
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrafast 2D-IR spectroscopy of [NiFe] hydrogenase from E. coli reveals the role of the protein scaffold in controlling the active site environment†
Solomon L. D.
Wrathall‡
a,
Barbara
Procacci‡
 a,
Marius
Horch
b,
Emily
Saxton
a,
Chris
Furlan
a,
Julia
Walton
a,
Yvonne
Rippers
b,
James N.
Blaza
a,
Gregory M.
Greetham
c,
Michael
Towrie
c,
Anthony W.
Parker
c,
Jason
Lynam
a,
Alison
Parkin
*a and
Neil T.
Hunt
*a
a,
Marius
Horch
b,
Emily
Saxton
a,
Chris
Furlan
a,
Julia
Walton
a,
Yvonne
Rippers
b,
James N.
Blaza
a,
Gregory M.
Greetham
c,
Michael
Towrie
c,
Anthony W.
Parker
c,
Jason
Lynam
a,
Alison
Parkin
*a and
Neil T.
Hunt
*a
aDepartment of Chemistry and York Biomedical Research Institute, University of York, York, YO10 5DD, UK. E-mail: neil.hunt@york.ac.uk; alison.parkin@york.ac.uk
bFreie Universität Berlin, Department of Physics, Ultrafast Dynamics in Catalysis, Arnimallee 14, 14195 Berlin, Germany
cSTFC Central Laser Facility, Research Complex at Harwell, Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
First published on 30th September 2022
Abstract
Ultrafast two-dimensional infrared (2D-IR) spectroscopy of Escherichia coli Hyd-1 (EcHyd-1) reveals the structural and dynamic influence of the protein scaffold on the Fe(CO)(CN)2 unit of the active site. Measurements on as-isolated EcHyd-1 probed a mixture of active site states including two, which we assign to Nir-SI/II, that have not been previously observed in the E. coli enzyme. Explicit assignment of carbonyl (CO) and cyanide (CN) stretching bands to each state is enabled by 2D-IR. Energies of vibrational levels up to and including two-quantum vibrationally excited states of the CO and CN modes have been determined along with the associated vibrational relaxation dynamics. The carbonyl stretching mode potential is well described by a Morse function and couples weakly to the cyanide stretching vibrations. In contrast, the two CN stretching modes exhibit extremely strong coupling, leading to the observation of formally forbidden vibrational transitions in the 2D-IR spectra. We show that the vibrational relaxation times and structural dynamics of the CO and CN ligand stretching modes of the enzyme active site differ markedly from those of a model compound K[CpFe(CO)(CN)2] in aqueous solution and conclude that the protein scaffold creates a unique biomolecular environment for the NiFe site that cannot be represented by analogy to simple models of solvation.
Introduction
The [NiFe] hydrogenases, which catalyze the interconversion of molecular hydrogen with protons and electrons (H2 ⇌ 2H+ + 2e−) are of considerable interest in the pursuit of sustainable H2 production technologies, because high turnover rates are achieved by an active site containing earth-abundant base metal atoms (Fig. 1).1 Furthermore, some of the Group-1 subclass of periplasmic membrane-bound [NiFe] hydrogenases, such as Hyd-1 from E. coli (EcHyd-1), can sustain H2-catalysis in the presence of O2, leading to an ‘O2-tolerant’ designation.2 The use of hydrogenase-inspired catalysts in sustainable energy supply, and the design of synthetic systems for photo-bio H2 production is however impeded by the lack of a complete understanding of the mechanisms of H2 activation and evolution.3–5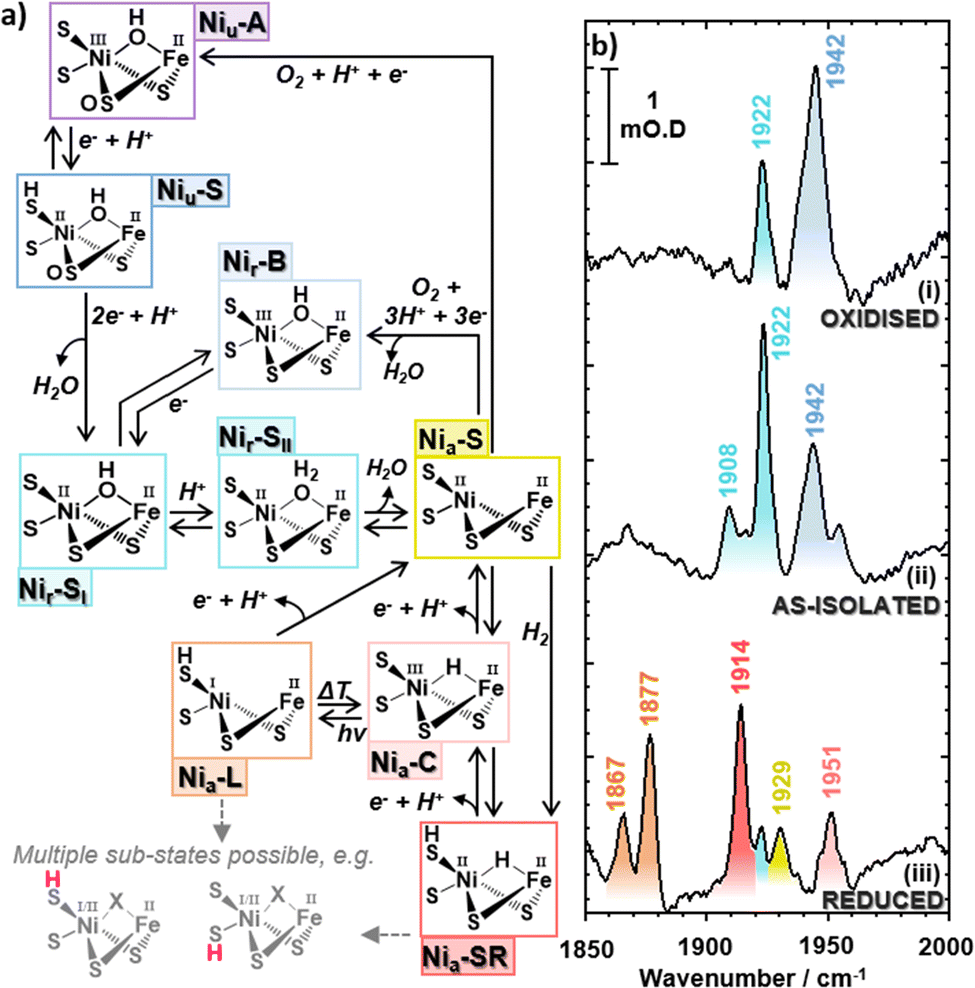 | ||
| Fig. 1 (a) Catalytic cycle for EcHyd-1 with truncated structures of the bimetallic active site. (b) IR absorption (FT-IR) spectra of EcHyd-1 in the νCO region of the spectrum showing the spectrum obtained under (i) H2-reduced and subsequently O2 oxidized, (ii) as-isolated (aerobically purified at pH 7.6), and (iii) H2 reduced conditions. Colored labels indicate the wavenumbers of bands due to carbonyl stretching modes and the color scheme is consistent with that in (a). The ‘a’ and ‘r’ subscripts used in state designations indicate ‘active’ and ‘ready’ active site states. | ||
The importance of achieving a deeper understanding of the hydrogenase active site is illustrated by the fact that, small molecules that mimic the biological reaction center have tended to be inferior H2 evolution catalysts5,6 indicating that the protein matrix plays an important role in controlling or defining the chemistry of the active site.7–10 Indeed, the synthetic systems that have approached, or exceeded, enzymatic levels of performance have all featured either a significant second coordination sphere surrounding the metal centers or synthetic subsites placed within a protein matrix.11–26 It follows that experimental techniques able to reveal the intermolecular interactions, structure and bonding of the hydrogenase active site, and quantify the modulation induced by the protein scaffold, will be valuable in determining the contributions of the second coordination sphere to H2 generation.
The hydrogenase-catalyzed H2 activation pathway involves a number of active site states,1 each of which features an FeII(CO)(CN)2 moiety (Fig. 1(a)). The frequencies of the stretching vibrational modes of these organometallic ligands (νCO, νCN) are sensitive to structural and redox changes of the active site. As such, IR spectroscopy has been central to the development of our current picture of hydrogenase chemistry, but a dynamic picture of the active site environment is still to fully emerge.27–33 As hydrogen bonds to the cyanide ligands are direct of points contact between the active site and the protein scaffold, spectroscopic methods that interrogate cyanide ligand structure and dynamics will provide particularly important source of information for validating quantum chemical simulations. In turn, the conclusions obtained will enable accurate predictions with which to guide our exploitation of the hydrogenase catalytic center, ideally leading to optimized synthetic systems.
To date numerous spectroscopy tools have provided valuable data on the hydrogenases. Recently, resonance Raman spectroscopy has been used to probe metal-ligand bonding,34–36 while nuclear resonance vibrational spectroscopy has allowed detection of bridging hydrides.37 Advanced time resolved photogating and potential jump spectroscopies have enabled investigation of proton-coupled electron transfer processes and short-lived intermediates along the catalytic pathway.38–45 Recently, ultrafast 2D-IR spectroscopy was used to investigate the active site of the regulatory hydrogenase from R. eutropha (ReRH). 2D-IR is an non-linear laser spectroscopy method that uses a sequence of ultrashort IR pulses to produce a two-dimensional map correlating excitation (pump) and detection (probe) frequencies.46–51 This 2D representation of the vibrational modes of a molecule effectively places the linear IR absorption spectrum along the diagonal of the 2D-plot while off-diagonal peaks identify coupled vibrational modes.47 The pump–probe nature of the 2D-IR experiment means that vibrational levels higher than v = 1 can be reached, giving information on the shape of vibrational potential energy surfaces, while pulse polarizations can be tailored to extract molecular structural information associated with mode specific orientation changes of excited modes relative to the general molecular frame. The sub-picosecond time resolution of 2D-IR means that structural and vibrational dynamics can be obtained revealing bond-specific insights into the rapid changes and interactions that occur during chemical processes.
When applied to ReRH, 2D-IR provided a detailed description of the νCO potential surface by accessing levels up to v = 4 and the associated vibrational energy relaxation mechanisms. A quantum beat phenomenon was used to identify the frequency difference between coupled vibrational modes of the two CN ligands.52ReRH has evolved to regulate the cellular production of other hydrogenases. It is purified in a single Ni(II)–Fe(II) active site state and is a relatively poor H2-catalyst. Here, we extend our approach to study a mixture of active site states using the group 1 [NiFe] hydrogenase EcHyd-1. This enzyme enables E. coli to use H2 as a fuel, with very high rates of catalytic turnover being reported (H2-oxidation coupled to methylene blue reduction rates of approximately 65 s−1 at pH 6.0, and 21 ± 4 s−1 at pH 4.5).53,54 We exploit the sensitivity and peak resolution of 2D-IR to probe both the νCO and νCN regions of the spectrum in detail while off-diagonal peaks enable pairs of νCN modes to be linked definitively with their νCO counterparts.27,55 We identify the vibrational signatures of a number of states, including the Nir-SI/II active site states previously un-reported in EcHyd-1. By determining vibrational energy levels up to and including two-quantum excited states for the νCO and νCN modes as well as their vibrational and structural dynamics, we have revealed significant deviations from measurements made on the organometallic model compound K[CpFe(CO)(CN)2] in solution. The results are discussed in terms of the role played by the protein scaffold in modulating the molecular environment of the active site and how 2D-IR can be employed to interrogate differences in active site states of hydrogenase enzymes to stimulate development of bio-H2 production methods.
Experimental
Enzyme preparation and characterization
The protocol for preparation of EcHyd-1, developed from a published protocol54 is described in the ESI.† The as-isolated enzyme was characterized using a methylene-blue H2-oxidation assay, protein gels, protein film electrochemistry and electron cryo-microscopy (cryo-EM). Full details of each method, including characterization data are given in the ESI† (Fig. S1–S6).Infrared spectroscopy
For all EcHyd-1 2D-IR spectroscopy experiments, the sample (270 μM, pH 7.6) was held in a gas-tight small volume (15 μL) sandwich cell featuring CaF2 windows and a PTFE spacer, giving an optical path length of 50 μm. For IRpump–IRprobe and 2D-IR spectra, the samples were held at a temperature of 10 °C. For gas-cycling IR experiments, the absorption spectra were recorded at room temperature.IR absorption spectra were recorded using a Bruker Vertex 70 spectrometer with a frequency resolution of 2 cm−1. Ultrafast spectroscopy experiments were performed using the ULTRA laser system based at the UK's STFC Central Laser Facility.56 Mid-IR pulses with a central frequency of 2000 cm−1, bandwidth >300 cm−1, 50 fs pulse duration and 10 kHz repetition rate were used in all cases. IRpump–IRprobe spectra were recorded with parallel and perpendicular pump–probe polarization geometries using methods reported previously.52 The pump–probe delay time (Tw) was scanned from −20 to 54 ps in increments of 250 fs. The spectra were acquired by frequency-dispersing the signal with a spectrograph followed by detection using 128-element Mercury–Cadmium–Telluride (MCT) array detectors, giving a frequency resolution of ∼2 cm−1.
2D-IR spectra were acquired using the pump–probe geometry.52 The pump pulse pair were created and the time delay between them scanned using a mid-IR pulse shaper applying a four-frame phase cycling method. 2D-IR spectra were recorded at values of Tw from 125 fs to 45 ps with parallel and perpendicular pump–probe polarization geometries. Fourier transformation of the data with respect to the time delay between the pump pulse pair was used to obtain the pump frequency axis of the 2D-IR spectrum. The probe frequency axis was generated by frequency-dispersal and detection of the signal as described for pump–probe measurements. Further details are given in the ESI.†
Results
As isolated EcHyd-1
Following previous IR absorption studies of EcHyd-1, the 1942 cm−1 band can be assigned to the νCO mode of the Nir-B state (Fig. 1(a and b), blue).33 The band at 1922 cm−1 (Fig. 1(b), turquoise) appears to coincide with previous observations of the Nia-SR state, though the presence of such a reduced state is unexpected in an as-isolated sample.2,57,59,60 The 1908 cm−1 band (Fig. 1(b), light turquoise) does not correspond to a previously reported state of EcHyd-1.
A series of additional experiments were performed to characterise the as-isolated EcHyd-1 sample in more detail and so guide the assignment of the observed νCO bands to individual active site states. Cryo-EM measurements (Fig. 2(a) and Fig. S3, ESI†) showed that the majority of enzyme molecules were present as dimers of heterodimers (Hya(AB)2). This correlated with native-PAGE H2-oxidation activity staining experiments which similarly showed that the EcHyd-1 sample had a molecular mass consistent with a dimer-of-dimers (Fig. S4, ESI†).
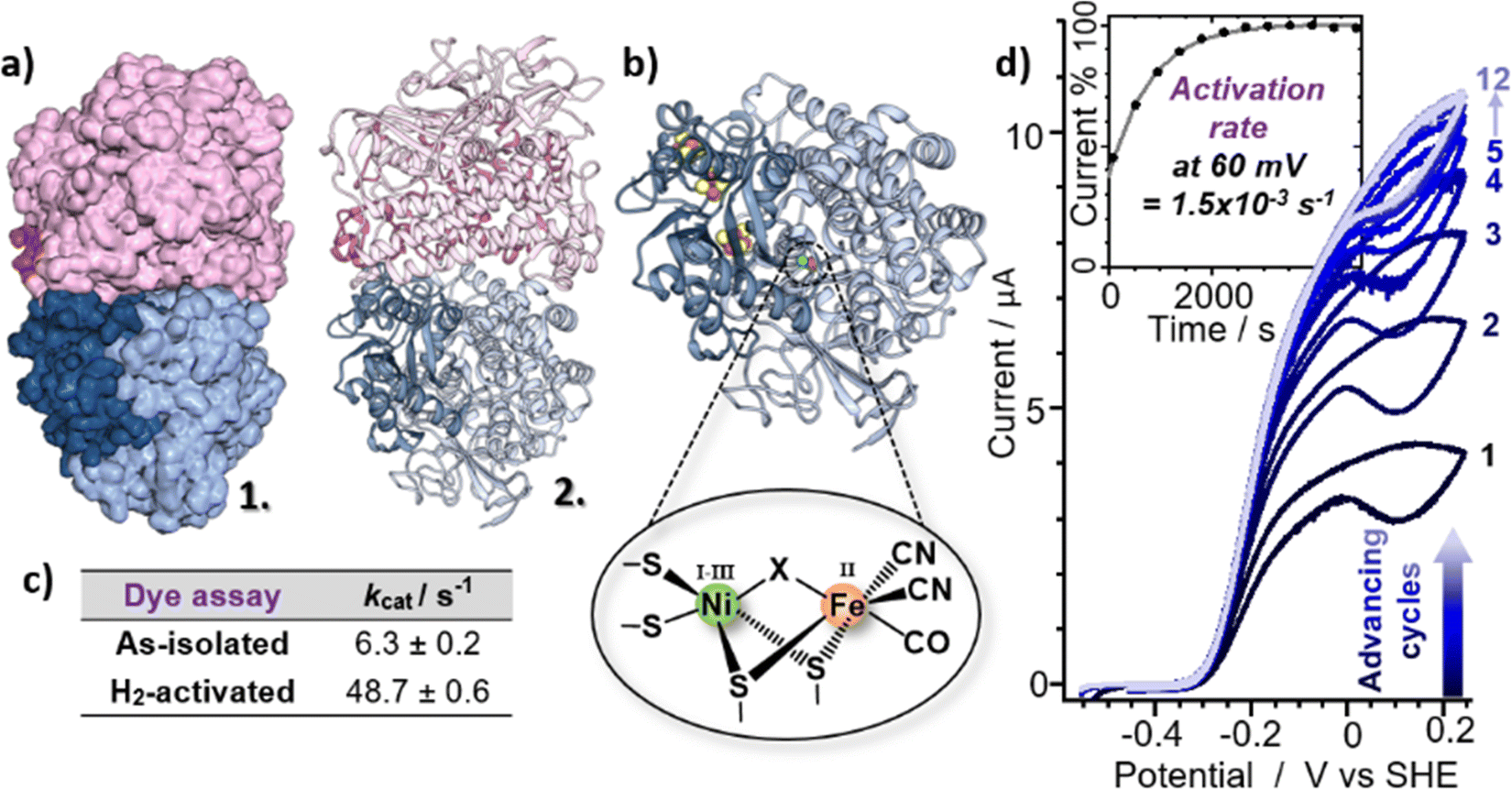 | ||
| Fig. 2 (a) Model for the EcHyd-1 preparation used here based on cryo-EM 2D class images and 6FPW,57 shown in surface mode (1) and ribbon format (2). (b) X-ray crystal structure of the monomer (PDB: 6FPW57) with pop-out schematic of the active site. (c) Table showing the H2-oxidising ability of EcHyd-1 determined via a methylene blue dye reduction assay (three replicates). (d) Cyclic voltammograms showing the activation of EcHyd-1 with inset current/time plot showing the activation rate at 60 mV. | ||
Further IR absorption spectroscopy experiments were performed following incubation of the as-isolated EcHyd-1 sample with 100% O2 (Fig. 1(b), panel i). O2 exposure resulted in an increase in intensity of the 1942 cm−1 band (blue) and a decrease of the 1908 and 1922 cm−1 bands (light turquoise). This is consistent with the assignment of the 1942 cm−1 band to the Nir-B state while the continued presence of the 1908 and 1922 cm−1 bands suggests that the associated states lie towards the oxidised end of the range of active site states (Fig. 1(a), blue to turquoise).33 In contrast, reduction of the as-isolated sample by incubation with 100% H2 (Fig. 1(b), panel iii) resulted in the loss of the 1908, 1922 and 1942 cm−1 bands, which were replaced by new bands at 1867 cm−1, 1877 cm−1, 1914 cm−1, 1929 cm−1 and 1951 cm−1. These new bands can be assigned by reference to previous work to the more reduced states of EcHyd-1: Nia-LIII, Nia-LII, Nia-SRIII, Nia-S and Nia-C states respectively (Fig. 1(a), orange, red, yellow, pink).33
Given that the behavior of the 1908 and 1922 cm−1 bands is consistent with assignment to more oxidised active site states, we hypothesise that they can be assigned to the un-reported Nir-SI/II states. The only other candidate, the Nia-S state has been reported to exhibit a νCO frequency of 1929 cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 while the corresponding νCN frequencies for the Nia-S state were in excess of 2077 cm−1, which does not match our observations (vide infra).33 Our assignment therefore highlights a possible coincidence of the νCO frequencies of the higher frequency Nir-SI/II state (1922 cm−1) and the Nia-SR state,33 though it is noted that both feature a NiII centre, which would suggest similar νCO frequencies.
33 while the corresponding νCN frequencies for the Nia-S state were in excess of 2077 cm−1, which does not match our observations (vide infra).33 Our assignment therefore highlights a possible coincidence of the νCO frequencies of the higher frequency Nir-SI/II state (1922 cm−1) and the Nia-SR state,33 though it is noted that both feature a NiII centre, which would suggest similar νCO frequencies.
To test the hypothesis that our as-isolated sample contained Nir-SI/II states rather than the more reduced Nia-SR state, the H2-oxidising activities of as-isolated and H2-activated EcHyd-1 were compared via methylene blue dye reduction assays (Fig. 2(c) and Fig. S5, ESI†). The activity (kcat) of as-isolated EcHyd-1 samples was found to increase by almost a factor of eight (from 6.3 ± 0.2 s−1 to 48.7 ± 0.6 s−1) following incubation with 100% H2 (Fig. 2). This observation is consistent with the as-isolated sample shifting from oxidised to reduced active states, as expected if Nir-SI/II, rather than Nia-SR, is present alongside Nir-B in the as-isolated mixture.
Protein film electrochemistry was used to monitor the activation kinetics of as-isolated EcHyd-1. Cyclic voltammetry (CV) experiments were performed in which as-isolated (i.e., air-purified) EcHyd-1 was applied to the working electrode under atmospheric conditions The working electrode was then fixed into an electrochemical cell which had been pre-equilibrated with an atmosphere of 100% H2. CV cycles were run until successive cycles resulted in no increase in positive current, indicating that all adsorbed enzyme molecules had been fully activated (Fig. 2(d) and Fig. S6, ESI†). Based on fitting the time-dependence of the change in current at 60 mV (Fig. 2(d) inset) to a monoexponential function values for kactivation of 1.5 × 10−3 s−1 were obtained. As the obtained kactivation value is an order of magnitude faster than previously reported for activation of Niu-A states formed by O2-sensitive hydrogenases the faster value is consistent with a reactivation process where the active site states change in a stepwise fashion transitioning through the intermediates with the same rate determining step.2,61 Taking this data together, we assign the νCO bands in the IR absorption spectrum of our as-isolated EcHyd-1 sample to a mixture of Nir-SI/II (1908, 1922 cm−1) and Nir-B (1942 cm−1) in which the population of the 1922 cm−1 state accounts for approximately 60% of the total.
Turning to the νCN region of the spectrum, we observe that the as-isolated sample features bands at 2050 and 2063 cm−1 (Fig. 3(a), black, Fig. S1 and S2, ESI†). It is noticeable that these too are very similar to the values reported for the Nia-SRII state, suggesting further coincidence with this state.33 However, it is not possible to directly link the νCO and νCN signals for the individual states using IR absorption spectroscopy and so we turn to 2D-IR spectroscopy for this as well as a more in-depth analysis of the spectroscopy, structure and dynamics of the states that we will henceforth refer to as Nir-SI/II. IR absorption measurements made as a function of pH showed that more basic conditions favoured the active site state with a νCO band at 1922 cm−1, which suggests that an assignment to the deprotonated Nir-SI state may be most likely. However, it is not possible to differentiate definitively between states labelled I or II and so we will refer them as Nir-SI/II and identify individual states by the νCO frequency if required.
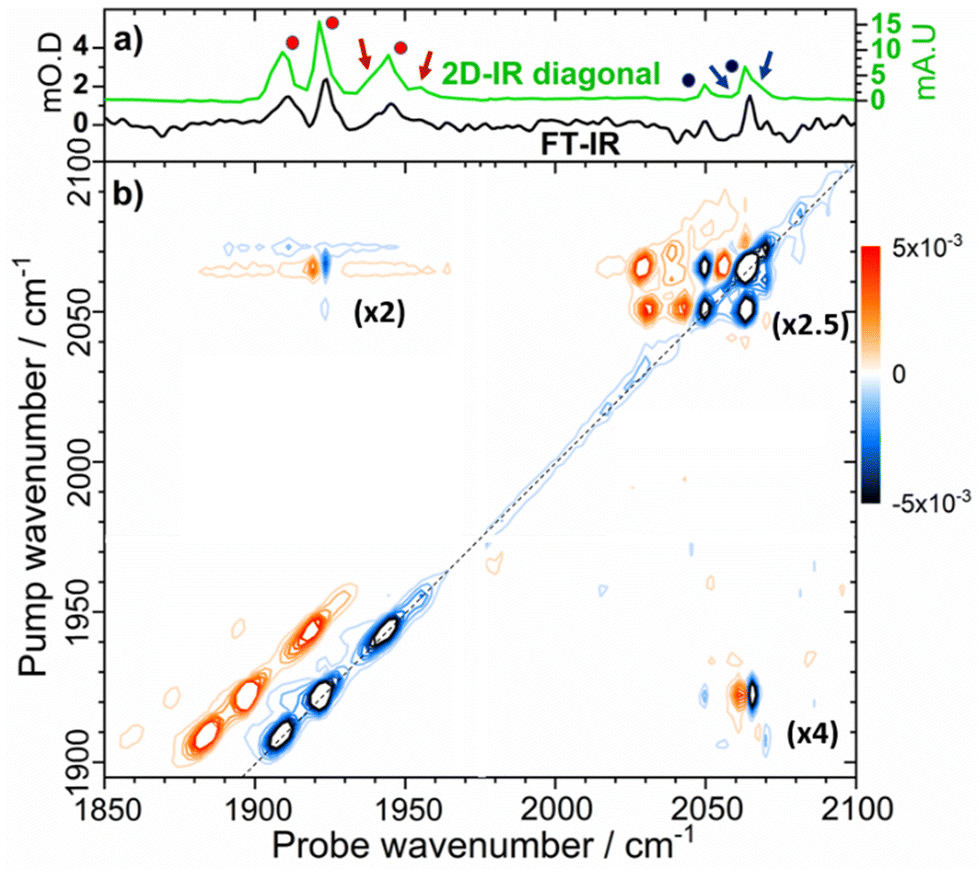 | ||
| Fig. 3 (a) IR absorption spectrum of as-isolated EcHyd-1 (black trace) and projection of the 2D-IR spectrum diagonal (green). The negative signals of the diagonal have been inverted for comparison with the IR absorption spectrum. (b) 2D-IR spectrum of as-isolated EcHyd-1 recorded at a waiting time (Tw) of 250 fs. The dashed line indicates the spectrum diagonal. Numbers in brackets indicate the magnification of the three quadrants of the 2D-IR spectrum containing peaks due to νCN modes in relation to the νCO region of the spectrum (1900–1950 cm−1), which contains the most intense peaks. See text for explanation of blue/red dots and arrows. | ||
In the νCN region of the 2D-IR spectrum, bands at 2050 and 2063 cm−1 are present (Fig. 3(a), blue circles). In addition, a high frequency shoulder on the 2063 cm−1 band is also clearly observed, with the indication of another weaker band between those at 2050 and 2063 cm−1 (minor bands are highlighted by blue arrows in Fig. 3(a)). We discuss these alongside other, less prominent, peaks in more detail below.
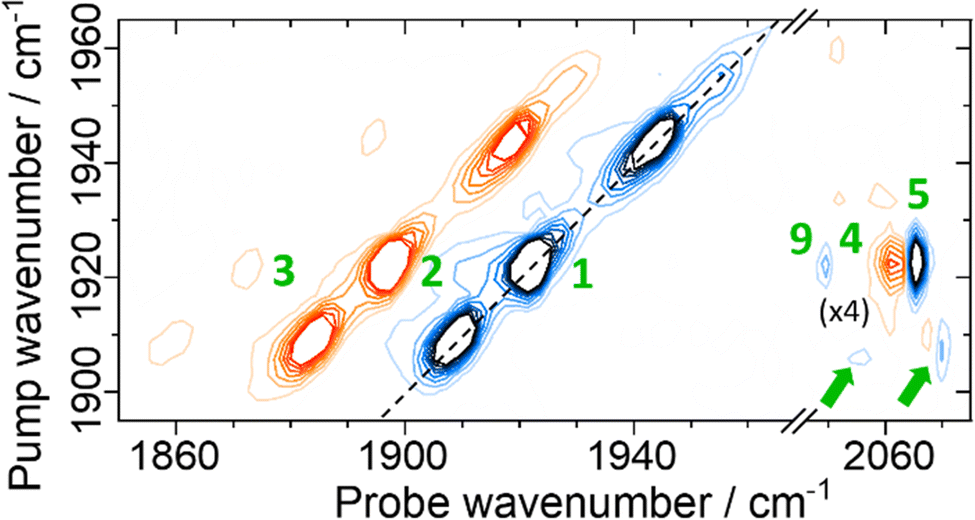 | ||
| Fig. 4 Magnification of the 2D-IR spectrum of EcHyd-1 shown in Fig. 3(b) for pump frequencies coinciding with νCO bands. The spectrum was measured with a Tw of 250 fs. Green numbers refer to peak assignments and green arrows highlight minor off diagonal peaks as discussed in the text. | ||
Each of the three main νCO diagonal peaks (1908, 1922 and 1942 cm−1) is accompanied by a strong positive (red) peak shifted by ∼25 cm−1 to lower probe frequency, with a second, much weaker positive peak shifted by the same amount again (Fig. 4). These are exemplified for the 1922 cm−1 (Nir-SI/II) diagonal peak by green peak labels 1, 2 and 3 in Fig. 4. The population of higher-lying vibrational levels by the pump pulse in the 2D-IR experiment allows the probe pulse to access transitions that are not observed using IR absorption methods at room temperature and so peaks 1–3 can be assigned to the v = 0–1, v = 1–2 and v = 2–3 transitions of the νCO mode respectively. The smaller diagonal peaks at 1955 and 1938 cm−1 also have an accompanying v = 1–2 transition, with similar anharmonicities to those of the larger bands suggesting that these too are assignable to νCO modes of two more active site states present in lower concentrations than those which give rise to the main bands.
The v = 0–1, v = 1–2 and v = 2–3 transition frequencies for each of the three most intense νCO modes were found to be well-represented by Morse anharmonic oscillator functions (Fig. S7 and S8, ESI†). The spectroscopic constants derived in each case are shown in Table S1 of the ESI.† The transitions and all peak assignments for the Nir-SI/II (νCO: 1922 cm−1) state of EcHyd-1 are summarized in a representative energy level diagram (Fig. 5) showing the νCO and νCN energy levels (vide infra). We confirm below that each observed state of EcHyd-1 is associated with one νCO and two νCN modes, as would be expected,65 and so we introduce vibrational state designations using the notation |νCOνCN1νCN2〉, where CN1 and CN2 indicate the higher and lower frequency νCN-modes, respectively. Using this notation peaks 1–3 are identified as: |000〉–|100〉; |100〉–|200〉; |200〉–|300〉.
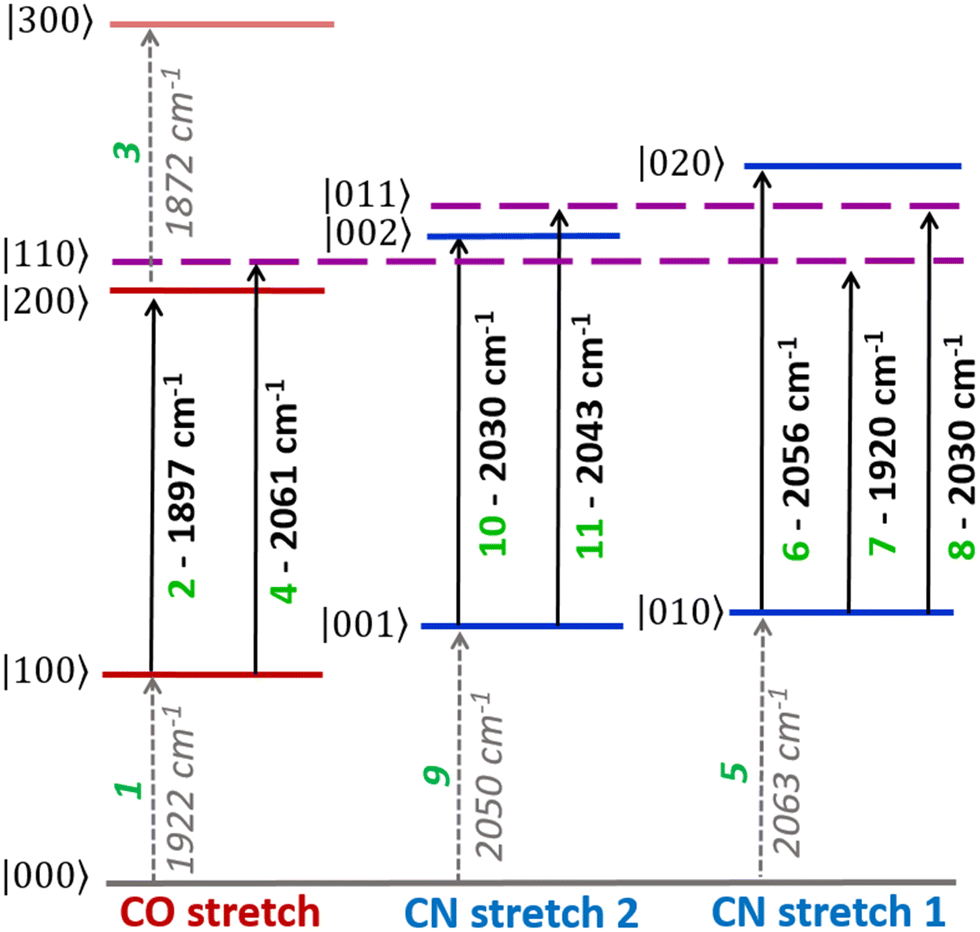 | ||
| Fig. 5 Energy level diagram showing vibrational energy levels |νCOνCN1νCN2〉 and transition wavenumbers of the νCO and νCN vibrational manifold, as detected for the active site state of EcHyd-1 with a νCO fundamental frequency of 1922 cm−1 (Nir-SI/II). Transitions are labelled with green numbers used to identify peak assignments in the 2D-IR spectra (see text). | ||
A further set of off-diagonal peaks with a pump frequency of 1922 cm−1 but with probe frequencies ranging between 2050 and 2063 cm−1 are present in the 2D-IR spectrum (Fig. 4, 4, 5 and 9). The most intense of these is a negative peak, 5, with frequency coordinates (pump, probe) = (1922, 2063 cm−1). The pump frequency matches one of the νCO bands assigned to Nir-SI/II while the probe frequency coincides with a diagonal peak in the νCN region of the spectrum. As the 2D-IR spectrum in Fig. 4 was obtained with a waiting time (Tw) of 250 fs, this off-diagonal peak indicates that these νCO and νCN modes are vibrationally coupled. A weaker negative peak at (1922, 2050 cm−1), 9, shows that the νCO band of this Nir-SI/II state is associated with a pair of νCN modes with frequencies of 2050 and 2063 cm−1. This enables an unambiguous assignment of a set of νCO and νCN mode frequencies to one of the Nir-SI/II active site states of EcHyd-1, consistent with the presence of a Fe(CO)(CN)2 unit.
The strong positive peak (4), which has the same pump frequency (1922 cm−1) but a probe frequency of 2061 cm−1 is assigned to a transition between the v = 1 level of the νCO mode (|100〉) populated by the pump pulse, and a combination state featuring one quantum of energy in both the νCO and the high frequency νCN stretching vibrations (|110〉). The separation of peaks 5 and 4 along the probe frequency axis in Fig. 4 indicates the mixed mode anharmonicity of the combination band.48,66,67 The mixed mode anharmonicity is the amount by which the combination band frequency is shifted relative to the sum of the fundamental transitions of the two participating modes and is a measure of coupling strength of the νCO and νCN modes. In this case, a shift of ≤2 cm−1 indicates weak νCO to νCN coupling. These assignments are shown in the energy level diagram in Fig. 5.
Closer examination of Fig. 4 shows a second pair of off-diagonal peaks (green arrows in Fig. 4) linking the Nir-SI/II νCO mode at 1908 cm−1 with the νCN modes at 2057 and 2070 cm−1. These peaks are weak, and the associated positive peaks are not clearly visible, but they indicate the set of coupled νCO and νCN modes that correspond to the Nir-SI/II state (νCO: 1908 cm−1) of EcHyd-1.
A number of off-diagonal peaks are present in the upper left quadrant of Fig. 3(b). These are the reverse of the peaks in the bottom right of the plot, which were discussed above (Fig. 3(b) and 4), in that the pump frequency now coincides with the νCN diagonal peaks, and the off-diagonal peaks link νCN and νCO modes. The peaks in this region include the peak marked 7 in Fig. 5 (2063, 1920 cm−1) and are consistent with the coupling patterns identified above, further confirming the linked sets of νCO and νCN modes.
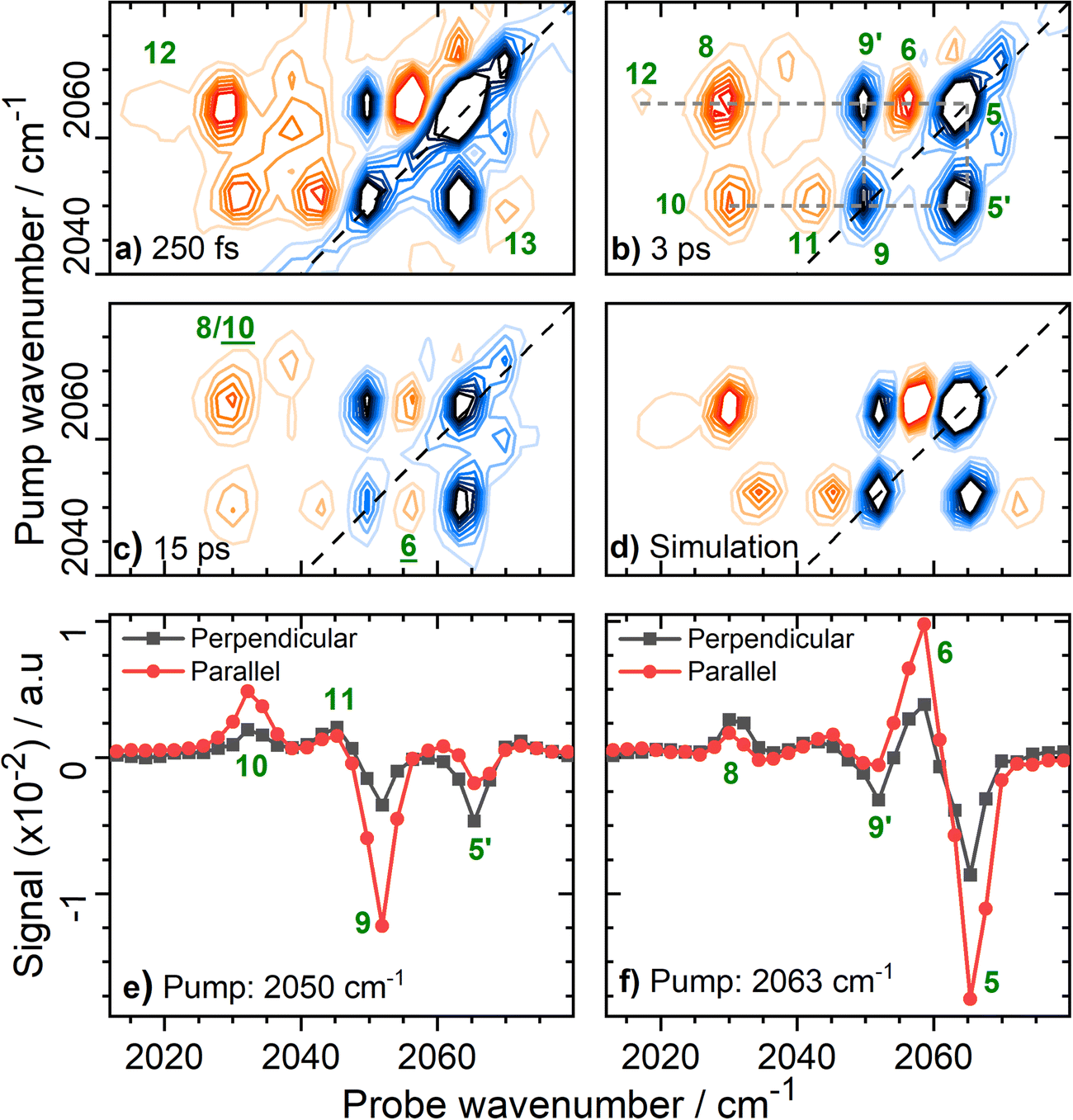 | ||
Fig. 6 Magnification of the 2D-IR spectrum of EcHyd-1 from Fig. 3(b) for pump frequencies coinciding with νCN bands at a series of values of Tw. (a) Tw = 250 fs with forbidden transitions, 12 and 13 highlighted; (b) Tw = 3 ps with full peak assignment for the major set of coupled νCN modes (dashed grey lines); (c) Tw = 15 ps showing energy transfer peaks, ![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) and and ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) (d) Simulated 2D-IR spectrum (see text) for the major pair of coupled CN modes with fundamental transition frequencies of 2050 and 2063 cm−1. (e and f) Slices through the 2D-IR spectrum shown in (b) under parallel (red) and perpendicular (black) polarization conditions. The pump frequency in each case is given in the legend. (d) Simulated 2D-IR spectrum (see text) for the major pair of coupled CN modes with fundamental transition frequencies of 2050 and 2063 cm−1. (e and f) Slices through the 2D-IR spectrum shown in (b) under parallel (red) and perpendicular (black) polarization conditions. The pump frequency in each case is given in the legend. | ||
The rich off-diagonal peak structure in the νCN region can be used to construct an energy level diagram for these modes up to states containing two quanta of vibrational energy (v = 2 or mixed mode combination states). We begin by focusing on the strongest νCN diagonal peak at 2063 cm−1 (Fig. 6(b), 5). When pumping the 2063 cm−1 mode (|000〉–|010〉), three clear off-diagonal peaks are visible (6, 8 and 9′; the prime indicates an off-diagonal peak arising from transition 9 in Fig. 5 to differentiate it from a diagonal peak which is identified by the same number below). The positive peak 6 is assigned to the v = 1–2 transition of the νCN vibrational mode (|010〉–|020〉). This shows a single mode anharmonic shift of 8 cm−1 relative to the fundamental transition (|000〉–|010〉).
Negative peak 9′ indicates that the νCN modes at 2063 cm−1 and 2050 cm−1 are vibrationally coupled, consistent with the identification above that these two modes arise from the same single Nir-SI/II state. It is important to note that the 2063 cm−1 diagonal peak only shows coupling to one other νCN mode, as would be expected for a Fe(CO)(CN)2 unit.
Peak 8 (positive) arises from a transition to the combination state featuring one quantum of excitation in each of the νCN modes at 2050 and 2063 cm−1 (|010〉–|011〉). These transitions are marked on the energy level diagram in Fig. 5. The large separation of peaks 8 and 9′ shows that the mixed mode anharmonicity of the |011〉 combination band is 20 cm−1, indicating that the two νCN modes are much more strongly coupled to each other than to the νCO modes.
The diagonal νCN peak at 2050 cm−1 (Fig. 6(b), 9) corresponding to the fundamental transition of the second νCN mode of the Nir-SI/II state, is also accompanied by three off-diagonal peaks. Peak 5′ reflects the coupling to the νCN mode at 2063 cm−1 as expected. Assignment of the positive peaks 10 and 11 is less straightforward. An initial assignment of peak 11 to the v = 1–2 transition of the 2050 cm−1 νCN mode might be expected. However, consideration of the energy level diagram (Fig. 5) shows that the unusually large mixed mode anharmonicity (20 cm−1) arising from the strong coupling of the two νCN modes leads to the positive partner expected near peak 5′ for a pair of coupled modes appearing on the opposite side of the spectrum diagonal from 5′. As a result, the correct assignment of peak 11 is to the transition to the combination state of the two νCN modes ((|001〉–|011〉), Fig. 5). This means that peak 10 is assigned to the v = 1–2 transition of the 2050 cm−1 νCN mode, (|001〉–|002〉), an anharmonic shift of 20 cm−1 that is significantly different to the value of 8 cm−1 identified for the 2063 cm−1 mode.
Our peak assignments in the νCN region are supported by examining the 2D-IR spectrum at longer waiting times. Fig. 6(c) shows an additional peak, (the underline is used to identify a peak appearing at later waiting times), which becomes visible at coordinates of (pump, probe) (2050, 2058 cm−1) at waiting times approaching ∼15 ps. The delayed arrival of this peak along with its position indicates that it is due to energy transfer from the v = 1 state of the excited (pumped) νCN mode at 2050 cm−1 (|001〉), to the v = 1 level of the νCN mode at 2063 cm−1 (|010〉). The probe pulse is then able to excite the (|010〉–|020〉) transition of the 2063 cm−1 mode, which lies at 2058 cm−1 (Fig. 5). Peak is thus assigned to the effects of energy transfer between the two νCN modes. The timescale is consistent with the relaxation dynamics of the v = 1 levels of the νCN modes (vide infra). The reverse peak, featuring energy transfer from the pumped 2063 cm−1 mode to the 2050 cm−1 mode followed by the probe exciting the (|001〉–|002〉) transition would be expected at (2063, 2030 cm−1). This position coincides almost exactly with peak 8, which is assigned to the effect of vibrational coupling. However, the persistence of peak 8 to a waiting time of at least 45 ps, in contrast to the other peak arising from coupling (11), is consistent with the presence of overlapping peaks, one of which arises from energy transfer (, Fig. S9, ESI†). The vibrational relaxation dynamics of the νCO and νCN modes are discussed in more detail below.
2D-IR measurements performed at different pump–probe pulse polarization geometries provide further support for our peak assignments in the νCN region of the spectrum as well as adding structural information relating to the EcHyd-1 active site in solution. Peaks 5/6 and 9/10 (Fig. 6(b)), which are assigned to v = 0–1 and v = 1–2 transitions of the high frequency and low frequency νCN modes of Nir-SI/II respectively, undergo significant reduction in amplitude upon changing from parallel to perpendicular relative pump–probe polarizations (Fig. 6(e) and (f)). This is as expected because the directions of the transition dipole moments associated with the v = 0–1 and v = 1–2 transitions of a given mode necessarily lie in the same direction. In contrast, the peaks arising from transitions involving combination states (8 and 11) show much weaker polarization dependence (Fig. 6(e) and (f)). This is consistent with a signal arising from pump and probe events, which interact with vibrational modes whose transition dipole moments lie at right angles to each other.
Polarization-dependent 2D-IR measurements can also be used to quantify the angular relationship between the two νCN modes by determining the anisotropy parameters for the pairs of diagonal and off-diagonal peaks labelled 9/5′ in Fig. 6(b).49 The values obtained, 0.45 (9) and −0.24 (5′) (Fig. S10, ESI†), are, within error, consistent with two vibrational modes with transition dipole moments oriented at 90° to each other, as expected for a cis geometry of the two cyanide ligands at the hexacoordinated Fe center.
The presence of strong coupling of the two νCN modes, as evidenced by a mixed mode anharmonicity (20 cm−1) greater than the mode separation (13 cm−1), would be expected to lead to the partial breakdown of vibrational selection rules (Δν = ±1), enabling transitions from the v = 1 level of one νCN mode to the v = 2 level of the second, (e.g. |001〉–|020〉), which would formally be forbidden.48 For the Nir-SI/II (νCO: 1922 cm−1) state these transitions are predicted by the energy level diagram in Fig. 5 to lie at (pump, probe) (2050, 2071 cm−1) and (2063, 2017 cm−1) and they can clearly be observed in Fig. 6(a), labelled 12 (|001〉–|020〉) and 13 (|010〉–|002〉) respectively. The presence of these features adds further weight to our assignments. The energy level diagram in Fig. 5 was used to construct a simulation of the νCN region of the 2D-IR spectrum based on 2D-Gaussian functions (see ESI,† for details). This is shown in Fig. 6(d) and the agreement with the experimental data is excellent.
In addition to the set of peaks arising from the pair of νCN modes at 2050 and 2063 cm−1, a second much less intense set of peaks can be observed linking the coupled pair of νCN modes due to the Nir-SI/II (νCO: 1908 cm−1) state at 2070 and 2058 cm−1. These are shown in Fig. 7, labelled (5)/(5′), (6), (8), (9)/(9′) and (10), showing that the νCN spectroscopy of the two different active site states is very similar.
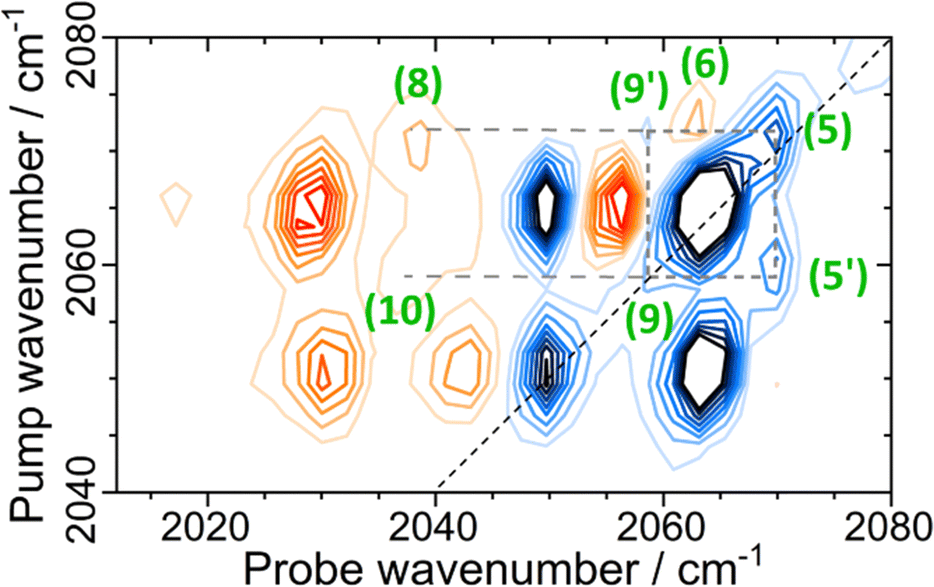 | ||
| Fig. 7 2D-IR spectrum of EcHyd-1 at Tw = 250 fs in the νCN region of the spectrum, with full peak assignment for the minor set (dashed lines) of coupled CN modes due to the Nir-SI/II state. | ||
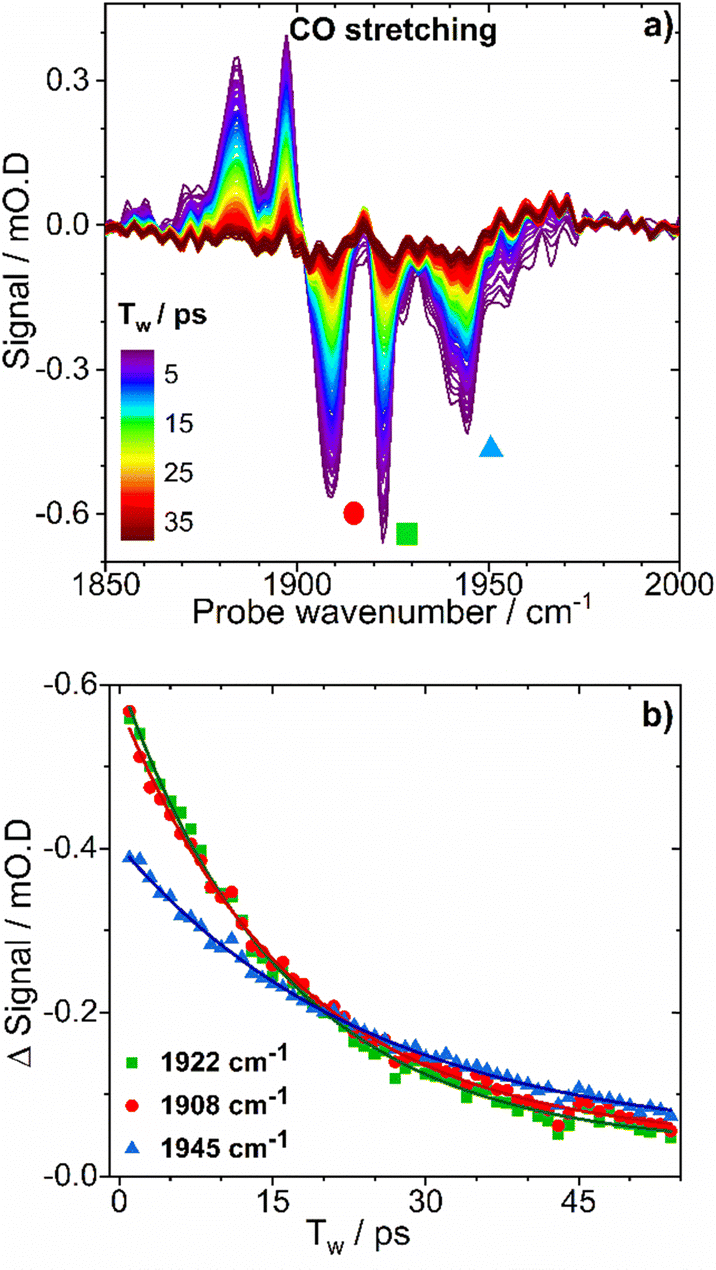 | ||
| Fig. 8 (a) IRpump–IRprobe spectra of as-isolated EcHyd-1in the νCO region of the spectrum. (b) Plots of band intensity versus time for selected bands from the spectra in (a), each identified by colored symbols. Solid lines show results of fitting experimental data with monoexponential decay functions. | ||
The vibrational lifetimes of the three most intense νCN modes, with fundamental transitions located at 2050, 2063 and 2070 cm−1 were 32 ± 2, 37 ± 2 and 29 ± 2ps respectively (Table S2 and Fig. S11, ESI†). These values of the vibrational lifetimes for the carbonyl and cyanide modes are consistent with peaks due to energy transfer between νCN modes, which appear in the 2D-IR spectra at waiting times of ∼15 ps (Fig. 6(c)). Similarly, a peak was observed in the 2D-IR spectrum at coordinates of (pump, probe) = (2063, 1897), which can be assigned to energy transfer from the pumped νCN (|010〉) level of Nir-SI/II (νCO: 1922 cm−1) to the νCO (|100〉, leading to the probe exciting the |100〉–|200〉 transition of the νCO mode (![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) , Fig. S12, ESI†).
, Fig. S12, ESI†).
In the case of EcHyd-1 the νCO, νCN1 and νCN2 bands exhibited ratios of the diagonal to antidiagonal full width half maximum values of 1.7, 1.6 and 1.5 respectively at a waiting time of 250 fs. These values were unchanged at a waiting time of 15 ps, indicating little in the way of structural dynamics occurring on timescales between 200 fs and 15 ps.
Examination of the dephasing time of the νCO mode of the 1922 cm−1 peak (4 ps) suggests an expected homogeneous linewidth of 3 cm−1. Together with the observed diagonal linewidth (5 cm−1) for this mode, this is consistent with the observed diagonal:antidiagonal width ratio of 1/7 (=5/3) and also indicates a very limited degree of inhomogeneous broadening.
The O2-oxidised sample of EcHyd-1 also gave rise to another set of νCO, νCN1 and νCN2 bands at 1948, 2098 and 2080 cm−1, which we tentatively assign to the Niu-S states (Fig. 1(a) and Fig. S14, ESI†). In the case of this set of bands, it is notable that the positions are somewhat different, with evidence for the single mode anharmonicity of the higher frequency νCN band being as high as 13 cm−1, while the mixed mode anharmonicity is reduced to 13 cm−1 (Table S3 and Fig. S14, ESI†). These changes may be attributable to the presence of a bridging sulphoxygenated cysteine residue in the active site structure (Fig. 1(a)).
Model compound: K[CpFe(CO)(CN)2]
To provide a direct insight into the influence of the protein scaffold on the spectroscopy of the enzyme active site, we measured the 2D-IR spectrum of the model compound K[CpFe(CO)(CN)2] (M1) in aqueous solution.68,69The results (Fig. 9(a)) show the spectral band patterns observed for M1 are broadly similar to those of the enzyme (reproduced in Fig. 9(b)) allowing similar assignments of the peaks to be made. This approach shows that the anharmonicity of the νCO mode of M1 is 24 cm−1 and that this mode is weakly coupled to the νCN modes (mixed mode anharmonicity: ≤5 cm−1). The two νCN modes of M1 show quite different single mode anharmonicities to one another (10 and 22 cm−1), while the mixed mode anharmonicity for the νCN modes of M1 was found to be 19 cm−1.
 | ||
| Fig. 9 2D-IR spectra of (a) compound M1 in H2O solution and (b) as isolated EcHyd-1 obtained for similar values of Tw. | ||
Examining the spectral linewidths and structural dynamics of the νCO and νCN modes shows that the νCO and νCN bands of M1 dissolved in H2O are much broader than those in the enzyme. The νCO band of M1 has a diagonal linewidth of 20 cm−1 as compared to 8, 5 and 12 cm−1 for the EcHyd-1 states at 1908, 1922 and 1942 cm−1 respectively. In the case of the νCN bands, widths of 12 and 14 cm−1 (M1) compare with 5 cm−1 for the enzyme, irrespective of the active site state. The 2D lineshapes of M1 in solution were observed to undergo rapid spectral diffusion, with the ratios of the diagonal to antidiagonal linewidths changing from 1.5, 1.2 and 1.5 (νCO, νCN1, νCN2) at short Tw (125 fs) to values of unity by a waiting time of 2 ps. This indicates that rapid structural fluctuations are present in solution that do not influence the 2D lineshapes of the enzyme. Finally, the vibrational relaxation dynamics of the νCO and νCN modes of M1 (4 and 6/7 ps respectively (Fig. S15, ESI†)) are significantly faster than those of EcHyd-1 Nir-SI/II (16–25 ps and 29–37 ps respectively).
Discussion
Applying 2D-IR spectroscopy alongside a suite of enzyme characterization measurements has enabled identification of νCO and νCN frequencies for two previously un-reported active site states of EcHyd-1, Nir-SI/II in addition to data on two other active site states. The 2D-IR spectra have also provided deeper insights into the vibrational potentials of the νCO and νCN bands of these states while the sub-picosecond time resolution has been exploited to reveal their vibrational and structural dynamics.Our observations show that the spectroscopy and dynamics of all of the active site states of EcHyd-1 studied so far are broadly similar, with the exception of some minor deviation for signals assigned to the Niu-S state. It is therefore interesting to consider whether this extends to other [NiFe] hydrogenases via our previous study of the ReRH enzyme.52 The vibrational relaxation and structural dynamics reported for ReRH were very similar to those observed for EcHyd-1, (νCO and νCN vibrational lifetimes were 18 and 30 ps respectively52) along with no evidence of significant structural changes occurring on this timescale as evidenced by spectral diffusion measurements. Although the νCN region of the 2D-IR spectrum of ReRH was less well-resolved than reported for the EcHyd-1 enzyme studied here, revisiting the ReRH 2D-IR spectroscopy in light of our new results allows a clearer picture to emerge. An early waiting time 2D-IR spectrum of the ReRH enzyme in the νCN region is presented (Fig. 10(a)) alongside a simulation based on a similar mode structure and energy level diagram (Fig. S16, ESI†) to that observed for EcHyd-1 (Fig. 10(b)). The agreement is excellent, allowing us to define single mode (8 and 18 cm−1) and mixed mode (24 cm−1) anharmonicities for the ReRH νCN modes (Table S3, ESI†) that are very similar to the values for EcHyd-1. This new information also allows complex time-evolution behavior of the peaks observed in the ReRH spectra52 to be assigned to the differential relaxation of combination band and energy transfer peaks as described for EcHyd-1 above (Fig. S17, ESI†). This result strongly suggests that the active sites of the two enzymes which perform quite different biological functions have common characteristics to their Fe(CO)(CN)2 units.
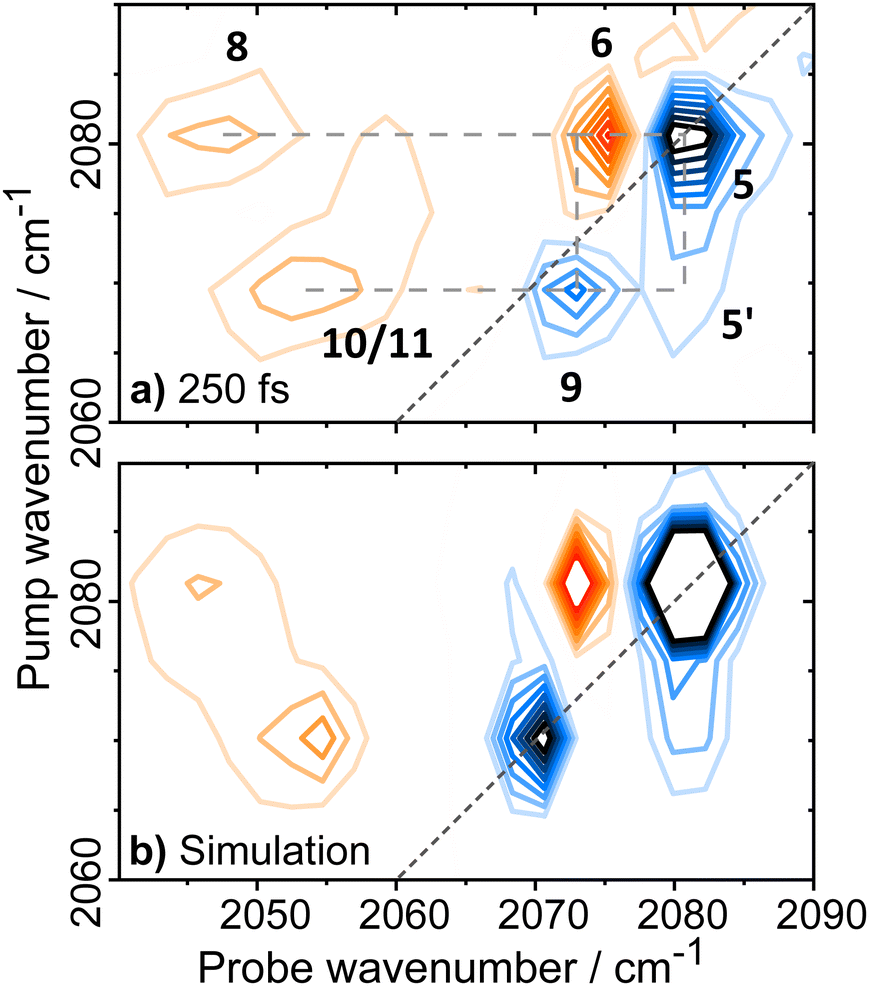 | ||
| Fig. 10 (a) 2D-IR spectrum of ReRH with pump frequencies resonant with νCN modes obtained with a Tw of 250 fs; (b) simulation of the ReRH 2D-IR spectrum in the νCN region based on a similar set of vibrational states as determined for EcHyd-1. | ||
We now proceed to consider how the results for the enzyme active site compare to those of a structural mimic of the Fe(CO)(CN)2 unit in aqueous solution. Our results for the enzyme show that while the spectroscopy of the νCO bands of the Nir-SI/II states are well-described by a Morse-oscillator model, the νCN bands show some unusual characteristics. In particular, the two νCN bands exhibit markedly different single mode anharmonicities, one with a value of 20 cm−1, while the other is much less anharmonic (8 cm−1). The vibrational coupling of the νCN modes is indicated by the observed mixed mode anharmonicity value of 20 cm−1. Although vibrational coupling of the νCN modes in hydrogenases has been detected previously,52,70 our results here suggest that it is very strong, and sufficient to cause a breakdown in harmonic oscillator selection rules that results in the observation of peaks 12 and 13 in Fig. 6(a). It is important to stress that these effects cannot be explained using a harmonic description of the potential energy surfaces of the CN bonds, rather, they arise from the higher order, cubic and quartic terms, the effects of which are not discernable using absorption spectroscopy.48,71–74
Our results obtained from M1 in solution show that some of the observations relating to the spectroscopy of the observed EcHyd-1 active site states can be attributed to the intrinsic nature of the Fe(CO)(CN)2 unit. For example the single mode anharmonicity of the low frequency νCN mode as well as the mixed-mode anharmonicity are consistent between M1 and the enzyme, the latter showing that the strong inter-νCN coupling is at least in part a feature of the Fe(CO)(CN)2 unit. Further evidence indicating that the observed spectroscopic patterns are an intrinsic property of the Fe(CO)(CN)2 can be found from other examples where single and mixed mode anharmonicities have been reported for organometallic cyanide-containing compounds, that also showed very different values to those of either M1 or EcHyd-1.75,76 However, the single mode anharmonicity observed for the high frequency νCN mode of EcHyd-1 is smaller than that of M1 in solution, suggesting a degree of environmental sensitivity in this parameter. The νCN mode frequency separation is much less in EcHyd-1 (ΔνCN–CN is 13 cm−1) than M1 (17 cm−1), a change which brings the mode separation and mixed mode anharmonicities closer together for M1. This implies that M1 approaches the weak coupling regime,48 such that the forbidden transitions observed in the enzyme do not appear in spectra of M1. These results suggest that our observed spectroscopic differences between M1 and the enzyme stem from the environment of the active site dictated by the protein scaffold. It is however important to note that, although the size and position of the Cp ring enables M1 to provide a good structural mimic of the Fe(CO)(CN)2 unit,68,69M1 does not replicate either the Ni or the terminal and bridging cysteine residues, which have been shown to strictly control the local geometry of the NiFe site, leading to asymmetry in the two cysteine bridges.9,10,35,77 These differences could in principle influence the vibrational potentials of the cyanide ligands that lie trans to them.
Although the impact of the protein scaffold upon spectroscopic parameters of the Fe(CO)CN)2 unit is subtle, the results from M1 reveal that it has a significant impact upon its dynamics. The data for M1 reveals vibrational relaxation times for the νCO and νCN modes of M1 on the order of a few picoseconds, in good agreement with previous studies of organometallic carbonyls and cyanides in aqueous solution.78,79 This rapid vibrational relaxation in water is attributed to spectral overlap of the νCN and νCO mode frequencies with the broad bend-libration combination band of H2O near 2100 cm−1 leading to an efficient energy dissipation pathway to the solvent.80 In contrast, much slower vibrational relaxation was observed for the νCN and νCO modes of EcHyd-1, which shows that bulk-like H2O does not interact with the active site. Instead, the observed vibrational relaxation times for EcHyd-1 are more consistent with observations of organometallic compounds in protic organic solvents. The fact that such solvents are capable of hydrogen bonding could imply a role for the H-bond links between the protein scaffold and Fe(CO)(CN)2 unit in determining the observed dynamics.76
Differences between the structural dynamics of the model compound and the enzyme were also observed. The 2D-IR diagonal peaks due to the νCN and νCO modes of M1 display inhomogeneously broadened lineshapes and fast (1–2 ps) spectral diffusion, while the enzyme modes show limited evidence of inhomogeneous broadening and no spectral diffusion on timescales up to 45 ps. The contrasting behavior of M1 can be explained by rapid fluctuations of the H-bonding environment around the ligands and is consistent with previous studies in aqueous solutions.81 Examination of the 2D-IR data shows that the νCO mode of M1 in solution exhibits a much shorter dephasing time than the enzyme (0.96 ps vs. 4 ps). This faster dephasing contributes to the increased linewidth observed for M1 compared to the enzyme, but would lead to a homogeneous linewidth of 12 cm−1, showing that the observed 20 cm−1 linewidth arises from a significant degree of inhomogeneous broadening. In contrast, the νCO mode of the enzyme (at 1922 cm−1) has a much narrower linewidth with little inhomogeneous contribution. These observations agree with previous work on ReRH.52
In the case of the enzyme, crystallography data indicates that the two cyanide ligands are H-bonded to the Thr532 and Arg509 residues (Fig. 11)57 which feature neutral and cationic side chains respectively. Fluctuations of the protein would therefore be expected to modulate the terminal charges on the CN ligands and so the νCN mode frequencies, leading to structural dynamics being manifest as inhomogeneous broadening and spectral diffusion of the νCN modes. By the same argument the CO ligand, which is not subject to H-bonds, could be expected to show different behavior to the CN ligand modes, but the behavior of the νCO and νCN bands is very similar, although fluctuations of the protein environment could impact upon the νCO mode frequency via an electrostatic, rather than specifically H-bonding, mechanism. It is instructive that the CO and CN ligands of M1 also demonstrate similar structural dynamics where again H-bonding would be expected to be stronger to the CN ligands, which would seem to support the hypothesis that the ligands detect similar environmental dynamics.
 | ||
| Fig. 11 Enlarged view of the EcHyd-1 active site Fe(CO)(CN)2 moiety and its local environment, displayed as a ball and stick schematic. The image is from an X-ray crystal structure (PDB: 6FPW57) of an (18 hour) H2-exposed sample of EcHyd-1. Distances to the hydrogen atoms are indicated. | ||
These observations could imply that the EcHyd-1 provides a very restricted environment with little fluctuation or that the fluctuations do not become manifest as spectral diffusion. However, these scenarios are not mutually exclusive and further study is required as it is not possible to be definitive from this data. An analysis of spectral linewidths and dephasing times performed on ReRH supported the presence of a dynamically rigid active site and the similarities between the data for ReRH and EcHyd-1 indicate that the same conclusions apply here.52 Such a scenario would entail a protein scaffold that behaves in a markedly different manner to those revealed by studies of spectral diffusion in other protein and enzyme structures, where few picosecond spectral diffusion dynamics have been widely observed.82–88 However, the role of the hydrogenases involves manipulating extremely small substrates as well as excluding slightly larger species such as water and so such a rigid architecture may be the biological solution to these complex problems. Another feasible explanation for the lack of observed spectral diffusion is that the H-bonding to the CN ligands is sufficiently weak that the cyanide ligand stretching frequencies do not act as an efficient reporter of protein motion. In this respect, the carbonyl ligand stretching mode frequency would appear to be similarly decoupled from protein fluctuations. If this were the case then it would be expected to have implications for the vibrational relaxation mechanism, the timescales of which are comparable with a protic, organic solvent. In the event of very weak hydrogen bonding, this could indicate that vibrational relaxation occurs via the dimetallic unit rather than through the protein.
Overall, we conclude that the enzyme active site dynamics, including both relaxation timescales and the lack of spectral diffusion, cannot be approximated by analogy to a single simple solvent model: the vibrational relaxation time points to a protic organic environment while the absence of spectral diffusion is more reminiscent of a non-interacting environment such as a non-polar organic solvent.89,90 As such, the data indicates that the NiFe active site is subject to complex, multifaceted influences but clearly communicates the importance of considering the protein scaffold as part of the active site. The evidence shows that the protein defines some aspects of the vibrational potential surfaces of the Fe(CO)(CN)2 unit but that its major contribution is in defining the dynamic environment by effectively isolating the Fe(CO)(CN)2 unit from the surrounding solvent and by either tightly controlling structural dynamics or limiting the impact of any fluctuations of the protein scaffold upon the active site.
Placing these results in context with the design of biomimetic hydrogen producing systems, the differences between our observations of the enzyme active site and measurements of model compound M1 in solution indicate that further work assessing the contribution of the secondary coordination spheres, self-assembled scaffolds and/or full apo-enzymes have in effective hydrogenase biomimetic systems would be beneficial in developing artificial systems. While the role that the protein scaffold plays in the enzyme mechanism is not clear, the conserved nature of the local active site environment suggests that it is an important component of the hydrogenase architecture. Our results show that 2D-IR spectroscopy provides a facile route to characterizing attempts to replicate the biological scenario, in particular solvent exclusion and the complex structural dynamics, with simpler scaffolds.11–18
Finally, we reflect on the coincidence of the νCO and νCN mode frequencies for the Nir-SI/II states determined here at 1922/2050/2063 cm−1 and 1908/2057/2070 cm−1 (Table S2, ESI†) with those previously assigned to the Ni-RII and Ni-RIII, (Nir-SRII/III using our notation) states which contain a hydride-bridged Ni(II)Fe(II) center (Fig. 1(a)).27 Although the νCO mode frequency of the Nir-SI/II state (νCO: 1922 cm−1) shows remarkable agreement with that assigned to Ni-RII, that of the other Nir-SI/II state (νCO: 1908 cm−1) differs from the literature values for the Ni-RIII state (νCO: 1914 cm−1).27 When added to the biochemical characterization data reported above, this discrepancy adds further weight to our assignment of the as-isolated EcHyd-1 spectrum to different states with coincident frequencies to one of the substates referred to as Ni-R.27 The frequency coincidence extends to the νCN modes, which for both the Ni-RII and Ni-RIII states were quoted as 2050 and 2067 cm−1,27 while a recent study in the crystalline phase identified four bands between 2049 and 2071 cm−1, though without direct assignment to the sub-states.91 Thus the fact that we identify a similar set of frequencies as being attributable to a particular state is consistent with the Nir-SI/II and Ni-RII and Ni-RIII states being spectroscopically very similar. There are in fact numerous reports of NiFe hydrogenases in distinct states exhibiting very similar IR signatures, for instance: Nir-B and Niu-A in Allochromatium vinosum MBH,29Desulfovibrio vulgaris Miyazaki F,92 and Desulfovibrio gigas MBH;93 and Nia-S and Nir-S in Ralstonia eutropha MBH94 and RH,95 and Nia-SRIII and Nia-SRII of Ralstonia eutropha SH.96 Finally, we note that the wavenumbers reported here for the Nir-SI/II states of EcHyd-1 fall within the range of reported frequencies for the same states of other enzymes (Fig. S18, ESI†).
Conclusions
We have reported the application of 2D-IR spectroscopy to study the active site of the EcHyd-1 enzyme, obtaining data for a number of active site states, including two previously un-reported Nir-SI/II states. By comparing the enzyme results to those of a model organometallic compound in aqueous solution, we reveal that the vibrational potential energy surfaces and dynamics of the Fe(CO)(CN)2 unit, particularly of the νCN stretching modes, offer a sensitive probe of the local environment. The vibrational potential surfaces of the two CN ligand stretching modes are found to be quite different in terms of their single mode anharmonicities and they are extremely strongly coupled. These observations are attributed partly to the nature of the Fe(CO)(CN)2 unit, but also to a specific influence of the protein scaffold, which creates a highly specialized local environment by eliminating solvent from the active site and leading to dynamics that cannot be replicated using solvent analogs. The indication is of a protein scaffold that interacts surprisingly weakly with the active site or which very tightly controls the local structure via cysteine links to the NiFe centre and asymmetric H-bonding arrangement between the protein scaffold and the cyanide ligands. Further experiments targeting the specific interactions of the amino acids near the Fe(CO)(CN)2 unit as well as investigating the potential role of vibrational relaxation phenomena such as IVR would be of benefit.97,98A similar set of vibrational parameters, albeit with some evidence for state-to-state variations, can be used to describe the active sites of other [NiFe] hydrogenases studied so far, leading to the conclusion that the spectroscopic observables are indicative of an evolutionarily selected active site environment that is central to the enzyme structure, if not directly to its function. These observations demonstrate the need for the inclusion of anharmonic effects in understanding the potential energy surfaces of the active site vibrational modes, its structure, and dynamics. They also appear to explain the need for complex secondary coordination spheres in promising biomimetic hydrogen producing catalysts and provide both a template and an approach to measuring important physical properties of novel biomimetic candidates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Robin Perutz for valuable discussions. The work was funded by the Leverhulme Trust (RPG-2018-188). SLDW acknowledges PhD studentship funding from the University of York. Access to the STFC Central Laser Facility (20130007) is gratefully acknowledged. This work (Y. R. and M. H.) was funded by the DFG under Germany's Excellence Strategy—EXC 2008–390540038—UniSysCat.References
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114(8), 4081–4148 CrossRef CAS PubMed.
- M. J. Lukey, A. Parkin, M. M. Roessler, B. J. Murphy, J. Harmer, T. Palmer, F. Sargent and F. A. Armstrong, How Escherichia coli Is Equipped to Oxidize Hydrogen under Different Redox Conditions, J. Biol. Chem., 2010, 285(6), 3928–3938 CrossRef CAS.
- J. A. Birrell, O. Rudiger, E. J. Reijerse and W. Lubitz, Semisynthetic Hydrogenases Propel Biological Energy Research into a New Era, Joule, 2017, 1(1), 61–76 CrossRef CAS.
- N. A. Eberhardt and H. R. Guan, Nickel Hydride Complexes, Chem. Rev., 2016, 116(15), 8373–8426 CrossRef CAS.
- P. W. Du and R. Eisenberg, Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: recent progress and future challenges, Energy Environ. Sci., 2012, 5(3), 6012–6021 RSC.
- S. Fukuzumi, Y. M. Lee and W. Nam, Thermal and photocatalytic production of hydrogen with earth-abundant metal complexes, Coord. Chem. Rev., 2018, 355, 54–73 CrossRef CAS.
- T. Kramer, M. Kamp, W. Lubitz, M. van Gastel and F. Neese, Theoretical Spectroscopy of the Ni-II Intermediate States in the Catalytic Cycle and the Activation of NiFe Hydrogenases, ChemBioChem, 2013, 14(14), 1898–1905 CrossRef.
- M. Kampa, W. Lubitz, M. van Gastel and F. Neese, Computational study of the electronic structure and magnetic properties of the Ni-C state in NiFe hydrogenases including the second coordination sphere, J. Biol. Inorg. Chem., 2012, 17(8), 1269–1281 CrossRef CAS PubMed.
- Y. Ilina, C. Lorent, S. Katz, J. H. Jeoung, S. Shima, M. Horch, I. Zebger and H. Dobbek, X-ray Crystallography and Vibrational Spectroscopy Reveal the Key Determinants of Biocatalytic Dihydrogen Cycling by NiFe Hydrogenases, Angew. Chem., Int. Ed., 2019, 58(51), 18710–18714 CrossRef CAS.
- M. Bruschi, M. Tiberti, A. Guerra and L. De Gioia, Disclosure of Key Stereoelectronic Factors for Efficient H-2 Binding and Cleavage in the Active Site of NiFe-Hydrogenases, J. Am. Chem. Soc., 2014, 136(5), 1803–1814 CrossRef CAS.
- N. T. Nguyen, Y. Mori, T. Matsumoto, T. Yatabe, R. Kabe, H. Nakai, K. S. Yoon and S. Ogo, A NiFe hydrogenase model that catalyses the release of hydrogen from formic acid, Chem. Commun., 2014, 50(87), 13385–13387 RSC.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100000 s(−1) for H-2 Production, Science, 2011, 333(6044), 863–866 CrossRef CAS PubMed.
- F. Wang, M. Wen, K. Feng, W. J. Liang, X. B. Li, B. Chen, C. H. Tung and L. Z. Wu, Amphiphilic polymeric micelles as microreactors: improving the photocatalytic hydrogen production of the FeFe-hydrogenase mimic in water, Chem. Commun., 2016, 52(3), 457–460 RSC.
- L. Z. Wu, B. Chen, Z. J. Li and C. H. Tung, Enhancement of the Efficiency of Photocatalytic Reduction of Protons to Hydrogen via Molecular Assembly, Acc. Chem. Res., 2014, 47(7), 2177–2185 CrossRef CAS PubMed.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Mimicking hydrogenases: from biomimetics to artificial enzymes, Coord. Chem. Rev., 2014, 270, 127–150 CrossRef.
- J. X. Jian, Q. Liu, Z. J. Li, F. Wang, X. B. Li, C. B. Li, B. Liu, Q. Y. Meng, B. Chen, K. Feng, C. H. Tung and L. Z. Wu, Chitosan confinement enhances hydrogen photogeneration from a mimic of the diiron subsite of FeFe-hydrogenase, Nat. Commun., 2013, 4, 2695 CrossRef PubMed.
- H. Y. Wang, W. G. Wang, G. Si, F. Wang, C. H. Tung and L. Z. Wu, Photocatalytic Hydrogen Evolution from Rhenium(I) Complexes to FeFe Hydrogenase Mimics in Aqueous SDS Micellar Systems: A Biomimetic Pathway, Langmuir, 2010, 26(12), 9766–9771 CrossRef CAS PubMed.
- F. Gloaguen and T. B. Rauchfuss, Small molecule mimics of hydrogenases: hydrides and redox, Chem. Soc. Rev., 2009, 38(1), 100–108 RSC.
- V. Firpo, J. M. Le, V. Pavone, A. Lombardi and K. L. Bren, Hydrogen evolution from water catalyzed by cobalt-mimochrome VI*a, a synthetic mini-protein, Chem. Sci., 2018, 9(45), 8582–8589 RSC.
- S. R. Soltau, P. D. Dahlberg, J. Niklas, O. G. Poluektov, K. L. Mulfort and L. M. Utschig, Ru-protein-Co biohybrids designed for solar hydrogen production: understanding electron transfer pathways related to photocatalytic function, Chem. Sci., 2016, 7(12), 7068–7078 RSC.
- I. Nath, J. Chakraborty and F. Verpoort, Metal organic frameworks mimicking natural enzymes: a structural and functional analogy, Chem. Soc. Rev., 2016, 45(15), 4127–4170 RSC.
- B. Kandemir, L. Kubie, Y. X. Guo, B. Sheldon and K. L. Bren, Hydrogen Evolution from Water under Aerobic Conditions Catalyzed by a Cobalt ATCUN Metallopeptide, Inorg. Chem., 2016, 55(4), 1355–1357 CrossRef CAS.
- B. Kandemir, S. Chakraborty, Y. X. Guo and K. L. Bren, Semisynthetic and Biomolecular Hydrogen Evolution Catalysts, Inorg. Chem., 2016, 55(2), 467–477 CrossRef CAS.
- S. C. Silver, J. Niklas, P. W. Du, O. G. Poluektov, D. M. Tiede and L. M. Utschig, Protein Delivery of a Ni Catalyst to Photosystem I for Light-Driven Hydrogen Production, J. Am. Chem. Soc., 2013, 135(36), 13246–13249 CrossRef CAS.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Nothl, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Spontaneous activation of FeFe-hydrogenases by an inorganic 2Fe active site mimic, Nat. Chem. Biol., 2013, 9(10), 607–609 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Biomimetic assembly and activation of FeFe-hydrogenases, Nature, 2013, 499(7456), 66–69 CrossRef CAS PubMed.
- R. Hidalgo, P. A. Ash, A. J. Healy and K. A. Vincent, Infrared Spectroscopy During Electrocatalytic Turnover Reveals the Ni-L Active Site State During H-2 Oxidation by a NiFe Hydrogenase, Angew. Chem., Int. Ed., 2015, 54(24), 7110–7113 CrossRef CAS.
- A. L. De Lacey, V. M. Fernandez, M. Rousset and R. Cammack, Activation and inactivation of hydrogenase function and the catalytic cycle: spectroelectrochemical studies, Chem. Rev., 2007, 107(10), 4304–4330 CrossRef CAS.
- B. Bleijlevens, F. A. van Broekhuizen, A. L. De Lacey, W. Roseboom, V. M. Fernandez and S. P. J. Albracht, The activation of the NiFe-hydrogenase from Allochromatium vinosum. An infrared spectro-electrochemical study, J. Biol. Inorg. Chem., 2004, 9(6), 743–752 CrossRef CAS.
- M. Y. Darensbourg, E. J. Lyon and J. J. Smee, The bio-organometallic chemistry of active site iron in hydrogenases, Coord. Chem. Rev., 2000, 206, 533–561 CrossRef.
- K. A. Bagley, E. C. Duin, W. Roseboom, S. P. J. Albracht and W. H. Woodruff, Infrared-detectable groups sense changes in charge-density on the nickel center in hydrogenase from Chromatium vinosum, Biochemistry, 1995, 34(16), 5527–5535 CrossRef CAS.
- K. A. Bagley, C. J. Vangarderen, M. Chen, E. C. Duin, S. P. J. Albracht and W. H. Woodruff, Infrared studies on the interaction of carbon-monoxide with divalent nickel in hydrogenase from Chromatium-vinosum, Biochemistry, 1994, 33(31), 9229–9236 CrossRef CAS.
- P. A. Ash, R. Hidalgo and K. A. Vincent, Proton Transfer in the Catalytic Cycle of NiFe Hydrogenases: insight from Vibrational Spectroscopy, ACS Catal., 2017, 7(4), 2471–2485 CrossRef CAS.
- S. Katz, J. Noth, M. Horch, H. S. Shafaat, T. Happe, P. Hildebrandt and I. Zebger, Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution, Chem. Sci., 2016, 7(11), 6746–6752 RSC.
- M. Horch, J. Schoknecht, M. A. Mroginski, O. Lenz, P. Hildebrandt and I. Zebger, Resonance Raman Spectroscopy on NiFe Hydrogenase Provides Structural Insights into Catalytic Intermediates and Reactions, J. Am. Chem. Soc., 2014, 136(28), 9870–9873 CrossRef CAS PubMed.
- E. Siebert, M. Horch, Y. Rippers, J. Fritsch, S. Frielingsdorf, O. Lenz, F. V. Escobar, F. Siebert, L. Paasche, U. Kuhlmann, F. Lendzian, M. A. Mroginski, I. Zebger and P. Hildebrandt, Resonance Raman Spectroscopy as a Tool to Monitor the Active Site of Hydrogenases, Angew. Chem., Int. Ed., 2013, 52(19), 5162–5165 CrossRef CAS PubMed.
- H. Ogata, T. Kramer, H. X. Wang, D. Schilter, V. Pelmenschikov, M. van Gastel, F. Neese, T. B. Rauchfuss, L. B. Gee, A. D. Scott, Y. Yoda, Y. Tanaka, W. Lubitz and S. P. Cramer, Hydride bridge in NiFe-hydrogenase observed by nuclear resonance vibrational spectroscopy, Nat. Commun., 2015, 6, 7890 CrossRef CAS PubMed.
- G. E. Vansuch, C. H. Wu, D. K. Haja, S. A. Blair, B. Chica, M. K. Johnson, M. W. W. Adams and R. B. Dyer, Metal-ligand cooperativity in the soluble hydrogenase-1 fromPyrococcus furiosus, Chem. Sci., 2020, 11(32), 8572–8581 RSC.
- M. L. K. Sanchez, S. E. Konecny, S. M. Narehood, E. J. Reijerse, W. Lubitz, J. A. Birrell and R. B. Dyer, The Laser-Induced Potential Jump: A Method for Rapid Electron Injection into Oxidoreductase Enzymes, J. Phys. Chem. B, 2020, 124(40), 8750–8760 CrossRef CAS PubMed.
- M. L. K. Sanchez, C. Sommer, E. Reijerse, J. A. Birrell, W. Lubitz and R. B. Dyer, Investigating the Kinetic Competency of CrHydA1 FeFe Hydrogenase Intermediate States via Time-Resolved Infrared Spectroscopy, J. Am. Chem. Soc., 2019, 141(40), 16064–16070 CrossRef CAS PubMed.
- B. L. Greene, G. E. Vansuch, B. C. Chica, M. W. W. Adams and R. B. Dyer, Applications of Photogating and Time Resolved Spectroscopy to Mechanistic Studies of Hydrogenases, Acc. Chem. Res., 2017, 50(11), 2718–2726 CrossRef CAS.
- B. L. Greene, G. J. Schut, M. W. W. Adams and R. B. Dyer, Pre-Steady-State Kinetics of Catalytic Intermediates of an FeFe-Hydrogenase, ACS Catal., 2017, 7(3), 2145–2150 CrossRef CAS.
- B. L. Greene, G. E. Vansuch, C. H. Wu, M. W. W. Adams and R. B. Dyer, Glutamate Gated Proton-Coupled Electron Transfer Activity of a NiFe-Hydrogenase, J. Am. Chem. Soc., 2016, 138(39), 13013–13021 CrossRef CAS.
- B. L. Greene, C. H. Wu, P. M. McTernan, M. W. W. Adams and R. B. Dyer, Proton-Coupled Electron Transfer Dynamics in the Catalytic Mechanism of a NiFe-Hydrogenase, J. Am. Chem. Soc., 2015, 137(13), 4558–4566 CrossRef CAS PubMed.
- B. L. Greene, C. A. Joseph, M. J. Maroney and R. B. Dyer, Direct Evidence of Active-Site Reduction and Photodriven Catalysis in Sensitized Hydrogenase Assemblies, J. Am. Chem. Soc., 2012, 134(27), 11108–11111 CrossRef CAS.
- N. T. Hunt, Transient 2D-IR spectroscopy of inorganic excited states, Dalton Trans., 2014, 43(47), 17578–17589 RSC.
- N. T. Hunt, 2D-IR spectroscopy: ultrafast insights into biomolecule structure and function, Chem. Soc. Rev., 2009, 38(7), 1837–1848 RSC.
- P. Hamm and M. T. Zanni, Concepts and methods of 2D infrared spectroscopy, Cambridge University Press, 2011 Search PubMed.
- M. Khalil, N. Demirdoven and A. Tokmakoff, Coherent 2D IR spectroscopy: molecular structure and dynamics in solution, J. Phys. Chem. A, 2003, 107(27), 5258–5279 CrossRef CAS.
- M. C. Asplund, M. T. Zanni and R. M. Hochstrasser, Two-dimensional infrared spectroscopy of peptides by phase-controlled femtosecond vibrational photon echoes, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(15), 8219–8224 CrossRef CAS.
- P. Hamm, M. H. Lim and R. M. Hochstrasser, Structure of the amide I band of peptides measured by femtosecond nonlinear-infrared spectroscopy, J. Phys. Chem. B, 1998, 102(31), 6123–6138 CrossRef CAS.
- M. Horch, J. Schoknecht, S. L. D. Wrathall, G. M. Greetham, O. Lenz and N. T. Hunt, Understanding the structure and dynamics of hydrogenases by ultrafast and two-dimensional infrared spectroscopy, Chem. Sci., 2019, 10(39), 8981–8989 RSC.
- H. Adamson, M. Robinson, J. J. Wright, L. A. Flanagan, J. Walton, D. Elton, D. J. Gavaghan, A. M. Bond, M. M. Roessler and A. Parkin, Retuning the Catalytic Bias and Overpotential of a NiFe-Hydrogenase via a Single Amino Acid Exchange at the Electron Entry/Exit Site, J. Am. Chem. Soc., 2017, 139(31), 10677–10686 CrossRef CAS.
- L. A. Flanagan, J. J. Wright, M. M. Roessler, J. W. Moir and A. Parkin, Re-engineering a NiFe hydrogenase to increase the H-2 production bias while maintaining native levels of O-2 tolerance, Chem. Commun., 2016, 52(58), 9133–9136 RSC.
- A. L. DeLacey, V. M. Fernandez, M. Rousset, C. Cavazza and E. C. Hatchikian, Spectroscopic and kinetic characterization of active site mutants of Desulfovibrio fructosovorans Ni-Fe hydrogenase, J. Biol. Inorg. Chem., 2003, 8(1-2), 129–134 CrossRef CAS.
- G. M. Greetham, P. Burgos, Q. A. Cao, I. P. Clark, P. S. Codd, R. C. Farrow, M. W. George, M. Kogimtzis, P. Matousek, A. W. Parker, M. R. Pollard, D. A. Robinson, Z. J. Xin and M. Towrie, ULTRA: A Unique Instrument for Time-Resolved Spectroscopy, Appl. Spectrosc., 2010, 64(12), 1311–1319 CrossRef CAS.
- R. M. Evans, P. A. Ash, S. E. Beaton, E. J. Brooke, K. A. Vincent, S. B. Carr and F. A. Armstrong, Mechanistic Exploitation of a Self-Repairing, Blocked Proton Transfer Pathway in an O-2-Tolerant NiFe-Hydrogenase, J. Am. Chem. Soc., 2018, 140(32), 10208–10220 CrossRef.
- S. F. A. Kettle, G. L. Aschero, E. Diana, R. Rossetti and P. L. Stanghellini, The vibrational spectra of the cyanide ligand revisited: terminal cyanides, Inorg. Chem., 2006, 45(13), 4928–4937 CrossRef CAS.
- K. G. V. Sigfridsson, N. Leidel, O. Sanganas, P. Chemev, O. Lenz, K. S. Yoon, H. Nishihara, A. Parkin, F. A. Armstrong, S. Dementin, M. Rousset, A. L. De Lacey and M. Haumann, Structural differences of oxidized iron-sulfur and nickel-iron cofactors in O-2-tolerant and O-2-sensitive hydrogenases studied by X-ray absorption spectroscopy, Biochim. Biophys. Acta, Bioenerg., 2015, 1847(2), 162–170 CrossRef CAS.
- A. Volbeda, C. Damault, A. Parkin, F. Sargent, F. A. Armstrong and J. C. Fontecilla-Camps, Crystal Structure of the O-2-Tolerant Membrane-Bound Hydrogenase 1 from Escherichia coli in Complex with Its Cognate Cytochrome b, Structure, 2013, 21(1), 184–190 CrossRef CAS.
- K. A. Vincent, A. Parkin, O. Lenz, S. P. J. Albracht, J. C. Fontecilla-Camps, R. Cammack, B. Friedrich and F. A. Armstrong, Electrochemical definitions of O-2 sensitivity and oxidative inactivation in hydrogenases, J. Am. Chem. Soc., 2005, 127(51), 18179–18189 CrossRef CAS.
- S. Hume, G. Hithell, G. M. Greetham, P. M. Donaldson, M. Towrie, A. W. Parker, M. J. Baker and N. T. Hunt, Measuring proteins in H2O with 2D-IR spectroscopy, Chem. Sci., 2019, 10(26), 6448–6456 RSC.
- E. B. Dunkelberger, M. Grechko and M. T. Zanni, Transition Dipoles from 1D and 2D Infrared Spectroscopy Help Reveal the Secondary Structures of Proteins: Application to Amyloids, J. Phys. Chem. B, 2015, 119(44), 14065–14075 CrossRef CAS PubMed.
- J. R. Zheng, K. Kwak, J. Asbury, X. Chen, I. R. Piletic and M. D. Fayer, Ultrafast dynamics of solute-solvent complexation observed at thermal equilibrium in real time, Science, 2005, 309(5739), 1338–1343 CrossRef CAS.
- R. P. Happe, W. Roseboom, A. J. Pierik, S. P. J. Albracht and K. A. Bagley, Biological activation of hydrogen, Nature, 1997, 385(6612), 126 CrossRef CAS PubMed.
- V. Cervetto, J. Helbing, J. Bredenbeck and P. Hamm, Double-resonance versus pulsed Fourier transform two-dimensional infrared spectroscopy: an experimental and theoretical comparison, J. Chem. Phys., 2004, 121(12), 5935–5942 CrossRef CAS.
- O. Golonzka, M. Khalil, N. Demirdoven and A. Tokmakoff, Coupling and orientation between anharmonic vibrations characterized with two-dimensional infrared vibrational echo spectroscopy, J. Chem. Phys., 2001, 115(23), 10814–10828 CrossRef CAS.
- C. H. Lai, W. Z. Lee, M. L. Miller, J. H. Reibenspies, D. J. Darensbourg and M. Y. Darensbourg, Responses of the Fe(CN)(2)(CO) unit to electronic changes as related to its role in NiFe hydrogenase, J. Am. Chem. Soc., 1998, 120(39), 10103–10114 CrossRef CAS.
- D. J. Darensbourg, J. H. Reibenspies, C. H. Lai, W. Z. Lee and M. Y. Darensbourg, Analysis of an organometallic iron site model for the heterodimetallic unit of NiFe hydrogenase, J. Am. Chem. Soc., 1997, 119(33), 7903–7904 CrossRef CAS.
- A. J. Pierik, W. Roseboom, R. P. Happe, K. A. Bagley and S. P. J. Albracht, Carbon monoxide and cyanide as intrinsic ligands to iron in the active site of NiFe-hydrogenases – NiFe(CN)(2)CO, biology's way to activate H-2, J. Biol. Chem., 1999, 274(6), 3331–3337 CrossRef CAS.
- M. H. Cho, Ultrafast vibrational spectroscopy in condensed phases, Phys. Chem. Commun., 2002, 5, 40–58 RSC.
- K. Park, M. H. Cho, S. Hahn and D. Kim, Two-dimensional vibrational spectroscopy. II. Ab initio calculation of the coherent 2D infrared response function of CHCl3 and comparison with the 2D Raman response function, J. Chem. Phys., 1999, 111(9), 4131–4139 CrossRef CAS.
- S. Hahn, K. Park and M. Cho, Two-dimensional vibrational spectroscopy. I. Theoretical calculation of the nonlinear Raman response function of CHCl3, J. Chem. Phys., 1999, 111(9), 4121–4130 CrossRef CAS.
- S. Hahn, D. Kim and M. H. Cho, Nonlinear optical properties of the linear quadrupolar molecule: structure-function relationship based on a three-state model, J. Phys. Chem. B, 1999, 103(39), 8221–8229 CrossRef CAS.
- K. M. Slenkamp, M. S. Lynch, B. E. Van Kuiken, J. F. Brookes, C. C. Bannan, S. L. Daifuku and M. Khalil, Investigating vibrational anharmonic couplings in cyanide-bridged transition metal mixed valence complexes using two-dimensional infrared spectroscopy, J. Chem. Phys., 2014, 140(8), 084505 CrossRef PubMed.
- S. Kaziannis, J. A. Wright, M. Candelaresi, R. Kania, G. M. Greetham, A. W. Parker, C. J. Pickett and N. T. Hunt, The role of CN and CO ligands in the vibrational relaxation dynamics of model compounds of the FeFe-hydrogenase enzyme, Phys. Chem. Chem. Phys., 2011, 13(21), 10295–10305 RSC.
- M. Horch, L. Lauterbach, M. A. Mroginski, P. Hildebrandt, O. Lenz and I. Zebger, Reversible Active Site Sulfoxygenation Can Explain the Oxygen Tolerance of a NAD(+)-Reducing NiFe Hydrogenase and Its Unusual Infrared Spectroscopic Properties, J. Am. Chem. Soc., 2015, 137(7), 2555–2564 CrossRef CAS PubMed.
- J. T. King, M. R. Ross and K. J. Kubarych, Water-Assisted Vibrational Relaxation of a Metal Carbonyl Complex Studied with Ultrafast 2D-IR, J. Phys. Chem. B, 2012, 116(12), 3754–3759 CrossRef CAS PubMed.
- D. Weidinger, G. M. Sando and J. C. Owrutsky, Vibrational dynamics of metal cyanides, Chem. Phys. Lett., 2010, 489(4-6), 169–174 CrossRef CAS.
- D. Czurlok, J. Gleim, J. Lindner and P. Vohringer, Vibrational Energy Relaxation of Thiocyanate Ions in Liquid-to-Supercritical Light and Heavy Water. A Fermi's Golden Rule Analysis, J. Phys. Chem. Lett., 2014, 5(19), 3373–3379 CrossRef CAS.
- S. Park and M. D. Fayer, Hydrogen bond dynamics in aqueous NaBr solutions, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(43), 16731–16738 CrossRef CAS.
- K. Adamczyk, N. Simpson, G. M. Greetham, A. Gumiero, M. A. Walsh, M. Towrie, A. W. Parker and N. T. Hunt, Ultrafast infrared spectroscopy reveals water-mediated coherent dynamics in an enzyme active site, Chem. Sci., 2015, 6(1), 505–516 RSC.
- C. Ranasinghe, P. Pagano, P. J. Sapienza, A. L. Lee, A. Kohen and C. M. Cheatum, Isotopic Labeling of Formate Dehydrogenase Perturbs the Protein Dynamics, J. Phys. Chem. B, 2019, 123(49), 10403–10409 CrossRef CAS PubMed.
- S. Ramos, R. E. Horness, J. A. Collins, D. Haak and M. C. Thielges, Site-specific 2D IR spectroscopy: a general approach for the characterization of protein dynamics with high spatial and temporal resolution, Phys. Chem. Chem. Phys., 2019, 21(2), 780–788 RSC.
- P. Pagano, Q. Guo, C. Ranasinghe, E. Schroeder, K. Robben, F. Hase, H. P. Ye, K. Wickersham, A. Aspuru-Guzik, D. T. Major, L. Gakhar, A. Kohen and C. M. Cheatum, Oscillatory Active-Site Motions Correlate with Kinetic Isotope Effects in Formate Dehydrogenase, ACS Catal., 2019, 9(12), 11199–11206 CrossRef CAS PubMed.
- C. Ranasinghe, Q. Guo, P. J. Sapienza, A. L. Lee, D. M. Quinn, C. M. Cheatum and A. Kohen, Protein Mass Effects on Formate Dehydrogenase, J. Am. Chem. Soc., 2017, 139(48), 17405–17413 CrossRef CAS PubMed.
- H. Ishikawa, S. Kim, K. Kwak, K. Wakasugi and M. D. Fayer, Disulfide bond influence on protein structural dynamics probed with 2D-IR vibrational echo spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(49), 19309–19314 CrossRef PubMed.
- H. Ishikawa, I. J. Finkelstein, S. Kim, K. Kwak, J. K. Chung, K. Wakasugi, A. M. Massari and M. D. Fayer, Neuroglobin dynamics observed with ultrafast 2D-IR vibrational echo spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(41), 16116–16121 CrossRef CAS.
- A. I. Stewart, I. P. Clark, M. Towrie, S. K. Ibrahim, A. W. Parker, C. J. Pickett and N. T. Hunt, Structure and vibrational dynamics of model compounds of the FeFe-hydrogenase enzyme system via ultrafast two-dimensional infrared spectroscopy, J. Phys. Chem. B, 2008, 112(32), 10023–10032 CrossRef CAS PubMed.
- G. M. Bonner, A. R. Ridley, S. K. Ibrahim, C. J. Pickett and N. T. Hunt, Probing the effect of the solution environment on the vibrational dynamics of an enzyme model system with ultrafast 2D-IR spectroscopy, Faraday Discuss., 2010, 145, 429–442 RSC.
- P. A. Ash, S. E. T. Kendall-Price, R. M. Evans, S. B. Carr, A. R. Brasnett, S. Morra, J. S. Rowbotham, R. Hidalgo, A. J. Healy, G. Cinque, M. D. Frogley, F. A. Armstrong and K. A. Vincent, The crystalline state as a dynamic system: IR microspectroscopy under electrochemical control for a NiFe hydrogenase, Chem. Sci., 2021, 12(39), 12959–12970 RSC.
- H. L. Tai, L. Y. Xu, S. Inoue, K. Nishikawa, Y. Higuchi and S. Hirota, Photoactivation of the Ni-SIr state to the Ni-SIa state in NiFe hydrogenase: FT-IR study on the light reactivity of the ready Ni-SIr state and as-isolated enzyme revisited, Phys. Chem. Chem. Phys., 2016, 18(32), 22025–22030 RSC.
- A. L. deLacey, E. C. Hatchikian, A. Volbeda, M. Frey, J. C. FontecillaCamps and V. M. Fernandez, Infrared spectroelectrochemical characterization of the NiFe hydrogenase of Desulfovibrio gigas, J. Am. Chem. Soc., 1997, 119(31), 7181–7189 CrossRef CAS.
- M. Saggu, I. Zebger, M. Ludwig, O. Lenz, B. Friedrich, P. Hildebrandt and F. Lendzian, Spectroscopic Insights into the Oxygen-tolerant Membrane-associated NiFe Hydrogenase of Ralstonia eutropha H16, J. Biol. Chem., 2009, 284(24), 16264–16276 CrossRef CAS PubMed.
- G. Caserta, C. Lorent, V. Pelmenschikov, J. Schoknecht, Y. Yoda, P. Hildebrandt, S. P. Cramer, I. Zebger and O. Lenz, In Vitro Assembly as a Tool to Investigate Catalytic Intermediates of NiFe-Hydrogenase, ACS Catal., 2020, 10(23), 13890–13894 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, M. Saggu, P. Hildebrandt, F. Lendzian, R. Bittl, O. Lenz and I. Zebger, Probing the Active Site of an O-2-Tolerant NAD(+)-Reducing NiFe-Hydrogenase from Ralstonia eutropha H16 by In Situ EPR and FTIR Spectroscopy, Angew. Chem., Int. Ed., 2010, 49(43), 8026–8029 CrossRef CAS PubMed.
- J. T. King, J. M. Anna and K. J. Kubarych, Solvent-hindered intramolecular vibrational redistribution, Phys. Chem. Chem. Phys., 2011, 13(13), 5579–5583 RSC.
- J. T. King, C. R. Baiz and K. J. Kubarych, Solvent-Dependent Spectral Diffusion in a Hydrogen Bonded “Vibrational Aggregate”, J. Phys. Chem. A, 2010, 114(39), 10590–10604 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the preparation and characterization of the enzyme samples along with supplemental figures supporting spectral assignment and analysis. See DOI: https://doi.org/10.1039/d2cp04188j |
| ‡ These authors contributed equally to the work. |
| This journal is © the Owner Societies 2022 |