
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond classical sulfone chemistry: metal- and photocatalytic approaches for C–S bond functionalization of sulfones
Javier
Corpas
 a,
Shin-Ho
Kim-Lee
a,
Pablo
Mauleón
*ab,
Ramón Gómez
Arrayás
*ab and
Juan C.
Carretero
*ab
a,
Shin-Ho
Kim-Lee
a,
Pablo
Mauleón
*ab,
Ramón Gómez
Arrayás
*ab and
Juan C.
Carretero
*ab
aDepartment of Organic Chemistry, Universidad Autónoma de Madrid, Cantoblanco 28049 Madrid, Spain. E-mail: pablo.mauleon@uam.es; ramon.gomez@uam.es; juancarlos.carretero@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Cantoblanco 28049 Madrid, Spain, and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Spain
First published on 15th July 2022
Abstract
The exceptional versatility of sulfones has been extensively exploited in organic synthesis across several decades. Since the first demonstration in 2005 that sulfones can participate in Pd-catalysed Suzuki–Miyaura type reactions, tremendous advances in catalytic desulfitative functionalizations have opened a new area of research with burgeoning activity in recent years. This emerging field is displaying sulfone derivatives as a new class of substrates enabling catalytic C–C and C–X bond construction. In this review, we will discuss new facets of sulfone reactivity toward further expanding the flexibility of C–S bonds, with an emphasis on key mechanistic features. The inherent challenges confronting the development of these strategies will be presented, along with the potential application of this chemistry for the synthesis of natural products. Taken together, this knowledge should stimulate impactful improvements on the use of sulfones in catalytic desulfitative C–C and C–X bond formation. A main goal of this article is to bring this technology to the mainstream catalysis practice and to serve as inspiration for new perspectives in catalytic transformations.
 Javier Corpas | Javier Corpas graduated with Honours in Chemistry from Universidad Autónoma de Madrid (UAM) in 2016. In 2017, he started his PhD studies under the supervision of Dr Ramón Gómez Arrayás and Dr Pablo Mauleón. In 2019 he carried out predoctoral work as a visiting student at Princeton University, USA, with Prof. Paul J. Chirik on the development of Fe-based catalysts for the selective H/D exchange of C–H bonds. His research interests deal with the development of metal and photocatalytic methods for selective functionalization of unsaturated C–C bonds. |
 Shin-Ho Kim-Lee | Dr Shin-Ho Kim-Lee graduated with Honours in Chemistry in 2015 and earned his PhD. Degree from Universidad Autónoma de Madrid (UAM) in 2021, working under the direction of Dr Ramón Gómez Arrayás and Dr Pablo Mauleón on organometallic catalysis. In 2018, he carried out predoctoral work at the Massachusetts Institute of Technology, USA, working with Prof. Alexander T. Radosevich on the development of novel phosphacatalytic reactions. Currently, he is working as a research consultant, bearing interests in the implementation machine learning tools in organic chemistry to assist in optimization of molecular design and new reaction discovery. |
 Pablo Mauleón | Dr Pablo Mauleón earned his PhD. Degree at UAM in 2006 working with Prof. Juan C. Carretero, and did postdoctoral studies from 2006 until 2011 (MICINN Postdoctoral Scholar with Prof. F. Dean Toste at the University of California, Berkeley, USA; Marie Curie Postdoctoral Fellow with Prof. Andreas Pfaltz at the University of Basel, Switzerland). In 2011 he returned to UAM as a Ramón y Cajal Researcher, and in 2017 was promoted to Associate Professor. His research interests span the stereo- and regioselective functionalization of unsaturated compounds combining organometallic and photocatalysis, non-metal hydrogenation processes, and more recently multicatalytic process and organic redox flow batteries. |
 Ramón Gómez Arrayás | Dr Ramón Gómez Arrayás graduated from UAM in 1992 with a BSc in Chemistry. Following his MSc (1994, Prof. José L. García Ruano and Prof. M. Carmen Carreño) and PhD (1999, Prof. Juan C. Carretero) at UAM, he enjoyed a postdoctoral spell at Emory University (Atlanta, 1999–2001, Prof. Lanny S. Liebeskind). He returned to the UAM as a Ramón y Cajal Researcher until he was appointed Associate Professor at the UAM in 2008. His research interest focusses on the development of innovative strategies in homogeneous transition-metal catalysis and photocatalysis. |
 Juan C. Carretero | Dr Juan Carlos Carretero completed his PhD at UAM in 1985 under the direction of Prof. José L. García Ruano. From 1985 to 1988 he was a postdoctoral fellow at the Université Catholique de Louvain (Belgium) with Prof. Leon Ghosez. He subsequently joined the Department of Organic Chemistry at UAM, where he became Associate Professor in 1988 and Professor in 2000. His current research interests are focused on metal-catalyzed reactions, C–H functionalization processes and catalytic asymmetric cycloadditions. |
1. Introduction
The fundamental role that sulfones play in Organic Chemistry is two-fold: firstly, they are present in target molecules of importance in Medicinal Chemistry and Materials Science;1 secondly, they are increasingly important in the development of novel synthetic methodologies for the creation of C–C bonds.2 In this regard, the discovery of activation modes of the C–S bond directed at developing new strategic applications of the SO2 group has emerged as an active and promising research area.3 From a synthetic viewpoint, the majority of examples concerning the use of sulfones in synthesis rely upon the intrinsic acidity of the H atoms at the α-position, followed by functionalization via electrophilic trapping and subsequent desulfonylation. This sequence enables the formation of a new C–C or C–X bond in the molecule. Recently, catalytic transformations that involve activation of the C–S bond have been reported in which new C–C or C–X are created directly via catalytic desulfonylation, thus circumventing the need for stoichiometric bases and two-step sequences. The known ability of sulfones, compared to sulfides and sulfoxides, to participate in C–S heterolytic elimination-type reactions could be ascribed at least in part in terms of the pKa of the resulting leaving group, the sulfinate group.4 In this regard, sulfinic acids (pKa around 2) are less acidic than sulfonic acids (pKa around −3), but significantly more acidic than thiols (pKa range 7–11) or sulfenic acids (pKa around 12). Therefore, the use of sulfones with very different electronic properties could offer ample opportunities for the development of orthogonal strategies based on sulfinate-type leaving groups.There are several aspects about the overall transformation that deserve consideration. One salient example is the control of the chemoselectivity during the C–S bond cleavage with a proper set of reaction conditions. As the sulfone moiety (R1–SO2–R2) usually bears two different carbon moieties, control in the activation of either C–S bond is crucial to the success of this chemistry in terms of selectivity. Additionally, control over the catalytically relevant species and the nature of the sulfur-based leaving group is of great importance. There are at least two different possibilities – the elimination of RSO2−vs. SO2 extrusion plus a carbon fragment – that may be critical to the selectivity and/or the efficiency of the catalytic cycle, due to their potential involvement in the recovery of the active catalyst or in undesired side-reactions.
This review aims to provide a critical assessment of the different paths for catalytic desulfonylative processes to create C–C and C–X bonds. In doing so, we discuss different concepts regarding the control of the selectivity as a crucial factor for the success of this chemistry and its application in synthesis. During the preparation of this review, elegant reviews by Crudden covering aspects of this chemistry have appeared in the literature.5
2. Catalytic C–C bond forming reactions
The most prevalent reaction in organic synthesis is the creation of C–C bonds in a regio- and stereoselective fashion. In this regard, sulfones can participate as C(sp3), C(sp2), and C(sp) synthon donors by catalytic scission of the C–S bond, either by metal-catalysed activation or photocatalytic induced radical additions. In this section, we present a rationalized discussion of catalytic methods for the creation of C–C bonds attending to the organic fragment of the sulfone that is transferred.2.1. Alkenylations
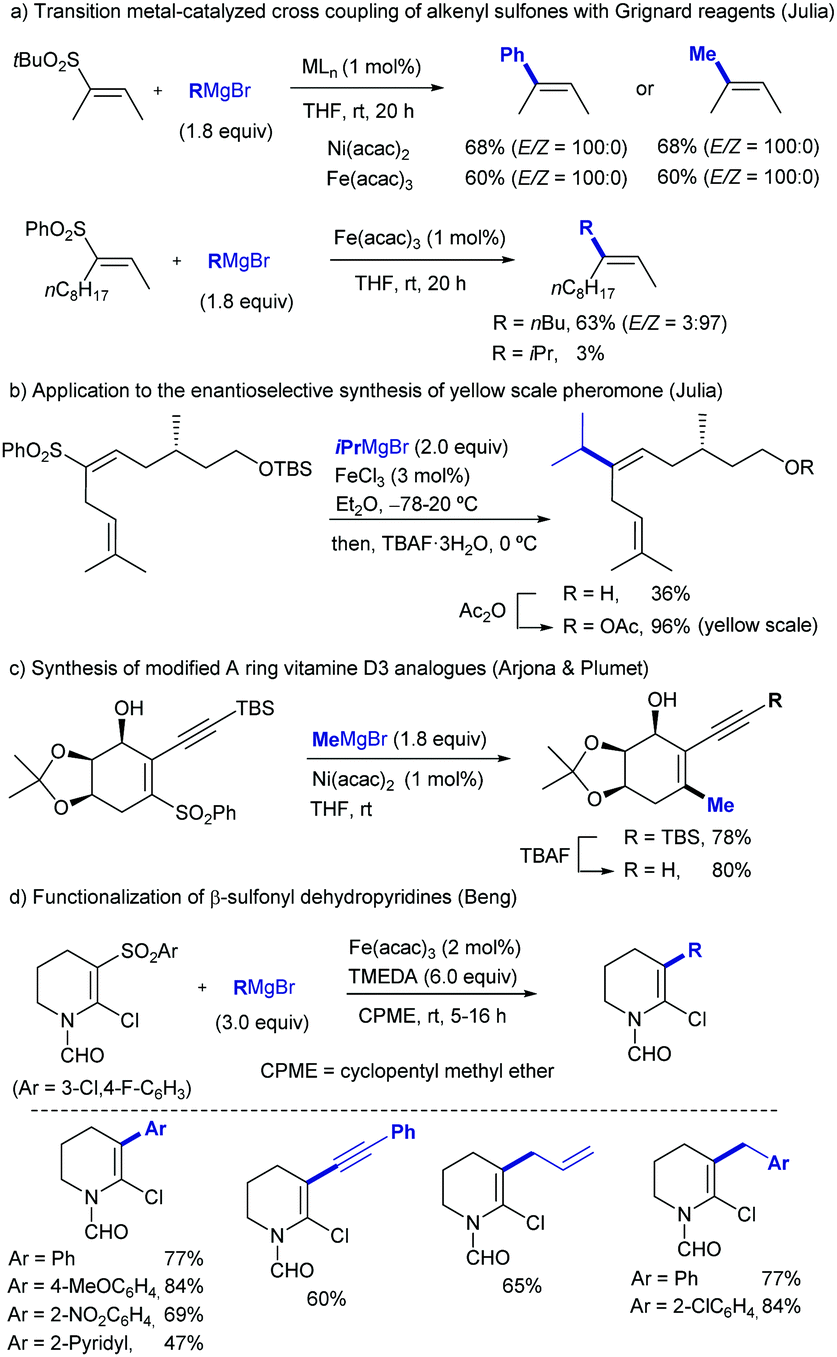 | ||
| Scheme 1 Cross coupling of alkenyl sulfones with Grignard reagents under Ni- or Fe-catalysis. | ||
Another early example of carbon-carbon bond formation by desulfonylation catalysed by a transition metal was reported by Kamigata in 1985. When E,E-bis(styryl)sulfones are heated in benzene at 150 °C in the presence of a catalytic amount of dichlorotris(triphenylphosphine)ruthenium(II), the corresponding (E,E)-l,4-diaryl-l,3-butadienes are formed in high yield (Scheme 2).12 The mechanistic implications of hydrogen chloride on the structure and catalytic property of the Ru(II) complex are not clear.
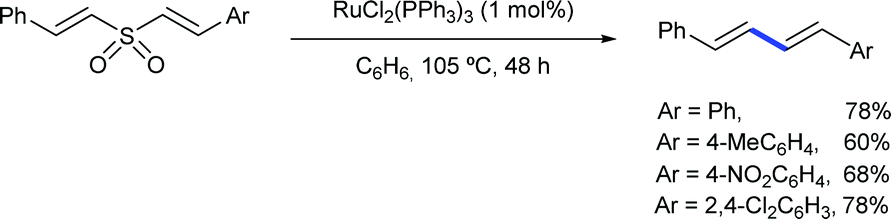 | ||
| Scheme 2 Ru-Catalysed synthesis of 1,4-diaryl-1,3-dienes from bis(styryl)sulfones. | ||
These initial reports have served as inspiration for recent development of more practical cross coupling processes involving more functional group-tolerant reagents. For example, in 2019 the group of Niu demonstrated that bench stable 1-sulfonyl glycals are competent electrophiles in Ni-catalysed Suzuki–Miyaura cross-coupling reactions with various aryl- and alkenylboron nucleophiles (Scheme 3).13 The C–sulfone bond in the α-oxo-vinylsulfone moiety is cleaved chemoselectively furnishing C-aryl glycals or acyclic vinyl ethers in high yields. The method was also extended to acyclic α-oxo-vinyl sulfones furnishing acyclic vinyl ethers. Boronic acid pinacol esters were similarly efficient as nucleophiles, whereas trifluoroborates led to almost no coupling product. A large variety of boronic acid derivatives took part in the reaction, including heteroaryl derivatives containing basic nitrogen. The synthetic utility of this method was showcased by its application to the synthesis of pharmaceutical agents and modified amino esters. Control experiments revealed the importance of the α-oxygen atom facilitating the oxidative insertion step, likely by precoordinating with the Ni(0) catalyst, which would also explain the high regioselectivity observed. A plausible mechanistic cycle was proposed that starts with a regioselective C–S bond insertion by Ni(0) to afford an alkenyl Ni(II) sulfinate complex, presumably facilitated by the transient coordination of the Ni(0) catalyst to the oxygen atom, which undergoes a transmetalation with the arylboron reagent followed by reductive elimination to give the coupling product while regenerating the active Ni(0) species. The facility with which this cross-coupling proceeds suggests that the transmetalation process could outcompete the desulfination step from the oxidative addition complex.
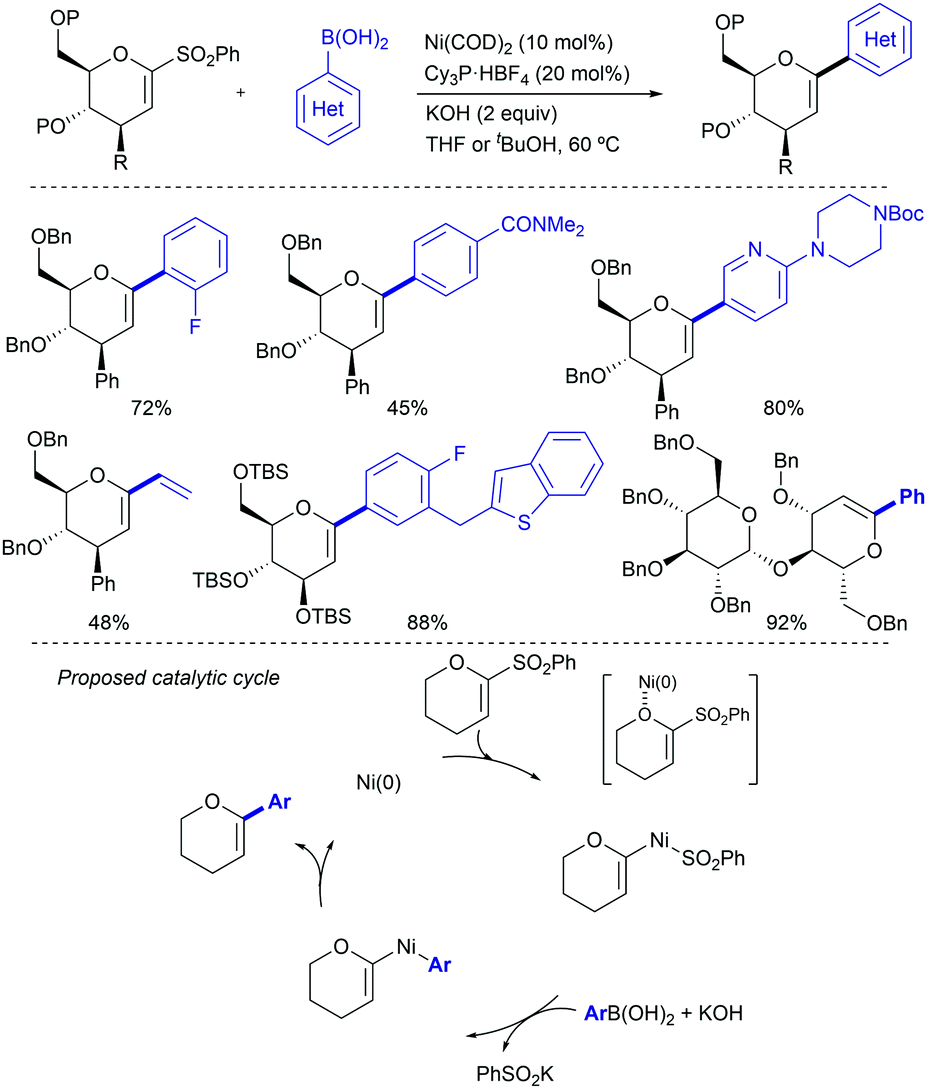 | ||
| Scheme 3 Ni-Catalysed coupling of 1-sulfonyl glycals with boronic acid derivatives. | ||
More recently, this group has extended the method to the stereoconvergent cross-coupling of acyclic α-oxo-vinylsulfones and employing alkyl zinc reagents as nucleophilic partners (Scheme 4).14 The method enables the access to Z-enol ethers with dialkyl substituion. High functional group compatibility was well tolerated for a wide range of vinyl sulfones and functionalized organozinc reagents.
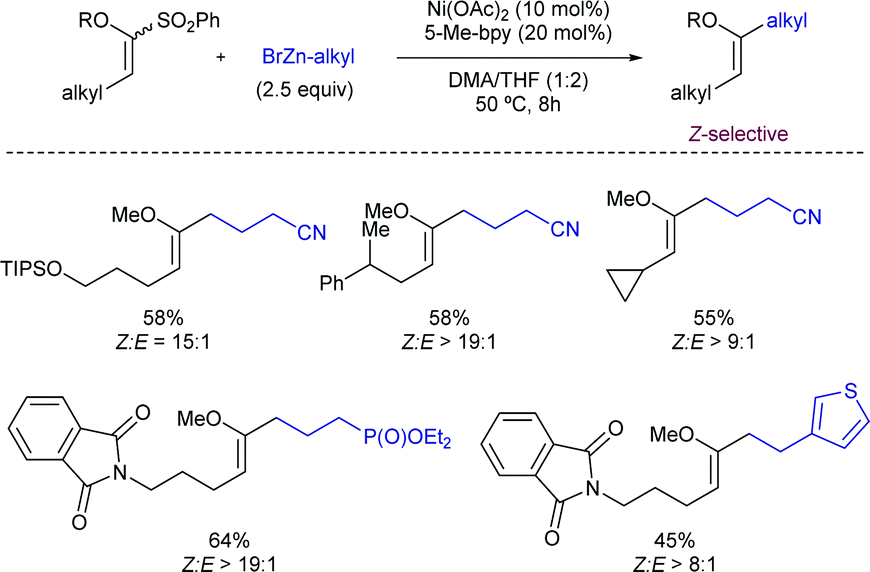 | ||
| Scheme 4 Ni-Catalysed coupling of α-oxovinylsulfones with organozinc reagents. | ||
In 2003, while investigating the rhodium-catalysed asymmetric arylation of α,β-unsaturated sulfones with aryltitanium reagents, the Hayashi group discovered that the conjugate addition product undergoes β-hydride elimination followed by reinsertion with opposite regioselectivity and β-elimination of the sulfone moiety to give the cine-substitution product (Scheme 5). This reaction pathway was supported by deuterium-labeling studies. The use of (S)-Binap as ligand led to high enantioselectivity (>99% ee) in some cases.
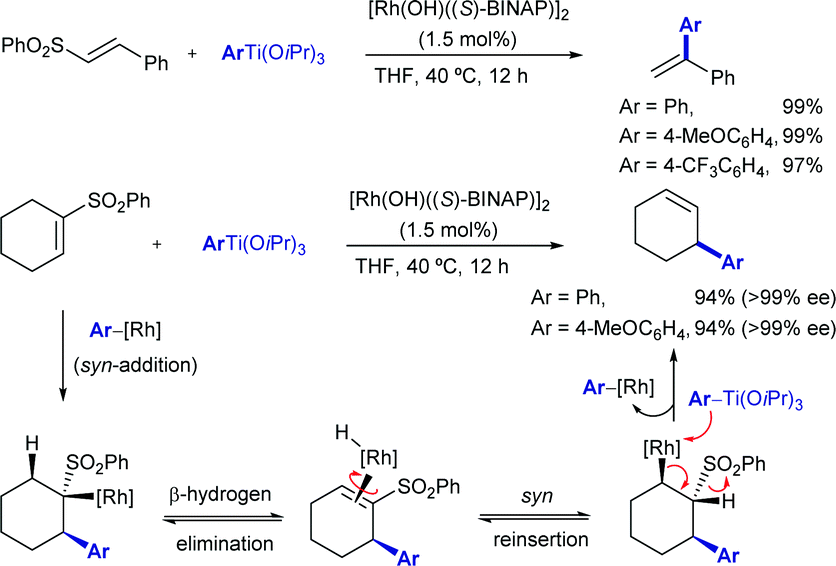 | ||
| Scheme 5 cine-Substitution of alkeny sulfones with aryltitanium reagents. | ||
Finally, sulfones have found excellent performance for dienylation reactions in presence of electrophilic bromoarenes. In particular, the Larionov group has reported15 the utilization of sulfolene as the 1,3-dienyl transfer reagent with the ability to access the two possible stereoisomers by ligand control (Scheme 6). The reaction starts with the base-promoted sulfolene ring-opening to furnish the dienylsulfinate intermediate which transmetalates with the in situ generated aryl-palladium(II). Further stereoselective reductive elimination yielding the Z-isomer closes the catalytic cycle and renders the required Pd(0) species. Both XantPhos and dppbz were found to be effective ligands for this transformation. However, for dppbz a further thermodynamically-driven Z–E isomerization was found to be productive for the generation of the E-selective dienylated product. Mechanistic experiments demonstrated the formation of nanoparticulated species by using the dppbz ligand, which were reactive in the isomerization event via the formation of Pd–H species.
 | ||
| Scheme 6 Stereodivergent ligand-controlled Pd-catalysed dienylation of aryl halides. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O generates a highly reactive oxyl radical species that induces homolytic cleavage of unactivated sp3 C–H bonds to provide the corresponding nucleophilic carbon radical intermediate (along with a ketyl radical), which undergoes addition to the electrophilic olefin partner and subsequent elimination of a sulfonyl radical to give the corresponding (E)-sulfonylalkenes. Reduction of the sulfinyl radical with the ketyl radical, most likely via a proton coupled electron transfer (PCET),19 would provide sulfinic acid along with the regeneration of benzophenone.
O generates a highly reactive oxyl radical species that induces homolytic cleavage of unactivated sp3 C–H bonds to provide the corresponding nucleophilic carbon radical intermediate (along with a ketyl radical), which undergoes addition to the electrophilic olefin partner and subsequent elimination of a sulfonyl radical to give the corresponding (E)-sulfonylalkenes. Reduction of the sulfinyl radical with the ketyl radical, most likely via a proton coupled electron transfer (PCET),19 would provide sulfinic acid along with the regeneration of benzophenone.
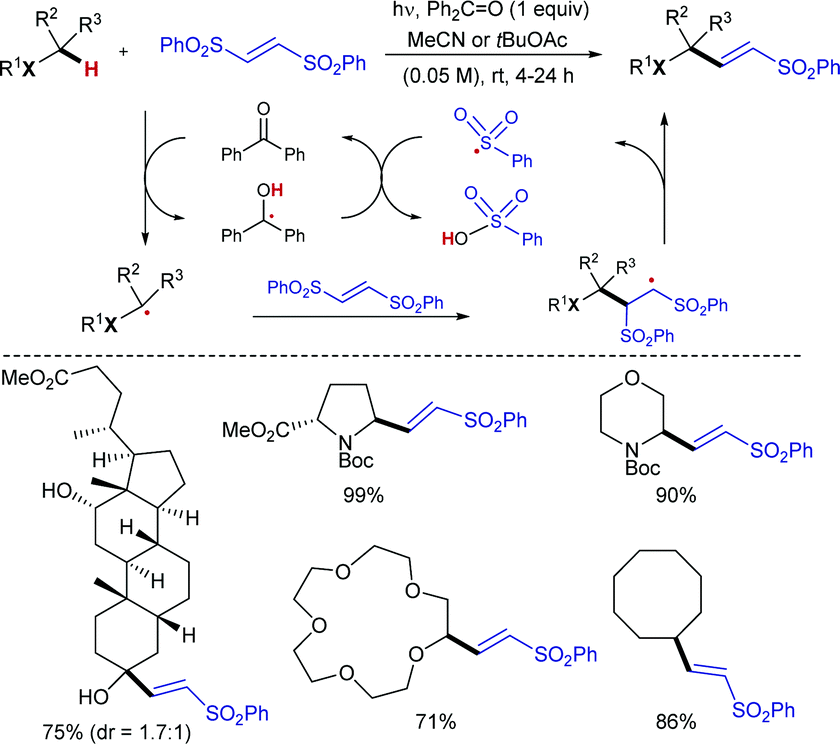 | ||
| Scheme 7 UV-induced radical sulfonylalkenylation of C(sp3)–H bonds. | ||
The C–H bond next to the heteroatom is selectively abstracted due to stabilization of the resulting radical by hyperconjugation with its lone pair. When there are available several C–H bonds adjacent to the heteroatom, the alkenylation occurred preferably at the most electron-rich one. The alkenylation products were typically obtained in yields over 70%, even when using more complex structures such as the steroidal methyl deoxycholate. The intermediacy of the carbon radical was confirmed by a radical-clock experiment with 1-cyclopropyl-1-ethanol, which afforded the ring-opened sulfonylalkene as the only product. Moreover, the product-determining step of the alkenylation was determined to be homolysis of the C–H bond, as suggested by a KIE value of 4 in a parallel experiment with cyclohexane and its fully deuterated analogue. The synthetic utility was further demonstrated by the conversion of the sulfonylalkenes into oxidized prenyl- and pyrrole-derivatives.
Simultaneously, the MacMillan group demonstrated that simple alkenylsulfones can also serve as efficient radical acceptors for photoredox-generated α-amino radicals from N-aryl tertiary amines or via the decarboxylation of N-Boc α-amino acids under visible light in the presence of iridium-based metal-to-ligand charge transfer catalysts (Scheme 8).18 In this alkenylation protocol, the photoexcited IrIII complex, formed by exposure to visible light irradiation, would undergo single-electron transfer (SET) with a tertiary amine to form a radical cation that upon deprotonation should deliver the α-amino radical (Scheme 8a). Alternatively, the α-amino radical can be generated via SET from the photoexcited IrIII complex with the deprotonated α-amino acid, followed by decarboxylation of the resulting carboxyl radical (Scheme 8b). Either way, the subsequent reaction of the α-amino radical with the α,β-unsaturated sulfone would generate a β-sulfonyl radical that can provide the corresponding alkene by β-elimination of the phenylsulfonyl radical. The reaction efficiency in terms of both reactivity and stereoselectivity was significantly impacted by the reducing ability of the Ir-catalyst, as well as the choice of base and solvent. These new C(sp3)–H alkenylation and decarboxylative alkenylation protocols proceed in high yield and with excellent olefin E-stereocontrol, leading to allylic amines of broad diversity.
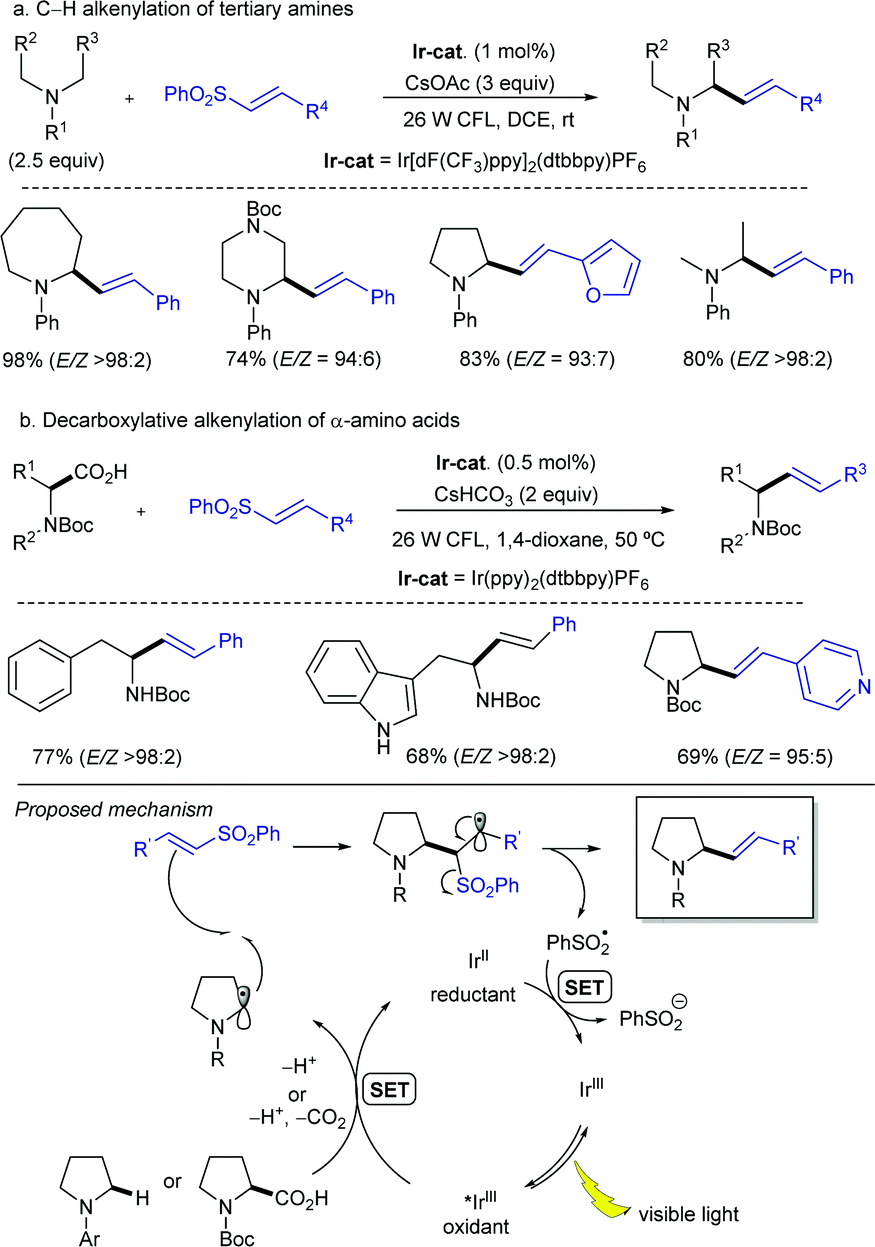 | ||
| Scheme 8 Visible light photoredox α-alkenylation of tertiary N-arylamines or α-amino acids. | ||
A more detailed mechanistic description of the SET events in this photoredox α-vinylation reaction mediated by iridium(III) catalysts was reported by Yang, Chen and Fang through theoretical electronic structure calculations along with the kinetic assessment of SETs and intersystem crossing.20 The presence of an electron-donating tert-butyl group in the bipyridine ligand of the Ir-catalyst notably reduces the energy level of the acceptor for metal-to-ligand charge transfer and promotes the decreased reorganization energy associated with the nuclear deformation of amine substrate planarization along the reductive quenching pathway of Ir(III) catalyst, thereby accelerating the rate-determining SET. Meanwhile, the presence of electron-withdrawing groups (F or CF3) in the two other ligands of the Ir complex facilitates the dispersion of excess negative charge when the reduction reaction of photocatalyst is triggered, thus producing the thermally stable Ir(II) catalyst in its reduced state. Importantly, the C–C bond construction was suggested to proceed in a concerted fashion with the proton transfer and the departure of the leaving group.
More recently, a radical vinylsulfonylations of α-amino carboxylic acids via direct decarboxylation through photoredox catalysis using (E)-1,2-bis(phenylsulfonyl)ethylene as radical acceptor has been reported by Opatz (Scheme 9).21 The use of (E)-3-phenylsulfonylprop-2-enenitrile as a radical trap leads to functionalized acrylonitriles. Since oxidative decarboxylations of aliphatic carboxylic acids are quite challenging (e.g., for N(nBu)4 salt of phenylacetic acid: E1/2(RCO2˙/RCO2−) = +1.27 V vs. SCE), to achieve the decarboxylative radical generation, the strongly oxidizing 9,10-dicyanoanthracene (DCA, E1/2(1[DCA]*/DCA˙−) = +1.99 V vs. SCE) was used as organic photocatalyst and biphenyl (BP) as a redox mediator. A variety of natural and unnatural α-amino acids with sensitive groups were converted into the corresponding vinyl sulfones. Neither sterically hindered tertiary radicals nor highly stabilized benzylic or allylic radicals are suitable reactions partners in the reaction. Simple alkyl radicals lacking the electron-donating and radical-stabilizing α-nitrogen atom were compatible, albeit with lower efficiency due to its decreased nucleophilicity. Substitution of the second phenylsulfonyl group on the resulting vinyl sulfones was not observed, which was ascribed to the lower reactivity and increased steric hindrance.
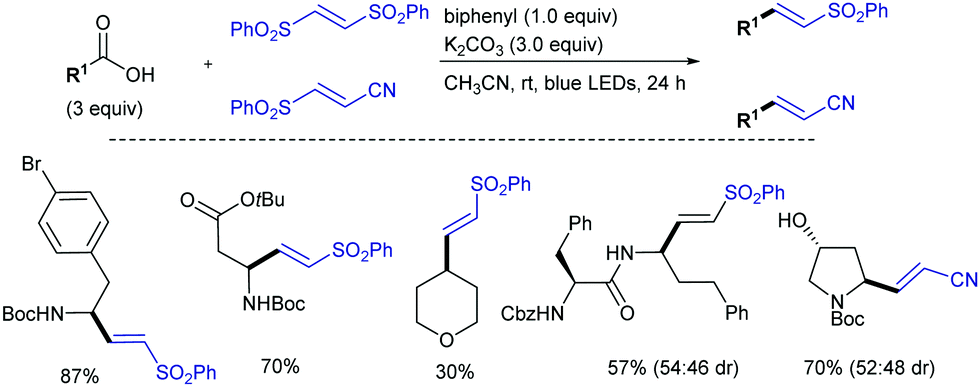 | ||
| Scheme 9 Photoredox decarboxylative alkenylation of carboxylic acids and peptides. | ||
Shortly after MacMillan's report, Molander described a metal-free version of this reaction using the inexpensive organic dye Eosin Y as photocatalyst and Boc-protected potassium α-pyrrolidinyltrifluoroborates as α-amino radical precursors (Scheme 10).22 This procedure exploits the lower oxidation potentials of potassium alkyltrifluoroborates (Ered1/2 = +0.78 V vs. SCE) compared to their corresponding cesium carboxylates (Ered1/2 = +0.95 V vs. SCE), the latter lying outside of Eosin Y's oxidation window (Ered1/2 = +0.83 V vs. SCE). A variety of allylic and homoallylic pyrrolidine derivatives having various substitution patterns at the alkene were produced in moderate yields with high E-selectivity. Unfortunately, this Eosin Y-catalysed alkenylation was found to be limited to secondary α-aminomethyltrifluoroborates; neither primary α-aminomethyl nor primary or secondary α-alkoxytrifluoroborates were compatible due to their higher oxidation potentials.
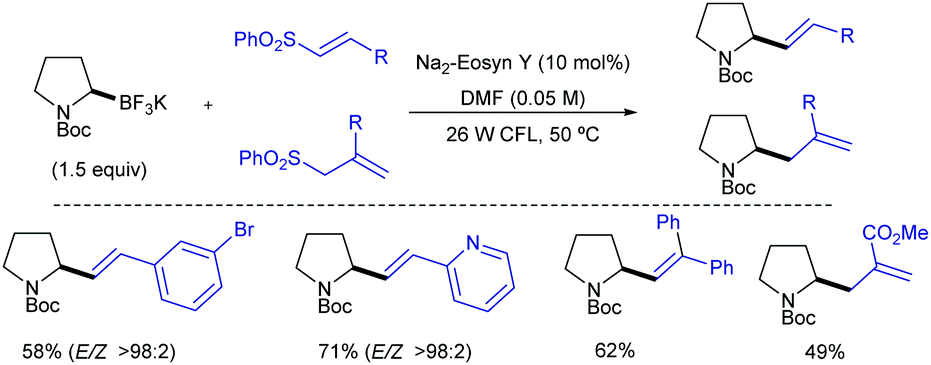 | ||
| Scheme 10 Metal-free visible light-photoredox catalysed alkenylation and allylation of potassium α-pyrrolidinyltrifluoroborates. | ||
More recently, the group of Wang has demonstrated that the more readily available alkyl boronic acids can also serve as an alternative radical source in the reaction with alkenyl sulfones (Scheme 11).23 Although alkylboronic acids have higher oxidation potentials (Ered = +2.5 V vs. SCE for secondary boronic acids) than the analogues trifluoroborate salts, it has been reported that the oxidation potential can be reduced by boron-activation with a Lewis base.24 Therefore, the authors hypothesized that benzenesulfinate ion released from the alkenyl sulfones during the coupling could effectively activate boronic acids, thus avoiding the need for an external Lewis base. On this basis, a protocol for the direct visible light-mediated alkenylation of a broad range of cyclic and acyclic primary and secondary alkyl boronic acids with various alkenyl sulfones at room temperature was devised. Evidences gained by analysis of the 1H NMR spectra of mixtures of phenethylboronic acid and benzenesulfinate at various concentrations in DMSO-d6, as well as cyclic voltammetry measurements to study the effect of the benzenesulfinate on the oxidation potential of cyclohexylboronic acid, led the authors to propose a mechanism involving benzenesulfinate-activated alkyl boronic acids. The mildness of the protocol makes it suitable for the late-stage functionalization of natural products and drug molecules.
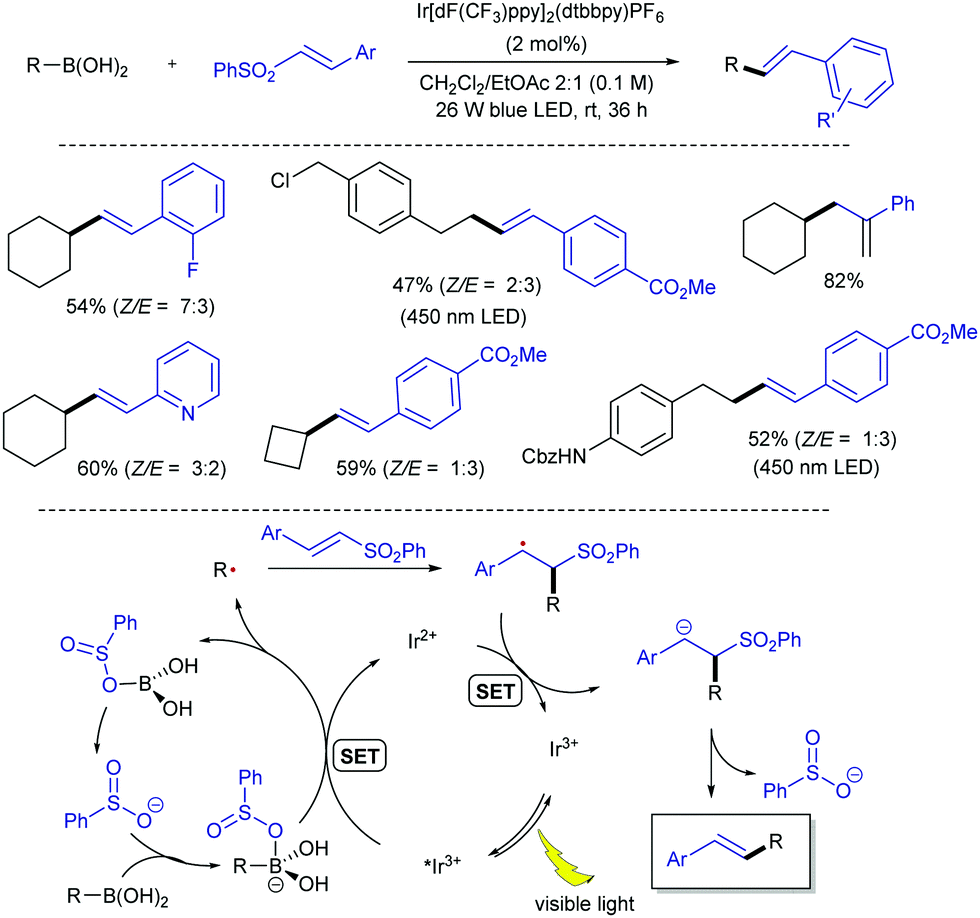 | ||
| Scheme 11 Alkylboronic acids as radical precursors in visible light-photoredox catalysed alkenylation with alkenyl sulfones. | ||
Paul and Guin later reported a greener visible light-mediated version of Kamijo and Inoue's protocol that addresses two major drawbacks from that study. First, stoichiometric amount of benzophenone is required; also, the use of high energy UV light, which can directly photolyze bonds in organic molecules, limits the functional group tolerance. This new method allows α-C–H coupling of ethers/amides with alkenyl- and alkynyl sulfones employing a catalytic amount of the HAT-promoter 4,4’-dichlorobenzophenone under a household fluorescent light bulb (Scheme 12).25 A broad range of synthetically useful functional groups, including the UV light sensitive aromatic halides can be incorporated, providing the corresponding allyl, homoallyl and propargyl products with excellent yield and E-stereoselectivity.
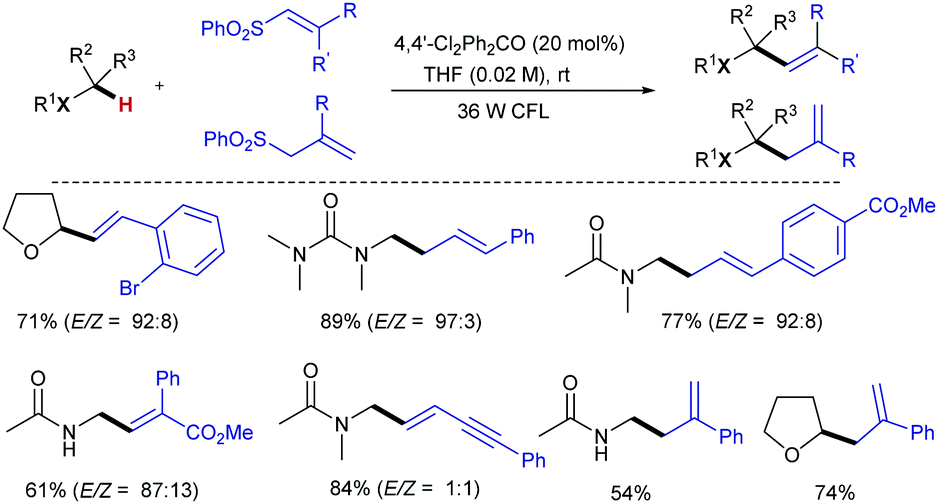 | ||
| Scheme 12 Visible light-photocatalytic C(sp3)–H α-alkenylation and allylation of ethers and amides. | ||
The redox-activated primary amine Katrizky pyridinium salts (i.e., 2,4,6-triphenylpyridinium salts) have been exploited by Gryko as radical precursors in this type of visible light-mediated addition-elimination reactions with alkenyl sulfones (Scheme 13).26 Considering reduction potential values of Katritzky salts (E1/2 = ca. −0.95 V vs. Ag/AgCl in MeOH) and guided by fluorescence quenching studies, eosin Y was chosen as photocatalyst, while DIPEA was used as sacrificial reductant in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MeOH/CH2Cl2 at room temperature. Under these mild conditions, trapping the radicals issued from secondary alkyl substituted pyridinium salts with alkenyl sulfones provides a broad range of functionalized aryl and heteroaryl-substituted olefins with excellent E-stereoselectivity. Stern–Volmer quenching experiments revealed a higher quenching constant for pyridinium salt compared with DIPEA, suggesting that the oxidative quenching cycle is favored. A mechanistic pathway was proposed in which photoexcited eosin Y (EY*) reduces the pyridinium salt via SET, followed by fragmentation to afford the alkyl radical along with the rearomatized pyridine, while EY+· is reduced to the initial form by DIPEA. Subsequent α-addition of the alkyl radical to the alkenyl sulfone followed by rapid elimination of arylsulfinyl radical affords the alkenylation product. The E-selectivity arises during the β-elimination step and is dictated by the thermodynamic and steric factors.
:1 mixture of MeOH/CH2Cl2 at room temperature. Under these mild conditions, trapping the radicals issued from secondary alkyl substituted pyridinium salts with alkenyl sulfones provides a broad range of functionalized aryl and heteroaryl-substituted olefins with excellent E-stereoselectivity. Stern–Volmer quenching experiments revealed a higher quenching constant for pyridinium salt compared with DIPEA, suggesting that the oxidative quenching cycle is favored. A mechanistic pathway was proposed in which photoexcited eosin Y (EY*) reduces the pyridinium salt via SET, followed by fragmentation to afford the alkyl radical along with the rearomatized pyridine, while EY+· is reduced to the initial form by DIPEA. Subsequent α-addition of the alkyl radical to the alkenyl sulfone followed by rapid elimination of arylsulfinyl radical affords the alkenylation product. The E-selectivity arises during the β-elimination step and is dictated by the thermodynamic and steric factors.
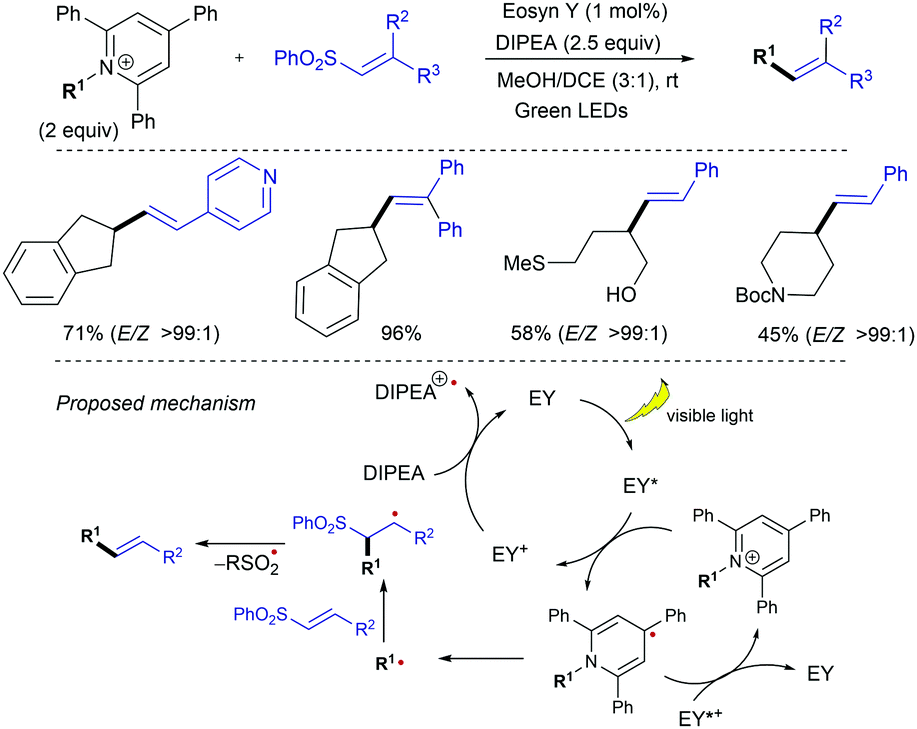 | ||
| Scheme 13 Redox activated amines as radical precursors in visible light-photoredox catalysed alkenylation with alkenyl sulfones. | ||
The exceptionally high redox potentials of unactivated alkyl halides (Ered [nBu–I] = −2.5 V vs. SCE in MeCN) makes very difficult the generation of alkyl radicals from this class of substrates under visible-light irradiation.27 Therefore, the generation of radical precursors from alkyl halides typically relies on the use of tin reagents or other stoichiometric initiators.28 Pd/light-initiated radical reactions,29 in which a SET reaction between alkyl iodides and Pd(0) under photoirradiation allows the generation of the initial alkyl radical is a practical way to solve this problem. Based on this design principle, Ryu and co-workers have devised a protocol for the alkenylation of alkyl iodides with alkenyl sulfones under Pd/photoirradiation (Xe, 800 W m−2) catalyst system (Scheme 14).30 Primary, secondary, and tertiary alkyl iodides, as well as variously substituted alkenyl sulfones, efficiently participated in the reaction to afford the corresponding alkenes in good to moderate yields. This reaction system is also applicable to the three-component coupling reactions including the radical carbonylative transformation. The reaction was suggested to proceed through initial SET from photoexcited Pd(0) to the alkyl iodide to generate an alkyl radical (R˙) and a PdI radical. Addition of the former to the α,β-unsaturated sulfone followed by β-elimination would produce the alkenylated product and sulfonyl radical (PhO2S˙). The combination of sulfonyl radical and Pd(I) generates sulfonyl palladium iodide, from which reductive elimination results in the formation of PhSO2I and Pd(0). The presence of water and Et3N in the catalytic cocktail was found to be beneficial, likely by promoting hydrolysis of PhSO2I, preventing the reverse oxidative addition step.
