to the images recorded using wide-field microscopy. The intensity profiles shown in Fig. 30ii(c, g and h) indicate a spatial resolution enhancement of ≈1.6-fold and 1.76-fold, respectively.
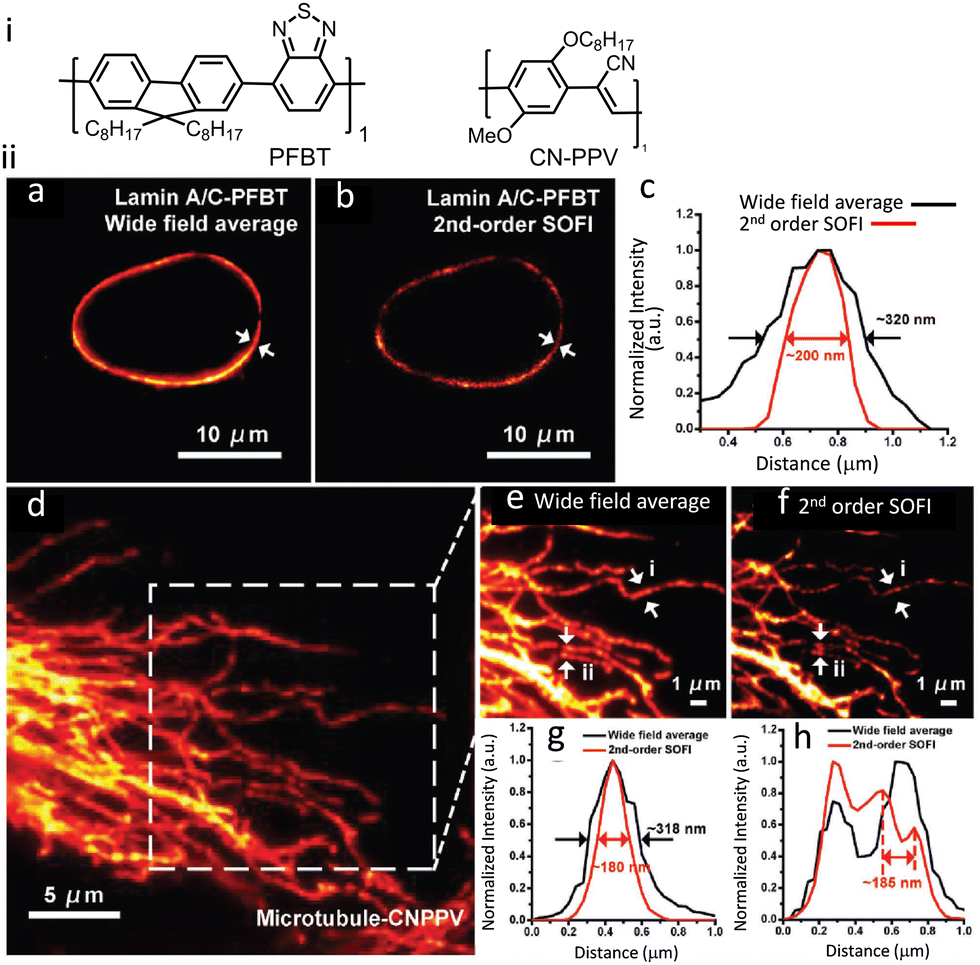 | ||
| Fig. 30 (i) Molecular structures that aggregate to form PFBT and CN-PPV PDots. (ii) SOFI nanoscopy of subcellular structures labelled with small photoblinking PDots: (a) wide-field image of Lamin A/C immunolabeled labelled PFBT PDots and (b) 2nd-order SOFI image generated by analyzing 1000 frames of raw data from panel (a). (c) The intensity profiles represent the white-arrow regions shown in panels (a) and (b). (d) Conventional wide-field imaging of tubulin labelled with CN-PPV PDots and the (e) magnified area in the square in panel (d). (f) 2nd-order SOFI image generated by analyzing 1000 frames of raw data from panel (e). (g and h) The intensity profiles of the white arrow regions are shown in panels (e) and (f). Scale bar: 10 μm in panels (a, b, i and j), 5 μm in (d) and 1 μm in (e and f). Reproduced with permission from ref. 128, Copyright 2017, Wiley-VCH. | ||
Soon after this report, the Xu research group reported a series of semiconducting polymers with varying donor-acceptor fractions.129 By using Pdots with an optimal ON-OFF ratio, they demonstrated a four-fold enhancement in resolution compared to wide-field microscopy. The FWHM value for the wide-field microscopy image (Fig. 31a) was mainly distributed around 400 nm and lacked the clarity required to observe details of microtubule structure. However, a substantial improvement in resolution was observed on increasing the cumulant SOFI order (Fig. 31c–e).
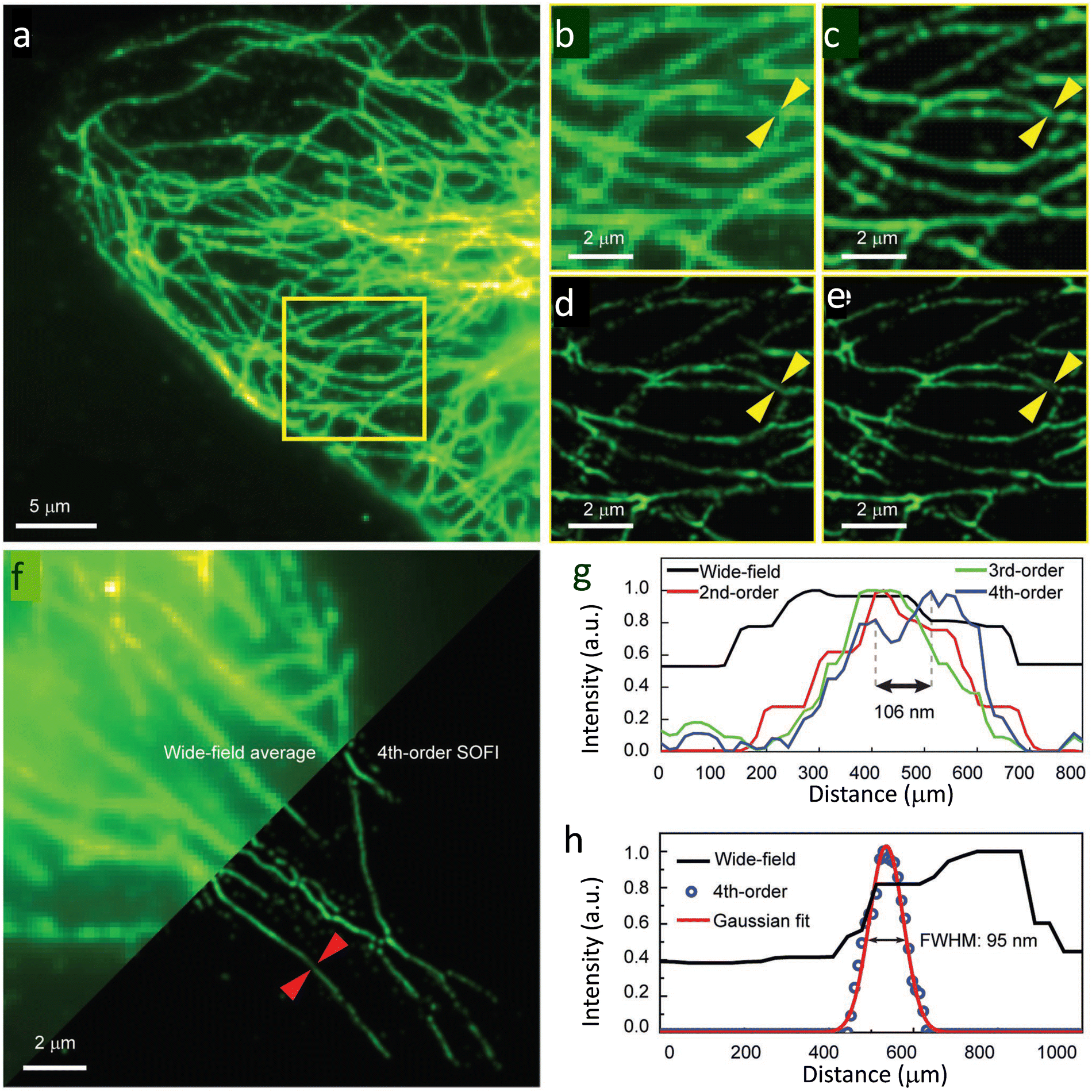 | ||
| Fig. 31 SOFI nanoscopy of microtubules labelled with PF10BT PDots: (a) wide-field image of microtubules in HeLa cells, and (b) a magnified view of the highlighted area (yellow box) in (a). (c–e) SOFI nanoscopy for the same area was reconstructed sequentially in the second, third, and fourth orders. (f) Comparison between the wide-field image (upper-left) and fourth-order balanced SOFI result (bottom-right). Profiles of the lines identified with yellow arrows in panels (b–e) shown in panel (g), while that for panel (f, red arrows) are shown in panel h before and after 4th-order SOFI analysis. Reproduced with permission from ref. 129, Copyright 2019, Wiley-VCH. | ||
Fourth-order balanced SOFI images provided sufficient clarity to observe the fine details of microtubule structures not observed in lower-order SOFI (Fig. 31g). The fourth-order spatiotemporal cross-cumulants offered an enhanced spatial resolution at both single-particle and subcellular levels.130
Fang and co-workers used semiconducting PDots derived from relatively large molecular weight (∼47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1000 Da) polymers, namely, poly[{9,9-dihexyl-2,7-bis(1-cyanovinylene) fluorene}-alt-co-{2,5-bis(N,N′-diphenylamino)-1,4-phenylene}] (PDFDP) for long-term STED imaging (Fig. 32a).131
1000 Da) polymers, namely, poly[{9,9-dihexyl-2,7-bis(1-cyanovinylene) fluorene}-alt-co-{2,5-bis(N,N′-diphenylamino)-1,4-phenylene}] (PDFDP) for long-term STED imaging (Fig. 32a).131
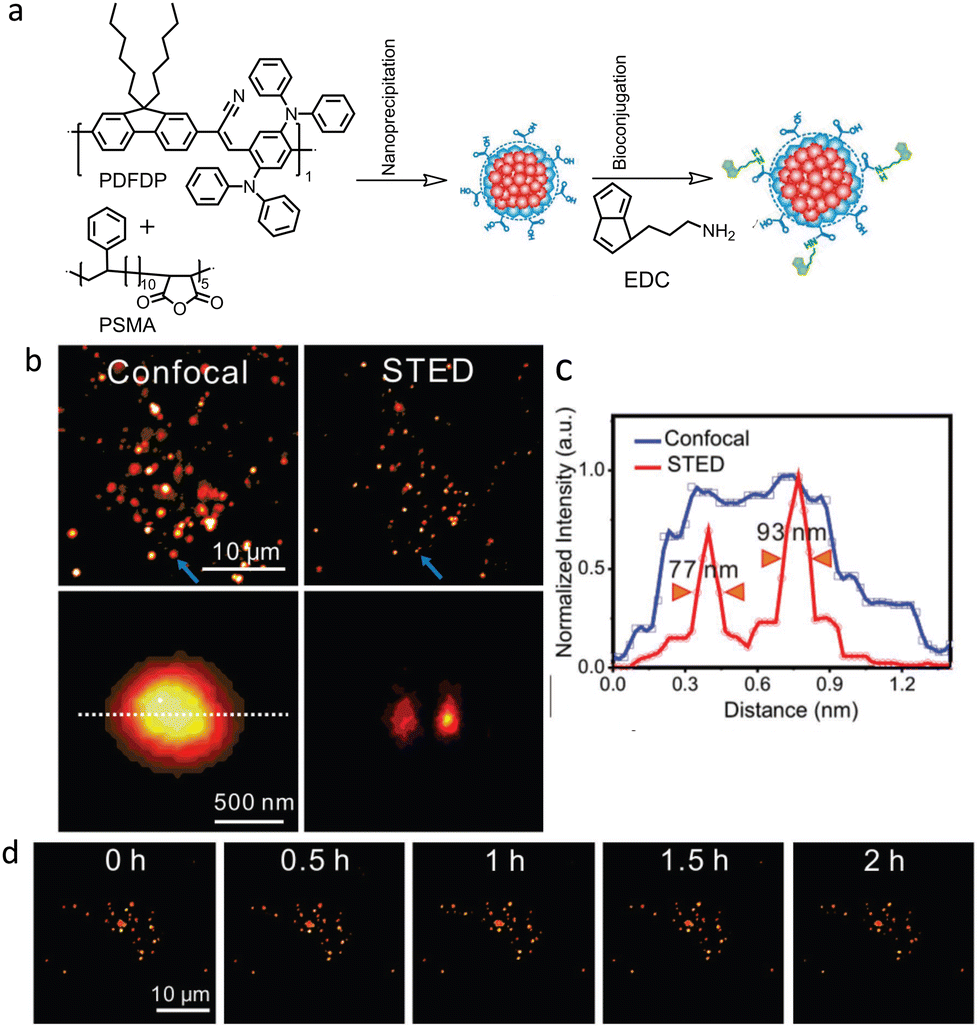 | ||
| Fig. 32 (a) Schematic illustration of PDots and biotin-conjugated polymer dots synthesis. (b) Confocal and STED images of biotin-polymer dots uptake by a HeLa cell. Two closely spaced NPs (indicated by blue arrows) were only distinguished in the STED image. (c) Respective intensity profiles of the polymer dots are indicated by the lines in (b). (d) Long-term STED imaging of biotin-polymer dots in HeLa cells under 2 h continues irradiation (field of view 35 × 35 μm2). STED beam power was kept at 3.3 MW cm−2. Reproduced with permission from ref. 131, Copyright 2018, Wiley-VCH. | ||
PDFDP-based PDots were chosen due to their structural rigidity and other favourable optical properties. A spatial resolution of 77 nm was achieved in STED nanoscopy (Fig. 32b and c) using these PDots, which enabled visualisation of the dynamic fusion and division of vesicles during a continuous 2 hours of imaging (Fig. 32d).
