
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A theoretical investigation of uranyl covalency via symmetry-preserving excited state structures†
Sapphire
Armstrong
 a,
Thomas
Malcomson
b and
Andy
Kerridge
*a
a,
Thomas
Malcomson
b and
Andy
Kerridge
*a
aDepartment of Chemistry, Faraday Building, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: a.kerridge@lancaster.ac.uk
bDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 21st October 2022
Abstract
Time dependent density functional theory (TDDFT) calculations have been performed on a series of symmetry-preserving excited states of the uranyl dication, UO22+. The simulated excited state electronic structures are compared to that of the ground state at both ground and excited state-optimised geometries. For the first time, the Quantum Theory of Atoms in Molecules (QTAIM) has been applied to the excited states electronic structures of uranyl in order to quantify the variation in bond covalency upon electronic excitation. QTAIM analysis of vertical excitations at the ground state geometry demonstrated an inverse relationship between the orbital mixing coefficient, λ, and the excitation energy. Furthermore, it was found that, for MOs with U 5f character, λ was more dependent on the metal–ligand Hamiltonian matrix element HML, whereas for those with U 6d character, λ became increasingly dependent on the difference in fragment orbital energy levels, ΔEML. Charge transfer from O to U reduced as the excitation energy increased, as did the degree of electron sharing between the centres. When considering the relaxed excited state geometries, a relationship between excitation energy and bond elongation was established, commensurate with the large magnitude of λ and its dependence on HML for MOs with U 5f character, and enhanced charge transfer otherwise.
1. Introduction
While covalency is well-established for the majority of the periodic table, understanding bonding interactions for actinide complexes is a continuously evolving area of research. Rationalising covalency in actinide complexes remains a challenging experimental and computational problem and deeper understanding is of great importance.1–3 From a fundamental perspective, understanding the bonding interactions within actinide complexes aids in the assessment of the viability of novel synthetic complexes,4,5 while in a practical application, variation in the bonding character is of particular importance for spent fuel reprocessing in the nuclear power industry. Indeed, selective complexation exploits the variation in the bonding character of complexes and is used in the chemical separation of the chemically similar trivalent actinides and lanthanides.6,7Covalency, as defined by Heitler and London,8 is fundamental to our understanding of chemical bonding. In transition metals, metal–ligand orbital mixing and overlap as descriptors of covalent character is well established; however, the prevalence of covalent bonding in actinides is heavily debated.9–11 Therefore, understanding the role of the 5f and 6d orbitals in covalent bonding of actinides remains an important goal in fundamental actinide chemistry.12,13 While the concept of covalency is well-established, there is no formal physical definition. At the theoretical ionic limit there is no mixing among the valence orbitals, deviations from this idealised picture can be considered through perturbation theory. The mixing between a metal (ϕM) and ligand (ϕL) orbital, with energies EM and EL, in a molecular orbital φ can be expressed as:
| φ(r) = ϕM(r) + λϕL(r) | (1) |
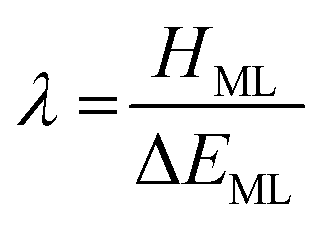 | (2) |
In uranyl, HML can be taken as a measure of the strength of interactions between the uranium 5f/6d and oxygen 2p orbitals, whereas, EML is the energy difference between the metal and ligand orbitals. The mixing coefficient (λ) is a measure of the covalent character of the bond. Values of λ are, in general, nonzero and maximum covalency is achieved when λ = 1, e.g. in a homonuclear diatomic at its equilibrium geometry.
Eqn (2) indicates that covalency can be driven by two factors:15
(i). Large values of HML, corresponding to overlap-driven covalency.
(ii). Small values of ΔEML, corresponding to (energy) degeneracy-driven covalency.
The inherent properties of actinides makes characterising the bonding interactions, and therefore the origins of covalency, challenging for both experimentalists and theorists. Actinide complexes exhibit strong relativistic effects, weak crystal fields and strongly correlated valence electronic structures.16,17 These strongly-correlated systems result in ambiguous orbital-based descriptions of the electronic structure and as such, analysis using orbital-based methods should be contextualised with other approaches.18–23 Analytical methods based on the experimentally observable electron density provide a robust and unambiguous alternative. In this contribution, the Quantum Theory of Atoms in Molecules (QTAIM), developed by Bader,24 will be utilised. QTAIM divides a molecule up into a contiguous set of space-filling atomic basins. This method provides insight into the bonding interactions via both topological and integrated properties of the electron density, which can be used to build up a picture of the covalency in a molecule. We direct the reader to a recent article which gives a detailed overview of this analysis.15
QTAIM has been successfully used to characterise the bonding interactions in many f-element complexes.21,23,25–34 One of the earliest applications of QTAIM analysis in f-element chemistry was conducted by Kaltsoyannis and co-workers.25,26 In these contributions, the covalency in AnCp4 and AnCp3 complexes (An = Th-Cm) were studied. The densities were generated using density functional theory (DFT) and the covalency was assigned using the magnitude of the topological QTAIM metric, ρBCP. This metric indicated that, in these organometallic complexes, the interaction between the actinide and coordinating carbon was mainly ionic in character and this ionic character increased with atomic number. This trend was also established by our own group32 in studies of AnCOT2 complexes (COT = η5–C8H8) where, in contrast to the previous contributions, the complete-active-space self-consistent-field (CASSCF) method was utilised to generate the simulated density. We also analysed the degree of electron sharing between the actinide and coordinating carbon species via the delocalisation index, an integrated QTAIM metric. Both integrated and topological metrics gave the same broad trend.
Of more direct relevance to the present study, QTAIM has also been utilised to study the covalency of uranyl and its complexes. Vallet et al.23 found that free uranyl (UO22+) has strongly covalent interactions between the uranium and oxygen as indicated by the large ρBCP and DI metrics. They also concluded that there is a significant decrease in the magnitude of these metrics (ρBCP and DI) after ligation to the equatorial plane, indicating a decrease in covalency. Comparable bonding characteristics were also established by our group4 using similar techniques. In this contribution, we investigate covalency in uranyl via the exploration of excitations which maintain symmetry of the electronic wavefunction, considering both ground and electronically excited state geometries. These excitations were chosen specifically to ensure that linearity of the uranyl unit is maintained since excitations are largely characterised as being from bonding to anti-bonding MOs of the same irreducible representation (Fig. 1), giving relatively simple excited state electronic structures. Throughout this work, singlet excitations comprised of transitions from bonding to antibonding orbitals of the same character (e.g. ) will be investigated, along with the optically accessible triplet excitation (σu → δu), which is allowed via spin–orbit coupling.35–39
) will be investigated, along with the optically accessible triplet excitation (σu → δu), which is allowed via spin–orbit coupling.35–39
 | ||
| Fig. 1 MOs involved in the excitations considered in this study. Irreps are those of the d2h point group. For degenerate MOs, only excitations between those of the same irrep were considered to ensure Ag symmetry was maintained in the electronic structure. | ||
The excitation energy, as well as the energy difference between a bonding (φ) and antibonding (φ*) MO of the same irrep can be linked with the terms in eqn (2). These energy differences would be expected to be large when either charge transfer character is large (implying a small value of λ) and/or HML is large (which implies a large value SML but doesn’t fully characterise λ).
2. Computational details
Initially, simulations employed version 7.3 of the TURBOMOLE code40 to explore the excitations of Ag symmetry within the d2h point group. Once excitations of interest were identified, Gaussian09 (Revision E.01)41 was employed and, unless explicitly stated, all presented results are derived using Gaussian. All simulations were performed at the hybrid Density Functional Theory (DFT) level, employing the B3LYP exchange-correlation functional.42,43 The aug-cc-pVDZ basis set44,45 was employed for oxygen along with Stuttgart RSC 1997 basis set and associated small core effective core potential for uranium.46–48 Geometry optimisations were performed for both ground and electronic excited state geometries and structures characterised as minima by vibrational frequency analysis. Electronic excitations were calculated at both the ground and excited state geometries employing the Tamm–Dancoff Approximation (TDA)49 to Time Dependent-DFT.50,51 Version 19.02.13 of AIMAll52 and version 3.6 of Multiwfn53 were used in the density-based analysis of the simulated electronic structures. The latter was also employed in the generations of density difference data.Throughout this contribution, variations in different properties (denoted δ/Δ) are evaluated by subtracting the ground state property from the excited state property. A schematic for the specific differences considered is shown in Fig. 2.
 | ||
| Fig. 2 Representation of comparative quantities considered in this contribution, along with nomenclature used to represent them. ΔGS compares ground and excited state properties at the ground state geometry, ΔES compares the same at the excited state geometry, and δ compares ground and excited state properties at their corresponding equilibrium geometries. | ||
3. Results and discussion
3.1 Electronic properties at the ground state geometry
Geometry optimisation of UO22+ produced a U-O bond length of 1.70 Å, in excellent agreement with previously reported values (1.68–1.72 Å).54–56 Electronic excitations in which the dominant orbital transitions were between MOs of the same irrep and same bonding character were identified and are summarised in Table 1. The π → π* excitations are comprised of two symmetry-equivalent transitions. The character of each excited state was assigned through a single dominant transition (>75%), with the exception of which, while the most dominant character, also presented significant (33%)
which, while the most dominant character, also presented significant (33%) 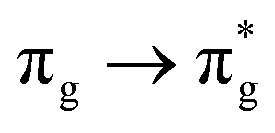 character.
character.
| Excitation | Δ GS E (eV) | Contribution (%) |
|---|---|---|
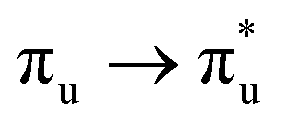
|
7.38 | 88 |

|
12.75 | 44 |
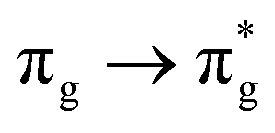
|
13.41 | 78 |
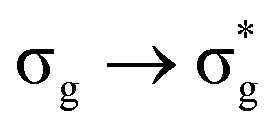
|
16.65 | 98 |
Excitations involving transitions between MOs of u (ungerade) symmetry, possessing U 5f character, are shown to occur at lower energies than those of g (gerade) symmetry, possessing U 6d character.
It then follows that transitions involving MOs with U 5f character maintain greater covalent character, as measured by λ, and/or smaller values HML when compared to those involving MOs with 6d character. Clark and co-workers12 investigated the covalency of actinide chlorides and concluded that the 6d-orbitals were available for orbital mixing to a larger extent than the 5f-orbitals. They also stated that this observation is consistent with other accounts,57–59 which all suggest that 5f-orbitals have a small HML and hence limited participation in covalent bonding.
In order to further rationalise the difference in U 5f/6d bond character, integrated properties of the electron density were evaluated within the Quantum Theory of Atoms in Molecules (QTAIM) framework. Specifically, the atomic charge (q), as well as the localisation (LI) and delocalisation (DI) indices were considered (Table 2).4,18,32 While the atomic charge is a one-electron property obtained via integration of the total electron density in an atomic basin, the localisation and delocalisation indices are formally two-electron properties. LI provides a measure of the number of electrons localised on a given atom while DI measures the number of electrons shared between two atoms and, in the absence of bond polarisation, acts as a bond order metric. Combined, these metrics can provide detailed information regarding the nature and magnitude of bonding interactions, highlighting variation in both charge transfer and covalency. Here, we consider the variation in a metric, M, by comparing its value in the excited state to that in the ground state, such that:
| ΔGSM = M(ES) − M(GS) | (3) |
| Excitation | ΔE (eV) | Δ GS q(U) (a.u.) | Δ GS q(O) (a.u.) | Δ GSLI(U) | Δ GSLI(O) | Δ GSDI(U,O) |
|---|---|---|---|---|---|---|

|
7.38 | −0.050 | +0.025 | +0.395 | +0.186 | −0.353 |

|
12.75 | −0.106 | +0.053 | +0.323 | +0.094 | −0.228 |

|
13.41 | −0.181 | +0.090 | +0.278 | −0.016 | −0.102 |
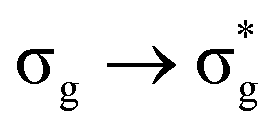
|
16.65 | +0.215 | −0.107 | −0.345 | −0.103 | +0.129 |
Atomic charge data shows that, as excitation energy increases, increasing electronic charge is transferred to the uranium. From an orbital perspective, this implies that the bonding MO has more oxygen character and the antibonding MO more uranium character. A partial exception to this trend is the high energy 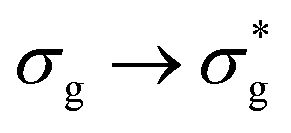 excitation, where, while the direction of charge transfer is reversed, the excitation has the largest magnitude charge transfer character, and so a correlation between charge transfer character and excitation energy is established. This is commensurate with the excitation energy data which suggest reduced orbital mixing, λ, with increasing excitation energy. This observation further verifies the analysis given that charge transfer would be expected to be minimal when λ = 1.
excitation, where, while the direction of charge transfer is reversed, the excitation has the largest magnitude charge transfer character, and so a correlation between charge transfer character and excitation energy is established. This is commensurate with the excitation energy data which suggest reduced orbital mixing, λ, with increasing excitation energy. This observation further verifies the analysis given that charge transfer would be expected to be minimal when λ = 1.
We have previously highlighted the fact that reliance on charge data alone can lead to incorrect interpretations with regard to variation in uranyl bonding.4 Here, while LI(U) qualitatively mirrors the atomic charge data, with ΔGSLI > 0 when charge is transferred to the uranium and ΔGSLI < 0 when charge is transferred from the uranium, the overall trend is not replicated, e.g. the increase in electron localisation is largest for the lowest energy  excitation. This is perhaps unsurprising, since the LI alone is insufficient to characterise the electron population, N, of an atom, which can be formally defined in QTAIM as:
excitation. This is perhaps unsurprising, since the LI alone is insufficient to characterise the electron population, N, of an atom, which can be formally defined in QTAIM as:
 | (4) |
A change in the LI of an atom can therefore be due to charge transfer from another atom, a change in the DI between it and other atoms, or a combination of the two. Given that the variation in DI(U,O) opposes that of LI(U) for all excitations and shows greater sensitivity to the excitation energy for the three lowest energy excitations, the atomic charge data can be rationalised. Charge transfer from O to U, as defined by the variation in LI, actually reduces with increased excitation energy, with the increase in ΔGSq(U) strongly affected by a smaller reduction in DI(U,O) (note that the  excitation shows qualitatively different behaviour, although the same general reasoning applies).
excitation shows qualitatively different behaviour, although the same general reasoning applies).
In a monodeterminantal wavefunction, the DI can be interpreted in terms of the overlap between MOs60 and, as such, ΔGSDI would be expected to be largest when λ is closest to 1. Since DI(U,O) reduces with increasing excitation energy, with the exception of the 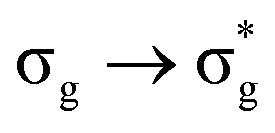 state, this further supports the assertion of the anticorrelation of λ with ΔE.
state, this further supports the assertion of the anticorrelation of λ with ΔE.
Returning our attention to the  excitation, it can be noted from Fig. 1 that the
excitation, it can be noted from Fig. 1 that the  MO has rather diffuse character with significant O contribution. This observation allows us to better understand the electronic structure data. As previously mentioned, the large excitation energy correlates with the substantial charge transfer character of the excitation, albeit with the charge being transferred to the oxygen centres. Here, both LI(U) and LI(O) reduce upon excitation and so the apparent charge transfer is explained by an increase in DI(U,O). As we have discussed above, this is unexpected behaviour for an excitation into a formally antibonding MO and so we suggest that the increase in electronic charge on the (already anionic) oxygen centres has a substantial secondary effect on the electronic structure, causing the doubly occupied σu and πg/u MOs to adopt more covalent character leading to the net increase in DI(U,O). This dramatic behaviour suggests that λ is smallest in σg, again strengthening the relationship between λ and ΔE that we have already identified.
MO has rather diffuse character with significant O contribution. This observation allows us to better understand the electronic structure data. As previously mentioned, the large excitation energy correlates with the substantial charge transfer character of the excitation, albeit with the charge being transferred to the oxygen centres. Here, both LI(U) and LI(O) reduce upon excitation and so the apparent charge transfer is explained by an increase in DI(U,O). As we have discussed above, this is unexpected behaviour for an excitation into a formally antibonding MO and so we suggest that the increase in electronic charge on the (already anionic) oxygen centres has a substantial secondary effect on the electronic structure, causing the doubly occupied σu and πg/u MOs to adopt more covalent character leading to the net increase in DI(U,O). This dramatic behaviour suggests that λ is smallest in σg, again strengthening the relationship between λ and ΔE that we have already identified.
In a simple two-level model, we would expect the energy difference between the bonding and antibonding MOs, and therefore the φ → φ* excitation energy, to increase monotonically with the magnitude of the Hamiltonian matrix element between the relevant metal and ligand levels. From our analysis above, this would imply an inverse relationship between λ and HML. However, eqn (2) states the opposite, leading us to deduce that ΔEML increasingly outweighs HML as the excitation energy increases, implying that ΔEML dominates in covalent interactions involving U 6d contributions, whereas HML dominates for covalent interactions involving U 5f orbitals. Since delocalisation indices themselves are unable to differentiate between overlap- and degeneracy-driven covalency,15 we turn our attention to topological properties of the electron density. ρBCP measures the electron density at the bond critical point (BCP) between two chemically bonded species and gives an indication of the charge accumulated at the BCP. This metric would be expected to reflect the degree of overlap-driven covalency, with a common rule of thumb being that ρBCP > 0.2 a.u. is indicative of a covalent interaction. However, this will only identify contributions due to σ-type interactions since π-interactions are characterised by a nodal plane in which the BCP typically lies. The Laplacian of ρBCP is a complementary metric which can further aid in the characterisation of a bonding interaction. Finally, the magnitude of the energy density, H, which is negative for interactions which have significant electron sharing, can reflect the covalency of an interaction.
The results presented in Table 3 are surprising in that ρBCP increases in the electronically excited state. Since the excitations are into formally antibonding MOs, one would expect ρBCP, which reflects the degree of covalent character, to decrease in the excited state. However, ρBCP also correlates strongly with bond length and so the small increases found here may simply be reflective of the constrained geometry associated with the vertical excitation (an analogous argument can also be made for H). We therefore delay our analysis of topological properties until excited state molecular structures have been considered.
| Excitation | Δ GS ρ BCP (a.u.) | Δ GS∇2ρBCP (a.u.) | Δ GS H (a.u.) |
|---|---|---|---|

|
0.026 | −0.217 | −0.048 |

|
0.028 | 0.036 | −0.048 |

|
0.032 | −0.293 | −0.057 |

|
0.006 | −0.148 | −0.010 |
3.2 Electronic properties at excited state geometries
Geometry optimisations were performed for each of the electronically excited states discussed in Section 3.1 and the results of these optimisations are presented in Table 4. Fig. 3 compares the density difference generated at both the ground and excited state geometries for each excitation.| Excitation | Δ ES E (eV) | Δr (Å) | Stokes shift (eV) | Contribution (%) |
|---|---|---|---|---|

|
5.55 | 0.135 | 1.83 | 47 |

|
10.16 | 0.166 | 2.59 | 23 |

|
9.91 | 0.194 | 3.50 | 66 |

|
16.37 | 0.051 | 0.28 | 98 |
 | ||
| Fig. 3 Comparison of density differences at ground (left) and excited (right) state geometries. Purple/green represents positive/negative electron density. | ||
Table 4 reveals an increase in bond length upon excitation, as expected. Again, the  state exhibits a quantitatively different behaviour, with only a modest bond elongation. Inspection of density differences (Fig. 3) reveals that the
state exhibits a quantitatively different behaviour, with only a modest bond elongation. Inspection of density differences (Fig. 3) reveals that the 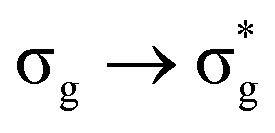 state exhibits a qualitatively different redistribution of charge and consequently a variation in the interplay between ionic and covalent bond character, providing a potential origin for the calculated bond length.
state exhibits a qualitatively different redistribution of charge and consequently a variation in the interplay between ionic and covalent bond character, providing a potential origin for the calculated bond length.
For the three excited states with qualitatively similar excitation character, the variation in bond length follows the same trend as found for the vertical excitation energies in Section 3.1 and provides further evidence that the reduction in λ is due to an increasingly dominant ΔEML contribution, rather than a decrease in HML, since only the latter would be expected to directly impact on bond stability. Diabatic excitations in which Δr is large imply that HML is also substantial as HML is approximately proportional to the overlap (SML) which is in turn correlated to bond length.
Excitation energies, along with corresponding orbital contributions, at the relevant excited state minima are presented in Table 4. This contribution remains unchanged for the  state but is significantly reduced for the other states, presumably reflecting the deviation from monodeterminantal character as the bond is stretched. The
state but is significantly reduced for the other states, presumably reflecting the deviation from monodeterminantal character as the bond is stretched. The  state has contributions from
state has contributions from  orbital transitions and both the
orbital transitions and both the  and
and  states include contributions from other π → π* transitions.
states include contributions from other π → π* transitions.
Excitation energies are all reduced in magnitude when compared to those at the ground state minima, indicating relative destabilisation of the ground state, and so the Stokes shift, calculated here as the difference between the vertical excitation energy at the excited and ground state geometry, describes a red shift for each excited state. The Stokes shift may be utilised to provide further bonding insight, e.g. a small Stokes shift may be indicative of the dependence of λ on ΔEML rather than HML, however, it also correlates strongly with variation in bond length (see Table 4), and for the states under consideration here this latter correlation restricts further interpretation in terms of electronic structure.
We now again turn our attention again to QTAIM metrics (Table 5), comparing the ground and excited state electronic structures at their corresponding equilibrium geometries (δ in Fig. 2). For completeness, corresponding energy differences (δE, see Fig. 2) at the excited state minima are also included. Trends in all metrics are qualitatively similar to those found at the ground state geometry (cf. Table 2) although the magnitudes vary. For example, the redistribution of charge is accentuated, with enhanced electron localisation as might be expected at longer bond lengths. Uranium localisation is substantial for the three lowest energy excited states and while this is accompanied by an increase in oxygen localisation for the excitations which involve MOs with U 5f character, commensurate with enhanced ionic character, δLI(O) is negative (and substantially larger than that seen at the ground state geometry) in the 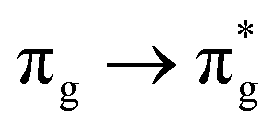 state, indicative of enhanced charge transfer character in the excitation.
state, indicative of enhanced charge transfer character in the excitation.
| Excitation | δE (eV) | δq(U) (a.u.) | δq(O) (a.u.) | δLI(U) | δLI(O) | δDI(U,O) |
|---|---|---|---|---|---|---|
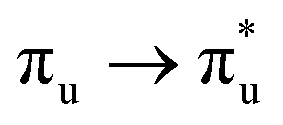
|
6.45 | −0.122 | +0.061 | +0.527 | +0.177 | −0.410 |

|
11.45 | −0.307 | +0.153 | +0.807 | +0.153 | −0.509 |

|
11.61 | −0.414 | +0.207 | +0.562 | −0.110 | −0.152 |

|
16.52 | +0.165 | −0.082 | −0.277 | −0.112 | +0.110 |
Table 6 summarises the variation in topological QTAIM parameters between ground and excited state electronic structures at their corresponding equilibrium geometries. In contrast to Table 3, ρBCP decreases and H increases in the excited states, as would be expected for excitations into formally antibonding MOs. The trends in these variations correlate strongly with the increase in bond length, with the strength of this correlation meaning that further bond characterisation is not possible.
| Excitation | δρBCP (a.u.) | δ∇2ρBCP (a.u.) | δH (a.u.) |
|---|---|---|---|

|
−0.066 | −0.196 | 0.130 |
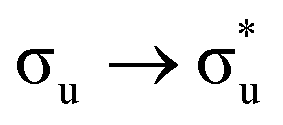
|
−0.082 | −0.082 | 0.159 |

|
−0.096 | −0.179 | 0.182 |

|
−0.032 | −0.119 | 0.064 |
These data allow us to make further observations regarding λ for the orbital excitations associated with each state. At the ground state geometry, it was deduced that for the πg MO, λ was strongly dependent on HML and relatively small. The  state provides the largest values of both Δr and the Stokes shift. This state also has the largest redistribution of charge, opposing localisation indices and a low variation in electron delocalisation. This provides further evidence that λ is indeed significantly dependent on HML in the πg MO and that there is a degree of charge transfer character in the excitation, particularly apparent when considering the electronic structure at the excited state minima. This latter point again supports the view of a small value of λ in the πg MO.
state provides the largest values of both Δr and the Stokes shift. This state also has the largest redistribution of charge, opposing localisation indices and a low variation in electron delocalisation. This provides further evidence that λ is indeed significantly dependent on HML in the πg MO and that there is a degree of charge transfer character in the excitation, particularly apparent when considering the electronic structure at the excited state minima. This latter point again supports the view of a small value of λ in the πg MO.
The 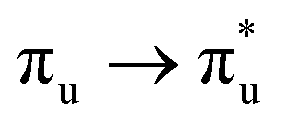 and
and  excited state minima give similar, albeit enhanced, changes in QTAIM metrics to those found at the ground state geometry. The variation in delocalisation index is largest in these two states and this, along with the small redistribution of charge, again supports the view that λ is largest for the πu and σu MOs. Since the excitation energies are lowest for these states, we tentatively suggest that HML is relatively small (although still substantial, as evidenced by the pronounced increase in bond lengths) and the large value of λ is due to the interplay between HML and ΔEML.
excited state minima give similar, albeit enhanced, changes in QTAIM metrics to those found at the ground state geometry. The variation in delocalisation index is largest in these two states and this, along with the small redistribution of charge, again supports the view that λ is largest for the πu and σu MOs. Since the excitation energies are lowest for these states, we tentatively suggest that HML is relatively small (although still substantial, as evidenced by the pronounced increase in bond lengths) and the large value of λ is due to the interplay between HML and ΔEML.
3.3. Symmetry decomposition of QTAIM metrics
The high symmetry of uranyl allows the DIs to be decomposed into their symmetry elements. We have previously employed such analysis to differentiate between 5f and 6d contributions to bonding interactions.15,18,32,61,62 These symmetry-decomposed DIs are used here to understand how electrons are shared in the ground state as the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is elongated. Table 7 orders the excited states in terms of increasing bond length and summarises the symmetry-decomposed DIs.
O bond is elongated. Table 7 orders the excited states in terms of increasing bond length and summarises the symmetry-decomposed DIs.
| State geometry | r (Å) | DI(U,O) | ||||
|---|---|---|---|---|---|---|
| Total | σu | πu | σg | πg | ||
| GS | 1.700 | 2.282 | 0.578 | 0.418 | 0.317 | 0.275 |

|
1.751 | 2.278 | 0.577 | 0.419 | 0.312 | 0.275 |

|
1.835 | 2.273 | 0.573 | 0.425 | 0.304 | 0.273 |
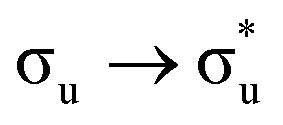
|
1.866 | 2.272 | 0.572 | 0.426 | 0.302 | 0.273 |

|
1.894 | 2.270 | 0.570 | 0.429 | 0.300 | 0.271 |
The total DI reduces with increasing bond length, as would be expected. This behaviour is mirrored in the σu and σg components, however the πu component increases with increasing bond length. Since orbital overlap (and therefore HML) necessarily decreases with increasing bond length, these data can be interpreted in terms of the relative positions of the energy levels of the fragment orbitals comprising the MO. We can therefore deduce that ΔEML decreases more rapidly for the πu MO in comparison to the others, suggesting that the fragment are brought more closely into resonance as the bond is elongated.
3.4 The optically active excited state
Previous computational studies35,37–39,56 have determined that the lowest lying optically active excited state in uranyl can be primarily described as a triplet excitation from the σu into the nonbonding 5fδ orbital, allowed via spin–orbit coupling. This excitation was simulated using the same model chemistry as that employed for our studies of symmetry-preserving excitations, and a vertical excitation energy of 2.43 eV was calculated, in reasonable agreement with the CASPT2 literature value of 2.79 eV calculated in the absence of spin–orbit coupling56 and our own CASSCF-calculated value of 2.95 eV. Relaxation of the excited state geometry produced an increase in bond length to 1.747 Å, again in reasonable agreement with the CASPT2 literature value of 1.765 Å, and an excited state energy of 2.18 eV at the excited state minimum. QTAIM metrics at both the ground and excited state geometries are summarised in Table 8.| Geometry | Δq(U) | Δq(O) | ΔLI(U) | ΔLI(O) | ΔDI(U,O) | ΔρBCP | Δ∇2ρBCP | ΔH |
|---|---|---|---|---|---|---|---|---|
| Ground state | −0.002 | +0.001 | −0.068 | −0.073 | +0.064 | +0.003 | −0.018 | −0.004 |
| Excited state | −0.049 | +0.025 | −0.013 | −0.103 | +0.055 | −0.035 | −0.041 | +0.068 |
All QTAIM metrics report little to no change in electronic structure upon excitation into the U based 5fδ orbital, which would only be expected if the σu MO were also entirely localised on the U centre. Since our previous analysis has revealed this not to be the case, with the σu MO having substantial contributions from both U and O centres, these data can therefore be interpreted as showing a reorganisation amongst the other MOs to accommodate the charge localised in the 5fδ orbital. Since this reorganisation must therefore involve charge transfer from the U centre, and noting that DI(U,O) increases upon excitation, we can deduce that λ must increase amongst the other MOs, although our methodology cannot be used to determine which specific MOs experience this increase.
4. Conclusions and outlook
Electron-density based analyses of f-element complexes have grown in utility in recent years.5,12,15,18,57,61,63 In this contribution, QTAIM analysis has been applied, for the first time, to probe the excited state electronic structure of a uranyl complex. This analysis has been shown to be able to characterise bond covalency in this compound, as well as its origins. Investigations of vertical excitations at the ground state geometry demonstrated an inverse relationship between the orbital mixing coefficient, λ, and the excitation energy. Furthermore, it was found that, for MOs with U 5f character (e.g. σu and πu), λ was more dependent on the metal–ligand Hamiltonian matrix element HML, whereas for those with U 6d character (e.g. σg and πg), λ became increasingly dependent on the difference in fragment orbital energy levels, ΔEML. Further analysis of the electronic structure showed that charge transfer from O to U reduced as the excitation energy increased, as did the degree of electron sharing between the two centres. The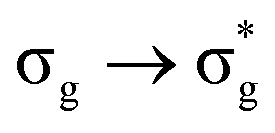 excited state often exhibited quantitatively different behaviour, and this was characterised as being due to a very low value of λ in the σg MO and a difference in the dominant atomic contribution to this MO, leading to significant redistribution of charge amongst the other orbitals in this state.
excited state often exhibited quantitatively different behaviour, and this was characterised as being due to a very low value of λ in the σg MO and a difference in the dominant atomic contribution to this MO, leading to significant redistribution of charge amongst the other orbitals in this state.
When considering the relaxed excited state geometries, a relationship between excitation energy and bond elongation was established. This was commensurate with the large magnitude of λ and its dependence on HML for the σu and πu MOs, however further analysis revealed that the large bond elongation associated with the  state could be understood by also recognising enhanced charge transfer character in the excitation. The
state could be understood by also recognising enhanced charge transfer character in the excitation. The  excitation was again an outlier, where the charge transfer character of the excitation was balanced by the redistribution of charge discussed above.
excitation was again an outlier, where the charge transfer character of the excitation was balanced by the redistribution of charge discussed above.
The dependence of λ on HML for excitations involving 5f MOs has also been investigated previously, in particular for actinide halides.12,61 Tanti et al.61 concluded that the 5f contributions to overlap-driven covalency were larger than 6d for uranyl/uranium halide complexes. Similar findings were also observed across the actinide series by Clark and co-workers;12 they suggest that as you traverse the actinides, “the positive effects from ΔEML outweigh negative impacts from HML and covalency increases.” This contribution complements these findings and suggests that these trends also extend into the excited state.
In contrast to previous literature however, here we conclude that excitations involving the U 5f orbitals have a larger λ than excitations involving U 6d orbitals.57–59 This is largely due to the anticorrelation between λ and excitation energy, with the large λ value due to the interplay between HML and ΔEML, as indicated previously.12
The calculated ground state delocalisation indices were decomposed into their symmetry-distinct components. It would be expected that delocalisation indices would decrease with increasing bond length, but the πu component exhibited the opposite behaviour, suggesting enhanced degeneracy-driven covalency with increasing bond length for this component.
Finally, the leading orbital contribution to the lowest energy optically accessible state in uranyl was investigated. QTAIM metrics revealed little difference between ground and excited state electronic structures, which could be interpreted in terms of the previously analysed states as an increase in covalent character amongst the bonding MOs which offset the charge localisation due to excitation into the non-bonding U 5fδ orbital.
This contribution has demonstrated that analysis of excited state electronic structures can be used to characterise properties of the ground state that would be otherwise challenging to access. Future studies will apply these techniques to more complicated f-element systems to better understand the relative contribution of overlap- and degeneracy-driven covalency to bonding in these compounds.
Conflicts of interest
There are no conflicts to declare.References
- A. Formanuik, A. M. Ariciu, F. Ortu, R. Beekmeyer, A. Kerridge, F. Tuna, E. J. L. McInnes and D. P. Mills, Actinide covalency measured by pulsed electron paramagnetic resonance spectroscopy, Nat. Chem., 2017, 9, 578–583 CrossRef CAS PubMed.
- L. R. Morss, N. Edelstein, J. Fuger and J. J. Katz, The Chemistry of the Actinide and Transactinide Elements, Springer, 4th edn, 2010 Search PubMed.
- L. S. Natrajan and M. H. L. Paden, F-block elements recovery, 2013, vol. 58 Search PubMed.
- P. Di Pietro and A. Kerridge, U-Oyl Stretching Vibrations as a Quantitative Measure of the Equatorial Bond Covalency in Uranyl Complexes: A Quantum- Chemical Investigation, Inorg. Chem., 2016, 55, 573–583 CrossRef CAS PubMed.
- P. Di Pietro and A. Kerridge, Assessing covalency in equatorial U-N bonds: Density based measures of bonding in BTP and isoamethyrin complexes of uranyl, Phys. Chem. Chem. Phys., 2016, 18, 16830–16839 RSC.
- H. H. Dam, D. N. Reinhoudt and W. Verboom, Multicoordinate ligands for actinide/lanthanide separations, Chem. Soc. Rev., 2007, 36, 367–377 RSC.
- F. W. Lewis, M. J. Hudson and L. M. Harwood, Development of highly selective ligands for separations of actinides from lanthanides in the nuclear fuel cycle, Synlett, 2011, 2609–2632 CAS.
- W. Heitler and F. London, Wechselwirkung Neutraler Atome Und Homöopolare Bindung Nach Der Quantenmechanik, Eur. Phys. J. A, 1927, 6–7, 455–472 Search PubMed.
- J. L. Krinsky, S. G. Minasian and J. Arnold, Covalent lanthanide chemistry near the limit of weak bonding: Observation of (CpSiMe3)3Ce-ECp* and a comprehensive density functional theory analysis of Cp3Ln-ECp (E = Al, Ga), Inorg. Chem., 2011, 50, 345–357 CrossRef CAS PubMed.
- F. A. Cotton, Chemical Applications of Group Theory, John Wiley & Sons, New York, 3rd edn, 2004 Search PubMed.
- K. N. Raymond and C. W. Eigenbrot, Structural Criteria for the Mode of Bonding of Organoactinides and Lanthanides and Related Compounds, Acc. Chem. Res., 1980, 13, 276–283 CrossRef CAS.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, Energy-Degeneracy-Driven Covalency in Actinide Bonding, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- R. M. Diamond, K. Street and G. T. Seaborg, An Ion-exchange Study of Possible Hybridized 5f Bonding in the Actinides, J. Am. Chem. Soc., 1954, 76, 1461–1469 CrossRef CAS.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. D. Conradson, S. A. Kozimor, R. L. Martin, D. E. Schwarz, D. K. Shuh, G. L. Wagner, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, Determining relative f and d orbital contributions to M-Cl covalency in MCl 62- (M = Ti, Zr, Hf, U) and UOCl 5- using Cl K-edge X-ray absorption spectroscopy and time-dependent density functional theory, J. Am. Chem. Soc., 2012, 134, 5586–5597 CrossRef CAS PubMed.
- A. Kerridge, Quantification of f-element covalency through analysis of the electron density: Insights from simulation, Chem. Commun., 2017, 53, 6685–6695 RSC.
- M. Dolg, Encyclopedia of Computational Chemistry, Wiley, Chichester, 2002 Search PubMed.
- M. Seth, P. Schwerdtfeger, M. Dolg and P. Fulde, Lanthanide and Actinide Contractions: Relativistic and Shell Structure Effects, J. Am. Chem. Soc., 1995, 117, 6597–6598 CrossRef CAS.
- R. Beekmeyer and A. Kerridge, Assessing covalency in cerium and uranium hexachlorides: A correlated wavefunction and density functional theory study, Inorganics, 2015, 3, 482–499 CrossRef CAS.
- M. Dolg, P. Fulde, W. Küchle, C. S. Neumann and H. Stoll, Ground state calculations of di-π-cyclooctatetraene cerium, J. Chem. Phys., 1991, 94, 3011–3017 CrossRef CAS.
- A. Kerridge, R. Coates and N. Kaltsoyannis, Is cerocene really a Ce(III) compound? all-electron spin–orbit coupled CASPT2 calculations on M(8-C8H8)2 (M = Th, Pa, Ce), J. Phys. Chem. A, 2009, 113, 2896–2905 CrossRef CAS PubMed.
- A. Kerridge, Oxidation state and covalency in f-element metallocenes (M = Ce, Th, Pu): A combined CASSCF and topological study, Dalton Trans., 2013, 42, 16428–16436 RSC.
- O. Mooßen and M. Dolg, Two interpretations of the cerocene electronic ground state, Chem. Phys. Lett., 2014, 594, 47–50 CrossRef.
- V. Vallet, U. Wahlgren and I. Grenthe, Probing the nature of chemical bonding in uranyl(VI) complexes with quantum chemical methods, J. Phys. Chem. A, 2012, 116, 12373–12380 CrossRef CAS PubMed.
- R. F. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- I. Kirker and N. Kaltsoyannis, Does covalency really increase across the 5f series? A comparison of molecular orbital, natural population, spin and electron density analyses of AnCp3 (An = Th-Cm; Cp = η5-C5H5), Dalton Trans., 2011, 40, 124–131 RSC.
- M. J. Tassell and N. Kaltsoyannis, Covalency in AnCp4 (An = Th–Cm): a comparison of molecular orbital, natural population and atoms-in-molecules analyses, Dalton Trans., 2010, 39, 6576–6588 RSC.
- P. L. Arnold, Z. R. Turner, N. Kaltsoyannis, P. Pelekanaki, R. M. Bellabarba and R. P. Tooze, Covalency in CeIV and UIV halide and N-heterocyclic carbene bonds, Chem. – Eur. J., 2010, 16, 9623–9629 CrossRef CAS PubMed.
- V. V. Zhurov, E. A. Zhurova, A. I. Stash and A. A. Pinkerton, Characterization of bonding in cesium uranyl chloride: Topological analysis of the experimental charge density, J. Phys. Chem. A, 2011, 115, 13016–13023 CrossRef CAS PubMed.
- M. B. Jones, A. J. Gaunt, J. C. Gordon, N. Kaltsoyannis, M. P. Neu and B. L. Scott, Uncovering f-element bonding differences and electronic structure in a series of 1:3 and 1:4 complexes with a diselenophosphinate ligand, Chem. Sci., 2013, 4, 1189–1203 RSC.
- A. C. Behrle, C. L. Barnes, N. Kaltsoyannis and J. R. Walensky, Systematic investigation of thorium(IV)- and uranium(IV)-ligand bonding in dithiophosphonate, thioselenophosphinate, and diselenophosphonate complexes, Inorg. Chem., 2013, 52, 10623–10631 CrossRef CAS PubMed.
- Q. Y. Wu, C. Z. Wang, J. H. Lan, C. L. Xiao, X. K. Wang, Y. L. Zhao, Z. F. Chai and W. Q. Shi, Theoretical investigation on multiple bonds in terminal actinide nitride complexes, Inorg. Chem., 2014, 53, 9607–9614 CrossRef CAS PubMed.
- A. Kerridge, F-Orbital covalency in the actinocenes (An = Th-Cm): Multiconfigurational studies and topological analysis, RSC Adv., 2014, 4, 12078–12086 RSC.
- Q. R. Huang, J. R. Kingham and N. Kaltsoyannis, The strength of actinide-element bonds from the quantum theory of atoms-in-molecules, Dalton Trans., 2015, 44, 2554–2566 RSC.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, Small molecule activation by uranium tris(aryloxides): Experimental and computational studies of binding of N2, coupling of CO, and deoxygenation insertion of CO2 under ambient conditions, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- K. Pierloot, E. Van Besien, E. Van Lenthe and E. J. Baerends, Electronic spectrum of UO22+ and [UO2Cl4]2- calculated with time-dependent density functional theory, J. Chem. Phys., 2007, 126, 194311 CrossRef PubMed.
- L. Gagliardi, B. O. Roos, P.-Å. Malmqvist and J. M. Dyke, On the Electronic Structure of the UO 2 Molecule, J. Phys. Chem. A, 2001, 105, 10602–10606 CrossRef CAS.
- P. Tecmer, A. S. P. Gomes, U. Ekström and L. Visscher, Electronic spectroscopy of UO22 +, NUO+ and NUN: An evaluation of time-dependent density functional theory for actinides, Phys. Chem. Chem. Phys., 2011, 13, 6249–6259 RSC.
- P. Tecmer, R. Bast, K. Ruud and L. Visscher, Reliable are Electronic Spectra from Relativistic Time-Dependent.
- P. Tecmer, N. Govind, K. Kowalski, W. A. De Jong and L. Visscher, Reliable modeling of the electronic spectra of realistic uranium complexes, J. Chem. Phys., 2013, 139, 034301 CrossRef PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, TURBOMOLE, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. J. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyez, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresmann, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, L. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09 (Revision E.01), Gaussian Inc., Wallingford CT, 2015.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- T. H. Dunning Jr., Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- R. G. Denning, Electronic Structure and Bonding in Actinyl Ions, 1992, pp. 217–276 Search PubMed.
- X. Cao, M. Dolg and H. Stoll, Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS.
- X. Cao and M. Dolg, Segmented contraction scheme for small-core actinide pseudopotential basis sets, J. Mol. Struct.: THEOCHEM, 2004, 673, 203–209 CrossRef CAS.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm–Dancoff approximation, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- E. Runge and E. K. U. Gross, No Title, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- E. K. U. Gross and W. Kohn, in Density Functional Theory of many-fermion systems, ed. P.-O. Löwdin, 1990, pp. 255–291 Search PubMed.
- T. A. Keith, AIMAll (Version 19.02.13), TK Gristmill Software, Overland Park KS, USA, 2019 (aim.tkgristmill.com).
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- F. Réal, S. P. Gomes, L. Visscher, V. Vallet and E. Ephraim, Benchmarking Electronic Structure Calculations on the Bare UO 22 + Ion: How Different are Single and Multireference Electron Correlation Methods?, J. Phys. Chem. A, 2009, 113, 12504–12511 CrossRef PubMed.
- F. Wei, G. Wu, W. H. E. Schwarz and J. Li, Geometries, electronic structures, and excited states of UN2, NUO +, and UO22 +: A combined CCSD(T), RAS/CASPT2 and TDDFT study, Theor. Chem. Acc., 2011, 129, 467–481 Search PubMed.
- K. Pierloot and E. Van Besien, Electronic structure and spectrum of UO22+ and UO2CI42-, J. Chem. Phys., 2005, 123, 204309 CrossRef PubMed.
- N. Kaltsoyannis, Does covalency increase or decrease across the actinide series? Implications for minor actinide partitioning, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed.
- M. Pepper and B. E. Bursten, The Electronic Structure of Actinide-Containing Molecules: A Challenge to Applied Quantum Chemistry, Chem. Rev., 1991, 91, 719–741 CrossRef CAS.
- H. M. Crosswhite, H. Crosswhite, W. T. Carnall and A. P. Paszek, Spectrum analysis of U3+:LaCI3, J. Chem. Phys, 1980, 72, 5103 CrossRef CAS.
- X. Fradera, M. A. Austen and R. F. W. Bader, The Lewis Model and Beyond, J. Phys. Chem. A, 1999, 103, 304–314 CrossRef CAS.
- J. Tanti, M. Lincoln and A. Kerridge, Decomposition of d- and f-Shell Contributions to Uranium Bonding from the Quantum Theory of Atoms in Molecules: Application to Uranium and Uranyl Halides, Inorganics, 2018, 6, 88 CrossRef.
- I. Fryer-Kanssen and A. Kerridge, Elucidation of the inverse: Trans influence in uranyl and its imido and carbene analogues via quantum chemical simulation, Chem. Commun., 2018, 54, 9761–9764 RSC.
- P. Di Pietro and A. Kerridge, U-Oyl Stretching Vibrations as a Quantitative Measure of the Equatorial Bond Covalency in Uranyl Complexes: A Quantum-Chemical Investigation, Inorg. Chem., 2016, 55, 573–583 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp02878f |
| This journal is © the Owner Societies 2022 |