
Enhanced internal ionic interaction of MFS efflux pump MdfA contributes to its elevated antibiotic export†
Ying
Li
and
Xizhen
Ge
 *
*
College of Biochemical Engineering, Beijing Union University, Beijing, 100023, China. E-mail: shtxizhen@buu.edu.cn; Fax: +86-10-52072337; Tel: +86-10-52072337
First published on 1st December 2022
Abstract
Infections caused by Gram-negative pathogens are difficult to manage due to their antibiotic resistance. Efflux pumps, which transport intracellular toxins out of the cytoplasm, play an important role in the detoxification of bacteria when treated with antibiotics. The major facilitator superfamily (MFS) is a kind of widely distributed efflux pumps and can actively export clinically important antibiotics such as ciprofloxacin, while the role of internal ionic interactions in regulating drug export remains poorly understood. Herein we used a representative MFS efflux pump MdfA to investigate the impact of internal ionic interactions on the antibiotic resistance of E. coli. First, we identified the internal salt bridges of MdfA and searched their natural variants across all the sequenced E. coli isolates. By constructing these variants, we discovered that extending the salt bridge on the cytoplasmic side (E136D) conferred an elevated antibiotic resistance level of E. coli, and the level was further enhanced by combining it with an artificial mutation K346R. By analyzing the trajectories of MdfA's variants in molecular dynamics (MD) simulations, we revealed that ionic interaction strengths on the two sides were proportionally enhanced, while the protein flexibility was not affected. Moreover, enhanced interactions resulted in a larger surface for MdfA's protonation, suggesting a higher possibility for its activation. Collectively, our data revealed the importance of internal interactions on the drug export of MdfA, offering insights for the development of novel inhibitors against MFS efflux pumps.
Introduction
Overuse and misuse of antibiotics have accelerated the spread of antibiotic resistant bacteria in recent years.1 Infections, especially caused by Gram-negative pathogens, are becoming more and more difficult to manage due to the improvements and genetic transfer of antibiotic resistance elements.2,3 Among the factors that contribute to bacterial antibiotic resistance, the active transport of antibiotics out of the cytoplasm by efflux pumps represents one of the most serious challenges for antibiotic therapy.4,5 The major facilitator superfamily (MFS) is the largest class of secondary transporters and is present in all kingdoms of life.6 Up to now, the roles of several MFS efflux pumps from Gram-negative pathogens have been characterized, such as MdfA from Escherichia coli, KmrA from Klebsiella pneumoniae and SmvA from Salmonella enterica.7–9 Different from the tripartite efflux system of the resistance/nodulation/cell division (RND) family, MFS efflux pumps are much smaller transporters embedded in the inner membrane of Gram-negative pathogens.10 However, these MFS efflux pumps are demonstrated to confer resistance against some clinically important antibiotics such as ciprofloxacin and chloramphenicol, posing potential risk clinically.11 With more and more MFS efflux pumps that transfer across the bacterial community through mobile genetic elements being discovered, there is an urgent need to understand the mechanism that favors their antibiotic transport.12,13MFS efflux pumps contain a 12-transmembrane helix core composed of two six-helix rigid domains (namely the N- and C-repeats) forming a central transmembrane channel (Fig. 1).14,15 A rocker-switch mechanism is proposed and is the most acceptable model for the description of this dynamic process.16 In this mechanism, the MFS efflux pump is believed to switch between its inward- and outward-facing conformations, which represent the accommodating and extruding modes, respectively.17 From the energy point of view, the inward-facing conformation is the excited state and the outward-facing one is the ground state. Previous studies deduced that the driving forces for the conformational transition of MFS efflux pumps are composed of several factors, such as the protonation of titratable residues, hydrophilic interaction with water, hydrophobic interaction with membranes, and ionic interaction (salt bridge) between the N- and C-repeats.6,18 For both the inward- and outward-facing conformations, salt bridges can be found on the closed side and these interactions are believed to be important for the activity of the transporter.19–21 However, the relationship between the ionic interaction strength and the transport efficiency still remains poorly understood.
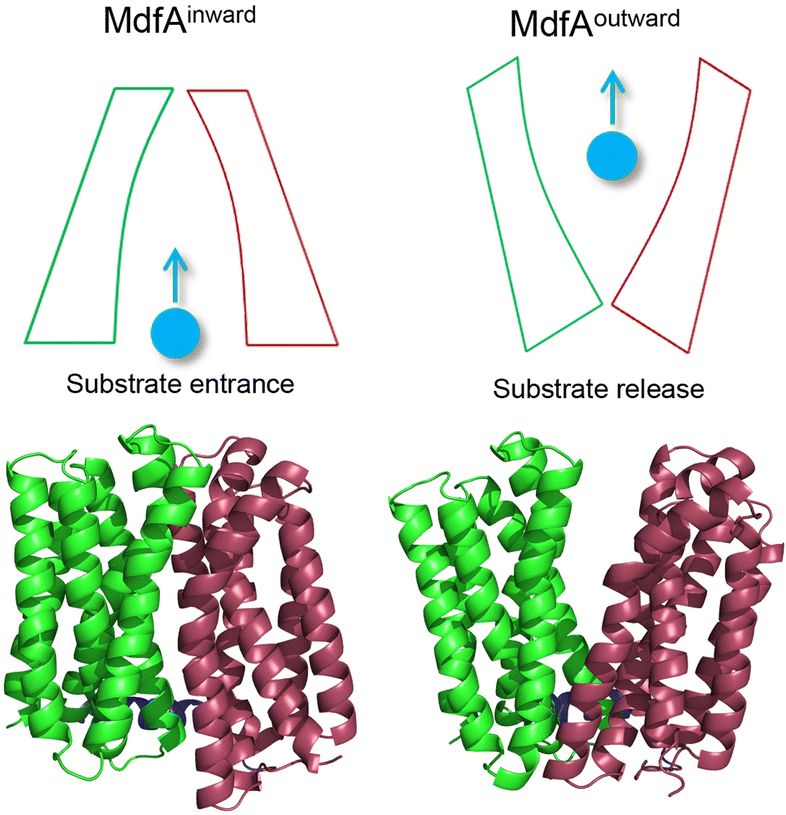 | ||
| Fig. 1 Schematic representation of the inward- and outward-facing conformations of MdfA. Structures of MdfA were visualized in Pymol. Green and dull red regions of MdfA indicated the N- and C-repeats, respectively. | ||
As a representative MFS efflux pump, MdfA from E. coli is well-characterized for which both the inward- and outward-facing conformations are observed.20,22 Moreover, the roles of several important residues have been investigated, including the residues for protonation and drug binding, while the effect of salt bridges on its transport efficiency is still unclear. Our recent study demonstrated that natural mutations near the periplasmic side of KmrA resulted in elevated antibiotic resistance of K. pneumoniae, revealing potential risk of natural mutations of MFS efflux pumps.23 Hence, in this work we seek to discover the amino acid variations near the salt bridges of MdfA to confirm the effect of ionic interaction strength on antibiotic resistance of E. coli. After identifying salt bridges of wild type MdfA via molecular dynamics (MD) simulations, we altered several residues based on their isoelectric points to enhance the ionic interactions. Our results indicated that stronger ionic interactions on the cytoplasmic side facilitated the elevated antibiotic resistance of E. coli, offering new details for the conformational transition of MFS efflux pumps.
Results
Original salt bridges of MdfAinward and MdfAoutward
In order to identify the major salt bridges in MdfAinward and MdfAoutward, MD simulations were conducted and the ionic interactions between the N- and C-repeats were calculated (Fig. 2). Within the simulation periods, one salt bridge of each simulation was detected. For MdfAinward, a salt bridge Asp52–Lys369 was detected on the periplasmic side, while the possibility was low for most of the simulation snapshots (Fig. 2A and B). For MdfAoutward, Glu136–Arg336 on the cytoplasmic side was observed. Different from that of MdfAinward, the ionic interaction strength of Glu136–Arg336 was evenly divided into two major parts (Fig. 2C and D). In nearly half of the snapshots, Glu136–Arg336 had a high interaction strength. These results were in accordance with the rocker-switch model of MdfA showing that the outward-facing conformation represented the ground state of MdfA.18 Stronger interaction was detected on the cytoplasmic side of the outward-facing conformation, making it stable before protonation.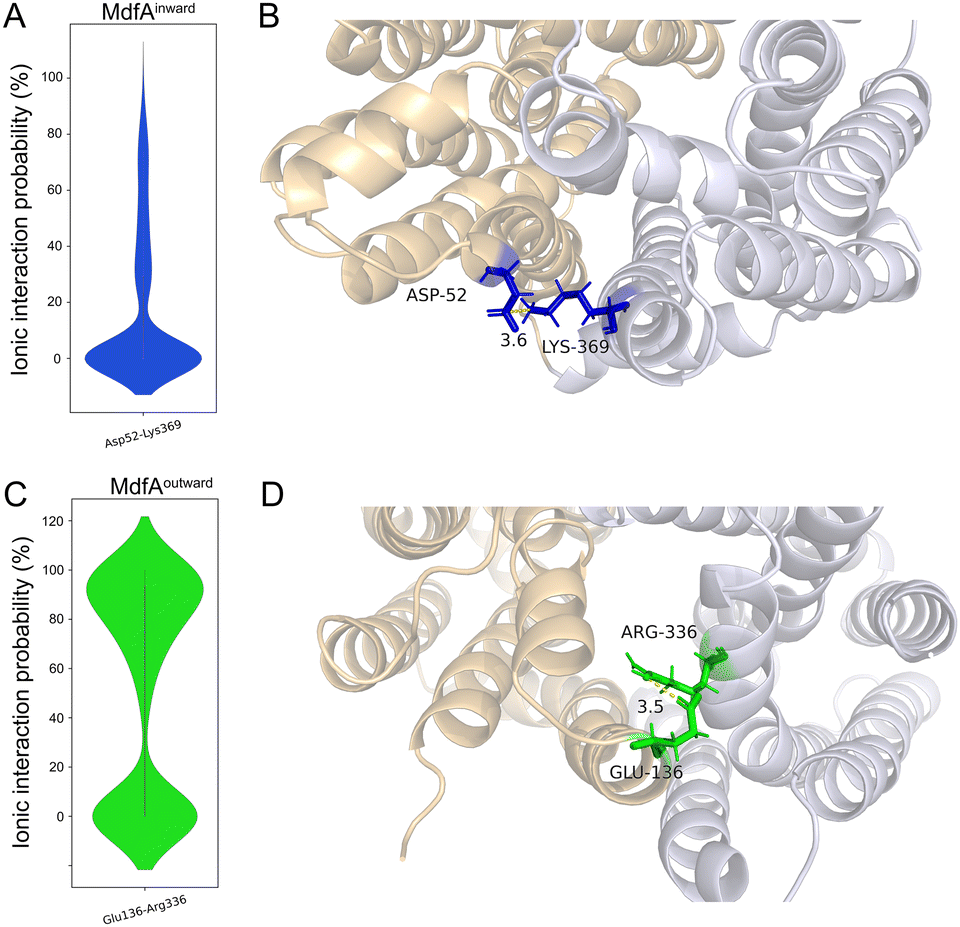 | ||
| Fig. 2 Original salt bridges between N- and C-repeats of MdfA. The probabilities of salt bridges were calculated across the MD simulations of MdfAinward (A) and MdfAoutward (C). Visualizations of the ionic interactions from periplasmic (B) and cytoplasmic (D) sides were conducted in Pymol. | ||
Natural and artificial alterations of salt bridges that confer elevated antibiotic resistance
Natural mutations play an underestimated role in the evolution of efflux pumps. Here we used our previously established methods to investigate the amino acid variations near the salt bridges across all the sequenced E. coli isolates.23 Within nearly 1500 reported MdfA paralogs, we identified several natural variations of MdfA that may confer enhanced ionic interactions (Glu to Asp and Lys to Arg, based on their isoelectric points, Table 1 and Table S2, ESI†). On the periplasmic side, no positive variation was discovered for both Asp52 and Lys369, while several variations of the adjacent residues were detected. In contrast, we detected a positive variation E136D on the cytoplasmic side, which has been deduced to confer stronger ionic interaction. According to the results, we constructed the MdfA variants and then expressed them in an E. coli bacterium in which the native mdfA gene was deleted. Meanwhile, we also made an artificial variant of K369R in order to enhance the interaction on the periplasmic side.According to the previous studies, overexpression of the mdfa gene in E. coli conferred the highest antibiotic resistance level against chloramphenicol and ciprofloxacin.22,24,25 Therefore, we first tested the chloramphenicol resistance levels of the recombinant strains in which MdfA or its variants were overexpressed (Fig. 3A and Fig. S1, ESI†). With similar expression level in the recombinant E. coli (Fig. S2, ESI†), overexpression of wild type MdfA resulted in increased viable cells in the presence of 2.5 μg mL−1 chloramphenicol compared to those on the vector control. Surprisingly, the E136D variant conferred further elevated chloramphenicol resistance compared to the wild type MdfA, while no differences were observed when ionic interaction on the periplasmic side (K346R) was enhanced. Since the preferred substrates of MdfA are chloramphenicol and ciprofloxacin, we subsequently tested the intracellular ciprofloxacin concentrations of the recombinant strains (Fig. 3B and Fig. S3, ESI†). As a result, the E136D variant significantly reduced the intracellular concentration compared to the wild type MdfA. Therefore, it can be concluded that enhanced ionic interaction on the cytoplasmic side of MdfA contributed to the elevated antibiotic resistance of E. coli.
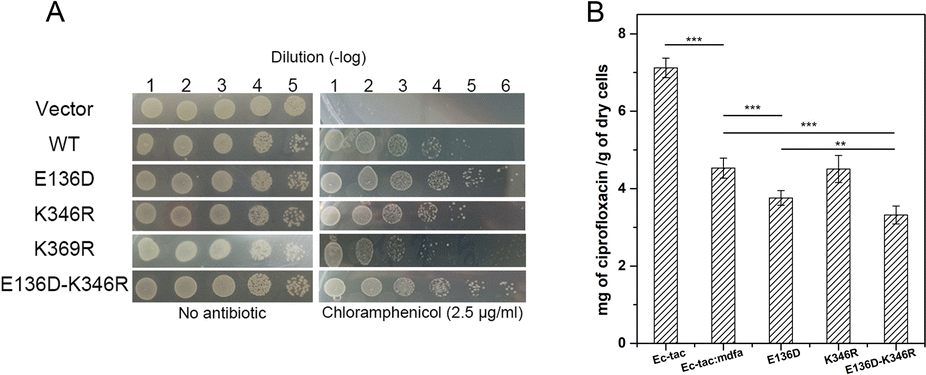 | ||
| Fig. 3 Amino acid variations of MdfA conferred elevated antibiotic resistance on E. coli. (A) Chloramphenicol resistance assay of the recombinant E. coli strains. Cultivations of the strains were 10-fold diluted and plated on solid LB medium containing 2.5 μg mL−1 chloramphenicol. (B) Intracellular ciprofloxacin concentrations were further decreased when MdfA variants were expressed (**: P < 0.01 and ***: P < 0.001). | ||
Simulations of MdfAE136D revealed altered salt bridges
Since the E136D variation resulted in elevated antibiotic resistance, we conducted the MD simulations of MdfAE136D with both of its inward- and outward-facing conformations, and the salt bridges were calculated (Fig. 4). In the inward-facing conformation, the interaction strength of Asp52–Lys369 remained similar to that of the wild type MdfA. However, the possibility of detecting a new salt bridge on the cytoplasmic side (Asp136–Lys346) was low (Fig. 4A). In contrast, for the outward-facing conformation of MdfAE136D, much stronger interaction was detected for Asp136–Arg336 on the cytoplasmic side (Fig. 4C).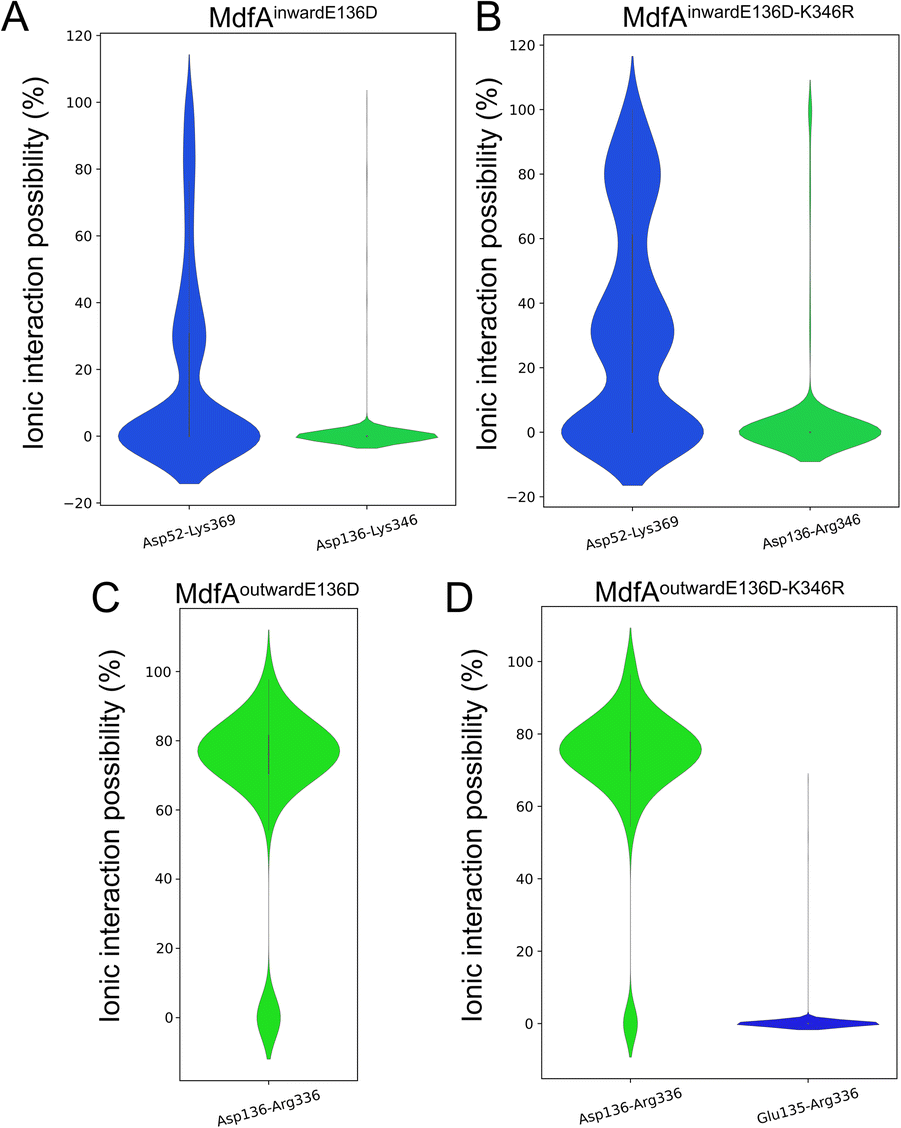 | ||
| Fig. 4 Salt bridges of MdfA variants were altered. The probabilities of salt bridges were calculated across the MD simulations of inward (A and B) and outward (C and D). | ||
With the emergence of a new salt bridge Asp136–Lys346 on the cytoplasmic side of MdfAinwardE136D, we suspected that K346R would further increase the ionic interaction strength. Therefore, we constructed the single and double variations of K346R from wild type MdfA and MdfAE136D. Chloramphenicol resistance assay indicated a moderate but not significant increase in viable cells of MdfAE136D–K346R compared to that of MdfAE136D, while the single variation K346R conferred slightly reduced resistance to chloramphenicol (Fig. 3A). For the intracellular ciprofloxacin assay, MdfAE136D–K346R further reduced intracellular concentration compared to that of single mutation (Fig. 3B).
Subsequently, we conducted MD simulations of MdfAE136D–K346R. In the inward-facing conformation, the ionic interactions on both sides were increased (Fig. 4B). However, the salt bridge Asp136–Arg346 was still only detected in the inward-facing conformation. For the outward-facing conformation, the Arg346 was not observed to form a salt bridge with the N-repeat. Compared to MdfAE136D, the strength of Asp136–Arg336 rarely changed. A slight difference was found that Arg336 also formed ionic interaction with Glu135 at a low level. Collectively, these data confirmed that ionic interactions on the cytoplasmic side were increased by altering residues on the basis of their isoelectric points. More importantly, these enhanced ionic interactions elevated the antibiotic resistance level of the host strain.
Opposite motion of the cytoplasmic mouth was increased with enhanced ionic interaction
Our previous work demonstrated that the opposite motion of the cytoplasmic mouth of KmrA was an important character for its export activity.21 With enhanced ionic interactions on the cytoplasmic side of MdfA, the opposite motion in the inward-facing conformation is anticipated to be elevated. Therefore, we calculated the DCCMs of MdfA and its variants to identify the regular trajectories (Fig. 5). Within the regions near the salt bridge Asp136–Arg346, the opposite motion was observed for all the three simulations at different levels (Fig. 5D). For the wild type MdfAinward, no ionic interaction was detected on the cytoplasmic side (Fig. 2A), and the DCCM value between Asp136 and Arg346 was around −0.2. In contrast, with the emergence of ionic interactions on the cytoplasmic side of the variants of MdfAinward (Fig. 4A and B), the DCCMs between these regions were reduced to −0.4 (Fig. 5B and D). These results indicated that increasing trends of opposite motion were reinforced for the variants of MdfA. Since the variants conferred elevated antibiotic resistance, these data suggested that ionic interaction and the opposite motion trends on the cytoplasmic side may have an underestimated role in MdfA's conformational transition back to its ground state.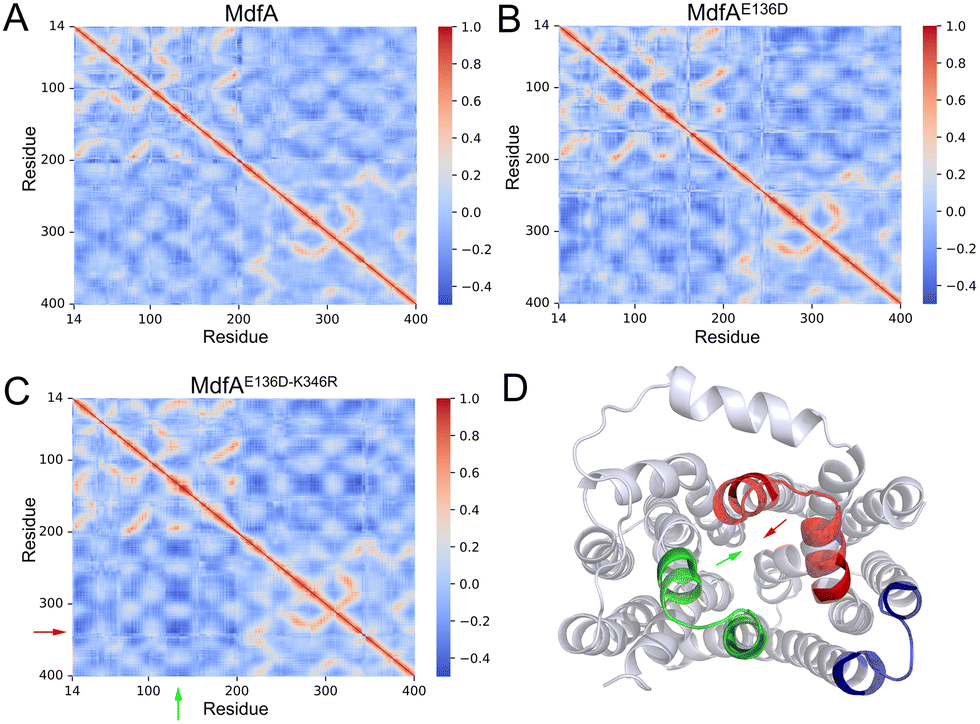 | ||
| Fig. 5 Opposite motions of the central helices were enhanced for the variants of MdfA in inward-facing conformations. DCCM was calculated to evaluate the relative motion of each residue (A–C). (D) Cytoplasmic view of regions that had opposite motions. Arrows indicate the region of the helices in which the motions were increased. Visualization was conducted in Pymol. | ||
Opening area of the cytoplasmic side was not reduced in inward-facing conformation
The inward-facing conformation of MdfA is the access mode for substrates.15,26 Therefore, the opening area of the cytoplasmic side is crucial for its ability to accommodate. We calculated the opening areas of MdfA and its variants to ensure the effect of ionic interaction on it (Fig. 6A). Even though stronger interactions were detected on the cytoplasmic side, the area of the opening was not decreased. The reason was deduced to be the simultaneously increased interaction on the periplasmic side (Asp52–Lys369, Fig. 4B). Next, we calculated MdfA's hydrophilic interaction with water in the inward-facing conformation (Fig. 6B, C and Fig. S4, ESI†). With stronger salt bridges being observed, the hydrophilic interactions of both residues 136 and 336 with water were enhanced, while those of residue 346 were not changed. Moreover, the total hydrophilic interaction of MdfA variants with water was significantly enhanced (Fig. 6C). Since the hydrophilic interaction on the cytoplasmic side was important for the entrance of some hydrophilic substrates,10,27 the elevated hydrophilic interaction was deduced to facilitate MdfA's binding to substrates.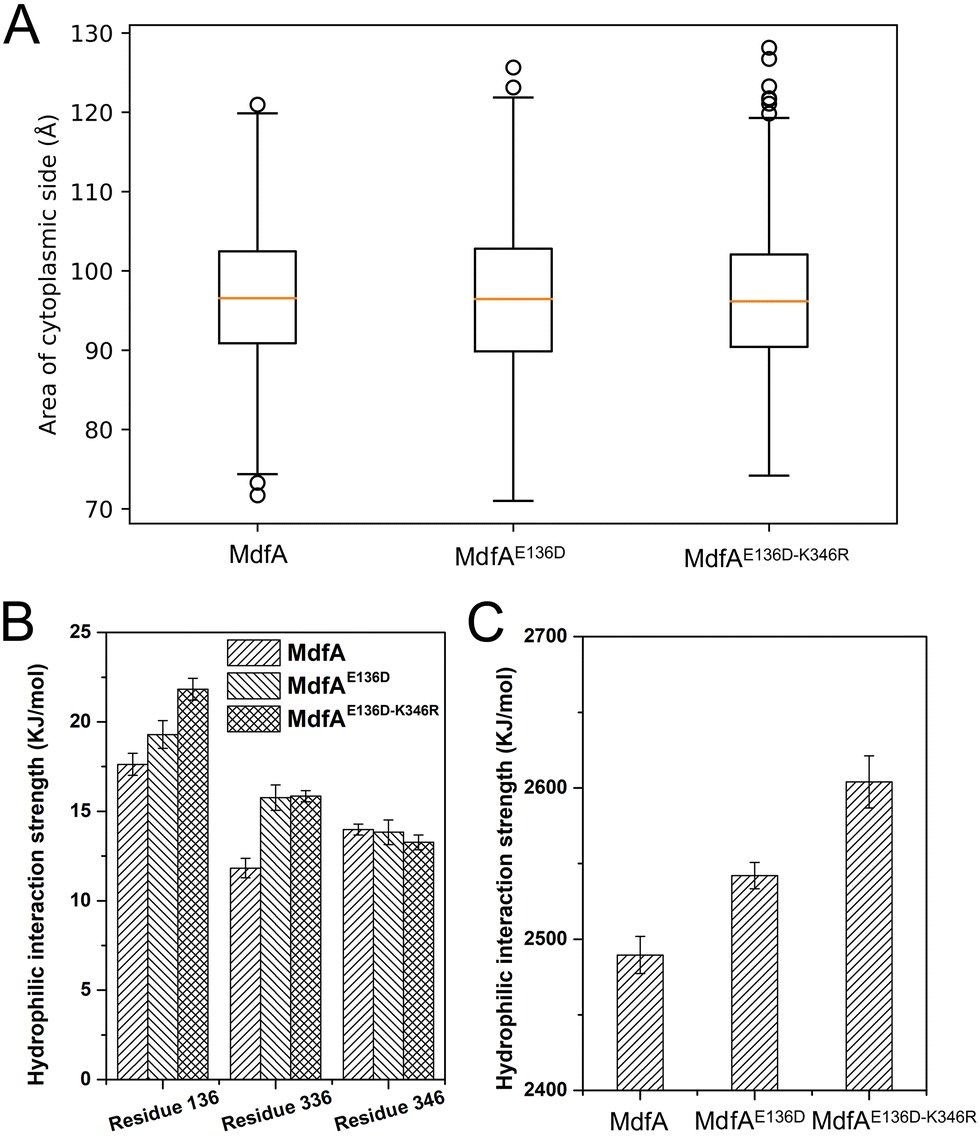 | ||
| Fig. 6 MdfA's variants had unchanged areas of the cytoplasmic opening in the inward-facing conformation (A), while the hydrophilic interactions with water were promoted (B and C). Area and hydrophilic interaction were calculated using the built-in function of the YASARA software suit. | ||
Solvent accessible surface of the outward-facing conformation was promoted
Different from the inward-facing conformation, the outward-facing conformation represents the ground state of MdfA, and the surface area of the periplasmic side is also crucial for the protonation of MdfA.28 Thus, we calculated the areas of the opening in their ground states (Fig. 7A). As we expected, enhanced ionic interactions on the cytoplasmic side (Fig. 4C and D) indeed increased the areas of the periplasmic surface. Then we calculated the solvent accessible surfaces of two titratable residues Glu26 and Asp34 (Fig. 7B), which were demonstrated to be important for the protonation of MdfA. The surface areas for Asp34 were significantly increased while only moderate increases were found for Glu26. Similar results were demonstrated for their hydrophilic interaction strengths with water (Fig. 7C). These data indicated the increased access of protons to the key residues in the outward-facing conformation, suggesting an increased possibility for MdfA's activation.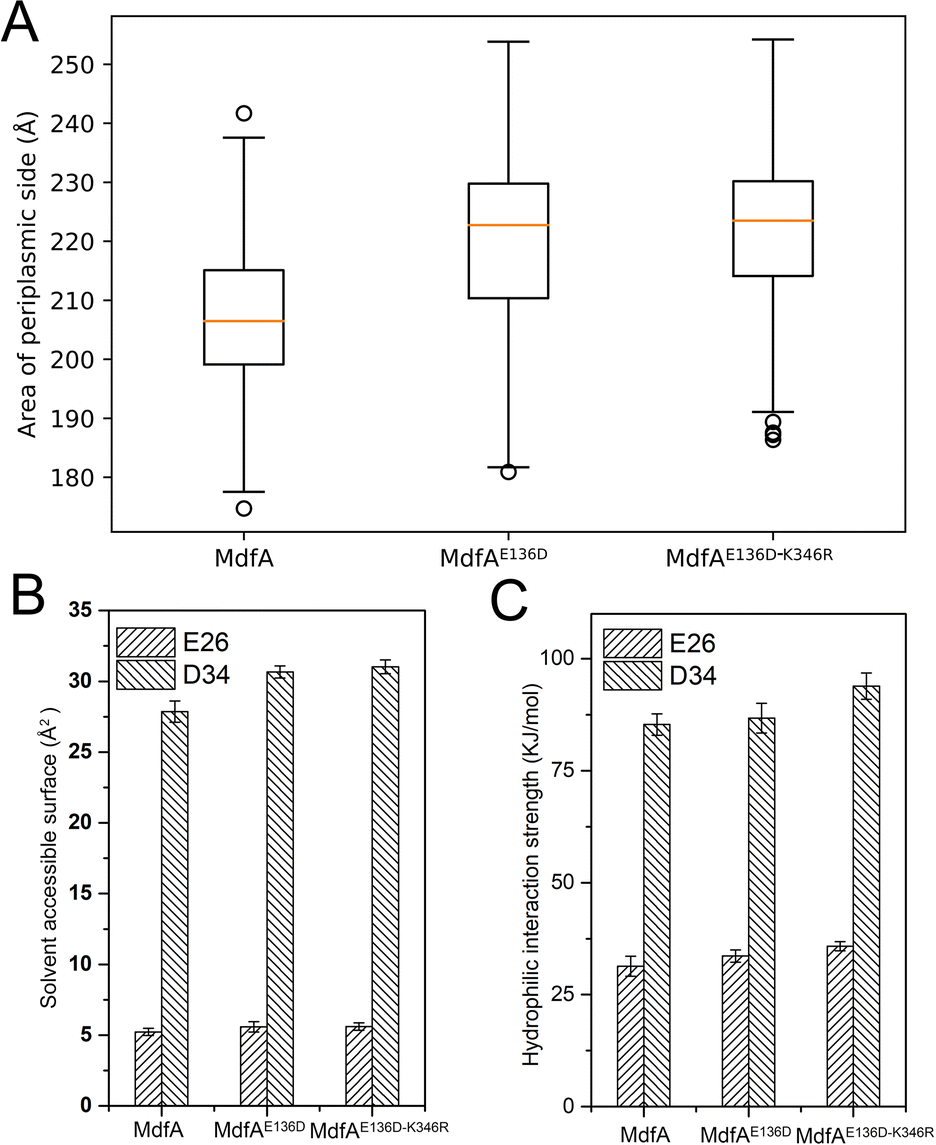 | ||
| Fig. 7 MdfA's variants indicated higher possibilities for activation in the ground state. (A) MdfA's variants had increased areas on the periplasmic side of the outward-facing conformations. The local solvent accessible surfaces and the hydrophilic interactions with water were both promoted (B and C). Areas and hydrophilic interaction were calculated with a built-in function of the YASARA software suit. | ||
Discussion
Antibiotic resistance poses serious threat to human health.29,30 With the overuse and misuse of antibiotics, efflux pumps are not only transferring through mobile genetic elements, but also evolving to have higher efficiency in transporting intracellular toxins.31,32 As a widely distributed efflux pump family in bacteria, MFS efflux pumps are demonstrated to confer antibiotic resistance against some clinically important antibiotics.33 Here our study investigated the role of internal ionic interactions in MdfA's export efficiency, providing insights into the regular of trajectory of this representative MFS efflux pump.The export mechanism of the MFS efflux pump is believed to occur through switching between its inward- and outward-facing conformations, and the rocker-switch model is the most acceptable one to describe this process. Based on this hypothesis, the MFS efflux pump is activated by protonation of Asp34 and Glu26, which are exposed to water at the ground state.22 Next, some internal electrostatic forces (e.g., hydrogen bonds and salt bridges) together with the fluidity of membranes drive the conformational transition to the excited state.6,17 During this process, some kinds of energy (e.g., negative-inside membrane potential) are supposed to be temporarily stored for the transition back to the ground state. After the opening of the cytoplasmic side, substrates are accommodated into the binding pocket and then extruded into the periplasm accompanied with conformational transition.34
In such a mechanism, salt bridges are deduced to have different roles in the two conformations. First, for the ground state of MdfA, the salt bridge Glu136–Arg336 might be important for the stability of MdfA since it is at a high level. In contrast, for the inward-facing conformation, this salt bridge is not detected within the simulation time. This result indicates that reducing the ionic interaction on the cytoplasmic side is a prerequisite for the activation of MdfA, and this process might be affected by the protonation of Asp34 and Glu26 because the distance between Asp34 and Arg336 is around 16 Å. By altering the pH of the central cavity, ionic interaction is reduced and MdfA is activated. There is also evidence that the MFS efflux pump is pH-dependent and is not active at either high or low pH level.35 On the other side, the Asp52–Lys369 on the periplasmic side of the inward-facing conformation is likely to be transient during the simulation process. Most importantly, no salt bridges are discovered on the cytoplasmic side of the inward-facing conformation, and the cytoplasmic side is wide open for substrates. Therefore, the transient Asp52–Lys369 might not affect MdfA's transition to the ground state, and this transition is believed more or less automatic.6
In our study, we revealed that MdfA's enhanced ionic interactions on the cytoplasmic side conferred increased antibiotic resistance to the recombinant E. coli. For the outward-facing conformation, the possibility of Asp136–Arg336 interaction of MdfA's variant is high, and higher energy is essential for the activation of MdfA. Surprisingly, with the enhanced interaction on the cytoplasmic side, the Asp52–Lys369 on the periplasmic side of MdfAinward also becomes stronger. More importantly, with a further increase in strength of interaction, a new salt bridge (Asp136–Lys346) on the cytoplasmic side of the inward-facing conformation is discovered, while this salt bridge is not present in the outward-facing conformation. Therefore, as a natural mutation, E136D conferred increased ionic interactions to both of MdfA's inward- and outward-facing conformations, but different positively-charged residues of the C-repeat are used for these interactions. These data imply that the strengths of salt bridges on different sides are proportional to the stability of MdfA.14,36 Meanwhile, even though the interactions between N- and C-repeats are promoted, the structural flexibilities are rarely altered, indicating that the reciprocal conformational transition of MdfA variants might not be affected (Fig. S5, ESI†).
In summary, our data offer important details on the role of salt bridges in regulating MdfA's activity. Since enhanced internal ionic interactions contribute to the elevated antibiotic resistance of E. coli, novel compounds that can eliminate these interactions are supposed to be used as novel MFS efflux pump inhibitors.
Experimental
Strains and genetic modifications
E. coli used in this work is a standard strain BL21, which is commonly used in protein expression. Ciprofloxacin (RPI) was dissolved in 0.15 M sodium hydroxide at 1 mg mL−1, and it was neutralized before addition to the medium. Chloramphenicol was formulated in ethanol at a concentration of 50 μg mL−1. LB medium (per liter: 10 g NaCl, 10 g peptone, 5 g yeast extract, and 15 g agar if necessary) was purchased from RPI (Wilmington, NC) and prepared as broth according to the instruction. All strains were grown overnight in a 37 °C shaking incubator (200 rpm) before initiating the experiment. The expression vector pET-28a was used for overexpression of mdfA. Gene deletion was conducted via an allelic exchange method with pmob-sacB plasmids.37 Briefly, 1000 bp of the upstream and downstream regions of the desired gene were amplified and then assembled with linear pmob-sacB plasmids through the Gibson assembly method. The recombinant plasmid was transformed into E. coli, and kanamycin (10 μg mL−1) was used to sustain the plasmid. After 12 h of cultivation, bacteria were spread on an LB plate, and colony PCR was performed to screen the genomic insertion of the kanamycin resistant gene. The elimination of the kanamycin resistant gene was carried out by cultivation of the recombinants in LB medium in the presence of 10% sucrose. A single colony after verification by second round of RCR was subjected to further study. Primers used in this study are listed in Table S1 (ESI†).Determination of intracellular ciprofloxacin concentration
The intracellular ciprofloxacin concentrations of the recombinant strains were determined as follows. Briefly, recombinant strains were cultured in LB broth overnight with shaking. Cultures were diluted 100-fold in fresh LB and then cultivated to the logarithmic phase. The cells were harvested by centrifugation and washed with PBS buffer (50 mM, pH 7.0), and resuspended in prewarmed LB medium at 37 °C with an OD600 of 1.0. Then 1 mL samples were used for intracellular ciprofloxacin assay with 5 μg mL−1 ciprofloxacin being provided. Samples were taken after cultivation for 30 min, and the cell dry weight was measured at the end of the experiment. Samples were taken and immediately diluted with 500 μL of cold PBS buffer. After centrifugation, cells were washed twice with cold PBS buffer and then resuspended in 0.1 M glycerin hydrochloride (pH 3.0) and shaken at 37 °C for 5 h. After centrifugation, the supernatants were used for HLPC analysis after filtration through a 0.22 μm filter. In the control groups, the MFS-specific inhibitor verapamil (50 μg mL−1) was added to reverse the transport of MdfA. HPLC analysis was conducted on a SHIMADZU LC-16 system equipped with a C18 column and a UV detector. The liquid phase was water/methanol/phosphate acid with a ratio of 70/30/0.01. The flow rate was set at 1.0 mL min−1 and the detective wavelength was set at 280 nm.Chloramphenicol resistance assay
Chloramphenicol resistance of the recombinant strain was evaluated with a viable cell that could survive in the LB plate containing 2.5 μg mL−1 chloramphenicol.22 Briefly, cells were cultivated overnight in the presence of 20 μg mL−1 kanamycin in a LB tube to sustain the plasmid. After measuring the number of viable cells and adjusting those of the different groups to the same level, cells were 10 times diluted in a 96-well plate with fresh LB medium. Next, 10 μL of each medium was dropped into a LB solid medium containing 2.5 μg mL−1 chloramphenicol. After cultivation at 37 °C for 24 h, the number of viable cells was recorded to assess the antibiotic resistance level.Structures and software
The inward (4zp0) and outward (6gv1) structures of MdfA were downloaded from the RCS PDB bank followed by deletion of ligands. After alignment of their amino acid sequences, we found that there existed one residue substitution (131Q for outward and 131R for inward). To make the simulations comparable, we made the swap R131Q of MdfAinward structure followed by energy minimization. Then the two structures were applied as the initial states of the two simulations. For the variation of MdfA, the relative residues were also swapped followed by energy minimization before molecular dynamics (MD) simulations. Structural modification and MD simulations were conducted in the commercial YASARA structure software suit (Version 21.6.17) since we have the license.MD simulation
All the MD simulations were carried out in the YASARA structure software unit using Amber 14 force field under periodic boundary conditions. The standard marco ‘md_run_membrane’ was applied for all the simulations. First, the PDB file of MdfA was cleaned and the ligand was removed. The simulation was set up automatically by first scanning the protein for exposed transmembrane helices. The major axis vectors of these helices were summed up to obtain the major axis of the protein, which was then oriented along the Y-axis, normally with respect to the plane of the membrane and the XZ-plane. The best shift of the membrane along this major axis was obtained by scanning the protein for the region with the largest number of exposed hydrophobic residues. Having placed an equilibrated membrane structure, the system was enclosed in a simulation cell of size [90 × 90 × 90] Å. The protein was temporarily scaled by 0.9 along the XZ-axes, and strongly clashing membrane lipids were deleted. The temporary protein scaling, which was needed to avoid the deletion of too many lipids around the protein, was then slowly removed during a short simulation at 298 K in vacuo. The force field was AMBER14 with Lipid17 parameters for non-standard residues. As soon as the protein had reached its original size again, the pKas of protein side-chains were predicted. The protonation state was assigned according to pH 7.4, and the simulation cell was filled with 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 140 water molecules, 0.9% NaCl and counterions. The main simulation was then run with PME and 8.0 Å cutoff for non-bonded real space forces, a 4 fs time-step, constrained hydrogen atoms, and at constant pressure and temperature (NPT ensemble). During the initial 250 picoseconds, the membrane was restrained to avoid distortions while the simulation cell adapted to the pressure exerted by the membrane. Each simulation was performed for 1000 ns at 310 K and 1 bar with two replicates. The snapshot was recorded every 0.1 ns. The marco used in MD simulation is available at https://www.yasara.org/md_runmembrane.mcr.
140 water molecules, 0.9% NaCl and counterions. The main simulation was then run with PME and 8.0 Å cutoff for non-bonded real space forces, a 4 fs time-step, constrained hydrogen atoms, and at constant pressure and temperature (NPT ensemble). During the initial 250 picoseconds, the membrane was restrained to avoid distortions while the simulation cell adapted to the pressure exerted by the membrane. Each simulation was performed for 1000 ns at 310 K and 1 bar with two replicates. The snapshot was recorded every 0.1 ns. The marco used in MD simulation is available at https://www.yasara.org/md_runmembrane.mcr.
Statistical analysis
Analysis of the trajectory of simulation was also conducted in YASARA with its built-in functions. Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were used to evaluate the total and local flexibilities. Dynamic cross-correlation matrix (DCCM) was used to evaluate the relative movement of each residue. Ionic interaction (salt bridge) was identified if the distance between positively and negatively charged residues was lower than 5 Å. Hydrophilic interaction strength and solvent accessible surfaces were calculated with the built-in function in the YASARA structure unit. All the plots were made in the Python ‘Matplotlib’ module and the structures were visualized in Pymol. Data of the growth curve were presented as means values ± standard errors of mean (SD). Statistical significances were indicated using P values analyzed by one-way ANOVA analysis.Conclusions
In this work we investigated the role of internal salt bridges in the activity of MdfA. Our data indicated that enhanced ionic interactions contributed to the elevated antibiotic resistance levels of E. coli. By using MD simulation approaches, we demonstrated that enhanced ionic interactions resulted in increased possibility for MdfA's activation and conformational transition. Our work offered insights for the development of novel MFS efflux pump inhibitors.Author contributions
YL and XG designed the research. YL conducted microbiological experiments and MD simulations. YL wrote the manuscript and XG submitted it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Project of Beijing Municipal Commission of Education (KZ202011417006).References
- G. Subramaniam and M. Girish, Antibiotic Resistance-A Cause for Reemergence of Infections, Indian J. Pediatr., 2020, 87, 937–944 CrossRef PubMed.
- G. Mancuso, A. Midiri, E. Gerace and C. Biondo, Bacterial Antibiotic Resistance: The Most Critical Pathogens, Pathogens, 2021, 10, 1310 CrossRef CAS PubMed.
- S. C. Forster, J. Liu, N. Kumar, E. L. Gulliver, J. A. Gould, A. Escobar-Zepeda, T. Mkandawire, L. J. Pike, Y. Shao, M. D. Stares, H. P. Browne, B. A. Neville and T. D. Lawley, Strain-level characterization of broad host range mobile genetic elements transferring antibiotic resistance from the human microbiome, Nat. Commun., 2022, 13, 1445 CrossRef CAS PubMed.
- H. Nikaido, Multiple antibiotic resistance and efflux, Curr. Opin. Microbiol., 1998, 1, 516–523 CrossRef CAS PubMed.
- M. A. Webber, The importance of efflux pumps in bacterial antibiotic resistance, J. Antimicrob. Chemother., 2003, 51, 9–11 CrossRef CAS PubMed.
- D. Jiang, Y. Zhao, X. Wang, J. Fan, J. Heng, X. Liu, W. Feng, X. Kang, B. Huang, J. Liu and X. C. Zhang, Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14664–14669 CrossRef CAS PubMed.
- S. Yang, Relative contributions of the AcrAB, MdfA and NorE efflux pumps to quinolone resistance in Escherichia coli, J. Antimicrob. Chemother., 2003, 51, 545–556 CrossRef CAS PubMed.
- Y. Li, H. Wen and X. Ge, Hormesis Effect of Berberine against Klebsiella pneumoniae is Mediated by Up-Regulation of the Efflux Pump KmrA, J. Nat. Prod., 2021, 84, 2885–2892 CrossRef CAS PubMed.
- M. E. Wand, S. Jamshidi, L. J. Bock, K. M. Rahman and J. M. Sutton, SmvA is an important efflux pump for cationic biocides in Klebsiella pneumoniae and other Enterobacteriaceae, Sci. Rep., 2019, 9, 1344 CrossRef PubMed.
- P. Hinchliffe, N. P. Greene, N. G. Paterson, A. Crow, C. Hughes and V. Koronakis, Structure of the periplasmic adaptor protein from a major facilitator superfamily (MFS) multidrug efflux pump, FEBS Lett., 2014, 588, 3147–3153 CrossRef CAS PubMed.
- I. Roca, S. Marti, P. Espinal, P. Martínez, I. Gibert and J. Vila, CraA, a Major Facilitator Superfamily Efflux Pump Associated with Chloramphenicol Resistance in Acinetobacter baumannii, Antimicrob. Agents Chemother., 2009, 53, 4013–4014 CrossRef CAS PubMed.
- K. Yamane, J. Wachino, S. Suzuki, K. Kimura, N. Shibata, H. Kato, K. Shibayama, T. Konda and Y. Arakawa, New Plasmid-Mediated Fluoroquinolone Efflux Pump, QepA, Found in an Escherichia coli Clinical Isolate, Antimicrob. Agents Chemother., 2007, 51, 3354–3360 CrossRef CAS PubMed.
- A. Nag and S. Mehra, A Major Facilitator Superfamily (MFS) Efflux Pump, SCO4121, from Streptomyces coelicolor with Roles in Multidrug Resistance and Oxidative Stress Tolerance and Its Regulation by a MarR Regulator, Appl. Environ. Microbiol., 2021, 87, e02238–20 CrossRef CAS PubMed.
- I. Ranaweera, U. Shrestha, K. C. Ranjana, P. Kakarla, T. M. Willmon, A. J. Hernandez, M. M. Mukherjee, S. R. Barr and M. F. Varela, Structural comparison of bacterial multidrug efflux pumps of the major facilitator superfamily, Trends Cell Mol. Biol., 2015, 10, 131–140 Search PubMed.
- D. Du, X. Wang-Kan, A. Neuberger, H. W. van Veen, K. M. Pos, L. J. V. Piddock and B. F. Luisi, Multidrug efflux pumps: structure, function and regulation, Nat. Rev. Microbiol., 2018, 16, 523–539 CrossRef CAS PubMed.
- M. J. Lemieux, Y. Huang and D.-N. Wang, The structural basis of substrate translocation by the Escherichia coli glycerol-3-phosphate transporter: a member of the major facilitator superfamily, Curr. Opin. Struct. Biol., 2004, 14, 405–412 CrossRef CAS PubMed.
- Y. Ural-Blimke, A. Flayhan, J. Strauss, V. Rantos, K. Bartels, R. Nielsen, E. Pardon, J. Steyaert, J. Kosinski, E. M. Quistgaard and C. Löw, Structure of Prototypic Peptide Transporter DtpA from E. coli in Complex with Valganciclovir Provides Insights into Drug Binding of Human PepT1, J. Am. Chem. Soc., 2019, 141, 2404–2412 CrossRef CAS PubMed.
- F. Zhou, D. Yao, B. Rao, L. Zhang, W. Nie, Y. Zou, J. Zhao and Y. Cao, Crystal structure of a bacterial homolog to human lysosomal transporter, spinster, Sci. Bull., 2019, 64, 1310–1317 CrossRef CAS.
- S. Dang, L. Sun, Y. Huang, F. Lu, Y. Liu, H. Gong, J. Wang and N. Yan, Structure of a fucose transporter in an outward-open conformation, Nature, 2010, 467, 734–738 CrossRef CAS PubMed.
- K. Nagarathinam, Y. Nakada-Nakura, C. Parthier, T. Terada, N. Juge, F. Jaenecke, K. Liu, Y. Hotta, T. Miyaji, H. Omote, S. Iwata, N. Nomura, M. T. Stubbs and M. Tanabe, Outward open conformation of a Major Facilitator Superfamily multidrug/H+ antiporter provides insights into switching mechanism, Nat. Commun., 2018, 9, 4005 CrossRef PubMed.
- Y. Li, H. Wen and X. Ge, Opposite motion of the Central Helices of efflux pump KmrA is important for its export efficiency, Microb. Pathog., 2022, 167, 105570 CrossRef CAS PubMed.
- J. Heng, Y. Zhao, M. Liu, Y. Liu, J. Fan, X. Wang, Y. Zhao and X. C. Zhang, Substrate-bound structure of the E. coli multidrug resistance transporter MdfA, Cell Res., 2015, 25, 1060–1073 CrossRef CAS PubMed.
- Y. Li and X. Ge, Discovering interrelated natural mutations of efflux pump KmrA from Klebsiella pneumoniae that confer increased multidrug resistance, Prot. Sci., 2022, 31, e4323 CAS.
- A. Schäfer, A. Tauch, W. Jäger, J. Kalinowski, G. Thierbach and A. Pühler, Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum, Gene, 1994, 145, 69–73 CrossRef PubMed.
- L. Lv, M. Wan, C. Wang, X. Gao, Q. Yang, S. R. Partridge, Y. Wang, Z. Zong, Y. Doi, J. Shen, P. Jia, Q. Song, Q. Zhang, J. Yang, X. Huang, M. Wang and J.-H. Liu, Emergence of a Plasmid-Encoded Resistance-Nodulation-Division Efflux Pump Conferring Resistance to Multiple Drugs, Including Tigecycline, in Klebsiella pneumoniae, mBio, 2020, 11, e02930–19 CrossRef CAS PubMed.
- T.-S. Huang, C. M. Kunin, H.-M. Wang, B.-S. Yan, S.-P. Huang, Y.-S. Chen, S. Shin-Jung Lee and W.-J. Syu, Inhibition of the Mycobacterium tuberculosis reserpine-sensitive efflux pump augments intracellular concentrations of ciprofloxacin and enhances susceptibility of some clinical isolates, J. Formosan Med. Assoc., 2013, 112, 789–794 CrossRef CAS PubMed.
- K. Wong, J. Ma, A. Rothnie, P. C. Biggin and I. D. Kerr, Towards understanding promiscuity in multidrug efflux pumps, Trends Biochem. Sci., 2014, 39, 8–16 CrossRef CAS PubMed.
- H. I. Zgurskaya and H. Nikaido, Multidrug resistance mechanisms: drug efflux across two membranes, Mol. Microbiol., 2000, 37, 219–225 CrossRef CAS PubMed.
- D. Du, H. W. van Veen and B. F. Luisi, Assembly and operation of bacterial tripartite multidrug efflux pumps, Trends Microbiol., 2015, 23, 311–319 CrossRef CAS PubMed.
- I. Roca, M. Akova, F. Baquero, J. Carlet, M. Cavaleri, S. Coenen, J. Cohen, D. Findlay, I. Gyssens, O. E. Heure, G. Kahlmeter, H. Kruse, R. Laxminarayan, E. Liébana, L. López-Cerero, A. MacGowan, M. Martins, J. Rodríguez-Baño, J.-M. Rolain, C. Segovia, B. Sigauque, E. Tacconelli, E. Wellington and J. Vila, The global threat of antimicrobial resistance: science for intervention, New Microbes New Infect., 2015, 6, 22–29 CrossRef CAS PubMed.
- M. Ferri, E. Ranucci, P. Romagnoli and V. Giaccone, Antimicrobial resistance: A global emerging threat to public health systems, Crit. Rev. Food Sci. Nutrit., 2017, 57, 2857–2876 CrossRef CAS PubMed.
- S. Nolivos, J. Cayron, A. Dedieu, A. Page, F. Delolme and C. Lesterlin, Role of AcrAB-TolC multidrug efflux pump in drug-resistance acquisition by plasmid transfer, Science, 2019, 364, 778–782 CrossRef CAS PubMed.
- L. H. Hansen, S. J. Sørensen, H. S. Jørgensen and L. B. Jensen, The Prevalence of the OqxAB Multidrug Efflux Pump amongst Olaquindox-Resistant Escherichia coli in Pigs, Microb. Drug Resist., 2005, 11, 378–382 CrossRef CAS PubMed.
- X. Jiang, T. Yu, P. Xu, X. Xu, S. Ji, W. Gao and L. Shi, Role of Efflux Pumps in the in vitro Development of Ciprofloxacin Resistance in Listeria monocytogenes, Front. Microbiol., 2018, 9, 2350 CrossRef PubMed.
- B. V. Bhaskar, T. M. C. Babu, N. V. Reddy and W. Rajendra, Homology modeling, molecular dynamics, and virtual screening of NorA efflux pump inhibitors of Staphylococcus aureus, Drug Des., Dev. Ther., 2016, 10, 3237–3252 CrossRef CAS PubMed.
- P. R. Steed, P. Zou, K. E. Trone and H. S. Mchaourab, Structure and pH-induced Structural Rearrangements of the Putative Multidrug Efflux Pump EmrD in Liposomes Probed by Site-Directed Spin Labeling, Biochemistry, 2013, 52, 7964–7974 CrossRef CAS PubMed.
- S. Jamshidi, J. M. Sutton and K. M. Rahman, An overview of bacterial efflux pumps and computational approaches to study efflux pump inhibitors, Future Med. Chem., 2016, 8, 195–210 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05059e |
| This journal is © the Owner Societies 2023 |