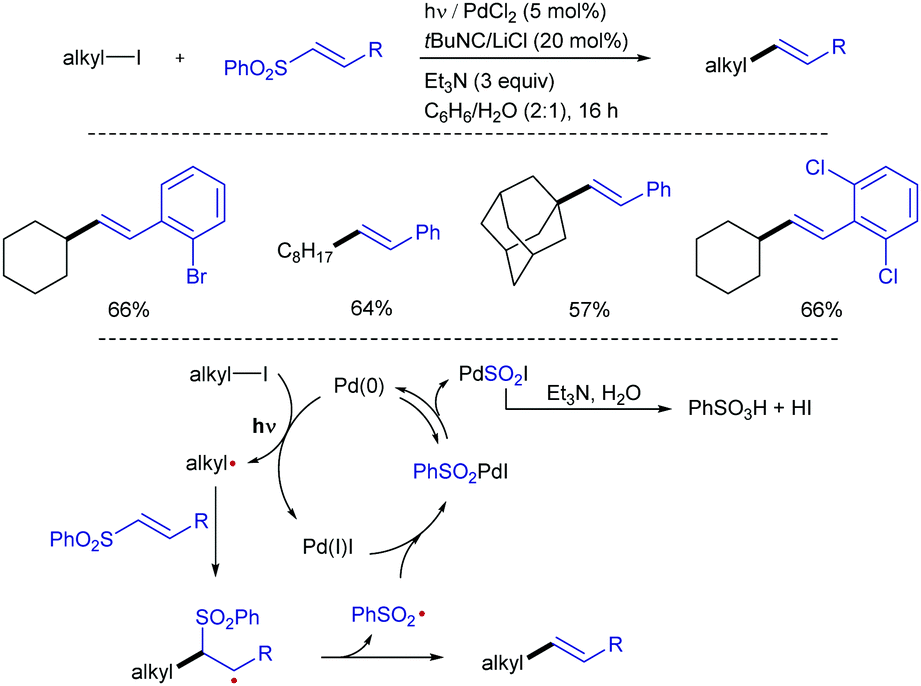 | ||
| Scheme 14 Palladium/light induced radical alkenylation of alkyl iodides with α,β-unsaturated sulfones. | ||
The direct use of unactivated alkyl bromides in alkenylation reactions through visible light photocatalysis remains a challenge compared to alkyl iodides.31 The group of König and Xiao managed to use unactivated alkyl bromides as efficient alkyl radical precursors to participate in alkenylation reactions with alkenyl phenyl sulfones through silicon-mediated radical debromination under visible light photocatalysis (Scheme 15).32 The combination of Ir(dF(CF3)ppy)2(dtbbpy)PF6 phatocatalyst with the capability of tris(trimethylsilyl)silane (TTMSS) as both potent hydrogen atom donor and competent halogen-atom transfer was exploited to activate the alkyl bromides. This protocol allows the preparation of a variety of alkenes from primary, secondary, and tertiary alkyl bromides with good reaction efficiency and high chemoselectivity under mild reaction conditions. The reaction can be scaled up to gram quantities while lowering the loading of the photocatalyst to 0.5 mol% without affecting the reaction efficiency. Mechanistically, the reaction was proposed to occur by reductive quenching of the excited state of the photocatalyst by bromide ion generated in small amounts upon elimination reaction promoted by the base. The formed bromine radical can abstract a hydrogen atom from (TMS)3SiH to afford a silyl radical that will abstract a bromine atom from the substrate to give an alkyl radical. This alkyl radical reacts with the alkenyl sulfone through the usual addition–elimination pathway to deliver the alkene product.
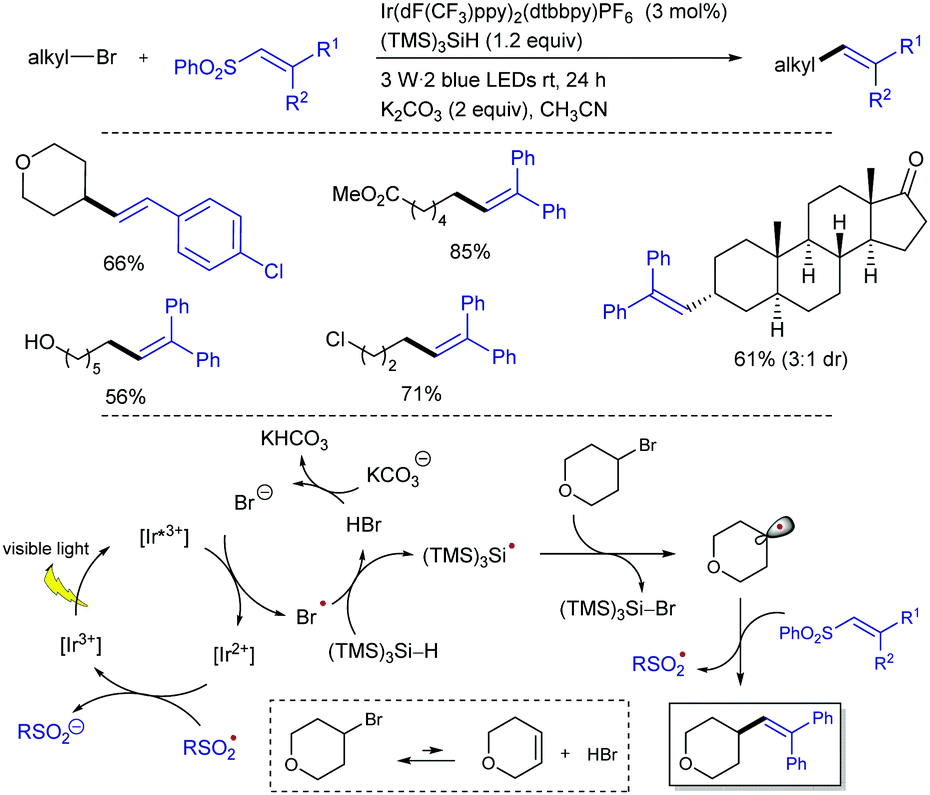 | ||
| Scheme 15 Photocatalytic alkenylation of alkyl bromides with vinyl phenyl sulfones. | ||
2.2. Arylations
Sulfones can also be employed as aryl source in metal catalysed cross-couplings; this is only logical, considering that alkenyl and aryl sulfones both bear C-sp2 centres, and must therefore behave similarly. Researchers have taken advantage of this pattern of reactivity and employed aryl sulfones as partners in cross-coupling reactions, among other transformations.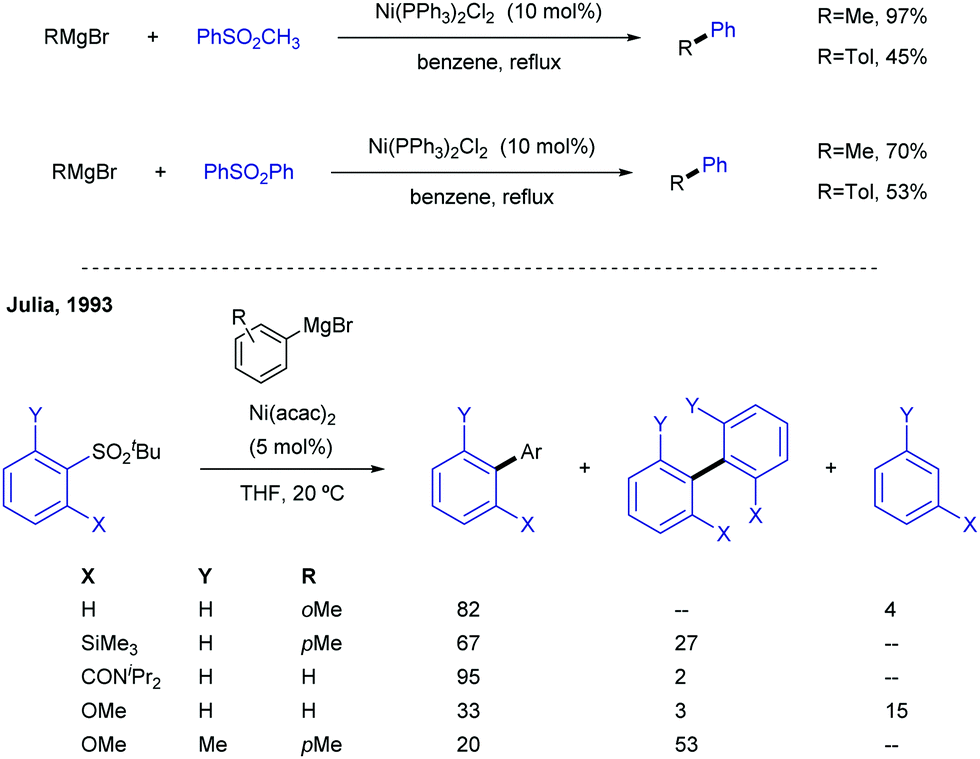 | ||
| Scheme 16 Ni-Catalysed Kumada cross-couplings of aryl sulfones. | ||
A more systematic study published two years later by the same research group35 threw the following conclusions: (i) NiCl2(PPh3)2 and Ni(acac)2 were the best catalysts for the transformation, outperforming other Ni salts and Fe(acac)3. (ii) The success of the reaction greatly depends on the nature of the organomagnesium reagent, with aryl Grignards providing much better results than MeMgBr. (iii) Use of Grignard reagents that possess β-hydrogen atoms resulted in large amounts of the corresponding reduction of the sulfone to form the corresponding reduced aryl ring. In fact, use of iPrMgBr turned the reduction path into the major reaction course. (iv) An excess of Grignard reagent (typically 2 equivalents) was necessary to bring the reaction to completion.
The Enthaler group found a well-defined Ni catalyst which, in combination with 1.5 equivalents of LiOtBu, can cleave the sp2 C–S bond in thioethers, sulfoxides, and sulfones to form biaryls in combination with aryl Grignards.36 The research team explained its reactivity in a model in which the sulphur derivative approaches the coordination sphere of Ni, and through single electron transfer (SET) oxidizes the complex to produce a radical. The oxidized complex can be oxidized at the metal center, or alternatively the electron can be stored in the ligand. After elimination of RSMgX, the radical recombines to form a Ni(IV) species that undergoes reductive elimination to release the active catalyst and Ar–R. The beneficial effect of LiOtBu was tentatively attributed to either de-aggregation of organomagnesium clusters or an increase in basicity of the Grignard reagent (Scheme 17).
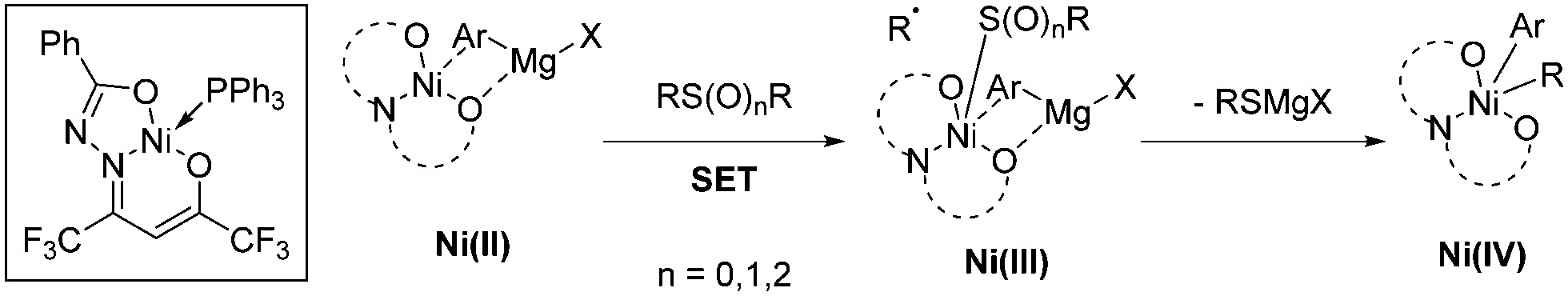 | ||
| Scheme 17 Ni-Catalysed cleavage of the sp2 C–S bond in thioethers, sulfoxides, and sulfones. | ||
In 2008, García observed that catalytic amounts of Ni complexes such as [Ni(dippe)H]2, [Ni(dcype)H]2, [Ni(dtbpe)H]2, [Ni(dippe)(Me)2], and NiCl2·6H2O reacted with a number of alkyl Grignard reagents and diethylzinc to effect the deoxydesulfurization of sulfones of dibenzothiophene (DBTO2), 4-methyl-dibenzothiophene (MeDBTO2) and 4,6-dimethyldibenzothiophene (Me2DBTO2) in very good yields (85–100% yield) when using a solvent mixture of toluene and THF.37 In this study, key intermediates were identified which are likely to participate in Kumada couplings involving ArSO2R compounds. In particular, the reaction of [Ni(dippe)H]2 with DBTO2 in THF yielded a complex in which the sulfone coordinated to Ni by the two oxygen atoms. Further studies on the thermal decomposition of said compound revealed several by-products derived from the cleavage of S–O and C–S bonds of the originally bound sulfone (Scheme 18).
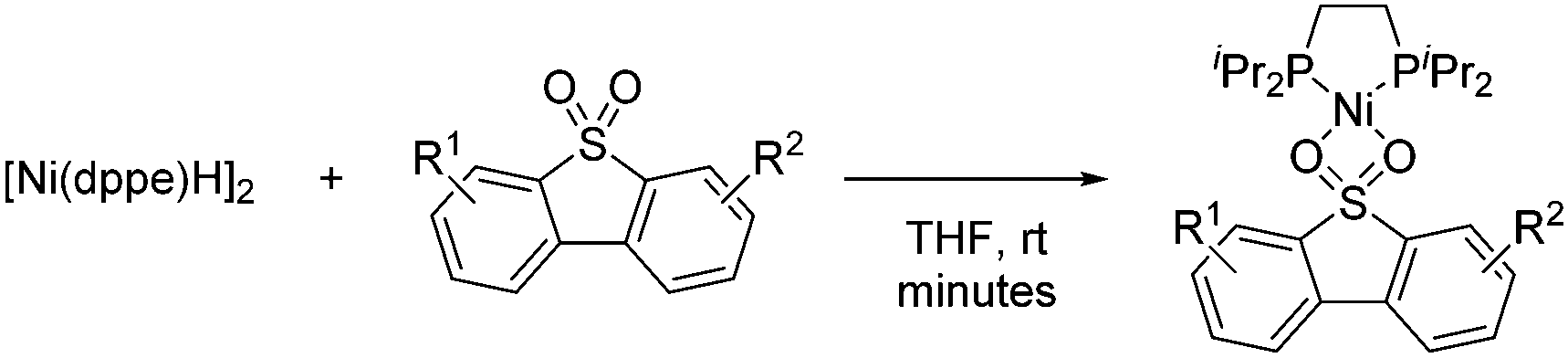 | ||
| Scheme 18 Deoxydesulfurization of sulfones: Ni-based intermediate. | ||
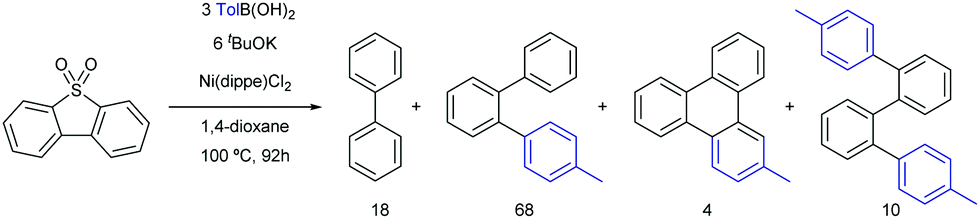 | ||
| Scheme 19 Ni-Catalysed Suzuki cross-couplings of dibenzothiophenesulfone. | ||
Heteroarylsulfones have also proven to participate as coupling partners in Suzuki–Miyaura reactions. In 2005, Robbins reported that purine sulfonyl derivatives can also undergo oxidative cleavage by Pd salts (Scheme 20, above).39 As a part of a broader study in which a fluoride and a thioether were also tested, a sulfonyl purine was arylated using 4-methoxyboronic acid in THF at 60 °C. This temperature was 30 °C lower than that required for the thioether analogue (90 °C). The ability of sulfonyl heterocycles to participate in Pd-catalysed Suzuki cross-couplings was further studied by researchers from AstraZeneca led by Hennessy.40 This team demonstrated that p-tolyl sulfonyl tetraazoles can be used as pronucleophiles with a range of boronic acids (Scheme 20, centre), as well as pinacolboronate esters and trifluoroborate salts. Key to this finding was the use of biaryl phosphines such as S-Phos, BrettPhos, and RuPhos, which provided high conversions to the products where traditional ligands such as PPh3 failed. Notably, the p-tolyl sulfonyl moiety remains intact when conducting Suzuki couplings in the presence of an aryl bromide (Scheme 20, below), opening the door to prior functionalization of compounds based on this scaffold.
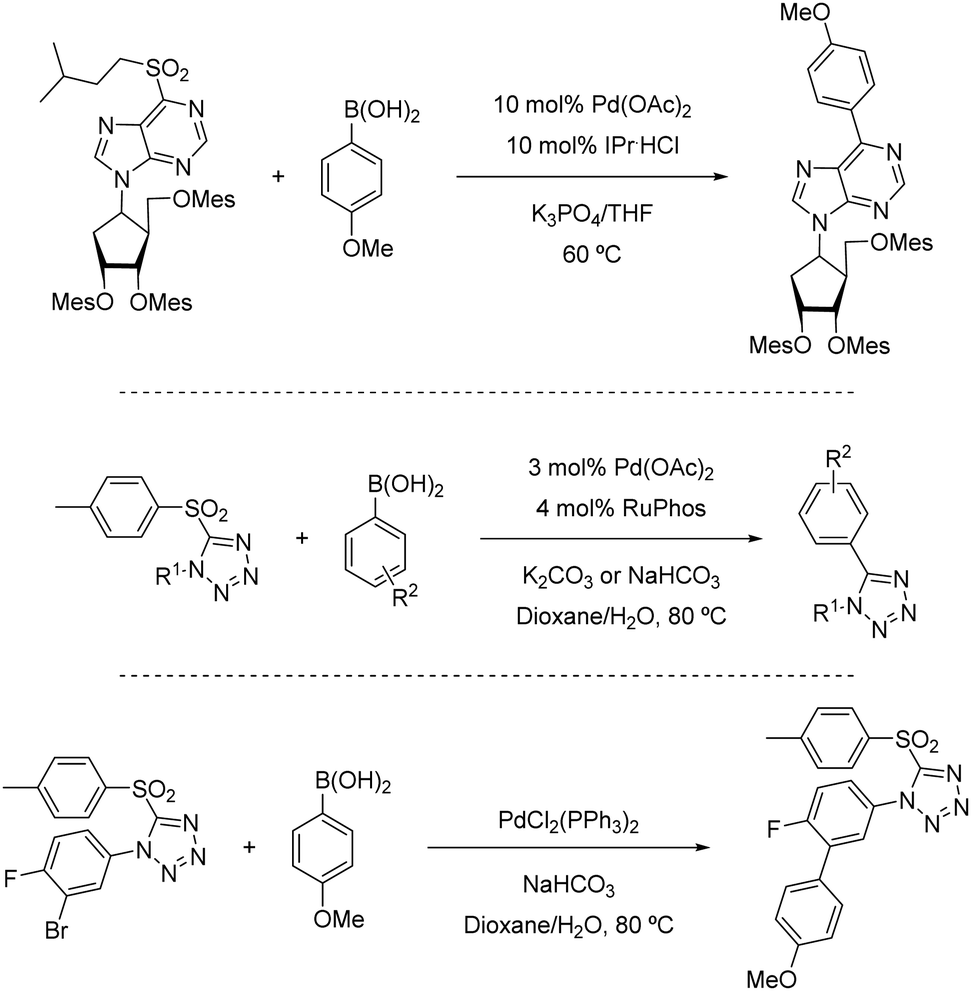 | ||
| Scheme 20 Heterocyclic sulfones in Suzuki cross-couplings. | ||
In 2019, Moran and co-workers explored in detail the ability of the CF3SO2 group to engage in Suzuki cross-couplings.41 Using a slightly higher than usual Pd(OAc)2 loading (5 mol%), a four-fold excess of Buchwald's RuPhos, and DMSO 1% (v/v) to solubilize the inorganic base (3 equivalents of K3PO4), the authors were able to couple multiple arylboronic acids and ArSO2CF3 partners with different electronic and steric properties. The only apparent limitation to the method were boronic acids bearing aldehydes, which in some cases showed modest yields (around 25%). The C(sp2)-S bond shows reactivity in between aryl chlorides (at least two orders of magnitude slower) and nitroarenes/aryl tosylates (at least two orders faster), which was elegantly used as a handle for sequential manipulation (Scheme 21). A series of experimental an DFT studies were conducted to shed light into the reaction mechanism and threw the following conclusions: (1) oxidative addition of the metal into the sulfone C–S bond to be the turnover-limiting step. (2) After this step, a Pd-sulfone bond is formed which quickly rearranges to form a slightly more stable Pd-sulfinate bond. (3) Unlike in the case of sulfonyl chlorides, for which oxidative addition is followed by decomposition to form chloride and sulphur dioxide, in this case the leaving group remains intact as CF3SO2. (4) Oxidative addition into the C–S bond is 5.8 kcal mol−1 higher for PhSO2Ph than it is for PhSO2CF3, which is consistent with the higher polarization of the C–S bond of the latter.
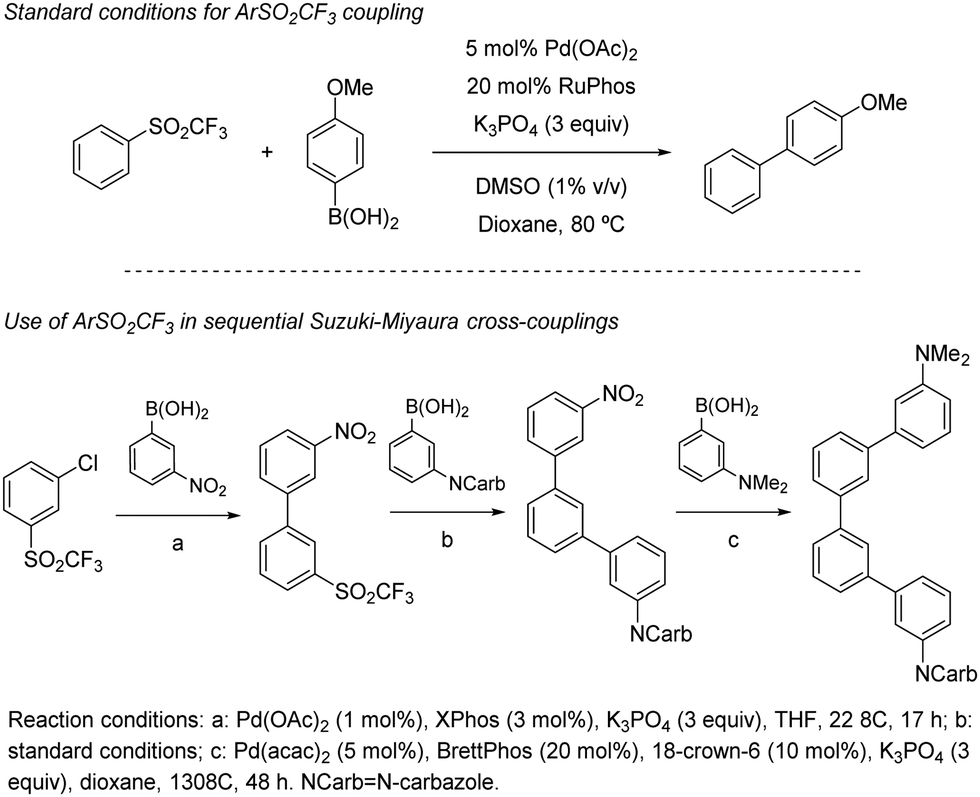 | ||
| Scheme 21 The CF3SO2 group in Suzuki cross-couplings with boronic acids. | ||
Also in 2019, Yorimitsu described an alternative set of conditions to use ArSO2CF3 as coupling partners in Suzuki–Miyaura cross-couplings with boronate esters using bimetallic Pd/Rh co-catalysis.42 While Pd-PEPPSI-IPr alone was able to promote the transformation, the high catalyst loading required (10 mol%) and yield obtained were inefficient, which led the team the explore the use of co-catalysts as a potential solution. After substantial optimization, it was found that a combination of a modified Pd-PEPPSI precatalyst and [Rh(cod)]2 was most effective at promoting the transformation, providing 91% yield for the model cross-coupling. A series of mechanistic studies allowed the formulation of a dual catalytic cycle (Scheme 22) with the following key points: (1) transmetalation between Pd and ArB(neop) would be the turn-over limiting step in the absence of Rh salts. (2) In the presence of [RhCl(cod)]2, however, the reaction was estimated to be almost zero-order in Rh and sulfone, and nearly first-order in Pd, which is consistent with reductive elimination prior to biaryl formation as the turn-over limiting step. This is in contrast with Moran's findings detailed above and was attributed by Yorimitsu to the use of an electron rich IPrNiPr2 carbene ligand that accelerated oxidative addition to the C–S bond.
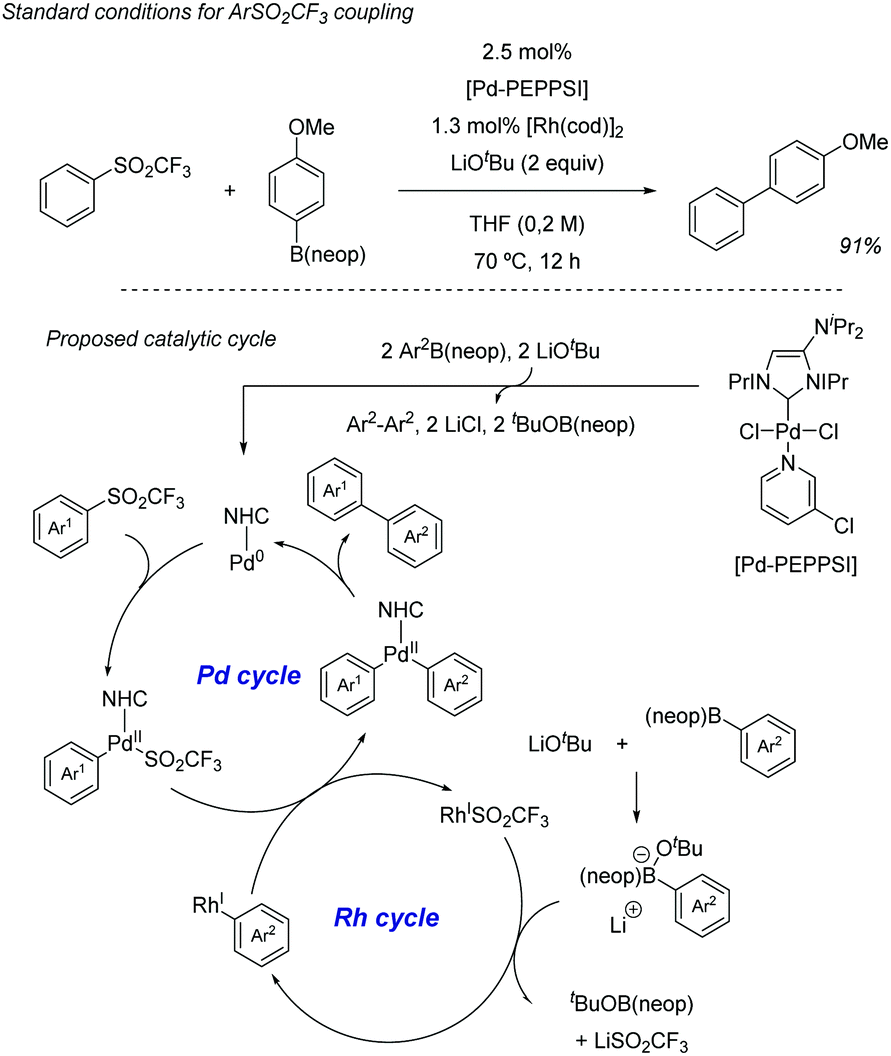 | ||
| Scheme 22 CF3SO2 group in Suzuki cross-couplings with boronate esters. | ||
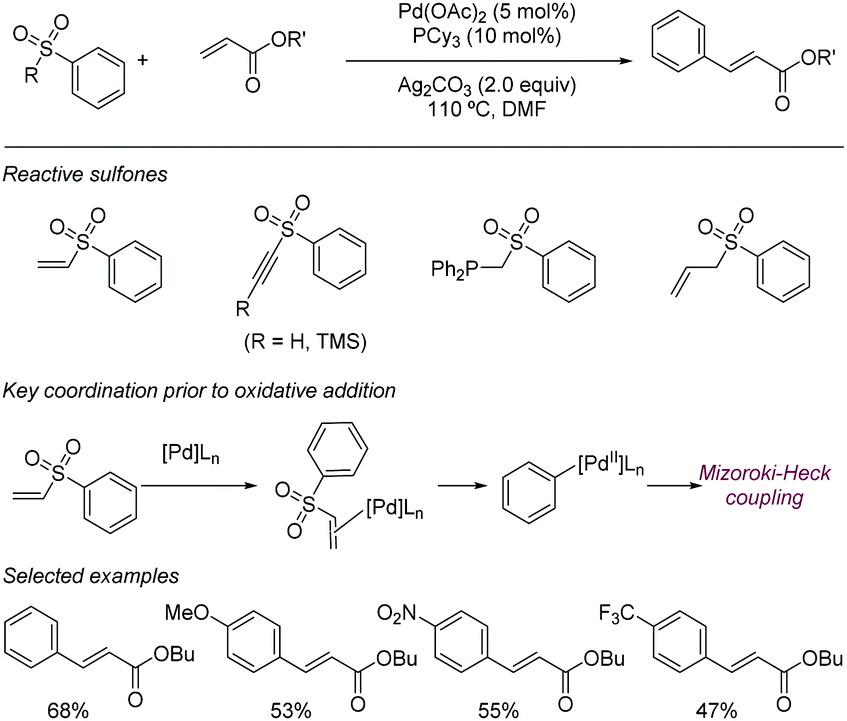 | ||
| Scheme 23 Vinyl sulfones as aryl donors in Mizoroki–Heck reactions. | ||
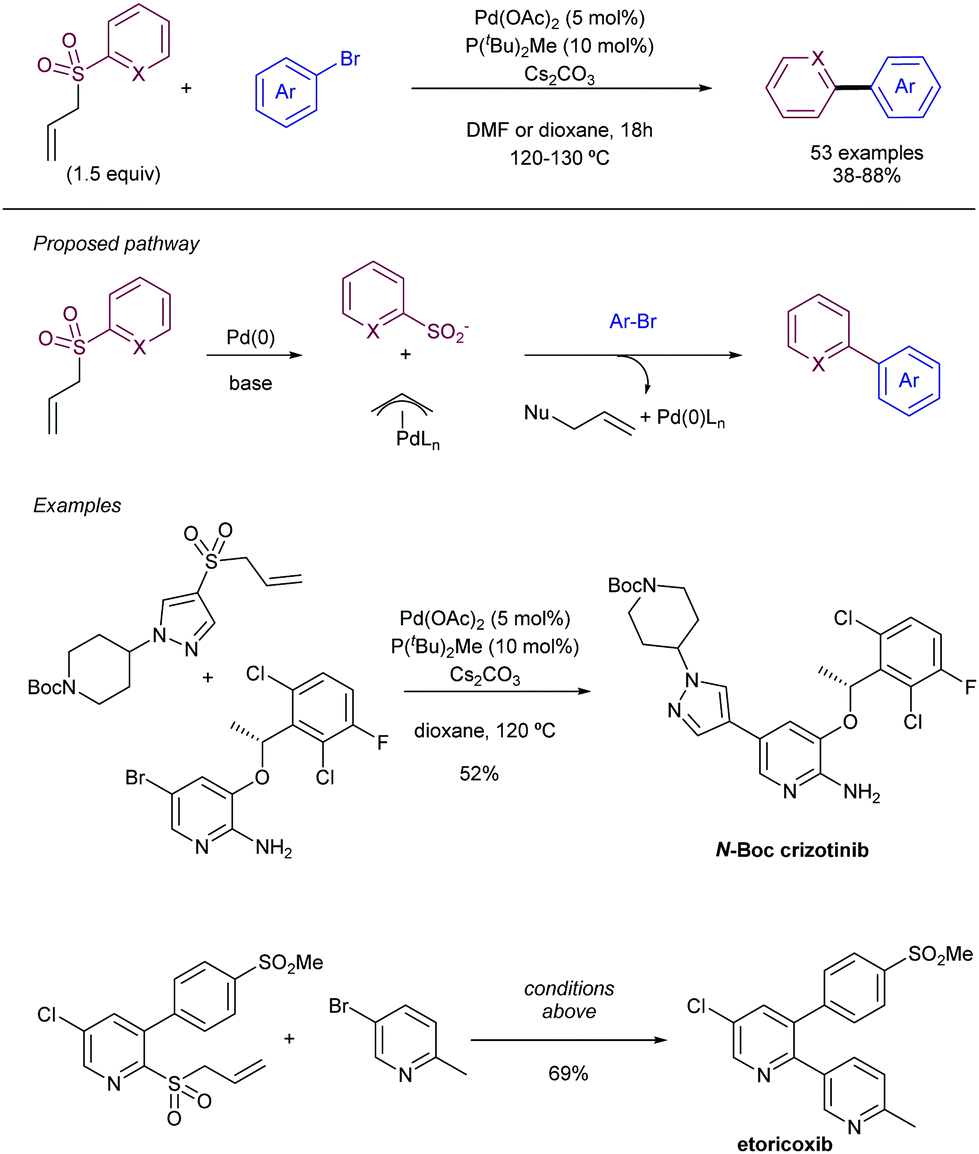 | ||
| Scheme 24 Heteroaryl allyl sulfones as latent sulfinates for cross-coupling with aryl electrophiles. | ||
More recently, the same group has reported on the use of β-nitrile and β-ester sulfones as precursors for the required heteroaryl sulfinate species (Scheme 25).48 In this case, there is a release of the active sulfinate via E1cb elimination under basic conditions. The corresponding cross-coupling product can be obtained under Pd-catalysed conditions in presence of aryl bromides by extrusion of SO2. This approach enables an alternative to the synthesis of allyl heteroaryl sulfones, which could be problematic in some instances. Interestingly, for the β-nitrile sulfones, the use of CataCXium A ligand (PAd2Bu) delivered a better performance in presence of K2CO3 as the base. The addition of acetic acid was found to be beneficial, likely because of the creation of a buffered system that enables the gradual release of the sulfinate species. On the contrary, when β-ester sulfones were employed, the reaction delivered better yields in the absence of the acid. In addition to pyridyl units, other heteroaromatic units such as pyrimidines, pyrazoles, imidazo[1,2-a]-pyrazines, and the electron-rich indoles and pyrazines were efficiently coupled with aryl bromides.
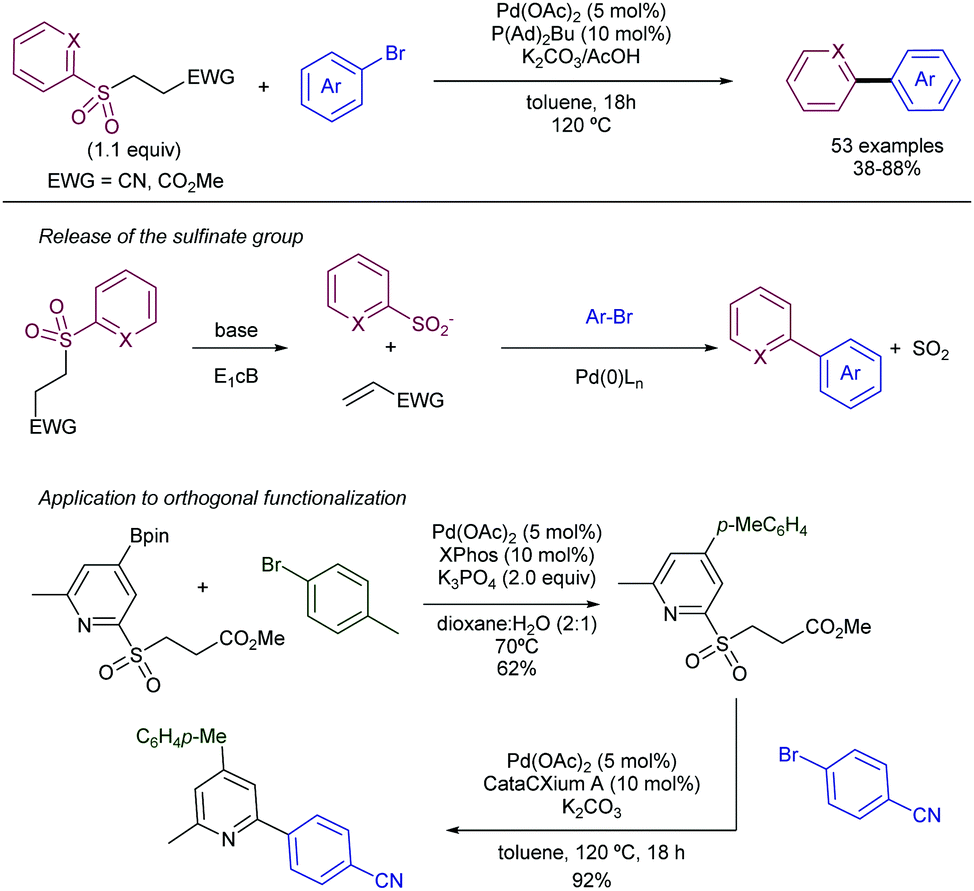 | ||
| Scheme 25 β-Nitrile and β-ester sulfones as precursors for heteroaryl sulfinates. | ||
As an alternative to this strategy, Hu and co-workers have reported on the use of difluoromethyl 2-pyridyl sulfones as electrophiles in the Ni-catalysed cross-coupling with iodoarenes in presence of Zn (Scheme 26).49 The method is based on a first oxidative addition of the fluoromethyl heteroaryl sulfone to an in situ generated Ni(0) complex. Simultaneously the iodoarene reacts with Zn to form an active aryl zinc reagent which transmetallates with the Ni(II) species to deliver the cross-coupling product after reductive elimination. When electron-poor substituents were present at the pyridyl unit, the 2-methyl sulfonyl analogue could be employed as a substrate. For plain pyridyl rings only the introduction of –CF2H and CFH2 groups promoted the reaction, while –CF3 and –CH3 groups were unreactive.
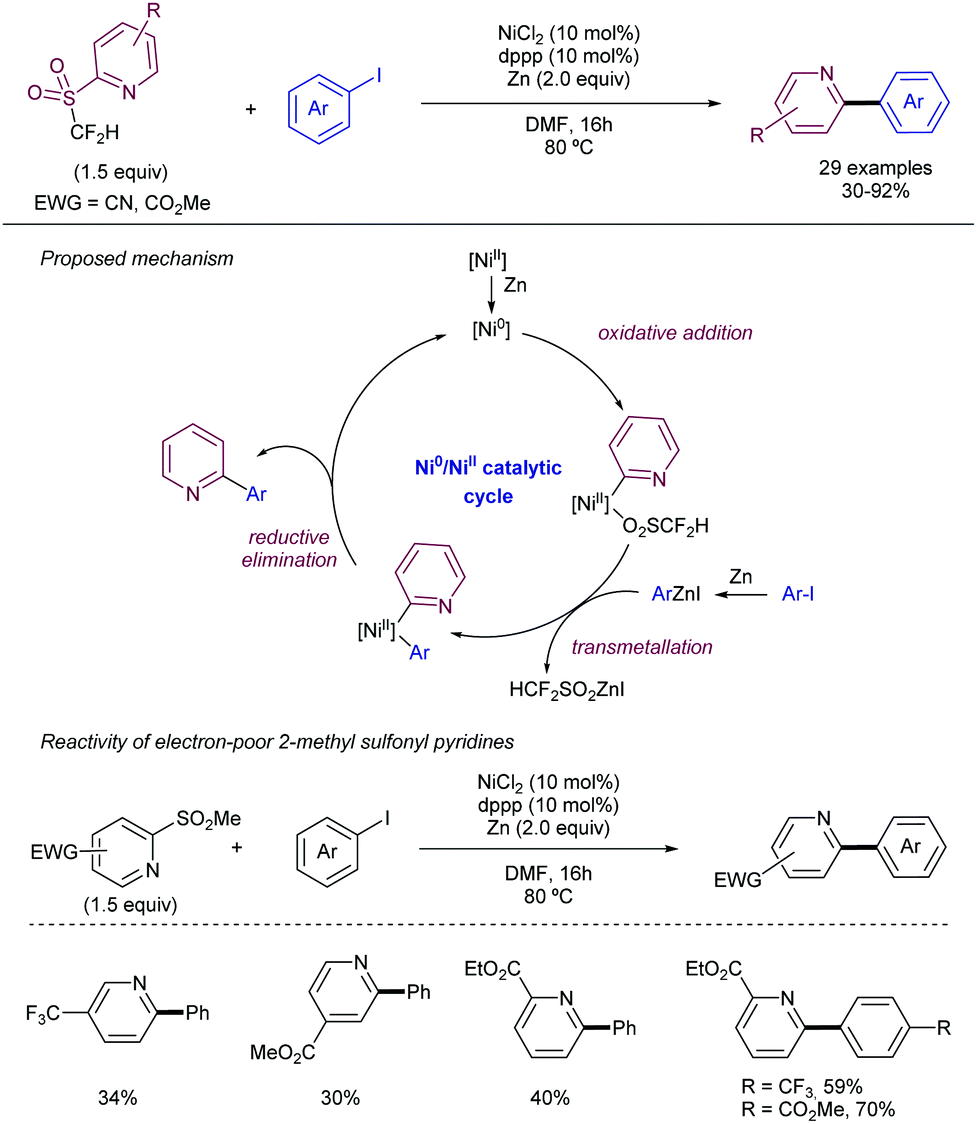 | ||
| Scheme 26 Difluoromethyl 2-pyridyl sulfones in Ni-catalysed cross-couplings. | ||
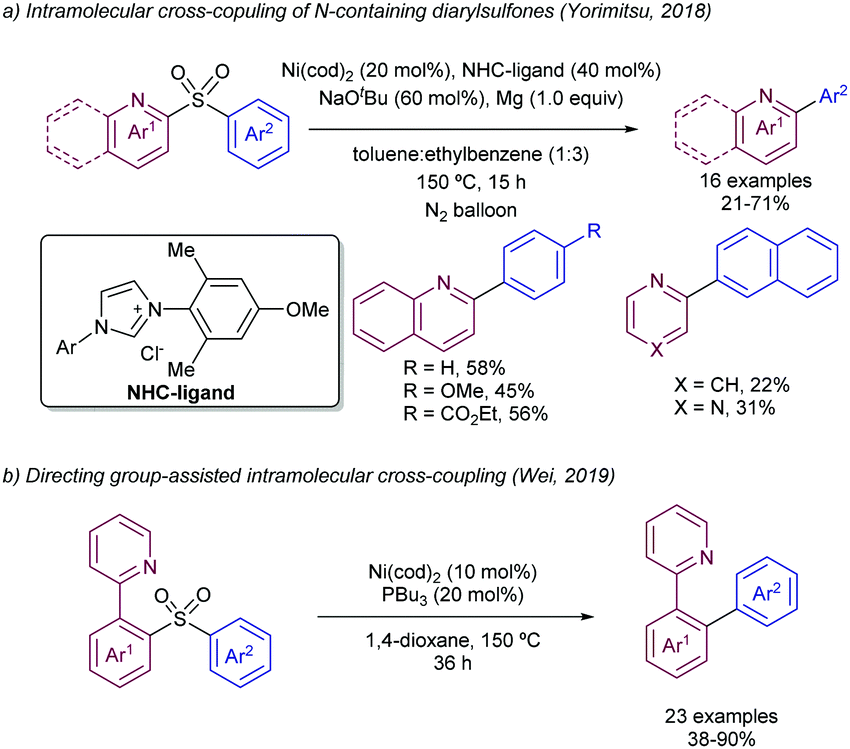 | ||
| Scheme 27 Ni-Catalysed desulfurization of diarylsulfones. | ||
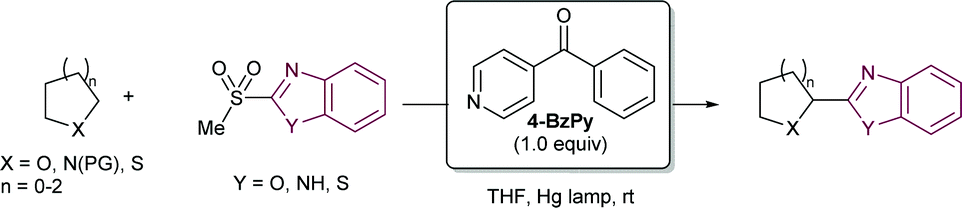 | ||
| Scheme 28 First report on heteroaromatic sulfones as radical acceptors. | ||
For instance, the Molander group pioneered the development of alkyl radical precursors that in combination with heteroaromatic methyl sulfones and a readily available photocatalyst delivered the desired arylated product with high efficiency (Scheme 29).54 The team reported that using alkyl silicates and [Ru(bpy)3]PF6 as photocatalyst in DMF under blue light irradiation, the corresponding Minisci-type products were obtained upon radical addition to methyl sulfonyl heteroarenes. Secondary and primary alkyl radicals were well tolerated, including those bearing potentially reactive functional groups, such as alkenes, esters, perfluoro ethers, alkyl chlorides, unprotected secondary amines, and pyrroles. As an alternative, the team also disclosed that alkyl 1,4-dihydropyridines (DHP) could be employed as surrogates of the alkyl silicates. In this case the use of 4-CzIPN as photosensitizer delivered better results with comparable functional group compatibility. To demonstrate the feasibility of the method the authors reported the selective heteroarylation of a saccharide-DHP derivative, yielding the expected product with complete stereoselectivity in 54% isolated yield. Mechanistic experiments demonstrated that the sulfone partner did not quench the excited photocatalyst, while a dynamic quenching was observed for the DHP. The measurement of redox potentials suggests that the formation of the alkyl radical from either the silicate or the DHP reagents is more favorable. For this reason, the authors proposed a mechanism that starts with the formation of the excited photocatalyst by light absorption, followed by SET to the alkyl radical precursor. The newly formed alkyl radical inserts into the heteroaryl sulfone at the α-position to the sulfone with the concomitant elimination of a sulfinate anion by SET between the transient aryl radical and the reduced photocatalyst. A related strategy was reported by Shirakawa55 using TBHP as radical initiator for the alpha-arylation of aliphatic amines using heteroaryl phenyl sulfones in a radical chain process.
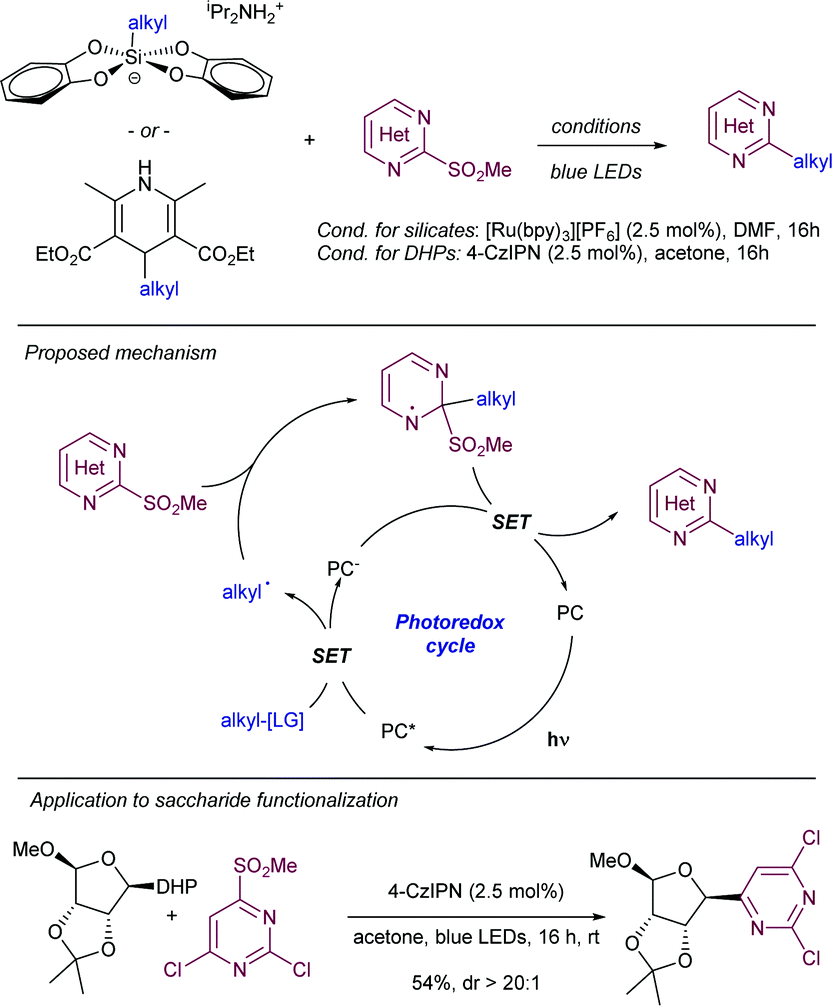 | ||
| Scheme 29 Molander's radical addition to methyl sulfonyl heteroarenes. | ||
More recently, the González–Gómez group has reported56 a similar strategy by the photocatalysed generation of radicals using carboxylic acids and 2-(phenylsulfonyl)benzothiazole. The use of riboflavin tetraacetate was employed as a suitable photosensitizer, thus yielding the corresponding α-oxo and α-aminoarylated products. A related metal-based strategy in which carboxylic acids are employed for the generation of the required radicals has been recently reported by Matsubara and co-workers (Scheme 30).57 The authors disclosed the employment of Ag(I) salts to promote the decarboxylative step. Interestingly, sulfonylated furoxan rings could be functionalized exclusively at the 3-position, even in the presence of disulfonylated substrates, which was rationalized in terms of the stability of the intermediate radicals. The addition of stoichiometric amounts of K2S2O8 was required as a terminal oxidant. The substrate scope was amenable to the introduction of 1°, 2°, and 3° alkyl radicals and those derived from α-stabilized radicals in presence of a heteroatom. Conversely, the method proved to be unsuccessful when α-ketocarboxylic and aryl carboxylic acids were studied.