The same research group reported another two PDots derived from poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-(1-cyanovinylene-1,4-pheny-lene)] (CNPPV having Mw = 17000 Da) and poly[{9,9-dihexyl-2,7-bis(1-cyanovinylene)fluorene}-alt-co-{2,5-bis(N,N′-diphenylamino)-1,4-phenylene}] (PDFDP having Mw = 47100 Da) with superior optical properties in terms of their stability towards photobleaching.132 These PDots were found to be suitable for the long-term STED imaging of subcellular organelles and their dynamic interactions (Fig. 33-i). A spatial resolution of 78 and 68 nm was achieved, respectively, for CNPPV-PDots and PDFDP-PDots. In STED microscopy, the streptavidin and the biotin antibody conjugated CNPPV-PDots and PDFDP-PDots could be utilized to image the membrane protein CD44 and microtubule filaments (Fig. 33-ii). Additionally, CNPPV-PDots and PDFDP-PDots were also utilized for STED nanoscopy to track the dynamic interactions of clathrin-derived endosomes and caveolin-1-positive endosomes in live cells.
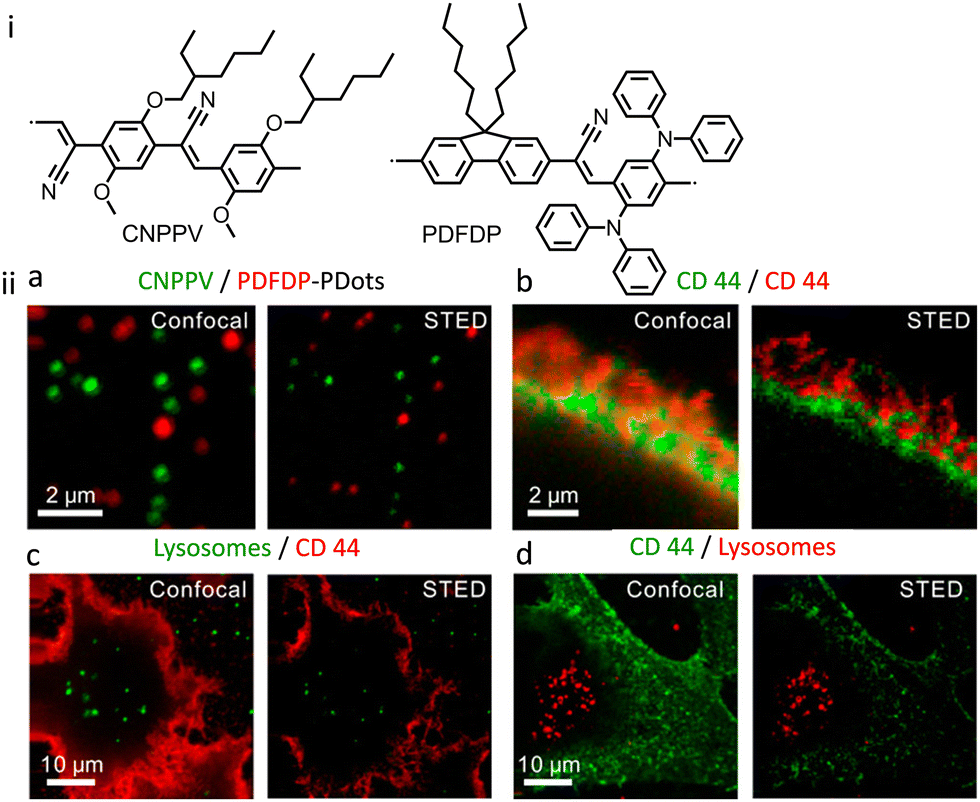 | ||
| Fig. 33 (i) Molecular structures of semiconducting PDots, CNPPV and PDFDP.(ii) Dual-color imaging of mixed particles and cellular samples after linear unmixing in confocal and STED mode: left to right (a) a mixture of CNPPV and PDFDP PDots, (b) CD 44 labelled with CNPPV and PDFDP PDots, (c) lysosomes labelled with CNPPV PDots and CD 44 labelled with PDFDP PDots, (d) lysosomes labelled with PDFDP PDots and CD 44 labelled with CNPPV PDots. Depletion power used was 10 MW cm−2 with a pixel dwell time of 1 ms; 200 × 200 pixels. The field of view was 7 × 7, 9 × 9, 45 × 45, and 45 × 45 μm2, separately. Reproduced with permission from ref. 1132, Copyright 2020, American Chemical Society. | ||
The Gianneschi research group has used SIM and nanoscale secondary ion mass spectrometry (NanoSI-MS) to investigate the sustained release of a drug from a nanocarrier.133 Without a requirement for any fluorophores, NanoSI-MS can be utilized to qualitatively visualize extracellular and intercellular processes directly with ∼100 nm resolution.134
The group synthesized a polymeric nanocarrier that encapsulated oxaliplatin. This purpose-built architecture incorporated a 15N-labeled monomer in the hydrophobic block of the polymer backbone and Cy5.5 (a NIR-active dye) in its hydrophilic block (Fig. 34a). The subcellular localization behaviour of this nanocarrier in HeLa cells was investigated using SIM and NanoSI-MS. Analysis of the data obtained from NanoSI-MS data, SIM nanoscopy (Fig. 34b), and endosomal tracking revealed that the nanocarriers were trapped in intracellular vesicles, and a sustained cytotoxic drug release from the internalized polymeric nanocarrier was observed.
 | ||
| Fig. 34 (a) Graphical illustration to represent the molecular structure of a self-assembled polymeric nanocarrier. (b) Overlaid NanoSIMS and SIM images to reveal the fluorescent tag (red) with 195Pt (blue) and 15N (green) after 4 (left) and 24 h (right) incubation of HeLa cells with Cy-15N-NP. The overlay of 195Pt and the fluorescent tag is shown in magenta, and the overlay of all three (195Pt, 15N, and fluorescence tag) is shown in white. Cell boundaries (white line) were delimited from the corresponding 12C14N-NanoSIMS ion images. Reproduced with permission from ref. 133, Copyright 2016, American Chemical Society. | ||
Combined, these techniques provide a useful tool to probe and track the composition, as well as the integrity, of the delivery system at each stage of the uptake and release process. In further studies, this nanocarrier was modified to achieve an enzyme-directed assembly by incorporating a peptide substrate into the hydrophilic block (Fig. 35a).135 This study helped in understanding the specific association of Pt with P-rich structures within cells such as DNA (Fig. 35b).
 | ||
| Fig. 35 (a) Molecular structures and self-assembly of a peptide decorated nanocarrier: Pep-Pt-P. L- and D-Amino acid peptide sequence “GPLGLAGGERDG” for L-Pep-Pt-NP and D-Pep-Pt-NP were used. Cleavage of the GL-sequence at “GPLGLAGGERDG” attributed to the release of the hydrophilic peptide sequence “LAGGERDG” which triggered the nano- to micrometre morphology change. (b) Quantitative analysis of 15N and 195Pt enrichment inside or outside of 15N- and 31P-rich region of interests: A: region of interests were defined according to 15N accumulation, as high 15N/14N (region of interests number 1–16) and low 15N enrichment (region of interests number 17–32). A.1: 15N region of interest on the L-Pep-Pt-NP hue-saturation intensity image. A.2: 15N region of interest on the D-Pep-Pt-NP hue-saturation intensity image. A.3: 15N/14N inside and outside of 15N-rich region of interests. A.4: 195Pt enrichment inside and outside of 15N-rich region of interests. (B) Region of interests were defined according to 31P signal, as high 31P (region of interests number A1–10) and low 31P (region of interest number B1–10). B.1: 31P region of interest on the L-Pep-Pt-NP 31P ion map image. B.2: 31P region of interest on the D-Pep-Pt-NP 31P ion map image. B.3: 15N/14N inside or outside of 31P-rich region of interests. B.4: 195Pt enrichment inside or outside of 31P-rich ROIs. Pt-was collected as 196Pt-for the saline sample. Pt counts on a saline region of interest were normalized to 195Pt by multiplying by 1.34, according to their isotopic abundance (33.8/25.2). Enrichment values were obtained from at least two independent images of each sample. The minimum significant difference was defined as a p-value < 0.05. Reproduced with permission from ref. 135, Copyright 2018, American Chemical Society. | ||
AIE-Luminescent material for super-resolution imaging
Organic luminophores typically favour π–π stacking interactions in their aggregated state and this leads to aggregation-induced quenching (AIQ) of fluorescence.124,136,137 However, another class of chromogenic molecules shows aggregation-induced emission (AIE) enhancement in their aggregated state, when present in a relatively high concentration, or in a non-solvent. In 2001, Tang and colleagues were first to report this phenomenon.138 In general, AIE luminophores are non-emissive or weakly emissive in dilute solution and become strongly emissive in their aggregated state.139 The restriction of intramolecular motion(s) (e.g. rotation and vibration) in the aggregated state favours radiative deactivation of the excited state of these luminophores (Fig. 36) and is ascribed as the primary reason for their observed AIE effect.140–143 Importantly, AIE-active molecules typically exhibit a large Stokes’ shift.144,145 Researchers have exploited AIE-induced phenomena for various super-resolution imaging, as discussed in the following section.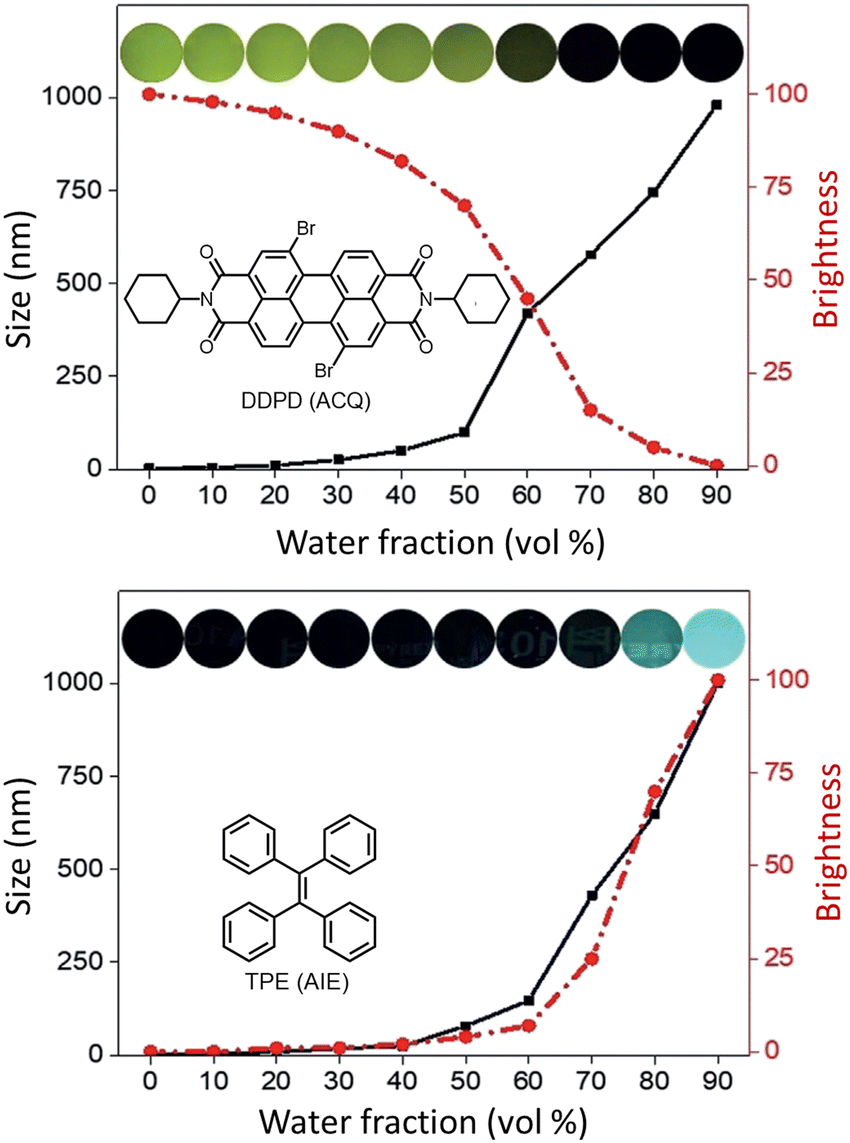 | ||
| Fig. 36 Images revealing size distribution (black curves) and fluorescence intensities (red curves) of (a) DDPD and (b) TPE in THF/water mixtures with different water contents upon UV illumination. Reproduced with permission from ref. 142, Copyright 2020, Wiley-VCH. | ||
In 2015 Qian, Tang and He and their co-workers demonstrated the efficacy of the AIE-active luminophore hexaphenylsilole (HPS) in super-resolution imaging using STED microscopy.146,147 Importantly, HPS molecules in their aggregated state were more stable towards photobleaching compared to Coumarin 102 during the STED process (Fig. 37) and also showed a much higher emission depletion efficiency than Coumarin 102 in organic solution. The same research group reported on a mitochondria-specific AIE-based molecular probe. Tetraphenylethylene derivative (o-TPE-ON+ with λex/em = 425/675) undergoes a novel photoactivation mechanism of photocyclodehydrogenation.148 Although it shows poor emission in aqueous solution, upon irradiation with visible light o-TPEON+ undergoes an photocyclodehydrogenation reaction to yield c-TPE-ON+ (λex/em = 503/580) that shows high emission quantum yield, as well as a long excitation wavelength (Fig. 38).
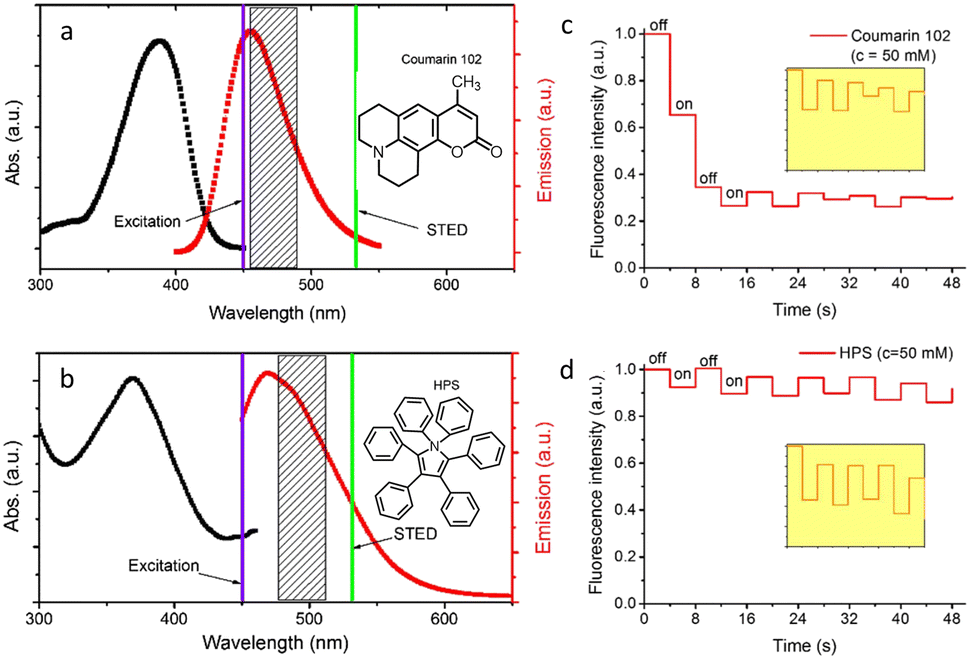 | ||
| Fig. 37 (a and b) Absorption (black curve) and emission spectra (red curve) of Coumarin 102 and AIE-1 (purple line: excitation laser; green line: STED laser). Photo-bleaching comparison between (c) coumarin 102 (d) and AIE-1 irradiated with a 25 mW STED beam ([c] = 50 μM). Reproduced with permission from ref. 146, Copyright 2015, Optica. | ||
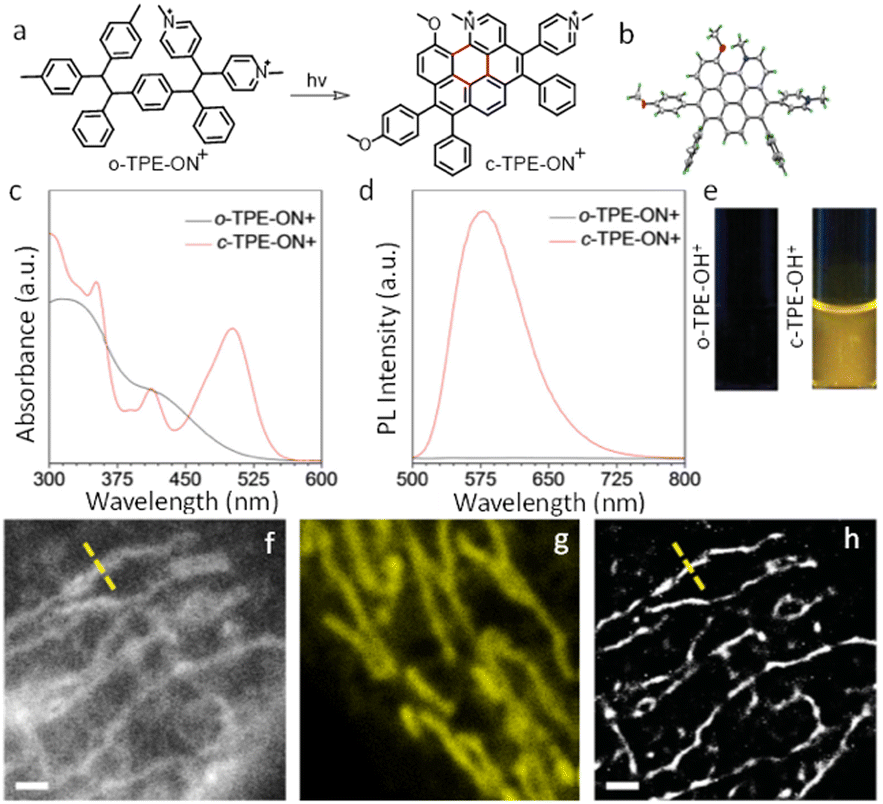 | ||
| Fig. 38 (a) The photo-induced cyclization associated with the simultaneous dehydrogenation process of o-TPE-ON+. (b) ORTEP diagram of c-TPE-ON+ at 50% probability (solvents and anion are omitted for clarity). (c) UV-Vis; (d) fluorescence spectra, and (e) image of a quartz cuvette containing o-TPE-ON+ (10 × 10−6 M) on 365 nm irradiation and c-TPE-ON+ (10 × 10−6 M) in PBS buffer solution (pH = 7.4, 10 × 10−3 M) using λext = 450 nm. (f) epifluorescent image, (g) confocal microscopy image, (h) super-resolution image of mitochondria in a fixed HeLa cell stained with o-TPE-ON+. Scale bar: 2 μm. Reproduced with permission from ref. 148, Copyright 2016, Wiley-VCH. | ||
The epifluorescent and confocal microscope image of treated mitochondria showed blurred structure with low resolution (Fig. 38f and g), while the STORM image of the same area showed distinct structural features of mitochondria (Fig. 38h). The transverse profiles of a single mitochondrion also revealed an FWHM of ∼104.5 nm, which was significantly narrower than that observed for an image obtained using an epifluorescence microscope (FWHM ∼ 697.1 nm) and confocal microscope (FWHM ∼ 467.54 nm). The results demonstrate the unique photoactivation characteristic of o-TPE-ON+, and the superiority of c-TPE-ON+ in effecting super-resolution imaging. In 2017, the same research group developed an AIEgen, 2,3-bis(4-(phenyl(4-(1,2,2-triphenylvinyl)phenyl)amino)phenyl) fumaronitrile (TTF), for STED imaging (Fig. 39-i).149
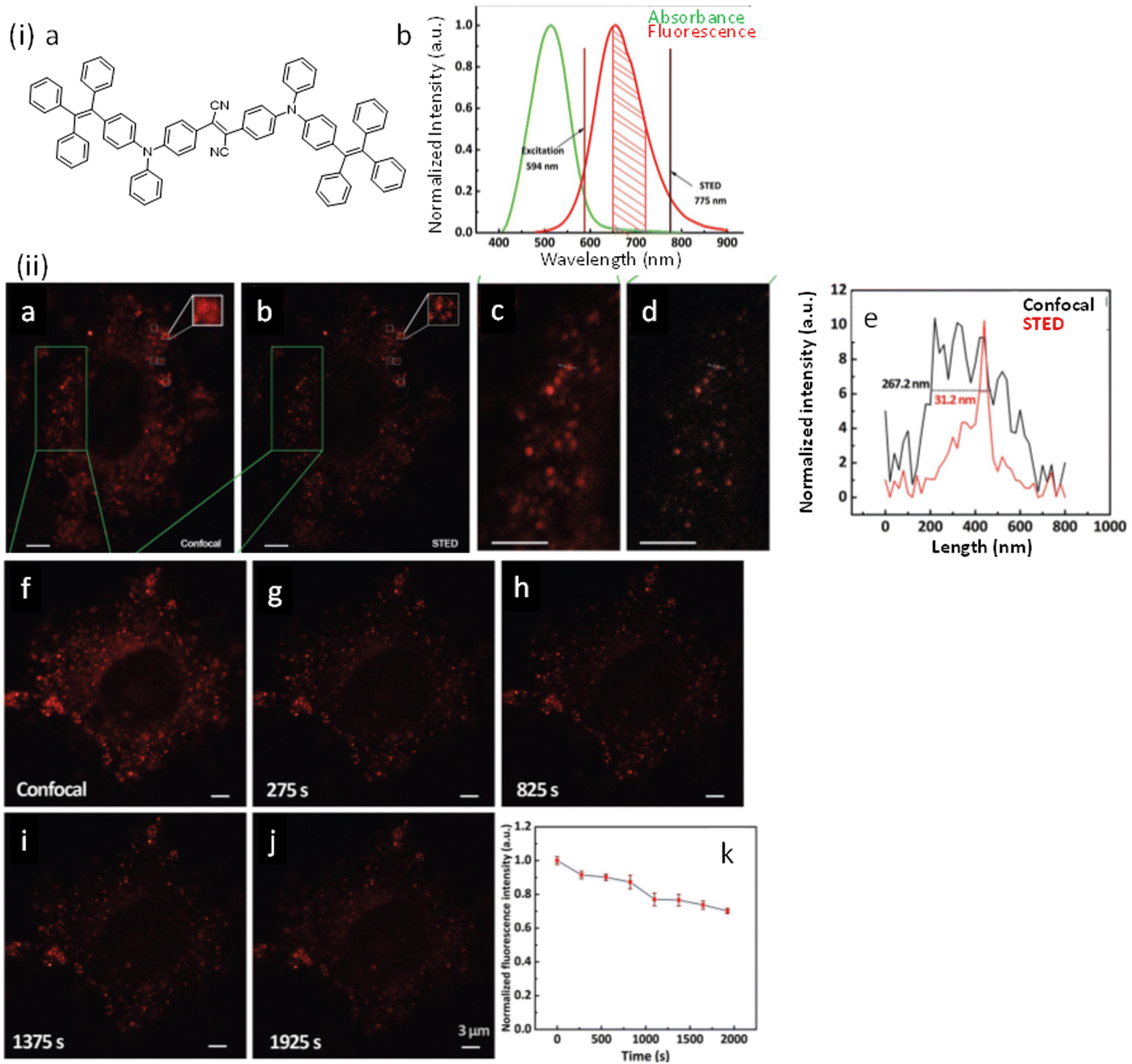 | ||
| Fig. 39 (i) Molecular structure, along with electronic/fluorescence spectra of TTF@SiO2 nanoparticles: (a) chemical structure of TTF; (b) absorption and fluorescence spectra of TTF@SiO2 nanoparticles. (ii) Comparison of confocal microscopy and STED nanoscopy imaging of HeLa cells: (a) confocal microscopy and (b) STED nanoscopy imaging of a HeLa cell, which was stained with TTF@SiO2 nanoparticles. (c and d) Are images of the green frames of panels (a) and (b). (e) Fluorescence intensity profiles along the dashed lines across the nanoparticles in panel (c) (black) and (d) (red), showing FWHM values of 267.2 nm (confocal microscopy) and 31.2 nm (STED nanoscopy). (f) A confocal image of the HeLa cell stained with TTF@SiO2 nanoparticles. (g–j) STED nanoscopy images of the HeLa cell taken at various time points, during 1925s continuous imaging. (k) Normalized fluorescence intensity of TTF@SiO2 nanoparticles in the HeLa cell at various time points, during 1925s continuous imaging. For confocal microscopy, the 594 nm excitation laser power was 2.5 mW. For STED nanoscopy, the 594 nm excitation laser power was 2.5 mW and the 775 nm STED laser power was 312.5 mW. Dwell time of each pixel: 20 μs. 1612 pixels × 1924 pixels for (a) and (b). 1486 pixels × 1542 pixels for (f)–(j). Reproduced with permission from ref. 139, Copyright 2017, Wiley-VCH. | ||
The stimulated emission efficiency of this TTF@SiO2 nanoparticles (NPs) was high, and fluorescence intensity after stimulated emission depletion was reduced to less than 30% of the original value. The Stokes’ shift of 150 nm helped in reducing the overlap of the absorption and fluorescence spectra of TTF@SiO2 NPs, as well as background fluorescence under the STED excitation beam. Additionally, these NPs were found to be stable against photo-bleaching under long-term (280 s) irradiation with a high-power (312.5 mW average power) STED beam, which also contributed to improving the STED image.
The STED nanoscopy imaging of TTF@SiO2-NPs with HeLa cells exhibited a high lateral resolution of 30 nm (Fig. 39-ii-e). On uninterrupted irradiation of the 594 nm excitation and 775 nm STED laser irradiation (312.5 mW power) for 30 min, the fluorescence intensity of internalized TTF@SiO2 NPs was only reduced by 30% (Fig. 39-ii-g–j) helping to achieve an improved temporal-spatial resolution.
Xu, et al. synthesized three types of oxetane-substituted AIE (AIE-OXE) molecules with blue, green, and red emissions (Fig. 40-i) for subcellular imaging using STED Nanoscopy.150 The Blue-AIE-OXE is a typical tetraphenylethylene derivative having a λMax of ∼480 nm (Fig. 40(i)). Structural modification provided extended conjugation that helped to provide a Green-AIE-OXE dye, while the synthesis of a Red-AIE-OXE involved the incorporation of appropriate electron donor and acceptor groups to alter the energies of the dye's frontier orbitals. All three luminophores showed stable and intense fluorescence in their respective aggregated state. The red-AIE–OXE nanodots were used to obtain STED images of microtubule structures with much better resolutions (FWHM ∼ 95 nm, STED power intensity: ≈100 MW cm−2) (Fig. 40-ii) compared to images obtained in confocal microscopy.
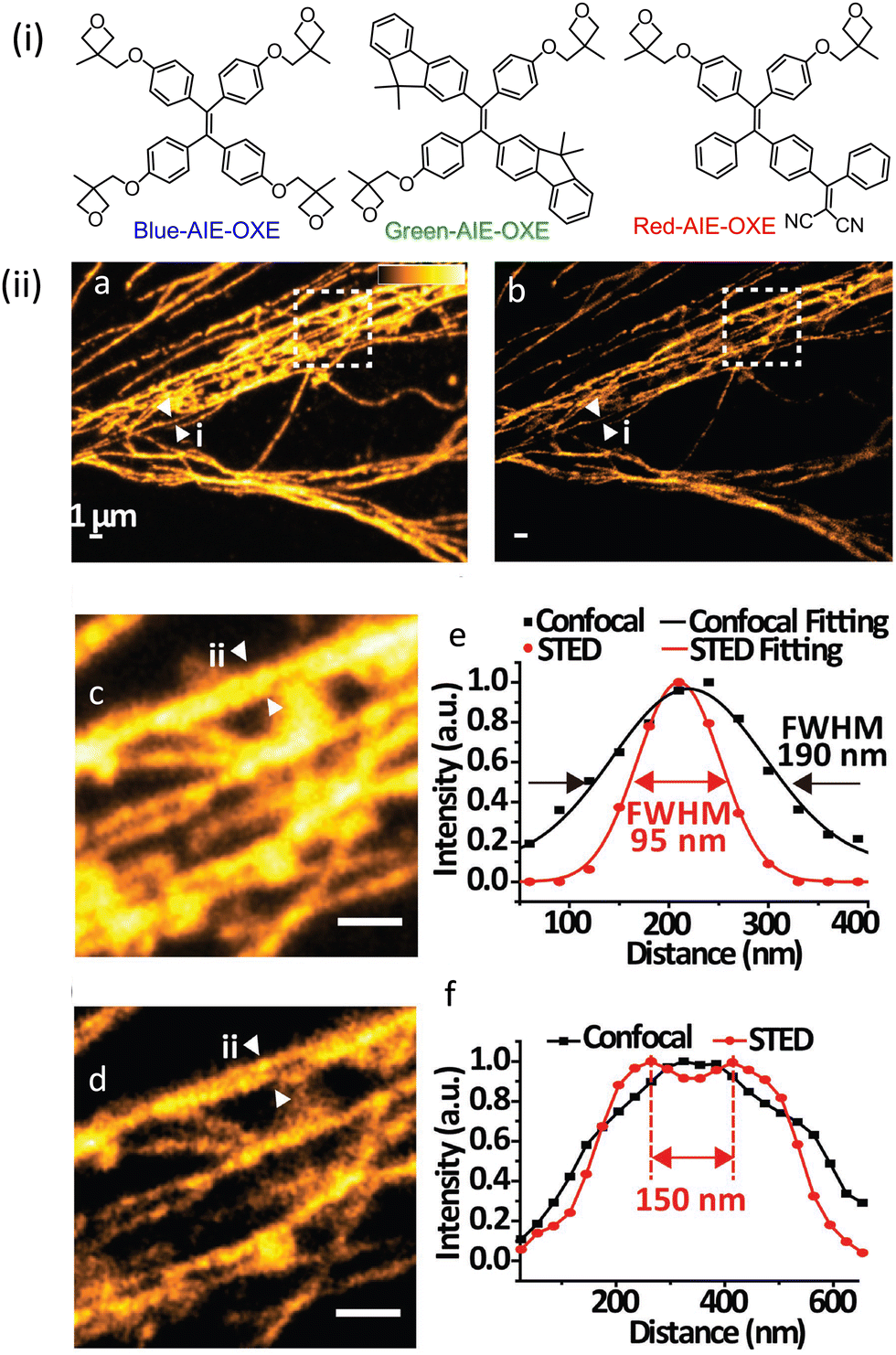 | ||
| Fig. 40 (i) Molecular structures of the Blue-AIE–OXE, Green-AIE–OXE, and Red-AIE–OXE. (ii) STED imaging of the microtubule structures labelled with the Red-AIE–OXE nanodots: (a) confocal and (b) STED images of the microtubules. (c and d) Magnified views of the box region marked in (a and b). (e and f) Intensity profiles of the position indicated by the arrowheads in (a–d), respectively. The scale bar in (a–d) represents 1 μm. Reproduced with permission from ref. 140, Copyright 2017, Wiley-VCH. | ||
Very recently, Tang and co-workers have reported a cationic AIEgen, DTPAP-P (Fig. 41-i) for organelle-specific imaging and dynamic tracking on a nanometer scale by utilizing anion-π+ interactions.151 These interactions helped to restrict intramolecular motions and radiative deactivation pathways, which resulted in a high fluorescence quantum yield of 35.04%. Favourable photophysical properties and the use of a low saturation power (26 mW) made the organelle-specific DTPAP-P ideally suited for STED nanoscopy and better-resolved image of the nucleus or mitochondrial regions of live cells were obtained. In STED, an FWHM of ∼165 nm (Fig. 41-ii) was obtained, which is far better than that observed in confocal microscopy (FWHM of ∼1028 nm). For fixed cells, DTPAP-P first stained mitochondria but then migrated to the nucleus on irradiation with 405 nm light. This dynamic migration was tracked in ultrahigh-resolution via STED nanoscopy (Fig. 41-iii) and eventually led a well-resolved image of the nucleus with FWHM of ∼184 nm.
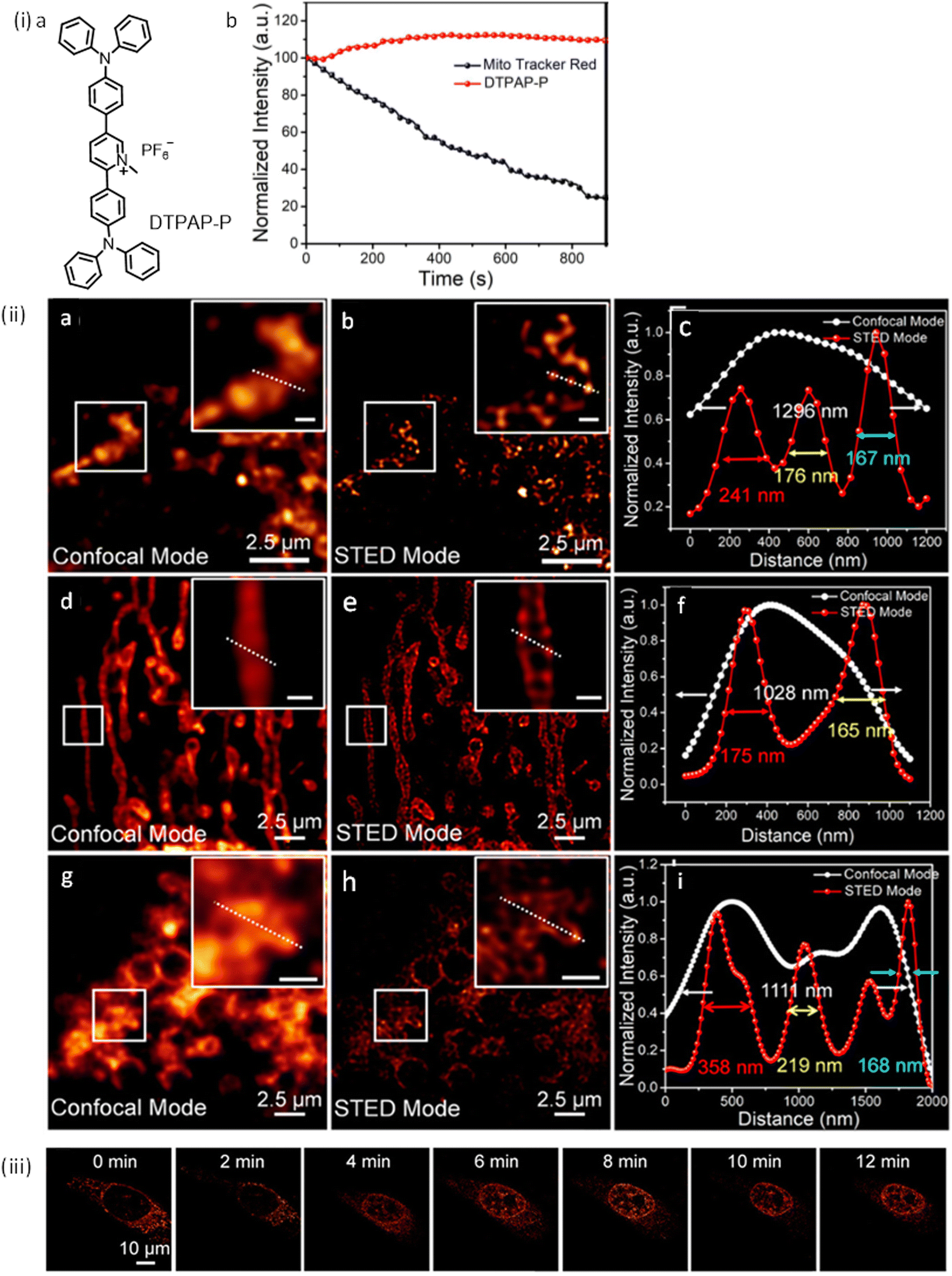 | ||
| Fig. 41 (i-a) Molecular structure of DTPAP-P. (i-b) Relative fluorescence intensity of Mito Tracker Red and DTPAP-P in HeLa cells after continuous irradiation by two lasers (475 nm and 560 nm) for 900 s. (ii) Fluorescence images of DTPAP-P stained mitochondria in fixed HeLa cells captured by (a) CLSM and (b) STED nanoscopy; insets: corresponding enlarged views (scale bar is 500 nm). (c) Fluorescence intensity profile along the white line in panels a and b. (d) Fluorescence images of DTPAP-P stained mitochondria (linear shape) in live cells recorded by CLSM and (e) STED nanoscopy; insets: corresponding enlarged views (scale bar is 500 nm). (f) Fluorescence intensity profile along the white line in panels d and e. Fluorescence images of DTPAP-P stained mitochondria (circular shape) in live cells were recorded by (g) CLSM and (h) STED nanoscopy. Insets: Enlarged views (scale bar is 500 nm). (i) Fluorescence intensity profile along the white line in panels g and h. (iii) Dynamic tracking of mitochondria–nucleus migration via STED nanoscopy. Time-dependent fluorescence images of HeLa cells stained by DTPAP-P. Reproduced with permission from ref. 151, Copyright 2022, American Chemical Society. | ||
Meng and co-workers recently prepared a red emissive AIE nanocrystal DTPA-BT-F (4,4′-(5,6-difuorobenzo[c][1,2,5]thiadiazole-4,7-diyl)bis(N,N-bis(4-methoxyphenyl)aniline) which was used for STED nanoscopy (Fig. 42-i and ii).152 DTPA-BT-F exhibits a bright deep-red emission in aggregated states (λAggre = 36.49%) which was attributed to a strong TICT (twisted intramolecular charge transfer) state with a large Stokes' shift (∼200 nm). These nanocrystals were found to be stable towards photobleaching even on continuous irradiation with a 775 nm STED depletion beam (600 mW, 25 min). This enabled visualizing the nanostructures in lysosomes in both fixed and live HeLa cells with superior lateral resolutions and narrower FWHM values (∼107 and 108 nm, respectively). Thus, the resolution achieved with STED microscopy with DTPA-BT-F was much better than that achieved with confocal microscopy (FWHM of 548 and 740 nm, respectively) (Fig. 42-iii). Importantly, 3D-STED imaging and real-time monitoring of lysosomal movements in live HeLa cells at high spatial resolution was possible thanks to this high depletion efficiency.
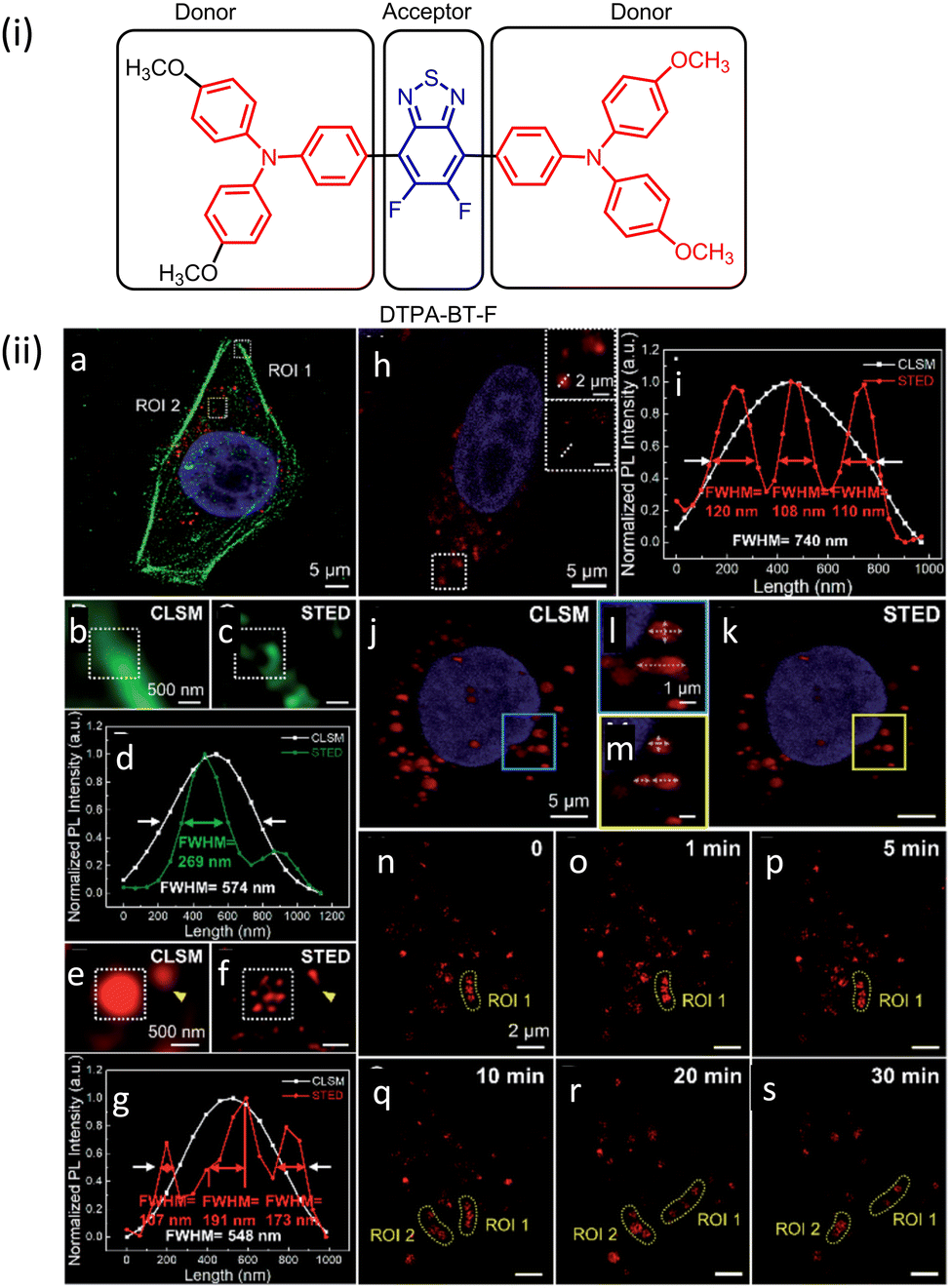 | ||
| Fig. 42 (i) Molecular structure of DTPA-BT-F. (ii) Fluorescence image of the fixed HeLa cell co-stained with DAPI (blue), Alexa Fluor488-phalloidin (green; depleted by a 592 nm STED laser, 96 mW) and DTPA-BT-F nanocrystals (red; depleted by a 775 nm STED laser, 96 mW) via STED nanoscopy: (a) fluorescence image, (b and e) magnified images with CLSM, (c and f) STED nanoscopy, and their corresponding photo luminiscence intensity curves (d and g) in ROI 1 (Alexa Fluor488-phalloidin, b–d) and ROI 2 (DTPA-BT-F NCs, e–g) in a; CLSM image of the live HeLa cell stained with Hoechst 33342 (blue) and DTPA-BT-F nanocrystals (red) (h, the insets are the magnified fluorescence images taken in the CLSM and STED mode), and the corresponding photo luminiscence intensity curves along the dashed line across the lysosome (i); 3D reconstructed images of the live HeLa cell by CLSM (j), STED nanoscopy (k), and their enlarged views (l and m); dynamic movements of lysosomes in HeLa cells captured by STED nanoscopy (n–s). All the STED images are processed on Huygens deconvolution software. Reproduced with permission from ref. 152, Copyright 2022, The Royal Society of Chemistry. | ||
Correlating fluorescence super resolution microscopy with cryo-electron microscopy
Electron microscopy, a powerful tool enabling the visualization at nanometer- and subnanometer resolution, has been the historical method of choice for probing ultrastructure in biological samples, that is, structures at scales smaller than the diffraction limit.153 In scanning electron microscopy (SEM) a focused beam of high-energy electrons scans across a sample surface to generate signals through electron-sample interactions,154 thereby offering sub-10 nm image resolution for structures close to the surface. Transmission electron microscopy (TEM) use a particle beam of electrons to prenetrate specimens and generate a highly-magnified image that achieves up to subnanometer resolution.155 Electron tomography (ET) is a standard technique to assess the three-dimensional (3D) morphology at very high-resolution (∼1–4 nm), by combining information from a tilted series of TEM images.156 In cryoEM and cryo-ET, samples are imaged under cryogenic conditions (<−150 °C) to bypass sample dehydration and, along with cryofixation, also circumvent chemical fixation which could otherwise disrupt or distort biological structures.156–158 Though immuno-EM (e.g., using antibodies conjugated nanoparticles) provides a means to localize specific targets in EM, its application is very limited by low labelling density and limited multitarget labelling strategies.159,160Subsequently, fluorescence microscopy has been successfully combined with EM to image the same sample and draw on the corresponding strengths of the two modalities, an approach often referred to as correlative light and electron microscopy (CLEM).161,162 Importantly, these combinatorial approaches add benefits over a single modality, and are particularly useful in studying events that comprise a sudden cellular change, such as cell division, shape conversion of lysosome-related organelles in antigen presenting cells, or synaptic vesicle release.162 Additionally, live-cell CLEM can provide insights into a range of dynamic events, such as organelle biogenesis, protein signalling, and cell adhesion.
Current advances in super resolution microscopy have radically improved the achievable optical resolution of light microscopy to ∼10 nm, to produce new standards that approach the resolution of EM. The closer match between resolutions of SRM and EM therefore allows more meaningful correlation with a much higher degree of structural detail when compared to previous CLEM efforts.
In 2006, Betzig and his research group demonstrated the potential of a super resolution-CLEM approach using photoactivated localization microscopy (PALM), in combination with TEM to image intracellular proteins at nanometer spatial resolution.23 The high degree of correlation between PALM and TEM resulting from fluorescently-tagged proteins on cryo-prepared thin sections of fixed cells helped validate PALM as a suitable technique for imaging intracellular proteins with subdiffraction-limit resolution (Fig. 43). Later Sauer, et al. combined dSTORM with SEM to map the position of proteins of nuclear pore complexes in isolated Xenopus laevis oocyte nuclear envelopes with molecular resolution in both imaging modes and also provide evidence for the reliability of dSTORM when applied in combination with highly specific antibodies for quantitative super-resolution imaging.163 The correlative images establish quantitative molecular labeling and localization of nuclear pore complex proteins by standard immunocytochemistry with primary and secondary antibodies and reveal that the nuclear pore complex is composed of eight gp210 protein homodimers (Fig. 44).
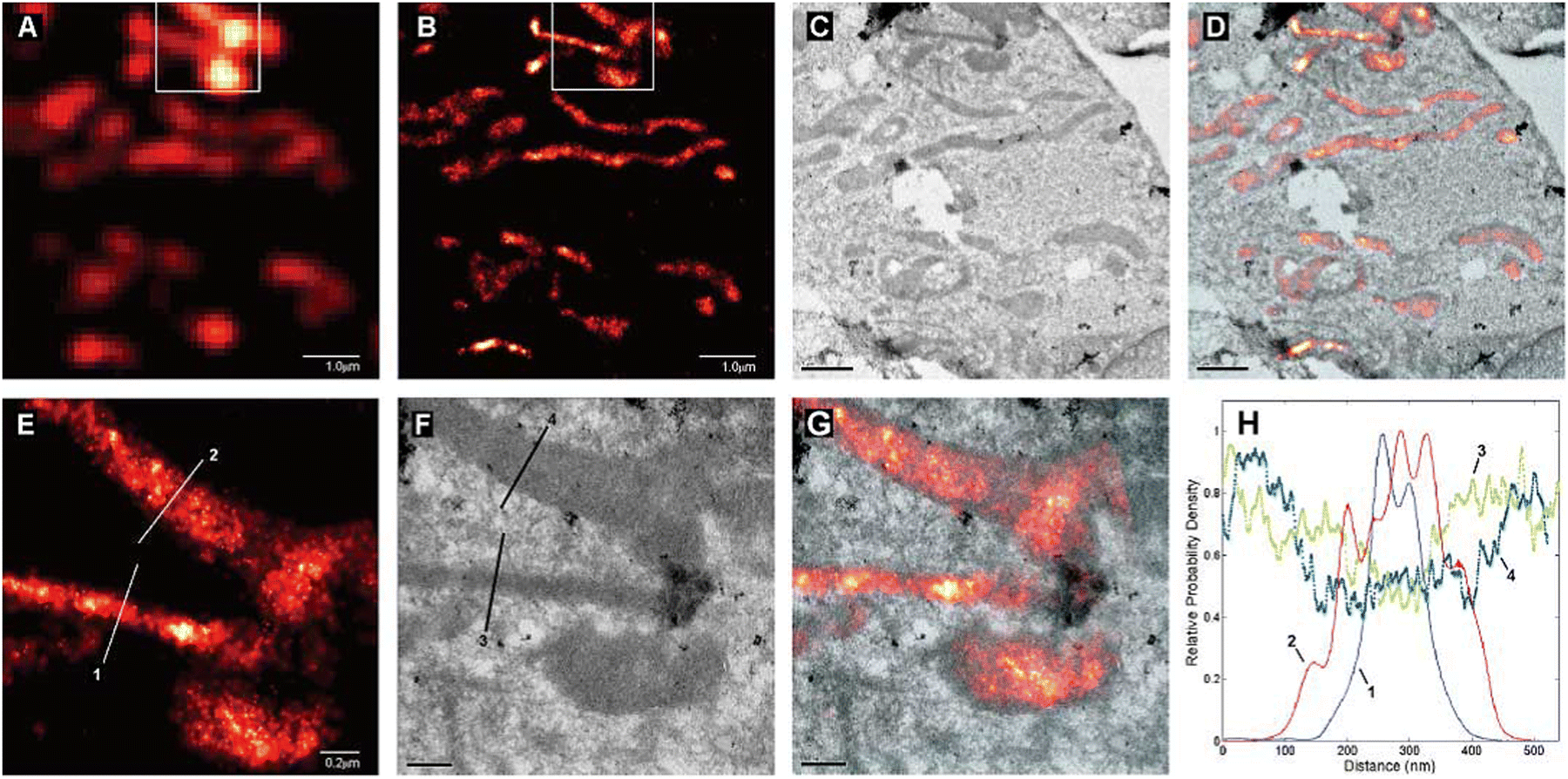 | ||
| Fig. 43 Comparative summed-molecule TIRF (A), PALM (B), TEM (C), and PALM/TEM overlay (D) images of mitochondria in a cryo-prepared thin section from a COS-7 cell expressing dEosFP-tagged cytochrome-C oxidase import sequence. Higher magnification PALM (E), TEM (F), and overlay (G) images within the box in (B) reveal that these matrix reporter molecules extend up to, but not into, the È20 nm outer mitochondrial membrane. The molecular distribution across two mitochondria along lines 1 and 2 in PALM image (E) are compared in (H) to the TEM signal along lines 3 and 4 in (F) across the same mitochondria. Scale bars: 1.0 mm in (A) to (D); 0.2 mm in (E) to (G). Reproduced with permission from ref. 23, Copyright 2006, AAAS. | ||
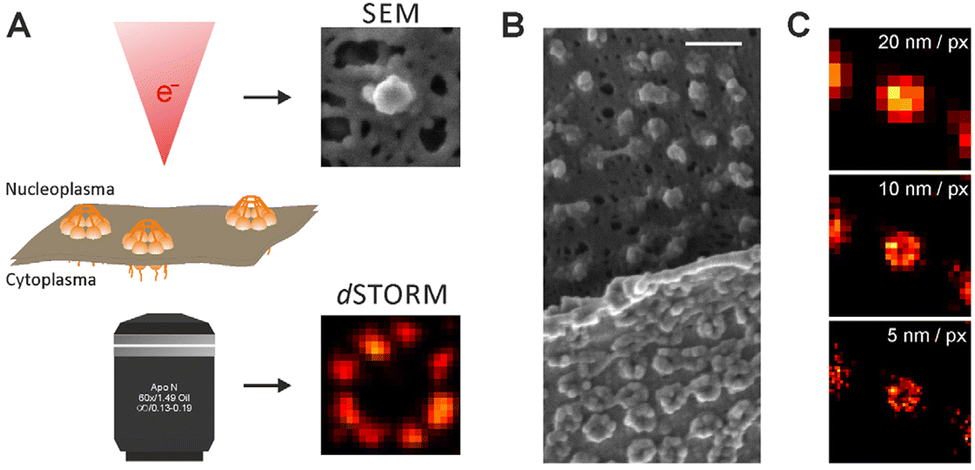 | ||
| Fig. 44 Imaging the nuclear envelope by SEM and dSTORM. (A) SEM imaging is restricted to the nucleoplasmatic, nuclear-basket-containing, side, whereas dSTORM imaging is performed from the cytoplasmatic side, where the anchoring proteins are oriented in the direction of the coverglass. Schematically, an electron beam and an objective are depicted to symbolize SEM and dSTORM imaging, respectively. (B) SEM image of a folded nuclear envelope. (C) Influence of the pixel binning used to reconstruct the super-resolved dSTORM image on resolution, that is, the visualization of the central channel of the NPC. Scale bar: 200 nm. Reproduced with permission from ref. 163, Copyright 2014, The Company of Biologists. | ||
Additionally, in combination with correlative focused ion beam scanning electron microscopy (FIB-SEM), dSTORM offers potential for 3D quantitative super-resolution imaging as the samples can be imaged with two-dimensional tiling and at multiple angles to create 3D tomograms in TEM.
Later the Xu group demonstrated the use of cryogenic single-molecule localization microscopy (cryo-PALM/SMLM/STORM) followed by cryo-electron tomography (cryo-ET) to precisely determine the spatial relationship between proteins and their native subcellular structures.164 They prepared a marker for the mitochondrial outer membrane by fusing Dronpa with the first 33 amino acids of TOM20, which are responsible for localizing TOM20 to the mitochondria. After high-pressure freezing and cryo-sectioning of HEK293 cells expressing TOM20-Dronpa they performed cryo-fluorescence super-resolution imaging first, followed by cryo-electron tomography (cryo-ET). Under this experimental condition, no charge-induced movement or radiation damage was observed. The mitochondrial outer membrane and cristae was clearly visualized by 3D reconstruction from cryo-ET (Fig. 45c), while super-resolution imaging of Tom20-Dronpa molecules showed correlated localization to the outer membrane at nanometer resolution (Fig. 45c and d). A 2–3-fold enhancement of resolution over similar studies was achieved due to the photon budget performance and optimized imaging conditions, even with thin sections which usually degrade structure resolution by lowering label density.
 | ||
| Fig. 45 3D correlative images from cryo-sections of HEK293 cells expressing TOM20-Dronpa. (a) Summed image from single-molecule data. Scale bar, 1 μm. (b) Correlative isosurface reconstruction of a PALM image with low magnification TEM data. (c) Each column in c was from a single layer of 3D PALM (top), single layer of cryo-ET (middle) and correlative representations (bottom), respectively. The structural resolution of the whole 3D data set was estimated to be 73.7 ± 5 nm employing the Fourier ring correlation method, corresponding to a density of 749 signals μm−2. Scale bar, 200 nm. (d) Correlative 3D segmentation of cryo-ET data with single-molecule localization of fluorescent proteins. Mitochondrial outer membrane and cristae are denoted by purple and blue, respectively. Dronpa molecules are denoted by green dots. For clear demonstration, we kept those signals that have higher localization precision (2.9/3.0 nm,mean per median), which rejects 90% of the signals. Scale bar, 200 nm. Reproduced with permission from ref. 164, Copyright 2015, Springer Nature. | ||
Drawbacks of using nanoparticles in cellular imaging applications
Semiconductor and graphene quantum dots offer outstanding photostability and brightness, which facilitates their use in STORM, STED and SIM imaging.78,165 However, the high duty cycles of their photoemission means they are not appropriate for use in single-molecule localization microscopy applications. Additionally, semiconductor quantum dots are very unstable under aqueous solution.53 Overcoming these drawbacks by lowering duty cycles requires surface engineering or the construction of hybrid systems. For example, nitrogen doping of graphene quantum dots can lower duty cycles to below 0.2%.166 Likewise, the blinking of semiconductor quantum dots can be controlled in hybrid core–shell systems.167 Considerably more work is required to render these systems into practical probes for single-molecule localization microscopy imaging applications.Polymer dots and modified upconversion nanoparticles are more flexible in this regard and they improve both the photoblinking and photostability characteristics of their guest fluorophores (e.g., organic dyes, metal complexes).168,169 These advantages are balanced by the vastly increased size of these nanoparticle systems in relatation to the luminophores on their own, and this creates limitations for biological applications.
However, AIE luminogens have shown excellent potential for STORM and STED imaging, because of their saturation behavior and large Stokes shifts.149,170 Some AIEs are photoactivatable; and therefore have potential for future application in SMLM imaging.171 Although a major disadvantage of these systems is again the problem of post functionalisation of the probes for targeted delivery.172
Strategies to improve the performance of nanocarriers for super-resolution imaging
For semiconductor quantum dots and graphene quantum dots, surface states play a leading role in their photoblinking and photoswitching property.167,173 For graphene quantum dots, emission intermittency comes from surface functionalities that provide energy wells for ejected electrons.167 The resultant electron transfer process leads to fluorescent on/off-states. The blinking characteristic of graphene quantum dots can thus be tuned through logical surface design, e.g., post grafting of the surface with electron donating or accepting agents.174,175 In the case of semiconductor quantum dots, nonradiative recombination via surface traps or charging-induced Auger recombination drastically affects the blinking behavior.176 Reduced on-to-off duty cycles can be attained via core passivation using thicker shell materials, the post grafting of ligands onto the semiconductor quantum dots surface, contacting semiconductor quantum dots with other nanoparticles, the construction of hybrid blinking systems, electrostatic gating, and irradiation with ultrafast mid-infrared pulses. For STORM and STED imaging, semiconductor quantum dots and graphene quantum dots could be improved, if multiphoton emission is effectively inhibited under lighting with high-power depletion lasers. On the one hand, supressing background signals from the STORM or STED laser in the first place is, of course, preferential to methods requiring background removal in a post-processing step.The upconversion nanocrystals are prepared by thermal decomposition and hydro(solvo)thermal methods with controllable size, crystalline phase, and morphology. In the co-precipitation method very high-purity stoichiometric upconversion nanocrystals can be synthesized. Additionally in this technique, surfactants can be incorporated to improve the water-solubility of the nanocrystals. Other strategies – such as surface passivation, host lattice manipulation, photonic crystal engineering, surface plasmon coupling, combination with other functionalities for the acceleration of energy transfer, and construction of organic–inorganic hybrid systems – are all fascinating approaches to enhance on present systems.177 For STED imaging, using dye-sensitized or Nd3+-sensitized upconversion nanocrystals irradiated at 800 nm instead of 980 nm, can solve the issue of overheating under 980 nm irradiation.178,179 javascript:void(0); AIE Luminogens are typically hydrophobic in nature. Their alteration with functional groups or post-grafting with hydrophilic functionalities will produce water-soluble AIE Luminogens for bioimaging applications. Linking various AIE moieties can also lead to red-shifted emission.180 javascript:void(0); furthermore, the donor–acceptor structure assembled by electron donating (e.g., amime or methoxy) or withdrawing (e.g., benzobisthiadiazole, benzothiadiazole) functionalities in AIE Luminogens can result photoluminescence systems that can be tunable from the visible to the NIR spectral regions.181
Conclusions and prospects
Since the inception of fluorescence microscopy, luminophores have played a vital part in improving spatial and lateral resolutions. The development of purpose-built luminophore molecules and materials has matched the continuous advancements in optics, electronics, mathematics in imaging technologies. For example, although SMLM and STED methods were described in the early 1990s,182 these techniques could not be fully developed until appropriate luminophores became available. STED, in particular, only became practical after the development of highly photostable probes with high brightness for stimulated emission.183 Similarly, implementation of SMLM was feasible only184,185 after the invention of carbocyanine dyes with photoswitching behaviour186 and nonlinear SIM and RESOLFT only became possible with the availability of related photoswitching luminophores.184,187 The development of new probes continues to support the advances in super-resolution microscopy and facilitate routes towards the ultimate goal of in-vivo structural biology.In this review, we have summarised the recent progress on luminophores used in super-resolution microscopy imaging, including quantum dots, up-conversion nanoscale materials, carbon dots, PDots, AIE luminogens, and inorganic materials. Owing to their superior photophysical properties, photostability and biocompatibility, inorganic material-based fluorophores, PDots and AIE luminogens, are expected to contribute to further lowering of resolution limits in super-resolution. Although the theoretical spatial resolution of various super-resolution microscopy techniques is unlimited.42,188 The challenge to routinely achieve resolution at molecular-scale – i.e., sub-10 nm level – still remains.
Resolution in some super-resolution imaging techniques can now approach a few nanometers through the use of superior localization schemes,189,190 large Stokes’ shift, luminophores with very high photostabilities, or accurate correction of sample drift.191,192 When the precision at which luminophores can be restricted is at nanometer scales, the linkage error provided by distances between the luminophore and the molecule of interest will become a barrier to any further resolution improvement.193 Thus, SR imaging-based techniques will benefit from minimizing fluorescent probe size and linkage distances. Although effective probe size can also be reduced by physically swelling the sample volume – so that linkage error of a pair of primary and secondary antibodies can be reduced from 15 to 7 nm after expansion194 – fundamentally, it would be best to increase the photostability of dyes without increasing probe size and also use small binders for proteins, such as photostable nanobodies, to reduce the dye-to-target linkage.28
As stated above, the ultimate goal of super-resolution microscopy is to understand the complex structural biology of live cells by resolving molecular structures in living cells. In recent years, the ground-breaking advances in cryo-electron microscopy may soon permit the visualisation of in situ structural biology at atomic resolution.195 However, due to the requirement of vacuum conditions and the high energy of electron beams, electron microscopy is incompatible with live cells, and super-resolution microscopy seems to be the best-suited technique to monitor the ultrastructural dynamics in real-time. Yet, live-cell super-resolution microscopy still requires improved luminophores with increased photostabilities and more reliable switching chemistries inside cells as well as more convenient/bio-orthogonal labelling protocols.196–198 Further improvements in photo stabilities and other photophysical characteristics of luminophores, like photon budgets and switching chemistries, may lead to breakthroughs to finally realize routine molecular-scale imaging in live cells.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is partially funded by CSIR-MLP 0045 (S. K. P.) and by SERB (India) grants (AD: CRG/2020/000492 & J. C. B./2017/000004). AD also acknowledges DRDO-DMRDE (GoI) CARS: TR/0569/CARS/136 for financial assistance in the form of a Grant-in-Aid Project. This manuscript bears a CSIR-CSMCRI PRIS no: 231/2022.Notes and references
- A. L. McGuire, S. Gabriel, S. A. Tishkoff, A. Wonkam, A. Chakravarti, E. E. M. Furlong, B. Treutlein, A. Meissner, H. Y. Chang, N. López-Bigas, E. Segal and J.-S. Kim, Nat. Rev. Genet., 2020, 21, 581–596 CrossRef CAS PubMed.
- N. Rajewsky, G. Almouzni, S. A. Gorski, S. Aerts, I. Amit, M. G. Bertero, C. Bock, A. L. Bredenoord, G. Cavalli, S. Chiocca, H. Clevers, B. De Strooper, A. Eggert, J. Ellenberg, X. M. Fernández, M. Figlerowicz, S. M. Gasser, N. Hubner, J. Kjems, J. A. Knoblich, G. Krabbe, P. Lichter, S. Linnarsson, J.-C. Marine, J. C. Marioni, M. A. Marti-Renom, M. G. Netea, D. Nickel, M. Nollmann, H. R. Novak, H. Parkinson, S. Piccolo, I. Pinheiro, A. Pombo, C. Popp, W. Reik, S. Roman-Roman, P. Rosenstiel, J. L. Schultze, O. Stegle, A. Tanay, G. Testa, D. Thanos, F. J. Theis, M.-E. Torres-Padilla, A. Valencia, C. Vallot, A. van Oudenaarden, M. Vidal, T. Voet, L. Alberi, S. Alexander, T. Alexandrov, E. Arenas, C. Bagni, R. Balderas, A. Bandelli, B. Becher, M. Becker, N. Beerenwinkel, M. Benkirame, M. Beyer, W. Bickmore, E. E. A. L. Biessen, N. Blomberg, I. Blumcke, B. Bodenmiller, B. Borroni, D. T. Boumpas, T. Bourgeron, S. Bowers, D. Braeken, C. Brooksbank, N. Brose, H. Bruining, J. Bury, N. Caporale, G. Cattoretti, N. Chabane, H. Chneiweiss, S. A. Cook, P. Curatolo, M. I. de Jonge, B. Deplancke, B. De Strooper, P. de Witte, S. Dimmeler, B. Draganski, A. Drews, C. Dumbrava, S. Engelhardt, T. Gasser, E. J. Giamarellos-Bourboulis, C. Graff, D. Grün, I. Gut, O. Hansson, D. C. Henshall, A. Herland, P. Heutink, S. R. B. Heymans, H. Heyn, M. Huch, I. Huitinga, P. Jackowiak, K. R. Jongsma, L. Journot, J. P. Junker, S. Katz, J. Kehren, S. Kempa, P. Kirchhof, C. Klein, N. Koralewska, J. O. Korbel, M. Kühnemund, A. I. Lamond, E. Lauwers, I. Le Ber, V. Leinonen, A. L. Tobon, E. Lundberg, A. Lunkes, H. Maatz, M. Mann, L. Marelli, V. Matser, P. M. Matthews, F. Mechta-Grigoriou, R. Menon, A. F. Nielsen, M. Pagani, R. J. Pasterkamp, A. Pitkänen, V. Popescu, C. Pottier, A. Puisieux, R. Rademakers, D. Reiling, O. Reiner, D. Remondini, C. Ritchie, J. D. Rohrer, A.-E. Saliba, R. Sanchez-Valle, A. Santosuosso, A. Sauter, R. A. Scheltema, P. Scheltens, H. B. Schiller, A. Schneider, P. Seibler, K. Sheehan-Rooney, D. Shields, K. Sleegers, A. B. Smit, K. G. C. Smith, I. Smolders, M. Synofzik, W. L. Tam, S. Teichmann, M. Thom, M. Y. Turco, H. M. M. van Beusekom, R. Vandenberghe, S. Van den Hoecke, I. Van de Poel, A. van der Ven, J. van der Zee, J. van Lunzen, G. van Minnebruggen, A. van Oudenaarden, W. Van Paesschen, J. van Swieten, R. van Vught, M. Verhage, P. Verstreken, C. E. Villa, J. Vogel, C. von Kalle, J. Walter, S. Weckhuysen, W. Weichert, L. Wood, A.-G. Ziegler, F. Zipp and G. LifeTime Community Working, Nature, 2020, 587, 377–386 CrossRef CAS PubMed.
- Y. Wu, X. Han, Y. Su, M. Glidewell, J. S. Daniels, J. Liu, T. Sengupta, I. Rey-Suarez, R. Fischer, A. Patel, C. Combs, J. Sun, X. Wu, R. Christensen, C. Smith, L. Bao, Y. Sun, L. H. Duncan, J. Chen, Y. Pommier, Y.-B. Shi, E. Murphy, S. Roy, A. Upadhyaya, D. Colón-Ramos, P. La Riviere and H. Shroff, Nature, 2021, 600, 279–284 CrossRef CAS PubMed.
- L. Möckl and W. E. Moerner, J. Am. Chem. Soc., 2020, 142, 17828–17844 CrossRef PubMed.
- B. R. Gallagher and Y. Zhao, Neurobiol. Dis., 2021, 154, 105362 CrossRef CAS PubMed.
- R. Schmidt, C. A. Wurm, S. Jakobs, J. Engelhardt, A. Egner and S. W. Hell, Nat. Methods, 2008, 5, 539–544 CrossRef CAS PubMed.
- S. W. Hell, E. H. Stelzer, S. Lindek and C. Cremer, Opt. Lett., 1994, 19, 222 CrossRef CAS PubMed.
- L. Ji, W. Yan, Y. Xia and D. Liu, J. Appl. Phys., 2018, 123, 183302 CrossRef.
- S. W. Hell, S. J. Sahl, M. Bates, X. Zhuang, R. Heintzmann, M. J. Booth, J. Bewersdorf, G. Shtengel, H. Hess, P. Tinnefeld, A. Honigmann, S. Jakobs, I. Testa, L. Cognet, B. Lounis, H. Ewers, S. J. Davis, C. Eggeling, D. Klenerman, K. I. Willig, G. Vicidomini, M. Castello, A. Diaspro and T. Cordes, J. Phys. D, 2015, 48, 443001 CrossRef.
- X. Chen, B. Zheng and H. Liu, Anal. Cell. Pathol., 2011, 34, 5–18 CrossRef.
- I. M. Khater, I. R. Nabi and G. Hamarneh, Patterns, 2020, 1, 100038 CrossRef CAS PubMed.
- B. Huang, H. Babcock and X. Zhuang, Cell, 2010, 143, 1047–1058 CrossRef CAS PubMed.
- G. M. R. De Luca, R. M. P. Breedijk, R. A. J. Brandt, C. H. C. Zeelenberg, B. E. de Jong, W. Timmermans, L. N. Azar, R. A. Hoebe, S. Stallinga and E. M. M. Manders, Biomed. Opt. Express, 2013, 4, 2644–2656 CrossRef PubMed.
- D. Rupsa, M. H. Tiffany, T. S. Joe, A. G. Amani and C. S. Melissa, J. Biomed. Opt., 2020, 25, 1–43 Search PubMed.
- S. Dhiman, T. Andrian, B. S. Gonzalez, M. M. E. Tholen, Y. Wang and L. Albertazzi, Chem. Sci., 2022, 13, 2152–2166 RSC.
- A. M. Szalai, C. Zaza and F. D. Stefani, Nanoscale, 2021, 13, 18421–18433 RSC.
- I. M. Pavlovetc, K. Aleshire, G. V. Hartland and M. Kuno, Phys. Chem. Chem. Phys., 2020, 22, 4313–4325 RSC.
- S. J. Sahl, S. W. Hell and S. Jakobs, Nat. Rev. Mol. Cell Biol., 2017, 18, 685–701 CrossRef CAS PubMed.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef PubMed.
- M. Hofmann, C. Eggeling, S. Jakobs and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17565–17569 CrossRef CAS PubMed.
- B. Hein, K. I. Willig and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14271–14276 CrossRef CAS PubMed.
- Y. Xu, R. Xu, Z. Wang, Y. Zhou, Q. Shen, W. Ji, D. Dang, L. Meng and B. Z. Tang, Chem. Soc. Rev., 2021, 50, 667–690 RSC.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- D.-R. Lee and J. Bewersdorf, Appl. Opt., 2021, 60, 5354–5359 CrossRef PubMed.
- D. T. Burnette, P. Sengupta, Y. Dai, J. Lippincott-Schwartz and B. Kachar, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 21081–21086 CrossRef CAS PubMed.
- J. Requejo-Isidro, J. Chem. Biol., 2013, 6, 97–120 CrossRef PubMed.
- R. R. M. Dijkstra, Master thesis, University of Twente, 2012.
- M. Lelek, M. T. Gyparaki, G. Beliu, F. Schueder, J. Griffié, S. Manley, R. Jungmann, M. Sauer, M. Lakadamyali and C. Zimmer, Nat. Rev. Methods Primers, 2021, 1, 39 CrossRef CAS PubMed.
- M. Pawlowska, R. Tenne, B. Ghosh, A. Makowski and R. Lapkiewicz, J. Phys.: Photonics, 2021, 4, 012002 Search PubMed.
- B. Huang, W. Wang, M. Bates and X. Zhuang, Science, 2008, 319, 810–813 CrossRef CAS PubMed.
- R. Heintzmann and T. Huser, Chem. Rev., 2017, 117, 13890–13908 CrossRef CAS PubMed.
- X. Chen, Y. Wang, X. Zhang and C. Liu, Biomater. Sci., 2021, 9, 5484–5496 RSC.
- M. Filius, T. J. Cui, A. N. Ananth, M. W. Docter, J. W. Hegge, J. van der Oost and C. Joo, Nano Lett., 2020, 20, 2264–2270 CrossRef CAS PubMed.
- G. Giannone, E. Hosy, F. Levet, A. Constals, K. Schulze, A. I. Sobolevsky, M. P. Rosconi, E. Gouaux, R. Tampé, D. Choquet and L. Cognet, Biophys. J., 2010, 99, 1303–1310 CrossRef CAS PubMed.
- Y. Wu and H. Shroff, Nat. Methods, 2018, 15, 1011–1019 CrossRef CAS PubMed.
- L. A. Masullo, F. Steiner, J. Zähringer, L. F. Lopez, J. Bohlen, L. Richter, F. Cole, P. Tinnefeld and F. D. Stefani, Nano Lett., 2021, 21, 840–846 CrossRef CAS PubMed.
- R. Schmidt, T. Weihs, C. A. Wurm, I. Jansen, J. Rehman, S. J. Sahl and S. W. Hell, Nat. Commun., 2021, 12, 1478 CrossRef CAS PubMed.
- J. K. Pape, T. Stephan, F. Balzarotti, R. Büchner, F. Lange, D. Riedel, S. Jakobs and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20607–20614 CrossRef CAS PubMed.
- F. Balzarotti, Y. Eilers, K. C. Gwosch, A. H. Gynnå, V. Westphal, F. D. Stefani, J. Elf and S. W. Hell, Science, 2017, 355, 606–612 CrossRef CAS PubMed.
- S. T. Hess, T. P. Girirajan and M. D. Mason, Biophys. J., 2006, 91, 4258–4272 CrossRef CAS PubMed.
- M. A. X. Born and E. Wolf, in Principles of Optics, ed. M. A. X. Born and E. Wolf, Pergamon, Sixth Edn, 1980, pp. 1–70 DOI:10.1016/B978-0-08-026482-0.50008-6.
- R. E. Thompson, D. R. Larson and W. W. Webb, Biophys. J., 2002, 82, 2775–2783 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Chem. Rev., 2019, 119, 1138–1192 CrossRef CAS PubMed.
- F. Pinaud, X. Michalet, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Iyer and S. Weiss, Biomaterials, 2006, 27, 1679–1687 CrossRef CAS PubMed.
- S. Ranjit, L. Lanzanò, A. E. Libby, E. Gratton and M. Levi, Nat. Rev. Nephrol., 2021, 17, 128–144 CrossRef CAS PubMed.
- E. Sapoznik, B.-J. Chang, J. Huh, R. J. Ju, E. V. Azarova, T. Pohlkamp, E. S. Welf, D. Broadbent, A. F. Carisey, S. J. Stehbens, K.-M. Lee, A. Marín, A. B. Hanker, J. C. Schmidt, C. L. Arteaga, B. Yang, Y. Kobayashi, P. R. Tata, R. Kruithoff, K. Doubrovinski, D. P. Shepherd, A. Millett-Sikking, A. G. York, K. M. Dean and R. P. Fiolka, eLife, 2020, 9, e57681 CrossRef CAS PubMed.
- S. Samanta, Y. He, A. Sharma, J. Kim, W. Pan, Z. Yang, J. Li, W. Yan, L. Liu, J. Qu and J. S. Kim, Chem, 2019, 5, 1697–1726 CAS.
- H. Singh, K. Tiwari, R. Tiwari, S. K. Pramanik and A. Das, Chem. Rev., 2019, 119, 11718–11760 CrossRef CAS PubMed.
- J. Kwon, J.-S. Park, M. Kang, S. Choi, J. Park, G. T. Kim, C. Lee, S. Cha, H.-W. Rhee and S.-H. Shim, Nat. Commun., 2020, 11, 273 CrossRef CAS PubMed.
- S. Sreedharan, R. Tiwari, D. Tyde, S. O. Aderinto, S. K. Pramanik, A. Das and J. A. Thomas, Mater. Chem. Front., 2021, 5, 1268–1282 RSC.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- S. Wilhelm, ACS Nano, 2017, 11, 10644–10653 CrossRef CAS PubMed.
- S. Silvi and A. Credi, Chem. Soc. Rev., 2015, 44, 4275–4289 RSC.
- M. D. Torelli, N. A. Nunn and O. A. Shenderova, Small, 2019, 15, 1902151 CrossRef CAS PubMed.
- J. Liu, R. Li and B. Yang, ACS Cent. Sci., 2020, 6, 2179–2195 CrossRef CAS PubMed.
- C. S. Palmer, J. Lou, B. Kouskousis, E. Pandzic, A. J. Anderson, Y. Kang, E. Hinde and D. Stojanovski, J. Cell Sci., 2021, 134, jcs252197 CrossRef CAS PubMed.
- M. Bruchez, Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef PubMed.
- W. C. Chan and S. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS PubMed.
- O. Yarema, M. Yarema and V. Wood, Chem. Mater., 2018, 30, 1446–1461 CrossRef CAS.
- T.-M. Liu, J. Conde, T. Lipiński, A. Bednarkiewicz and C.-C. Huang, NPG Asia Mater., 2016, 8, e295 CrossRef CAS.
- A. T. Frawley, V. Wycisk, Y. Xiong, S. Galiani, E. Sezgin, I. Urbančič, A. Vargas Jentzsch, K. G. Leslie, C. Eggeling and H. L. Anderson, Chem. Sci., 2020, 11, 8955–8960 RSC.
- W. R. Algar, K. Susumu, J. B. Delehanty and I. L. Medintz, Anal. Chem., 2011, 83, 8826–8837 CrossRef CAS PubMed.
- S. J. Lim, M. U. Zahid, P. Le, L. Ma, D. Entenberg, A. S. Harney, J. Condeelis and A. M. Smith, Nat. Commun., 2015, 6, 8210 CrossRef CAS PubMed.
- A. Foubert, N. V. Beloglazova, A. Rajkovic, B. Sas, A. Madder, I. Y. Goryacheva and S. De Saeger, TrAC, Trends Anal. Chem., 2016, 83, 31–48 CrossRef CAS.
- R. Mahle, P. Kumbhakar, D. Nayar, T. N. Narayanan, K. Kumar Sadasivuni, C. S. Tiwary and R. Banerjee, Dalton Trans., 2021, 50, 14062–14080 RSC.
- E. Petryayeva, W. R. Algar and I. L. Medintz, Appl. Spectrosc., 2013, 67, 215–252 CrossRef CAS PubMed.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- M. Sousa de Almeida, E. Susnik, B. Drasler, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, Chem. Soc. Rev., 2021, 50, 5397–5434 RSC.
- P. Liu, X. Mu, X.-D. Zhang and D. Ming, Bioconjugate Chem., 2020, 31, 260–275 CrossRef CAS PubMed.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- S. Sarkar, P. Le, J. Geng, Y. Liu, Z. Han, M. U. Zahid, D. Nall, Y. Youn, P. R. Selvin and A. M. Smith, J. Am. Chem. Soc., 2020, 142, 3449–3462 CrossRef CAS PubMed.
- P. J. Bosch, I. R. Corrêa, Jr., M. H. Sonntag, J. Ibach, L. Brunsveld, J. S. Kanger and V. Subramaniam, Biophys. J., 2014, 107, 803–814 CrossRef CAS PubMed.
- S. E. Irvine, T. Staudt, E. Rittweger, J. Engelhardt and S. W. Hell, Angew. Chem., Int. Ed., 2008, 47, 2685–2688 CrossRef CAS PubMed.
- H. C. Ishikawa-Ankerhold, R. Ankerhold and G. P. C. Drummen, Molecules, 2012, 17, 4047–4132 CrossRef CAS PubMed.
- J. Hanne, H. J. Falk, F. Görlitz, P. Hoyer, J. Engelhardt, S. J. Sahl and S. W. Hell, Nat. Commun., 2015, 6, 7127 CrossRef CAS PubMed.
- J. Xu, K. F. Tehrani and P. Kner, ACS Nano, 2015, 9, 2917–2925 CrossRef CAS PubMed.
- X. Yang, K. Zhanghao, H. Wang, Y. Liu, F. Wang, X. Zhang, K. Shi, J. Gao, D. Jin and P. Xi, ACS Photonics, 2016, 3, 1611–1618 CrossRef CAS.
- Y. Chang, D.-H. Kim, K. Zhou, M. G. Jeong, S. Park, Y. Kwon, T. M. Hong, J. Noh and S. H. Ryu, Exp. Mol. Med., 2021, 53, 384–392 CrossRef CAS PubMed.
- J. M. Urban, W. Chiang, J. W. Hammond, N. M. B. Cogan, A. Litzburg, R. Burke, H. A. Stern, H. A. Gelbard, B. L. Nilsson and T. D. Krauss, J. Phys. Chem. B, 2021, 125, 2566–2576 CrossRef CAS PubMed.
- T. Wang, G. Li, D. Wang, F. Li, D. Men, T. Hu, Y. Xi and X.-E. Zhang, Nanoscale, 2019, 11, 18224–18231 RSC.
- R. Wang and F. Zhang, Near-infrared Nanomaterials: Preparation, Bioimaging and Therapy Applications, The Royal Society of Chemistry, 2016, pp. 1–39 10.1039/9781782623939-00001.
- Q. Yang, H. Ma, Y. Liang and H. Dai, Acc. Mater. Res., 2021, 2, 170–183 CrossRef CAS.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- F. Wang, J. Wang and X. Liu, Angew. Chem., Int. Ed., 2010, 49, 7456–7460 CrossRef CAS PubMed.
- Q. Liu, Y. Sun, T. Yang, W. Feng, C. Li and F. Li, J. Am. Chem. Soc., 2011, 133, 17122–17125 CrossRef CAS PubMed.
- Y.-W. Zhang, X. Sun, R. Si, L.-P. You and C.-H. Yan, J. Am. Chem. Soc., 2005, 127, 3260–3261 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC.
- M. Ding, D. Chen, D. Ma, J. Dai, Y. Li and Z. Ji, J. Mater. Chem. C, 2016, 4, 2432–2437 RSC.
- V. Chugh, K. Vijaya Krishna and A. Pandit, ACS Nano, 2021, 15, 17080–17123 CrossRef CAS PubMed.
- H. Chu, T. Cao, G. Dai, B. Liu, H. Duan, C. Kong, N. Tian, D. Hou and Z. Sun, RSC Adv., 2021, 11, 35472–35488 RSC.
- S. A. Hilderbrand, F. Shao, C. Salthouse, U. Mahmood and R. Weissleder, Chem. Commun., 2009, 4188–4190, 10.1039/B905927J.
- Y. Liu, Y. Lu, X. Yang, X. Zheng, S. Wen, F. Wang, X. Vidal, J. Zhao, D. Liu, Z. Zhou, C. Ma, J. Zhou, J. A. Piper, P. Xi and D. Jin, Nature, 2017, 543, 229–233 CrossRef CAS PubMed.
- Q. Zhan, H. Liu, B. Wang, Q. Wu, R. Pu, C. Zhou, B. Huang, X. Peng, H. Ågren and S. He, Nat. Commun., 2017, 8, 1058 CrossRef PubMed.
- W. Ren, S. Wen, S. A. Tawfik, Q. P. Su, G. Lin, L. A. Ju, M. J. Ford, H. Ghodke, A. M. van Oijen and D. Jin, Chem. Sci., 2018, 9, 4352–4358 RSC.
- F. Wang, S. Wen, H. He, B. Wang, Z. Zhou, O. Shimoni and D. Jin, Light: Sci. Appl., 2018, 7, 18007 CrossRef CAS PubMed.
- S. K. Pramanik, S. Sreedharan, H. Singh, N. H. Green, C. Smythe, J. A. Thomas and A. Das, Chem. Commun., 2017, 53, 12672–12675 RSC.
- H. Singh, S. Sreedharan, E. Oyarzabal, T. S. Mahapatra, N. Green, Y.-Y. I. Shih, M. Das, J. A. Thomas, S. K. Pramanik and A. Das, Chem. Commun., 2020, 56, 7945–7948 RSC.
- N. Soni, S. Singh, S. Sharma, G. Batra, K. Kaushik, C. Rao, N. C. Verma, B. Mondal, A. Yadav and C. K. Nandi, Chem. Sci., 2021, 12, 3615–3626 RSC.
- X. Liu, S.-Y. Chen, Q. Chen, X. Yao, M. Gelléri, S. Ritz, S. Kumar, C. Cremer, K. Landfester, K. Müllen, S. H. Parekh, A. Narita and M. Bonn, Angew. Chem., Int. Ed., 2020, 59, 496–502 CrossRef CAS PubMed.
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744–4822 CrossRef CAS PubMed.
- P. Lesani, A. H. Mohamad Hadi, Z. Lu, S. Palomba, E. J. New and H. Zreiqat, Commun. Mater., 2021, 2, 108 CrossRef CAS.
- P. Tian, L. Tang, K. S. Teng and S. P. Lau, Mater. Today Chem., 2018, 10, 221–258 CrossRef CAS.
- G. Leménager, E. De Luca, Y.-P. Sun and P. P. Pompa, Nanoscale, 2014, 6, 8617–8623 RSC.
- A. M. Chizhik, S. Stein, M. O. Dekaliuk, C. Battle, W. Li, A. Huss, M. Platen, I. A. T. Schaap, I. Gregor, A. P. Demchenko, C. F. Schmidt, J. Enderlein and A. I. Chizhik, Nano Lett., 2016, 16, 237–242 CrossRef CAS PubMed.
- W. Vandenberg and P. Dedecker, Sci. Rep., 2017, 7, 44665 CrossRef PubMed.
- H. He, X. Liu, S. Li, X. Wang, Q. Wang, J. Li, J. Wang, H. Ren, B. Ge, S. Wang, X. Zhang and F. Huang, Anal. Chem., 2017, 89, 11831–11838 CrossRef CAS PubMed.
- B. Zhi, Y. Cui, S. Wang, B. P. Frank, D. N. Williams, R. P. Brown, E. S. Melby, R. J. Hamers, Z. Rosenzweig, D. H. Fairbrother, G. Orr and C. L. Haynes, ACS Nano, 2018, 12, 5741–5752 CrossRef CAS PubMed.
- J. Shen, J.-H. Zhang, H. Xiao, J.-M. Wu, K.-M. He, Z.-Z. Lv, Z.-J. Li, M. Xu and Y.-Y. Zhang, Cell Death Dis., 2018, 9, 81 CrossRef PubMed.
- W. M. Saxton and P. J. Hollenbeck, J. Cell Sci., 2012, 125, 2095–2104 CAS.
- X.-W. Hua, Y.-W. Bao, J. Zeng and F.-G. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 32647–32658 CrossRef CAS PubMed.
- G. Han, J. Zhao, R. Zhang, X. Tian, Z. Liu, A. Wang, R. Liu, B. Liu, M.-Y. Han, X. Gao and Z. Zhang, Angew. Chem., Int. Ed., 2019, 58, 7087–7091 CrossRef CAS PubMed.
- H. Singh, S. Sreedharan, K. Tiwari, N. H. Green, C. Smythe, S. K. Pramanik, J. A. Thomas and A. Das, Chem. Commun., 2019, 55, 521–524 RSC.
- C. He, X. Lin, Y. Mei, Y. Luo, M. Yang, Y. Kuang, X. Yi, W. Zeng, Q. Huang and B. Zhong, Front. Chem., 2022, 10, 905475 CrossRef CAS PubMed.
- H. He, X. Chen, Z. Feng, L. Liu, Q. Wang and S. Bi, Nano Lett., 2021, 21, 5689–5696 CrossRef CAS PubMed.
- J. Yang, X. Zhang, Y.-H. Ma, G. Gao, X. Chen, H.-R. Jia, Y.-H. Li, Z. Chen and F.-G. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 32170–32181 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- Y. Jiang, M. Novoa, T. Nongnual, R. Powell, T. Bruce and J. McNeill, Nano Lett., 2017, 17, 3896–3901 CrossRef CAS PubMed.
- K. Sun, Y. Tang, Q. Li, S. Yin, W. Qin, J. Yu, D. T. Chiu, Y. Liu, Z. Yuan, X. Zhang and C. Wu, ACS Nano, 2016, 10, 6769–6781 CrossRef CAS PubMed.
- H. Shi, X. Ma, Q. Zhao, B. Liu, Q. Qu, Z. An, Y. Zhao and W. Huang, Adv. Funct. Mater., 2014, 24, 4823–4830 CrossRef CAS.
- X. Qin, H. Chen, H. Yang, H. Wu, X. Zhao, H. Wang, T. Chour, E. Neofytou, D. Ding, H. Daldrup-Link, S. C. Heilshorn, K. Li and J. C. Wu, Adv. Funct. Mater., 2018, 28, 1704939 CrossRef PubMed.
- H. Chen, J. Zhang, K. Chang, X. Men, X. Fang, L. Zhou, D. Li, D. Gao, S. Yin, X. Zhang, Z. Yuan and C. Wu, Biomaterials, 2017, 144, 42–52 CrossRef CAS PubMed.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed.
- X. Chen, R. Li, Z. Liu, K. Sun, Z. Sun, D. Chen, G. Xu, P. Xi, C. Wu and Y. Sun, Adv. Mater., 2017, 29, 1604850 CrossRef PubMed.
- Z. Sun, Z. Liu, H. Chen, R. Li, Y. Sun, D. Chen, G. Xu, L. Liu and C. Wu, Adv. Opt. Mater., 2019, 7, 1900007 CrossRef.
- S. K. Kaiser, I. Surin, A. Amorós-Pérez, S. Büchele, F. Krumeich, A. H. Clark, M. C. Román-Martínez, M. A. Lillo-Ródenas and J. Pérez-Ramírez, Nat. Commun., 2021, 12, 4016 CrossRef CAS PubMed.
- Y. Wu, H. Ruan, R. Zhao, Z. Dong, W. Li, X. Tang, J. Yuan and X. Fang, Adv. Opt. Mater., 2018, 6, 1800333 CrossRef.
- Y. Wu, H. Ruan, Z. Dong, R. Zhao, J. Yu, X. Tang, X. Kou, X. Zhang, M. Wu, F. Luo, J. Yuan and X. Fang, Anal. Chem., 2020, 92, 12088–12096 CrossRef CAS PubMed.
- M. T. Proetto, C. R. Anderton, D. Hu, C. J. Szymanski, Z. Zhu, J. P. Patterson, J. K. Kammeyer, L. G. Nilewski, A. M. Rush, N. C. Bell, J. E. Evans, G. Orr, S. B. Howell and N. C. Gianneschi, ACS Nano, 2016, 10, 4046–4054 CrossRef CAS PubMed.
- J. F. Frisz, K. Lou, H. A. Klitzing, W. P. Hanafin, V. Lizunov, R. L. Wilson, K. J. Carpenter, R. Kim, I. D. Hutcheon, J. Zimmerberg, P. K. Weber and M. L. Kraft, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E613–E622 CrossRef CAS PubMed.
- M. T. Proetto, C. E. Callmann, J. Cliff, C. J. Szymanski, D. Hu, S. B. Howell, J. E. Evans, G. Orr and N. C. Gianneschi, ACS Cent. Sci., 2018, 4, 1477–1484 CrossRef CAS PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- K. Li, T.-B. Ren, S. Huan, L. Yuan and X.-B. Zhang, J. Am. Chem. Soc., 2021, 143, 21143–21160 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741, 10.1039/B105159H.
- G. R. Suman, M. Pandey and A. S. J. Chakravarthy, Mater. Chem. Front., 2021, 5, 1541–1584 RSC.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- M. Li, Y. Gao, Y. Yuan, Y. Wu, Z. Song, B. Z. Tang, B. Liu and Q. C. Zheng, ACS Nano, 2017, 11, 3922–3932 CrossRef CAS PubMed.
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS PubMed.
- S. Wang, W. Wu, P. Manghnani, S. Xu, Y. Wang, C. C. Goh, L. G. Ng and B. Liu, ACS Nano, 2019, 13, 3095–3105 CrossRef CAS PubMed.
- S. Liu, X. Zhou, H. Zhang, H. Ou, J. W. Y. Lam, Y. Liu, L. Shi, D. Ding and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 5359–5368 CrossRef CAS PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- J. Yu, X. Sun, F. Cai, Z. Zhu, A. Qin, J. Qian, B. Tang and S. He, Opt. Lett., 2015, 40, 2313–2316 CrossRef CAS PubMed.
- C. Kuang, W. Zhao and G. Wang, Rev. Sci. Instrum., 2010, 81, 053709 CrossRef PubMed.
- X. Gu, E. Zhao, T. Zhao, M. Kang, C. Gui, J. W. Y. Lam, S. Du, M. M. T. Loy and B. Z. Tang, Adv. Mater., 2016, 28, 5064–5071 CrossRef CAS PubMed.
- D. Li, W. Qin, B. Xu, J. Qian and B. Z. Tang, Adv. Mater., 2017, 29, 1703643 CrossRef PubMed.
- X. Fang, X. Chen, R. Li, Z. Liu, H. Chen, Z. Sun, B. Ju, Y. Liu, S. X.-A. Zhang, D. Ding, Y. Sun and C. Wu, Small, 2017, 13, 1702128 CrossRef PubMed.
- Y. Xu, D. Dang, N. Zhang, J. Zhang, R. Xu, Z. Wang, Y. Zhou, H. Zhang, H. Liu, Z. Yang, L. Meng, J. W. Y. Lam and B. Z. Tang, ACS Nano, 2022, 16, 5932–5942 CrossRef CAS PubMed.
- R. Xu, D. Dang, Z. Wang, Y. Zhou, Y. Xu, Y. Zhao, X. Wang, Z. Yang and L. Meng, Chem. Sci., 2022, 13, 1270–1280 RSC.
- F. S. Yasin, T. R. Harvey, J. J. Chess, J. S. Pierce, C. Ophus, P. Ercius and B. J. McMorran, Nano Lett., 2018, 18, 7118–7123 CrossRef CAS PubMed.
- N. Asano, S. Asahina, J. Lu, J. Xu, Y. Shen, Z. Qin and S. Mintova, Inorg. Chem. Front., 2022, 9, 4225–4231 RSC.
- Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153–1175 CrossRef CAS.
- W. Albrecht and S. Bals, J. Phys. Chem. C, 2020, 124, 27276–27286 CrossRef CAS.
- M. Elbaum, S. Seifer, L. Houben, S. G. Wolf and P. Rez, Acc. Chem. Res., 2021, 54, 3621–3631 CrossRef CAS PubMed.
- Y. Li, W. Huang, Y. Li, W. Chiu and Y. Cui, ACS Nano, 2020, 14, 9263–9276 CrossRef CAS PubMed.
- A. L. Koh, C. M. Shachaf, S. Elchuri, G. P. Nolan and R. Sinclair, Ultramicroscopy, 2008, 109, 111–121 CrossRef CAS PubMed.
- J.-H. Tao-Cheng, V. Crocker, S. L. Moreira and R. Azzam, Mol. Brain, 2021, 14, 86 CrossRef CAS PubMed.
- S. Mohammadian, A. V. Agronskaia, G. A. Blab, E. G. van Donselaar, C. de Heus, N. Liv, J. Klumperman and H. C. Gerritsen, Ultramicroscopy, 2020, 215, 113007 CrossRef CAS PubMed.
- F. Lange, P. Agüi-Gonzalez, D. Riedel, N. T. N. Phan, S. Jakobs and S. O. Rizzoli, PLoS One, 2021, 16, e0240768 CrossRef CAS PubMed.
- A. Löschberger, C. Franke, G. Krohne, S. van de Linde and M. Sauer, J. Cell Sci., 2014, 127, 4351–4355 Search PubMed.
- B. Liu, Y. Xue, W. Zhao, Y. Chen, C. Fan, L. Gu, Y. Zhang, X. Zhang, L. Sun, X. Huang, W. Ding, F. Sun, W. Ji and T. Xu, Sci. Rep., 2015, 5, 13017 CrossRef CAS PubMed.
- J. Owen and L. Brus, J. Am. Chem. Soc., 2017, 139, 10939–10943 CrossRef CAS PubMed.
- H. Bian, Q. Wang, S. Yang, C. Yan, H. Wang, L. Liang, Z. Jin, G. Wang and S. Liu, J. Mater. Chem. A, 2019, 7, 5740–5747 RSC.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Díaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536–711 CrossRef CAS PubMed.
- W. Li, G. S. Kaminski Schierle, B. Lei, Y. Liu and C. F. Kaminski, Chem. Rev., 2022, 122, 12495–12543 CrossRef CAS PubMed.
- M. S. Arai and A. S. S. de Camargo, Nanoscale Adv., 2021, 3, 5135–5165 RSC.
- Y. Liu, Z. Peng, X. Peng, W. Yan, Z. Yang and J. Qu, Front. Chem., 2021, 9, 641330 CrossRef CAS PubMed.
- A. L. McEvoy, D. Greenfield, M. Bates and J. Liphardt, BMC Biol., 2010, 8, 106 CrossRef PubMed.
- W. H. De Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS PubMed.
- S. Zhu, J. Shao, Y. Song, X. Zhao, J. Du, L. Wang, H. Wang, K. Zhang, J. Zhang and B. Yang, Nanoscale, 2015, 7, 7927–7933 RSC.
- M. J. Molaei, RSC Adv., 2019, 9, 6460–6481 RSC.
- M. Li, T. Chen, J. J. Gooding and J. Liu, ACS Sens., 2019, 4, 1732–1748 CrossRef CAS PubMed.
- T. Ahmed, S. Seth and A. Samanta, ACS Nano, 2019, 13, 13537–13544 CrossRef CAS PubMed.
- H. Zhang, M. Zhao, I. M. Ábrahám and F. Zhang, Front. Bioeng. Biotechnol., 2021, 9, 692075 CrossRef PubMed.
- S. Wanninger, V. Lorenz, A. Subhan and F. T. Edelmann, Chem. Soc. Rev., 2015, 44, 4986–5002 RSC.
- X. Xie, Z. Li, Y. Zhang, S. Guo, A. I. Pendharkar, M. Lu, L. Huang, W. Huang and G. Han, Small, 2017, 13, 1602843 CrossRef PubMed.
- G. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404–1414 CrossRef CAS PubMed.
- J. Liu, C. Chen, S. Ji, Q. Liu, D. Ding, D. Zhao and B. Liu, Chem. Sci., 2017, 8, 2782–2789 RSC.
- E. Betzig, Opt. Lett., 1995, 20, 237–239 CrossRef CAS PubMed.
- S. Jakobs, T. Stephan, P. Ilgen and C. Brüser, Annu. Rev. Biophys., 2020, 49, 289–308 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- R. M. Dickson, A. B. Cubitt, R. Y. Tsien and W. E. Moerner, Nature, 1997, 388, 355–358 CrossRef CAS PubMed.
- M. Heilemann, E. Margeat, R. Kasper, M. Sauer and P. Tinnefeld, J. Am. Chem. Soc., 2005, 127, 3801–3806 CrossRef CAS PubMed.
- M. G. L. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13081–13086 CrossRef CAS PubMed.
- F. Balzarotti, Y. Eilers, C. Gwosch Klaus, H. Gynnå Arvid, V. Westphal, D. Stefani Fernando, J. Elf and W. Hell Stefan, Science, 2017, 355, 606–612 CrossRef CAS PubMed.
- M. Weber, M. Leutenegger, S. Stoldt, S. Jakobs, T. S. Mihaila, A. N. Butkevich and S. W. Hell, Nat. Photonics, 2021, 15, 361–366 CrossRef CAS PubMed.
- S. Coelho, J. Baek, S. Graus Matthew, M. Halstead James, R. Nicovich Philip, K. Feher, H. Gandhi, J. J. Gooding and K. Gaus, Sci. Adv., 2020, 6, eaay8271 CrossRef CAS PubMed.
- M. Dai, R. Jungmann and P. Yin, Nat. Nanotechnol., 2016, 11, 798–807 CrossRef CAS PubMed.
- R. P. Moore and W. R. Legant, Nat. Methods, 2018, 15, 659–660 CrossRef CAS PubMed.
- F. U. Zwettler, S. Reinhard, D. Gambarotto, T. D. M. Bell, V. Hamel, P. Guichard and M. Sauer, Nat. Commun., 2020, 11, 3388 CrossRef CAS PubMed.
- S. Pfeffer and J. Mahamid, Curr. Opt. Struct. Biol., 2018, 52, 111–118 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, H. Choi, A. K. Muthusamy, B. P. Mehl, P. Dong, T. A. Brown, J. Lippincott-Schwartz, Z. Liu, T. Lionnet and L. D. Lavis, Nat. Methods, 2016, 13, 985–988 CrossRef CAS PubMed.
- M. Sunbul, J. Lackner, A. Martin, D. Englert, B. Hacene, F. Grün, K. Nienhaus, G. U. Nienhaus and A. Jäschke, Nat. Biotechnol., 2021, 39, 686–690 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |