
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
syn-Cryptophanes: macrocyclic compounds with optimized characteristics for the design of 129Xe NMR-based biosensors†
Estelle
Léonce
a,
Thierry
Brotin
b and
Patrick
Berthault
 *a
*a
aNIMBE, CEA, CNRS, Université de Paris Saclay, CEA Saclay, 91191 Gif-sur-Yvette, France. E-mail: patrick.berthault@cea.fr
bENSL, CNRS, Laboratoire de Chimie UMR 5182, 46 allée d’Italie, 69364 Lyon, France
First published on 30th September 2022
Abstract
A new water-soluble xenon host system with great promise for the 129Xe NMR-based biosensing approach is presented: the syn-cryptophane-222-hexacarboxylate. It compares favorably with its already known anti diastereomer, on the one hand, and with cucurbit[6]uril, on the other hand, in particular in terms of xenon binding constant and xenon in–out exchange, a key parameter for the efficiency of the most sensitive HyperCEST method.
Introduction
In an approach pioneered by the group of Pines in 2001,1 spin hyperpolarized xenon is encapsulated in molecular systems functionalized with ligands in order to target specific biological receptors. Xenon reversibly complexed in these host molecules has a specific chemical shift and an in–out exchange that gives the method a high sensitivity. All this combines to constitute a powerful NMR molecular imaging tool.2,3Since the first studies in this field, cryptophanes, cage molecules consisting of two cyclotribenzylene (CTB) groups linked by three alkoxy chains, have been most often used in this purpose. A wide range of applications was covered, concerning the targeting of biological receptors4–7 (here fluorescence detection usually accompanies 129Xe NMR) or small molecules such as metal cations,8–10 biothiols,11 detection of temperature12 or pH variations.13,14 Water-soluble cryptophanes have been synthesized, based on the structure of anti-cryptophane-A, among which is compound 1 shown in Fig. 1.15 But the use of cryptophane derivatives in this approach was questioned by some researchers, due to the difficulty of their synthesis, their poor solubility in aqueous media, the presence of enantiomers and also the fact that the xenon in–out exchange rate was lower than with other host systems. Indeed this dynamic parameter is key in the sensitivity of the detection method based on Chemical Exchange Saturation Transfer (Hyper-CEST).16 For instance, cucurbit[6]uril derivatives were preferred to cryptophanes despite the difficulty of functionalizing them with biological ligands.17–20 Native cucurbit[6]uril (compound 3 of Fig. 1) has even been chosen as a proof-of-concept for in vivo detection.21,22
 | ||
| Fig. 1 Xenon hosts used in this study. 1: anti-cryptophane-222 hexa carboxylate; 2: syn-cryptophane-222-hexacarboxylate. 3: cucurbit[6]uril. | ||
However, the synthesis of syn-cryptophane-B (the diastereomeric molecule of cryptophane-A) has recently been achieved.23 Interestingly, this compound opens up new possibilities for designing water-soluble molecular receptors for xenon after appropriate modification of its backbone. Thus, from the synthesis of syn-cryptophane-B, many different derivatives can be produced. The water-soluble compound 2, shown in Fig. 1, is an example among others and is the diastereomer of compound 1 which has a good affinity for xenon in aqueous solution. The synthetic pathway for cryptophane 2 and its characterization as well as this of the intermediates is given in the ESI† (Fig. S1–S11). Unlike other syn-cryptophanes (except for those containing nitrogen groups on the linkers or as aromatic substituents24,25), xenon in the presence of compound 2 exhibits a slow exchange situation at 11.7 T and room temperature, i.e. the xenon exchange in and out of the cryptophane cavity is slower than the xenon resonance frequency difference between these environments.
Fig. 2 displays one-scan 129Xe NMR spectra recorded with laser-polarized gas dissolved in a quasi-equimolar solution mixture of compounds 1 and 2. After calibration of the free dissolved xenon signal at 196 ppm, while xenon caged in 1 (Xe@1) resonates near 65 ppm in agreement with ref. 15, a third signal at 116 ppm shows the encapsulation of xenon in 2 (Xe@2, as confirmed in Fig. S12, ESI†). The larger linewidth for Xe@2 than for Xe@1 is a first indication of a higher xenon in–out exchange rate. This prompted us to evaluate comparatively with other xenon hosts the performance of this new member of the syn-cryptophane family as a basis for 129Xe NMR-based biosensors.
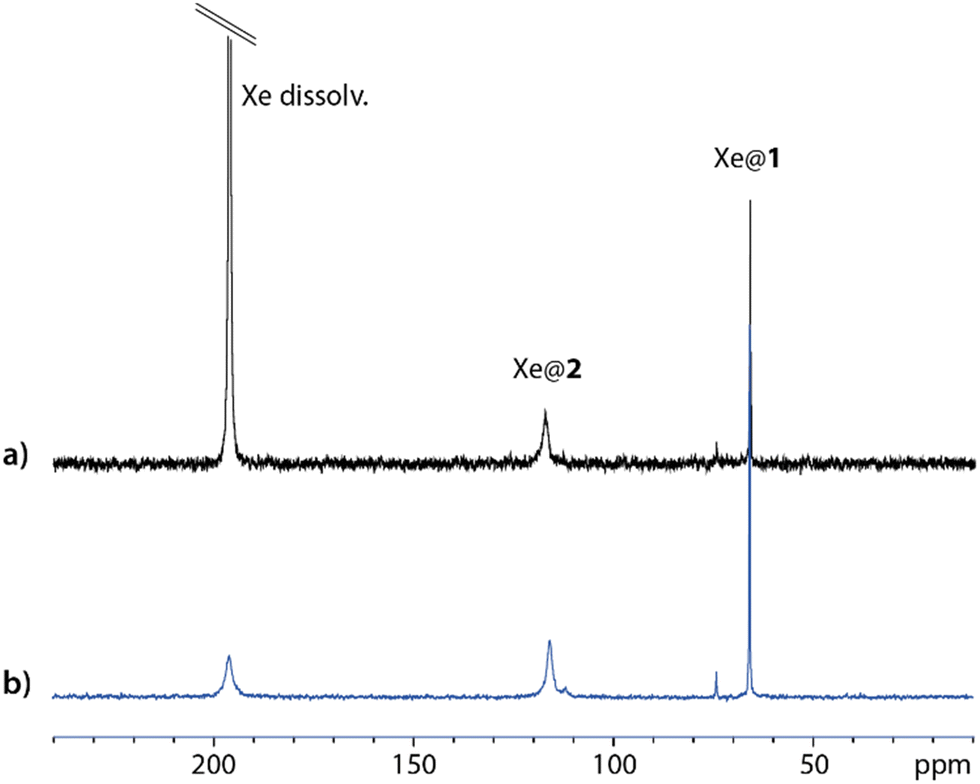 | ||
| Fig. 2 One-scan 129Xe NMR spectra obtained on a mixture of compounds 1 (0.51 mM) and 2 (0.49 mM) in D2O + εNaOD, at (a) high xenon concentration, (b) low xenon concentration (sum of three experiments). B0 = 11.7 T; T = 298 K. | ||
Results and discussion
Introducing low amounts of xenon inside the solution – the situation displayed in Fig. 2b – avoids saturation of the xenon cages, and thus opens the way to estimation of the affinity constant. In this purpose, comparison is made with cryptophane 1, whose binding constant with xenon has been reported to 6800 M−1 at 298 K.15Fig. 2b displays the hyperpolarized 129Xe spectrum for a mixture with equimolar concentrations of cryptophanes. The respective integrals of the caged xenon signals are very close, revealing that the affinity constants are similar. In any case they are far higher than the binding constant of cucurbit[6]uril with xenon, reported to 210 M−1.17 This parameter dictates the instantaneous occupancy rate of the host molecule in xenon, and such high values for compounds 1 and 2 ensure that there are not too many guest competitors in solution. This competition effect can be detrimental for in vivo experiments, as already noticed by McHugh et al. in the case of cucurbituril derivatives.22But other factors play into the performance of a xenon host for potential 129Xe NMR-based biosensors. When dissolving as is cryptophane 1 in water, the canonical crown–crown conformation is not the only form present as it coexists with crown-saddle forms (see Fig. S13b of the ESI† and ref. 15). Strong bubbling during several hours (for instance with helium) or pressurization of the NMR sample with several bars of xenon during days was shown necessary to recover the canonical form.
This shows that the non-canonical forms are not prone to encapsulate xenon when it is present in low concentration, which can be detrimental to the biosensing approach. At the contrary, cryptophane 2 dissolved in water exhibits a very simple 1H spectrum (see Fig. S13a of the ESI†), expected for the canonical form of C3h symmetry.
Note that non-canonical forms are also observed in the preparation of intermediates required for the synthesis of the anti-1 compound. This therefore considerably complicates the synthesis and purification of derivatives of anti configuration and biosensors made from these compounds. Thus, the absence of non-canonical form with syn-2 and its congeners seems to be an asset for the preparation of new NMR-based biosensors.
Also, from the comparison of the 1H NMR spectra of an equimolar mixture of cryptophanes 1 and 2 recorded just after dissolution, after solution degassing and after introduction of xenon (Fig. 3), several conclusions can be drawn. First, the signals that are the most affected – the most sharpened – by the degassing belong to cryptophane 1. This effect, particularly observable on the aromatic protons, indicates that cryptophane 1 must complex paramagnetic oxygen molecules dissolved in water. To check this hypothesis, in a separate experiment we have added pure oxygen gas into NMR tubes containing degassed solutions of either cryptophane 1, cryptophane 2 or a mixture of cryptophanes 1 and 2 (Fig. S14 and S15 of the ESI†). Comparison of the 1H spectra confirms the specific complexation of dissolved oxygen by cryptophane 1. This is neither to be the case for 2, not for cucurbit[6]uril 3, while cucurbit[5]uril has been shown to slowly but strongly trap oxygen.26
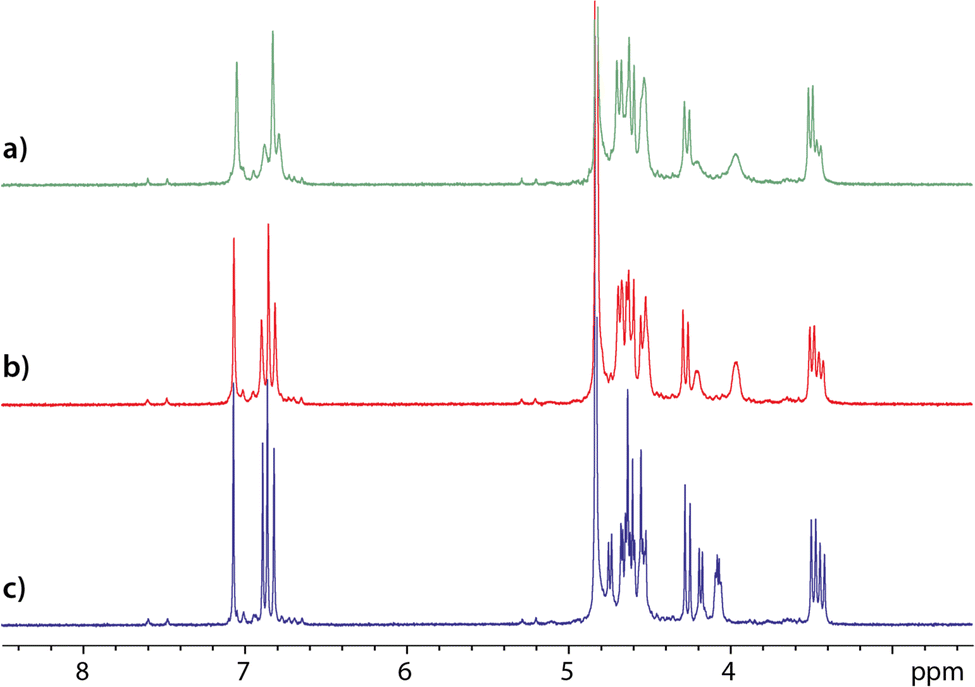 | ||
| Fig. 3 1H NMR spectrum of a mixture of cryptophanes 1 and 2 in D2O (B0 = 11.7 T; T = 298 K). (a) as is; (b) after having degassed the solution; (c) after addition of ca. 5 mM of xenon. Concentration of 1 = 51 μM; concentration of 2 = 49 μM. Small signals visible on spectra (a–c) are characteristic of the ‘non-canonical’ forms of compound 1 (see text). | ||
The addition of xenon in solution further sharpens the 1H NMR signals, but in an equivalent way for both cryptophanes. In the last spectrum displayed in Fig. 3c, thus even after solution degassing and addition of xenon, the relative integrals of the aromatic protons reveal that only ca. 75% of the cryptophane 1 is in the canonical crown–crown conformation. On this spectrum, notice also the narrowing of the linker proton signals (near 4 ppm) for both cryptophanes when xenon is present. This undoubtedly indicates a modification of the dynamics of these linkers and an adaptation of the host molecule structure to xenon.
Using a dedicated pulse sequence, we have assessed the detectability of each compound via indirect detection based on the xenon in–out exchange. In the so-called HyperCEST approach,27 the intensity of the signal of free xenon is observed after saturation at a given frequency. If the saturation is applied near the resonance of the caged xenon frequency, the magnetization decreases quickly due to the exchange, and depletion of the main signal is observed. With a saturation of strength ω1 = γB1 applied exactly on-resonance, the depolarization rate is given by:28,29
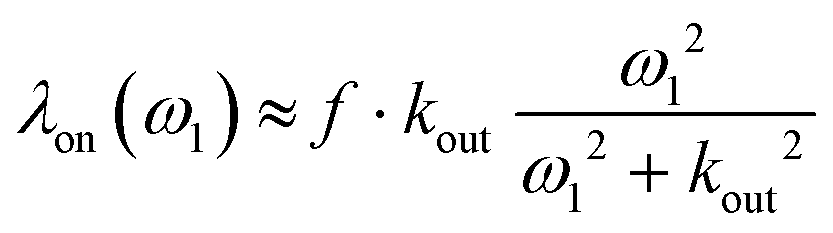 | (1) |
But directly quantifying the detectability of a host through the HyperCEST depolarization rate is a difficult task. Indeed in the case of cryptophanes and cucurbiturils the xenon in–out exchange rate depends on the guest/host concentration ratio, as a degenerate or kick-out exchange has been shown to occur.30 The concentration of dissolved xenon being tricky to precisely determine, a large uncertainty may be introduced. Thus, we have preferred to use competition experiments with xenon host mixtures, and a dedicated pulse sequence using a fixed B1 saturation simply placed at various frequency offsets (see Experimental). The limitation of this method is the prior knowledge of the Xe@host resonance frequencies (observable on 129Xe spectra only at the highest concentrations), since at these low B1 field amplitudes the saturation offset effect is important. Obviously a more sophisticated and accurate method such as the one proposed recently by Mitschang et al.31 could have been used, but here the purpose is just a comparison between xenon hosts.
Fig. 4 displays the depolarization curves obtained for cryptophanes 1 and 2 with concentrations ranging from 5 μM to 50 nM in D2O, using a saturation field strength of B1 = 4.2 μT and a temperature of 298 K.
 | ||
| Fig. 4 Example of evolution curves of the 129Xe magnetization after saturation at the frequency of Xe@2 (left) and Xe@1 (right) as a function of saturation time, for different cryptophane concentrations: (a) 5 μM, (b) 500 nM, (c) 50 nM, in D2O. (a) Left: Note that due to a very fast magnetization loss with 2 at 5 μM, only the 28 first data points have been kept. In (c)-left, the data points obtained with off-resonance saturation (see text) and the corresponding best-fit curve are superimposed in blue. | ||
In order to perform the experiments in a more biological medium new cryptophane solutions have been prepared in PBS (phosphate buffer saline). Table 1 gives the corresponding xenon depolarization rates for various concentrations of hosts 1 and 2. For each concentration, the depolarization rate is faster for Xe@2 than for Xe@1 by a factor between 3.5 and 5.9.
| Concentration (nM) | Depolarization rate Xe@1 (s−1) | Depolarization rate Xe@2 (s−1) |
|---|---|---|
| 5000 | 0.3552 ± 0.0190 | 0.8236 ± 0.0658 |
| 2500 | 0.1146 ± 0.0021 | 0.4074 ± 0.0231 |
| 500 | 0.0373 ± 0.0030 | 0.2453 ± 0.0422 |
| 250 | 0.0435 ± 0.0042 | 0.2559 ± 0.0161 |
| 50 | 0.0268 ± 0.0004 | 0.1069 ± 0.0032 |
In a last step, we aimed to assess the detectability via129Xe NMR of cucurbit[6]uril 3, using the same study protocol. Xenon in cucurbit[6]uril resonates at 127 ppm (see Fig. S16 of the ESI†), so it was not obvious to compare it with the syn cryptophane 2 because of the proximity of the resonance frequencies of xenon inside these two hosts (462 Hz). We instead turned to a 1–3 comparison. But we encountered several difficulties. First, while cryptophanes solubilize in water at slightly basic pH, cucurbiturils require the addition of cations such as Na2SO4.32 Our first attempts to use a mother solution and successively dilute in D2O gave unexpected results that precluded any quantitative interpretation. Fig. S17 of the ESI† shows the low field region of the 1H NMR spectrum of a mixture of compound 1 (3.9 μM) and compound 3 (3.3 μM) and after dilution in D2O by a factor 2. Clearly, while as expected a ratio of 2 is observed for the cryptophane, the 1H signals of the curcurbituril vanish. This is probably due to the subsequent dilution of Na2SO4 and a too low ionic strength which favors the formation of cucurbit[6]uril aggregates.
Keeping constant the concentration of Na2SO4 in order to maintain a sufficiently high ionic strength for cucurbit[6]uril was questionable, as carboxylic acid (also present with carboxylate forms in the experimental conditions used) and sulfate groups are known to interact,33 which could become problematic for the cryptophane. Thus, we turned to solutions in PBS, as previously for comparison between 1 and 2. This solved the problem of the cucurbituril solubility and thereby enabled us to prepare solutions of equimolar concentrations of 1 and 3 at different dilutions (ESI,† Fig. S18).
While for comparison between the cryptophanes 1 and 2 the cage occupancy by xenon (the f term in eqn (1)) is almost the same, and thus λon directly derives from kout, it is not the same situation for comparison between 1 and 3. The xenon binding constant lower with cucurbit[6]uril than for cryptophanes makes that the occupancy factors can be quite different.
When the xenon host concentration diminishes, the f factor diminishes, while, due to the degenerate exchange, kout increases. From eqn (1), Kunth and co-workers34 distinguish two limit cases:
| Smatrong saturation (ω1 ≫ f·kout): λon ≈ f·kout | (2) |
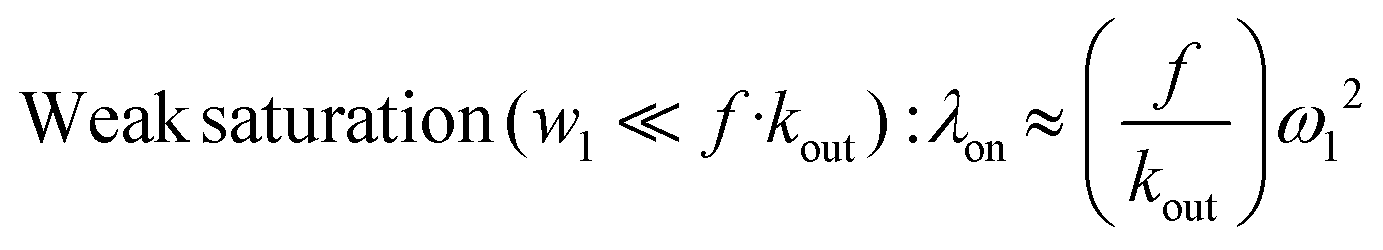 | (3) |
Table 2 gives the depolarization rates measured for xenon in anti-cryptophane 1 and cucurbit[6]uril 3 at different concentrations in PBS. Systematically, the depolarization rate is higher for Xe@3 than for Xe@1 by a factor between 1.8 and 2.6.
| Concentration (nM) | Depolarization rate Xe@1 (s−1) | Depolarization rate Xe@3 (s−1) |
|---|---|---|
| 4000 | 0.3479 ± 0.0243 | 0.6175 ± 0.0349 |
| 800 | 0.0601 ± 0.0113 | 0.1366 ± 0.0248 |
| 400 | 0.0428 ± 0.0062 | 0.1112 ± 0.0136 |
| 200 | 0.0206 ± 0.0011 | 0.0508 ± 0.0029 |
Experimental
Synthesis of compound syn-2
The compound syn-2 was prepared in four steps from the known syn-cryptophane-4 (C3-symmetry) decorated with three hydroxyl functions on one CTB cap and three protected hydroxyl groups on the other CTB cap (see Fig. S1 of the ESI†). Three ester functions were first introduced by reacting compound syn-4 with an excess of methyl bromoacetate at 60 °C in the presence of a base in DMF to give rise to compound syn-5 in 68% yield. Removal of the three benzyl groups was then achieved by reacting syn-5 in the presence of a palladium catalyst with hydrogen gas in a CH2Cl2/CH3OH mixture. The procedure provided compound syn-6 in quantitative yield. Three additional ester groups were then grafted on the cryptophane skeleton with 82% yield by reacting syn-6 in the presence of an excess of methyl bromoacetate as reported for the preparation of compound syn-5. Finally, the resulting syn-7 derivative was subjected to hydrolysis under basic conditions followed by acidification with concentrated HCl to give the expected syn-2 compound in 79% yield (isolated). It was noticed that compound syn-2 shows lower solubility in DMSO and water than its congener anti-1 under similar conditions. Compound syn-2 (C3h symmetry) was fully characterized by 1H, 13C NMR spectroscopy (see Fig. S2–S4 of the ESI†) and HRMS. 1H NMR (500 MHz, D2O, 298 K): d 7.04 (s, 6H), 6.83 (s, 6H), 4.61 (d, 6H, J = 14 Hz), 4.57 (d, 6H, J = 15 Hz), 4.50 (q, 6H, J = 10 Hz, J = 4 Hz), 4.19 (d, 6H, J = 15 Hz), 4.01 (q, 6H, J = 10 Hz, J = 4 Hz), 3.43 (d, 6H, J = 14 Hz). 13C{1H} NMR (125.7 MHz, D2O, 298 K): d 178.0 (6C), 148.2 (6C), 146.0 (6C), 136.3 (6C), 134.7 (6C), 119.7 (6C), 116.0 (6C), 69.5 (6C), 69.2 (6C), 36.5 (6C). HRMS (ESI) m/z: [M + Na]+ calcd for C60H54NaO24: 1181.2897; found: 1181.2892.Preparation of the xenon host solutions for NMR
For the 1H–13C HSQC and HMBC experiments, 4.48 mg of syn-cryptophane 2 were dissolved in 600 μL D2O and 20 μL NaOD 0.1 M. In a second step, for the competition experiments in water, stock solutions were prepared. For the first one, 0.71 mg of anti-cryptophane 1 was dissolved in 600 μL D2O with 3 μL NaOD 0.1 M. For the second one, 0.69 mg of syn-cryptophane 2 was dissolved in 600 μL D2O with 3 μL NaOD 0.1 M. 300 μL of each solution was then added in a tube to provide the mother solution of the cryptophane mixture. Then successive dilutions gave the concentrations wished for the 129Xe depolarization experiments. Thirdly, for the study of the xenon hosts in a more biological medium, they have been dissolved in a PBS buffer made of 20 mM sodium phosphate + 150 mM NaCl in D2O for a pH of 7.4. 0.81 mg of anti-cryptophane 1 was dissolved in 600 μL of the same buffer, and 0.84 mg of syn-cryptophane 2 was dissolved in 600 μL of PBS. Then successive dilutions gave the concentrations wished for the 129Xe depolarization experiments.For the competition experiments with cucurbit[6]uril 3, a mother solution was prepared by dissolving 0.42 mg of this molecule in 600 μL of PBS buffer.
Introduction of hyperpolarized xenon
All the 129Xe NMR spectra and all the 129Xe exchange experiments were performed with hyperpolarized xenon. Laser-polarized xenon was produced in the batch mode, using our home-built apparatus described in ref. 35. Due to issues related to the laser source the current average polarization was only ca. 5%. Then it was transported in the frozen state to the NMR spectrometer and transferred via a vacuum line in the fringe field of the magnet to the NMR tube equipped with a screw-cap and a J. Young valve. In this purpose the NMR tube was previously degassed and a cold point was created by the use of a dedicated hollow spinner. In this way, xenon condensed on top of the solution, without significantly cooling it. The NMR tube was then vigorously shaken in order to speed up the xenon dissolution. A delay of 10 seconds before the acquisition was systematically set. The amount of xenon inside the NMR tube was estimated by weighting the tube after degassing and after xenon introduction.1H, 13C and 129Xe NMR experiments
Except otherwise indicated, all NMR experiments were performed at 11.7 T and 298 K with a Bruker 5 mm-broadband inverse probehead equipped with z gradient. The radiofrequency field strength on the 129Xe channel was calibrated using a reference tube containing molar concentration of xenon in dodecane.Measurement of the 129Xe depolarization rates
In this purpose a specific NMR pulse sequence, depicted in ESI† (Fig. S19), was conceived. This pseudo-2D sequence starts by a read pulse of small flip angle θ. Then after detection, which provides the first data point at time 0, saturation occurs at a chosen frequency. Saturation is achieved by one hundred repetitions (n = 100) of D-Snob pulses of 10 ms duration. Then, detection is achieved after a small flip angle read pulse. The pulse program loops 48 times (td1 = 48) on the ‘read pulse – detection – saturation’ sequence. For the sake of speed, there is no interscan delay and the data are only written on the disk at the end of the sequence. In order to enable a short acquisition time and thus a short interscan delay (34 ms), a magnetic field gradient of 2 mT m−1 is applied during detection, and a purge gradient of random amplitude is applied between each loop. According to the xenon host concentration, the saturation rf strength is adjusted between ν1 = 12.5 Hz (B1sat = 1.05 μT) and ν1 = 50 Hz (B1sat = 4.2 μT).Rigorously, three effects should be considered to account for the magnetization loss during the sequence: relaxation, rf pulse and exchange. Dealing with relaxation, considering that xenon relaxes in a similar rate in 1 and in 2, a term in exp(−t/T1eff) occurs, with 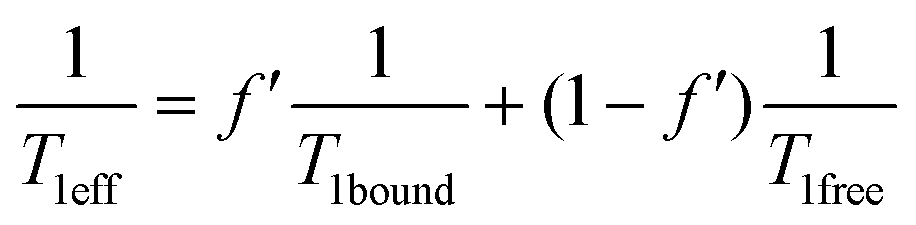 , where f′ is the fraction of bound xenon:
, where f′ is the fraction of bound xenon: 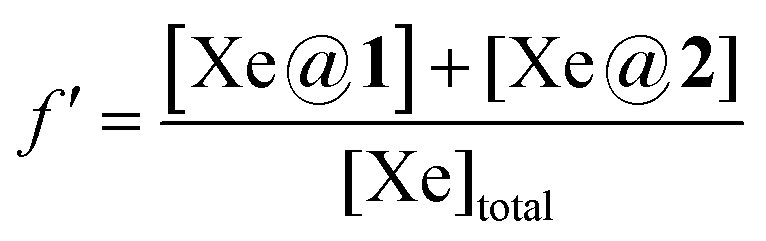 .
.
For the effect of the rf pulse, the theoretical curve for a pulse of 4° flip angle is a straight line of slope s = −0.00257 (cf. Fig. S20 of the ESI†). The exchange term finally is expressed as an exponential decay exp(−kext), where kex is the exchange rate. But one can consider that at current cryptophane concentrations used for the exchange experiments (10−5 to 10−8 M), and given the concentration of dissolved xenon (in the millimolar range), one can neglect the relaxation. Thus for the depolarization experiments the data points obey a recursive law in S(x) = (S(x − 1) + s)·exp(−kexΔt) with S(0) = 1. A value of 10958 Hz separates the frequency of xenon caged in 2 from that of free xenon (Δν2 = δfree − δXe@2 = −10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 958 Hz) and 17989 Hz separate the frequency of xenon caged in 1 from it (Δν1 = −17989 Hz). For each of the cryptophane concentration, in order to check that no cross-over effect can perturb the result, an ‘off-resonance’ experiment was performed in which saturation is applied at an offset symmetrical to Δν2 with respect to the frequency of free xenon (Δνoff = +10958 Hz). The same protocol (fit etc.) was used for comparison between cucurbit[6]uril 3 and anti-cryptophane 1. 9480 Hz separate the frequency of xenon complexed with 3 from that of free xenon (Δν3 = −9480 Hz). The depolarization rates given in Tables 1 and 2 are the means of three measures, with standard deviations.
958 Hz) and 17989 Hz separate the frequency of xenon caged in 1 from it (Δν1 = −17989 Hz). For each of the cryptophane concentration, in order to check that no cross-over effect can perturb the result, an ‘off-resonance’ experiment was performed in which saturation is applied at an offset symmetrical to Δν2 with respect to the frequency of free xenon (Δνoff = +10958 Hz). The same protocol (fit etc.) was used for comparison between cucurbit[6]uril 3 and anti-cryptophane 1. 9480 Hz separate the frequency of xenon complexed with 3 from that of free xenon (Δν3 = −9480 Hz). The depolarization rates given in Tables 1 and 2 are the means of three measures, with standard deviations.
Conclusions
Until now the synthesis of syn-cryptophanes with small cavities was challenging. But the advent of new methods, such as the use of 1,1,1,3,3,3-hexafluoroisopropanol as co-solvent and the recent synthesis of functionalized syn-cryptophanes with C3-symmetry,23,36 has facilitated their production and opens new doors, in particular for the conception of 129Xe NMR-based biosensors. Interestingly, the second approach seems well suited for the design of these biosensors since the starting material used for the synthesis of syn-cryptophane 2 has lower symmetry and reactive phenolic functions that can be directly engaged to graft water-soluble groups and ligands aimed at recognizing a biological target.This represents a major asset. Indeed, 129Xe NMR-based biosensors made from anti-cryptophanes usually require additional chemical transformations to introduce water-solubilizing substituents. Here, compounds 4 and 6 (see Fig. S1, ESI†) appear as ideal host molecules to prepare rapidly biosensors. Progress is underway to further improve the synthesis of these syn intermediates.
The newly synthesized syn-cryptophane 2 is the first member of a family of water-soluble xenon hosts with very singular characteristics. Its properties, as well as those of its complex with xenon, make it a very attractive candidate for 129Xe NMR-based biosensing. Firstly, as the resonance frequency of xenon in 2 differs drastically from that of xenon in 1 – more than 50 ppm (or 7000 Hz at 11.7 Tesla) – this can allow interesting multiplexing experiments,37 even at low magnetic field.38 Secondly, from a structural point of view, when dissolved in water it is exempt of forms not able to encapsulate xenon. In that it is superior to anti-1 and to several anti-cryptophanes in general. Thirdly, it does not complex dissolved oxygen, which represents an advantage over cryptophane anti-1. Fourthly, it shows a high binding constant with xenon, which means that there are less competitors for encapsulation in solution. For instance, although this has not been extensively tested, at the difference of cucurbit[6]uril 3, it does not complex anions. Note that while cryptophane 2 is slightly soluble in water at neutral pH, cucurbit[6]uril needs the addition of salts to become soluble. Last but not least, the xenon in–out exchange with syn-cryptophane 2 is faster than with anti-1, by a factor ca. 6 at low cryptophane concentration. Even if it was not a question here of trying to reach the lowest detection threshold (at lower cryptophane concentration we could use a stronger rf saturation without crossover effect), we have shown that its molar detectivity is much higher. All together the syn-2 compound and the syn-congeners in general appear to be an excellent basis for designing new 129Xe NMR-based biosensors with superior characteristics to its anti-congener, in particular an efficient xenon turnover. Obviously other systems such as gas vesicles are performing well for sensitive xenon detection,39 but with a view to building bioprobes composed of a xenon host and a recognition antenna, syn cryptophanes are very promising. In the context of the search for an ultimate detection they could constitute the elementary brick of dendrimers for example.40
Author contributions
T. B. synthesized the cryptophanes 1 and 2. P. B. conceived the 129Xe NMR approach to evaluate the exchange rate and wrote the pulse sequence. E. L. prepared the xenon host solutions. E. L. and P. B. performed the 129Xe NMR experiments and processed the data. P. B. wrote the initial version of the manuscript. All authors have given their approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The French National Research Agency (ANR) is acknowledged for financial support (Project ANR19-CE19 0024 PHOENIX).Notes and references
- M. M. Spence, S. M. Rubin, I. E. Dimitrov, E. J. Ruiz, D. E. Wemmer, A. Pines, S. Q. Yao, F. Tian and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10654–10657 CrossRef CAS PubMed.
- P. Berthault, G. Huber and H. Desvaux, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 55, 35–60 CrossRef CAS.
- J. Jayapaul and L. Schröder, Molecules, 2020, 25, 4627–4722 CrossRef CAS PubMed.
- C. Boutin, A. Stopin, F. Lenda, T. Brotin, J.-P. Dutasta, N. Jamin, A. Sanson, Y. Boulard, F. Leteurtre, G. Huber, A. Bogaert-Buchmann, N. Tassali, H. Desvaux, M. Carrière and P. Berthault, Bioorg. Med. Chem., 2011, 19, 4135–4143 CrossRef CAS PubMed.
- G. K. Seward, Y. Bai, N. S. Khan and I. J. Dmochowski, Chem. Sci., 2011, 2, 1103–1110 RSC.
- C. Witte, V. Martos, H. M. Rose, S. Reinke, S. Klippel, L. Schröder and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 2806–2810 CrossRef CAS.
- G. Milanole, B. Gao, A. Paoletti, G. Pieters, C. Dugave, E. Deutsch, S. Rivera, F. Law, J.-L. Perfettini, E. Mari, E. Léonce, C. Boutin, P. Berthault, H. Volland, F. Fenaille, T. Brotin and B. Rousseau, Bioorg. Med. Chem., 2017, 25, 6653–6660 CrossRef CAS.
- N. Tassali, N. Kotera, C. Boutin, E. Léonce, Y. Boulard, B. Rousseau, E. Dubost, F. Taran, T. Brotin, J.-P. Dutasta and P. Berthault, Anal. Chem., 2014, 86, 1783–1788 CrossRef CAS PubMed.
- K. Jeong, C. C. Slack, C. C. Vassiliou, P. Dao, M. D. Gomes, D. J. Kennedy, A. E. Truxal, L. J. Sperling, M. B. Francis, D. E. Wemmer and A. Pines, Chem. Phys. Chem., 2015, 16, 3573–3577 CrossRef CAS.
- Q. Guo, Q. Zeng, W. Jiang, X. Zhang, Q. Luo, X. Zhang, L.-S. Bouchard, M. Liu and X. Zhou, Chem. – Eur. J., 2016, 22, 3967–3970 CrossRef CAS PubMed.
- S. Yang, W. Jiang, L. Ren, Y. Yuan, B. Zhang, Q. Luo, Q. Guo, L.-S. Bouchard, M. Liu and X. Zhou, Anal. Chem., 2016, 88, 5835–5840 CrossRef CAS.
- F. Schilling, L. Schröder, K. K. Palaniappan, S. Zapf, D. E. Wemmer and A. Pines, Chem. Phys. Chem., 2010, 11, 3529–3533 CrossRef CAS PubMed.
- E. Léonce, J.-P. Dognon, D. Pitrat, J.-C. Mulatier, T. Brotin and P. Berthault, Chem. – Eur. J., 2018, 24, 6534–6537 CrossRef PubMed.
- P. Berthault, H. Desvaux, T. Wendlinger, M. Gyejacquot, A. Stopin, T. Brotin, J.-P. Dutasta and Y. Boulard, Chem. – Eur. J., 2010, 16, 12941–12946 CrossRef CAS.
- G. Huber, T. Brotin, L. Dubois, H. Desvaux, J.-P. Dutasta and P. Berthault, J. Am. Chem. Soc., 2006, 128, 6239–6246 CrossRef CAS PubMed.
- L. Schroder, T. J. Lowery, C. Hilty, D. E. Wemmer and A. Pines, Science, 2006, 314, 446–449 CrossRef PubMed.
- M. El Haouaj, M. Luhmer, Y. H. Ko, K. Kim and K. Bartik, J. Chem. Soc., Perkin Trans. 2, 2001, 804–807 RSC.
- B. S. Kim, Y. H. Ko, Y. Kim, H. J. Lee, N. Selvapalam, H. C. Lee and K. Kim, Chem. Commun., 2008, 2756–2758 RSC.
- J. A. Finbloom, C. C. Slack, C. J. Bruns, K. Jeong, D. E. Wemmer, A. Pines and M. B. Francis, Chem. Commun., 2016, 52, 3119–3122 RSC.
- Y. Wang and I. J. Dmochowski, Chem. Commun., 2015, 51, 8982–8985 RSC.
- F. T. Hane, T. Li, P. Smylie, R. M. Pellizzari, J. A. Plata, B. DeBoef and M. S. Albert, Sci. Rep., 2017, 7, 41027 CrossRef CAS.
- C. T. McHugh, M. Kelley, N. J. Bryden and R. T. Branca, Magn. Reson. Med., 2022, 87, 1480–1489 CrossRef PubMed.
- T. Brotin, E. Jeanneau, P. Berthault, E. Léonce, D. Pitrat and J.-C. Mulatier, J. Org. Chem., 2018, 83, 14465–14471 CrossRef CAS PubMed.
- M. Doll, P. Berthault, E. Léonce, C. Boutin, T. Buffeteau, N. Daugey, N. Vanthuyne, M. Jean, T. Brotin and N. De Rycke, J. Org. Chem., 2021, 86, 7648–7658 CrossRef CAS PubMed.
- M. Doll, P. Berthault, E. Léonce, C. Boutin, E. Jeanneau, T. Brotin and N. De Rycke, J. Org. Chem., 2022, 87, 2912–2920 CrossRef.
- G. Huber, F.-X. Legrand, V. Lewin, D. Baumann, M.-P. Heck and P. Berthault, Chem. Phys. Chem., 2011, 12, 1053–1055 CrossRef CAS PubMed.
- L. Schroder, T. J. Lowery, C. Hilty, D. E. Wemmer and A. Pines, Science, 2006, 314, 446–449 CrossRef.
- M. Zaiss, M. Schnurr and P. Bachert, J. Chem. Phys., 2012, 136, 144106 CrossRef.
- M. Kunth, C. Witte and L. Schröder, J. Chem. Phys., 2014, 141, 194202 CrossRef CAS PubMed.
- S. Korchak, W. Kilian, L. Schröder and L. Mitschang, J. Magn. Reson., 2016, 265, 139–145 CrossRef CAS PubMed.
- L. Mitschang, S. Korchak, W. Kilian and T. Riemer, Anal. Chem., 2022, 94, 2561–2568 CrossRef CAS PubMed.
- Y.-M. Jeon, J. Kim, D. Whang and K. Kim, J. Am. Chem. Soc., 1996, 118, 9790–9791 CrossRef CAS.
- A. Hopkins and A. Williams, J. Org. Chem., 1982, 47, 1745–1750 CrossRef CAS.
- M. Kunth, C. Witte and L. Schröder, NMR Biomed., 2015, 28, 601–606 CrossRef CAS.
- C. Chauvin, L. Liagre, C. Boutin, E. Mari, E. Léonce, G. Carret, B. Coltrinari and P. Berthault, Rev. Sci. Instrum., 2016, 87, 016105 CrossRef CAS PubMed.
- O. Della-Negra, Y. Cirillo, T. Brotin, J.-P. Dutasta, P.-L. Saaidi, B. Chatelet and A. Martinez, Chem. Commun., 2022, 58, 3330–3333 RSC.
- P. Berthault, A. Bogaert-Buchmann, H. Desvaux, G. Huber and Y. Boulard, J. Am. Chem. Soc., 2008, 130, 16456–16457 CrossRef CAS.
- K. Chighine, E. Léonce, C. Boutin, H. Desvaux and P. Berthault, Magn. Reson., 2021, 2, 409–420 CrossRef CAS.
- M. G. Shapiro, R. M. Ramirez, L. J. Sperling, G. Sun, J. Sun, A. Pines, D. V. Schaffer and V. S. Bajaj, Nat. Chem., 2014, 6, 629–634 CrossRef CAS.
- J. L. Mynar, T. J. Lowery, D. E. Wemmer, A. Pines and J. M. J. Fréchet, J. Am. Chem. Soc., 2006, 128, 6334–6335 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetical pathway to cryptophane 2, 1H and 13C spectra of the intermediates, HSQC, HMBC of 2, 129Xe spectra of compounds 1 and 3, 1H spectra comparing 1 and 2, 1 and 3 in different experimental conditions, pulse sequence to measure the depolarization rate, simulation of the effect of the rf pulse. See DOI: https://doi.org/10.1039/d2cp03714a |
| This journal is © the Owner Societies 2022 |