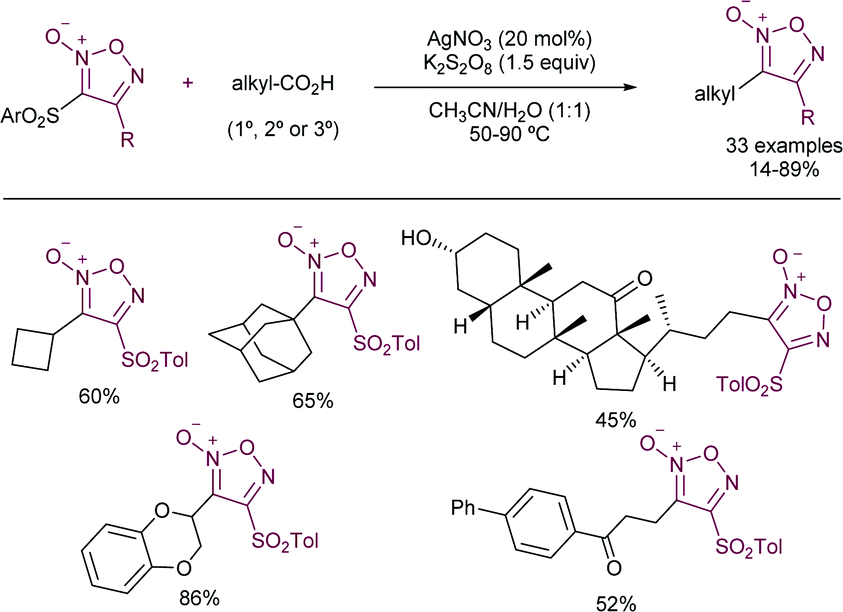 | ||
| Scheme 30 Ag-Based-generation of alkyl radicals from carboxylic acids. | ||
2.3. Alkynylations
Whereas alkynylsulfones have been used for several decades as readily available reagents to install alkynes, efficient catalytic methods have only been recently explored. Traditionally, alkynylsulfones were frequently employed in conjugate additions58 and cycloadditions59 in resemblance to the behavior of alkynoates, owing to the electron-withdrawing effect conferred by the sulfonyl group (Scheme 31a). However, the uniquely pronounced nucleofugacity of the sulfone has also given rise to a wide range of α-carbon substitution reactions, often called “anti-Michael” reactions (Scheme 31b).60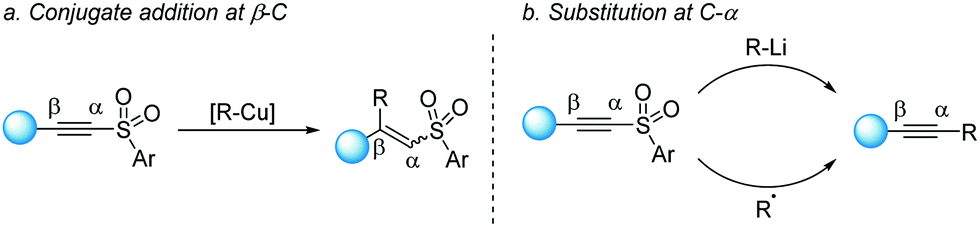 | ||
| Scheme 31 Traditional use of alkynylsulfones in conjugate additions. | ||
While this approach opened an interesting alternative for alkynylation of nucleophiles, the requirement of using strongly reactive organolithium reagents has posed an important limitation to its application. In this regard, in 2013 Xie described a cross-coupling reaction between alkynyl magnesium reagents and alkynyl-SO2pTol by activating the C–S bond through nickel catalysis (Scheme 32).61 Although the reaction was somewhat limited to alkynes with aryl substitution, it provided a promising prospective on the use of milder conditions to make use of sulfones as alkynylating reagents.
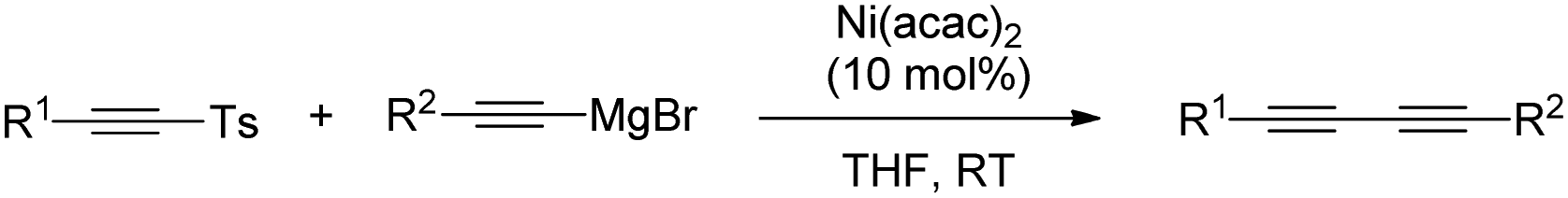 | ||
| Scheme 32 Cross-coupling between alkynyl magnesium reagents and alkynyl-SO2pTol. | ||
Nonetheless, alkynyl transfer reactions via polar mechanisms have received much less attention compared to the undoubtedly more advantageous radical pathways. Since the early works by Fuchs,62 alkynylsulfones have been widely used for its facility to undergo radical fragmentation by releasing sulfinyl radicals, which can promote chain reactions.63 Thus, numerous alkynylation methods have been described to date relying on thermally activated radical initiators, among which AIBN is the most commonly used reaction trigger. Experimental evidence, such as isotopic labelling techniques conducted by several groups,64 have suggested that these reactions take place by a fragmentation mechanism as detailed in Scheme 33.
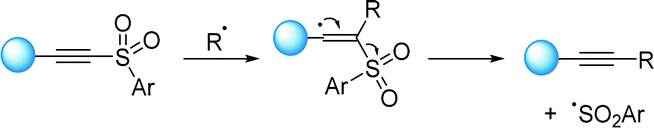 | ||
| Scheme 33 Radical fragmentation with concomitant release of sulfinyl radicals. | ||
In this vein, new catalytic approaches have provided opportunities to improve the efficiency and extend the scope of alkynylation reactions using sulfones. Among those, functionalization of C–C and C–H bonds by attachment of the versatile alkynyl group has received much attention in recent years. In 2016, Zhu's group described a Mn-catalysed oxidative ring-opening reaction of cyclobutanols to afford 1,5-ynones (Scheme 34).65 The method not only rendered high yields for a wide variety of substituents at the cyclobutyl unit, but was compatible with other sulfonyl reagents, namely allyl-SO2Ph and TsCN. The proposed mechanism involves a single electron transfer from the cyclobutanol to the Mn/bipy catalytic system, resulting in an unstable alkoxy radical which undergoes rapid C–C cleavage. As the newly formed radical proceeds to the fragmentation of the alkynylsulfone by releasing a sulfinyl radical, the stoichiometric oxidant ensures catalyst regeneration and oxidation of the latter byproduct.
 | ||
| Scheme 34 Mn-Catalysed oxidative ring-opening reaction of cyclobutanols. | ||
Conversely, the use of reducing conditions leading to alkoxy radical intermediates was reported by Liu and coworkers in the functionalization of readily accessible alkyl peroxides into 1,5-ynol products (Scheme 35).66 In this case, the reaction is initiated by an oxygen atom transfer from the peroxide substrate to an iron-containing porphyrin catalyst. The resulting alkoxy radical then undergoes intramolecular 1,5-hydrogen atom transfer, followed by reaction with the radical acceptor, while LiBH4 reduces the oxygenated iron complex to allow catalytic turnover. The method proved to be effective for all prim-, sec-, and tert-alkyl peroxides in remarkably short reaction times using alkynyl triflones.
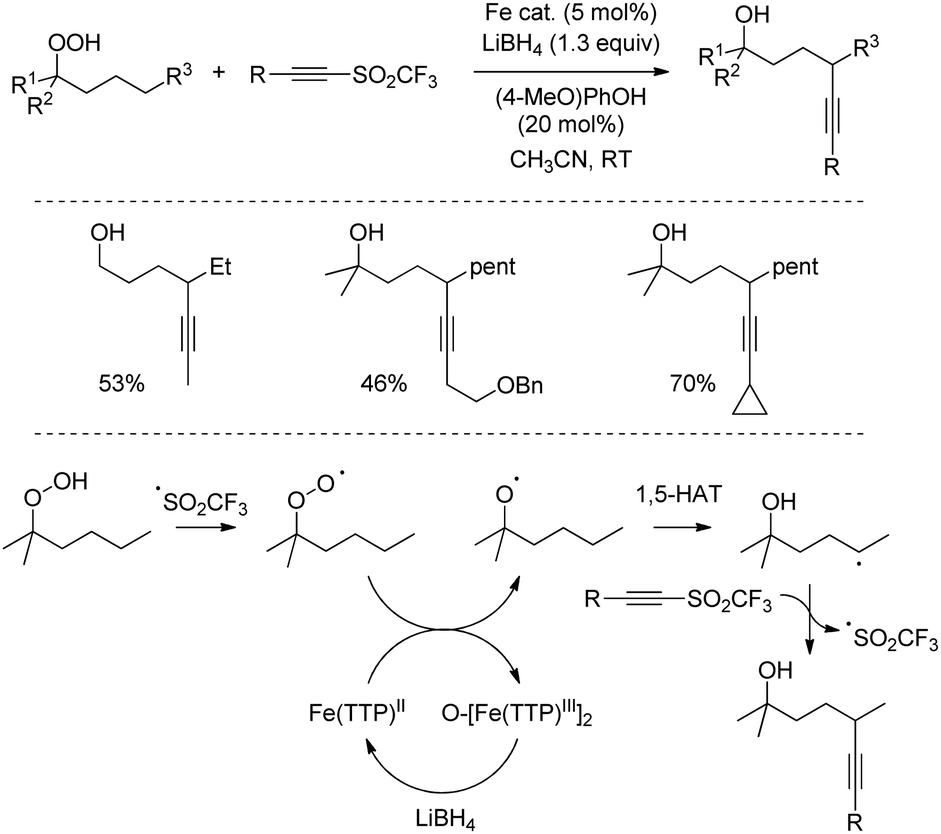 | ||
| Scheme 35 Fe-Catalysed alkynylation of alkyl peroxides into 1,5-ynols. | ||
The versatility of remote alkynylation by 1,5-hydrogen atom transfer was further illustrated by Wu's group in 2019 (Scheme 36).67 By using a Cu/bathocuproine catalyst, authors described a practical reaction of N-fluorinated alkylamines with alkynyl triflones to furnish alkyne appended amines. Optimal conditions for the reaction were found by using inexpensive ethyl acetate as solvent and LiOtBu, although milder bases such as Li2CO3 also provided practical yields. Other aryl sulfones were also suitable for the reaction as leaving group, whereas alkynyl iodide, a common electrophilic alkyne, only afforded 30% of alkynylation product, highlighting the advantage of using alkynylsulfones.
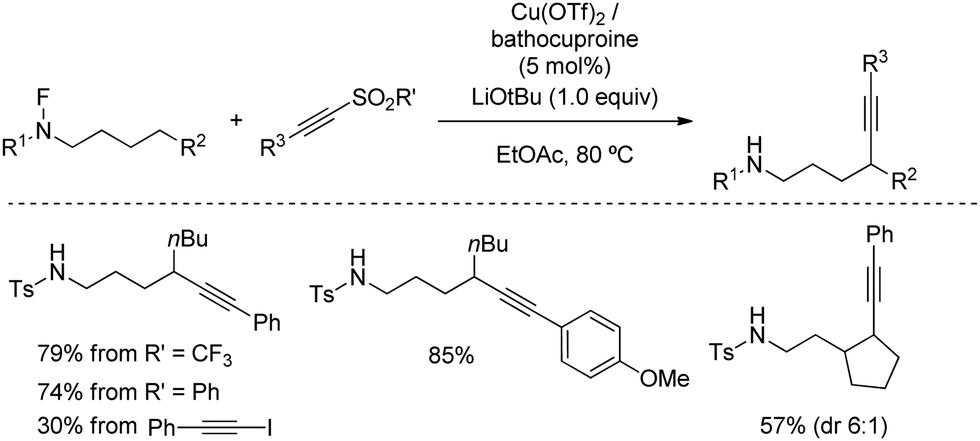 | ||
| Scheme 36 Remote alkynylation by HAT N-fluorinated alkylamines with alkynyl triflones. | ||
Several alkynylation via radical additions based on sulfone reagents have also been recently studied by modern techniques. In 2017, Li's group described a metal-free catalytic tri-component 1,2-trifluoromethylalkynylation of alkenes by reaction with tosylalkynes and Togni's reagent as the source of –CF3 (Scheme 37).68 The catalyst, 2,4,6-trimethylpyridine (TMP), was proposed to activate I(III) reagent through the formation of a donor-acceptor complex, which readily undergoes homolytic cleavage at the reaction temperature to generate the CF3 radical. The reaction then proceeds with a radical addition of CF3 to the alkene, followed by coupling with the alkynylsulfone via addition/elimination. The described protocol tolerated a wide range of cyclic and acyclic alkenes equipped with useful functional groups, including gem-disubstituted alkenes leading to the formation of quaternary propargylic carbon in high yields.
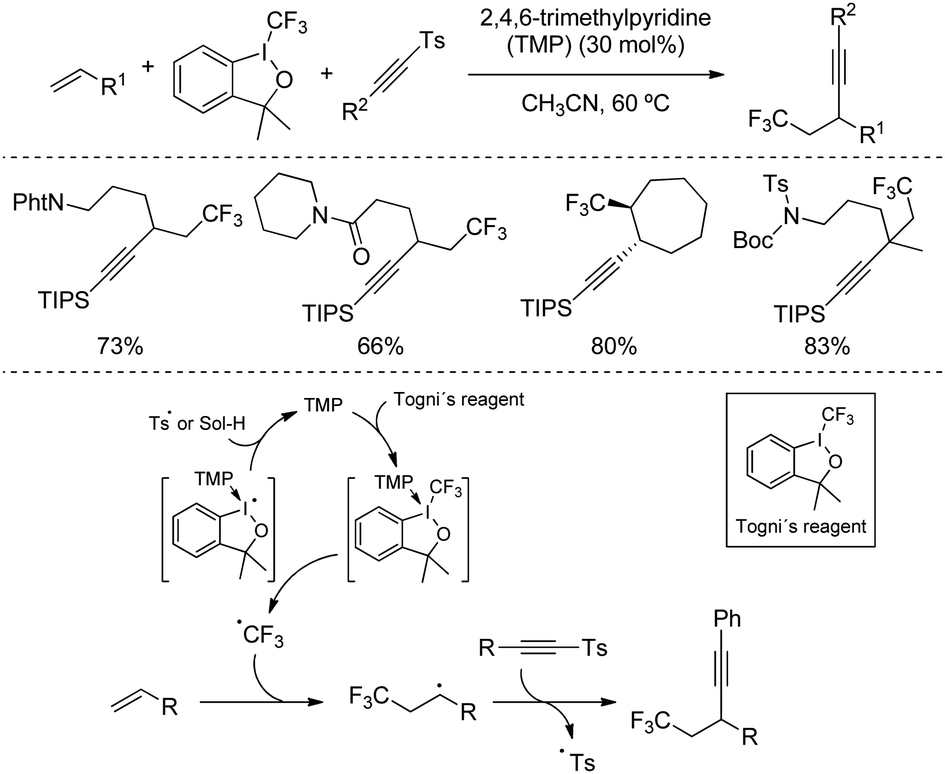 | ||
| Scheme 37 Metal-free catalytic tri-component 1,2-trifluoromethylalkynylation of alkenes. | ||
The arylsulfinyl radical byproduct is a relatively stable species, which is believed to be either consumed in catalyst regeneration or involved in the propagation of chain reactions. On the contrary, sulfinyl radicals bearing halogenated substituents are less stable and tend to decompose into SO2 and an alkyl radical species. As a result, alkynyl sulfones bearing CF3 groups at sulfur have shown the potential to transfer both substituents. Concurrent to Li's work described above, Yu and coworkers reported an alkene trifluoromethylalkynylation method based on a similar Lewis base activation (Scheme 38).69 By leveraging the ability of alkynyl triflones to transfer both functionalities, the reaction took place in high yields using a catalytic amount of the Togni's reagent.
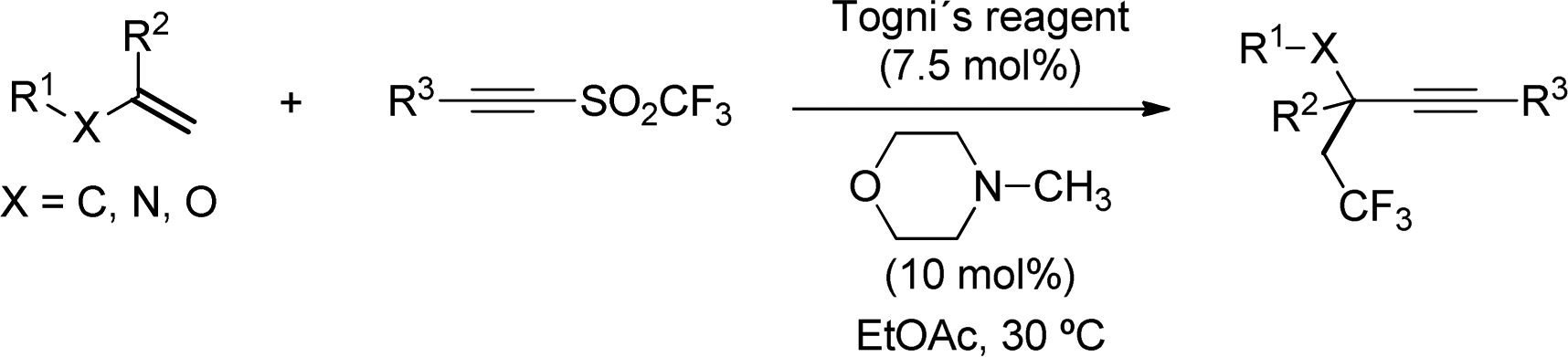 | ||
| Scheme 38 Lewis-base catalysed trifluoromethylalkynylation of alkenes. | ||
The transference of both substituents from an alkynyl triflone was further exploited by Qiang Zhu and coworkers for the 1,1-trifluoromethylalkynylation of isocyanides (Scheme 39a).70 Among other metal catalysts, Cu(OAc)2 turned to be the most effective at reducing the sulfone bond to release the CF3 radical and initiate the radical addition. Control experiments suggested the presence of isocyanide was critical to trigger the SET from the copper catalyst. The reaction displayed compatibility with a number of N-alkyl substitutions, as well as a wide scope of aryl alkynes. Furthermore, its synthetic utility was demonstrated by the preparation of the commercial anti-inflammatory drug Celecoxib in a one-pot procedure (Scheme 39b).
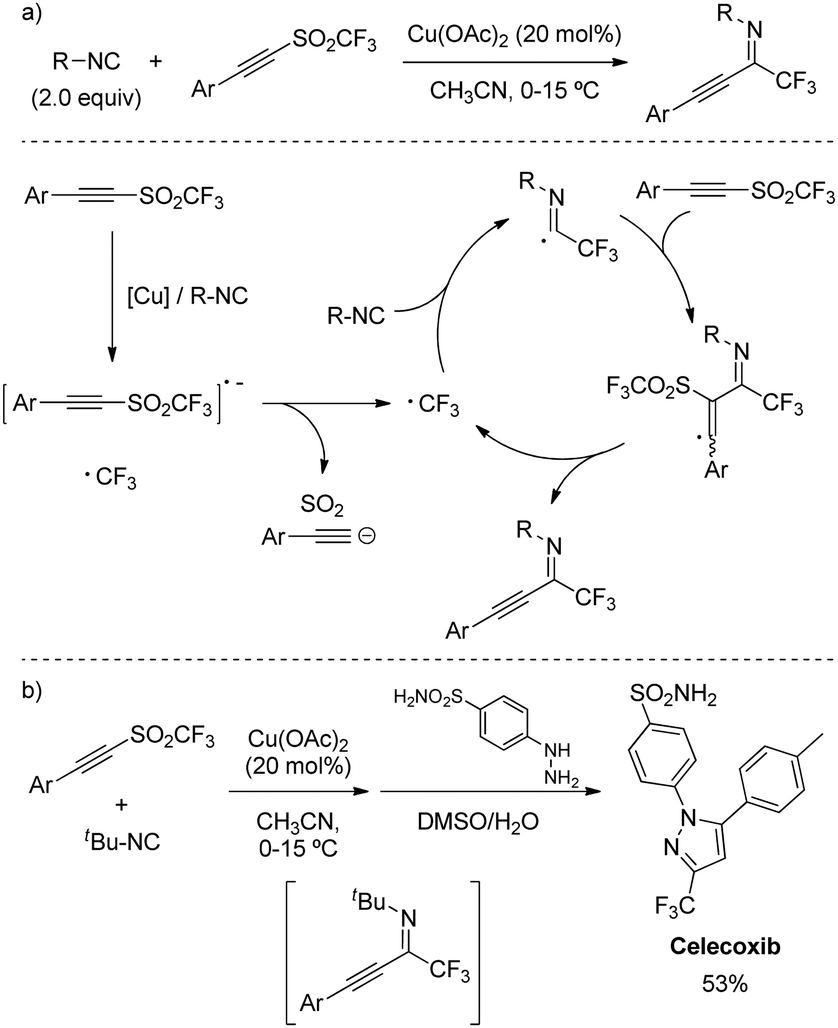 | ||
| Scheme 39 1,1-Trifluoromethylalkynylation of isocyanides from alkynyl triflones. | ||
More recently, a Cu-catalysed alkene 1,2-difunctionalization protocol was disclosed by Chen Zhu and coworkers to introduce both the alkynyl and a dihalocarboxylate group from a single sulfone reagent (Scheme 40).71 Although the reaction was limited to monosubstituted alkenes, a useful scope with ample variety of styrenes and alkyl olefins were provided in good yields. This included an impressive array of alkenes appended to bioactive structures which were found to successfully undergo radical addition. Interestingly, the C–X substitution at the product depended on the halogen atom used in the solvent molecule, offering a facile means of product divergency. The mechanism of the reaction was proposed to start through a halogen atom transfer via Cu(I/II) oxidation. Addition of the resulting α-carbonyl radical to the alkene followed an intramolecular alkynylation and extrusion of SO2 would result in a second α-carbonyl radical intermediate, which undergoes a halogen atom transfer with the solvent.
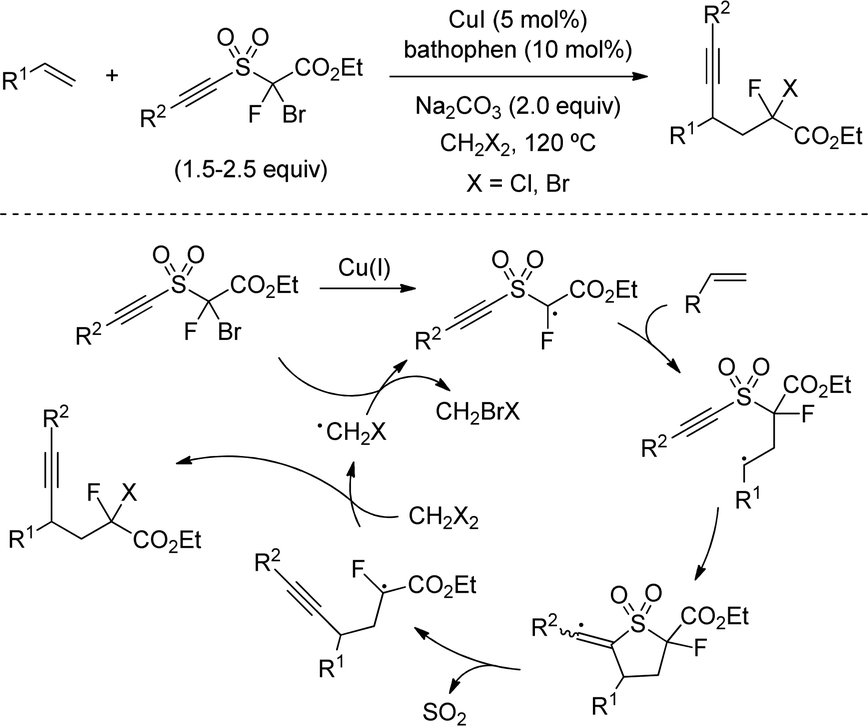 | ||
| Scheme 40 Cu-Catalysed alkene 1,2-difunctionalization using alpha-haloalkyl alkynyl sulfones. | ||
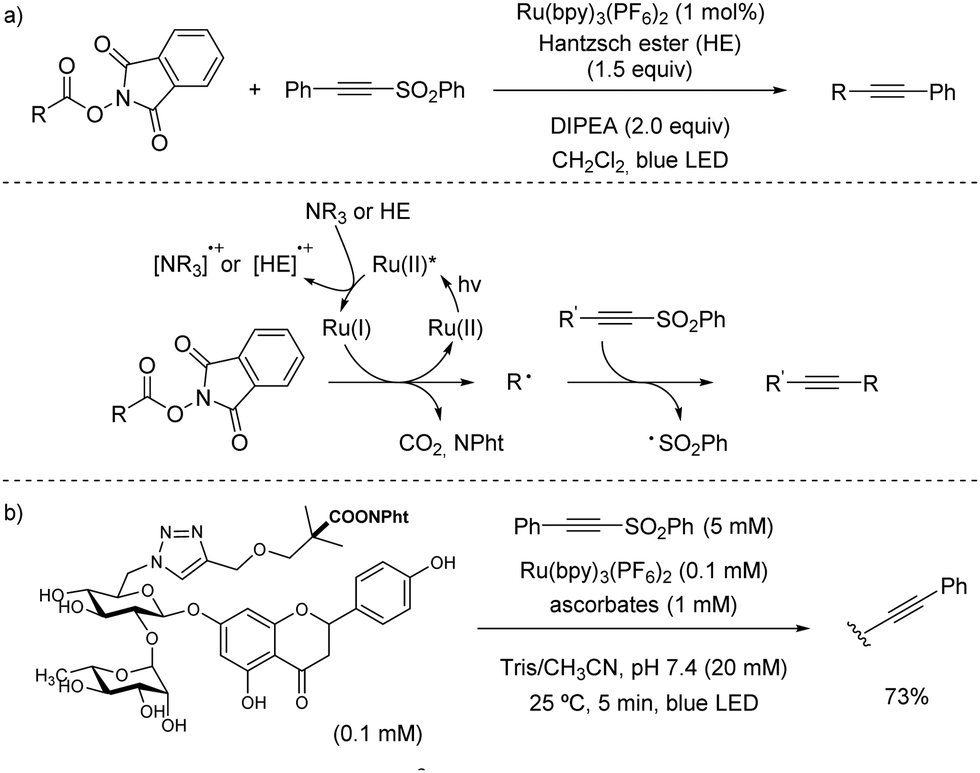 | ||
| Scheme 41 Ru-Catalysed C(sp3)–C(sp) bond coupling reaction through decarboxylation of N-acyloxyphthalimides. | ||
A similar decarboxylation strategy based on Ru-catalysis was employed by Fu for the alkynylation of aspartic and glutamic acid derivatives activated by NHPI.73 In addition, the same group described a procedure for the synthesis of cyclopentenes by using redox active diadipates as 1,4-butyl diradical synthons (Scheme 42).74 Although the cyclization could be achieved with practical yields, the delivery of trisubstituted alkenes generally resulted in equal mixtures of E/Z isomers.
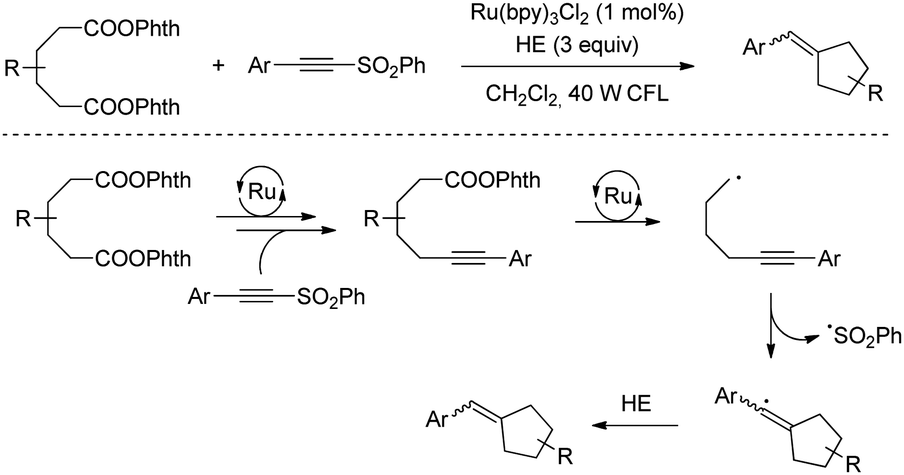 | ||
| Scheme 42 Redox active diadipates as 1,4-butyl diradical synthons synthesis of cyclopentenes by using. | ||
Alternatively, König and Schwarz reported the use of eosin Y as a photosensitizer to catalyse the decarboxylative alkynylation focusing on biomolecules (Scheme 43).75 The reaction scope encompassed amino acids, fatty acids, and steroid derivatives, which underwent alkynylation from moderate to high yields, serving as an effective means to valorize widely available biomass-derived compounds.
 | ||
| Scheme 43 Eosyn Y catalysed decarboxylative alkynylation of bioactive compounds. | ||
Redox active oxalates are useful synthetic devices to trigger dehydroxylation following a closely related strategy to decarboxylation. In 2016 Fu's group described a protocol for the alkynylation of tertiary alcohols activated in the form of N-phthalimidoyl oxalates. By using a Ru(bpy)32+ catalyst and CFL as the source of light, a number of highly congested tertiary alkyls were successfully alkynylated in good yields (Scheme 44).76
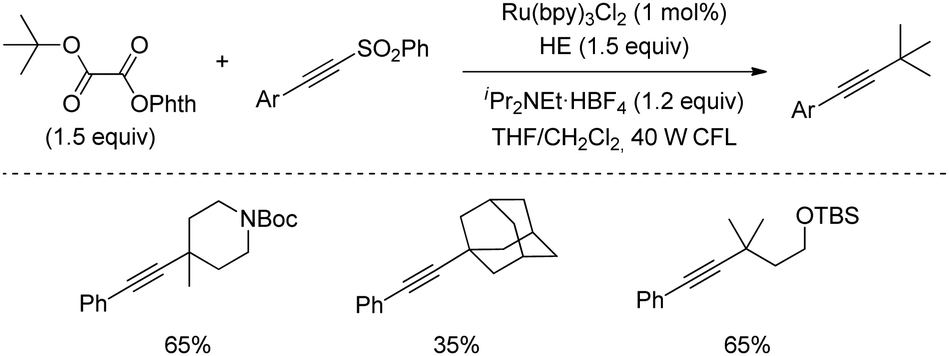 | ||
| Scheme 44 Alkynylation of tertiary alcohols using redox active oxalates. | ||
Alkylamines, on the other hand, have been used for C–C coupling with alkynylsulfones through derivatization into redox active Katritzky's pyridinium salts. In 2018, an eosin Y-catalysed method was disclosed by Gryko featuring a robust and scalable reaction (Scheme 45).77 Secondary alkyls and benzylic groups rendered the alkyne product with excellent yields being compatible with substituents of increasingly structural complexity, however, primary alkyl pyridinium salts displayed low yields. A detailed mechanistic study confirmed an oxidative quenching cycle was operative between the eosin Y catalyst and the pyridinium salt. Furthermore, the reaction rate was highly dependent on the stability of the radical intermediate, which explains the lower coupling efficiency with primary alkyls.
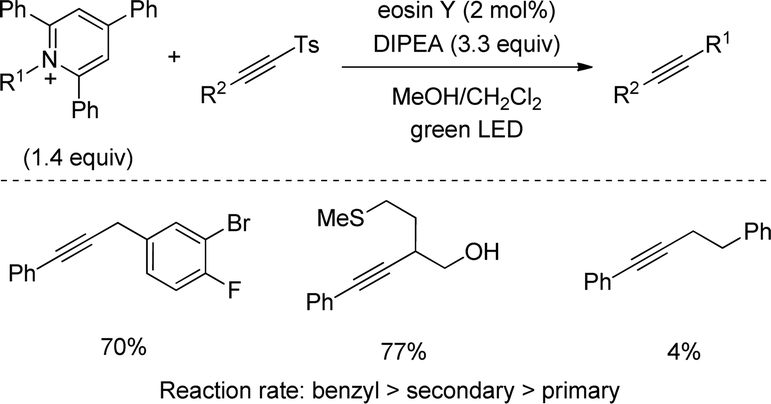 | ||
| Scheme 45 C–C coupling with alkynylsulfones and redox active Katritzky's pyridinium salts. | ||
In 2013, Inoue reported on the use of benzophenone to photochemically induce cross coupling of alkynylsulfones with amines and ethers by hydrogen atom abstraction at the alpha-carbon.78 Although the photosensitizer operated catalytically, 0.5 to 1 equivalent of benzophenone was required for the reaction to take place optimally, in addition to the use of UV light. In this regard, Paul and Guin improved the efficiency of this reaction by employing 4,4′-dichlorobenzophenone as catalyst under CFL light (Scheme 46).79 Authors proposed that a high yielding reaction despite the low absorption of the catalyst in the visible region was possible owing to the competitive radical chain mechanism caused by the residual sulfinyl radical.
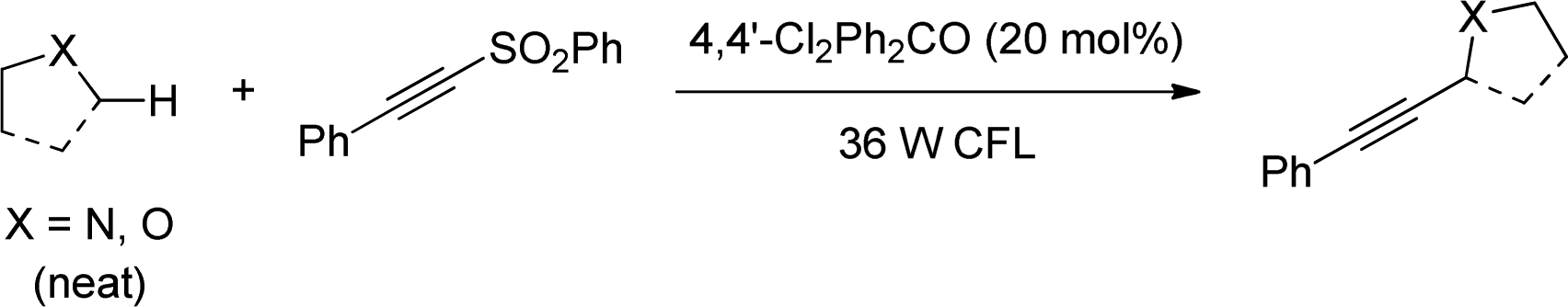 | ||
| Scheme 46 Cross coupling of alkynylsulfones with amines and ethers by hydrogen atom abstraction. | ||
In a very recent work from Capaldo, a closely related C–H alkynylation was reported driven by a decatungstate catalyst (TBADT) and 390 nm LED light. Contrary to previous examples, this method proceeds with lower quantities of the H-donor and a broader scope, including aldehydes and non-stabilized sec- and tert-alkyl groups (Scheme 47).80
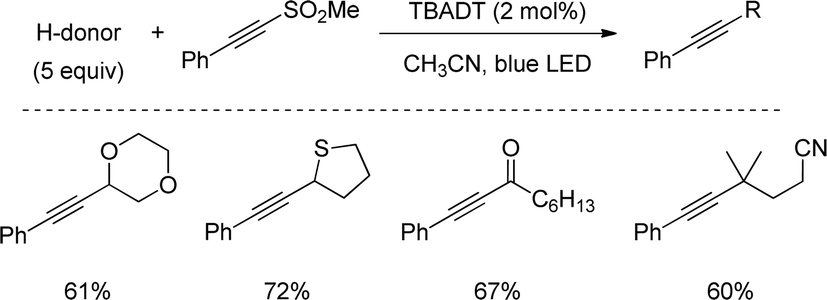 | ||
| Scheme 47 Decatungstate catalysed C–H alkynylations. | ||
2.4. Allylations
The introduction of allyl-type functionalities in a rapid and controlled manner is a highly desirable goal in synthetic chemistry, because of its chemical versatility.81 For instance, the allylic fragment imparts the ability to create a new chemical space upon selective modifications of this group, such as the access to electrophilic functions by oxidation. This chemistry is exemplified by the rapid access to carbonyl groups and their extensive and transformative chemistry. Other important processes include the post-modification of allyl fragments via alkene-transformative reactions. A salient example of this reactivity pertains to the emergence of metathesis, in which a newly unsaturated system can be conceived by employing two previously existing double bonds.82 These two important examples – among others – highlight the constant interest in developing novel synthetic approaches for the introduction of allyl groups in a versatile, modulable, and selective fashion. This section is devoted to the use of allyl sulfones for this purpose including both metal and photocatalytic methods.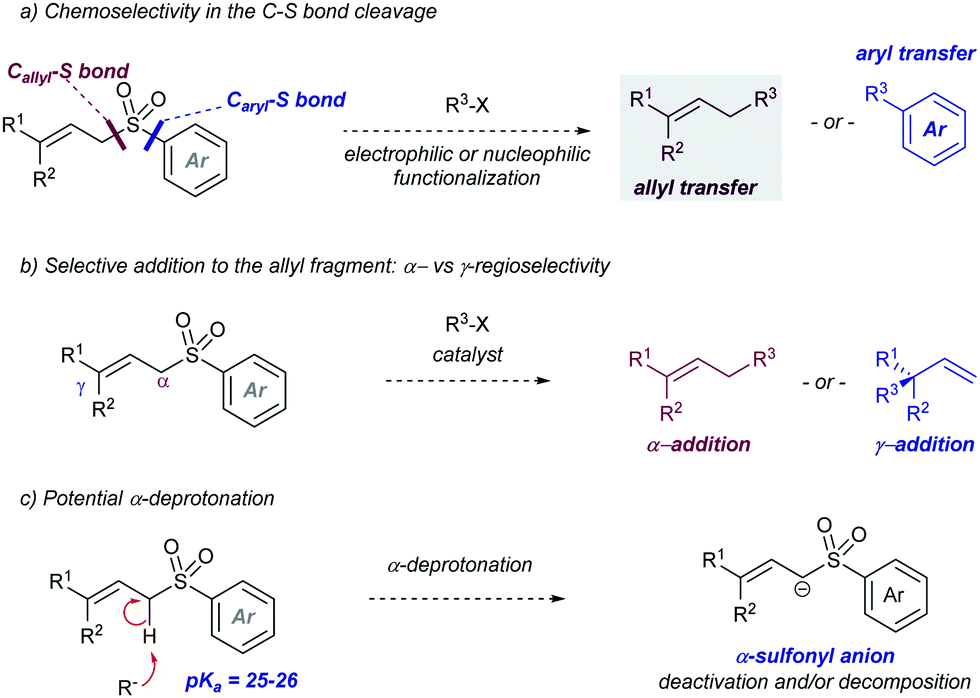 | ||
| Scheme 48 Selectivity issues to address when using allyl aryl sulfones as allylating partners. | ||
2.4.1.1. Cu-Catalysed allylic substitution of sulfones. The use of allylsulfones as electrophilic allylating reagents was firstly described by Julia and co-workers83 employing copper catalysis in presence Grignard reagents as nucleophiles. One important feature of this reaction pertains to the effect of the α- and/or γ-substitution using allyl phenylsufones, and the stereochemistry of the C–C double bond as well. From a global point of view, the reaction is generally controlled by steric factors, resulting in the allylation of the Grignard reagent at the less sterically hindered position of the allyl fragment. As expected, the double substitution at the γ-position typically results in a depletion of the reactivity. However, further studies developed by Bäckvall and co-workers84 revealed a dependence between the nuclearity of the intermediate copper species and the observed α- or γ-attack for a different arrange of allylic substrates. Regarding the reactivity of allylsulfones (Table 1) the authors demonstrated that under reactions conditions for the generation of mono-alkylcopper reactive species [RCuX]−, the γ-addition was favored. In contrast, when di-alkylcuprate species of the type [R2Cu]− are the productive intermediates, the α-addition is facilitated.85 This fact explains that under catalytic conditions, in which a Grignard reagent is mixed with the substrate with catalytic amounts of copper, the γ-product is typically observed which results from the addition of [RCuX]−. This behavior has also been explored for other Cu-catalysed allylic substitution reactions.86
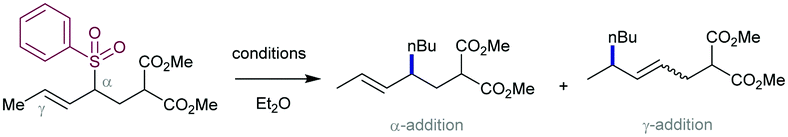

The stereoselectivity of the newly C–C bond formed in the reaction depends on the α- or γ-attack by the Grignard reagent (Scheme 49a). Under catalytic conditions for promoting the α-attack (entries 1 and 2), the E/Z ratios of the final products were higher than those obtained for the γ-attack (entries 3 and 4). However, Trost and co-workers87 have reported that high levels of acyclic diastereocontrol can be achieved when an existing element of stereocontrol is present in the molecule. For instance, the reaction between silylated γ-hydroxy allyl phenylsulfones with either alkyl or phenyl Grignard reagents under CuCN catalysis affords the corresponding products with complete γ-regio- and E-stereoselectivity (Scheme 49b). These conditions have been also applied to prepare an advanced intermediate in the synthesis of an alkyl side-chain of α-tocoferol.88
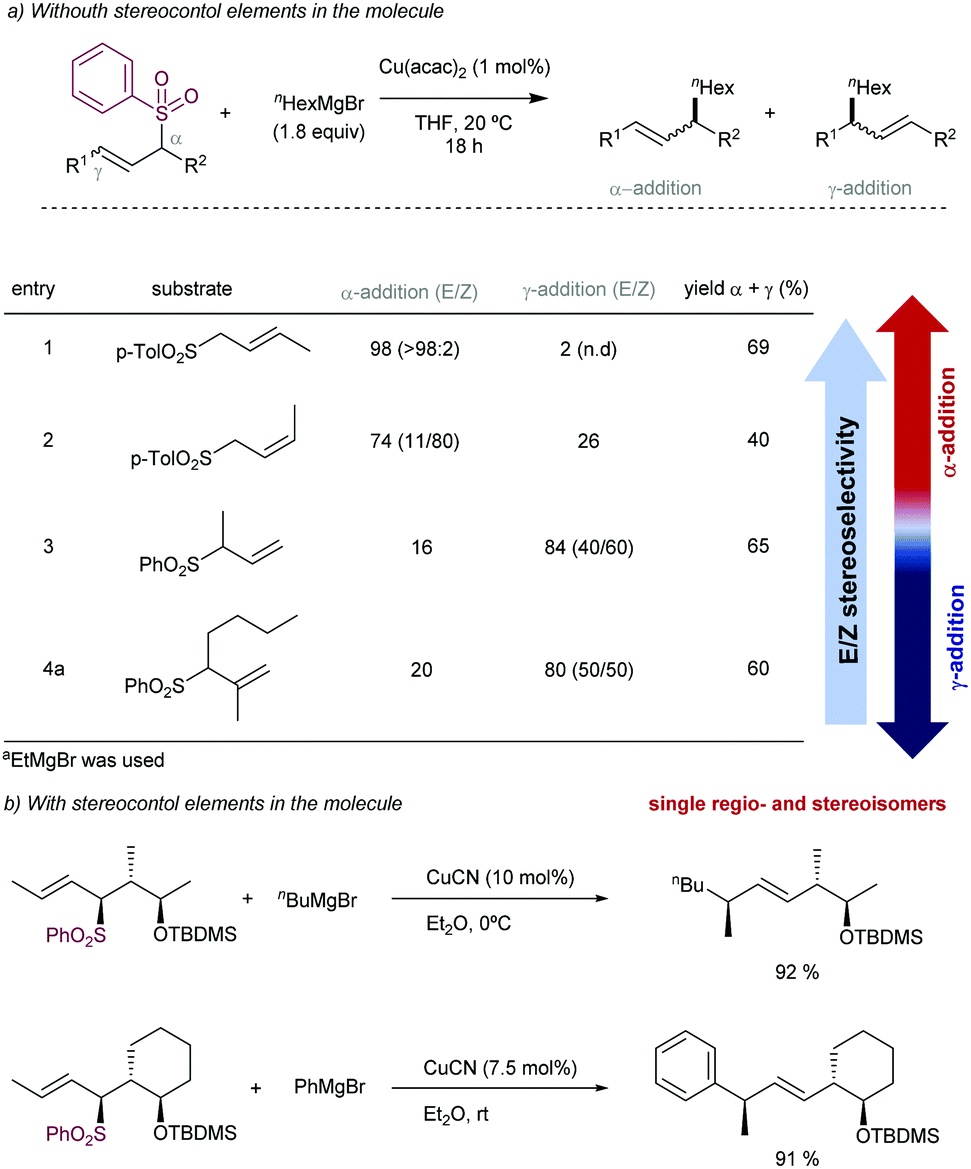 | ||
| Scheme 49 Effect of the stereoselectivity in the Cu-catalysed allylic substitution of allyl sulfones and Grignard reagents. | ||
As mentioned above, the acidity of allylsulfones is relatively high (pKa(DMSO) = 25–26), which can be a potential problem in presence of Grignard reagents. Thus, an important factor to be addressed in this reaction is the competition between deprotonation at the α-position of the allyl sulfone and the cross-coupling event. To stablish conditions to avoid the negative impact of the deprotonation, several factors were studied in combination with a variety of allyl phenylsulfones and different alkyl Grignard reagents under copper catalysis. The effect of the counter-anion in the Grignard reagent was important, with more electronegative chloride being more reactive than bromide. The catalytic loading was also important since an increased amount of copper catalyst accelerates the cross-coupling reaction. This side-reaction was deemed responsible for the lower yields obtained using allyl sulfones with acidic protons at the α-position, as the reaction with α,α-disubstituted allyl sulfones proceeds typically with higher yields.
In addition to Grignard reagents, Dzhemilev and co-workers89 described the use of dialkylmagnesium compounds as nucleophilic partners in this reaction (Scheme 50). Copper salts demonstrated superior catalytic activity over other metals salts (including Fe, Mn, Ni, Ti, Zr, Pd and Cr), in combination with PPh3 as ligand (N-donor ligands such as 2,2′-bipyridine, and other P-donor ligands such as trialkylphosphines and phosphites reduced the efficiency). High reaction yields were obtained for different symmetrical dialkylmagnesium compounds (90–98%). Unsymmetrical aryl alkylmagnesium reagents delivered the corresponding allylated product with high yield, but mixtures of aryl- and alkyl- allylated products were formed, favoring the transfer of the alkyl chain (Scheme 50b). Additionally, trialkylalanes or dialkylaluminium chlorides have been studied as nucleophiles in this reaction.90
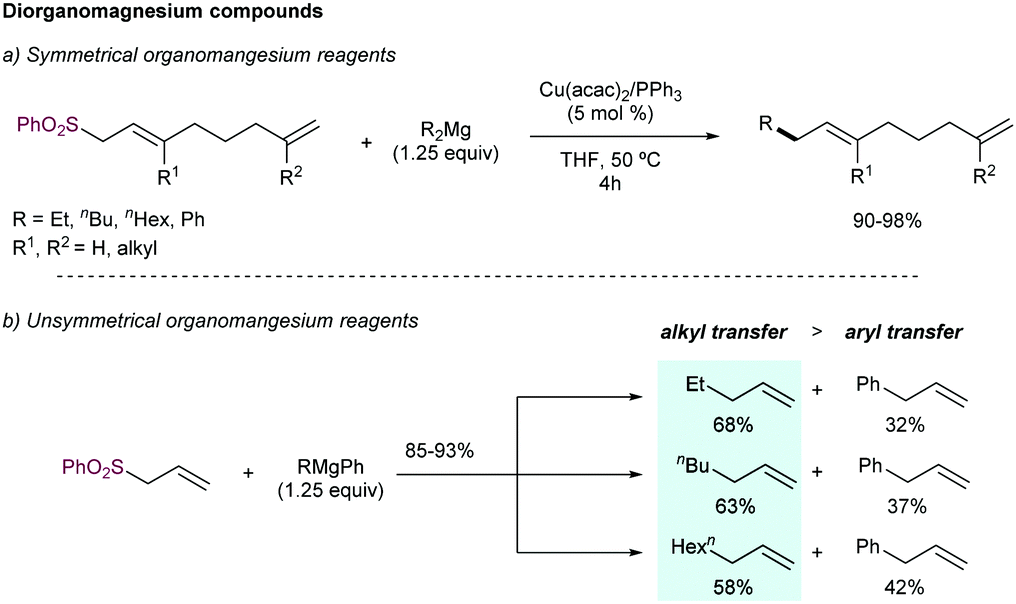 | ||
| Scheme 50 Use of diorganomagnesium reagents as nucleophiles in Cu-catalysed allylic substitution of allyl sulfones. | ||
The examples above show that steric hindrance around the C–C double bond of the allylic fragment negatively impacts the reaction outcome, likely because the coordination of copper species to the alkene is hindered. In order to solve this problem, our research group91 described in 2004 that the presence of a coordinating 2-pyridylsulfonyl (SO2Py) group is responsible of an increased reactivity in the Cu-catalysed substitution of allyl sulfones with Grignard reagents (Scheme 51). The presence of the sulfonyl group at the 2-position of the pyridine ring allows for reversible coordination to the copper center, enabling high catalytic performance, and reducing the entropic penalty due to the coordination effect. This behavior was remarkable in comparison with other sulfonyl groups since the phenyl sulfonyl counterpart did not promote any reaction. Recently, we have extended the use of the SO2Py as a leaving group in the Cu-catalysed allylic substitution of acyclic substrates with both alkyl- and aryl Grignard reagents, yielding the corresponding product with complete α-regioselectivity and no erosion of the E:Z stereoselectivity (Scheme 51b).92
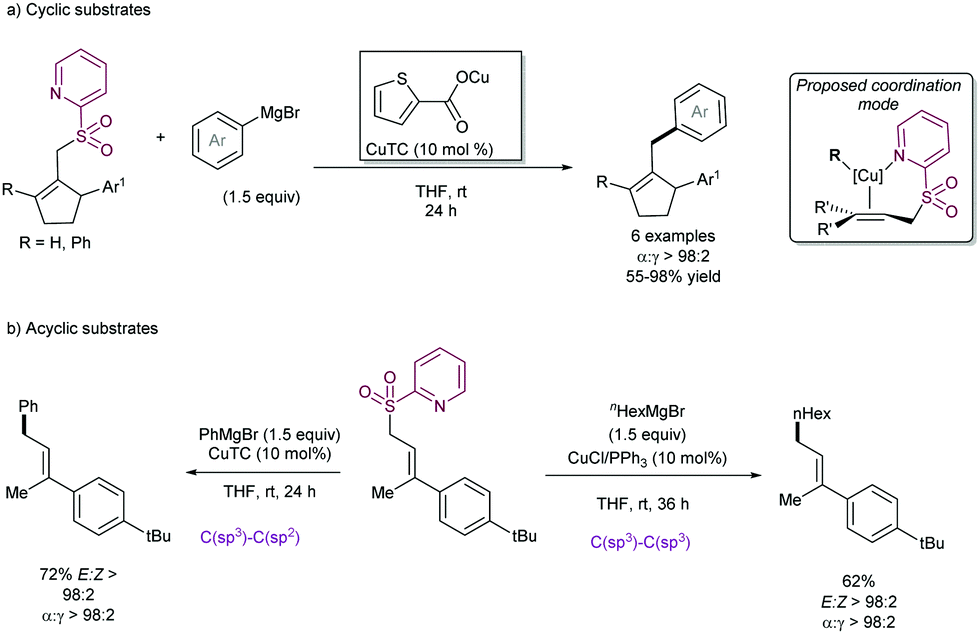 | ||
| Scheme 51 2-Pyridyl sulfonyl as a leaving group in Cu-catalysed allylic substitutions. | ||
Following the interest of the SO2Py moiety as a leaving group in Cu-catalysed allylic substitution, our group studied the presence of potential sensitive functional groups in the allyl sulfone moiety, such as boronic esters93 and silyl groups,94 which would highly impart chemical versatility for further transformations via cross-coupling or homologation strategies. These studies were performed in the context of the use of this sulfone group as a regiocontroller in hydrofunctionalization of unsymmetrical di-alkylalkynes. After a first copper-catalysed hydroboration (Scheme 52a) or hydrosilylation (Scheme 52b), the SO2Py was rapidly substituted by the corresponding Grignard reagent to furnish the desired alkenylboronates or alkenylsilanes, respectively. Interestingly, the high steric hindrance of the SiMe2Ph group is likely to be the reason for the divergent regioselectivity in the allylic substitution step.
 | ||
| Scheme 52 2-Pyridil sulfonyl group in Cu-catalysed allylic substitution of boronates and silylated alkenes. | ||
The mechanism proposed for this reaction follows the generally accepted catalytic cycle postulated for most of the Cu-catalysed allylic substitutions with Grignard reagents (Scheme 53).95 After a first formation of mono-alkylcopper species by reaction of the pre-catalyst and the Grignard reagent, the coordination of the allylsulfone to the electron-rich copper intermediates promotes an intramolecular oxidative addition to generate π-allyl-Cu(III) species A with concomitant elimination of the arylsulfinate group. Further reductive elimination yields the γ- or α-regioisomer depending on the reaction conditions (mono- vs. dialkylcuprate species) and/or steric factors.
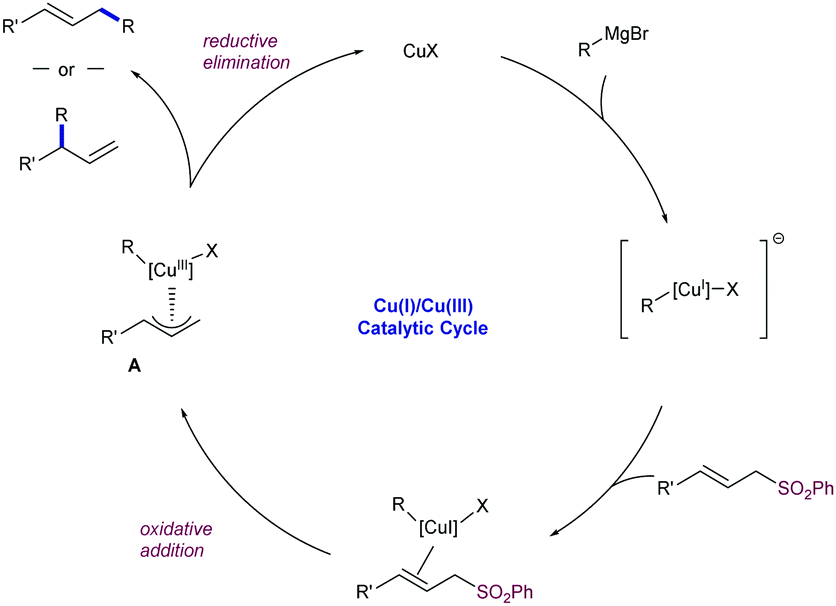 | ||
| Scheme 53 Generally accepted mechanism for the Cu-catalysed allylic substitution. | ||
2.4.1.2. Other metal-catalysed allylic substitution of sulfones: the Tsuji–Trost and Suzuki–Miyaura reactions. Allyl sulfones have been also used as electrophiles in the Tsuji–Trost reaction. Initially described by Trost96 in 1980, this approximation toward the catalytic desulfonylation of allyl sulfones allows the direct displacement of the phenylsulfonyl moiety by an array of C-centered nucleophiles. The reaction displays the typical features of the traditional Tsuji–Trost transformation employing more conventional electrophiles, such as allylic acetates or phosphonates. Other versions of the reaction have been reported using Ni catalysis,97 and it has been applied in the synthesis of different natural products.98
The regioselectivity of the new C–C bond is controlled by steric factors and occurs with net retention of the configuration, as a consequence of the intermediacy of a π-allyl-Pd species.99 Thus, the allylation typically occurs at the less hindered position. This strategy has been used as the last step in sequential functionalization reactions in which a last C–C bond forming reaction is performed under desulfonylative conditions (Scheme 54). These strategies include the prior use of the sulfone as a nucleophile (via α-deprotonation/alkylation, Scheme 54a) as reported by Trost in the synthesis of a precursor en route to a sex pheromone of the Monarch butterfly. The reversed strategy, in which electrophilic functionalization precedes the desulfonylative step has been also employed.100
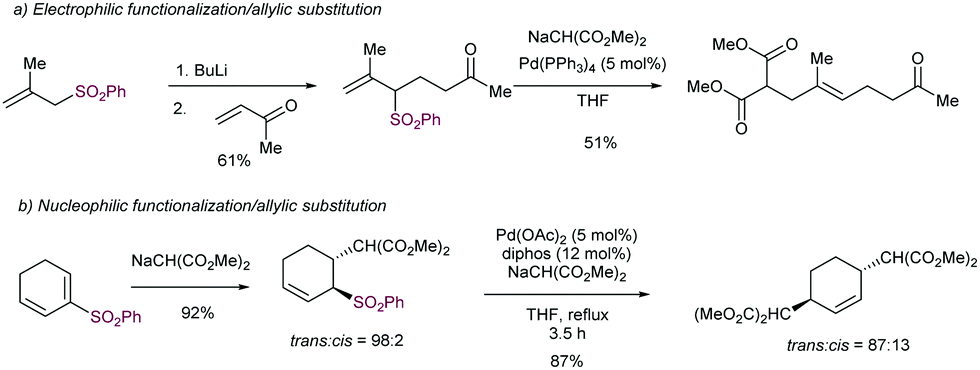 | ||
| Scheme 54 Sequential functionalization of sulfones involving a final desulfonylative Tsuji–Trost coupling. | ||
Regarding the regioselectivity, efforts to invert the typical pathway with a proper choice of reaction conditions can serve to modulate the α- or γ-substitution under kinetic or thermodynamic conditions, but mixtures of both regioisomers are usually obtained.101 In this context, the ability of molybdenum catalysts to invert the regioselectivity in the catalytic allylic substitution is well-known. This behavior is believed to occur because of the high coordination number present in π-allylmolibdenum complexes.102 Additionally, depending on the ligand, the charge over the two-different carbon atoms of the allylic system bound to the Mo-catalyst can be different, favoring a positive charge at the most substituted position,103 which would entail a crucial factor in the regioselectivity of the reaction. To reverse the regiochemical outcome of the reaction in the case of allyl sulfones, Trost described the Mo-catalysed substitution of allyl sulfones (Scheme 55a).104 This strategy allowed for the creation of quaternary centers by desulfonylative alkylation at the α-position. However, when very sterically demanding nucleophiles are employed, this strategy typically fails, and the γ-attack becomes the major pathway. Later, Matsunaga and co-workers105 described the allylic alkylation of malonates under cobalt and organophotoredox catalysis (Scheme 55b). In this case, a photochemically generated Co(I) intermediate is the reactive species, which undergoes oxidative addition to the allyl sulfone to generate a π-allyl cobalt(III) intermediate. Further nucleophilic attack occurred with complete α-regioselectivity. This approximation complements other versions of the reaction, as it is more efficient than previous allylic substitutions performed with Rh106 or Ir107 precious metals.
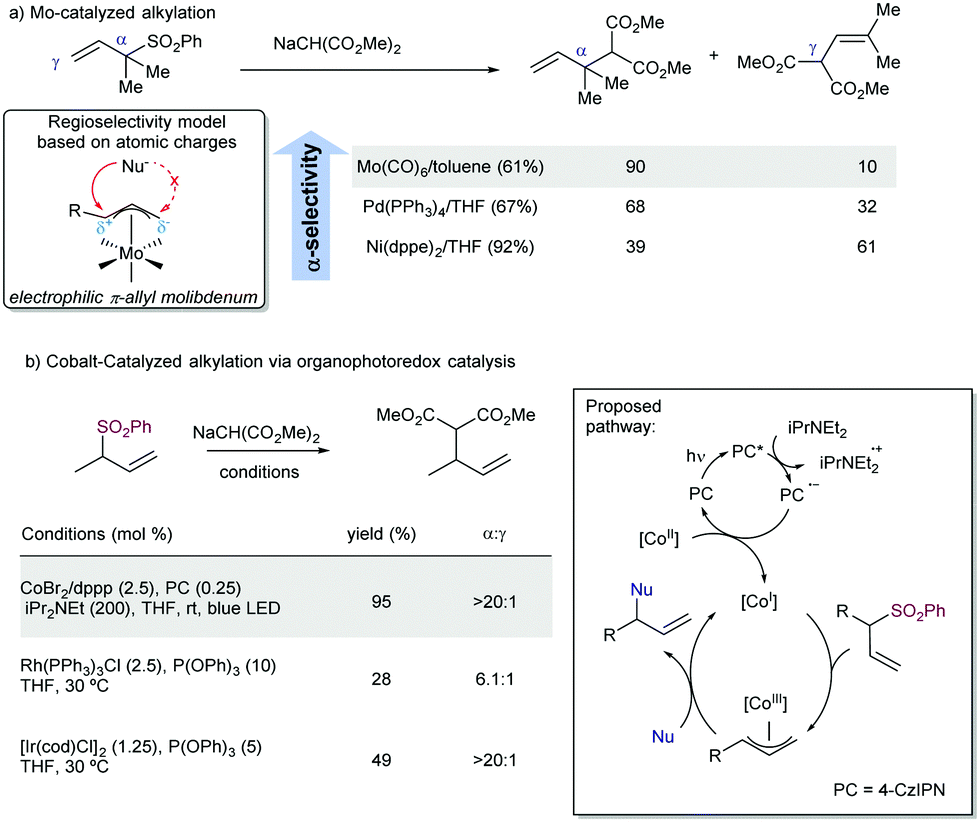 | ||
| Scheme 55 Typical approaches toward α-regioselective alkylation of allyl sulfones by: (a) Mo catalysis and (b) photoinduced Co(I) catalysis. 4-CzIPN = 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene. | ||
Organotitanium reagents are also suitable nucleophiles in the Pd-catalysed substitution of allylic sulfones, as described by Lomakina and co-workers.108 Notably, chlorine substitution at the aryl moiety of the sulfonyl group was crucial for the reactivity in some instances, as opposed to the use of phenylsulfone as a leaving group which inhibited the reaction.
Apart from the Tsuji–Trost reaction, the use of other potential nucleophiles has been also studied for the development of highly α-regioselective allylic substitution using sulfones as electrophiles. In this regard, the Crudden group has developed a Suzuki–Miyaura-type methodology for the use of tertiary allyl phenylsulfones as electrophiles in the Ni-catalysed Suzuki–Miyaura cross-coupling reaction using arylboroxines as nucleophiles (Scheme 56).109 The reaction tolerated a wide range of arylboroxines, with the exception of electron-deficient heteroaryl derivatives. P-Donor ligands were screened, with a dialkyl aryl phosphine ligand reported by Doyle110 found to be broadly applicable for all the structural scope. The nature of the ligand was crucial for the reactivity, as other P-donor ligands resulted in no reactivity in comparison with Doyle's ligand for electron-deficient and heteroaromatic substrates. Importantly, no loss of stereochemistry was observed, along with complete regioselectivity to the α-position.
 | ||
| Scheme 56 Ni-Catalysed α-regioselective Suzuki–Miyaura reaction between allylsulfones and aryl boroxines. | ||
In this vein, the use of boronic acids instead of the corresponding aryl boroxines leads to lower yields, albeit maintaining excellent levels of regioselectivity. This reactivity profile has been recently exemplified by Khan and co-workers in the Ni-catalysed formal synthesis of (±)-hinokirenisol (Scheme 57).111 Although the reaction occurred with low yield, the use of a Pd catalyst was found beneficial in comparison with Ni(cod)2.
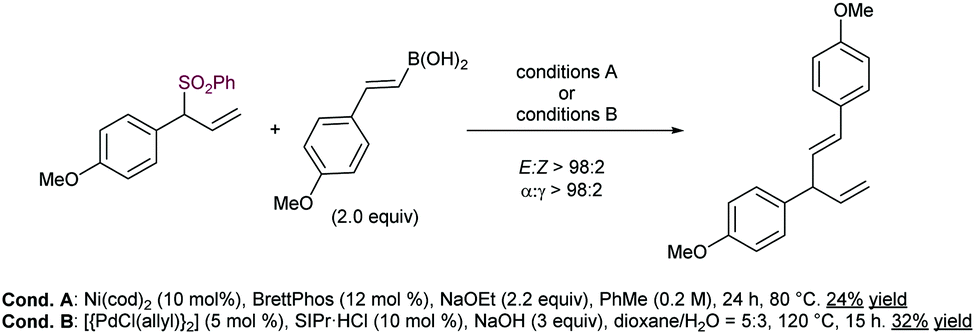 | ||
| Scheme 57 Boronic acids in Suzuki–Miyaura type cross coupling of allyl sulfones. | ||
More recently, Song112 has described an alternative approach for the use of allyl sulfones as allylating reagents for electrophiles. The team reported on a reductive Ni-catalysed cross-coupling with α-chloroboronates that rendered the corresponding homoallylic boronates mediated by the presence of Zn (Scheme 58). This strategy was amenable to a variety of different substituted boronates, thus enabling the synthesis of primary and secondary homoallylic boronates. Different functional groups were also well tolerated at the substituent of the allyl sulfone. The reaction conditions did not suffer loss of efficiency when the reaction was scaled up to 5 mmol, thus highlighting its potential for large scale application. The intermediacy of radicals in the process was demonstrated by the addition of TEMPO, which inhibited the reaction. Thus, the postulated catalytic cycle proposed by the authors starts with the reduction of the nickel pre-catalyst to generate a Ni(0) species which undergoes SET to the chloroboronate to generate the corresponding α-boryl radical. This species then adds to the allylic sulfone, rendering a β-sulfonyl radical. The radical intermediate further collapses with the Ni(I) forming a transient alkyl-nickel(II) species which furnishes the desired product via β-sulfonyl elimination. Reduction of the in situ generated sulfinyl-nickel(II) by Zn regenerates the active nickel catalyst.
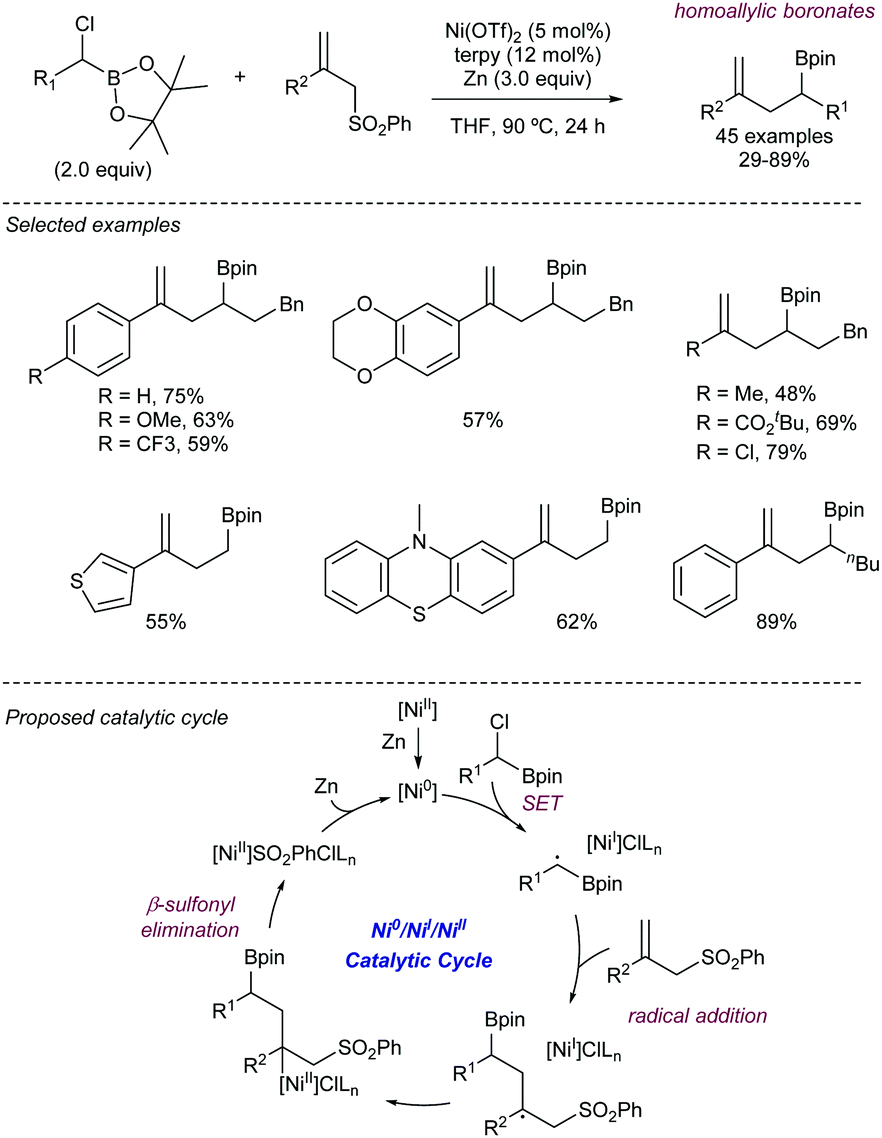 | ||
| Scheme 58 Stereodivergent ligand-controlled Pd-catalysed dienylation of aryl halides. | ||
2.4.1.3. Allyl aryl sulfones as latent nucleophiles. Julia and Clayden113 reported that allyl aryl sulfones can be used in the allylation of different carbonyl compounds as allyl anion equivalents (Scheme 59a). This strategy is based on a first oxidative addition of the allyl sulfone to a Pd(0) center with the concomitant elimination of the phenylsulfonyl group. Further transmetalation with Et2Zn forms a mixed diorganozinc reagent in situ, which is able to transfer the allyl group to an electrophile, such as aldehydes and ketones. The reaction proceeded with high regioselectivity, but the substitution at the γ-position slows down the reaction. Important contributions to this chemistry have been described by Cohen and co-workers exploiting this strategy for the generation of allylzincs in an intramolecular fashion via a Pd-catalysed Zinc-ene cyclization (Scheme 59b).114 Different synthetically useful intermediates toward natural products synthesis such as (−)-erythrodiene and (−)-kainic acid have been used employing this approximation.
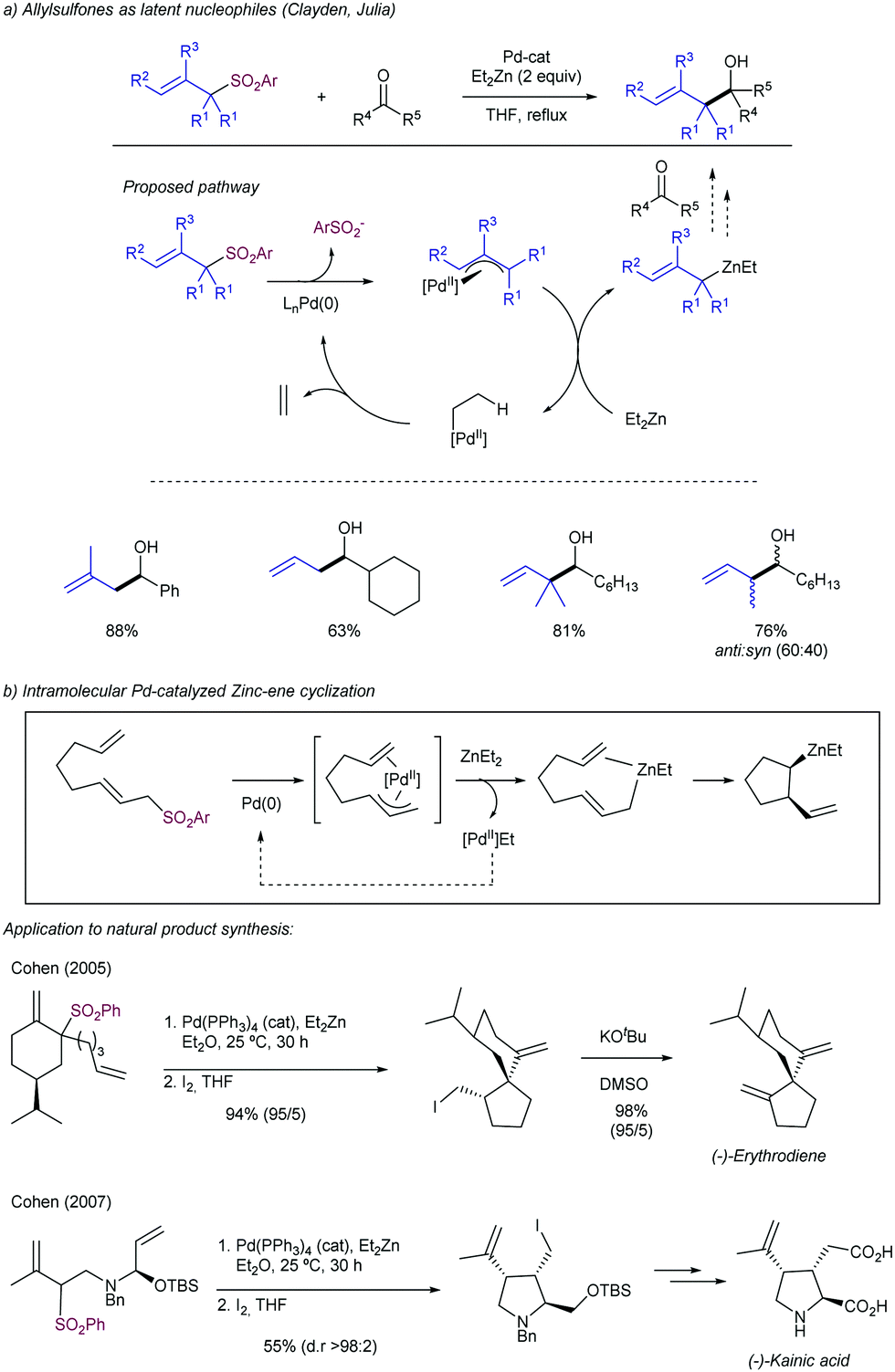 | ||
| Scheme 59 Allyl sulfones as latent nucleophiles by a Pd-catalysed Et2Zn-mediated reaction. | ||
2.4.2.1. Modern radical allylation involving transition metal catalysis. Li and co-workers have described the decarboxylative Ag-catalysed allylation of free aliphatic carboxylic acids under aqueous conditions employing K2S2O8 as an external oxidant (Scheme 60).118,119 The reaction is initiated by decarboxylation of the corresponding carboxylic acid by reaction with a Ag+ catalyst, with the concomitant elimination of CO2 and the generation of the aliphatic radical, which is trapped in an intermolecular fashion with an allyl sulfone. The allylation reaction delivered the corresponding product high reaction yields and excellent chemoselectivity for a wide range of secondary and tertiary carboxylic acids, including α-oxy or α-amino acids as well. As expected, primary substrates reacted with lower efficiency, but still gave the desired product. Interestingly, the reaction was successful for alkene-containing aliphatic carboxylic acids, promoting a cascade process in which after a first radical formation and cyclization, the resulting radical was intercepted with the allylsulfone. A variant of this reaction has been recently described by Yao and co-workers for benzylic α,α-difluorocarboxylic acids.120
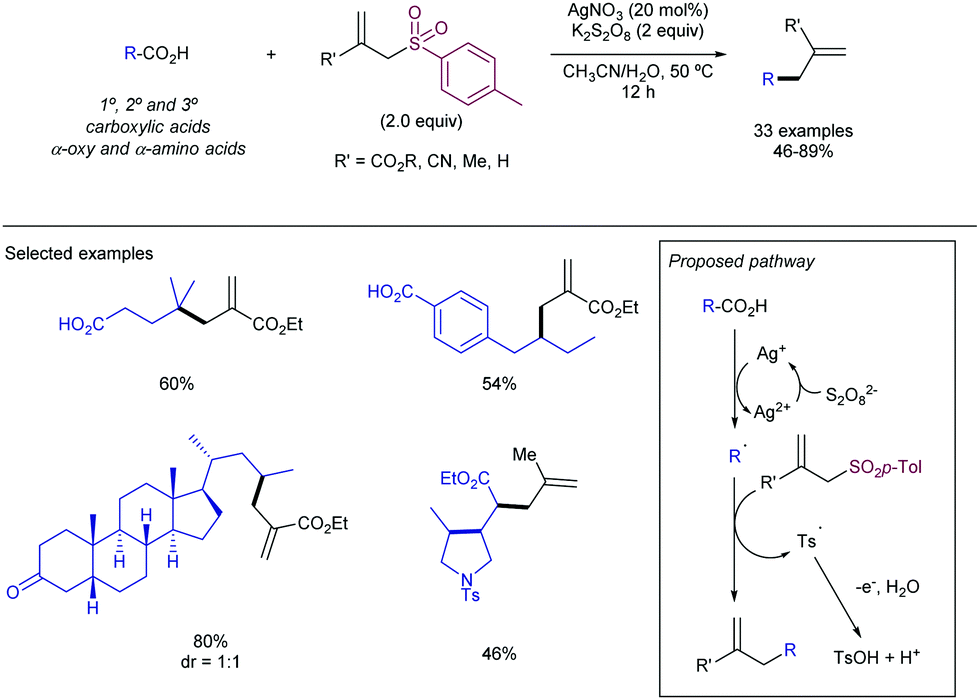 | ||
| Scheme 60 Ag-Catalysed decarboxylative allylation. | ||
In this regard, Guo and co-workers have described the use of allyl phenylsulfones for the allylation of C(sp3) centers via Fe(II)-catalysed C–C bond breaking in cyclobutanone ester oxime derivatives (Scheme 61).121 Formally, the reaction can be catalogued as the γ-allylation of alkyl nitriles. Remarkably, different allyl sulfones furnished the corresponding product, including those bearing esters, cyano, alkyl-, aryl-, and plain allylsulfones. Mechanistic studies suggest that the Fe(II) species promotes N–O bond cleavage of the cyclobutanone oxime, generating a N-centered radical. This radical evolves toward a C-centered radical upon ring-opening step, which generates an intermediate radical at the γ-position of the nitrile. Further reaction with the allyl sulfone generates the allylated product, along with the concomitant formation of a phenylsulfonyl radical. Finally, the previously formed Fe(III) species enables the oxidation of the sulfonyl radical to give phenylsulfonic acid and the re-generation of the catalytically active Fe(II) species.
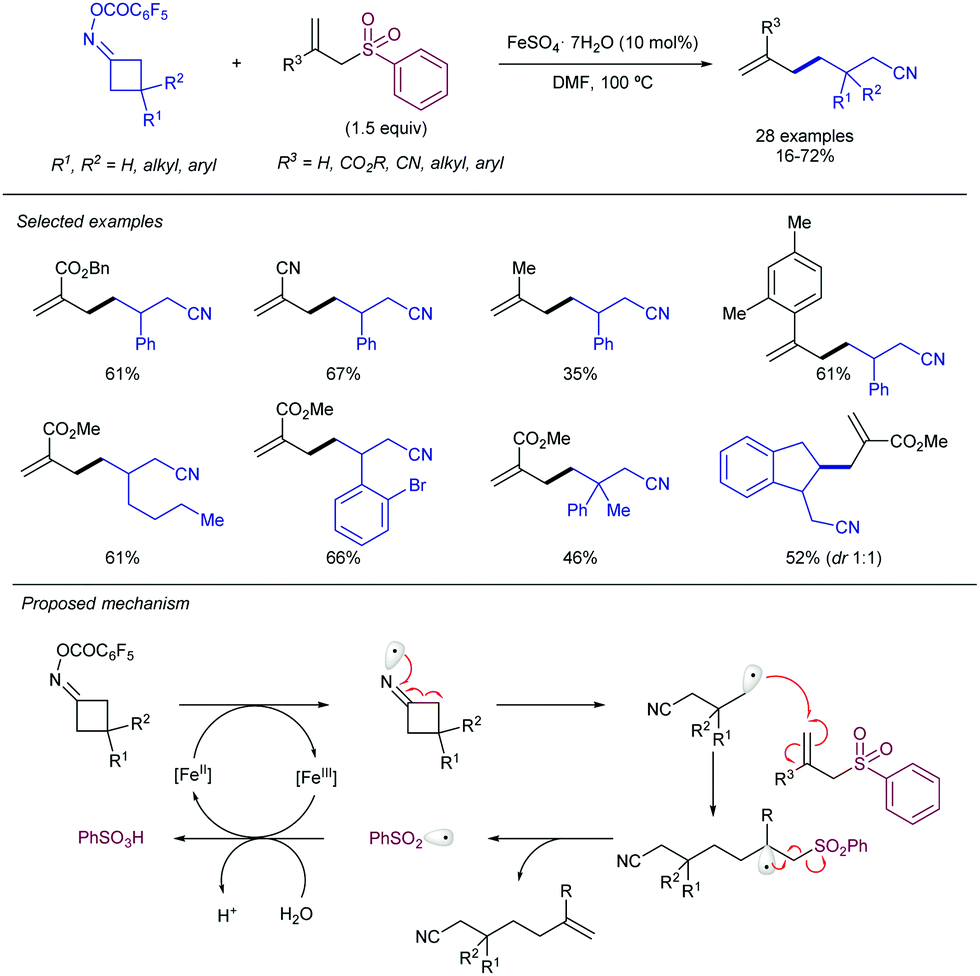 | ||
| Scheme 61 Fe-Catalysed allylation via ring-opening of cyclobutanone oximes. | ||
2.4.2.2. Allylation of alkyl radical precursors by photocatalytic methods. One of the advantageous uses of photocatalysis to induce the allylation of different simple alkyl chains is the high number of precursors for this purpose (Scheme 62). In 2015, Chen introduced the use of N-acyloxyphtalimide-protected carboxylic acids for the photogeneration of alkyl radicals using a Ru-based photocatalyst (entry 1).122 The reaction performed well for primary, secondary, and tertiary substrates. Additionally, both aryl and alkyl bromides and iodides were tolerated under the reaction conditions. Other potential sensitive groups such as aldehydes, nitriles and azides demonstrated no interference in the allylation process. This reaction could be performed under aqueous conditions, as demonstrated by the allylation of the oligosaccharide naringin with an N-acyloxiphthalimide derivative (Scheme 63). The same year Olivier and Fensterbank described the use of silicates as a latent alkyl radical precursor (Scheme 62, entry 2).123 The reaction was conducted in DMF using an Ir-based photocatalyst under blue LED irradiation. Different primary, secondary, and tertiary alkyl chains could be allylated. In addition, silicates bearing α-heteroatoms such as O, N and Cl were also suitable partners. The generation of C-centered radicals from C–H bonds in a direct fashion was accomplished by Kamijo in 2016.124 The treatment of the corresponding alkane (in a large excess, typically 10 eq./eq. allylsulfone) in the presence of a pentacenetetrone organophotocatalyst in CH2Cl2 delivered the corresponding allylated product with high chemoselectivity and moderate reactivity. Other protocols have been also described for the allylation of alkyl radicals, including deaminative strategies125 (Scheme 62, entry 4) and the use of alkyl iodides using Pd-catalysis under light irradiation (Scheme 62, entry 5).126
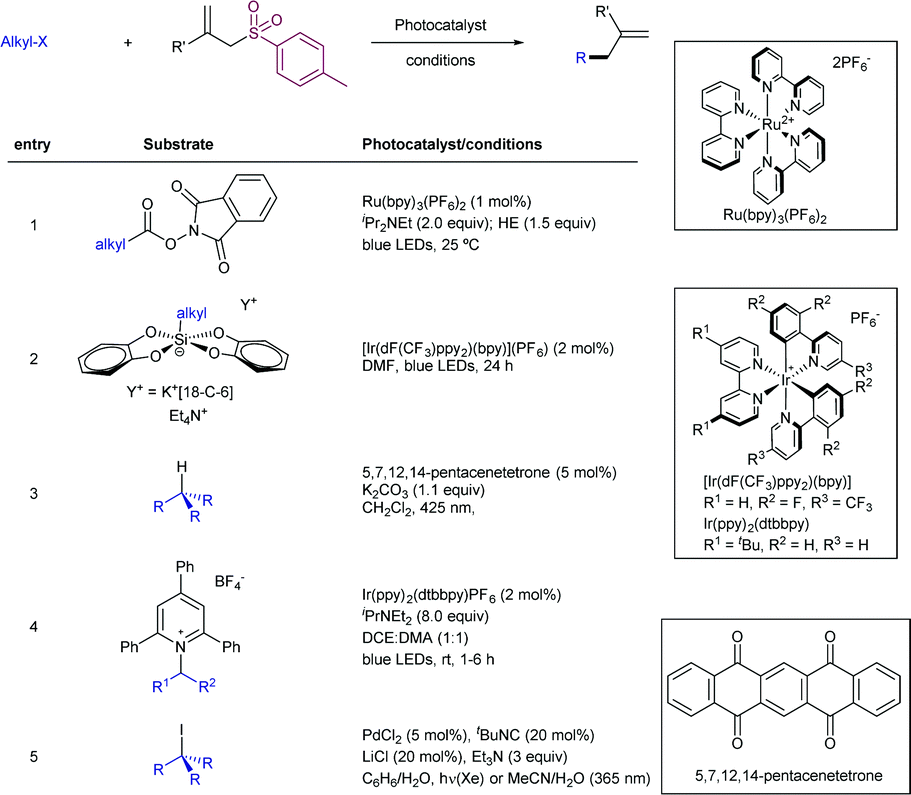 | ||
| Scheme 62 General alkyl radical precursors using photocatalytic conditions. | ||
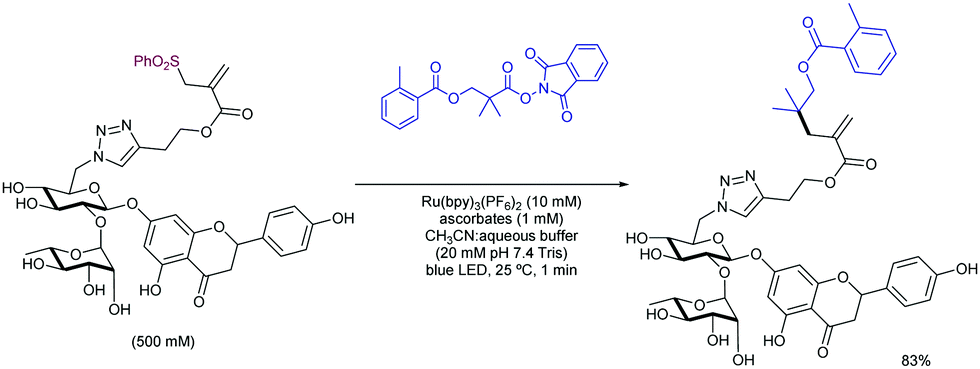 | ||
| Scheme 63 General alkyl radical precursors using photocatalytic conditions. | ||
Another important family of alkyl radical precursor is related to those molecules which render an α-heteroatom substituted radical, which stabilizes the intermediate and facilitates the formation of these species. In this regard, Molander and co-workers127 took advantage of the ability of α-amino alkyl tetrafluoroborates to obtain 2-allyl pyrrolidines using eosin Y as organophotocatalyst (Table 2, entry 1). In addition, Zhu described the formation of α-amino radicals via decarboxylation of glicyne derivatives employing an Ir photocatalyst (entry 2).128 This decarboxylative strategy has also been exploited by González–Gómez, extending the method to the presence of O- and S-heteroatoms at the α-position using flavin tetraacetate as organophotocatalyst (entry 3).129 Finally, it is noteworthy to highlight the work by Ryu which employed α,α-difluoro alkyl halides in presence of a Ru-based photocatalyst (entry 4).130 This method enables the access to fluorinated allylic-containing molecules, and can be coupled to an intermolecular addition of alkenes, resulting in a tri-component reaction. Analogously, methods based on the generation or radicals bearing an electron-withdrawing group at the α-position have been described, which allows the access to α-allylated ketones and related groups.131
|
|
|||
|---|---|---|---|
| Entry | Substrate | Photocatalyst/conditions | |
| 1 |
|
Na2Eosin Y (2 mol%), DMF, 26 W CFL, 50 °C |
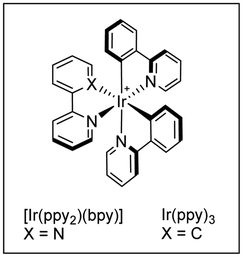
|
| 2 |
|
Ir(ppy)2(bpy)(PF6) (1 mol%), Cs2CO3 (1.0 equiv.), Ar CH3CN:H2O, 36 W CFL, 25 °C |
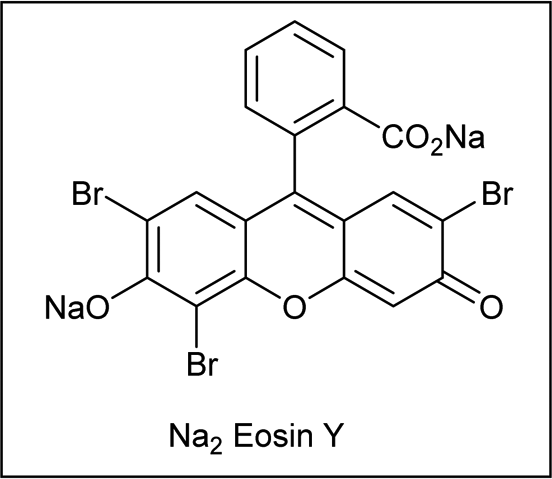
|
| 3 |
|
Riboflavin tetraacetate (5 mol%), CH3CN, blue LEDs, rt, 22 h |
|
| 4 |
|
Ru(bpy)3Cl2 (1 mol%). HE (0.6 equiv.), iPr2NEt (2.0 equiv.), CH3CN, white LEDs, 2 h | |
Recently, Sawamura, Shimizu and co-workers described132 the direct α-allylation of carboxylic acids employing allyl sulfones. The strategy is based on the formation of a boron-endiolate using a boron catalyst bearing a π-extended ligand to ensure visible light absorption. These intermediates are excited under blue light irradiation to promote a SET towards the allyl sulfone (Scheme 64).
 | ||
| Scheme 64 General precursors and conditions for the access to α-heteroarom substituted alkyl radicals. | ||
The ability to access α-heteroatom radicals via photocatalytic activation has enabled the use of photochemistry for the direct allylation of carbonyl and imine groups via an umpolung strategy. In 2016 Chen and co-workers studied the ability of aldehydes and imines to form this type of radicals (Scheme 65).133 Using an Ir catalyst and the Hantzsch ester as a H-donor, the authors were able to trigger the formation of α-heteroatom radicals, which easily reacted with the corresponding allyl sulfone. This method allowed for the direct allylation of aryl- and α-heteroatom substituted aldehydes. In addition, imines were also suitable partners for this transformation, leading to the corresponding allylated amines. Mechanistically, this methodology is based on the direct excitation of an Ir(III) photocatalyst which is reductively quenched by the Hantzsch ester (HE) to form a highly reducing Ir(II) species. Therefore, the carbonyl or imine group associated with the oxidized HE by hydrogen bonding can be easily reduced to the corresponding α-O- or α-N-radical intermediate. Finally, the newly formed radical collapses with the allyl sulfone furnishing the desired product. In this context, Dixon has described the allylation of imines photocatalysed by eosin Y.134 The reaction proceeded via a tricomponent system in which the addition of the corresponding aldehyde in combination with the desired amine allowed in situ formation of the corresponding imine which further suffers the allylation step. This procedure was crucial for the allylation of aliphatic imines formed because of problems associated with isolation of said species.
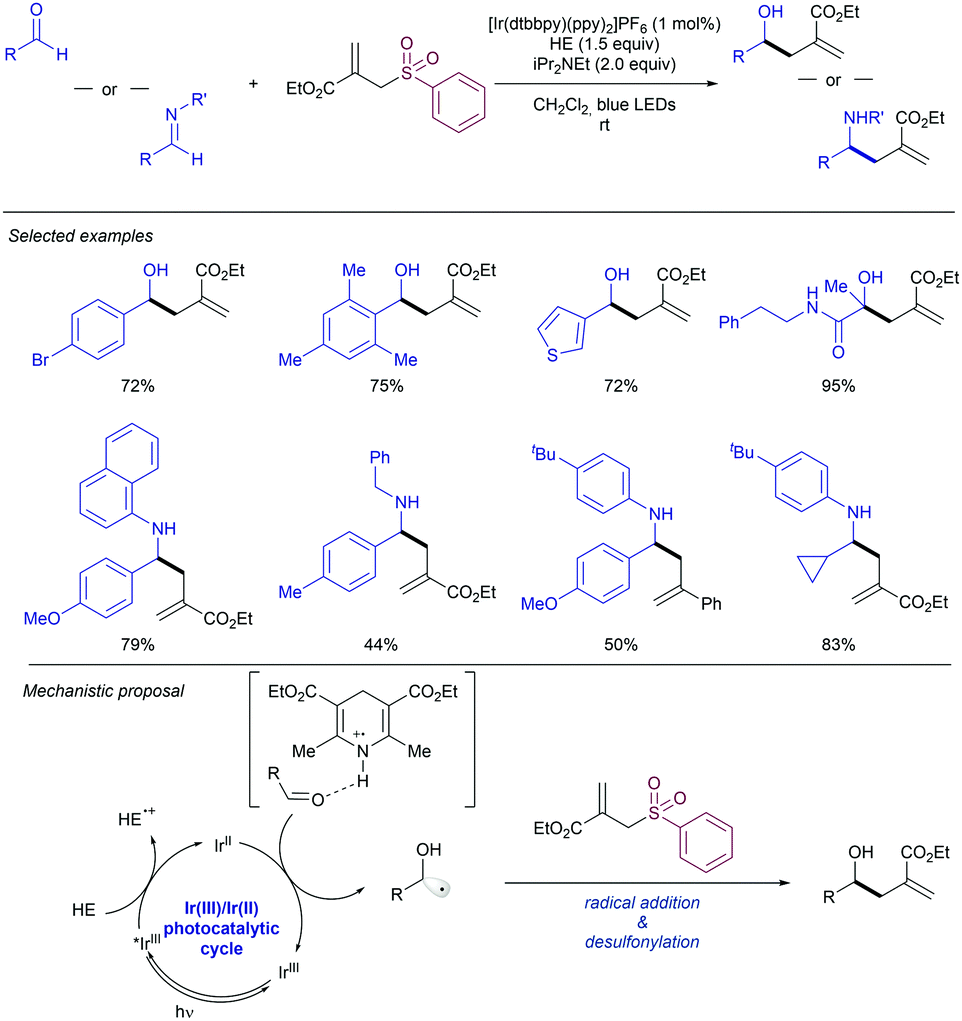 | ||
| Scheme 65 Photocatalytic allylation of aldehydes and imines with allyl sulfones. | ||
A related important class of precursors are those in which the radical formation occurs via a first N–O bond cleavage, forming a heteroatom-centered radical. Further 1,5-HAT (hydrogen atom transfer) enables the formation of a new C-centered radical, which is the reactive species. In 2016 the Chen group introduced N-alkoxyphthalimides a radical precursor (Scheme 66a).135 Following this strategy, tertiary, secondary and α-hetereoatom positions were successfully functionalized in presence of ethyl 2-((phenylsulfonyl)methyl)acrylate. The reaction showed excellent functional group tolerance, and it could be scaled up to gram scale. In this regard, Flechsig and Wang introduced the use of aryloxyamides as a radical precursor.136 After initial cleavage of the N–O bond – promoted by a photoexcited eosin catalyst- and subsequent 1,5-HAT, the corresponding C-allylated products were isolated with high yield and broad substrate compatibility (Scheme 66b). This method enables the remote allylation of aliphatic amines in which the resulting C-centered radical must be tertiary or secondary. Other strategies based on a first N-centered radical generation followed by the formation of a C-centered radical which undergoes allylation has also been described, such as the synthesis of dihydropyrazoles and tetrahydropyridazines developed by Xiao and co-workers in 2017.137
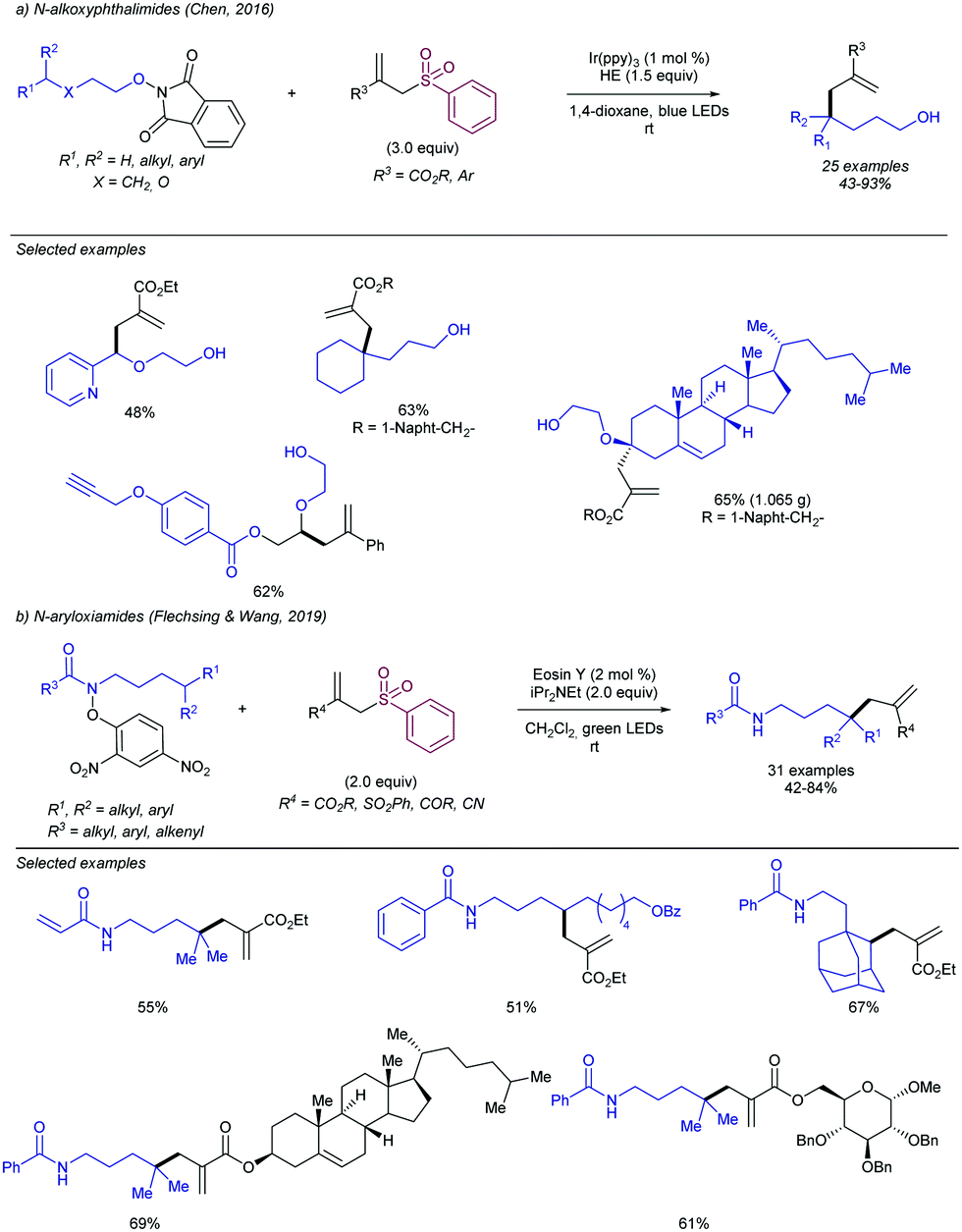 | ||
| Scheme 66 Remote C(sp3)–H allylation by photoinduced N–O bond breaking and further 1,5-HAT. | ||
Another activation approach has been recently reported by the Feng lab in the photocatalytic-promoted fluoride-ring opening of difluoromethylene cyclopropanes in presence of allylsulfones (Scheme 67).138 This method enables for the elaboration of CF3-containing organic scaffolds, which are highly embedded in Medicinal Chemistry areas. The reaction proceeds via a SET oxidation of phenyl difluorocyclopropanes by the intermediacy of an excited Ir catalyst, thus enhancing its electrophilic properties and allowing the fluoride-promoted ring-opening step. The newly formed radical species is further trapped by the allylsulfone.
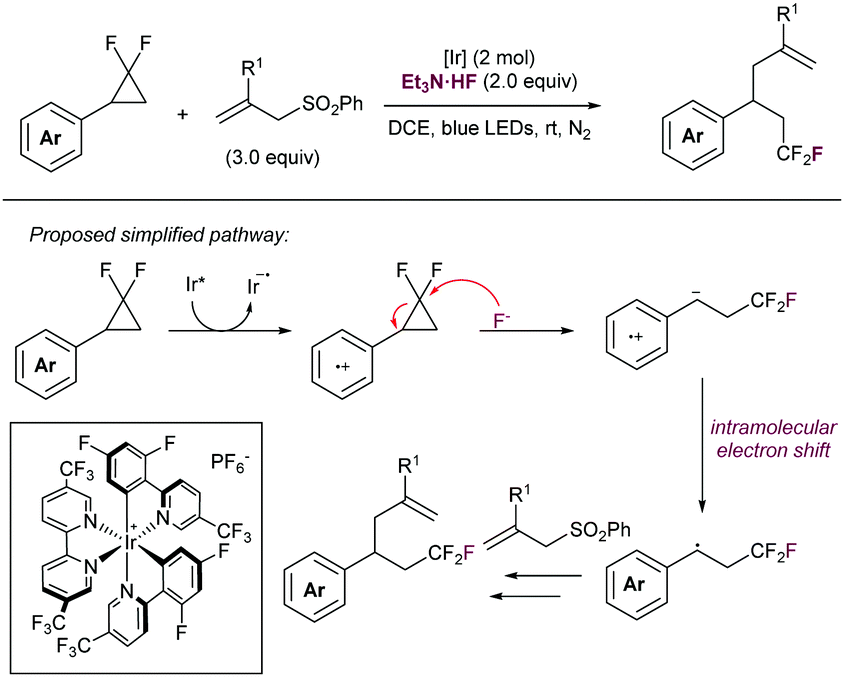 | ||
| Scheme 67 Photocatalytic ring-opening of difluorocyclopropanes. | ||
2.4.2.3. Allylation of aryl radical precursors by photocatalytic methods. The construction of C(sp2)-allyl fragments is also a highly desirable transformation which can be addressed by using aryl radical precursors under photocatalytic conditions. In 2013 Ollivier and co-workers described the allylation of aryl units using diaryliodonium salts as the radical precursors (Scheme 68a). The reaction was catalysed by a photoactive [Cu(dpp)2][PF6] catalyst, which enabled the mild formation of aryl radicals at room temperature. The same group reported the use of triarylsulfonium salts as a suitable radical generator using Ru(bpy)3Cl2 as the photoredox catalyst (Scheme 68b). Despite of the existence of these precedents, the allylation of aryl units under photocatalytic conditions employing allyl sulfones is an underdeveloped area, mainly limited by a narrow substrate scope and the use of large amounts of the allyl sulfone partner.
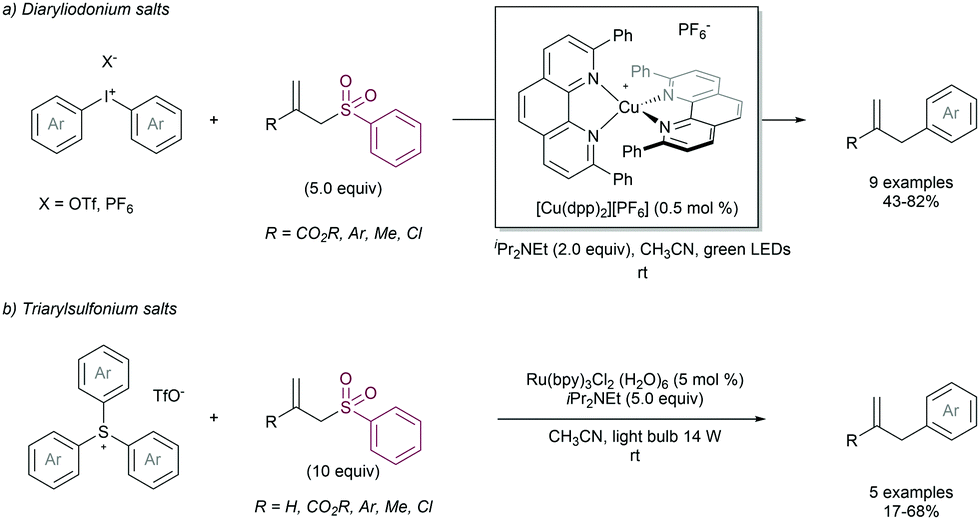 | ||
| Scheme 68 Photocatalytic approaches for the allylation of aryl groups. | ||
2.4.2.4. Enantioselective processes. Attempts to develop enantioselective version of the radical allylation have been performed by the Meggers group (Scheme 69).139 In this case the generation of the radical was approached upon coordination of an α,β-unsaturated carbonyl compound to a chiral-at-metal Rh-complex. This coordination enabled the formation of a C-centered radical at the β-position using the Hantzsch ester as a photoredox mediator under visible light irradiation. The newly formed radical undergoes desulfonylative allylation with the concomitant formation of the corresponding phenylsulfonyl radical. Interestingly, the authors found that the sulfonyl radical is also engaged in the addition to the unsaturated ketone in an enantioselective fashion, a rarely mode of reactivity in this chemistry.
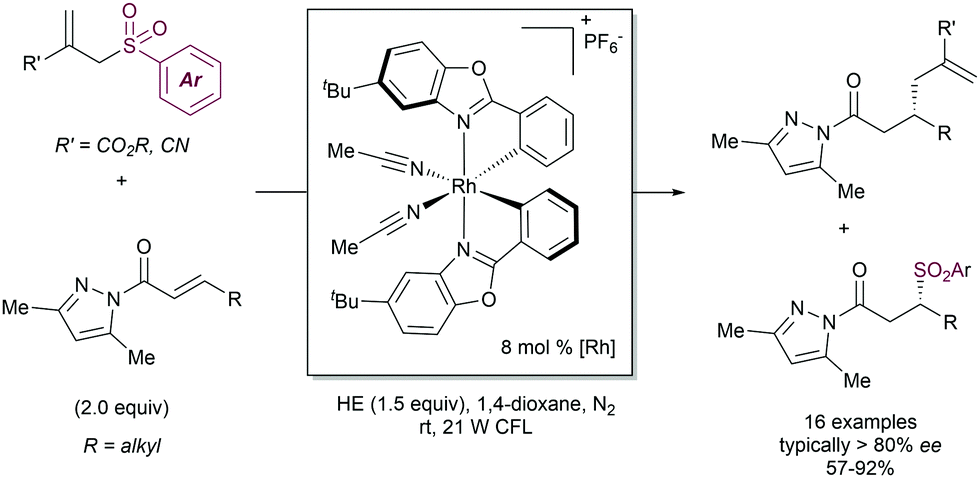 | ||
| Scheme 69 Enantioselective allylation of unsaturated compounds. | ||
2.5. Alkylations
One of the milestones in cross-coupling chemistry is the development of efficient methods using alkyl electrophiles as coupling partners. Difficulties with this substrate class include the facility of the intermediate metal-complexes to undergo smooth β-hydrogen elimination. Another factor is related to the activation of the C–X bond via oxidative addition pathways, which would entail a crucial factor for the successful of the cross-coupling for unactivated alkyl electrophiles. Research in this area has enabled the development of this approach, broadening the toolbox to applications in challenging synthetic problems. Among different strategies to avoid undesired side-reactions, one is the use of suitable electrophiles that allow for novel activation outcomes and/or new mechanistic features, unlocking mild and selective transformations with alkyl partners. In this context, alkyl sulfones have surfaced as promising reagents. Different activation modes of the C–S bond, along with the effect of different catalytic systems, are discuss in this section. | ||
| Scheme 70 Fe-Catalysed Kumada cross-coupling using alkyl phenylsulfones. | ||
It should be noted that some features of this reaction remain unsolved in terms of substrate scope compatibility. Even though this methodology is still underexplored, the use of other nucleophiles has allowed its satisfactory employment in the development of useful reactions. A prominent example has been recently reported by Hu and co-workers in the context of difluoromethylation of diarylzinc reagents (Scheme 71).143 The reaction proceeded via a Fe-catalysed Negishi-type cross-coupling using difluoromethyl 2-pyridylsulfone as electrophile. Interestingly, the use of Grignard reagents did not afford the desired product with synthetically useful yields. Remarkably, the use of TMEDA as additive was also essential for obtaining high yields, albeit lower quantities were needed in comparison with Denmark's work.140 The reaction was compatible with a broad range of diarylzinc reagents, including O-protected alcohols, heteroaromatic rings, and potential sensitive groups such as –CN or –F. Mechanistically, the authors demonstrated that the reaction proceeded via a radical-type pathway in which a low-valent Fe species would undergo a SET process with the sulfone, thus promoting the radical-induced desulfonylative process to generate a difluoromethyl radical and the corresponding 2-pyridylsulfinate, which rapidly recombines with the iron center. Further C–C reductive elimination furnishes the cross-coupling product. After transmetalation of the resulting iron sulfinate species with the diaryl zinc reagent, the catalytically active Fe species is recovered.
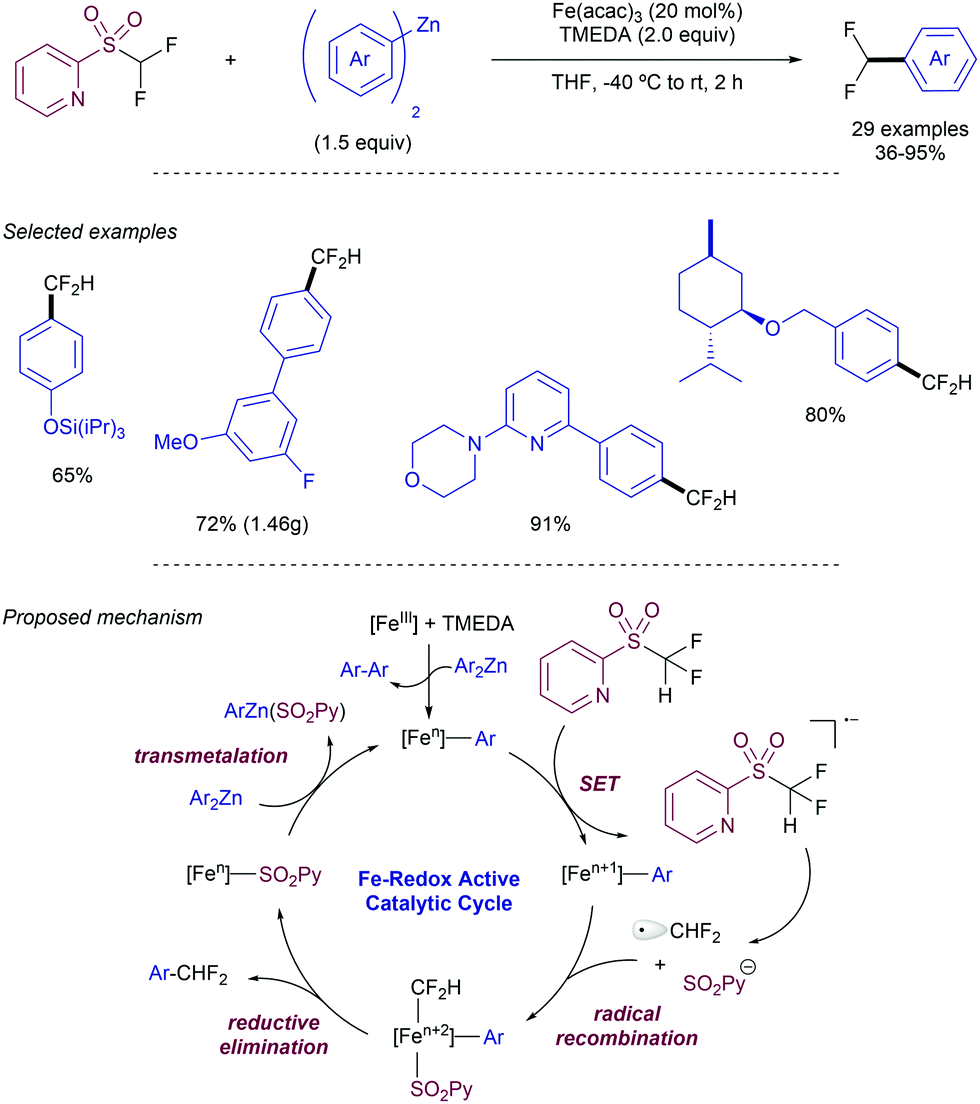 | ||
| Scheme 71 Fe-Catalysed Negishi-type difluoromethylation of diaryl zinc reagents. | ||
A related process reported by Baran disclosed a broadly applicable use of alkyl sulfones as electrophiles in cross-coupling chemistry.144 Using a Ni catalyst in combination with the N,N-bidentate 2,2′-bipyridine ligand, the authors studied systematically the potential activation of the sulfone considering different aromatic and heteroaromatic groups. Among the seven tested aromatic groups, only the redox-active 1-phenyl-1H-tetrazol-5-yl (PT) group shown reactivity in presence of aryl zinc chlorides as the nucleophilic partner (Scheme 72). This example is unique between the methods developed so far intended to the use of alkyl sulfones as electrophiles because of its reactive features. For instance, this method tolerates an exceptional variety of sulfone substrates, including primary, secondary, and benzylic ones in combination with potential sensitive functional groups in nickel chemistry, such as alkyl chlorides. In addition, it is also useful for a wide range of nucleophilic partners, delivering high reactivity for sterically demanding substrates — such as those presenting ortho-substitution — and electrophilic functionalities like C–X (X = halide) bonds. As in the case of Hu,142 Baran's method enables the introduction of fluorinated groups (CFH2, CF2H and CF2R) and aryl zinc chlorides, which are more atom-economical reagents than diaryl zinc species. Remarkably, the use of alkenyl sulfones bearing the PT group permits to use them in a prior Michael- or Giese-type addition, followed by the Ni-catalysed desulfonylative Negishi protocol, enabling the iterative functionalization of this family of substrates. Although moderate yields were obtained in most cases (about 50–60%), several target compounds could be obtained significantly reducing the number of steps for their obtention. Additionally, this strategy has been extensively exploited for the synthesis of relevant biologically active and functional molecules, showing the practicability and utility of the transformation.
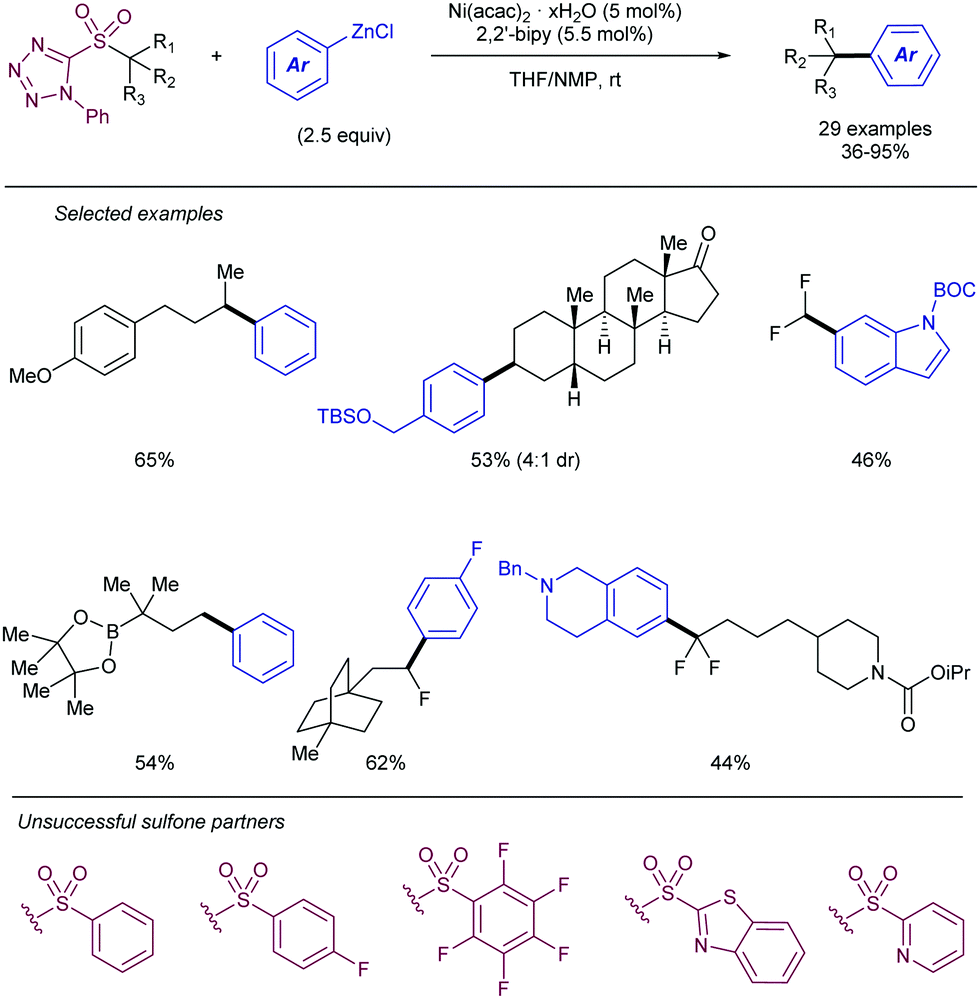 | ||
| Scheme 72 Ni-Catalysed Negishi-type cross-coupling with PT-alkyl sulfones. | ||
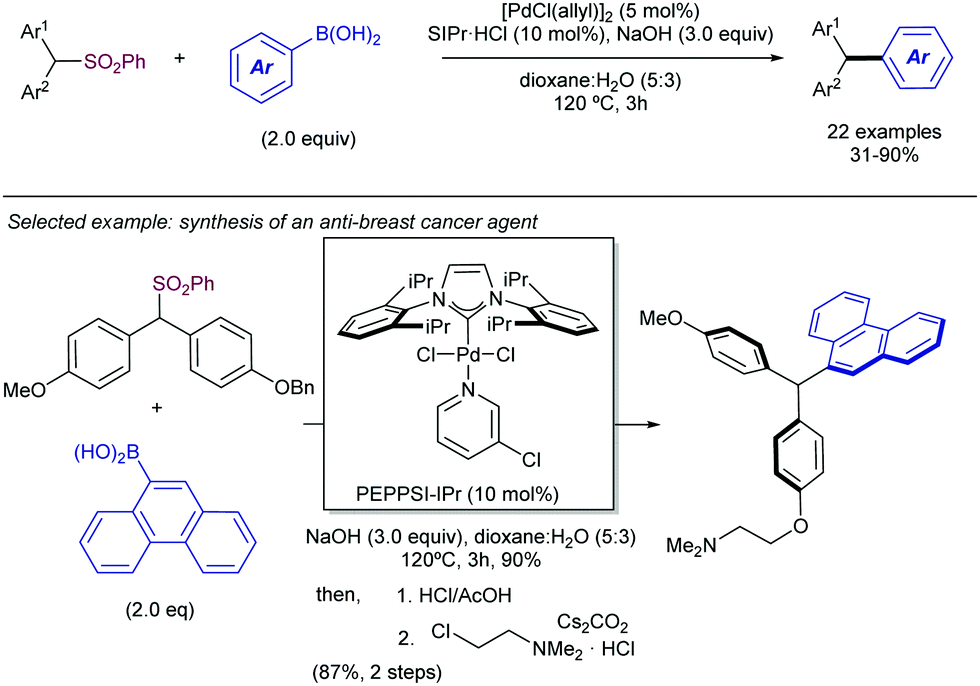 | ||
| Scheme 73 Pd-Catalysed Suzuki–Miyaura reaction with benzylic sulfones. | ||
The same group reported in 2017 a new family of aryl benzylic sulfones as a highly reactive electrophiles for the Suzuki–Miyaura reaction with aryl boronic acids under Pd-catalysis (Scheme 74a).146 The key-point in the method consisted in the use of an electron-poor, 3,5-bis(trifluoromethyl)phenyl group attached at the sulfonyl group, allowing the use of fluorinated sulfones as competent electrophiles in this important synthetic transformation. Apart from an exceptionally varied structural scope, both regarding the sulfone and the boronic acid counterparts, the method showed high applicability in gram-scale experiments. Importantly, the authors reported the iterative cross-coupling of halide- and sulfone-containing substrates, illustrating that different reaction conditions can be manipulated in an orthogonal manner to selectively afford the functionalization product from each reactive functional group (Scheme 74b). The utility of the method was further highlighted in the short-step synthesis of the thyroid hormone receptor β-selective analogue GC-24, with properties against arteriosclerosis and obesity (Scheme 74c).
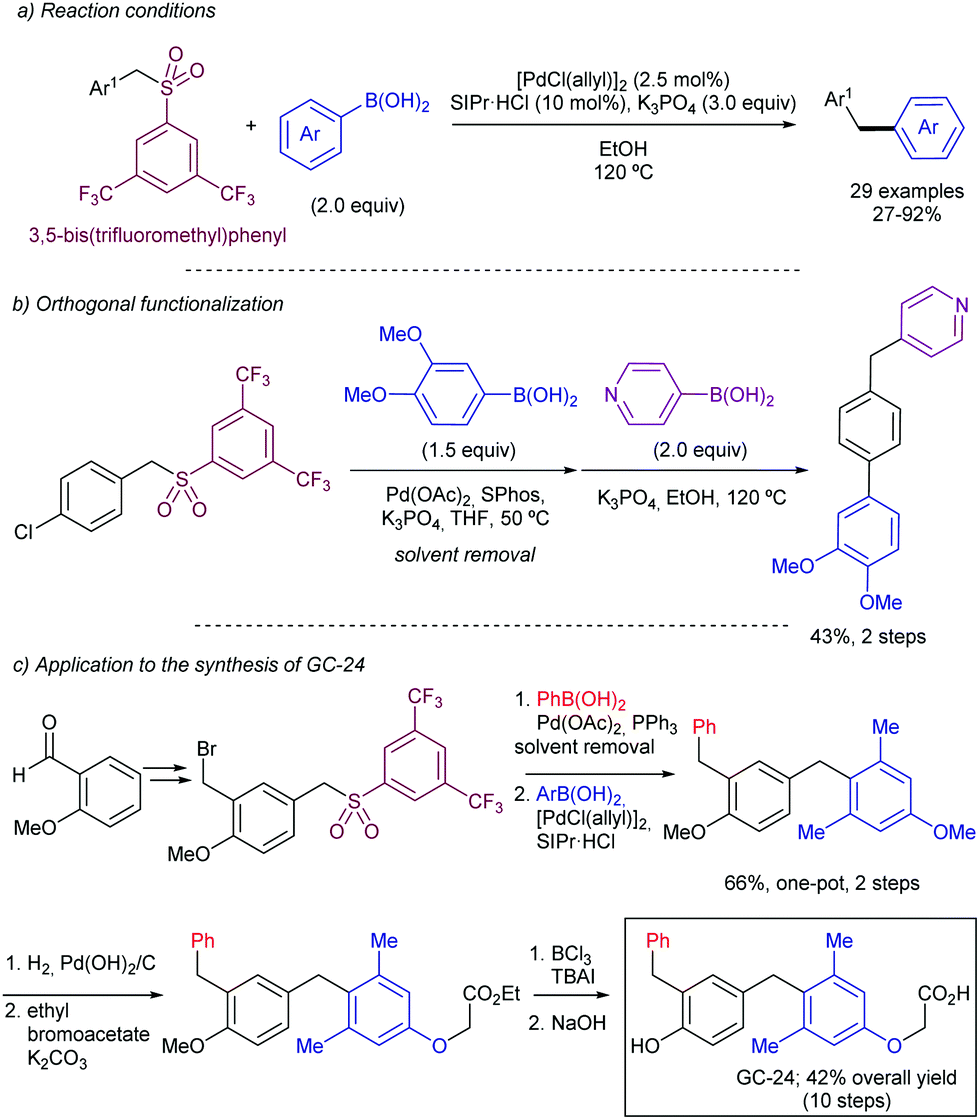 | ||
| Scheme 74 3,5-Bis(trifluoromethyl)phenylsulfones as electrophiles in Pd-catalysed Suzuki–Miyaura reactions. | ||
The reactivity of the 3,5-bis(trifluoromethyl)phenyl sulfonyl group has been exploited in other functionalization reactions upon activation of the C–S bond under Pd catalysis. In this regard, Crudden reported in 2017 that these sulfones undergo desulfonylative cross-coupling in presence of an acidic 1,3-oxazole in combination with a base (Scheme 75a).147 In this case the nucleophilic partner is the deprotonated 1,3-oxazole. This reaction furnished unsymmetrical triarylmethanes in which one of the aromatic rings is the N,O-heterocyclic unit. The strategy has been also described under Cu catalysis, in which the reaction is believed to proceed via an intermediate carbene species (Scheme 75b).148 This intermediate could proceed from the formation of the α-sulfonyl anion which transmetalates with copper to generate an α-copper sulfonyl species. Further desulfonylation is induced by the formation of the carbene, which is subsequently attacked by the deprotonated oxazole generating the final product via protodemetalation.
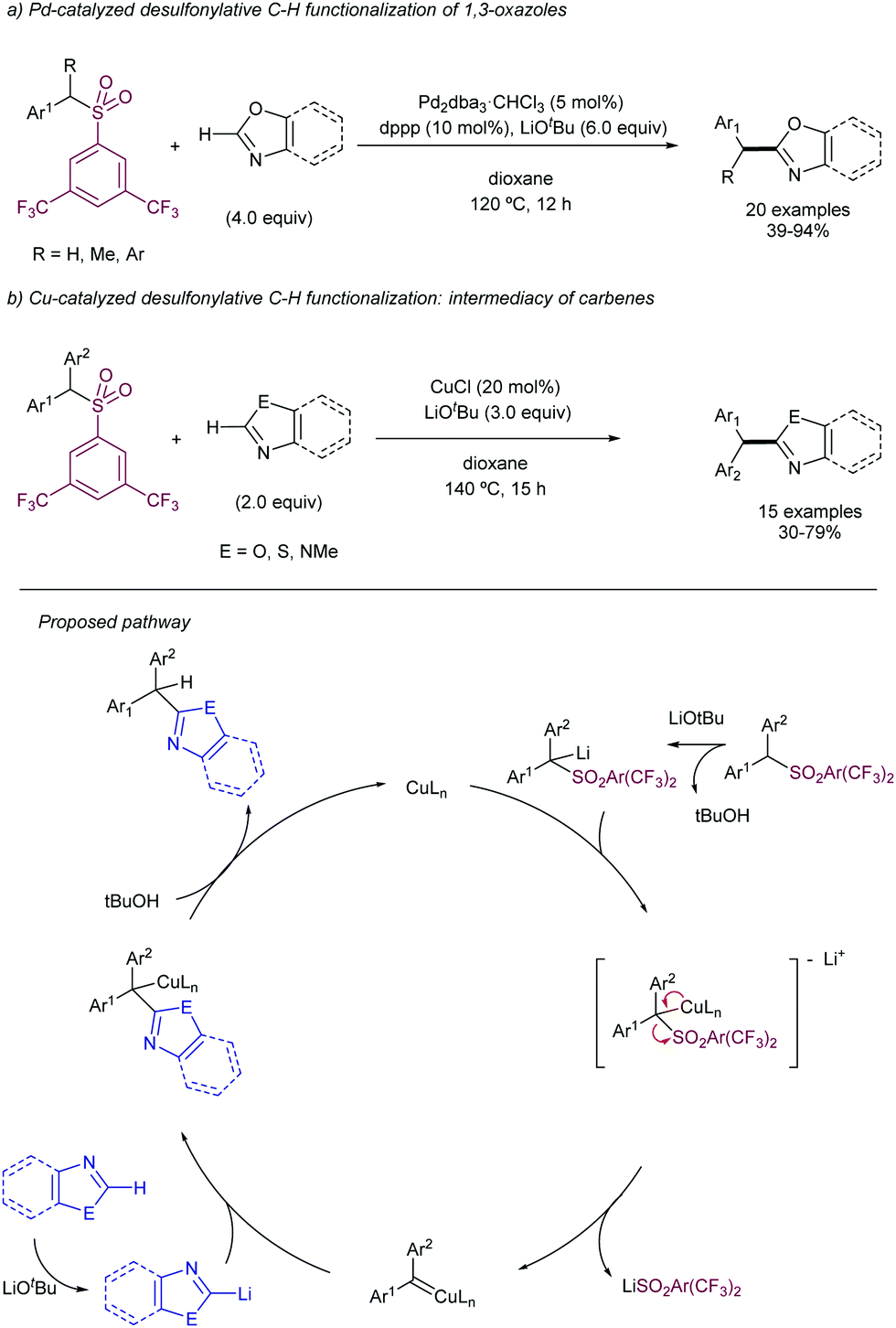 | ||
| Scheme 75 3,5-Bis(trifluoromethyl)phenylsulfones in C–H Functionalization of aromatic rings. | ||
Other fluorinated, electron-poor groups have shown high reactivity in the Pd-catalysed desulfonylative Suzuki–Miyaura reaction. Crudden has reported the use of a CF3-containing sulfone as a versatile functionality.149 This reactivity was exemplified using α-fluoroalkylaryl trifluoromethyl sulfones as electrophiles, facilitating the introduction of fluorine-containing groups in aryl structures without unstable organozinc reagents (Scheme 76). Other groups typically employed for the activation of the C–S bond, such as phenyl, 3,5-bis(trifluoromethyl)phenyl, 2-pyridyl, benzothiazole, and 2-phenyl tetrazole, did not afford the cross-coupling product. Thus, the effect of the CF3 group in the reaction mechanism was studied by means of theoretical calculations. The results indicated that the oxidative addition step is driven by a prior coordination of the aryl group present in the sulfone substrate, and kinetically facilitated by the CF3 group in comparison with the rest of the aromatic groups. Additionally, the CF3 group stabilizes thermodynamically the resulting Pd(II)-product as a result of the oxidative addition, explaining the high, and preferential reactivity of CF3-containing sulfones in comparison with other substituents. This unique feature permits its use in the orthogonal functionalization of readily available substrates, with high functional group compatibility including other sulfonyl groups.
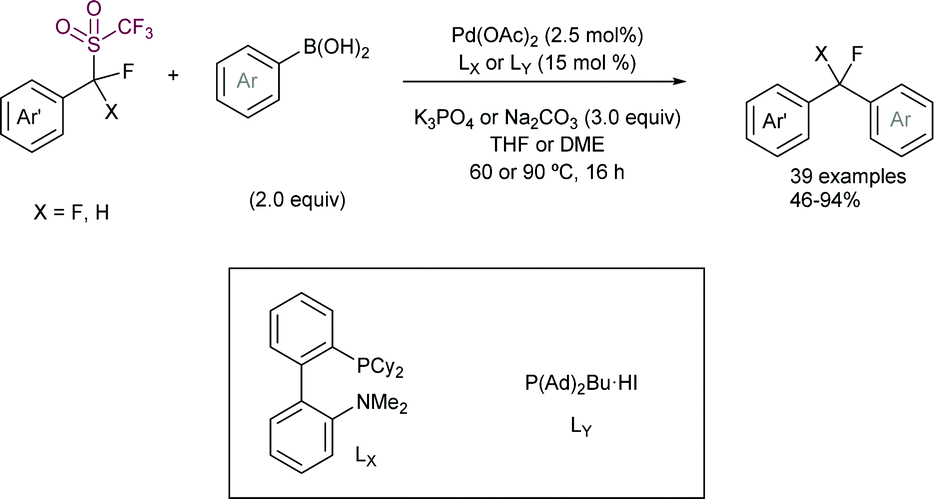 | ||
| Scheme 76 α-Fluoroalkylaryl trifluoromethyl sulfones as electrophiles. | ||
Additionally, Crudden has reported the construction of all-carbon quaternary centers by using tertiary sulfones as electrophilic substrates via a Ni-catalysed Suzuki–Miyaura reaction (Scheme 77a).150 The use of the one of the ligands developed by Doyle was necessary for high reactivity, as the use of other P-donor ligands resulted in a negligible or no reactivity. Poor reactivity was found for electron-poor aryl boroxines, in contrast with electron-rich partners, which gave the desired product in high yields. The applicability of this method was demonstrated in the synthesis of a vitamin D receptor modulator in which two of the key steps involved reactivity imposed by the presence of a sulfone (Scheme 77b). Firstly, in the α-functionalization of the alkyl sulfone via deprotonation/alkylation strategy, and then in a second Ni-catalysed desulfonylative cross coupling to install the required all-carbon quaternary center. Further derivatizations of the newly aryl ring led to the final structure of the target molecule.
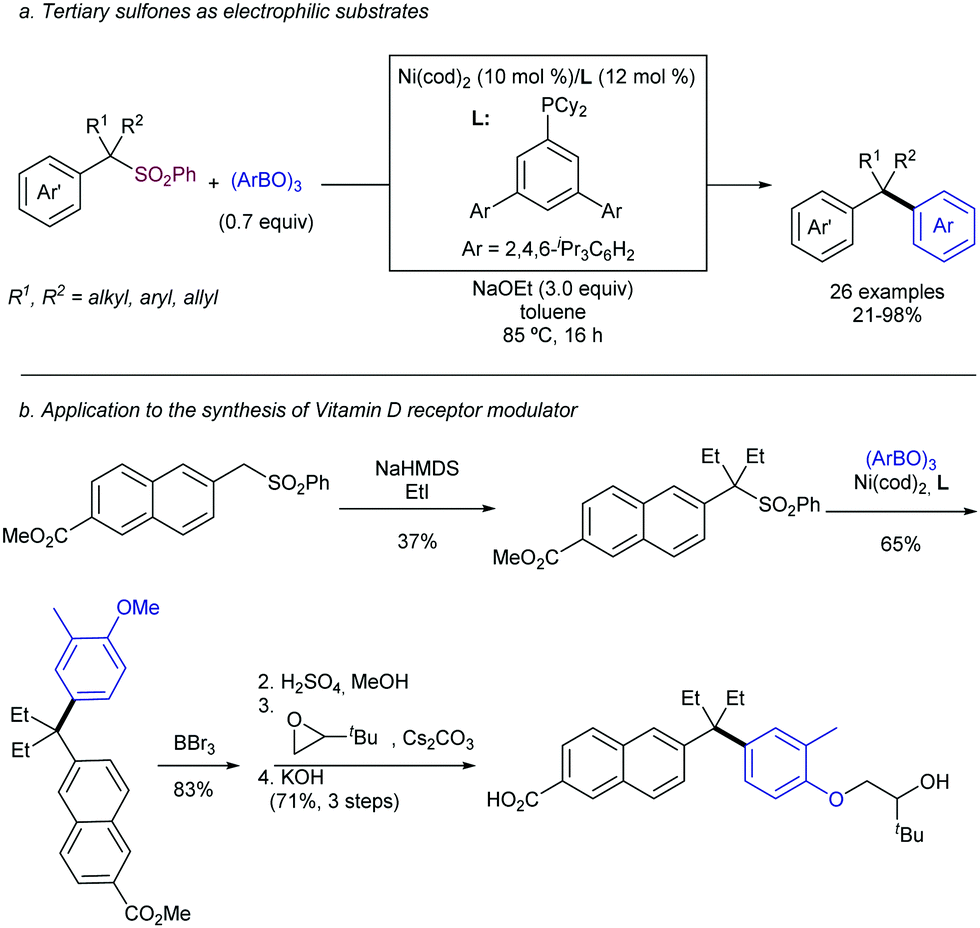 | ||
| Scheme 77 Construction of all-carbon quaternary stereocenters via Ni-catalysed Suzuki Miyaura with benzylic sulphones as electrophiles. | ||
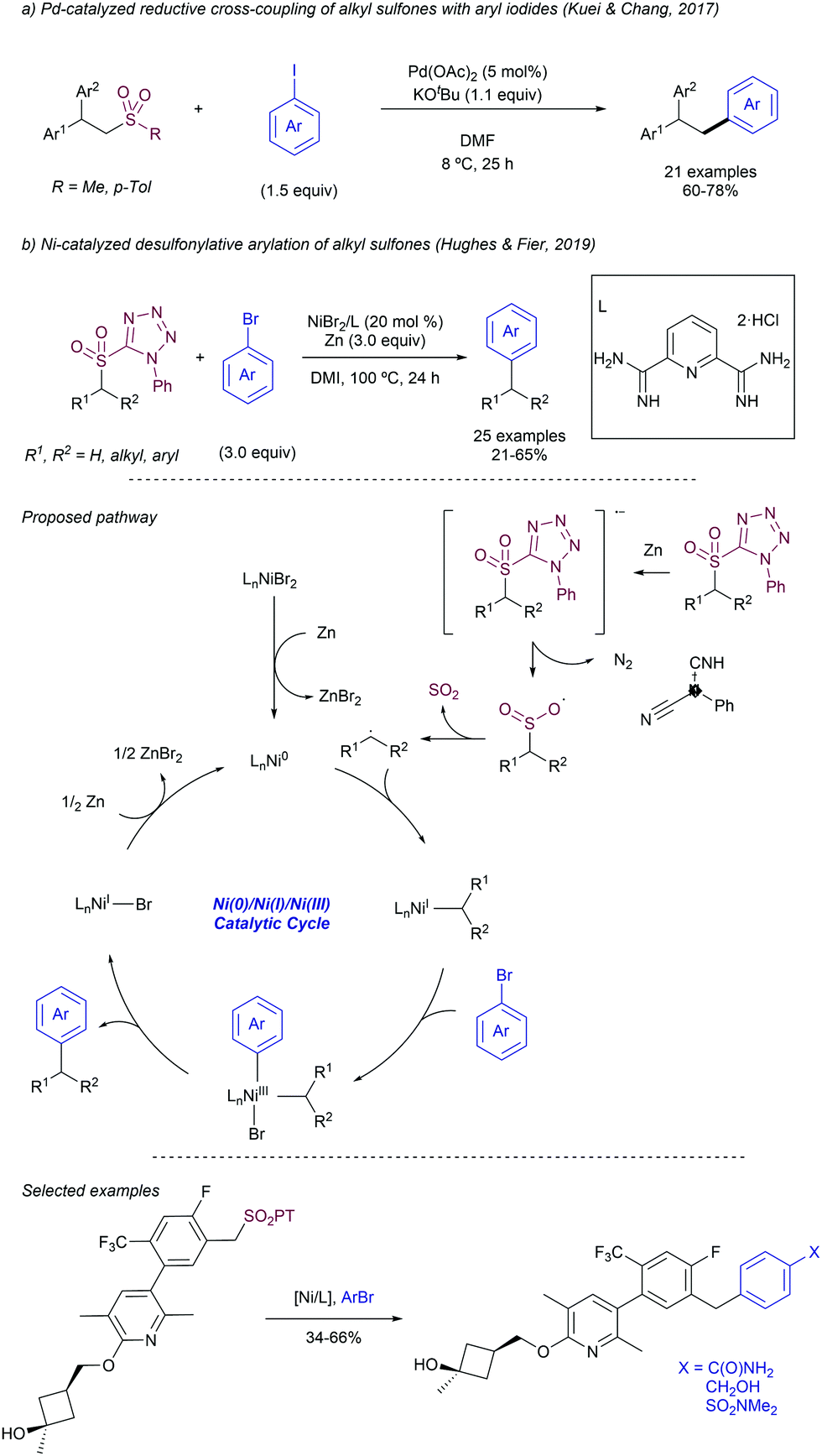 | ||
| Scheme 78 Alkyl sulfones in reductive cross-coupling chemistry. | ||
Another example of this reactivity has been recently reported by Hughes and Fier harnessing the ability of 1-phenyl tetrazole sulfones to generate alkyl radicals (Scheme 78b).154 In presence of Zn as a reductant, the PT sulfone substrate is reduced via SET generating a radical anion. This species then evolves via N2 liberation to form a radical sulfinate, which generates the corresponding alkyl radical upon extrusion of SO2. Once the alkyl radical is formed, a Ni(0) species undergoes one-electron oxidative addition with the radical to generate a Ni(I)-alkyl intermediate. In presence of the aryl bromide, the Ni(I) is oxidized to a Ni(III), which further undergoes reductive elimination to generate the coupled product, with concomitant formation of a new Ni(I) species which is reduced in situ in presence of Zn closing the catalytic cycle. However, a major problem of this reaction is the over-reduction of the alkyl sulfonyl radical from the PT sulfone, which renders an inactive alkyl sulfinate. Nevertheless, due to the high functional group compatibility, and the creation of C(sp2)–C(sp3) bonds in a direct fashion, the methodology could be highly useful as a key step for the assembly of complex molecules. In a related work Chen, Qian, and Sun155 exploited the Ni-catalysed reductive cross-coupling of sulfonyl-containing peptides with C(sp2)-tosylates using Zn as terminal reductant (Scheme 79). This strategy enables the post-synthetic modifications of peptides, thus enabling the access to unnatural amino acids on demand. The use of NiCl2(Py)4 in combination with dtBBPy as pre-catalyst system gave the best results. Addition of TABI was found to promote the reaction, likely by formation of intermediate alkyl iodides.
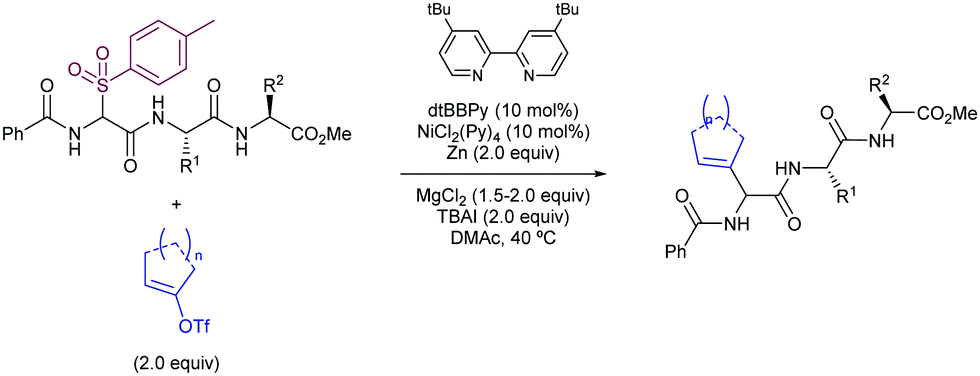 | ||
| Scheme 79 Modification of peptides via reductive cross-coupling. | ||
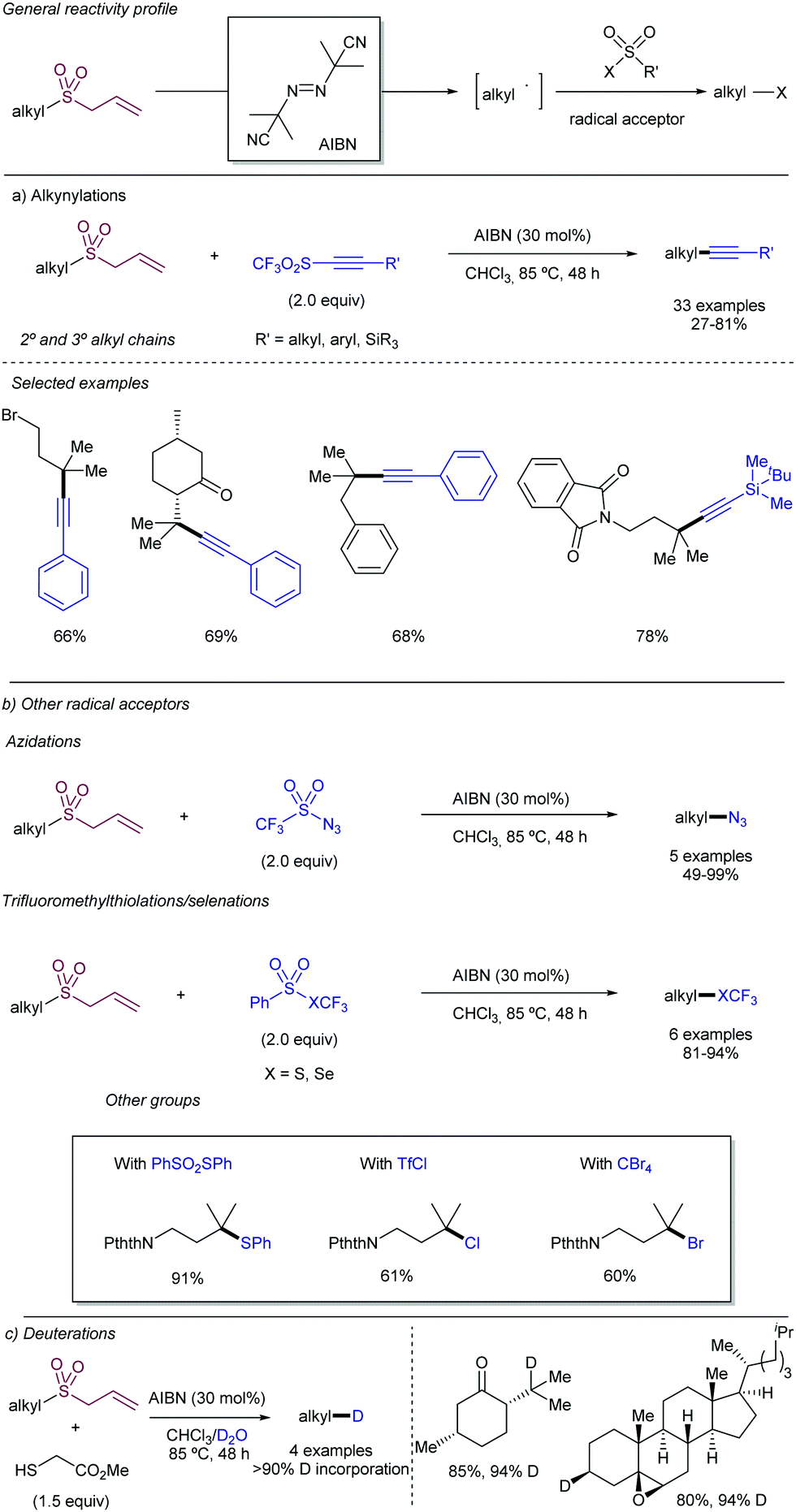 | ||
| Scheme 80 Radical-mediated transformations employing alkyl sulfones. | ||
As shown above, alkyl sulfones serve as a readily and valuable alkyl radical precursors. A powerful application of this strategy is the use of fluorinated aryl sulfones as sources of fluorinated alkyl radicals, which undergo insertion into a high variety of functionalities, thus rendering a simple method for the direct fluorination of organic molecules. Such a transformation is highly desirable in drug discovery and related applications in medicinal chemistry because of the intrinsic interest of fluorinated molecules in this area. Ni and Hou described the activation of RF–SO2Ar bonds by means of photocatalysis, in which the benzothiazole aryl group demonstrated superior reactivity in comparison with other aromatic and heteroaromatic groups (Scheme 81a).157,158 The homolytic scission of the RF–S bond was possible using Ru(bpy)3Cl2 as photocatalyst, promoting the generation of the corresponding RF˙ radical, which rapidly underwent insertion into isocyanides to deliver fluoroalkylated phenantridine derivatives. This approach has been used for the introduction of fluorinated moieties into different organic structures. For instance, Fu reported the oxydifluoromethylation of olefinic amides by addition of the corresponding RF˙ radical and intramolecular cyclization (Scheme 81b),159 thus forming six- and five-membered heterocycles with a –CF2H group. Zou also reported a similar strategy for the introduction of –CF2H or –CF3 groups by arylfluoromethylation of a related substrates toward the synthesis of fluorinated isoquinolinediones (Scheme 81c).160 Other important applications of this strategy include the synthesis of fluorinated oxindoles from the corresponding N-arylacrylamides reported by Zhu in 2019,161 and the introduction of radioactive 18F isotopes in relevant biologically active molecules by Lemos, Genicot, and Luxen.162
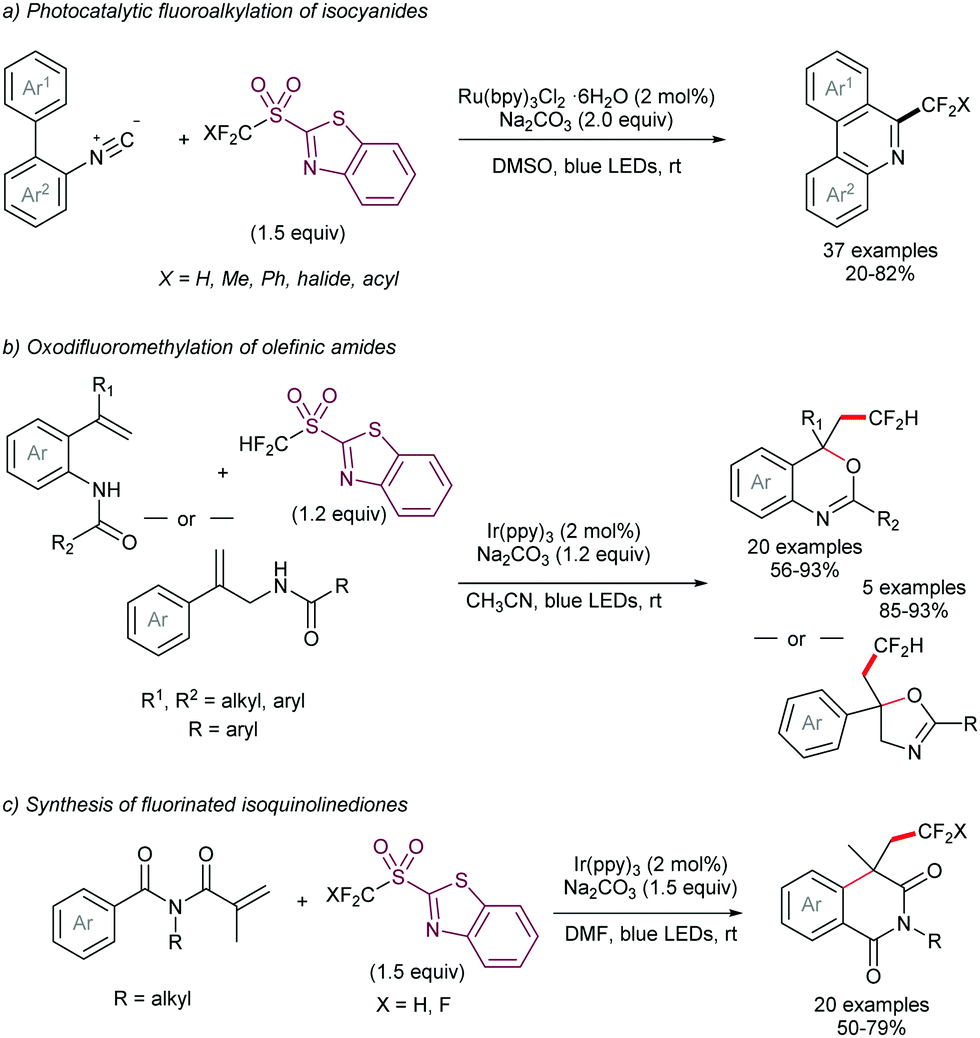 | ||
| Scheme 81 Photocatalytic approaches toward fluoromethylation using benzothiophenesulfones. | ||
3. Catalytic C–C double bond formation by Julia-type transformations
Olefins are essential building blocks found in functional and biological molecular structures,163 and are synthetically useful functional groups in organic synthesis as a consequence of their exceptional reactivity.164 Therefore, the development of synthetic tools for the creation of C–C double bonds in a straightforward manner is a central interest in synthetic chemistry.165 In this regard, the use of sulfones as olefination partners for the construction of the unsaturated π bond has found tremendous momentum in organic synthesis. In this section we discuss the development of alkenylation methods enabled by the intrinsic reactivity of sulfones under catalytic conditions. Early examples of catalytic alkenylations were reported by Julia in the 1980's. After α-metalation of allyl phenyl sulfones employing BuLi, catalytic amounts of Ni(acac)2 delivered alkenylated product as a mixture of stereoisomers.166 Further studies demonstrated that Cu-, Pd-, and Fe-based catalysts could perform the same transformation. However, because of the requirement of strong bases and the obtention of symmetrical, non-stereoselective alkenes, this approach has not found wide application. As an alternative to homocoupling of sulfones, the olefination of carbonyl-containing groups by reaction with metalated sulfones (the Julia-Lythgoe olefination) is one of the most extended methods for the stereoselective construction of C–C double bonds. A modified version of this reaction involves heteroaromatic sulfones that undergo desulfonylation by in situ Smiles rearrangement without the need to isolate the β-alkoxysulfone intermediate.Although this transformation is practical, its catalytic version is much more appealing and opens new reactivity scenarios. In this regard, the first catalytic olefination employing sulfones was reported by Milstein in 2014 (Scheme 82).167 The team employed a Ru-catalysed acceptorless dehydrogenative approach to convert benzylic alcohols into the corresponding olefin upon treatment with dimethyl sulfone and KOtBu that enabled the direct use of alcohols instead of their oxidized form for the creation of the C–C double bond. Remarkably, the reaction was highly selective toward the formation of the olefination product and H2 liberation. With regard the sulfone partner, both electron-poor and rich- benzylic sulfones reacted with good yields, allowing the access to (E)-1,2-disubstituted olefins. Attempts to isolate key reactive intermediates led to the obtention of the Ru catalyst as a polymeric complex, in which the ruthenium atom is bound to the sulfone through the deprotonated C–H bond, along with dearomatization of the pincer ligand (Scheme 82a). This last feature could be important, as these complexes potentially activate alcohols via metal-ligand cooperation. As in the Julia reaction, it is expected that the corresponding aldehyde from the benzylic alcohol were involved in the process, but the reaction between benzaldehyde and dimethylsulfone under the standard conditions led to low quantities (10% yield) of the desired styrene (Scheme 82b). Additionally, potential intermediate β-hydroxysulfone gave only traces of the product. Thus, the authors propose that a typical Julia-type mechanism is not likely to be operative under the reaction conditions, and that non-free, metal-coordinated aldehyde seems to be necessary (Scheme 82b). This approach has been exploited in other metal-catalysed dehydrogenative processes in which the sulfone group is maintained, giving rise to an interesting family of sulfonylated building blocks.168
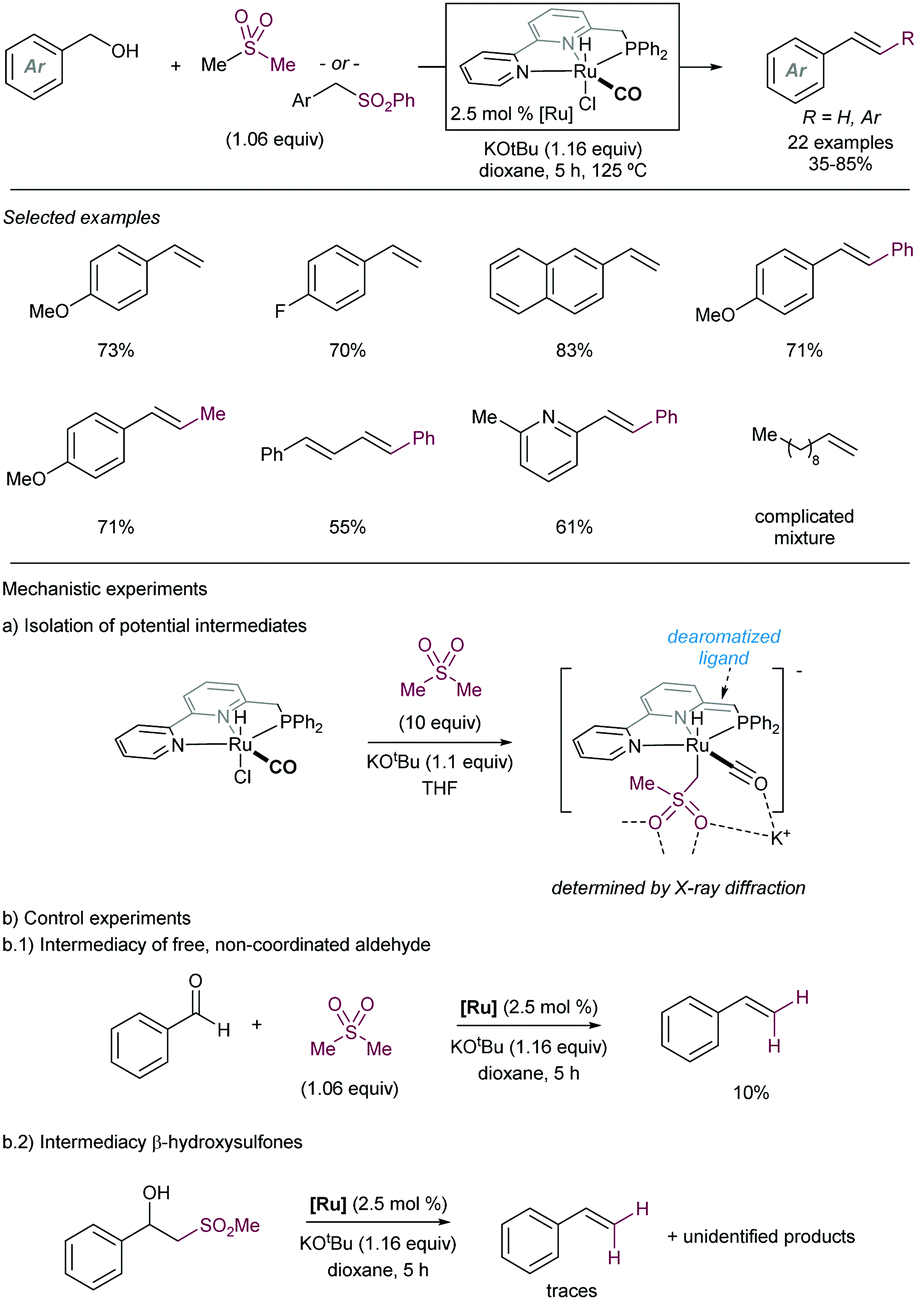 | ||
| Scheme 82 Ru-Catalysed acceptorless dehydrogenative olefination of benzylic alcohols. | ||
Later, Shimizu studied the direct olefination of alcohols employing sulfones as alkenylating reagents under heterogeneous catalysis.169 The authors screened several supported Pt catalysts pre-reduced with a H2 atmosphere at 300 °C to evaluate their potential activity in the olefination of benzylic alcohol and dimethylsulfone employing KOtBu (Scheme 83). Carbon-supported Pt nanoparticles (Pt/C) demonstrated superior activity in comparison with other Pt-based materials. Additionally, other transition metals supported on carbon also resulted in a lower catalytic activity (Pt > Rh > Ir > Ru > Pd > Re > Cu > Ni > Co). Interestingly, the reaction could be carried out under O2 atmosphere without apparent negative influence on the yield, suggesting that O2 is not the hydrogen acceptor of the system. One important feature of this protocol pertains to the possible recovering of the catalyst after each catalytic cycle. Indeed, the Pt/C catalyst was used and recovered up to five cycles with no marked impact on the reaction yield. Several substrates were studied, furnishing the corresponding olefin with excellent yields and superior catalytic activity compared to the Ru-catalysed conditions, as exemplified by the high value of the TONs for different substrates at 10 mmol scale. Another important contribution of this work refers to the use of alkyl and allylic alcohols, which gave complicated mixtures using the ruthenium catalyst developed by Milstein.167 Again, when the reaction was carried out using an aldehyde instead of the alcohol, low yields of the olefin product were obtained. Additionally, a KIE value of 2.2 was obtained by using the corresponding benzylic alcohol and the α-deutero benzylic alcohol. Thus, the dehydrogenation of the alcohol was deemed to be involved in the rate-limiting step. In accordance with these results, the reaction orders for each component were found to be positive for the benzylic alcohol and the KOtBu, 1.2 and 1.5, respectively. On the contrary, a negative value of −0.6 was found for dimethylsulfone, suggesting that this species is not involved in the rate-limiting step. Thus, the authors claimed to the importance of having metal-coordinated intermediates and their role to drive the reaction until completion. In this regard, the postulated mechanism involves a first, rate-determining step dehydrogenation of the corresponding alcohol by the Pt units present in the Pt/C catalyst. This dehydrogenative pathway leads to the corresponding carbonyl compound adsorbed into the catalyst, which is likely the reason behind the excellent reactivity profile. The carbonylic substrate is then attacked by the α-sulfonyl anion derived from the deprotonation of the parent sulfone by the KOtBu, thus giving rise to a transient β-hydroxysulfone. Final elimination of a sulfonate salt furnishes the desired olefin.
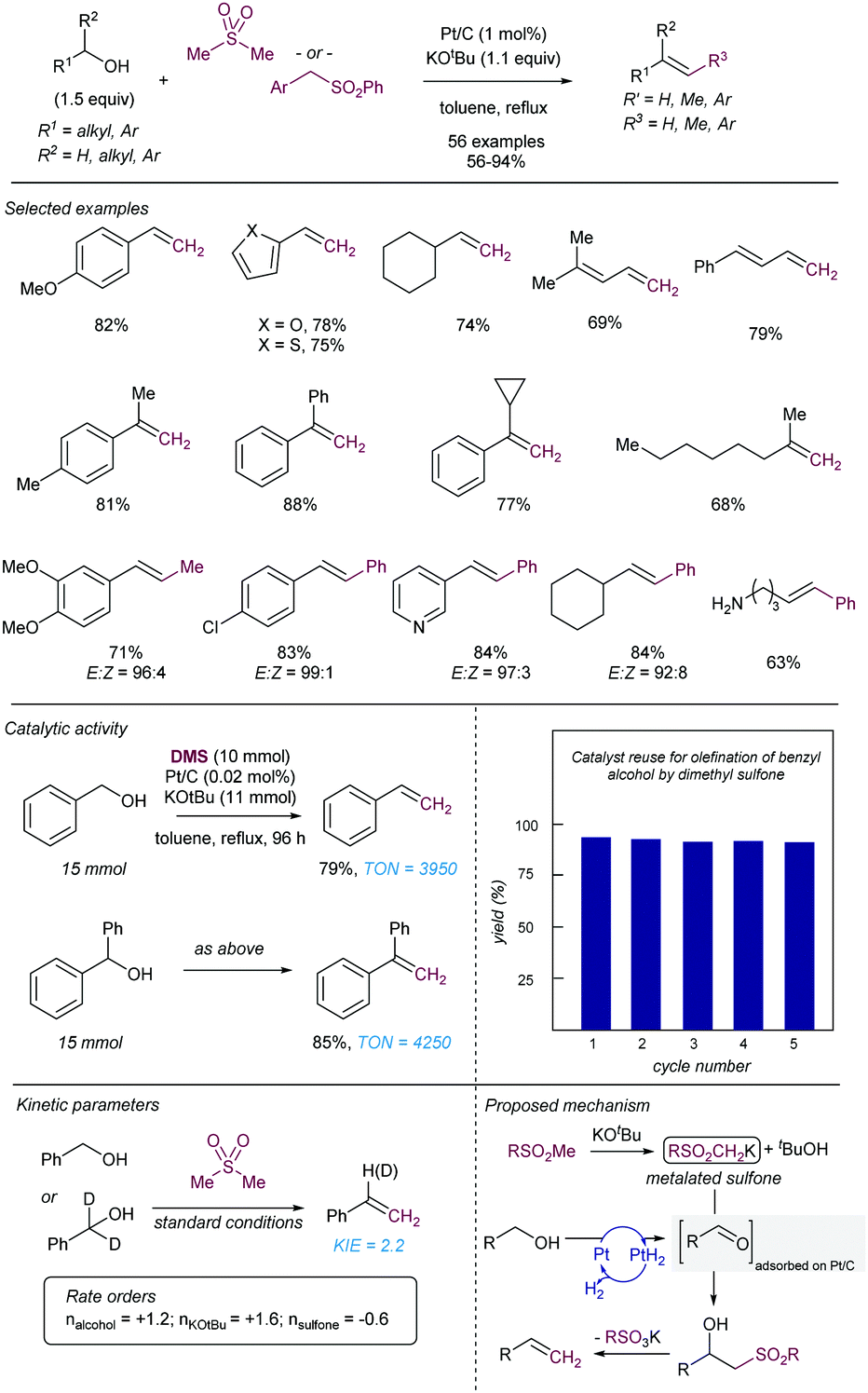 | ||
| Scheme 83 Heterogeneous olefination of alcohols catalysed by Pt/C. | ||
The last years have witnessed a renewed interest in developing more sustainable, environmentally friendly processes involving the olefination of alcohols by using earth-abundant catalysts based on first-row transition metals. In this regard, the group of Balaraman reported in 2019 a method that enables the use of a well-defined Ni catalyst bearing a pincer ligand as an alternative to precious metals-based catalyst (Scheme 84).170 This method furnishes the corresponding olefin upon treatment of the benzylic alcohol with a sulfone in combination with KOtBu as a base and a Ni complex. Both electron-rich and poor aromatic rings as substituents for the benzylic alcohol were tolerated for this transformation, including halogens such as F, Cl and Br. Regarding the substitution at the sulfone moiety, when DMS was replaced by phenyl benzylsulfones, the corresponding benzylic group acted as the olefination partner, with different substituents at the aromatic groups of diverse electronic nature, including π-deficient and π-excessive rings. This approach allows the formation of (E)-1,2-disubstituted styrenes. However, when plain aryl alkylsulfones were studied no olefination product was observed, along with lack of reactivity when disubstituted benzylic alcohols were employed, thus representing the main limitation of this chemistry. An elegant application of this reaction system was illustrated by the synthesis of the biologically active Resveratrol (Scheme 84). In contrast with previous results observed using Ru and Pt catalysts, under these Ni-catalysed conditions, the use of benzaldehyde as a substrate furnished the olefination product in 70% yield. Indeed, when the benzyl alcohol was treated with the Ni catalyst under the standard conditions in the absence of the sulfone, the benzaldehyde was obtained in 80% yield, along with the liberation of H2 gas. Additionally, the authors support the potential participation of transient Ni–H species, likely generated in the dehydrogenation step. Albeit attempts to isolate or detect the corresponding hydridic species in the reaction media were unsuccessful, the use of a more stable (PCy3)2NiBrH complex as a pre-catalyst gave the corresponding aldehyde in 23% yield.
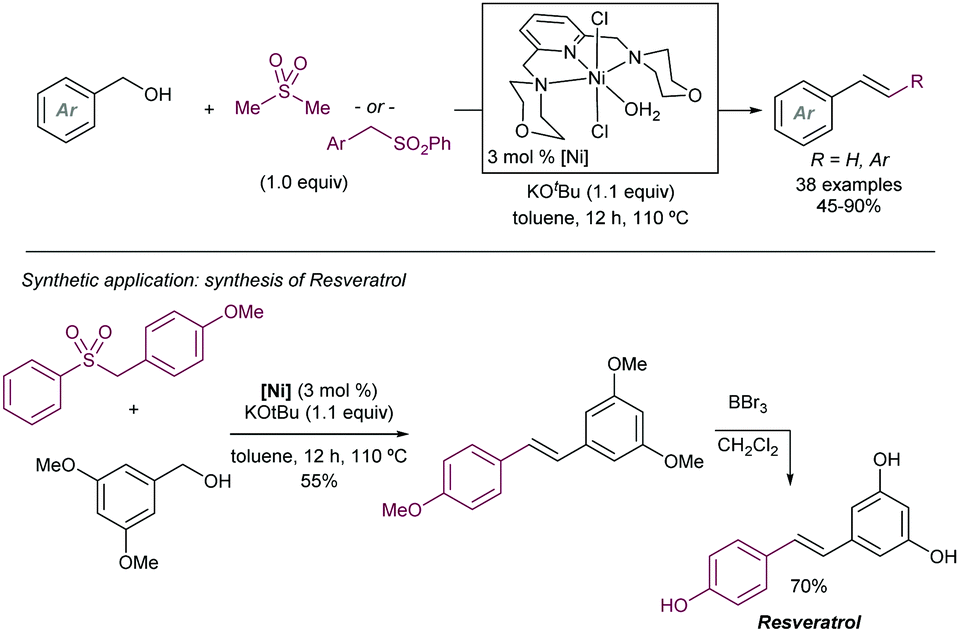 | ||
| Scheme 84 Ni-Catalysed olefination of benzylic alcohols with alkyl and benzyl sulfones. | ||
Simultaneously, Maji described the same reaction employing the combination of NiBr2/neocuproine as a catalytic system (Scheme 85).171 Mechanistic insights revealed that different KIE values were obtained depending on the substrate employed. When an α-deutero benzylic alcohol was studied in comparison with the non-deuterated counterpart, a KIE value of 3.1 was obtained (Scheme 85, mechanistic experiments). On the contrary, performing the experiment with the deuterated sulfone led to a KIE of 1. These results demonstrated that the dehydrogenation process is involved in the rate-determining step, while the sulfone must be involved in a fast base-mediated Aldol-type reaction with the aldehyde formed in situ, generating an intermediate β-hydroxysulfone. In accordance with Balaraman's work, the use of (PCy3)2NiBrH as a pre-catalyst also led to corresponding product with comparable efficiency, which is in accord with the intermediacy of Ni–H species (Scheme 85, intermediacy of Ni–H). Finally, alkenyl sulfones originated from base-mediated elimination of the OH group from the corresponding β-hydroxysulfone mentioned above could be likely intermediates in this process. In this vein, treatment of an alkenyl sulfone under the reaction conditions using the (PCy3)2NiBrH pre-catalyst delivered an olefin in 85% yield (Scheme 85, intermediacy of alkenylsulfones). Also, when an alkenyl sulfone was reacted with an α-deutero benzylic alcohol under the optimized reaction conditions, an olefin was isolated with 14% D incorporation at the position coming from the sulfone, and 62% D incorporation from the alcohol. Additionally, ArSO2K salts were isolated and identified by 1H NMR spectroscopy and HRMS analysis.
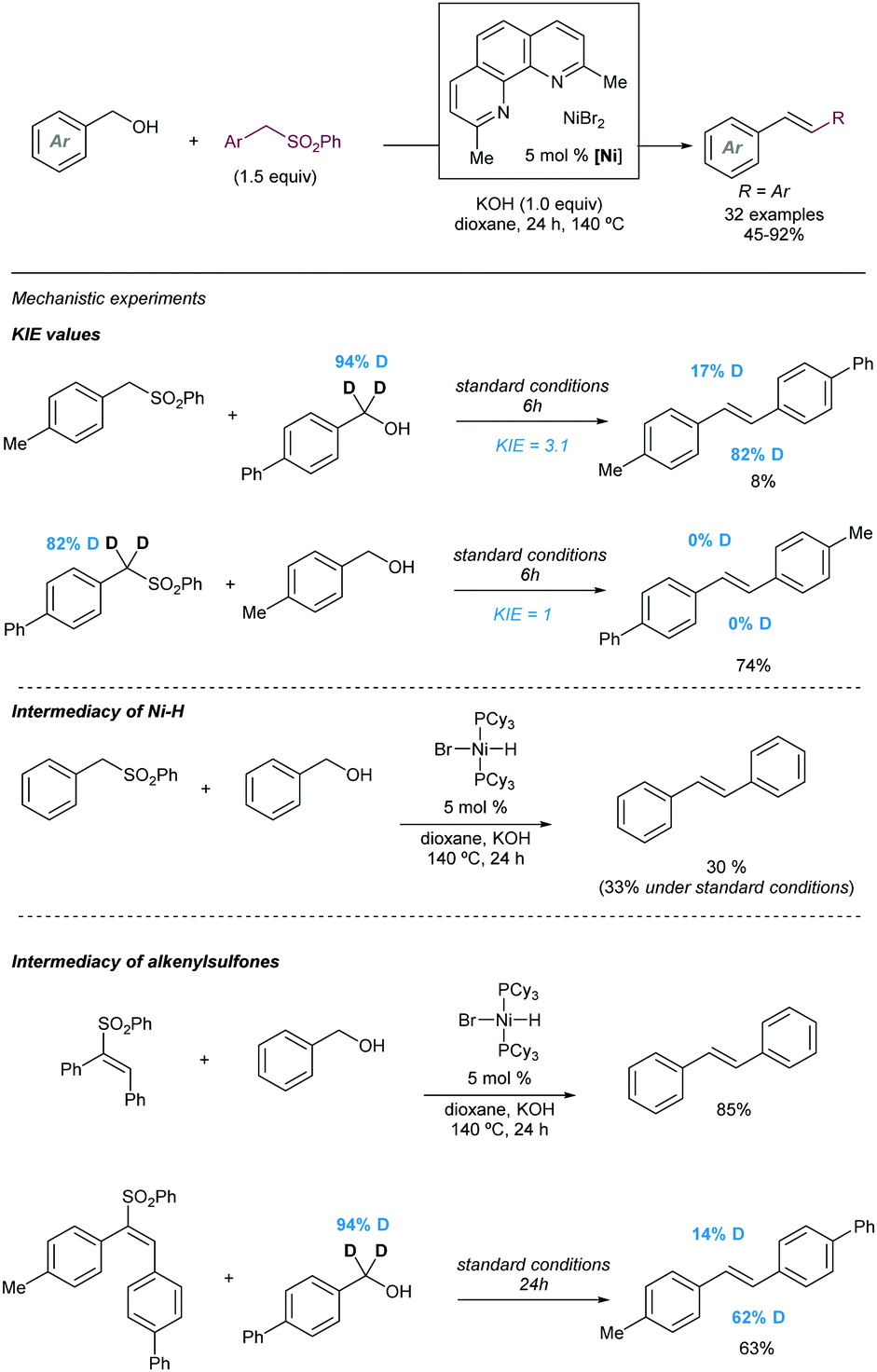 | ||
| Scheme 85 Ni-Catalysed olefination of alcohols reported by Maji. | ||
With these results, the authors suggested a mechanistic scenario in which the nickel catalyst is involved in an auto-tandem catalytic cycle (Scheme 86). Initially, the Ni-species is involved in the oxidation of the benzylic alcohol via a dehydrogenative pathway, to form the corresponding aldehyde and a Ni–H intermediate. The aldehyde then undergoes Aldol-type addition by the metalated sulfone with the concomitant elimination of water to deliver the alkenylsulfone. Finally, the initially formed Ni–H species would reduce the alkenylsulfone via a hydrogenation process, leading to an alkylsulfone which evolves into the alkenylated product via a base-mediated desulfonylation process.
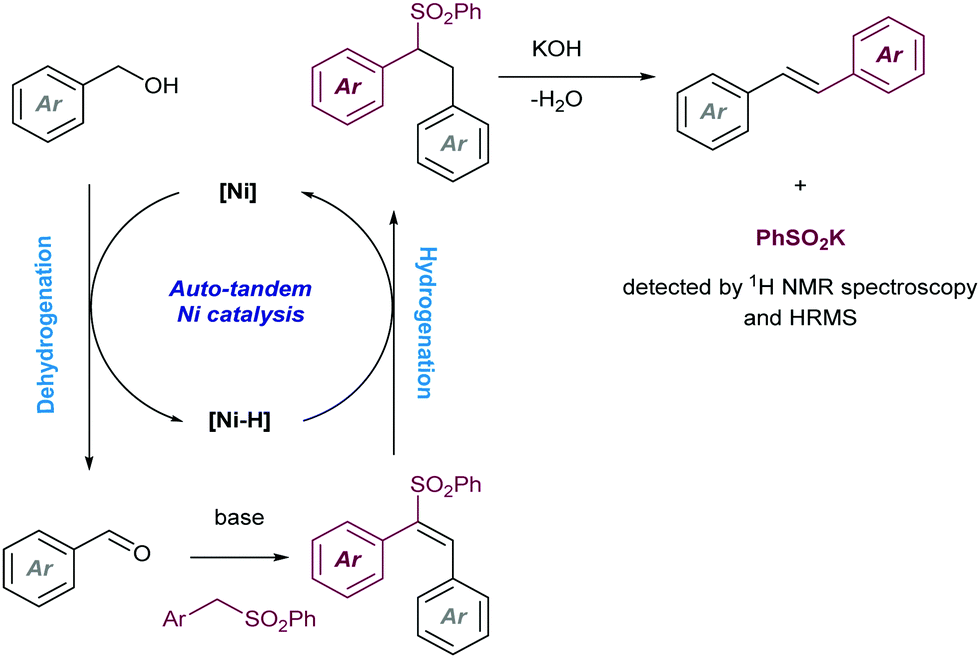 | ||
| Scheme 86 Proposed auto-tandem mechanism for the Ni-catalysed olefination of benzylic alcohols. | ||
More recently, Balaraman, Gupta and co-workers have broadened the catalytic arsenal for the olefination of benzylic alcohols employing a more sustainable metal complexes based on a Fe salt.172 The strategy follows the aforementioned features for other acceptorless dehydrogenative coupling sequences developed so far for this transformation, in which the use of FeCl2 in combination with the readily available 1,10-phenantroline as catalytic system delivered the desired olefin in medium to good yields (Scheme 87a). Both primary and secondary alcohols were found to react under this reaction conditions, in which the 1,1-diaryl alkene product was the major regioisomer when using secondary alcohols. Regarding this second substrate class, the use of angularly fused cyclic secondary alcohols in combination with dimethyl sulfone led to a different array of 1-methylnaphtalene compounds via a tandem acceptorless dehydrogenative cross-coupling followed by a dehydrogenative isomerization (Scheme 87b). Mechanistic experiments (Scheme 87c) revealed that benzylic alcohol is oxidized to the corresponding carbonyl compound in presence of the iron catalyst, along with the liberation of hydrogen gas. In addition, the corresponding ketone evolves toward the naphthalene derivatized upon treatment with dimethyl sulfone under the standard reaction conditions with catalytic amounts of the starting material. The reaction did not take place when the catalyst or the base were not present. Interestingly, when the ketone was treated only with dimethylsulfone, the base and the catalytic system a low yield of the desired product was observed. These results highlight the potential intermediacy on O-coordinated species that might enhance the catalytic performance of this reaction. Furthermore, the potential intermediates II and III led to the aromatized product in presence of the Fe-catalyst (Scheme 87c). Kinetic analysis of deuterated and non-deuterated alcohols provided a KIE value of 2.51, indicating that the dehydrogenative oxidation of the alcohol is the rate-determining step.
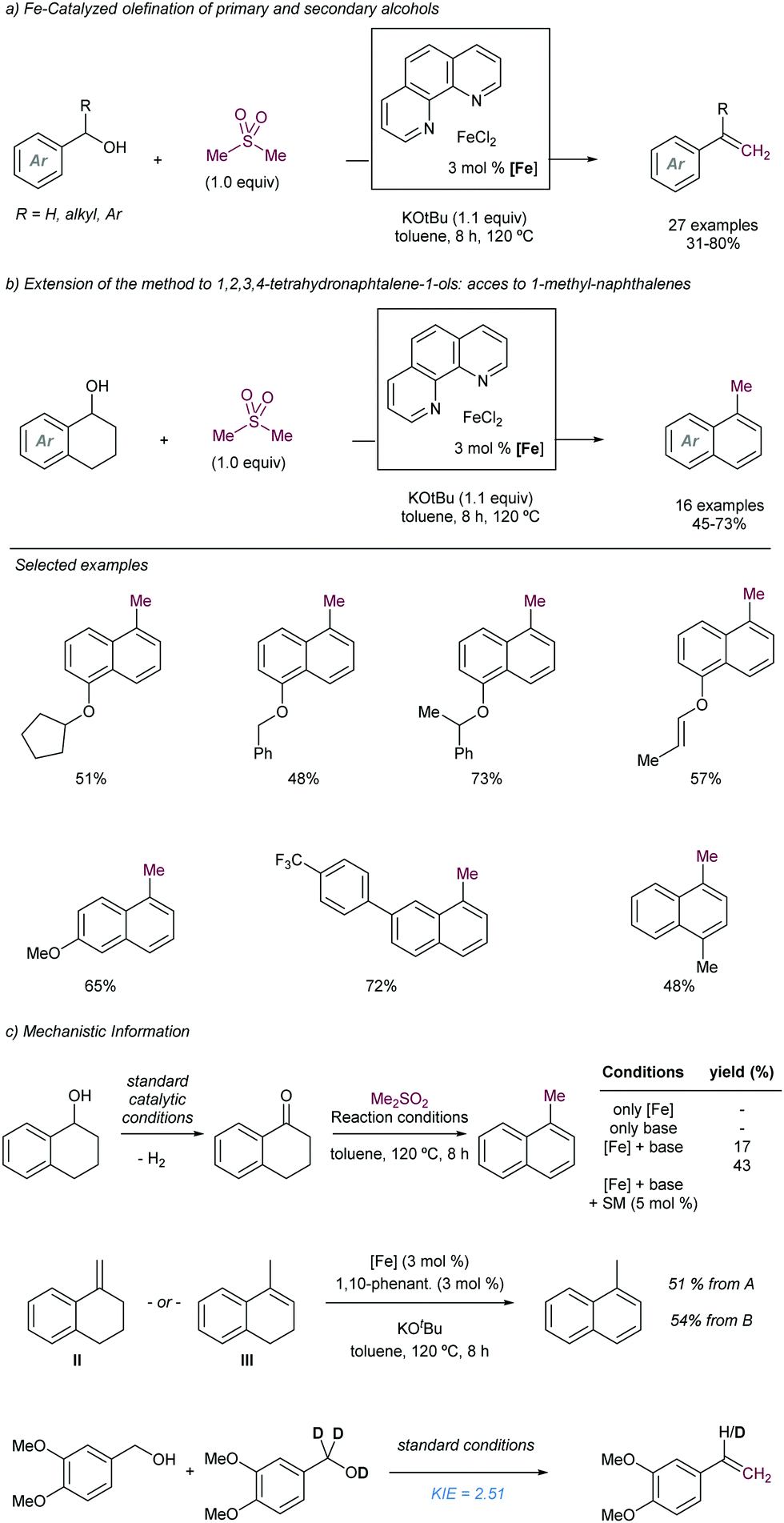 | ||
| Scheme 87 Fe-Catalysed dehydrogenative cross-coupling between benzylic alcohols and sulfones. | ||
Olefination protocols employing different types of sulfones have been also described for other substrates other than benzylic alcohols. For instance, Jang and co-workers discovered that thiols can be transformed into the corresponding alkene employing copper catalysis (Scheme 88).173 In this case the reaction was amenable to the use of α-sulfonylketones, leading to the α,β-unsaturated ketone with good yields. Different Cu(II)-based pre-catalyst worked well, with the readily available CuBr2 showing better catalytic performance. Interestingly the use of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) base was particularly effective, as other bases typically employed for this transformation such as KOtBu led to poor yields. However, in this case O2 was essential to gain synthetically useful yields, suggesting that either the dehydrogenative pathway needs a hydrogen acceptor or the active copper species need to be re-oxidized at some point of the reaction mechanism. Although the reaction delivered the corresponding unsaturated enone for a representative family of substrates, the scope including secondary benzylic thiols or alkyl derivatives for this transformation was not studied. A potential reaction mechanism for this transformation would commence with the copper-mediated oxidation of the thiol. This step is believed to occur via the deprotonation of an in situ formed dibenzyldisulfide, which is form from the thiol.174 In this regard, the copper catalyst in combination with O2 can accelerate the reaction, as the absence of copper led to the formation of the product, albeit in lower yields. After that, the thiobenzaldehyde can react in a Julia-type reaction with the α-sulfonylketone furnishing the corresponding enone, and phenyl thiosulfinate a by-product.
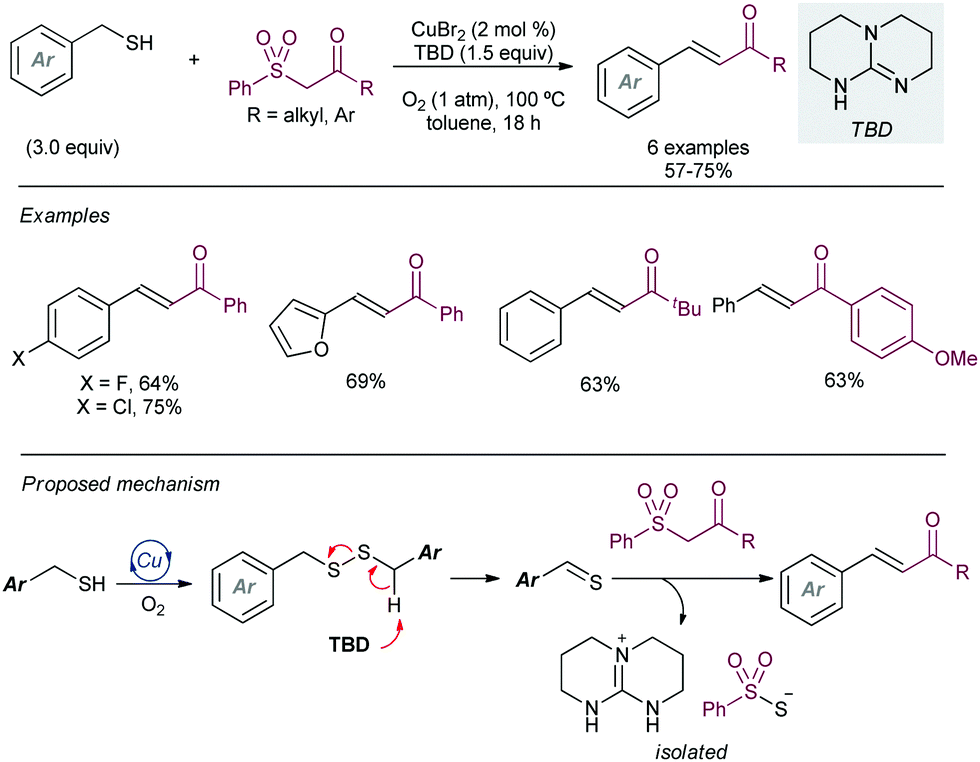 | ||
| Scheme 88 Cu-Catalysed olefination of benzylic thiols. | ||
Wang and co-workers reported the use of N-sulfonylhydrazones as suitable partners for the olefination reaction with sulfones employing copper catalysis (Scheme 89).175 The reaction was catalysed by CuI in presence of LiOtBu as base, yielding the alkenylated product with synthetically useful yields. Remarkably, N-sulfonyl hydrazones derived from alkyl-, aryl-, disubstituted-, and α,β-unsaturated ketones or aldehydes were found to be excellent reagents for this transformation, thus improving considerably the reaction scope, as this method surmounts different incompatibilities previously found with alcohol derivatives. In this regard, hydrazones derived from diarylketones reacted in excellent yields, which was important because diarylketones were found to be unreactive on previous reports with Julia–Kociensky-type protocols, likely because of their lower reactivity toward carbon nucleophiles.176 Notably, when N-tosylhydrazones were substituted by N-mesylhydrazones the authors found a slightly increased reactivity, allowing the use of lower temperatures and obtaining better yields. However, in some cases minor by-product formation was detected in the reaction media, derived from products generated by a Shapiro-type elimination.177 Since diazo compounds also gave the corresponding alkenylated product, the authors proposed the intermediacy of carbene-like intermediates derived from the N-sulfonylhydrazone in combination with the base. Thus, a catalytic cycle was suggested in which the reaction starts with the metalation of the sulfone upon reaction with LiOtBu, followed by transmetalation with copper to generate a Cu intermediate (Scheme 89, mechanistic proposal). This intermediate can react with the in situ formed carbene from the reaction between the N-sulfonylhydrazone with the base. The carbene then inserts into the C–Cu bond via migratory insertion, and final β-S elimination furnishes the alkenylated product along with a copper(I) sulfinate, which regenerates the α-cuprated sulfone.
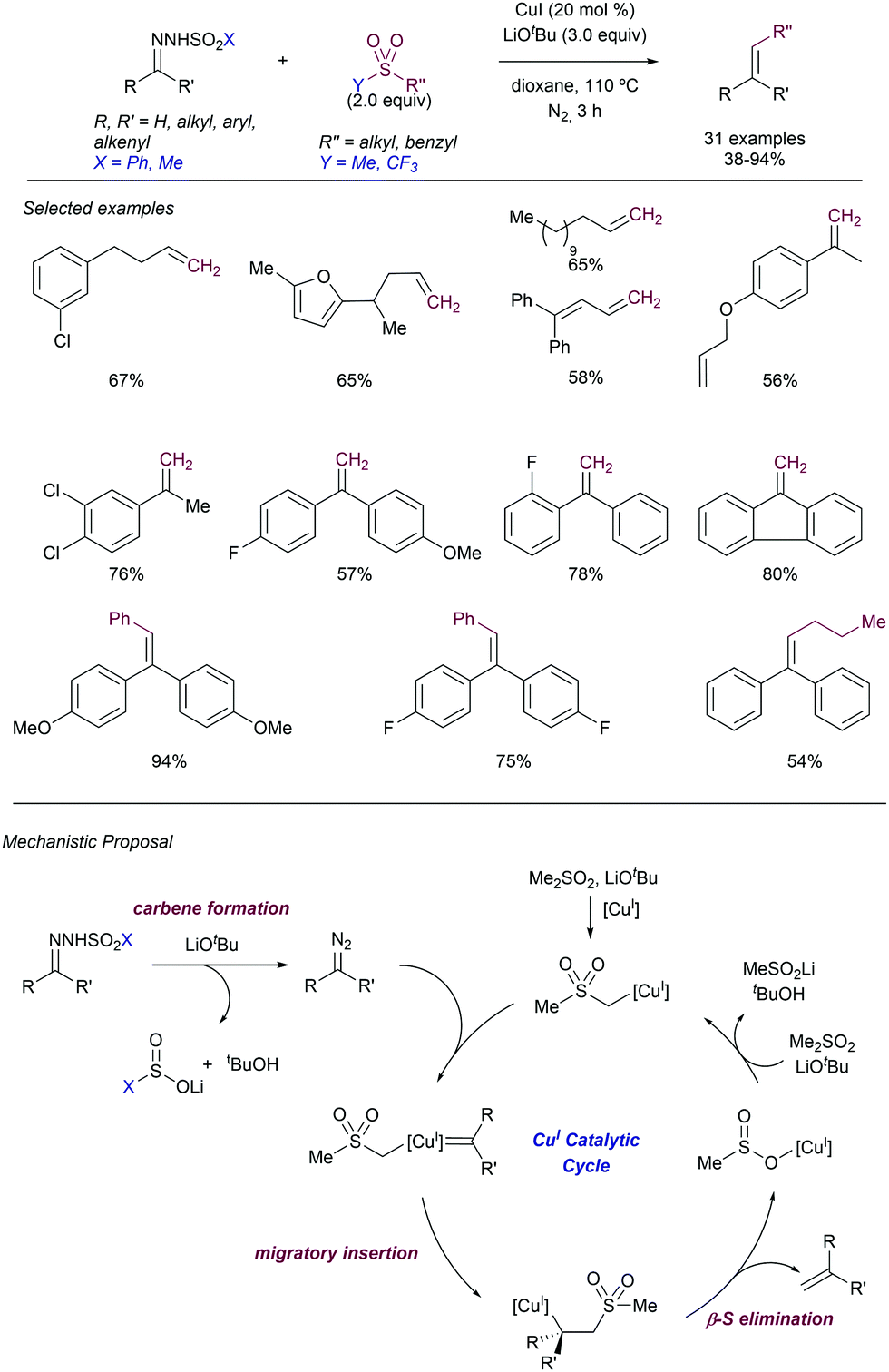 | ||
| Scheme 89 Cu-Catalysed alkenylation of N-sulfonylhydrazones with sulfones. | ||
4. Sulfones as cyanating reagents
The nitrile functionality (–CN) is a valuable functional group in organic chemistry because of its remarkable synthetic versatility. The introduction of this motif in chemical structures allows for the access to new chemical space due to the availability of transformative methods for this group.178 They are also good candidates in an array of reactions involving metal-mediated C–CN bond activation,179 and participate as key functionalities in the synthesis of an extensive family of heterocycles.180 Additionally, nitriles are found in biologically active molecules181 and represent important scaffolds en route to functional materials.182 Hence, the introduction of cyano groups in organic chemistry is an active area in which, despite of the availability of cyanation protocols, the development of novel strategies for this purpose is a growing field.One of the main drawbacks related to the introduction of –CN into molecules pertains to the toxicity and negative environmental impact of the cyanating reagents typically employed, such as MCN (M = K, Na, Cu, Zn), TMSCN, Bu4NCN, DDQ, cyanhidrines and malononitriles. Therefore, the discovery and application of new, non-toxic cyanide sources is extremely appealing in order to expand the synthetic utility of nitrile-containing substrates.183 In this context, ArSO2CN reagents have been catalogued as convenient and relatively non-toxic cyanating compounds with electrophilic properties that can be exploited under catalytic desulfonylative conditions.184 The most extended method for the activation of the C–S bond in ArSO2CN reagents uses radical conditions in which the TsCN reagent can be used either as a radical trap or as a radical promoter upon activation with an appropriate catalyst or radical initiator. In this section we discuss several strategies aimed at the use of such reagents for the synthesis of nitriles under catalytic conditions.
4.1. Cyanation of pre-functionalized substrates
In 2006, Renaud disclosed the first example on the use of B-alkylcatecholborane as a source of radicals in combination with TsCN using catalytic amounts of di-tert-butylhiponitrite (tBuON = NOtBu) or oxygen as a radical initiator (Scheme 90).186 The reaction begins with deboronative radical formation by effect of the radical initiator, which then collapses with the electrophilic –CN group to eliminate a p-TolSO2· radical. The authors employed alkenes as substrates for a first hydroboration with catechol borane (HBCat), which then undergoes cyanation. The protocol demonstrated good reactivity for primary and secondary radicals.
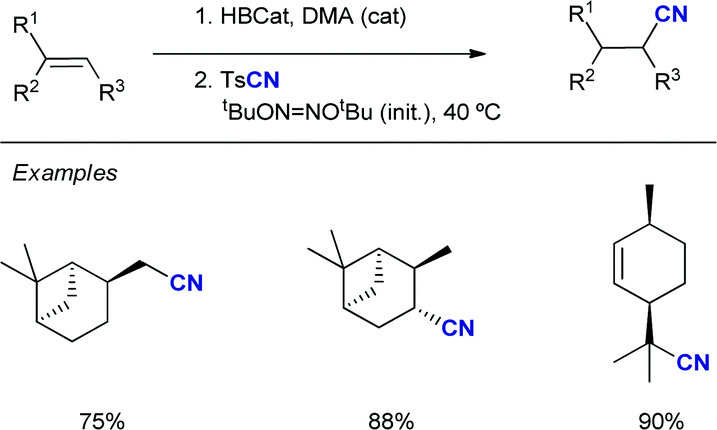 | ||
| Scheme 90 First reports on using TSCN as cyanating reagent. | ||
More recently, this conceptual challenge has been tackled in its catalytic version by Xu and co-workers employing photoredox catalysis.187 The treatment of the corresponding alkyl trifluoroborate salts with the electrophilic TsCN reagent employing [Ru(bpy)3](PF6)2 as photocatalyts under blue light irradiation delivered the corresponding nitrile with high efficiency (Scheme 91). The authors found that the use of mild oxidants such as iodosobenzene (PhIO), 1-hydroxy-1,2-benziodoxol-3-(1H)-one (BI-OH) or its acetoxylated version (BI-OAc), was crucial to obtain high yields. Conversely, the use of strong oxidants (K2S2O8 or PhI(OAc)2) delivered low yields. Additionally, the use of acidic additives such as trifluoroacetic acid (TFA) allowed for the obtaining of high yields. Although the role of the acid is not clear, the authors suggested that it may help to prevent competitive quenching processes of the catalyst with other species. Interestingly, the use of boronic acids or pinacol boronic ester was found to give very poor results in comparison with the use of trifluoroborate salts. In terms of the structural scope, the reaction was compatible with a diverse array of functional groups, including ethers, amides, cyano, esters, alkynes, heterocycles, and even halides such as bromine and chlorine atoms. Regarding the alkyl precursor, primary, secondary, and tertiary alkyl trifluoroborate salts could be employed with satisfactory results. However, for secondary and tertiary substrates the amount of TsCN was increased, along with the elimination of the TFA additive to attain reasonable yields.
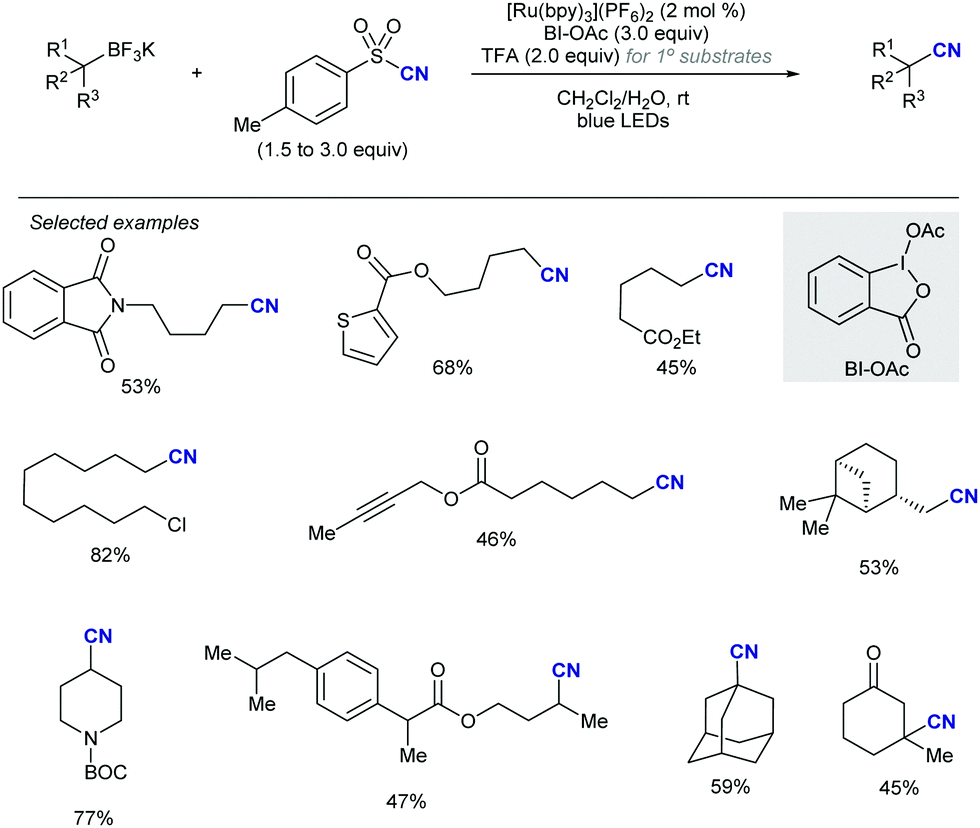 | ||
| Scheme 91 Cyanation of trifluoroborate salts. | ||
The same year Molander described a organophotoredox-catalysed cyanation of potassium alkyl trifluoroborate salts using (MesAcr)ClO4 as photocatalyst (Scheme 92).188 Under these conditions, cyanation occurred with medium to high yields and without the need for external oxidants or acidic additives. The corresponding α-, β-, and γ-amino nitriles derived from different N-heterocycles were easily synthesized from the corresponding alkenes via a hydroboration/cyanation protocol. Additionally, cyanohydrins were also obtained using these reaction conditions. The mechanism of the cyanation reaction under MesAcrClO4 photocatalyst is proposed to occur as shown in Scheme 92. After irradiation of the photocatalyst under visible light, the MesAcr+ species is excited to a singlet excited state which rapidly evolves to a long-lived triplet state via intersystem crossing (ISC). The alkyl trifluoroborate salt can be oxidized to the corresponding alkyl radical, which then undergoes addition to the TsCN reagent to deliver a new C–C bond with the elimination of the aryl sulfinate radical p-TolSO2˙. Finally, the reduction of the sulfinate radical by the photocatalyst intermediate enables to close the catalytic cycle, in which the sulfonyl moiety acts as both radical acceptor and oxidant.
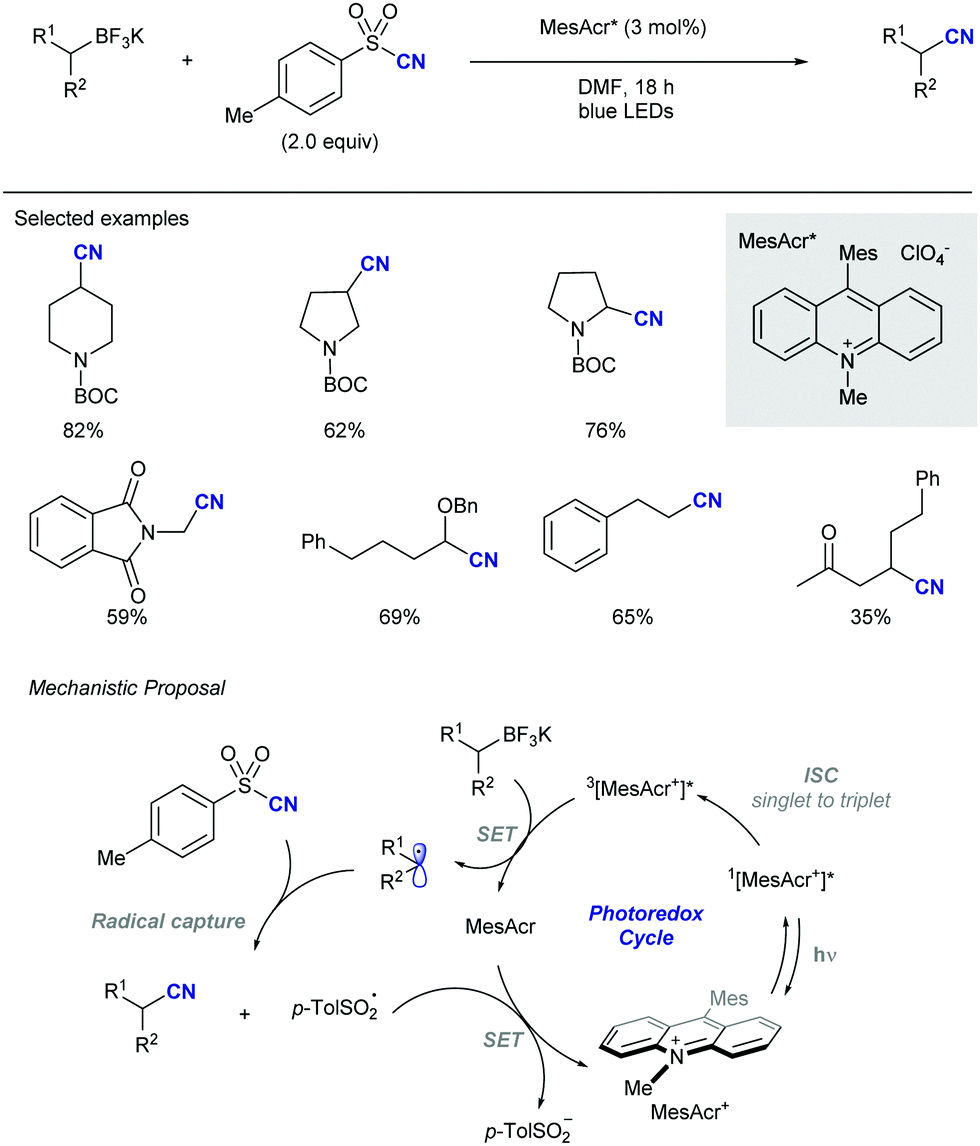 | ||
| Scheme 92 Photocatalytic cyanation of trifluoroborate salts. | ||
These deboronative transformations have been extended to other compounds and/or explored under different reaction conditions. Molander explored a photoredox-catalysed cyanation of trifluoroborate salts employing 2,1-borazaronaphthalenes (Scheme 93a), giving rise to a new family of interesting borylated compounds bearing a B–N bond.189 In this regard, Renaud reported a strategy that enables the direct use of alkylboronic pinacol esters in the deboronative cyanation protocol, which are challenging substrates in alkyl radical formation reactions due to their reduced Lewis acidity.190 The key of the method relies in the use of a catalytic amount of catechol methyl borate, which in combination with the alkylboronic pinacol ester forms the reactive catecholboronic ester by boron-transesterification (Scheme 93b). In the presence of a radical initiator such as tBuONNOtBu, catechol reacts with PhSO2CN, leading to the cyanation product and PhSO2BCat. This last intermediate is catalytically competent in the subsequent B-transesterification process, enabling the use of the catechol methyl borate in catalytic quantities.
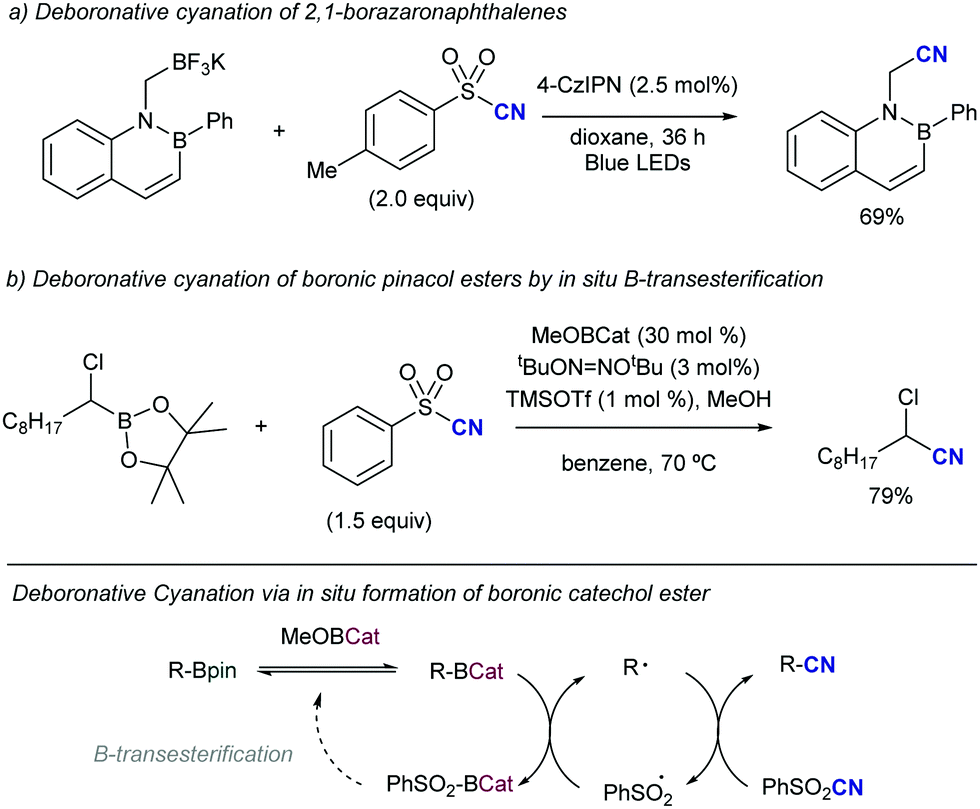 | ||
| Scheme 93 Extension of cyanation protocols employing organoboron compounds and TsCN. | ||
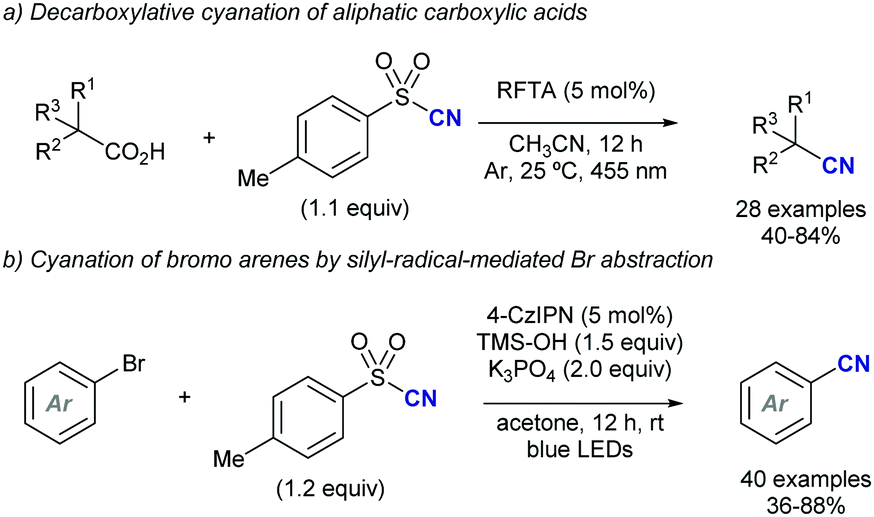 | ||
| Scheme 94 Pre-functionalized substrates for the in situ generation of radicals. | ||
An interesting transformation that enables distal δ-cyanation of N-fluoro-N-sulfonylamides has been recently described by Zhang and co-workers.193 Using a Cu(I) salt in combination with a N,N-coordinating ligand such as bathocuproine, initial N–F bond cleavage to generate the N-centered radical and the Cu(II)–F species occurs. Then, a 1,5-HAT process delivers the C-centered radical at the δ-position, which is intercepted with the TsCN to deliver the cyanation product along with the sulfinyl radical p-TolSO2˙. This radical reduces the Cu(II)–F complex by fluorine abstraction closing the catalytic cycle (Scheme 95). This work represents the first cyanation protocol to achieve high selectivity in the δ-cyanation of N-fluorotosylamides by virtue of a Hofmann–Löffler–Freytag reaction intercepting the TsCN reagent. An exceptional degree of flexibility was found by using different substrates, which rendered the corresponding product with high fidelity. Notably, this protocol was applied in late-stage functionalization of different natural products with acceptable yields and maintaining high levels of diastereoselectivity.
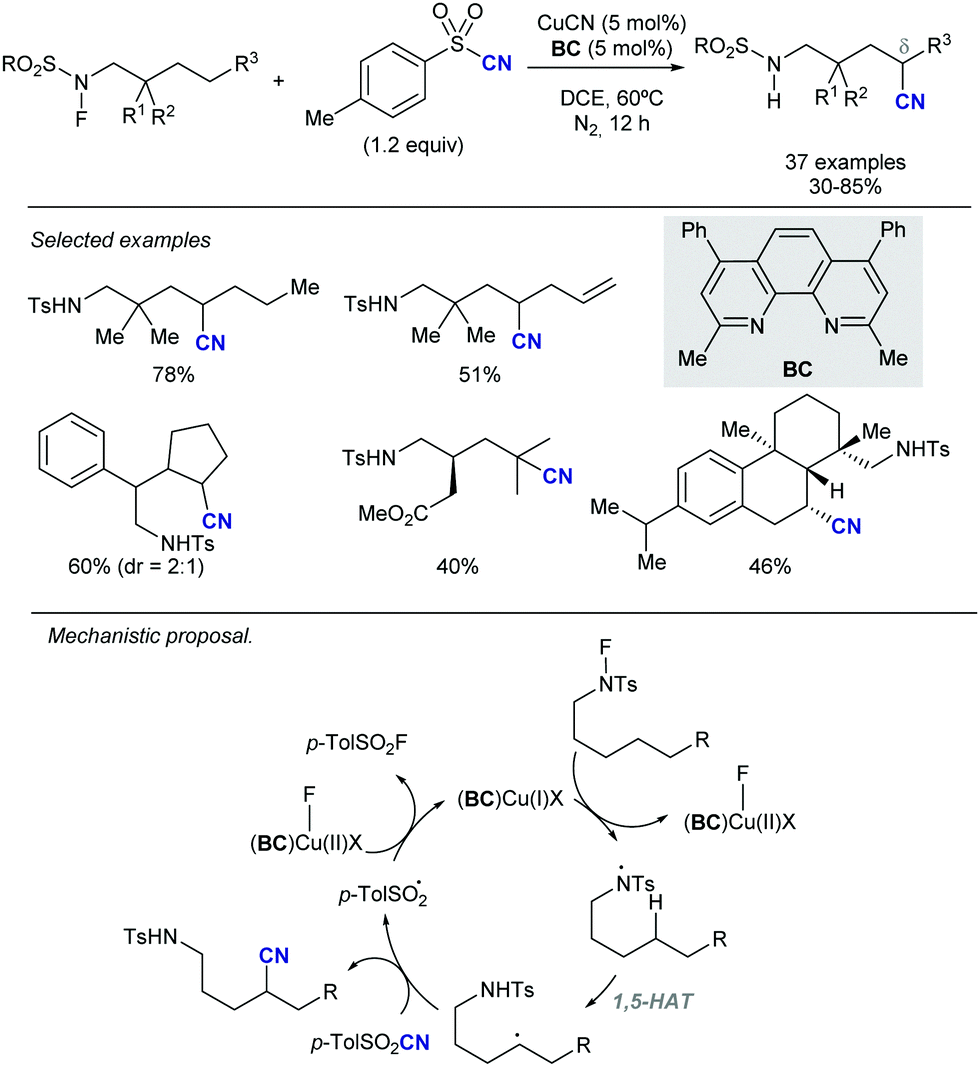 | ||
| Scheme 95 Distal δ-cyanation of N-fluoro-N-sulfonylamides. | ||
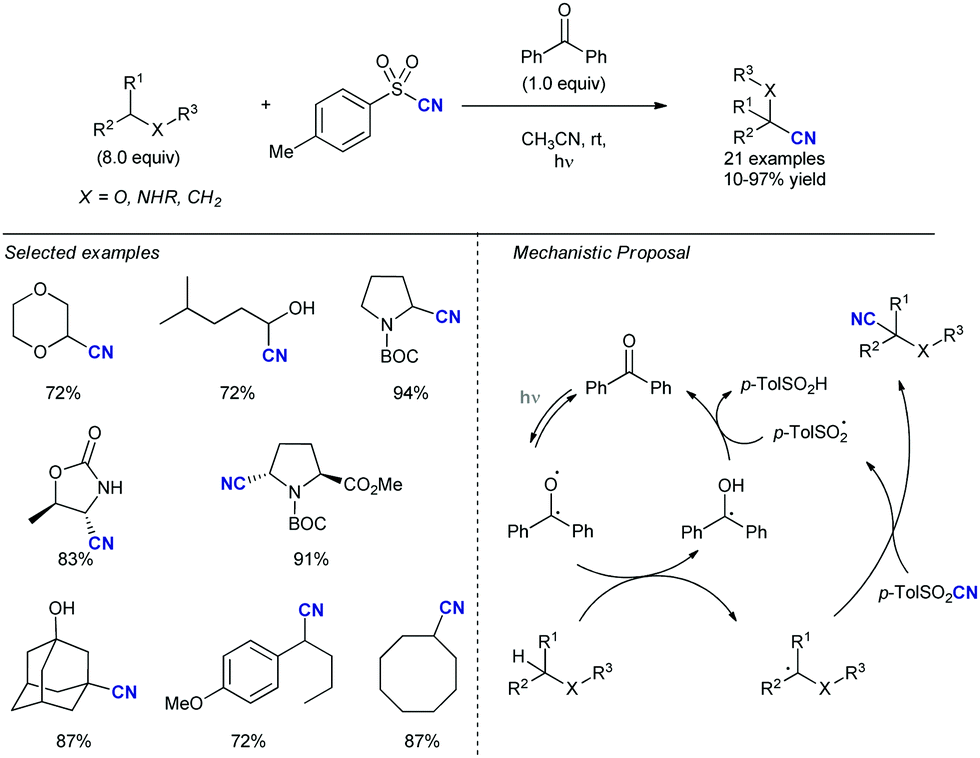 | ||
| Scheme 96 C–H cyanation of unactivated C(sp3)–H by photochemical generation of radicals. | ||
In 2018 Oisaki and Kanai described the first photocatalytic C–H cyanation of C(sp3)–H bonds at the α-position with respect to a heteroatom using TsCN (Scheme 97a).196 In this case the use of an iridium photocatalyst is involved in the single-electron photooxidation of a catalytic amount of a phosphate salt, generating a phosphate radical (Scheme 97b). This radical is active in the generation of the C-centered radical through HAT. In such process, the corresponding nitrile product was isolated employing different N-, O-, and S-containing substrates. Wu and co-workers have described a similar protocol employing eosin Y as a photocatalyst with the ability to promote the required HAT to selectively form the alkyl radical.197
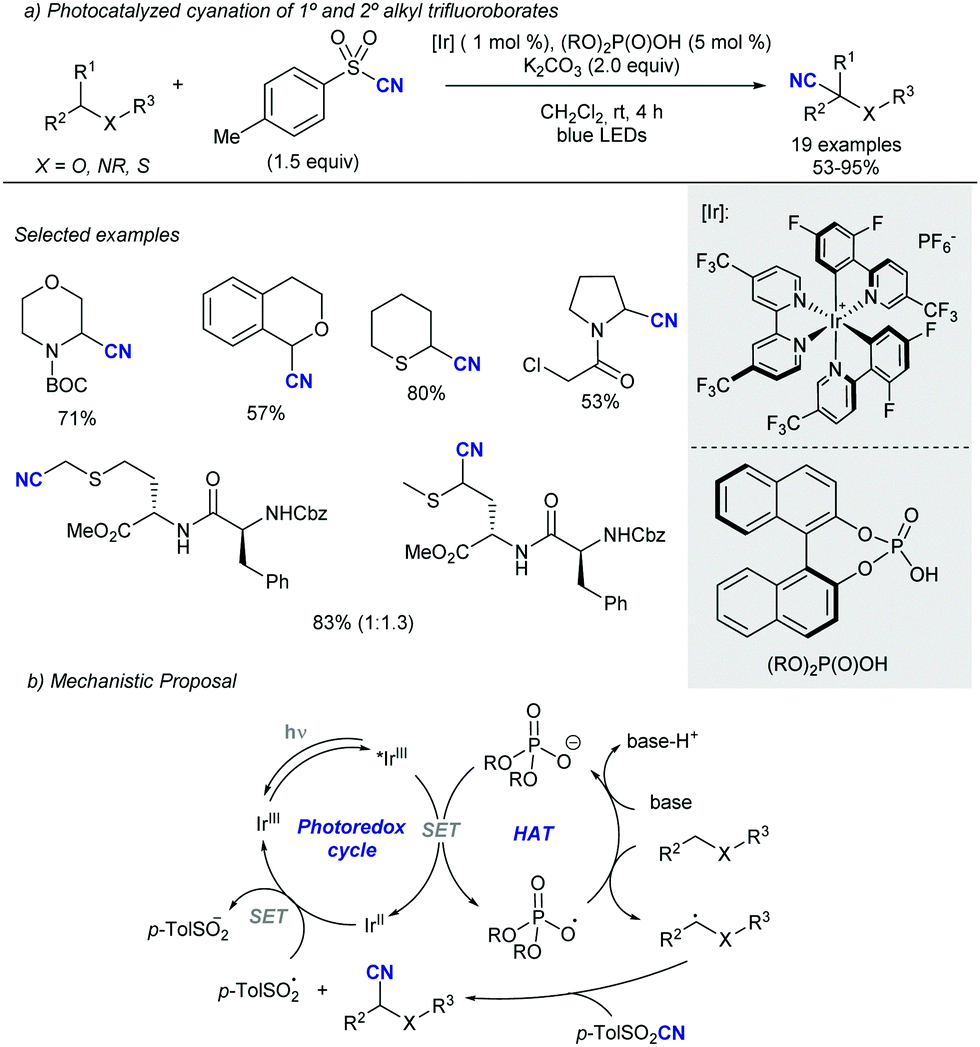 | ||
| Scheme 97 Photocatalytic C–H cyanation of C(sp3)–H α to a heteroatom. | ||
The same year, Hamashima reported the redox-neutral C–H cyanation of tetrahydroisoquinolines with TsCN using catalytic amounts of Ir(ppy)3 under blue light irradiation (Scheme 98).198 The authors propose a mechanism in which a photoexcited Ir(III) species reduces the TsCN reagent by SET and generates a nucleophilic cyanide species and a highly oxidant Ir(IV). This intermediate can oxidize the α-amino-C–H bond generating an iminium species, which is attacked by the cyanide anion previously formed. Interestingly, electron-rich substrates reacted slower than the electron-poor counterparts. Thus, prolonged reaction times and higher amounts of TsCN (2.0 equiv.) were necessary to obtain practical yields.
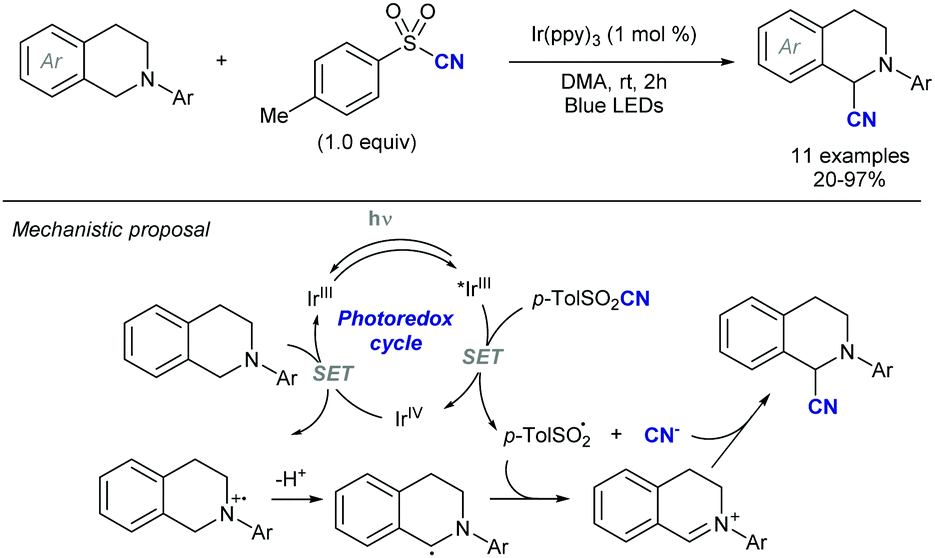 | ||
| Scheme 98 Redox-neutral C–H cyanation of tetrahydroisoquinolines. | ||
4.2. 1,2-Difunctionalization of alkenes
Alkenes are ideal platforms to trigger radical-addition processes in which more than one C–C bond is created in a single step. The ability of CC bonds to act as radical acceptors has enabled the discovery of new disconnection pathways to readily access complex organic scaffolds by hydro- or 1,2-difunctionalization of the π-system.199 ArSO2CN reagents have found applicability in 1,2-difunctionalization of alkenes for the introduction of CN, along with the sulfonylation of the substrate, which then can be transformed into another group. Stoichiometric reagents for the generation of the radical, or radical initiators to trigger radical chain reactions have been used in cyanations of π-systems in heterocarbo- or dicarbofunctionalization processes.200 However, the discovery of catalytic conditions to activate the C–S bond in ArSO2CN reagents to promote a radical addition was not known until recently. In 2018, Chu found that eosin Y in its excited state is quenched by TsCN, presumably to induce the C–S bond scission via SET process.201 This activation mode enabled in situ generation of a cyanide anion and a sulfonyl radical, which inserts into a C–C double bond. Then, the C-centered radical formed by addition to the alkene is oxidized by the photocatalyst, leading to a carbocation that is intercepted by the cyanide group (Scheme 99a). The reaction delivered the corresponding 1-sulfonyl-2-cyanofunctionalized products with high yields and a broad functional group compatibility. Halide atoms (Cl, Br), esters, ethers, ketones, boronic acid esters and amines could be used for synthetic purposes. Stern-Vomer analysis and light-dark experiments suggest that the reaction proceeds via a radical-polar crossover mechanism photocatalysed by eosin Y, instead of a radical chain progress. The same year, Landais reported a related process in which both aryl- and alkyl sulfonyl cyanides underwent 1,2-photocatalysed addition to unactivated alkenes employing eosin Y as photocatalyst (Scheme 99b).202 In this case, the authors performed an extensive mechanistic study that suggested the intermediacy of cyanated eosin photocatalyst from the sulfonyl cyanide reagents and the importance of sulfonyl radicals in the addition to the π-system. This method was applied to the synthesis of a metalloproteinase inhibitor in which a first sulfonyl cyanation is the key step.
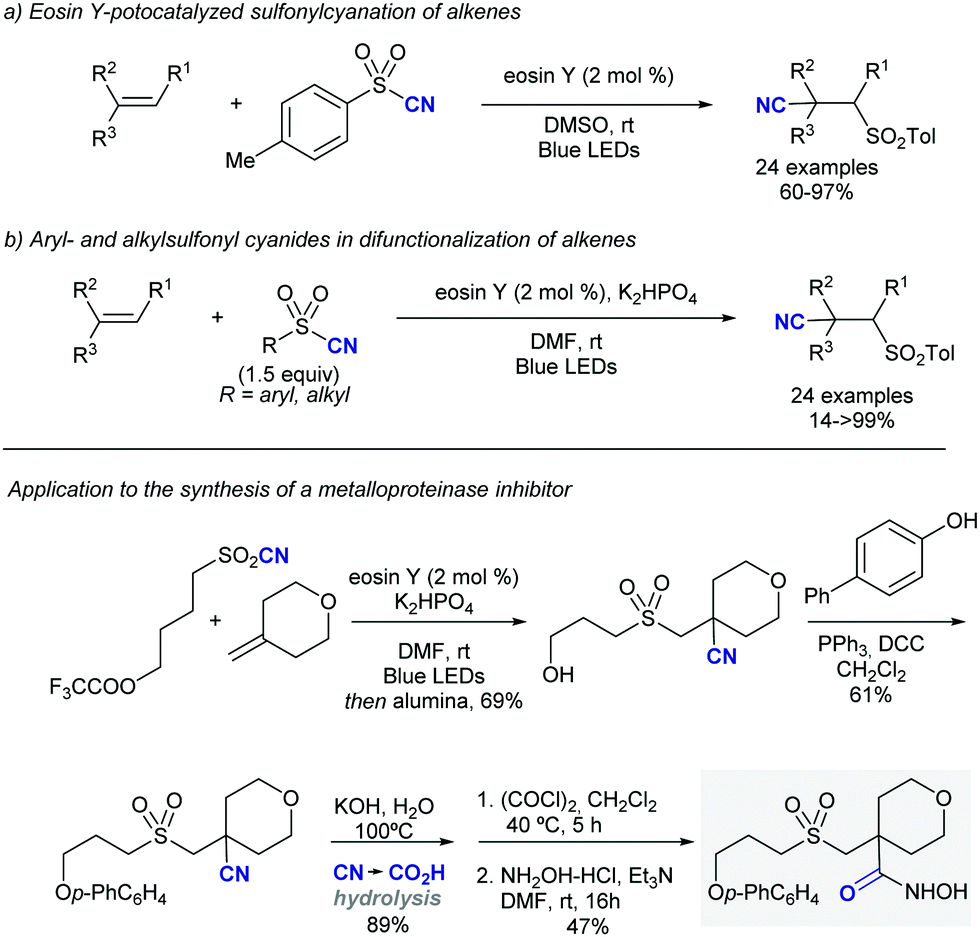 | ||
| Scheme 99 Catalytic conditions to activate the C–S bond in ArCN reagents. | ||
More recently, Landais has reported an activation mode of the C–S bond in organosulfonyl cyanides via triplet photosensitization using p-anisaldehyde as photocatalyst, enabling the sulfonylcyanation of alkenes via atom transfer radical addition (ATRA).203 This reactivity mode was applied to the sulfonylcyanation of chiral cyclobutenes, which typically exhibit a limited reactivity in radical-involved transformations (Scheme 100). The success of the method relies on the triplet sensitization of the organosulfonyl cyanide by the photocatalyst via energy transfer process. After that, homolytic scission of the RSO2CN bond is expected to occur, forming an electrophilic sulfonyl radical with capability to insert into the π-system of the cyclobutene. At this point, the authors propose a radical chain propagation sequence in which the new C-centered radical abstracts the cyano group from another RSO2CN molecule. Different chiral cyclobutenes bearing an oxazolidinone moiety were screened, and the reaction delivered the corresponding sulfonylcyanation product with decent yields and very high stereoselectivity. This reaction sequence enables post-modification of the chiral cyclobutane core to access chiral cyclobutanes and cyclobutenes. A salient application pertains to the SmI2-promoted ring opening of the cyclobutane core that enables access to useful chiral enantiopure carbonyl derivatives bearing an all-carbon γ-quaternary center with a cyano substituent.
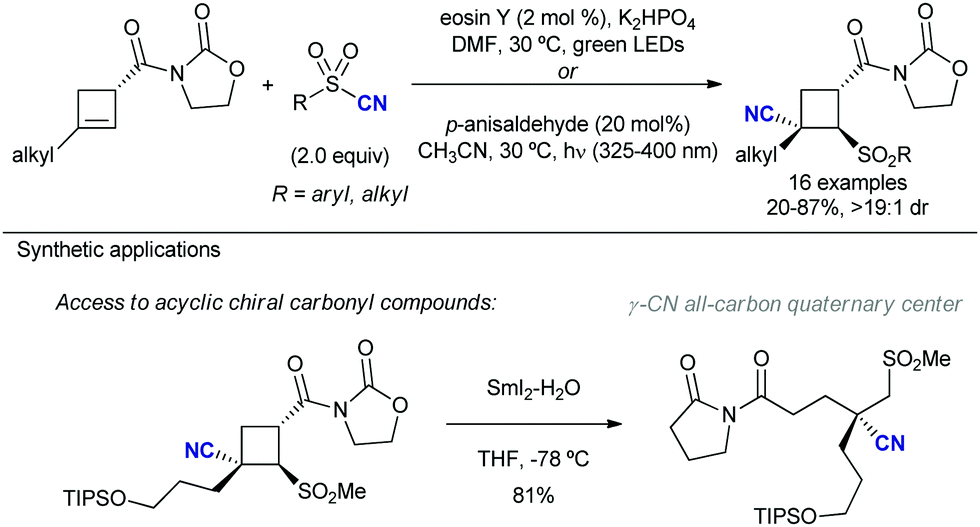 | ||
| Scheme 100 Activation mode of the C–S bond in organosulfonyl cyanides via triplet photosensitization. | ||
4.3. Transition metal-catalysed cyanation
Carreira described in 2007 a prominent transformation involving the cyanation of unsaturated systems using TsCN under cobalt catalysis (Scheme 101).204 This transformation involves the hydrocyanation of non-activated olefins by the intermediacy of a Co–H species generated in situ from a Co-complex incorporating a salen ligand in combination with PhSiH3 as a reductant. After the hydrometallation step, the resulting cobalt species can react with TsCN, likely by formation of intermediate alkyl radicals. To enhance the catalytic performance, tBuO2H was employed to reduce the induction period for activation of the catalyst. An exceptional functional group compatibility was found for this catalytic system, in which monosubstituted, 1,1-disubstituted, and trisubstituted alkenes exhibited excellent Markovnikov-type selectivity. However, this protocol failed when α,β-unsaturated esters or non-activated 1,2-disubstituted olefins were studied. The method is exceptionally robust and finds notable application as a key step in several total syntheses. Some examples are related to the synthesis of the tricyclic skeleton of taxanes,205 guanosine modification en route to therapeutic agents,206 and the synthesis of brasilicardines207 or andrastins208 among others. Later, Nishida applied this methodology to the hydrocyanation of enamines employing the same catalytic system.209 The authors found exclusive formation of the α-aminonitrile product, in which the presence of the nitrogen atom seems to impart higher reactivity.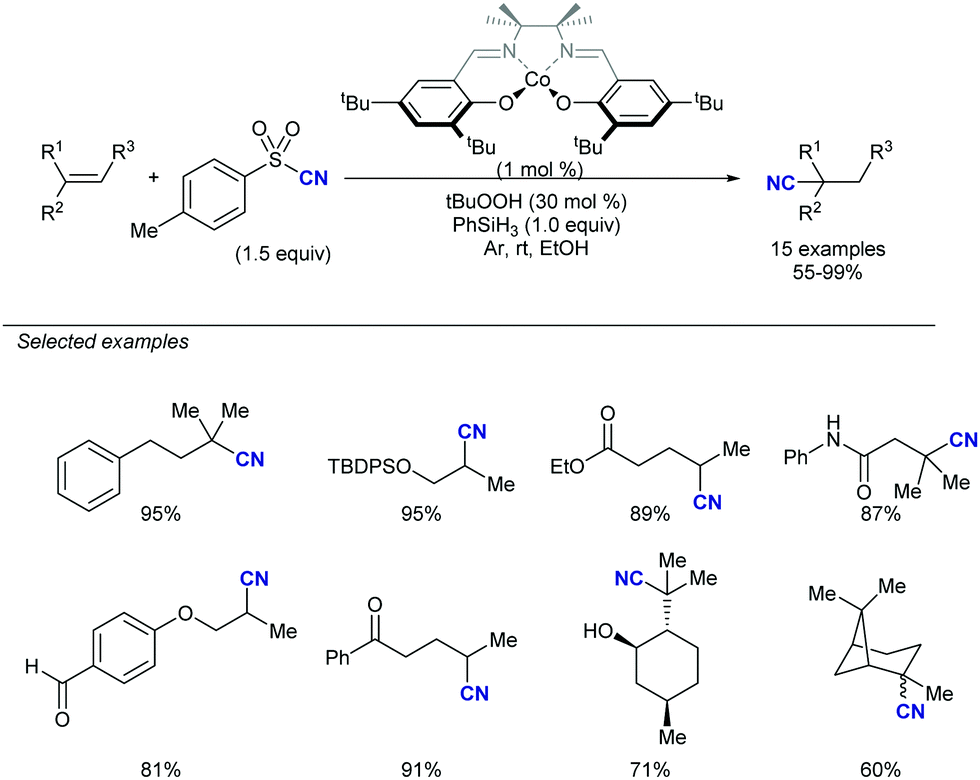 | ||
| Scheme 101 Cyanation of unsaturated systems using TsCN under cobalt catalysis. | ||
Apart from alkene-hydrocyanation, transition metal catalysis has been explored in the search of other reactivity modes toward the synthesis of nitriles. In 2016, Zhu capitalised on the high reactivity of cyclobutanol to synthesize of γ-cyanoketones using Mn catalysis (Scheme 102). The process begins with C–C bond cleavage of the cyclobutanol core via SET by a Mn(V) catalyst, generating a ketone with a C-centered γ-radical species along with a Mn(IV) intermediate. This C-center radical abstracts the cyano group of TsCN via C–S bond cleavage with concomitant formation of a sulfonyl radical, which is then oxidized by the Mn(IV) intermediate to form a Mn(III) complex. The newly generated sulfonyl cation further evolves to a sulfonate product by acetate addition. Finally, the Mn(III) species is re-oxidized using an external oxidant to achieve turnover and regenerate the active Mn(V) catalyst. The reaction proceeded with high reactivity, delivering the corresponding γ-cyanoketone, which is an important structural motif difficult to prepare by other methods.
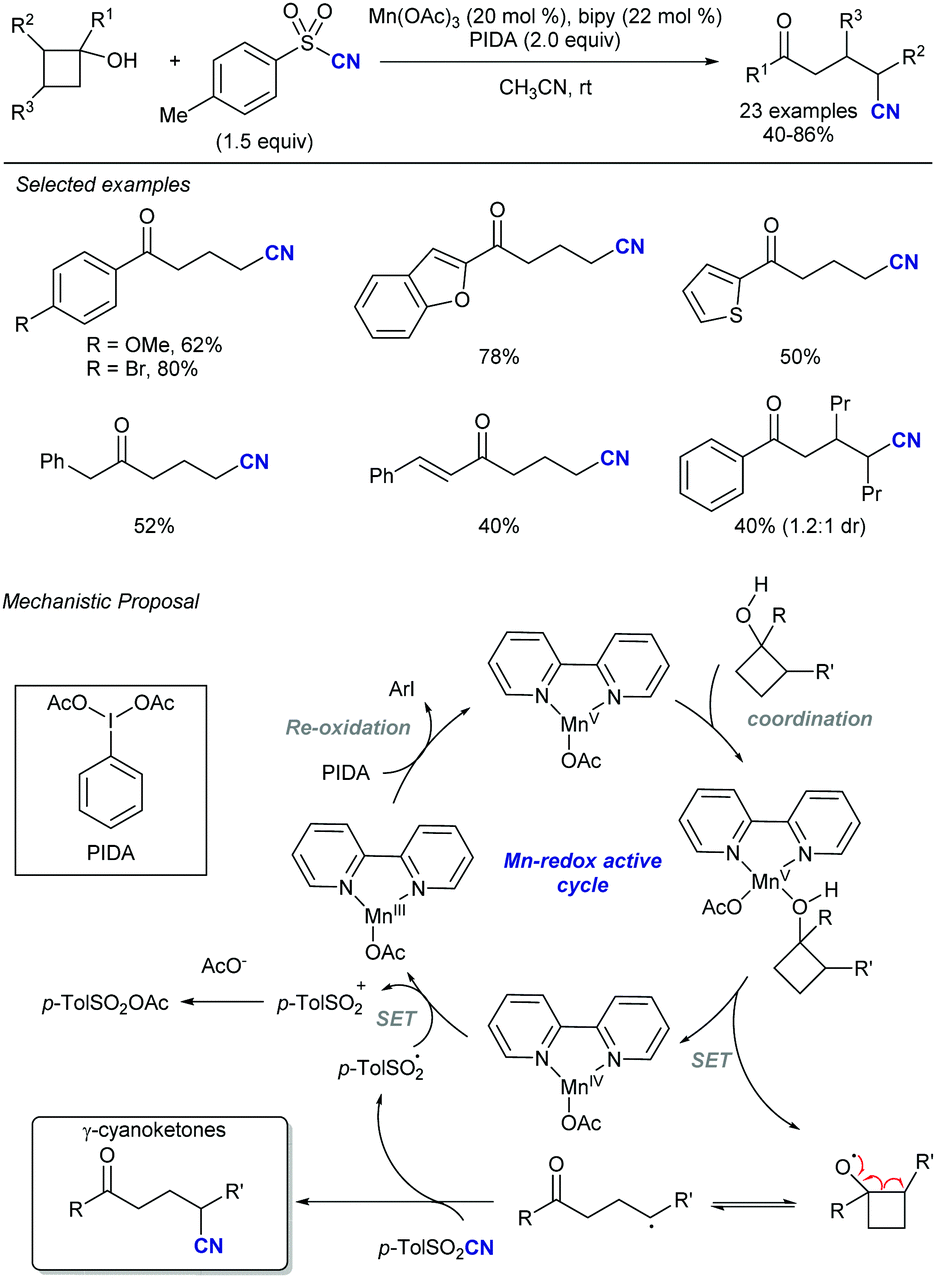 | ||
| Scheme 102 Mn-Catalyzed synthesis of γ-cyanoketones. | ||
Recently, Shi has developed a C–H cyanation process by Rh(III)-catalysed C–H functionalization.210 Using pyrimidines or pyridynes as directing groups, and [Cp*Rh(CH3CN)3](SbF6)2 as a pre-catalyst in combination with TsCN, the reaction delivered the corresponding nitriles with excellent levels of ortho-regioselectivity for a different array of indoles, pyrroles and phenyl groups attached to the directing group (Scheme 103). After deuteration and isolation of key reaction intermediates, the authors proposed a mechanistic scenario starting with ortho-C–H activation by the electrophilic Rh(III) species, leading to a five-membered rhodacycle. After that, the cyano group of TsCN coordinates to the Rh(III) metal center, promoting a subsequent 1,2-insertion of the C–N triple bond into the C–Rh bond. Final elimination of a rhodium sulfinate intermediate delivers the cyanated product, and the Rh(III) active catalyst re-enters the catalytic cycle after protonation of the sulfinate.
 | ||
| Scheme 103 Rh(III)-Catalysed C–H cyanations. | ||
5. Catalytic desulfonylative C–X formation
Sulfones have been traditionally utilized as removable functionalities to introduce substituents at remote positions by exploiting their strength as electron withdrawing groups.211 However, leveraging sulfones as coupling partners for carbon-heteroatom (C–X) bond formations is still rare: an early application of this strategy was reported by Fuchs when he developed Pd-catalysed aminations of allylic sulfones towards the synthesis of cephalotaxine (Scheme 104).212 Unfortunately, the reaction appeared to be rather substrate specific and little progress has been made since this report. Only recently, developments in transition metal catalysis have contributed in expanding the versatility of sulfones not only toward C–C cross coupling reactions, but also for the conversion into useful C-heteroatom bonds. | ||
| Scheme 104 Pd-Catalysed desulfonative aminations of allylic sulfones. | ||
In 2016, Han disclosed in 2016 the possibility of transforming sulfenyl and sulfonyl groups into arylphosphine oxides by using Ni catalysis (Scheme 105).213 In both cases, aryl groups were selectively phosphinylated over alkyl groups, enabling the use of readily accessible methyl thioethers and methylsulfonyl fragments. Thioethers generally underwent phosphinylation in good yields, although the reaction was not effective when electron-deficient thioethers were involved. Sulfones, in comparison, were found to possess higher reactivity, rendering the corresponding phosphine oxides with excellent yields for different types of aryl substitution. Furthermore, phosphonylation with (iPrO)2P(O)H was found to be compatible under the reaction conditions exclusively on sulfones. The proposed catalytic cycle involved oxidative addition of the sulfone or thioether to Ni(0) followed by reductive elimination of a phosphide ligand, which is generated upon reaction of the phosphine oxide and a base. Interestingly, electron-deficient arylsulfones (i.e., substituted with 4-CN or 4-pyrimidinyl) underwent high conversions to phosphine oxides without requiring the catalyst, suggesting a direct substitution pathway for strongly activated sulfones. Direct substitution of alkynyl, alkenyl and allyl sulfones with phosphine oxides under transition metal-free conditions was further examined bt Xiao,214 thus improving the phosphonylation of alkynylsulfones previously studied by Takeda.215
 | ||
| Scheme 105 Ni-Catalysed transformation of sulfenyl and sulfonyl groups into arylphosphine oxides. | ||
Boronic acids are another class of substrates with high synthetic potential. In recent years, numerous strategies have been developed to incorporate C(sp3)–B bonds into highly functional molecules. In 2017, Yorimitsu and coworkers reported a Pd-catalysed borylation of diphenylsulfone within the context of borylation of sulfoxides (Scheme 106).216 The reaction was thought to proceed via two parallel catalytic cycles, one involving the initial diphenylsulfone, and the second cycle taking place on the resultant phenylsulfinyl residue. This served to introduce an interesting approach of double borylation, however, diphenylsulfone only provided a moderate yield being the sole example in the study.
 | ||
| Scheme 106 A Pd-catalysed borylation of diphenylsulfone. | ||
While most of such methods rely on the use of transition metal catalysts, Crudden reported a pyridine-catalysed desulfonylative borylation using B2pin2 reagent (Scheme 107).217 Based on the 4-phenylpyridine catalysis described by Zhang and Jiao,218 the team established an effective protocol that was applicable to several benzylic and dibenzylic sulfones using 4-anthracenylpyridine as the optimal catalyst. Limitations of the reaction were thoroughly examined, revealing incompatibility with aryl groups substituted with electron withdrawing groups. Moreover, tertiary substitution with alkyl groups at the benzylic position resulted in undesirable β-elimination. However, when applied to cyclic sulfones, the reaction rendered the corresponding sulfinate intermediate, which could be trapped in situ by adding various types of electrophiles to deliver sulfones. On the basis of previous studies, the formation of borate complex IV is believed to trigger the initial SET to reduce the sulfone and generate the benzylic radical. While the use of radical clocks failed to confirm the participation of radical intermediates, addition of TEMPO to the reaction resulted in complete inhibition of the borylation process. The catalytic cycle continues with the formation of a benzylic carbanion by single electron oxidation of complex V, which results in regeneration of the pyridine catalyst. The borylated product is then obtained by reaction with MeOBpin.
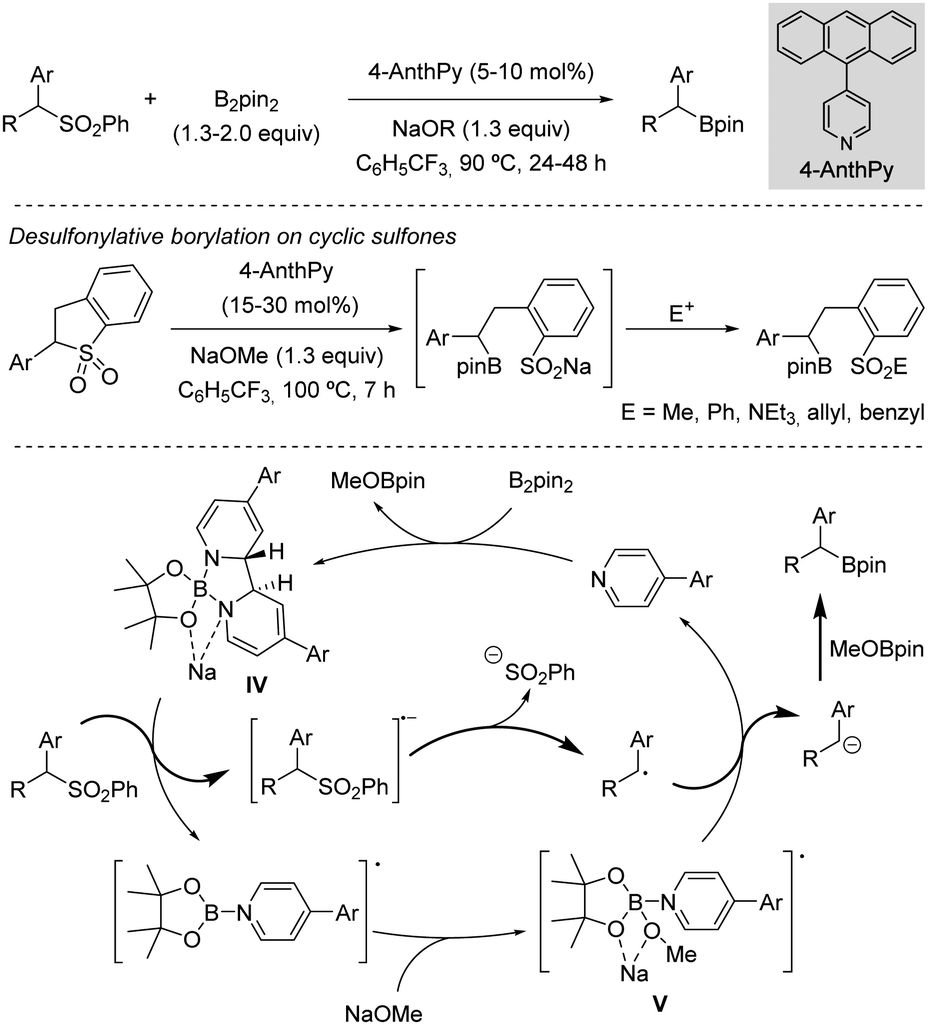 | ||
| Scheme 107 Pyridine-catalysed desulfonylative borylations using B2pin2. | ||
Later that year, Crudden studied the potential of sulfones to be employed as precursors of C–N bonds. Problems associated to the use of unpractical halide derivatives in the synthesis of benzhydryl amines were addressed by utilizing the more stable and accessible benzhydryl sulfones. Using 3,5-bis(trifluoromethyl)phenylsulfones to facilitate C–S cleavage, a Cu-catalysed procedure allowed the authors to couple a wide variety of primary and secondary amines in good yields (Scheme 108).219 Interestingly, the use of typical ligands such as bpy or NHC turned to be detrimental, although coupling reaction with an aniline benefitted from adding DPEphos to avoid the formation of oxidation byproducts. To further demonstrate the utility of the reaction, an orthogonal di-amination was conducted by sequential SN2 amination and Cu-catalysed desulfonylative amination. Control experiments in the absence of the amine showed the appearance of olefination products, suggesting the involvement of Cu-carbenes during the reaction. The formation of carbenes was rationalized by the α-elimination of the sulfonyl group after α-lithiation of the sulfone and transmetalation with Cu.
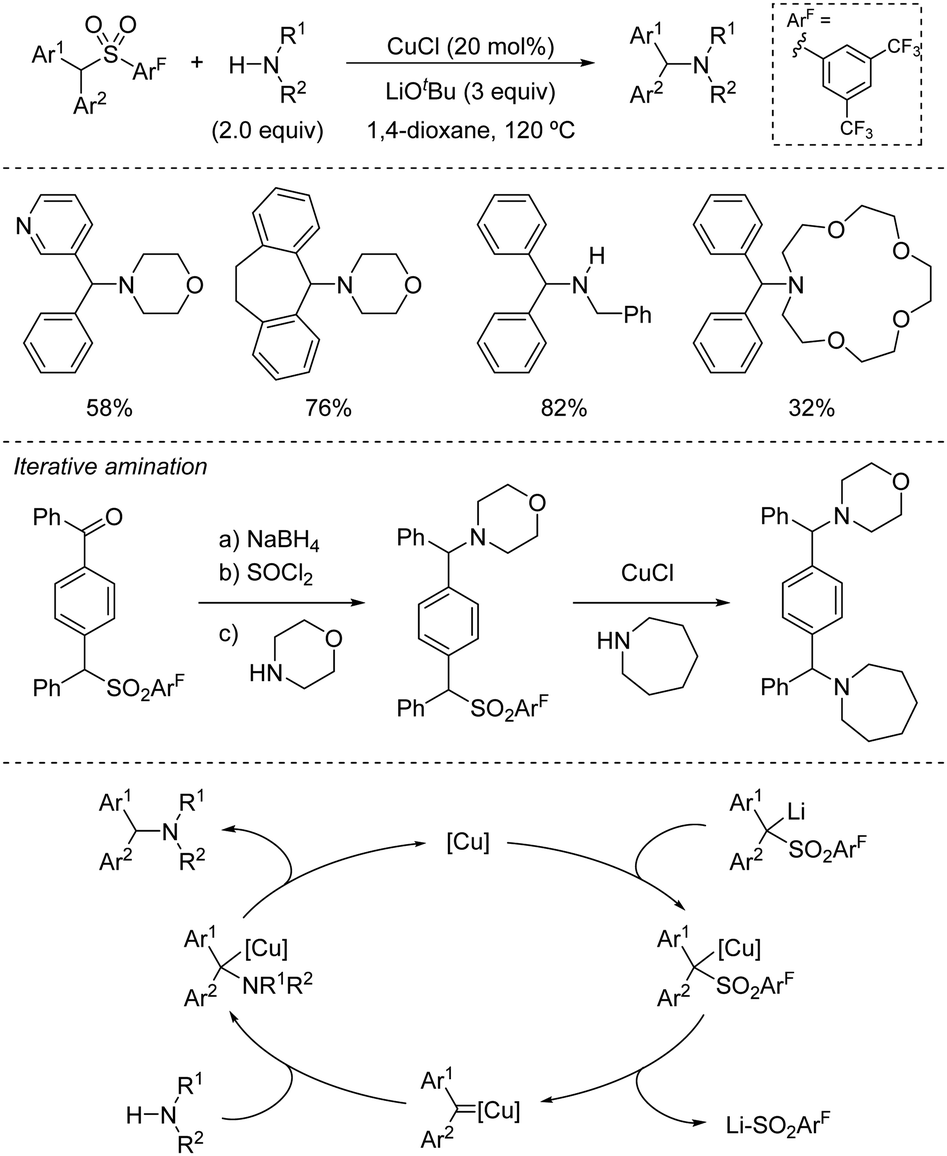 | ||
| Scheme 108 Cu-Catalysed C–N bond formation with primary and secondary amines. | ||
Finally, an electrochemical method for the allylic substitution of sulfones with tosylamides was developed by Sun.220 The reaction was set up in an undivided cell at constant current using a Pt electrode to form allylic amines in good yield (Scheme 109). Other leaving groups such as bromide, acetate and carbonate led to the desired product in poorer yields than sulfones. Allylic sulfones, however, were required to be activated by the presence of an ester at the β-carbon. A radical substitution mechanism was proposed for this reaction, in which a radical amine is generated with intermediacy of an N-iodoamine. This was supported by the inhibition effect observed upon addition of BHT, which trapped the amine radical in 70% yield. Furthermore, since introducing I2 as oxidant without electric current did not render the amination product, a higher oxidation state of iodine was thought to be involved.
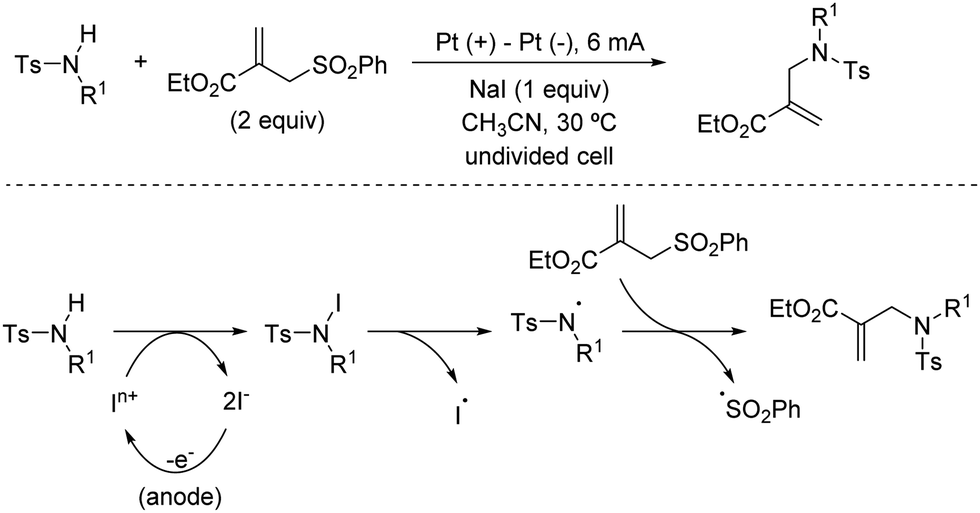 | ||
| Scheme 109 Electrochemical method for the allylic substitution of sulfones with tosylamides. | ||
6. Concluding remarks and future directions
The goal of this Review is to provide a comprehensive account of useful catalytic transformations in which organosulfones can be transformed by cleavage of the C–S bond. This is a particularly useful tool in organic synthesis because of the wide availability of methods to prepare sulfones and their well-established benchtop stability. Moreover, their reactivity can be tuned by altering the nature of the substituents on the other end of the sulfone moiety, which in comparison with other leaving groups such as halogens adds value to this group. However, although the C–S bond has already shown tremendous potential in organic synthesis, researchers still face challenges that will unquestionably fuel future investigations in the field. Among them are the following:1. Enantioselective transformations are notably absent from the toolbox of reactions available to the organic chemist.
2. The replacement of S by heteroatoms such as O, N, P, or B would significantly expand the utility of these transformations.
3. The generalized use of heteroaryl coupling partners will increase and extend the applicability of these reactions in industrial settings.
4. Also of interest in a non-academic environment, the transition from methods that use noble metals such as Rh or Pd to those that can operate under cheap base-metal catalysts would be of great interest.
5. An improved mechanistic understanding of the cleavage of the carbon–sulfone bond should eventually lead to the extension of these methods to the activation of other types of C–S bonds, including sulfoxides, sulfonates, and naturally occurring, abundant thiols.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministerio de Ciencia e Innovación (MICINN) and Fondo Europeo de Desarrollo Regional (FEDER, UE) for financial support (Agencia Estatal de Investigación/Project PGC2018-098660-B-I00). J. C. and S.-H. K.-L. thank Ministerio de Educación, Cultura y Deporte (MECD) for FPU predoctoral fellowships (FPU2016/03642 and FPU2015/04287, respectively).References
- (a) Q. A. Acton, Sulfones—Advances in Research and Application, ScholaryEditions™, Atlanta, 2013 Search PubMed; (b) S. Liang, S. Shaaban, N.-W. Liu, K. Hofman and G. Manolikakes, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, San Diego, CA, USA, 2018, vol. 69, pp. 135–207 CrossRef CAS; S. Liang, S. Shaaban, N.-W. Liu, K. Hofman and G. Manolikakes, Adv. Organomet. Chem., 2018, 69, 135–207 CrossRef CAS; (c) D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793–814 CrossRef CAS PubMed; (d) I. Ahmad and Shagufta, Int. J. Pharm. Pharm. Sci., 2015, 7, 19–27 CAS.
- (a) S. Patai, Z. Rappoport and C. J.-M. Stirling, The Chemistry of Sulphones and Sulphoxides, Wiley, New York, 1988 CrossRef; (b) B. M. Trost and C. A. Kalnmals, Chem. – Eur. J., 2019, 25, 11193–11213 CrossRef CAS.
- (a) Y. M. Markitanov, V. M. Timoshenko and Y. G. Shermolovich, J. Sulfur Chem., 2014, 35, 188–236 CrossRef CAS; (b) E. Marcantoni, A. Palmieri and M. Petrini, Org. Chem. Front., 2019, 6, 2142–2182 RSC; (c) F. Takahashi, K. Nogi and H. Yorimitsu, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 742–745 CrossRef CAS; (d) J. Lou, Q. Wang, P. Wu, H. Wang, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2020, 49, 4307–4359 RSC; (e) X.-Q. Chu, D. Ge, Y.-Y. Cui, Z.-L. Shen and C.-J. Li, Chem. Rev., 2021, 121, 12548–12680 CrossRef CAS PubMed.
- D. B. Boyd, J. Org. Chem., 1985, 50, 886–888 CrossRef CAS.
- (a) M. Nambo and C. M. Crudden, Chem. Rec., 2021, 21, 3978–3989 CrossRef CAS PubMed; (b) M. Nambo, Y. Maekawa and C. M. Crudden, ACS Catal., 2022, 12, 3013–3032 CrossRef CAS.
- J.-L. Fabre, M. Julia and J.-N. Verpeaux, Tetrahedron Lett., 1982, 23, 2469–2472 CrossRef CAS.
- E. Alvarez, T. Cuvigni, C. Herve du Penhoat and M. Julia, Tetrahedron, 1988, 44, 111–118 CrossRef CAS.
- E. Alvarez, T. Cuvigni, C. Herve du Penhoat and M. Julia, Tetrahedron, 1988, 44, 119–126 CrossRef CAS.
- O. Arjona, F. Iradier, J. Plumet, M. P. Martinez-Alcfizar, F. Hernández-Cano and I. Fonseca, Tetrahedron Lett., 1998, 39, 6741–6744 CrossRef CAS.
- S. E. Denmark and A. J. Cresswell, J. Org. Chem., 2013, 78, 12593–12628 CrossRef CAS PubMed.
- T. K. Beng and D. P. Bassler, Tetrahedron Lett., 2014, 55, 6662–6664 CrossRef CAS.
- (a) N. Kamigata, J.-i Ozaki and M. Kobayashi, J. Org. Chem., 1985, 50, 5045–5050 CrossRef CAS . See also: ; (b) M. V.-R. Reddy, A. B. Manjubhashini, S. Reddy, P. V.-R. Reddy and D. B. Reddy, Synth. Commun., 1991, 21, 1589–1596 CrossRef CAS.
- L. Gong, H.-B. Sun, L.-F. Deng, X. Zhang, J. Liu, S. Yang and D. Niu, J. Am. Chem. Soc., 2019, 141, 7680–7686 CrossRef CAS PubMed.
- L. Gong, Q. Zhang, D. Xie, W. Zhang, S. Xu, X. Zhang and D. Niu, Chem. Commun., 2021, 57, 12273–12276 RSC.
- (a) V. T. Nguyen, H. T. Dang, H. H. Pham, V. D. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2018, 140, 8434–8438 CrossRef CAS PubMed; (b) H. T. Dang, V. D. Nguyen, H. H. Pham, H. D. Arman and O. L. Larionov, Tetrahedron, 2019, 75, 3258–3264 CrossRef CAS PubMed; (c) H. T. Dang, V. D. Nguyen, G. C. Haug, N. T.-H. Vuong and H. D. Arman, ACS Catal., 2021, 11, 1042–1052 CrossRef CAS PubMed.
- For selected examples, see: (a) N. Miyamoto, D. Fukuoka, K. Utimoto and H. Nozaki, Bull. Chem. Soc. Jpn., 1974, 47, 503–504 CrossRef CAS; (b) G. A. Russell, P. Ngoviwatchai, H. Tashtoush, A. Pla-Dalmau and R. K. Khanna, J. Am. Chem. Soc., 1988, 110, 3530–3538 CrossRef CAS; (c) J. Xiang, W. Jiang, J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1997, 119, 4123–4129 CrossRef CAS; (d) F. Bertrand, B. Quiclet-Sire and S. Z. Zard, Angew. Chem., Int. Ed., 1999, 38, 1943–1946 CrossRef CAS; (e) A.-P. Schaffner, V. Darmency and P. Renaud, Angew. Chem., Int. Ed., 2006, 45, 5847–5849 CrossRef CAS PubMed; (f) V. Liautard, F. Robert and Y. Landais, Org. Lett., 2011, 13, 2658–2661 CrossRef CAS PubMed; (g) R. Beniazza, V. Liautard, C. Poittevin, B. Ovadia, S. Mohammed, F. Robert and Y. Landais, Chem. – Eur. J., 2017, 23, 2439–2447 CrossRef CAS PubMed.
- Y. Amaoka, M. Nagatomo, M. Watanabe, K. Tao, S. Kamijo and M. Inoue, Chem. Sci., 2014, 5, 4339–4345 RSC.
- A. Noble and D. W.-C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed.
- (a) K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef CAS PubMed; (b) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed; (c) D. C. Miller, K. T. Tarantino and R. R. Knowles, Top. Curr. Chem., 2016, 374, 30 CrossRef PubMed.
- W. Yang, X. Chen and W. Fang, ACS Catal., 2018, 8, 7388–7396 CrossRef CAS.
- L. M. Kammer, B. Lipp and T. Opatz, J. Org. Chem., 2019, 84, 2379–2392 CrossRef CAS PubMed.
- D. R. Heitz, K. Rizwan and G. A. Molander, J. Org. Chem., 2016, 81, 7308–7313 CrossRef CAS PubMed.
- F. Yue, J. Dong, Y. Liu and Q. Wang, Org. Lett., 2021, 23, 2477–2481 CrossRef PubMed.
- (a) F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085–14089 CrossRef CAS PubMed; (b) F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. Van der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef CAS PubMed.
- S. Paul and J. Guin, Green Chem., 2017, 19, 2530–2534 RSC.
- M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362–11367 CrossRef CAS.
- Y. Shen, J. Cornella, F. Julia-Hernández and R. Martin, ACS Catal., 2017, 7, 409–412 CrossRef CAS.
- (a) B. Ovadia, F. Robert and Y. Landais, Chimia, 2016, 70, 34–42 CrossRef CAS PubMed; (b) A. Chaambi, G. Kurtay, R. Abderrahim, F. Robert and Y. Landais, Helv. Chim. Acta, 2019, 102, e1900140 CrossRef.
- For selected examples: (a) K. S. Bloome, R. L. McMahen and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 20146–20148 CrossRef CAS PubMed; (b) H. Liu, Z. Qiao and X. Jiang, Org. Biomol. Chem., 2012, 10, 7274–7277 RSC; (c) B. M. Monks and S. P. Cook, Angew. Chem., Int. Ed., 2013, 52, 14214–14218 CrossRef CAS PubMed; (d) Y. Zou and J. Zhou, Chem. Commun., 2014, 50, 3725–3728 RSC; (e) X. Wu, J. W.-T. See, K. Xu, H. Hirao, J. Roger, J.-C. Hierso and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13573–13577 CrossRef CAS PubMed; (f) A. R.-O. Venning, P. T. Bohan and E. J. Alexanian, J. Am. Chem. Soc., 2015, 137, 3731–3734 CrossRef CAS PubMed; (g) Z. Li, A. García-Domínguez and C. Nevado, J. Am. Chem. Soc., 2015, 137, 11610–11613 CrossRef CAS PubMed.
- S. Sumino, M. Uno, H.-J. Huang, Y.-K. Wu and I. Ryu, Org. Lett., 2018, 20, 1078–1081 CrossRef CAS PubMed.
- (a) J. D. Nguyen, E. M. D’Amato, J. M.-R. Narayanam and C. R.-J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed; (b) D. Fernandez Reina, A. Ruffoni, Y. S.-S. Al-Faiyz, J. J. Douglas, N. S. Sheikh and D. Leonori, ACS Catal., 2017, 7, 4126–4130 CrossRef CAS; (c) D. Kurandina, M. Rivas, M. Radzhabov and V. Gevorgyan, Org. Lett., 2018, 20, 357–360 CrossRef CAS PubMed.
- Q.-Q. Zhou, S. J.-S. Düsel, L.-Q. Lu, B. König and W.-J. Xiao, Chem. Commun., 2019, 55, 107–110 RSC.
- E. Wenkert, T. W. Ferreira and E. L. Michelotti, J. Chem. Soc., Chem. Commun., 1979, 637–638 RSC.
- J. Clayden and M. Julia, J. Chem. Soc., Chem. Commun., 1993, 1682–1683 RSC.
- J. Clayden, J. J.-A. Cooney and M. Julia, J. Chem. Soc., Perkin Trans. 1, 1995, 7–14 RSC.
- C. I. Someya, M. Weidauer and S. Enthaler, Catal. Lett., 2013, 143, 424–431 CrossRef CAS.
- A. Oviedo, J. Torres-Nieto, A. Arévalo and J. J. García, J. Mol. Catal. A: Chem., 2008, 293, 65–71 CrossRef CAS.
- R. Gutiérrez-Ordaz and J. J. García, Polyhedron, 2018, 154, 373–381 CrossRef.
- J. Liu and M. J. Robins, Org. Lett., 2005, 7, 1149–1151 CrossRef CAS PubMed.
- E. J. Hennessy, M. Cornebise, L. Gingipalli, T. Grebe, S. Hande, V. Hoesch, H. Huynh, S. Throner, J. Varnes and Y. Wu, Tetrahedron Lett., 2017, 58, 1709–1713 CrossRef CAS.
- P. Chatelain, A. Sau, C. N. Rowley and J. Moran, Angew. Chem., Int. Ed., 2019, 58, 14959–14963 CrossRef CAS PubMed.
- J.-i Fukuda, K. Nogi and H. Yorimitsu, Org. Lett., 2019, 21, 8987–8991 CrossRef CAS PubMed.
- (a) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CrossRef CAS; (b) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 14, 2320–2322 CrossRef.
- J. L.-G. Ruano, J. Alemán and C. G. Paredes, Org. Lett., 2006, 8, 2683–2686 CrossRef CAS PubMed.
- (a) P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS PubMed; (b) P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed.
- (a) T. Markovic, B. N. Rocke, D. C. Blakemore, V. Mascitti and M. C. Willis, Chem. Sci., 2017, 8, 4437–4442 RSC; (b) T. Markovic, B. N. Rocke, D. C. Blakemore, V. Mascitti and M. C. Willis, Org. Lett., 2017, 19, 6033–6035 CrossRef CAS PubMed; (c) For a review of sulfinates in coupling chemistry, see D. H. Ortgies, A. Hassanpour, F. Chen, S. Woo and P. Forgione, Eur. J. Org. Chem., 2016, 408–425 CrossRef CAS.
- T. Markovic, P. R.-D. Murray, B. N. Rocke, A. Shavnya, D. C. Blakemore and M. C. Willis, J. Am. Chem. Soc., 2018, 140, 15916–15923 CrossRef CAS PubMed.
- X. A.-F. Cook, L. R.-E. Pantaine, D. C. Blakemore, I. B. Moses, N. W. Sach, A. Shavnya and M. C. Willis, Angew. Chem., Int. Ed., 2021, 60, 22461–22468 CrossRef CAS PubMed.
- W. Miao, C. Ni, P. Xiao, R. Jia, W. Zhang and J. Hu, Org. Lett., 2021, 23, 711–715 CrossRef CAS PubMed.
- (a) L. Ramberg and B. Bäcklund, Arkiv. Kemi Mineral. Geol., 1940, 13A, 1–50 Search PubMed . For selected reviews, see: ; (b) L. A. Paquette, Acc. Chem. Res., 1968, 1, 209–216 CrossRef CAS; (c) R. J.-K. Taylor, Chem. Commun., 1999, 217–227 RSC; (d) L. A. Paquette, Synlett, 2001, 1–12 CrossRef; (e) R. J.-K. Taylor and G. Casy, Org. React., 2003, 62, 359–475 Search PubMed.
- F. Takahashi, K. Nogi and H. Yorimitsu, Org. Lett., 2018, 20, 6601–6605 CrossRef CAS PubMed.
- T.-Y. Yu, Z.-J. Zheng, J.-H. Bai, H. Fang and H. Wei, Adv. Synth. Catal., 2019, 361, 2020–2024 CrossRef CAS.
- S. Kamijo, K. Kamijo and T. Murafuji, Synthesis, 2019, 3859–3864 CAS.
- Z.-J. Wang, S. Zheng, J. Matsui, Z. Lu and G. A. Molander, Chem. Sci., 2019, 10, 4389–4393 RSC.
- Y. Ikeda, R. Ueno, Y. Akai and E. Shirakawa, Chem. Commun., 2018, 54, 10471–10474 RSC.
- R. Matsubara, H. Kim, T. Sakaguchi, W. Xie, X. Zhao, Y. Nagoshi, C. Wang, M. Tateiwa, A. Ando, M. Hayashi, M. Yamanaka and T. Tsuneda, Org. Lett., 2020, 22, 1182–1187 CrossRef CAS PubMed.
- R. Matsubara, H. Kim, T. Sakaguchi, W. Xie, X. Zhao, Y. Nagoshi, C. Wang, M. Tateiwa, A. Ando, M. Hayashi, M. Yamanaka and T. Tsuneda, Org. Lett., 2020, 22, 1182–1187 CrossRef CAS PubMed.
- Thomas G. Back, Tetrahedron, 2001, 57, 5263–5301 CrossRef CAS.
- Thomas G. Back, Kristen N. Clary and Detian Gao, Chem. Rev., 2010, 110, 4498–4553 CrossRef CAS PubMed.
- J. L. García Ruano, J. Alemán, A. Parra and L. Marzo, Eur. J. Org. Chem., 2014, 1577–1588 CrossRef.
- K. Fang, M. Xie, Z. Zhang, P. Ning and G. Shu, Tetrahedron Lett., 2013, 54, 3819–3821 CrossRef CAS.
- (a) J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 4486–4487 CrossRef CAS; (b) J. Xiang, W. Jiang and P. L. Fuchs, Tetrahedron Lett., 1997, 38, 6635–6638 CrossRef CAS; (c) J. Xiang and P. L. Fuchs, Tetrahedron Lett., 1998, 39, 8597–8600 CrossRef CAS.
- Applications in total synthesis: (a) J. Bian, M. Van Wingerden and J. M. Ready, J. Am. Chem. Soc., 2006, 128, 7428–7429 CrossRef CAS PubMed; (b) S. Yoshioka, M. Nagatomo and M. Inoue, Org. Lett., 2015, 17, 90–93 CrossRef CAS PubMed.
- (a) J. Lei, X. Wu and Q. Zhu, Org. Lett., 2015, 17, 2322–2325 CrossRef CAS PubMed; (b) H. Jiang, Y. He, Y. Cheng and S. Yu, Org. Lett., 2017, 19, 1240–1243 CrossRef CAS PubMed.
- R. Ren, Z. Wu, Y. Xu and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2866–2869 CrossRef CAS PubMed.
- H. Guan, S. Sun, Y. Mao, L. Chen, R. Lu, J. Huang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 11413–11417 CrossRef CAS PubMed.
- Z. Yin, Y. Zhang, S. Zhang and X.-F. Wu, Adv. Synth. Catal., 2019, 361, 5478–5482 CrossRef CAS.
- S. Zhou, T. Song, H. Chen, Z. Liu, H. Shen and C. Li, Org. Lett., 2017, 19, 698–701 CrossRef CAS PubMed.
- H. Jiang, Y. He, Y. Cheng and S. Yu, Org. Lett., 2017, 19, 1240–1243 CrossRef CAS PubMed.
- J. Lei, X. Wu and Q. Zhu, Org. Lett., 2015, 17, 2322–2325 CrossRef CAS PubMed.
- M. Wang, H. Zhang, J. Liu, X. Wu and C. Zhu, Angew. Chem., Int. Ed., 2019, 58, 17646–17650 CrossRef CAS PubMed.
- J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275–5278 RSC.
- M. Jiang, Y. Jin, H. Yang and H. Fu, Sci. Rep., 2016, 6, 26161–26168 CrossRef CAS PubMed.
- J. Li, H. Tian, M. Jiang, H. Yang, Y. Zhao and H. Fu, Chem. Commun., 2016, 52, 8862–8864 RSC.
- J. Schwarz and B. König, ChemPhotoChem, 2017, 1, 237–242 CrossRef CAS.
- C. Gao, J. Li, J. Yu, H. Yang and H. Fu, Chem. Commun., 2016, 52, 7292–7294 RSC.
- M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362–11367 CrossRef CAS.
- T. Hoshikawa, S. Kamijo and M. Inoue, Org. Biomol. Chem., 2013, 11, 164–169 RSC.
- S. Paul and J. Guin, Green Chem., 2017, 19, 2530–2534 RSC.
- L. Capaldo and D. Ravelli, Org. Lett., 2021, 23, 2243–2247 CrossRef CAS PubMed.
- For selected reviews on allylating strategies, see: (a) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 22072293 CrossRef; (b) M. Yus, J. C. Gonzalez-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595–5698 CrossRef CAS PubMed.
- For selected reviews on olefin metathesis, see: (a) Handbook of Metathesis, ed. R. H. Grubbs, A. G. Wenzel, D. J. O’Leary and E. Khosravi, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) A. H. Hoveyda, S. J. Malcolmson, S. J. Meek and A. R. Zhugralin, Angew. Chem., Int. Ed., 2010, 49, 34–44 CrossRef CAS PubMed; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS PubMed.
- (a) M. Julia, A. Righini and J.-N. Verpeaux, Tetrahedron Lett., 1979, 26, 2393–2396 CrossRef; (b) M. Julia, A. Righini-Tapie and J.-N. Verpeaux, Tetrahedron, 1983, 39, 3283–3287 CrossRef CAS.
- J. E. Bäckvall, M. Sellén and B. Grant, J. Am. Chem. Soc., 1990, 112, 6615–6621 CrossRef.
- (a) M. van Klaveren, E. S.-M. Persson, D. M. Grove, J.-E. Bäckvall and G. van Koten, Tetrahedron Lett., 1994, 35, 5931–5934 CrossRef CAS; (b) J.-E. Bäckvall, Acta Chem. Scand., 1996, 50, 661–665 CrossRef; (c) W. Sheng, M. Wang, M. Lein, L. Jiang, W. Wei and J. Wang, Chem. – Eur. J., 2013, 19, 14126–14142 CrossRef CAS PubMed.
- (a) E. Nakamura and S. Mori, Angew. Chem., Int. Ed., 2000, 39, 3750–3771 CrossRef CAS; (b) N. Yoshikai, S.-L. Zhang and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 12862–12863 CrossRef CAS PubMed; (c) N. Yoshikai and E. Nakamura, Chem. Rev., 2012, 112, 2339–2372 CrossRef CAS PubMed.
- B. M. Trost and C. A. Merlic, J. Am. Chem. Soc., 1998, 110, 5216–5218 CrossRef.
- K. Takabe, Y. Uchiyama, K. Okisaka, T. Yamada, T. Katagiri, T. Okazaki, Y. Oketa, H. Kumobayashi and S. Akutagawa, Tetrahedron Lett., 1985, 26, 5153–5154 CrossRef CAS.
- A. G. Ibragimov, D. L. Minsker, R. A. Saraev and U. M. Dzhemilev, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1983, 32, 2104–2107 CrossRef.
- U. M. Dzhemilev, A. G. Ibragimov and D. L. Minsker, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1984, 33, 625–627 CrossRef.
- T. Llamas, R. G. Arrayás and J. C. Carretero, Adv. Synth. Catal., 2004, 346, 1651–1654 CrossRef CAS.
- J. Corpas, M. T. Quirós, P. Mauleón, R. G. Arrayás and J. C. Carretero, ACS Catal., 2019, 9, 10567–10574 CrossRef CAS.
- (a) A. L. Moure, R. G. Arrayás, D. J. Cárdenas, I. Alonso and J. C. Carretero, J. Am. Chem. Soc., 2012, 134, 7219–7222 CrossRef CAS PubMed; (b) A. L. Moure, P. Mauleón, R. G. Arrayás and J. C. Carretero, Org. Lett., 2013, 15, 2054–2057 CrossRef CAS PubMed.
- A. García-Rubia, J. A. Romero-Revilla, P. Mauleón, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2015, 137, 6857–8665 CrossRef PubMed.
- L. M. Stanley and J. F. Hartwig, Copper-Catalyzed Allylic Substitution, Chemistry Publications, 2010 Search PubMed.
- B. M. Trost, N. R. Schmuff and M. J. Miller, J. Am. Chem. Soc., 1980, 102, 5981–5983 CrossRef.
- T. Cuvigny and M. Julia, J. Organomet. Chem., 1983, 250, C21–C24 CrossRef CAS; T. Cuvigny and M. Julia, J. Organomet. Chem., 1986, 317, 383–408 CrossRef.
- M. B.-T. Thuong, S. Sottocornola, G. Prestat, G. Broggini, D. Madec and G. Poli, Synlett, 2007, 1521–1524 CAS.
- R. A. Fernandes and J. L. Nallasivam, Org. Biomol. Chem., 2019, 17, 8647–8672 RSC.
- J.-E. Bäckvall and S. K. Juntunen, J. Am. Chem. Soc., 1987, 109, 6396–6403 CrossRef.
- Y. I.-M. Nilsson, P. G. Adersson and J.-E. Bäckvall, J. Am. Chem. Soc., 1993, 115, 6609–6613 CrossRef CAS.
- B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1982, 104, 5543–5545 CrossRef CAS.
- B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1983, 105, 3343–3344 CrossRef CAS.
- B. M. Trost and C. A. Merlic, J. Org. Chem., 1990, 55, 1129–1132 CrossRef.
- K. Takizawa, T. Sekino, S. Sato, T. Yoshino, M. Kojima and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 9199–9203 CrossRef CAS PubMed.
- P. A. Evans and J. D. Nelson, J. Am. Chem. Soc., 1998, 120, 5581–5582 CrossRef CAS.
- (a) R. Takeuchi and M. Kashio, Angew. Chem., Int. Ed. Engl., 1997, 36, 262–265 CrossRef; (b) G. A. Tolstikov, N. R. Popod'ko, A. A. Pozdeeva, R. I. Khusnutdinov, S. I. Zhdanov, O. K. Galeev and U. M. Dzhemilev, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1984, 33, 1186–1189 CrossRef.
- A. N. Kasatkin, A. N. Kulak, G. A. Tostikov and S. I. Lomakina, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1988, 37, 1939–1945 CrossRef.
- Z. T. Ariki, Y. Maekawa, M. Nambo and C. M. Crudden, J. Am. Chem. Soc., 2018, 140, 78–81 CrossRef CAS PubMed.
- K. Wu and A. G. Doyle, Nat. Chem., 2017, 9, 779–784 CrossRef CAS PubMed.
- Y. Xu, M. Salman, S. Khan, J. Zhang and A. Khan, J. Org. Chem., 2020, 85, 11501–11510 CrossRef CAS PubMed.
- Y. Lou, J. Qiu, K. Yang, F. Zhang, C. Wang and Q. Song, Org. Lett., 2021, 23, 4564–4569 CrossRef CAS PubMed.
- J. Clayden and M. Julia, J. Chem. Soc., Chem. Commun., 1994, 1905–1906 RSC.
- J. M. Chalker, A. Yang, K. Deng and T. Cohen, Org. Lett., 2007, 9, 3825–3828 CrossRef CAS PubMed.
- V. I. Timokhin, S. Gastaldi, M. P. Bertrand and C. Chatgilialoglu, J. Org. Chem., 2003, 68, 3532–3537 CrossRef CAS PubMed.
- C. Gorsche, M. Griesser, G. Gescheidt, N. Moszner and R. Liska, Macromolecules, 2014, 47, 7327–7336 CrossRef CAS.
- (a) M. P. Bertrand, Org. Prep. Proced., 1994, 26, 257–290 CrossRef CAS; (b) B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 1996, 118, 1209–1210 CrossRef CAS; (c) B. Ovadia, F. Robert and Y. Landais, Chimia, 2016, 70, 34–42 CrossRef CAS PubMed.
- L. Cui, H. Chen, C. Liu and C. Li, Org. Lett., 2016, 18, 2188–2191 CrossRef CAS PubMed.
- S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085–5144 CrossRef CAS.
- X. Li, R. Zhang, X. Zhang, P. Zhu and T. Yao, Chem. – Asian J., 2020, 15, 1175–1179 CrossRef CAS PubMed.
- J.-F. Zhao, P. Gao, X.-H. Duan and L.-N. Guo, Adv. Synth. Catal., 2018, 360, 1775–1779 CrossRef CAS.
- C. Hu and Y. Chen, Org. Chem. Front., 2015, 2, 1352–1355 RSC.
- V. Corcé, L.-M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414–11418 CrossRef PubMed.
- S. Kamijo, K. Kamijo, K. Maruoka and T. Murafuji, Org. Lett., 2016, 18, 6516–6519 CrossRef CAS PubMed.
- M.-Miao Zhang and F. Liu, Org. Chem. Front., 2018, 5, 3443–3446 RSC.
- S. Sumino, M. Uno, H.-J. Huang, Y.-K. Wu and I. Ryu, Org. Lett., 2018, 20, 1078–1081 CrossRef CAS PubMed.
- D. R. Heitz, K. Rizwan and G. A. Molander, J. Org. Chem., 2016, 81, 7308–7313 CrossRef CAS PubMed.
- Y. Duan, M. Zhang, R. Ruzi, Z. Wu and C. Zhu, Org. Chem. Front., 2017, 4, 525–528 RSC.
- N. P. Ramirez, T. Lana-Villareal and J. C. Gonzalez-Gomez, Eur. J. Org. Chem., 2020, 1539–1550 CrossRef CAS.
- M. Uno, S. Sumino, T. Fukuyama, M. Matsuura, Y. Kuroki, Y. Kishikawa and I. Ryu, J. Org. Chem., 2019, 84, 9330–9338 CrossRef CAS PubMed.
- (a) M.-H. Larraufie, R. Pellet, L. Fensterbank, J.-P. Goddard, E. Lacôte, M. Malacria and C. Ollivier, Angew. Chem., Int. Ed., 2011, 50, 4463–4466 CrossRef CAS PubMed; (b) M. Daniel, L. Fensterbank, J.-P. Goddard and C. Ollivier, Org. Chem. Front., 2014, 1, 551–555 RSC.
- K. Sun, M. Ueno, K. Imaeda, K. Ueno, M. Sawamura and Y. Shimizu, ACS Catal., 2021, 11, 9722–9728 CrossRef CAS.
- L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef CAS PubMed.
- A. L.-F. de Arriba, F. Urbitsch and D. J. Dixon, Chem. Commun., 2016, 52, 14434–14437 RSC.
- J. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872–1875 CrossRef CAS PubMed.
- K. Wu, L. Wang, S. Colón-Rodríguez, G.-U. Fleschsig and T. Wang, Angew. Chem., Int. Ed., 2019, 58, 1774–1778 CrossRef CAS PubMed.
- Q.-Q. Zhao, J. Chen, D.-M. Yan, J.-R. Chen and W.-J. Xiao, Org. Lett., 2017, 19, 3620–3623 CrossRef CAS PubMed.
- H. Liu, Y. Li, D.-X. Wang, M.-M. Sun and C. Feng, Org. Lett., 2020, 22, 8681–8686 CrossRef CAS PubMed.
- X. Huang, S. Luo, O. Burghaus, R. D. Webster, K. Harms and E. Meggers, Chem. Sci., 2017, 8, 7126–7131 RSC.
- J.-C. Wu, L. B. Gong, Y. Xia, R.-J. Song, Y.-X. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2012, 51, 9909–9913 CrossRef CAS PubMed.
- S. E. Denmark and A. J. Cresswell, J. Org. Chem., 2013, 78, 12593–12628 CrossRef CAS PubMed.
- Primary sulfones suffered from poor yields because of the competing α-deprotonation reaction by the Grignard reagent. Tertiary sulfones underwent β-elimination pathways.
- W. Miao, Y. Zhao, C. Ni, B. Gao, W. Zhang and J. Hu, J. Am. Chem. Soc., 2018, 140, 880–883 CrossRef CAS PubMed.
- R. R. Merchant, J. T. Edwards, T. Qin, M. M. Kruszyk, C. Bi, G. Che, D.-H. Bao, W. Qiao, L. Sun, M. R. Collins, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, P. Nuhant and P. S. Baran, Science, 2018, 360, 75–80 CrossRef CAS PubMed.
- M. Nambo and C. M. Crudden, Angew. Chem., Int. Ed., 2014, 53, 742–746 CrossRef CAS PubMed.
- M. Nambo, E. C. Keske, J. P.-G. Rygus, J. C.-H. Yim and C. M. Crudden, ACS Catal., 2017, 7, 1108–1112 CrossRef CAS.
- J. C.-H. Yim, M. Nambo and C. M. Crudden, Org. Lett., 2017, 19, 3715–3718 CrossRef CAS PubMed.
- J. C.-H. Yim, M. Nambo, Y. Tahara and C. M. Crudden, Chem. Lett., 2019, 48, 975–977 CrossRef CAS.
- M. Nambo, J. C.-H. Yim, L. B.-O. Freitas, Y. Tahara, Z. T. Ariki, Y. Maekawa, D. Yokogawa and C. M. Crudden, Nat. Commun., 2019, 10, 4528 CrossRef PubMed.
- Z. T. Ariki, Y. Maekawa, M. Nambo and C. M. Crudden, J. Am. Chem. Soc., 2018, 140, 78–81 CrossRef CAS PubMed.
- C. E.-I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. J. von Wangelin, Chem. – Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed.
- T. Moragas, A. Correa and R. Martin, Chem. – Eur. J., 2014, 20, 8242–8258 CrossRef CAS PubMed.
- Y.-S. Lin, Y.-C. Kuo, C.-H. Kuei and M.-Y. Chang, Tetrahedron, 2017, 73, 1275–1282 CrossRef CAS.
- J. M.-E. Hughes and P. S. Fier, Org. Lett., 2019, 21, 5650–5654 CrossRef CAS PubMed.
- X. Tao, G. Ma, Y. Song, Y. Chen, Q. Qian, D. Sun and H. Gong, Org. Lett., 2021, 23, 7418–7422 CrossRef CAS PubMed.
- Y. Xia and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9836–9840 CrossRef CAS PubMed.
- J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743–2747 CrossRef CAS PubMed.
- For similar examples using 2-pyridyl sulfones for the introduction of CF2SAr moieties, see: J. Wei, D. Gu, S. Wang, J. Hu, X. Dong and R. Sheng, Org. Chem. Front., 2018, 5, 2568–2572 RSC.
- W. Fu, X. Han, M. Zhu, C. Xu, Z. Wang, B. Ji, X.-Q. Hao and M.-P. Song, Chem. Commun., 2016, 52, 13413–13416 RSC.
- G. Zou and X. Wang, Org. Biomol. Chem., 2017, 15, 8748–8754 RSC.
- M. Zhu, Q. You and R. Li, J. Fluor. Chem., 2019, 228, 109391 CrossRef CAS.
- A. L.-P. Lemos, L. Trump, B. Lallemand, P. Pasau, J. Mercier, C. Lemaire, J.-C. Monbaliu, C. Genicot and A. Luxen, Catalysts, 2020, 10, 275 CrossRef CAS.
- (a) K. Itami and J.-i Yoshida, Bull. Chem. Soc. Jpn., 2006, 79, 811–824 CrossRef CAS; (b) J. Liu, X. Liu, K. Wu and C.-C. Li, Chem, 2020, 6, 579–615 CrossRef CAS.
- For selected reviews, see: (a) H. H. Voge and C. R. Adams, Adv. Catal., 1967, 17, 151–221 CAS; (b) L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479–1494 CrossRef CAS PubMed; (c) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130–2169 CrossRef CAS PubMed; (d) R. Remy and C. G. Bochet, Chem. Rev., 2016, 116, 9816–9849 CrossRef CAS PubMed; (e) S. W.-M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed; (f) S. Kraft, K. Ryan and R. B. Kargbo, J. Am. Chem. Soc., 2017, 139, 11630–11641 CrossRef CAS PubMed; (g) J. V. Obligacion and P. J. Chirik, Nat. Rev. Commun., 2018, 2, 15–34 CAS; (h) O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC; (i) M. R. Becker, R. B. Watson and C. S. Schindler, Chem. Soc. Rev., 2018, 47, 7867–7881 RSC; (j) R. K. Dhungana, R. R. Sapkota, D. Niroula and R. Giri, Chem. Sci., 2020, 11, 9757–9774 RSC.
- (a) J. Wang and I. Chataigner, Stereoselective alkene synthesis, Springer Verlag, Heidelberg, New York, 2012 CrossRef; (b) T. Takeda, Modern Carbonyl Olefination: Methods and Applications, WileyVCH, Weinheim, 2004 Search PubMed.
- M. Julia and J.-N. Verpeaux, Tetrahedron Lett., 1982, 23, 2457–2460 CrossRef CAS.
- D. Srimani, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 11092–11095 CrossRef CAS PubMed.
- (a) T. C. Jankis, R. R. Fayzullin and E. Khaskin, Organometallics, 2018, 37, 2609–2617 CrossRef; (b) S. Waiba, M. K. Barman and B. Maji, J. Org. Chem., 2019, 84, 973–982 CrossRef CAS PubMed; (c) T. Vojkovsky, S. Deolka, S. Stepanova, M. C. Roy and E. Khaskin, ACS Catal., 2020, 10, 6810–6815 CrossRef CAS.
- S. M.-A. H. Siddiki, A. S. Touchy, K. Kon and K.-I. Shimizu, Chem. – Eur. J., 2016, 22, 6111–6119 CrossRef CAS PubMed.
- V. G. Landge, V. Yadav, M. Subaramanian, P. Dangarh and E. Balaraman, Chem. Commun., 2019, 55, 6130–6133 RSC.
- S. Waiba, A. Das, M. K. Barman and B. Maiji, ACS Omega, 2019, 4, 7082–7087 CrossRef CAS PubMed.
- V. G. Landge, R. babu, V. Yadav, M. Subaramanian, V. Gupta and E. Balaraman, J. Org. Chem., 2020, 85, 9876–9886 CrossRef CAS PubMed.
- P. K. Shyam, C. Lee and H.-Y. Jang, Bull. Korean Chem. Soc., 2015, 36, 1824–1827 CrossRef CAS.
- S. Lim, M. Ji, X. Wang, C. Lee and H.-Y. Jang, Eur. J. Org. Chem., 2015, 591–595 CrossRef CAS.
- S. Xu, Y. Gao, R. Chen, K. Wang, Y. Zhang and J. Wang, Chem. Commun., 2016, 52, 4478–4480 RSC.
- (a) J. B. Baudin, G. Hareau, S. A. Julia and O. Ruel, Tetrahedron Lett., 1991, 32, 1175–1178 CrossRef CAS; (b) P. A. Blakemore, W. J. Cole, P. J. Kocienski and A. Morley, Synlett, 1998, 26–28 CrossRef CAS; (c) P. J. Kocienski, A. Bell and P. R. Blakemore, Synlett, 2000, 365–366 CAS; (d) D. Mirk and J.-M. Grassot, Synlett, 2006, 1255–1259 CAS; (e) C. Aïssa, J. Org. Chem., 2006, 71, 360–363 CrossRef PubMed; (f) K. Ando, T. Kobayashi and N. Uchida, Org. Lett., 2015, 17, 2554–2557 CrossRef CAS PubMed.
- R. H. Shapiro, Org. React., 1976, 23, 405–507 CAS.
- For selected reviews, see: (a) R. A. Michelin, M. Mozzon and R. Bertani, Coord. Chem. Rev., 1996, 147, 299–338 CrossRef CAS; (b) V. Y. Kukushkin and A. J.-L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef CAS PubMed; (c) S. J. Collier and P. Langer. Science of Synthesis, Georg Thieme, Stuttgart, 2004, vol. 19, pp. 403–425 Search PubMed; (d) M. X. Wang, Top. Catal., 2005, 35, 117–130 CrossRef CAS.
- Y. Nakao, Chem. Rev., 2020, 121(1), 327–344 CrossRef PubMed.
- For representative examples, see: (a) A. M. Negm, F. M. Abdelrazek, M. H. Elnagdi and L. H. Shaaban, Arch. Pharm. Res., 1994, 17, 411–414 CrossRef CAS PubMed; (b) A. Z.-A. Hassanien and Z. E. Kandeel, J. Chem. Res., 2003,(S), 687–688 CrossRef CAS; (c) K. Rad-Moghadam and L. Samavi, J. Heterocyclic Chem., 2006, 43, 913–916 CrossRef CAS; (d) Z. Wei, L. Zheng, Q. Dang and X. Bai, J. Heterocyclic Chem., 2009, 46, 1425–1429 CrossRef CAS; (e) M. Anzini, S. Valenti, C. Braile, A. Cappelli, S. Vomero, S. Alcaro, F. Ortuso, L. Marinelli, V. Limongelli, E. Novellino, L. Betti, G. Giannaccini, A. Lucacchini, S. Daniele, C. Martini, C. Ghelardini, L. D.-C. Mannelli, G. Giorgi, M. P. Mascia and G. Biggio, J. Med. Chem., 2011, 54, 5694–5711 CrossRef CAS PubMed; (f) R. D. Taylor, M. MacCoss and A. D. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- For some examples, see: (a) M. Capron, S. Loiseau, J. P. Papin, S. Robertson and A. Capron, Int. Arch. Allergy Immunol., 1998, 116, 140–146 CrossRef CAS PubMed; (b) P. A.-J. Janssen, P. J. Lewi, E. Arnold, F. Daeyaert, M. de Jonge, J. Heeres, L. Koymans, M. Vinkers, J. Guillemont, E. Pasquier, M. Kukla, D. Ludovici, K. Andries, M.-P. de Béthune, R. Pauwels, K. Das, A. D. Clark, Jr., Y. V. Frenkel, S. H. Hughes, B. Medaer, F. De Knaep, H. Bohets, F. De Clerck, A. Lampo, P. Williams and P. Stof, J. Med. Chem., 2005, 48, 1901–1909 CrossRef CAS PubMed; (c) I. Laponogov, M. K. Sohi, D. A. Veselkov, X. S. Pan, R. Sawhney, A. W. Thompson, K. E. McAuley, L. M. Fisher and M. R. Sanderson, Nat. Struct. Mol. Biol., 2009, 16, 667–669 CrossRef CAS PubMed; (d) M. E. Jung, S. Ouk, D. Yoo, C. L. Sawyers, C. Chen, C. Tran and J. Wongvipat, J. Med. Chem., 2010, 53, 2779–2796 CrossRef CAS PubMed.
- C. G. Claessens, D. Gonzalez-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed.
- A. M. Nauth and T. Opatz, Org. Biomol. Chem., 2019, 17, 11–23 RSC.
- X. Fei, Synlett, 2013, 2021–2022 Search PubMed.
- For earlier examples of radical-mediated cyanations employing TsCN, see: (a) J.-M. Fang and M.-Y. Chen, Tetrahedron Lett., 1987, 28, 2853–2856 CrossRef CAS; (b) D. H.-R. Barton, J. C. Jaszberenyi and E. A. Theodorakis, Tetrahedron, 1992, 48, 2613–2626 CrossRef CAS . For recent extensions of this chemistry, see: ; (c) S. Kim and H.-J. Song, Synlett, 2002, 2110–2112 CrossRef CAS; (d) S. Kim, C. H. Cho, S. Kim, Y. Uenoyama and I. Ryu, Synlett, 2005, 3160–3162 CrossRef CAS; (e) W. Shao, M. Lux, M. Breugst and M. Klussmann, Org. Chem. Front., 2019, 6, 1796–1800 RSC.
- A.-P. Schaffner, V. Darmency and P. Renaud, Angew. Chem., Int. Ed., 2006, 45, 5847–5849 CrossRef CAS PubMed.
- J.-J. Dai, W.-M. Zhang, Y.-J. Shu, Y.-Y. Sun, J. Xu, Y.-S. Feng and H.-J. Xu, Chem. Commun., 2016, 52, 6793–6796 RSC.
- D. R. Heitz, K. Rizwan and G. A. Molander, J. Org. Chem., 2016, 81, 7308–7313 CrossRef CAS PubMed.
- X. Wang, G. H.-M. Davies, A. Koschitzcky, S. R. Wisniewski, C. B. Kelly and G. A. Molander, Org. Lett., 2019, 21, 2880–2884 CrossRef CAS PubMed.
- E. André-Joyaux, A. Kuzovlev, N. D.-C. Tappin and P. Renaud, Angew. Chem., Int. Ed., 2020, 59, 13859–13864 CrossRef PubMed.
- N. P. Ramirez, B. König and J. C. Gonzalez-Gomez, Org. Lett., 2019, 21, 1368–1373 CrossRef CAS PubMed.
- M. Shee, S. Shah and N. D.-P. Singh, Chem. Commun., 2020, 56, 4240–4243 RSC.
- H. Zhang, Y. Zhou, P. Tian and C. Jiang, Org. Lett., 2019, 21, 1921–1925 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Ping, Q. Ding and Y. Peng, ACS Catal., 2016, 6, 5989–6005 CrossRef CAS; (b) B. L. Elbert, A. J.-M. Farley, T. W. Gorman, T. C. Johnson, C. Genicot, B. Lallemand, P. Pasau, J. Flasz, J. L. Castro, M. MacCoss, R. S. Paton, C. J. Schofield, M. D. Smith, M. C. Willis and D. J. Dixon, Chem. – Eur. J., 2017, 23, 14733–14737 CrossRef CAS PubMed; (c) L.-Y. Liu, K.-S. Yeung and J.-Q. Yu, Chem. – Eur. J., 2019, 25, 2199–2202 CrossRef CAS PubMed; (d) D. Zhao, P. Xu and T. Ritter, Chem, 2019, 5, 97–107 CrossRef CAS; (e) X. Wang, M. Makha, S.-W. Chen, H. Zheng and Y. Li, J. Org. Chem., 2019, 84, 6199–6206 CrossRef CAS PubMed.
- (a) S. Kamijo, T. Hoshikawa and M. Inoue, Org. Lett., 2011, 13, 5928–5931 CrossRef CAS PubMed; (b) T. Hoshikawa, S. Yoshioka, S. Kamijo and M. Inoue, Synthesis, 2013, 0874–0877 CAS.
- T. Wakaki, K. Sakai, T. Enomoto, M. Kondo, S. Masaoka, K. Oisaki and M. Kanai, Chem. – Eur. J., 2018, 24, 8051–8055 CrossRef CAS PubMed.
- X.-Z. Fan, J.-W. Rong, H.-L. Wu, Q. Zhou, H.-P. Deng, J. D. Tan, C.-W. Xue, L.-Z. Wu, H.-R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed.
- T. Ide, K. Shimizu, H. Egami and Y. Hamashima, Tetrahedron Lett., 2018, 59, 3258–3261 CrossRef CAS.
- For selected examples on the use of alkenes as radical acceptors and radical 1,2-difunctionalization, see: (a) B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753–764 CrossRef; (b) R. W. Hoffmann, Chem. Soc. Rev., 2016, 45, 577–583 RSC; (c) X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821–5851 CrossRef CAS; (d) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef CAS; (e) Z. L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC; (f) D. C.-S. Costa, Arab. J. Chem., 2020, 13, 799–834 CrossRef.
- For selected examples, see: (a) R. K. Quinn, V. A. Schmidt and E. J. Alexanian, Chem. Sci., 2013, 4, 4030–4034 RSC; (b) N. S. Dange, F. Robert and Y. Landais, Org. Lett., 2016, 18, 6156–6159 CrossRef CAS PubMed; (c) H. Hassan, V. Pirenne, M. Wissing, C. Khiar, A. Hussain, F. Robert and Y. Landais, Chem. – Eur. J., 2017, 23, 4651–4658 CrossRef CAS PubMed; (d) R. Hara, C. Khiar, N. S. Dange, P. Bouillac, F. Robert and Y. Landais, Eur. J. Org. Chem., 2018, 4058–4063 CrossRef CAS.
- J. Sun, P. Li, L. Guo, F. Yu, Y.-P. He and L. Chu, Chem. Commun., 2018, 54, 3162–3165 RSC.
- V. Pirenne, G. Kurtay, S. Voci, L. Bouffier, N. Sojic, F. Robert, D. M. Bassani and Y. Landais, Org. Lett., 2018, 20, 4521–4525 CrossRef CAS PubMed.
- V. Pirenne, I. Traboulsi, L. Rouvière, J. Lusseau, S. Massip, D. M. Bassani, F. Robert and Y. Landais, Org. Lett., 2020, 22, 575–579 CrossRef CAS PubMed.
- B. Gaspar and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 4519–4522 CrossRef CAS PubMed.
- R. Hanada, K. Mitachi and K. Tanino, Tetrahedron Lett., 2014, 55, 1097–1099 CrossRef CAS.
- V. Girijavallabhan, A. Arasappan, F. Bennett, K. Chen, Q. Dang, Y. Huang, A. Kerekes, L. Nair, D. Pissarnitski, V. Verma, C. Alvarez, P. Chen, D. Cole, S. Esposite, Y. Huang, Q. Hong, Z. Liu, W. Pan, H. Pu, R. Rossman, Q. Truong, B. Vibulbhan, J. Wang, Z. Zhao, D. Olsen, A. Stamford, S. Bogen and F. G. Njoroge, Nucleosides, Nucleotides Nucleic Acids, 2016, 35, 277–294 CrossRef CAS PubMed.
- F. Yoshimura, R. Itoh, M. Torizuka, G. Mori and K. Tanino, Angew. Chem., Int. Ed., 2018, 57, 17161–17167 CrossRef CAS PubMed.
- F. Yoshimura, T. Abe, Y. Ishioka and K. Tanino, J. Antibiot., 2019, 72, 384–388 CrossRef CAS PubMed.
- S. Arai, Y. Sato, N. Ito and A. Nishida, Tetrahedron Lett., 2019, 60, 151314 CrossRef CAS.
- M. Liu, E. You, W. Cao and J. Shi, Asian J. Org. Chem., 2019, 8, 1850–1853 CrossRef CAS.
- D. A. Alonso and C. Nájera, Organic Reactions, 2008, vol. 72, pp. 367–656 Search PubMed.
- Z. Jin and P. L. Fuchs, Tetrahedron Lett., 1996, 37, 5253–5256 CrossRef CAS.
- J. Yang, J. Xiao, T. Chen, S.-F. Yin and L.-B. Han, Chem. Commun., 2016, 52, 12233–12236 RSC.
- H.-M. Guo, Q.-Q. Zhou, X. Jiang, D.-Q. Shi and W.-J. Xiao, Adv. Synth. Catal., 2017, 359, 4141–4146 CrossRef CAS.
- Y. Yatsumonji, A. Ogata, A. Tsubouchi and T. Takeda, Tetrahedron Lett., 2008, 49, 2265–2267 CrossRef CAS.
- H. Saito, K. Nogi and H. Yorimitsu, Synthesis, 2017, 4769–4774 CAS.
- Y. Maekawa, Z. T. Ariki, M. Nambo and C. M. Crudden, Org. Biomol. Chem., 2019, 17, 7300–7303 RSC.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607–610 CrossRef CAS PubMed.
- M. Nambo, Y. Tahara, J. C.-H. Yim and C. M. Crudden, Chem. – Eur. J., 2019, 25, 1923–1926 CrossRef CAS PubMed.
- C.-C. Sun, F. Lian, K. Xu, C.-C. Zeng and B.-G. Sun, Adv. Synth. Catal., 2019, 361, 4141–4146 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |