In silico capture of noble gas atoms with a light atom molecule†
Received
2nd June 2022
, Accepted 15th August 2022
First published on 15th August 2022
Abstract
Noble gas atoms (Ng = He, Ne, Ar, and Kr) can be captured in silico with a light atom molecule containing only C, H, Si, O, and B atoms. Extensive density functional theory (DFT) calculations on series of peri-substituted scaffolds indicate that confined spaces (voids) capable to energy efficiently encapsulate and bind Ng atoms are accessible by design of a tripodal peri-substituted ligand, namely, [(5-Ph2B-xan-4-)3Si]H (xan = xanthene) comprising (after hydride abstraction) four Lewis acidic sites within the cationic structure [(5-Ph2B-xan-4-)3Si]+. The host (ligand system) thereby provides an adoptive environment for the guest (Ng atom) to accommodate for its particular size. Whereas considerable chemical interactions are detectable between the ligand system and the heavier Ng atoms Kr and Ar in the host guest complex [(5-Ph2B-xan-4-)3Si·Ng]+, the lighter Ng atoms Ne and He are rather tolerated by the ligand system instead of being chemically bound to it, nicely highlighting the gradual onset of (weak) chemical bonding along the series He to Kr. A variety of real-space bonding indicators (RSBIs) derived from the calculated electron and pair densities provides valuable insight to the situation of an “isolated atom in a molecule” in case of He, uncovering its size and shape, whereas minute charge rearrangements caused by polarization of the outer electron shell of the larger Ng atoms results in formation of polarized interactions for Ar and Kr with non-negligible covalent bond contributions for Kr. The present study shows that noble gas atoms can be trapped by small light-atom molecules without the forceful conditions necessary using cage structures such as fullerenes, boranes and related compounds or by using super-electrophilic sites like [B12(CN)11]− if the chelating effect of several Lewis acidic sites within one molecule is employed.
Introduction
Noble gas (Ng) atoms resist chemical interactions due to their closed electron shells.1 The large size of Kr and Xe, however, allows polarization of the outer electron shell and thus, chemical bonding under specific conditions. Pioneering work by Bartlett, Hoppe and others in the 1960th revealed that “Xe+[PtF6]−“ (actually a mixture of [XeF]+[PtF5]−, [XeF]+[Pt2F11]−, and [Xe2F3]+[PtF6]−), XeF2, XeF4, XeF6, KrF2, XeO3 and XeO4 comprise Xe and Kr compounds in higher oxidation states.2 Prior to these landmark discoveries, it was well recognized that noble gases can undergo formation of host guest complexes, such as clathrates (e.g. in ice structures)3,4 or inclusion complexes (e.g. in hydroquinone or cyclodextrine),5,6 in which the noble gas in the oxidation state 0 fills a void in the crystal lattice. Xenon(0) can also serve as ligand in the gold complexes, such as [AuXe4]2+.7–10 At a supramolecular level, π- and σ-hole bonding between Xe and electron rich atoms is known to guide crystal packing,11,12 but the smaller Ar and Ne are basically inert. Few complexes with actinides such as CUO(Ng)n13–15 and [UO2(Ngn)]+16 and transition metals17–21 are known so far, as well as low-valent insertion compounds such as HNg(Ng)X (Ng = Xe, Kr, Ar; X = F, Cl, Br, I, NC, CN, CCCN).22–27 Recent studies showed that heavier Ng atoms can be attached to super-electrophiles like [B12(CN)11]−.28–34 A recent DFT study investigated the binding towards MCp (M = Be–Ba) half-sandwich complexes.35 No bonding interaction was ever observed involving a He atom, disregarding the heavily debated and still hypothetical He@adamantane case.36–39 Besides the many attempts to actively bind Ng atoms, they can alternatively be inserted into fullerenes, boron cages and clathrates (confinement strategy), from which they don’t escape due to kinetic stabilization,40–51 trapped in ionic solid-state-compounds or metals,52–55 or adsorbed in metal organic frameworks (MOFs).56,57 Numerous computational studies analyzed the capability of other (mostly hypothetical) cage structures, such as closo-borane derivatives, BN-clusters, etc., to carry one or more Ng atom.58–63 The approach applied here combines both strategies, confinement and bonding, in order to find a class of molecules or ideally a single molecule (denoted as the ligand system in the following), which potentially captures noble gas atoms. For the smaller Ng atoms, He and Ne, the focus is set towards confinement, i.e. the ligand system is ought to provide a void (a physical space) in which the Ng atom can reside without exerting repulsive forces to its environment, whereas for the larger Ng atoms, Ar and Kr, chemical interactions between ligand system and Ng atom should come into play to further stabilize the adduct. A prerequisite was that the ligand system should in principle be assessable by means of chemical synthesis. The conceptual design employs peri-substituted ligand systems consisting of organic spacer-fragments, which are typically used to enforce atom–atom contacts.64–72 Linking three of such spacer fragments, e.g. acenaphthalene, biphenylene, (thio)xanthene moieties together by a central HSi group (tripodal ligand design) provides a dynamically confined space, the size of which depends on the spacer type and the substituent in the peri-position (“starting state”). After hydride abstraction the silyl cation serves as Lewis acidic coordination site for the Ng atom (“active state”). In addition, we chose the group 13 elements B or Ga as additional Lewis acidic coordination sites. Scheme 1 shows the systems analyzed in this study.
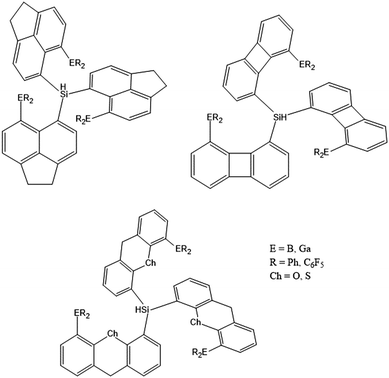 |
| | Scheme 1 Design of neutral tripodal ligands employing different peri-substituted spacer types, such as 5,6-acenaphthalene (ace; upper left), 1,8-biphenylene (bip; upper right), 4,5-(thio)xanthene (txn, xan; bottom). Models employing Al showed inferior results and are not further considered. | |
Ligand systems included 5,6-acenaphthalene (ace) with DFT calculated H⋯H peri-distance (d(H,H)) of 2.7 Å in the non-substituted species, 1,8-biphenylene (bip; d(H,H) = 3.9 Å), 4,5-xanthene (xan, d(H,H) = 4.9 Å), and 4,5-thioxanthene (txn, d(H,H) = 5.4 Å) as spacer fragments covering a large range of peri-distances and according void sizes, and phenyl or C6F5 as substituents attached to the atom E (B or Ga) in peri-position aiming at optimized electronic effects, resulting in 16 potential molecules; 11 of which have explicitly been calculated, see Table 1 and Tables S1 and S2 (ESI†). By this screening approach, computational chemistry can help to pinpoint most promising candidates for subsequent synthesis in the laboratory, which is a relevant topic for sustainable science in general.
Table 1 Relative molecular energies (ΔE in kJ mol−1) of [(R2E-spacer-)3Si]H, [(R2E-spacer-)3Si]+ and [(R2E-spacer-)3Si·Ng]+
| L |
E |
R |
State |
ΔE |
L |
E |
R |
State |
ΔE |
| Ng = noble gas atom, L = spacer fragment, E = B or Ga, R = Ph or C6F5. The ΔE values listed next to state “start” refer to the difference between the desired starting state (void-forming pre-configuration) and unfavorable orientation of the three ligand arms (dead-end state). The ΔE values listed next to state “Ng” refer to the difference between the sum of active state and Ng atom versus the Ng-adduct. All Ng-adduct energies were corrected for basis-set superposition error (BSSE) using the following routine: since BSSE-calculations could not be performed for the large number of structures, it was calculated only for the most promising ligand system, [(5-Ph2B-xan-4-)3Si]H (2a), [(5-Ph2B-xan-4-)3Si]+ (2c) and [(5-Ph2B-xan-4-)3Si·Ng]+, applying the counterpoise correction with Gaussian16, leading to add-on values of 4.5 kJ mol−1 (He, 2d), 5.7 kJ mol−1 (Ne, 2e), 4.2 kJ mol−1 (Ar, 2f), and 60.2 kJ mol−1 (Kr, 2g). The BSSE-corrected 6-31+G* ΔE values are given in italic. ΔE estimation including BSSE-correction was repeated using the 6-311+G(2df,p) basis-set on the 6-31+G* relaxed geometry, resulting in similar values (lower right part of the table). Consequently, the add-on numbers were then added to the ΔE values of all other ligand systems, which should serve as suitable estimate for the BSSE. For further details, see Tables S1 and S2 (ESI). The lower left part of the table (state designated as “no Ng”) refer to single-point calculations on the “empty” ligand systems after geometry optimization (Ng atom removed); the corresponding ΔE values compare their energies to those of the freely optimized ligand systems without Ng atom (2c). |
|
ace
|
B |
C6F5 |
start |
−34.8 |
xan
|
B |
C6F5 |
start |
−33.4 |
|
ace
|
B |
C6F5 |
He |
52.1 |
xan
|
B |
C6F5 |
He |
52.2 |
|
ace
|
B |
C6H5 |
start |
−21.4 |
xan
|
B |
C6F5 |
Ne |
48.6 |
|
ace
|
B |
C6H5 |
He |
31.8 |
xan
|
B |
C6F5 |
Ar |
27.1 |
|
ace
|
Ga |
C6F5 |
start |
83.9 |
xan
|
B |
C6F5 |
Kr |
1.6 |
|
ace
|
Ga |
C6F5 |
He |
30.1 |
xan
|
B
|
C
6
H
5
|
start
|
−37.5
|
|
bip
|
B |
C6H5 |
start |
−7.1 |
xan
|
B
|
C
6
H
5
|
He
|
−0.4
|
|
bip
|
B |
C6H5 |
He |
13.2 |
xan
|
B
|
C
6
H
5
|
Ne
|
−5.3
|
|
bip
|
Ga |
C6F5 |
start |
53.2 |
xan
|
B
|
C
6
H
5
|
Ar
|
−31.1
|
|
bip
|
Ga |
C6F5 |
He |
25.2 |
xan
|
B
|
C
6
H
5
|
Kr
|
−53.7
|
|
bip
|
Ga |
C6H5 |
start |
33.9 |
xan
|
Ga |
C6F5 |
start |
−15.4 |
|
bip
|
Ga |
C6H5 |
He |
6.5 |
xan
|
Ga |
C6F5 |
He |
105.3 |
|
txn
|
B |
C6F5 |
start |
−7.0 |
xan
|
Ga |
C6F5 |
Ne |
112.1 |
|
txn
|
B |
C6F5 |
He |
10.4 |
xan
|
Ga |
C6F5 |
Ar |
89.6 |
|
txn
|
B |
C6F5 |
Kr |
14.5 |
xan
|
Ga |
C6F5 |
Kr |
68.9 |
|
txn
|
Ga |
C6F5 |
start |
−3.9 |
|
|
|
|
|
|
txn
|
Ga |
C6F5 |
He |
22.8 |
|
|
|
|
|
|
txn
|
Ga |
C6F5 |
Kr |
67.8 |
|
|
|
|
|
|
|
|
|
|
|
xan
|
B |
C6H5 |
start |
−37.3 |
|
xan
|
B |
C6H5 |
no He |
−0.3 |
xan
|
B |
C6H5 |
He |
−0.6 |
|
xan
|
B |
C6H5 |
no Ne |
−0.2 |
xan
|
B |
C6H5 |
Ne |
−5.2 |
|
xan
|
B |
C6H5 |
no Ar |
0.3 |
xan
|
B |
C6H5 |
Ar |
−34.9 |
|
xan
|
B |
C6H5 |
no Kr |
3.2 |
xan
|
B |
C6H5 |
Kr |
−58.1 |
Limitations of the method: the neutral tripodal ligand is prone to rotational isomerism because the three spacer fragments are interconnected to the central Si atom by a single bond. Since alternative isomers, which do not exhibit a [{(C6F5)2E-spacer-}3Si]+ (spacer = ace, bip, xan, txn; E = B, Ga) “active site” after hydride abstraction potentially capable of adduct formation with Ng atoms (denoted as “dead-end state” in the following), might be lower in energy than those with the right configuration (denoted as “starting state” in the following), two different neutral states were calculated for each ligand system in order to discriminate different ligand systems for their potential use. It is of course a prerequisite for successful ligand synthesis in the laboratory that the {(C6F5)2E-spacer-}3SiH precursor has the correct orientation prior to hydride abstraction in order to facilitate formation of the charged active state. The molecular energy of the active state and the isolated Ng atom ([{(C6F5)2E-spacer-}3Si]+ + Ng) was compared to the different [{(C6F5)2E-spacer-}3Si·Ng]+ adducts. Since the computational costs were too large to include all four Ng atoms for all ligand systems, only the He (or He and Kr) case(s) was (were) considered in most cases. Accordingly, a minimum of four states had to be calculated for each ligand system. All in all, 55 DFT optimizations (each ligand system with more than 120 atoms, see ESI† for further details) were conducted, which prompted us to keep the DFT at the b3pw91-D3/6-31+G* level. To compensate for that, single-point calculations at the b3pw91-D3/6-311+G(2df,p) were performed for the b3pw91-D3/6-31+G* optimized coordinates of the best ligand systems in addition. Solvation effects were not taken into account. Vibrational frequency calculations (normal mode analysis) necessary to extract the Gibbs free energy (ΔG) could only be performed for the most promising cases, vide infra.
To understand the different modes of bonding between the ligand system and the Ng atom along the series, He–Kr, the electronic characteristics of the most promising Ng adducts were determined computationally employing a variation of real-space bonding indicators (RSBIs) derived from the Atoms-In-Molecules (AIM73), Non-Covalent Interactions index (NCI74), and Electron Localizability Indicator (ELI-D75) approaches. Topological analysis of the electron density (ED, ρ(r)) is a more and more commonly used method to characterize complex bonding scenarios, e.g. cage structures and secondary interactions, for which sole inspection of molecular orbitals might not be clearly interpretable. The resulting AIM bond paths motif refers to the chemical structure but transcends the conventional Lewis picture as it also includes secondary contacts. In addition, AIM provides atomic and fragmental charges, volumes and shapes. The NCI, which employs the reduced ED gradient, provides contact patches between non-covalently interacting atoms, even if those interactions are very weak, e.g. too weak to give rise to the formation of a bond critical point (bcp) within the AIM framework. The analysis is complemented by the ELI-D, which provides basins of paired electrons, and is thus closely related to covalent bonding aspects. Notably, NCI and ELI-D show spatial complementary. Overlapping ELI-D basins with adjacent AIM atomic basins is a measure for bond polarities (Raub-Jansen-Index, RJI76). The combined use of those RSBI facilitates a comprehensive picture of the weak chemical interactions between the ligand system and the Ng atoms, being rather of “physical nature” for He and Ne, and of “chemical nature” for Ar and Kr.
DFT calculations
Structural optimizations were conducted for 55 compounds by density functional theory (DFT) at the b3pw91-D3/6-31+G*77,78 level of theory using Gaussian1679 at the curta super-computer system of the Freie Universität Berlin. London dispersion was modelled using Grimme's GD3BJ parameters (b3pw91-D3).80 Normal mode (or frequency) analysis was performed for the best models to get ΔG values (at 298.15 K and 1 atm). The wavefunction (wfn) files of the best models were used for a topological analysis of the electron density and AIM basin integrations according to the Atoms-In-Molecules (AIM73) space-partitioning scheme using AIM200081 and AIMALL,82 whereas DGRID-5-183 was used to generate and analyze the Electron-Localizability-Indicator (ELI-D75) related real-space bonding descriptors applying a grid step size of 0.05 a.u. using the formatted checkpoint files (fchk). For ELI-D figures, additional grids of 0.15 a.u. step size were computed. NCI74 grids were generated with NCIplot84 with a grid step-size of 0.12 a.u. Analyses of the reduced density gradient, s(r) = [1/2(3π2)1/3]|∇ρ|/ρ4/3, according to the NCI method is used to visualize non-covalent bonding aspects. An estimation of different non-covalent contact types according to steric/repulsive (λ2 > 0, red-colored), van der Waals-like (λ2 ≈ 0, green-colored), and attractive (λ2 < 0, blue-colored) is facilitated by mapping the ED times the sign of the second eigenvalue of the Hessian (sign(λ2)ρ) on the iso-surfaces of s(r). Structures are displayed with ChemCraft,85 bond paths are displayed with AIM2000, NCI figures are displayed with VMD,86 and ELI-D figures are displayed with MolIso.87
Results and discussion
Structures and energies
The here applied screening approach to find a suitable ligand system, which on the one hand is flexible enough to “open and close” like a fly trap to give access to a Ng atom and encapsulate or even bind it then, and to provide an intra-molecular void of the right spatial extension to carry all atoms from He to Kr, but on the other hand is not too flexible, so that unfavorable rotational isomers (dead-end or quenched states) become dominant, generated a vast number of potential structures. In order to keep the discussion of the results as clear-cut as possible, we will go one-by-one through the different classes of compounds (based on the spacer fragment). All structures are compiled as xyz-files and figures in the ESI.† Relevant examples will be presented here.
4,5-Acenaphthalene (ace)
Since ace is the smallest peri-spacer molecule, it provides the smallest void, so only He was tested. With quite bulky –ER2 groups (E = B, Ga; R = C6F5, Ph) in peri-position, there is little structural flexibility in general. Notably, if E = B the unfavorable starting state is higher in energy by about 20–35 kJ mol−1 (see Table 1), than the favorable one, which is desired, although it forms a short Si–H⋯B interaction (d(B,H) = 1.575 Å) in case of [{6-(C6F5)2B-ace-5-}3Si]H (compare Fig. 1(a) and (b) for 1a and 1b). In contrast, due to the formation of two strong R2Ga⋯C(ace) interactions in [{5-(C6F5)2Ga-ace-6-}3Si]H (d(Ga,Cace) = 2.376 and 2.640 Å) it is lower by 84 kJ mol−1 if E = Ga, disqualifying the latter, because in chemical synthesis only small amounts of the neutral starting material would then be oriented in a way to form a proper void after hydride abstraction. Fig. 1(c) displays the active state of [{6-(C6F5)2B-ace-5-}3Si]+ (1c), unraveling another central problem of the ligand design, that is, the ortho-F atoms of the –C6F5 rings tend to stick with the Si cation in the center (d(Si,F) = 1.953 Å). This interaction is strong enough to push the He atom out of the void in [{6-(C6F5)2B-ace-5-}3Si·He]+ (1d), resulting in a positive/repulsive overall binding energy of 52 kJ mol−1, see Fig. 1(d). Energy efficient Ng binding is only possible if the Ng atom is in close proximity to the silyl center as well as to all three group 13 elements (B or Ga), also resulting in complete encapsulation of the guest atom by the host molecule, thereby (almost) exhibiting C3-symmetry.
 |
| | Fig. 1 Relaxed gas-phase geometries of (a) Neutral starting state in the preferred orientation {6-(C6F5)2B-ace-5-}3SiH (1a). (b) Neutral starting state 1b in the unfavorable orientation with a short Si–H⋯B interaction (d(B,H) = 1.575 Å). (c) Active state, [{6-(C6F5)2B-ace-5-}3Si]+ (1c) which is quenched by a short C–F⋯Si interaction (d(Si,F) = 1.953 Å). (d) He-adduct [{6-(C6F5)2B-ace-5-}3Si·He] (1d). Unfavorable Lewis acid Lewis base contacts are highlighted by black arrows. | |
The problem may seemingly be solved by exchanging C6F5 with phenyl rings in the BR2 fragments, however, the binding energy, although less strongly, remains positive (32 kJ mol−1), which finally disqualifies all ace-containing structures. Apparently, the void size is as crucial for effective adduct formation as the electronic effect of the four Lewis acidic sites are.
1,8-Biphenylene (bip)
Like for the ace-series, the unfavorable starting state is higher in energy if E = B, but lower in energy if E = Ga, and adduct formation with He results in less positive numbers if phenyl instead of C6F5 is employed as R group in [(R2E-bip-)3Si·He]+, see Table 1. Disregarding those conserved trends, it is apparent that He binding in general becomes less repulsive in the bip-series compared to the ace-series, again highlighting the central role of the void size. With 6.5 kJ mol−1 it is already almost energy neutral for the [(1-R2Ga-bip-8-)3Si·He]+ case. However, in the corresponding unfavorable starting (or dead-end) state, next to the short Si–H⋯Ga interaction (d(H,Ga) = 2.030 Å) the onset of R2Ga⋯C(bip) interactions (d(Cbip,Ga) = 2.680 and 2.777 Å) is still detectable between the ligand arms, although much less pronounced than in the ace-variants, pushing the energy of the dead-end state below the level of the preferred orientation. As a consequence, all bip-containing structures are disqualified as well. The spacer fragments need to be still larger to provide proper voids for the Ng atoms (including the larger ones) and ideally prevent the formation of unfavorable secondary Si–H⋯E, (spacer)C⋯E, and F⋯Si contacts. See Fig. S2 (ESI†) for the [(8-R2B-bip-1-)3SiH, [(8-R2B-bip-1-)3Si]+, and [(8-R2B-bip-1-)3Si·He]+ case.
4,5-Thioxanthene (txn)
In contrast to ace and bip, txn is not only larger but brings inherent structural flexibility as it can bend in a butterfly motion along the central S⋯C axis. For this ligand system He and Kr binding was tested accordingly. However, it turned out that in case of txn the increased structural flexibility and the fact that the spacer fragment contains a hetero-atom rather generates new problems than serving old ones. In the active state of [{5-(C6F5)2B-txn-4-}3Si]+ short S⋯B (d(S,B) = 2.162 Å) and S⋯Si (d(S,Si) = 2.360 Å) contacts are formed, whereas in [{4-(C6F5)2Ga-txn-5-}3Si]+ short S⋯Ga (d(S,Ga) = 2.506 Å) and F⋯Ga (d(F,Ga) = 2.139 Å) contacts are formed, see Fig. S3 (ESI†). Moreover, in three of the four tested Ng-adducts the Ng atom moved far away from the center position, although all optimizations were started with a C3-symmetrical structure. Consequently, txn-containing ligand systems are also not suitable for capturing Ng atoms.
4,5-Xanthene (xan)
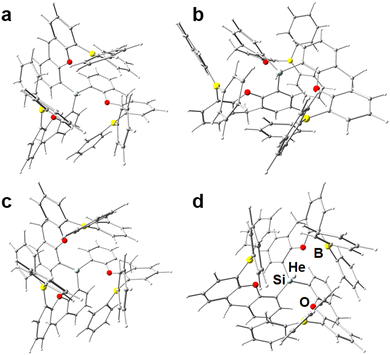
Using xan as spacer fragment results in lower energies for the preferred starting state for all tested variants (−37 to −15 kJ mol−1), which is desired, but for those variants including the E(C6F5)2 group, Ng binding becomes strongly unfavored, again due to the formation of a short and strong F⋯Si contact in the active state (2.040 Å in [{5-(C6F5)2B-xan-4-}3Si]+ and 1.923 Å in [{4-(C6F5)2Ga-xan-5-}3Si]+, see Table 1. To our own surprise, the quite simple and synthetically potentially available ligand system [(5-Ph2B-xan-4-)3Si]+ (2c) shows none of the above mentioned issues, but instead is capable to form adducts (by means of ΔE values) with all Ng atoms from He (−0.4 kJ mol−1, 2d) to Ne (−5.3 kJ mol−1, 2e), Ar (−31.1 kJ mol−1, 2f), and Kr (–53.7 kJ mol−1, 2g), see Fig. 2. Normal mode analysis was performed subsequently for those models, providing corresponding thermodynamic properties, e.g. differences in Gibbs free energy (ΔG), which (after BSSE correction, see Table S3, ESI†) suggests that adduct formation is slightly endergonic for He–Ar (24.5, 16,2, and 7.5 kJ mol−1), but becomes exergonic for Kr (−13.4 mol−1). Since the number of moles is changing, Ng-adduct formation comes with another expense of 7.9 kJ mol−1 to correct for the conversion of the 1 atm standard state (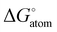 ) to the 1 M standard state (
) to the 1 M standard state (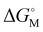 ).88 Comparable results were obtained in computational studies on C70 fullerenes49 as well as on star-shaped super-alkali clusters (C5Li7+ and O2Li5+)52 or for series of dianionic [NgBeB11(CN)11]2− and [NgB12(X)11]2− (X = F–I, CN) superelectrophiles.29,33 Whereas on the one hand fullerenes rely solely on the confinement approach, they have to overcome considerable Ng atom insertion barriers. The proposed super-alkali clusters and superelectrophiles on the other hand rely solely on direct bonding and can’t benefit from stabilizing confinement effects. Our conceptual design of an “open dynamic cage” with a direct bonding partner inside the void attempts to overcome both limitations. An estimate of the energy requirements for the distortion of the ligand system (which also might be considered as “lattice distortions”) necessary to accommodate for the size of the individual Ng atom was obtained by single-point calculations on the optimized coordinates of each adduct after removing the Ng atom (in other word the free ligand system), see lower left part of Table 1. For He, Ne, and Ar, the energy of the ligand system is within a ±0.3 kJ mol−1 range identical to the energy of the optimized structure of the active state (2c), for Kr only 3.2 kJ mol−1 had to be afforded, which confirms that the ligand system is clearly able to fully encapsulate all tested Ng atoms with little to no energy cost. However, this is only the steady-state (permanent) energetic cost after adduct formation, so in order to estimate the costs of the dynamic process of adduct formation, a potential energy scan was performed with fixed Si⋯Kr distances of 3, 4, 5, 7.5, and 10 Å, see Fig. 3. Kr (2g) was chosen, because it is the biggest Ng atom, providing the maximum cost estimate. Starting from 3 Å to larger Si⋯Kr distances, the ΔE curve rises steeply and levels off at about 85 kJ mol−1 for 7.5–10 Å. On the one hand, this means, it will not be easy to get the Ng atoms into the void experimentally against this kinetic barrier, on the other hand this means, once the Ng atom is in the void, losing the Ng atom again is strongly hindered, adding to the thermodynamic gain to the kinetic barrier. Dispersion plays a crucial role in molecules including large organic groups. Accordingly, large contributions of the Grimme dispersion correction of more than 1250 kJ mol−1 were obtained for all xan-models, see Table S4 and Fig. S1 (ESI†), and the values rise along the Ng series, as expected. The averaged value for all calculations of the active ligand (active- and “no Ng”-states) was subtracted from the respective Ng-adducts to get an estimate of the effect of dispersion to the ligand–Ng interaction, revealing it to be the dominant force of Ng binding (−6.3 to −84.3 kJ mol−1), compare italic-style values in Table 1 with Table S4 (ESI†). All in all, the energetic considerations indicate that adduct formation is potentially feasible also experimentally for most if not all of the four Ng atoms. [(5-Ph2B-xan-4-)3Si]+ (2c) can also trap gaseous N2 (ΔE = −82.1 kJ mol−1, see also Fig. S7, ESI†), so any experimental attempt to trap Ng atoms should include the use of pure Ng gases. With d(Si–N) = 1.988 Å, d(B⋯N) > 3.3 Å, and d(N
).88 Comparable results were obtained in computational studies on C70 fullerenes49 as well as on star-shaped super-alkali clusters (C5Li7+ and O2Li5+)52 or for series of dianionic [NgBeB11(CN)11]2− and [NgB12(X)11]2− (X = F–I, CN) superelectrophiles.29,33 Whereas on the one hand fullerenes rely solely on the confinement approach, they have to overcome considerable Ng atom insertion barriers. The proposed super-alkali clusters and superelectrophiles on the other hand rely solely on direct bonding and can’t benefit from stabilizing confinement effects. Our conceptual design of an “open dynamic cage” with a direct bonding partner inside the void attempts to overcome both limitations. An estimate of the energy requirements for the distortion of the ligand system (which also might be considered as “lattice distortions”) necessary to accommodate for the size of the individual Ng atom was obtained by single-point calculations on the optimized coordinates of each adduct after removing the Ng atom (in other word the free ligand system), see lower left part of Table 1. For He, Ne, and Ar, the energy of the ligand system is within a ±0.3 kJ mol−1 range identical to the energy of the optimized structure of the active state (2c), for Kr only 3.2 kJ mol−1 had to be afforded, which confirms that the ligand system is clearly able to fully encapsulate all tested Ng atoms with little to no energy cost. However, this is only the steady-state (permanent) energetic cost after adduct formation, so in order to estimate the costs of the dynamic process of adduct formation, a potential energy scan was performed with fixed Si⋯Kr distances of 3, 4, 5, 7.5, and 10 Å, see Fig. 3. Kr (2g) was chosen, because it is the biggest Ng atom, providing the maximum cost estimate. Starting from 3 Å to larger Si⋯Kr distances, the ΔE curve rises steeply and levels off at about 85 kJ mol−1 for 7.5–10 Å. On the one hand, this means, it will not be easy to get the Ng atoms into the void experimentally against this kinetic barrier, on the other hand this means, once the Ng atom is in the void, losing the Ng atom again is strongly hindered, adding to the thermodynamic gain to the kinetic barrier. Dispersion plays a crucial role in molecules including large organic groups. Accordingly, large contributions of the Grimme dispersion correction of more than 1250 kJ mol−1 were obtained for all xan-models, see Table S4 and Fig. S1 (ESI†), and the values rise along the Ng series, as expected. The averaged value for all calculations of the active ligand (active- and “no Ng”-states) was subtracted from the respective Ng-adducts to get an estimate of the effect of dispersion to the ligand–Ng interaction, revealing it to be the dominant force of Ng binding (−6.3 to −84.3 kJ mol−1), compare italic-style values in Table 1 with Table S4 (ESI†). All in all, the energetic considerations indicate that adduct formation is potentially feasible also experimentally for most if not all of the four Ng atoms. [(5-Ph2B-xan-4-)3Si]+ (2c) can also trap gaseous N2 (ΔE = −82.1 kJ mol−1, see also Fig. S7, ESI†), so any experimental attempt to trap Ng atoms should include the use of pure Ng gases. With d(Si–N) = 1.988 Å, d(B⋯N) > 3.3 Å, and d(N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) = 1.103 Å, N2 is confined in the void but not activated. A crucial step is the initial formation of the active state by hydride abstraction from the preferred neutral state (2a + (+)CPh3 → 2c + CPh3H). Hydride abstractions from silanes are typically achieved using trityl salts of weakly coordinating anions. In order to properly assign this, both states were reoptimized applying the dielectric constant of dichloromethane within the COSMO solvation model, resulting in a slightly endergonic reaction (ΔG = 29.2 kJ mol−1), see Table S5 (ESI†).
N) = 1.103 Å, N2 is confined in the void but not activated. A crucial step is the initial formation of the active state by hydride abstraction from the preferred neutral state (2a + (+)CPh3 → 2c + CPh3H). Hydride abstractions from silanes are typically achieved using trityl salts of weakly coordinating anions. In order to properly assign this, both states were reoptimized applying the dielectric constant of dichloromethane within the COSMO solvation model, resulting in a slightly endergonic reaction (ΔG = 29.2 kJ mol−1), see Table S5 (ESI†).
 |
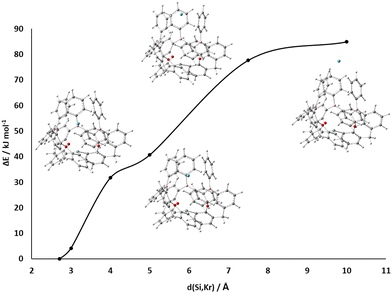
| | Fig. 2 Relaxed gas-phase geometries of (a) Neutral starting state in the preferred orientation [(5-Ph2B-xan-4-)3Si]H (2a). (b) Neutral starting state in the unfavorable orientation (2b). (c) Active state [(5-Ph2B-xan-4-)3Si]+ (2c). (d) He-adduct [(5-Ph2B-xan-4-)3Si·He]+ (2d). No unfavorable Lewis acid Lewis base contacts are present in starting and active states. | |
 |
| | Fig. 3 Si⋯Kr distance constrained relaxed gas-phase geometries of [(5-Ph2B-xan-4-)3Si·Kr]+ (potential energy scan of 2g). The unconstrained optimization is set to 0 kJ mol−1. Color code: Si (light gray), O (red), B (yellow), Kr (light blue). The black line is inserted to guide the eye. | |
In order to exploit the electronic effect of the C6F5 residue but not to suffer from quenching the Si+ site by the ortho F-atoms (F–Si bond formation), two variants of the [(5-Ph2B-xan-4-)3Si]+ ligand system with R = C6H3(m-CF3)2 or C6H2F3 were also tested for He capturing, see Fig. S4 (ESI†). The impact, however, was quite small, with ΔE = −1.1 kJ mol−1 for the former and −0.3 kJ mol−1 for the latter. The impact may be larger for the heavier congeners like Kr and Xe, but since this gain comes along with significantly increased steric crowding, the kinetic barrier for the Ng atom to enter the void of the host molecule will also be much larger. Accordingly, this approach was not followed further.
Relevant bond distances of the DFT optimized structures of [(5-Ph2B-xan-4-)3Si·Ng]+ (2d–g) are compiled in Table 2 (see Table S6, ESI† for more details). %![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (C,V) is the relative position of the DFT atom–atom distance within the range spanned by the difference between sum of covalent radii and sum of van der Waals radii, i.e. a %(C,V) value larger than 100% indicates no considerable interaction, a %(C,V) value between 50 and 100% a rather weak electrostatic or van der Waals like interaction, and a %(C,V) value below 50% a polarized interaction with non-negligible covalent bond contributions. The DFT Ng–Si and Ng–B distances are all shorter than the sum of the respective van der Waals radii (%(C,V) < 100). Especially small %(C,V) values below 50 are obtained for the Si–Ar and Si–Kr interactions, strongly suggesting polar-covalent bonding aspects, see next section. With about 40%, the Si–Ar interaction is at the borderline between medium and weak contacts. Notably, for [(5-Ph2B-xan-4-)3Si·Kr]+ also the Ng–O interactions are within the regime of van der Waals contacts, increasing the coordination number from four to seven. The average Si⋯B and B⋯B distances are hardly affected by Ng-adduct formation (compare with the active state, 2c), especially for [(5-Ph2B-xan-4-)3Si·He]+ (2d) to [(5-Ph2B-xan-4-)3Si·Ar]+ (2f), confirming the energetic considerations, vide supra. This underlines the idea that the ligand system is capable to capture all noble gas atoms from He to Kr, the smaller ones by physical confinement, the larger ones by confinement and in addition by chemical bonding.
(C,V) is the relative position of the DFT atom–atom distance within the range spanned by the difference between sum of covalent radii and sum of van der Waals radii, i.e. a %(C,V) value larger than 100% indicates no considerable interaction, a %(C,V) value between 50 and 100% a rather weak electrostatic or van der Waals like interaction, and a %(C,V) value below 50% a polarized interaction with non-negligible covalent bond contributions. The DFT Ng–Si and Ng–B distances are all shorter than the sum of the respective van der Waals radii (%(C,V) < 100). Especially small %(C,V) values below 50 are obtained for the Si–Ar and Si–Kr interactions, strongly suggesting polar-covalent bonding aspects, see next section. With about 40%, the Si–Ar interaction is at the borderline between medium and weak contacts. Notably, for [(5-Ph2B-xan-4-)3Si·Kr]+ also the Ng–O interactions are within the regime of van der Waals contacts, increasing the coordination number from four to seven. The average Si⋯B and B⋯B distances are hardly affected by Ng-adduct formation (compare with the active state, 2c), especially for [(5-Ph2B-xan-4-)3Si·He]+ (2d) to [(5-Ph2B-xan-4-)3Si·Ar]+ (2f), confirming the energetic considerations, vide supra. This underlines the idea that the ligand system is capable to capture all noble gas atoms from He to Kr, the smaller ones by physical confinement, the larger ones by confinement and in addition by chemical bonding.
Table 2 Relevant bond distances (in Å) of [(5-Ph2B-xan-4-)3Si·Ng]+
| State |
Ng–Si |
%(C,V) |
Ng–B |
%(C,V) |
Ng–O |
%(C,V) |
Ø(Si–B) |
Ø(B–B) |
Δ(Si–B) |
Δ(B–B) |
| act. (2c) |
|
|
|
|
|
|
4.883 |
5.661 |
|
|
| He (2d) |
3.149 |
76 |
3.323 |
99 |
3.543 |
133 |
4.893 |
5.697 |
0.01 |
0.04 |
| Ne (2e) |
3.119 |
66 |
3.332 |
92 |
3.535 |
126 |
4.893 |
5.705 |
0.01 |
0.04 |
| Ar (2f) |
2.890 |
41 |
3.384 |
82 |
3.428 |
106 |
4.877 |
5.739 |
−0.01 |
0.08 |
| Kr (2g) |
2.707 |
18 |
3.379 |
63 |
3.340 |
79 |
4.819 |
5.674 |
−0.06 |
0.01 |
Real-space bonding indicators
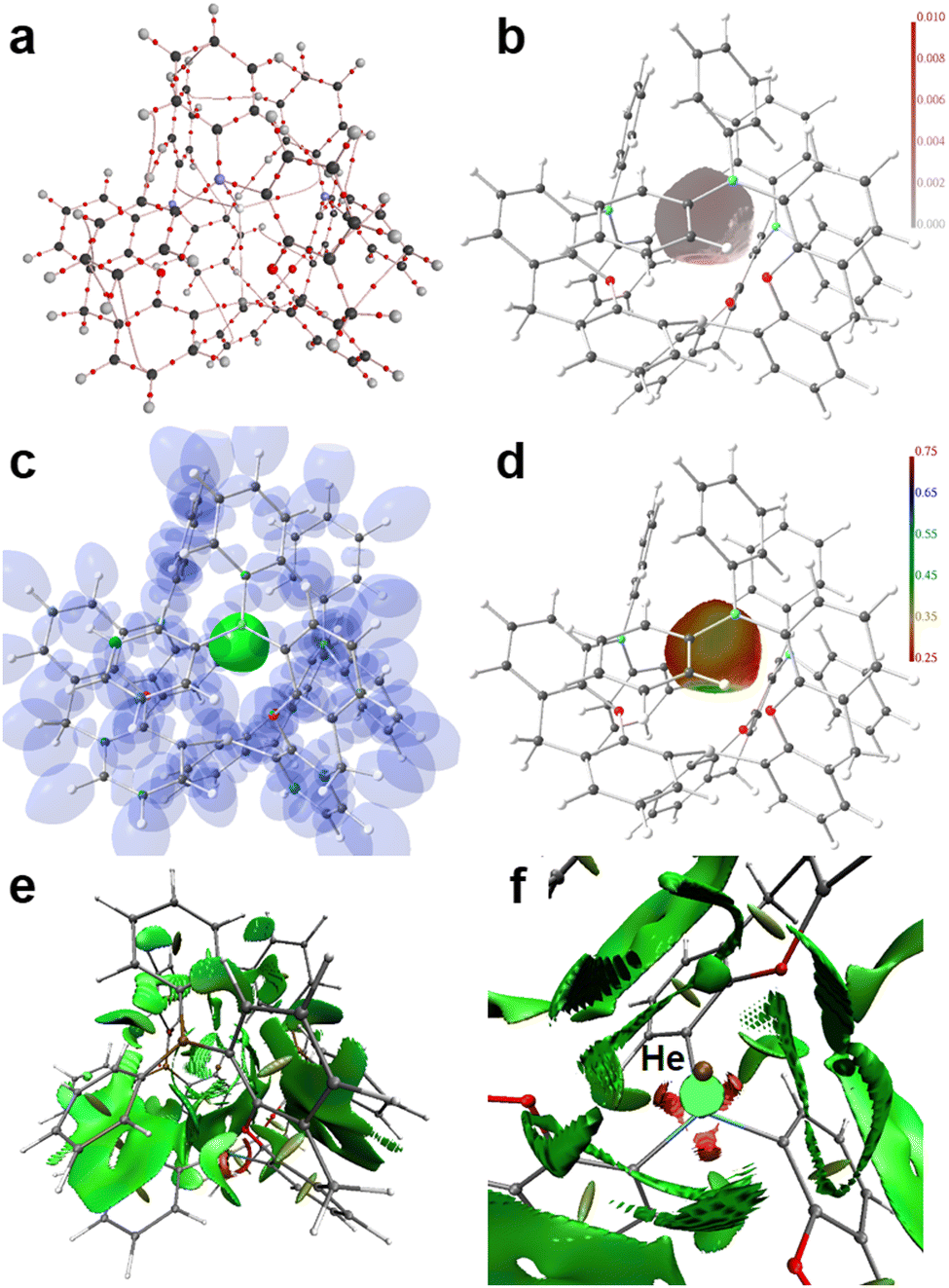
Applying real-space bonding indicators (RSBIs) derived from the Atoms-In-Molecules (AIM), Electron Localizability Indicator (ELI-D), and Non-Covalent Interactions (NCI) index methods to the series [(5-Ph2B-xan-4-)3Si·Ng]+ (2d–g, Ng = He, Ne, Ar, Kr) nicely depicts the onset of weak permanent-polarized atom–atom contacts or in other words the transformation from physical to chemical interactions, with a focus on the Ng–Si contacts. We will not step into the question whether or not the formation of a bond-critical point (bcp) between a pair of atoms within the AIM toolkit is a necessary and sufficient condition to denote such an atom–atom contact as “chemical bond”, as we are aware that the NCI already shows contact patches in cases of very weak H⋯H contacts, which do not form a bcp,89 and that DFT-results are dependent on the used level of theory. Nevertheless, we think that trends are typically more reliable than single numbers, and that the combined use of a variety of RSBIs provide a comprehensive picture of chemical bonding, even if it (seemingly) may contain contradictions in some details. Fig. 4 and 5 and Fig. S8, S9 (ESI†) display the calculated AIM bond topology (part a), the electron density (ED, ρ(r)) distribution mapped on the AIM atomic basin of the Ng atom (part b), an iso-surface representation of the ELI-D (part c), the ELI-D distribution mapped on the core (He) or valence (Ne–Kr) basins of the Ng atoms (part d), the NCI-distribution (part e, side view), and a NCI zoom-in around the Ng atom (part f, top view). For He and Ne, four AIM bond paths are observed, one Ng–Si and three Ng–C(B)/B paths (in three of the four cases, 2d–g, the “Ng–B” bond path is bending towards the C atom, which connects the B atom to the xan spacer group, so by means of AIM topology, these contacts are rather Ng–C than Ng–B contacts, but for convenience we discuss these sluggish Ng⋯B/C contacts as Ng–B interactions in the following). The electronic characteristics at the bcps are listed in Table 3. We note, that all adduct structures approach C3 symmetry and might be optimized also with fixed symmetry. Accordingly, averaged values are given for the Ng–B and Ng–O contacts. The shortest and strongest contacts are the Ng–Si interactions, as expected. The distances decrease down the Ng-column from 3.15 Å (He) to 2.71 Å (Kr), despite increasing Ng atomic radii. The bond paths are not curved, but straight, resulting in identical distances for d1 + d2 (d1 and d2 are the distance of the respective atom to the bcp). In case of the small He and Ne atoms the bcp is closer to the Ng atom than to the Si atom, but closer to Si for Ar and Kr. The ED at the Ng–Si bcp is close to zero for He and Ne, suggesting weak electrostatic interactions, which is supported by the large positive number of the kinetic energy density over ED ratio (G/ρ(r) > 0.7), the fact that the total energy density over ED ratio, which is typically negative and a sign for the presence of covalent bonding aspects, is also considerably positive (H/ρ(r) ≈ 0.2) and delocalization index (DI) values close to zero, the latter indicating the number of shared electron pairs.
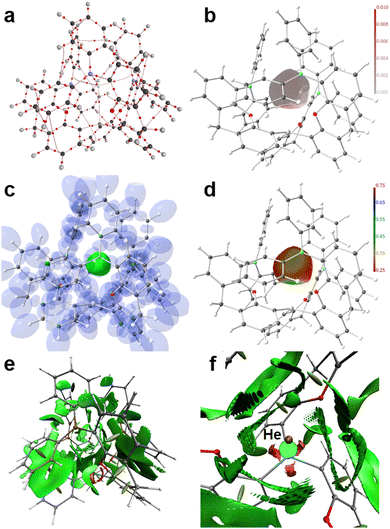 |
| | Fig. 4 RSBI analysis of [(5-Ph2B-xan-4-)3Si·He]+(2d): (a) AIM bond paths motif, (b) ED distribution (in a.u.) mapped on the AIM atomic basin of He, (c) ELI-D localization domain representation at an iso-value of 1.35, (d) ELI-D distribution mapped on the He ELI-D bonding basin, (e) NCI iso-surface at s(r) = 0.5, (f) magnification of the binding site; top view. | |
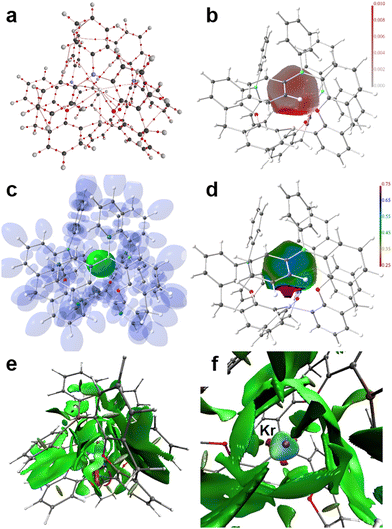 |
| | Fig. 5 RSBI analysis of [(5-Ph2B-xan-4-)3Si·Kr]+ (2g): (a) AIM bond paths motif, (b) ED distribution (in a.u.) mapped on the AIM atomic basin of He, (c) ELI-D localization domain representation at an iso-value of 1.35, (d) ELI-D distribution mapped on the He ELI-D bonding basin, (e) NCI iso-surface at s(r) = 0.5, (f) magnification of the binding site; top view. | |
Table 3 Topological and integrated AIM properties of the Ng–Si, Ng–B, and Ng–O contacts in [(5-Ph2B-xan-4-)3Si·Ng]+
| State |
Contact |
d [Å] |
d
1 + d2 [Å] |
Δd [Å] |
d
1/d |
ρ(r) [e Å3] |
∇2ρ(r)bcp [e Å−5] |
ε
|
G/ρ(r)bcp [a.u.] |
H/ρ(r)bcp [a.u.] |
DI |
|
ρ(r)bcp: ED at the bcp, ∇2ρ(r)bcp: Laplacian, ε: bond ellipticity, d1: distance atom(1)-bcp, G/ρ(r)bcp, H/ρ(r)bcp: kinetic and total energy density over ρ(r)bcp ratios, DI: delocalization index. |
| He |
He–Si |
3.149 |
3.149 |
0.000 |
0.42 |
0.02 |
0.06 |
0.00 |
0.72 |
0.18 |
0.01 |
| Ne |
Ne–Si |
3.119 |
3.119 |
0.000 |
0.47 |
0.03 |
0.12 |
0.00 |
0.83 |
0.22 |
0.02 |
| Ar |
Ar–Si |
2.890 |
2.890 |
0.000 |
0.55 |
0.11 |
0.18 |
0.00 |
0.48 |
−0.03 |
0.10 |
| Kr |
Kr–Si |
2.707 |
2.707 |
0.000 |
0.59 |
0.21 |
0.13 |
0.00 |
0.39 |
−0.21 |
0.20 |
|
|
| He |
He–B |
3.323 |
3.349 |
0.026 |
0.43 |
0.01 |
0.04 |
2.64 |
0.77 |
0.29 |
0.01 |
| Ne |
Ne–B |
3.332 |
3.349 |
0.018 |
0.47 |
0.02 |
0.07 |
2.38 |
0.89 |
0.39 |
0.00 |
| Ar |
Ar–B |
3.384 |
3.406 |
0.022 |
0.52 |
0.03 |
0.10 |
1.70 |
0.67 |
0.19 |
0.02 |
| Kr |
Kr–B |
3.379 |
3.413 |
0.034 |
0.54 |
0.04 |
0.12 |
1.50 |
0.61 |
0.13 |
0.03 |
|
|
| Ar |
Ar–O |
3.428 |
3.439 |
0.011 |
0.52 |
0.03 |
0.12 |
1.40 |
0.81 |
0.20 |
0.02 |
| Kr |
Kr–O |
3.340 |
3.344 |
0.004 |
0.53 |
0.05 |
0.16 |
0.61 |
0.76 |
0.12 |
0.04 |
The situation changes gradually for Ar–Si, which shows the bond characteristics of H⋯H contacts, that is an ED value about 0.1 e Å−3, G/ρ(r) still dominating over H/ρ(r), but the latter being already slightly negative. ED values of about 0.2 e Å−3 for Kr–Si are reminiscent of conventional hydrogen bonds, but in contrast to them, the modulus of H/ρ(r) is much closer to G/ρ(r), indicating comparable non-covalent and covalent contributions to the (still weak) Kr–Si contact.90 The Ng–B and Ng–O contacts vary in the shallow range of 3.32–3.43 Å with ED values of 0.01–0.05 e Å−3 and positive H/ρ(r) values, and are readily characterized as very weak electrostatic. Particularly the Ng–B bond paths are considerably curved, resulting in up to 0.03 Å longer d1 + d2 values (as reflected in Δd), as well as very large bond ellipticities – ε is a measure for the deviation from cylindrical symmetry or in other words “electron smearing”. Although much weaker, the same trend towards increased covalent bond contributions becomes apparent for the Ng–B and Ng–O contacts along the Ng series. Notable, Ng–O bonding is equally relevant as Ng–B bonding for Ar and Kr.
It falls in the same line of evidence that starting with Ar, an explicit Ng–Si disynaptic bonding basin is formed within the ELI-D framework, which, however, is very small (0.1–0.2 Å3) and contains only 0.8–1.0 e, see Table 4 and Fig. S14 (ESI†). For Ar–Si, the Raub–Jansen–Index (RJI) is still 100%, i.e. 100% of the electron population within the Ar–Si bonding basin is located within the AIM atomic basin of Ar, suggesting negligible electron sharing. This number drops to 93.7% for Kr–Si, a value typically found for dative and ionic bonds. In contrast, the RJI of the weaker Ng–B contacts remains basically 100% in all considered cases. The trend is also visible in the AIM atomic and fragmental charges, see Table 5. Whereas no considerable charge transfer between Ng atom and ligand system is visible for He–Ar (ΔQAIM < ±0.2 e) compared to the active state, which carries no Ng atom, small amounts of charge are expelled from the Kr (0.07 e) atom and transferred to the ligand system. Parts b or d of Fig. 4 and 5 and Fig. S8, S9 (ESI†) show the full AIM atomic or ELI-D-core/outer-valence basins of the Ng atoms. AIM and ELI-D basin are almost identical in shape and size for each Ng atom within the series He–Ar due to the lack of electron sharing with other atoms. However, even for the small Ng atoms, the basin shapes are considerably flattened towards the Si+ center, and the ED or ELI-D values are increased along the Ng–Si axes, confirming the onset of electrostatic interactions. In case of Kr a bulge is formed in the ELI-D (not in AIM) along the Ng–Si axes, representing the onset of polarized-covalent bonding interactions, as mentioned above. Parts e or f of 4 and 5 and Fig. S8 and S9 (ESI†) show a total side view or zoomed-in top view of the NCI, uncovering the formation of a small disc-shaped and green colored Ng–Si NCI-basin already in case of He, which becomes larger, thicker, and increasingly bluish along the Ng column, confirming the increased relevance of covalent (ELI-D) and non-covalent (NCI) bonding aspects for Kr. NCI surrounds the Ng atoms with its pattern of contact patches, giving another impression of the atomic size and shape. Whereas the small and blurry NCI basins in case of He hardly indicate relevant He-atom contacts other than He–Si, the picture changes also down the Ng column, as localized disc-shaped basins point towards the B/C and O atoms for the heavier Ng atoms. AIM atomic volumes (VAIM) are given in Table 6, and they vary considerably from 13–33 Å. From VAIM corresponding radii (rAIM) were extracted, assuming the atomic shape would be spherical. Also, d1, which in every AIM atomic basin is the shortest distance between atom center and surface, can be considered as a radius descriptor. We chose here the Ng–B bcp values, because these interactions are the weakest. The volumes derived from d1 as radius (Vd1) are of course smaller than the volumes of the basins, and the ratio of both (Vd1/VAIM) gives an impression about deviation of the atomic shape from globularity. As expected, the deviation from 1 (spherical) is smallest for He (0.95), followed by Ne (0.86) and the others (0.79).
Table 4 Topological and integrated ELI-D properties in [(5-Ph2B-xan-4-)3Si·Ng]+
| State |
Contact |
N
ELI [e] |
V
ELI [Å3] |
γ
ELI
|
RJI [%] |
| Ar |
Ar–Si |
0.78 |
0.12 |
1.58 |
100.0 |
| Kr |
Kr–Si |
0.98 |
0.15 |
1.46 |
93.7 |
|
|
| Ar |
Ar–B |
2.37 |
0.35 |
1.61 |
100.0 |
| Kr |
Kr–B |
2.40 |
0.36 |
1.53 |
100.0 |
Table 5 Atomic and fragmental AIM charges (in e) in [(5-Ph2B-xan-4-)3Si·Ng]+
|
|
Active |
He |
Ne |
Ar |
Kr |
| Ng |
|
−0.01 |
−0.02 |
0.02 |
0.07 |
| Si |
2.69 |
2.70 |
2.70 |
2.71 |
2.71 |
| R1 |
−0.56 |
−0.56 |
−0.56 |
−0.57 |
−0.59 |
| R2 |
−0.57 |
−0.56 |
−0.56 |
−0.57 |
−0.59 |
| R3 |
−0.57 |
−0.56 |
−0.56 |
−0.58 |
−0.60 |
Table 6 Ng atomic radii (in Å) and volumes (in Å3) in [(5-Ph2B-xan-4-)3Si·Ng]+
|
|
d1AIM |
r
AIM
|
V
d1
|
V
AIM
|
V
d1/VAIM |
|
d1AIM: distance Ng atom center to Ng–B/C bcp, rAIM: radii extracted from VAIM, assuming the latter to be a sphere, Vd1: Volume extracted from d1AIM as radii, VAIM: atomic volume resulting from basin integration with AIM2000. |
| He |
1.436 |
1.458 |
12.4 |
13.0 |
0.95 |
| Ne |
1.566 |
1.645 |
16.1 |
18.6 |
0.86 |
| Ar |
1.781 |
1.923 |
23.7 |
29.8 |
0.79 |
| Kr |
1.840 |
1.989 |
26.1 |
33.0 |
0.79 |
Conclusion
Single noble gas atoms can energy efficiently be trapped – at least in silico – with simple organic light atom molecules, which neither consist of closed cage structures or carry super-electrophilic centers, by combing the confinement strategy with attempts to establish direct chemical bonding for the heavier congeners. Tripodal ligand design provides a flexible, but not too flexible intra-molecular space (a void), to take up and contain all Ng atoms from He to Kr. In addition, this confinement space offers four Lewis acidic sites, one silyl cation and three borane atoms, to involve the Ng atoms in weakly attractive atom–atom interactions, which for He and Ne are almost weak enough to consider them as negligible, but become increasingly relevant for Ar and Kr. A variety of real-space bonding indicators derived from the DFT calculated electron and electron pair densities, following the AIM, NCI, and ELI-D approaches, unraveled localized polarized(-covalent) Ng–Si and non-localized purely electrostatic Ng–B or Ng–O interactions, and provided atomic charges and volumes for the Ng atoms within the organic ligand system. Considerable efforts have been made to find a ligand system, for which the starting state remains in the preferred orientation, for which the active state will not be quenched by nucleophilic side groups, and for which the intra-molecular void has the proper size. With [(5-Ph2B-xan-4-)3Si·Ng]+ such a system was finally found. However, with up to 85 kJ mol−1 (for Kr), the barrier to enter the void is still quite high and it will be the task for experimental chemistry to overcome this issue. Nevertheless, the current study shows that with the aid of hypothetical chemistry (computational chemistry of so far unknown compounds) fundamental tasks, like trapping single Ng atoms with a small molecule, can be approached in a sustainable fashion.
Author contributions
S.M.: conceptual design, DFT calculations and analysis, manuscript writing. J. B. Conceptual design, critical revision of the article.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
Dr Emanuel Hupf is greatly acknowledged for software support. The DFG is acknowledged for funding.
References
- E. G. Hope, Coordination chemistry of the noble gases and noble gas fluorides, Coord. Chem. Rev., 2013, 257(5–6), 902–909 CrossRef CAS.
- W. Grochala, Atypical compounds of gases, which have been called ‘noble’, Chem. Soc. Rev., 2007, 36(10), 1632–1655 RSC.
-
E. D. Sloan and C. A. Koh, Clathrate Hydrates of Natural Gases, CRC Press, 2007 Search PubMed.
- P. Villard, Combination of argon with water, C. R. Hebd. Seances Acad. Sci., Ser. D, 1896, 123 Search PubMed.
- H. M. Powell, The Structure of molecular compounds 8. The compound of krypton and quinol, J. Chem. Soc., 1950, 300–301 RSC , Resumed.
- W. Saenger and M. Noltemeyer, X-ray structure-analysis of alpha-cyclodextrin-krypton inclusion complex – noble-gas in an organic matrix, Angew. Chem., Int. Ed. Engl., 1974, 13(8), 552–553 CrossRef.
- N. Bartlett, Xenon hexafluoroplatinate(V) Xe+ [PtF6]−, Proc. Chem. Soc. Lond., 1962, 218 CAS.
- S. Seidel and K. Seppelt, Xenon as a complex ligand: The tetra xenono gold(II) cation in AuXe42 + (Sb2F11-)(2), Science, 2000, 290(5489), 117–118 CrossRef CAS PubMed.
- L. Graham, O. Graudejus, N. K. Jha and N. Bartlett, Concerning the nature of XePtF6. Coordin, Chem. Rev., 2000, 197, 321–334 CAS.
- T. Drews, S. Seidel and K. Seppelt, Gold - Xenon complexes, Angew. Chem., Int. Ed., 2002, 41(3), 454–456 CrossRef CAS PubMed.
- A. Frontera, Noble gas bonding interactions involving xenon oxides and fluorides, Molecules, 2020, 25, 15 CrossRef PubMed.
- A. Bauza and A. Frontera, Sigma/pi-hole noble gas bonding interactions: Insights from theory and experiment. Coordin, Chem. Rev., 2020, 404 Search PubMed.
- B. Y. Liang, L. Andrews, J. Li and B. E. Bursten, Noble gas-actinide compounds: Evidence for the formation of distinct CUO(Ar)(4-n)(Xe)(n) and CUO(Ar)(4-n)(Kr)(n) (n = 1, 2, 3, 4) complexes, J. Am. Chem. Soc., 2002, 124(31), 9016–9017 CrossRef CAS PubMed.
- B. Y. Liang, L. Andrews, J. Li and B. E. Bursten, Bonding of multiple noble-gas atoms to CUO in solid neon: CUO(Ng)(n) (Ng = Ar, Kr, Xe; n = 1, 2, 3, 4) complexes and the singlet-triplet crossover point, Chem. – Eur. J., 2003, 9(19), 4781–4788 CrossRef CAS PubMed.
- L. Andrews, B. Y. Liang, J. Li and B. E. Bursten, Noble gas-actinide complexes of the CUO molecule with multiple Ar, Kr, and Xe atoms in noble-gas matrices, J. Am. Chem. Soc., 2003, 125(10), 3126–3139 CrossRef CAS PubMed.
- X. F. Wang, L. Andrews, J. Li and B. E. Bursten, Significant interactions between uranium and noble-gas atoms: Coordination of the UO2+ cation by Ne, Ar, Kr, and Xe atoms, Angew. Chem., Int. Ed., 2004, 43(19), 2554–2557 CrossRef CAS PubMed.
- D. C. Grills and M. W. George, Transition metal-noble gas complexes, Adv. Inorg. Chem., 2001, 52, 113–150 CrossRef CAS.
- I. C. Hwang, S. Seidel and K. Seppelt, Gold(I) and mercury(II) xenon complexes, Angew. Chem., Int. Ed., 2003, 42(36), 4392–4395 CrossRef CAS PubMed.
- Y. Y. Zhao, G. J. Wang, M. H. Chen and M. F. Zhou, Noble gas-transition metal complexes: Coordination of ScO+ by multiple Ar, Kr, and Xe atoms in noble gas matrixes, J. Phys. Chem. A, 2005, 109(30), 6621–6623 CrossRef CAS PubMed.
- Y. Y. Zhao, Y. Gong, M. H. Chen, C. F. Ding and M. F. Zhou, Coordination of ScO+ and YO+ by multiple Ar, Kr, and Xe atoms in noble gas matrixes: A matrix isolation infrared spectroscopic and theoretical study, J. Phys. Chem. A, 2005, 109(51), 11765–11770 CrossRef CAS PubMed.
- Y. Y. Zhao, Y. Gong, M. H. Chen and M. F. Zhou, Noble gas-transition-metal complexes: Coordination of VO2 and VO4 by Ar and Xe atoms in solid noble gas matrixes, J. Phys. Chem. A, 2006, 110(5), 1845–1849 CrossRef CAS PubMed.
- L. Khriachtchev, M. Pettersson, N. Runeberg, J. Lundell and M. Rasanen, A stable argon compound, Nature, 2000, 406(6798), 874–876 CrossRef CAS PubMed.
- L. Khriachtchev, A. Lignell, H. Tanskanen, J. Lundell, H. Kiljunen and M. Rasanen, Insertion of noble gas atoms into cyanoacetylene: An ab initio and matrix isolation study, J. Phys. Chem. A, 2006, 110(42), 11876–11885 CrossRef CAS PubMed.
- L. Khriachtchev, K. Isokoski, A. Cohen, M. Rasanen and R. B. Gerber, A small neutral molecule with two noble-gas atoms: HXeOXeH, J. Am. Chem. Soc., 2008, 130(19), 6114–6118 CrossRef CAS PubMed.
- L. Khriachtchev, M. Rasanen and R. B. Gerber, Noble-gas hydrides: New chemistry at low temperatures, Acc. Chem. Res., 2009, 42(1), 183–191 CrossRef CAS PubMed.
- C. Oscar, C. Jimenez-Halla, I. Fernandez and G. Frenking, Is it possible to synthesize a neutral noble gas compound containing a Ng–Ng bond? A theoretical study of H–Ng–Ng–F (Ng = Ar, Kr, Xe), Angew. Chem., Int. Ed., 2009, 48(2), 366–369 CrossRef PubMed.
- S. J. Grabowski, J. M. Ugalde, D. M. Andrada and G. Frenking, Comparison of hydrogen and gold bonding in [XHX](−), [XAuX](−), and isoelectronic [NgHNg](+), [NgAuNg](+) (X = Halogen, Ng = noble gas), Chem. – Eur. J., 2016, 22(32), 11317–11328 CrossRef CAS PubMed.
- M. Mayer, V. van Lessen, M. Rohdenburg, G. L. Hou, Z. Yang, R. M. Exner, E. Apra, V. A. Azov, S. Grabowsky, S. S. Xantheas, K. R. Asmis, X. B. Wang, C. Jenne and J. Warneke, Rational design of an argon-binding superelectrophilic anion, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(17), 8167–8172 CrossRef CAS PubMed.
- M. Joshi and T. K. Ghanty, Quantum chemical prediction of a superelectrophilic dianion and its binding with noble gas atoms, Chem. Commun., 2019, 55(95), 14379–14382 RSC.
- M. Mayer, M. Rohdenburg, V. van Lessen, M. C. Nierstenhofer, E. Apra, S. Grabowsky, K. R. Asmis, C. Jenne and J. Warneke, First steps towards a stable neon compound: observation and bonding analysis of [B12(CN)11Ne]−, Chem. Commun., 2020, 56(33), 4591–4594 RSC.
- M. M. Zhong, H. Fang, Deepika and P. Jena, Super-electrophiles of tri- and tetra-anions stabilized by selected terminal groups and their role in binding noble gas atoms, Phys. Chem. Chem. Phys., 2021, 23(38), 21496–21500 RSC.
- M. Mayer, M. Rohdenburg, S. Kawa, F. Horn, H. Knorke, C. Jenne, R. Tonner, K. R. Asmis and J. Warneke, Relevance of π-Backbonding for the Reactivity of Electrophilic Anions [B12X11]− (X = F, Cl, Br, I, CN), Chem. – Eur. J., 2021, 27(40), 10274–10281 CrossRef CAS PubMed.
- K. Wohner, T. Wulf, N. Vankova and T. Heine, Strong binding of noble gases to [B12X11](−): A theoretical study, J. Phys. Chem. A, 2021, 125(22), 4760–4765 CrossRef PubMed.
- H. Fang, D. Deepika and P. Jena, Binding of noble gas atoms by superhalogens, J. Chem. Phys., 2021, 155(1), 014304 CrossRef CAS PubMed.
- R. Saha, S. Pan and P. K. Chattaraj, NgMCp(+): Noble gas bound half-sandwich complexes (Ng = He–Rn, M = Be–Ba, and Cp = eta(5)-C5H5), J. Phys. Chem. A, 2017, 121(18), 3526–3539 CrossRef CAS PubMed.
- T. Strenalyuk and A. Haaland, Chemical bonding in the inclusion complex of He in adamantane (He@adam): The origin of the barrier to dissociation, Chem. – Eur. J., 2008, 14(33), 10223–10226 CrossRef CAS PubMed.
- M. von Hopffgarten and G. Frenking, Chemical bonding in the inclusion complex of He in adamantane, He@adame: Antithesis and complement, Chem. – Eur. J., 2008, 14(33), 10227–10231 CrossRef CAS PubMed.
- S. G. Wang, Y. X. Qiu and W. H. E. Schwarz, Bonding or nonbonding? description
or explanation? “Confinement Bonding” of He@adamantane, Chem. – Eur. J., 2009, 15(24), 6032–6040 CrossRef CAS PubMed.
- P. I. Dem’yanov and P. M. Polestshuk, Forced Bonding and QTAIM Deficiencies: A Case Study of the Nature of Interactions in He@Adamantane and the Origin of the High Metastability, Chem. – Eur. J., 2013, 19(41), 13619 CrossRef.
- M. Saunders, R. J. Cross, H. A. JimenezVazquez, R. Shimshi and A. Khong, Noble gas atoms inside fullerenes, Science, 1996, 271(5256), 1693–1697 CrossRef CAS.
- R. Shimshi, A. Khong, H. A. JimenezVazquez, R. J. Cross and M. Saunders, Release of noble gas atoms from inside fullerenes, Tetrahedron, 1996, 52(14), 5143–5148 CrossRef CAS.
- D. E. Giblin, M. L. Gross, M. Saunders, H. JimenezVazquez and R. J. Cross, Incorporation of helium into endohedral complexes of C-60 and C-70 containing noble-gas atoms: A tandem mass spectrometry study, J. Am. Chem. Soc., 1997, 119(41), 9883–9890 CrossRef CAS.
- V. V. Albert, J. R. Sabin and F. E. Harris, Simulated structure and energetics of endohedral complexes of noble gas atoms in buckminsterfullerene, Int. J. Quantum Chem., 2007, 107(15), 3061–3066 CrossRef CAS.
- H. Yan, S. P. Yu, X. Wang, Y. He, W. Huang and M. L. Yang, Dipole polarizabilities of noble gas endohedral fullerenes, Chem. Phys. Lett., 2008, 456(4-6), 223–226 CrossRef CAS.
- Y. M. Chen, J. Shi, L. Rui and Q. X. Guo, Theoretical study on C-32 fullerenes and their endohedral complexes with noble gas atoms, J. Mol. Struct., 2009, 907(1-3), 104–108 CrossRef CAS.
- S. G. Balasubramani, D. Singh and R. S. Swathi, Noble gas encapsulation into carbon nanotubes: Predictions from analytical model and DFT studies, J. Chem. Phys., 2014, 141, 18 CrossRef PubMed.
- S. Pan, S. Kar, R. Saha, E. Osorio, X. Zarate, L. L. Zhao, G. Merino and P. K. Chattaraj, Boron nanowheels with axles containing noble gas atoms: Viable noble gas bound M (c) B-10(−) clusters (M = Nb, Ta), Chem. – Eur. J., 2018, 24(14), 3590–3598 CrossRef CAS PubMed.
- S. Pan, M. Ghara, S. Kar, X. Zarate, G. Merino and P. K. Chattaraj, Noble gas encapsulated B-40 cage, Phys. Chem. Chem. Phys., 2018, 20(3), 1953–1963 RSC.
- S. Gomez and A. Restrepo, Noble gas dimers confined inside C-70, Phys. Chem. Chem. Phys., 2019, 21(28), 15815–15822 RSC.
- J. Q. Zhong, M. G. Wang, N. Akter, J. D. Kestell, T. C. Niu, A. M. Boscoboinik, T. Kim, D. J. Stacchiola, Q. Wu, D. Y. Lu and J. A. Boscoboinik, Ionization-facilitated formation of 2d (alumino)silicate-noble gas clathrate compounds, Adv. Funct. Mater., 2019, 29(20), 1806583 CrossRef.
- R. Pal and P. K. Chattaraj, Possible effects of fluxionality of a cavitand on its catalytic activity through confinement, Phys. Chem. Chem. Phys., 2021, 23(30), 15817–15834 RSC.
- S. Pan, M. Contreras, J. Romero, A. Reyes, P. K. Chattaraj and G. Merino, C5Li7+ and O2Li5+ as noble-gas-trapping agents, Chem. – Eur. J., 2013, 19(7), 2322–2329 CrossRef CAS PubMed.
- C. Jiang, Y. F. Zhang, Y. P. Gao and J. Gan,
Ab initio theory of noble gas atoms in bcc transition metals, Phys. Chem. Chem. Phys., 2018, 20(25), 17048–17058 RSC.
- Q. Qin, B. Wan, B. M. Yan, B. Gao, Q. Y. Hu, D. Z. Zhang, H. Hosono and H. Y. Gou, Potential interaction of noble gas atoms and anionic electrons in Ca2N, J. Phys. Chem. C, 2020, 124(23), 12213–12219 CrossRef CAS.
- A. J. Lloyd, B. R. Hester, S. J. Baxter, S. Y. Ma, V. B. Prakapenka, S. N. Tkachev, C. Park and A. P. Wilkinson, Hybrid double perovskite containing helium: [He-2][CaZr]F-6, Chem. Mater., 2021, 33(9), 3132–3138 CrossRef.
- J. Liu, D. M. Strachan and P. K. Thallapally, Enhanced noble gas adsorption in Ag@MOF-74Ni, Chem. Commun., 2014, 50(4), 466–468 RSC.
- O. V. Magdysyuk, D. Denysenko, I. Weinrauch, D. Volkmer, M. Hirscher and R. E. Dinnebier, Formation of a quasi-solid structure by intercalated noble gas atoms in pores of Cu-I-MFU-4l metal-organic framework, Chem. Commun., 2015, 51(4), 714–717 RSC.
- O. P. Charkin, N. M. Klimenko, D. Moran, A. M. Mebe, D. O. Charkin and P. V. Schleyer, Theoretical study of icosahedral closo-borane, -alane, and -gallane dianions (A(12)H(12)(2−); A = B, Al, Ga) with endohedral noble gas atoms (Ng = He, Ne, Ar, and Kr) and their lithium salts (Li[Ng@A(12)H(12)](−) and Li-2[Ng@A(12)H(12)]), Inorg. Chem., 2001, 40(27), 6913–6922 CrossRef CAS PubMed.
- E. Cerpa, A. Krapp, R. Flores-Moreno, K. J. Donald and G. Merino, Influence of Endohedral Confinement on the Electronic Interaction between He atoms: A He-2@C20H20 Case Study, Chem. – Eur. J., 2009, 15(8), 1985–1990 CrossRef CAS PubMed.
- M. Khatua, S. Pan and P. K. Chattaraj, Confinement induced binding of noble gas atoms, J. Chem. Phys., 2014, 140(16), 164306 CrossRef PubMed.
- S. Pan, S. Mandal and P. K. Chattaraj, Cucurbit[6]uril: A possible host for noble gas atoms, J. Phys. Chem. B, 2015, 119(34), 10962–10974 CrossRef CAS PubMed.
- D. Chakraborty and P. K. Chattaraj, Confinement induced binding in noble gas atoms within a BN-doped carbon nanotube, Chem. Phys. Lett., 2015, 621, 29–34 CrossRef CAS.
- R. Pino-Rios, E. Chigo-Anota, E. Shakerzadeh and G. Cardenas-Jiron, B12N12 cluster as a collector of noble gases: A quantum chemical study, Physica E, 2020, 115 Search PubMed.
- J. Beckmann, T. G. Do, S. Grabowsky, E. Hupf, E. Lork and S. Mebs, Peri-interactions in 8-diphenylphosphino-1-bromonaphthalene, 6-diphenylphosphino-5-bromoacenaphthene, and derivatives, Z. Anorg. Allg. Chem., 2013, 639(12-13), 2233–2249 CrossRef CAS.
- J. Beckmann, E. Hupf, E. Lork and S. Mebs, Peri-substituted (Ace)naphthylphosphinoboranes. (Frustrated) Lewis Pairs, Inorg. Chem., 2013, 52(20), 11881–11888 CrossRef CAS PubMed.
- E. Hupf, E. Lork, S. Mebs and J. Beckmann, Intramolecularly coordinated (6-(diphenylphosphino)acenaphth-5-yl)stannanes. repulsion vs. attraction of P- and Sn-containing substituents in the peri positions, Organometallics, 2014, 33(10), 2409–2423 CrossRef CAS.
- E. Hupf, E. Lork, S. Mebs and J. Beckmann, Sterically congested 5-diphenylphosphinoacenaphth-6-yl-silanes and -silanols, Organometallics, 2015, 34(15), 3873–3887 CrossRef CAS.
- S. Furan, E. Hupf, E. Lork, S. Mebs and J. Beckmann, Insights into frustrated and regular peri-substituted (Ace-)naphthylaminoboranes and (Ace-)naphthylphosphinoboranes, Eur. J. Inorg. Chem., 2017,(27), 3302–3311 CrossRef CAS.
- E. Hupf, M. Olaru, C. I. Rat, M. Fugel, C. B. Hubschle, E. Lork, S. Grabowsky, S. Mebs and J. Beckmann, Mapping the trajectory of nucleophilic substitution at silicon using a peri-substituted acenaphthyl scaffold, Chem. – Eur. J., 2017, 23(44), 10568–10579 CrossRef CAS PubMed.
- F. Kutter, A. Denhof, E. Lork, S. Mebs and J. Beckmann, 1,8-Bis(diphenylphosphino)biphenylene. A new ligand for late transition metal complexes, Z. Kristallogr. – Cryst. Mater., 2018, 233(9–10), 627–639 CrossRef CAS.
- F. Kutter, E. Lork, S. Mebs and J. Beckmann, Intramolecular P–H center dot center dot center dot H–Si dihydrogen bonding in the 5-dimethylsily1-9,9-dimethylxanthen-4-yl-diphenylphosphonium cation, Organometallics, 2018, 37(22), 4287–4296 CrossRef CAS.
- D. Duvinage, P. Bottke, M. Wark, E. Lork, S. Mebs and J. Beckmann, The effect of donor additives on the stability and structure of 5-diphenylphosphinoacenaphth-6-yllithium, Eur. J. Inorg. Chem., 2019,(5), 712–720 CrossRef CAS.
- R. F. W. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18(1), 9–15 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. T. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132(18), 6498–6506 CrossRef CAS PubMed.
- M. Kohout, A measure of electron localizability, Int. J. Quantum Chem., 2004, 97(1), 651–658 CrossRef CAS.
- S. Raub and G. Jansen, A quantitative measure of bond polarity from the electron localization function and the theory of atoms in molecules, Theor. Chem. Acc., 2001, 106(3), 223–232 Search PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, molecules, solids, and surfaces – applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46(11), 6671–6687 CrossRef CAS PubMed.
- A. D. Becke, A new mixing of Hartree-Fock and local density-functional theories, J. Chem. Phys., 1993, 98(2), 1372–1377 CrossRef CAS.
-
M. J. T. Frisch, G. W. Schlegel, H. B. Scuseria, G. E. Robb, M. A. Cheeseman, J. R. Scalmani, G. Barone, V. Petersson, G. A. Nakatsuji, H. Li, X. Caricato, M. Marenich, A. V. Bloino, J. Janesko, B. G. Gomperts, R. Mennucci, B. Hratchian, H. P. Ortiz, J. V. Izmaylov, A. F. Sonnenberg, J. L. Williams-Young, D. Ding, F. Lipparini, F. Egidi, F. Goings, J. Peng, B. Petrone, A. Henderson, T. Ranasinghe, D. Zakrzewski, V. G. Gao, J. Rega, N. Zheng, G. Liang, W. Hada, M. Ehara, M. Toyota, K. Fukuda, R. Hasegawa, J. Ishida, M. Nakajima, T. Honda, Y. Kitao, O. Nakai, H. Vreven, T. Throssell, K. Montgomery, J. A. Jr.; J. E. Peralta; F. Ogliaro; M. J. Bearpark; J. J. Heyd; E. N. Brothers; K. N. Kudin; V. N. Staroverov; T. A. Keith; R. Kobayashi; J. Normand; K. Raghavachari; A. P. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; J. M. Millam; M. Klene; C. Adamo; R. Cammi; J. W. Ochterski; R. L. Martin; K. Morokuma; O. Farkas; J. B. Foresman and D. J. Fox, Gaussian 16, C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- F. Biegler-Konig, J. Schonbohm and D. Bayles, Software news and updates – AIM2000 – A program to analyze and visualize atoms in molecules, J. Comput. Chem., 2001, 22(5), 545–559 CrossRef.
-
T. A. Keith, AIMALL, 19.10.12, TK Gristmill Software, Overland Park KS, USA, 2019 Search PubMed.
-
M. Kohout, DGRID, 4.6, 2015 Search PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. T. Yang, NCIPLOT: A program for plotting noncovalent interaction regions, J. Chem. Theory Comput., 2011, 7(3), 625–632 CrossRef CAS PubMed.
-
G. A. Andirenko, ChemCraft, 1.8, CrowdStrike, https://www.chemcraftprog.com Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics Modell., 1996, 14(1), 33–38 CrossRef CAS.
- C. B. Hübschle and P. Luger, MolIso – a program for colour-mapped iso-surfaces, J. Appl. Crystallogr., 2006, 39, 901–904 CrossRef.
-
C. J. Cramer, Essentials of Computational Chemistry: Theories and Models. 2nd edn, Wiley & Sons Ltd, 2004 Search PubMed.
- J. R. Lane, J. Contreras-Garcia, J. P. Piquemal, B. J. Miller and H. G. Kjaergaard, Are bond critical points really critical for hydrogen bonding?, J. Chem. Theory Comput., 2013, 9(8), 3263–3266 CrossRef CAS PubMed.
- S. Borocci, F. Grandinetti, F. Nunzi and N. Sanna, Classifying the chemical bonds involving the noble-gas atoms, New J. Chem., 2020, 44(34), 14536–14550 RSC.
|
| This journal is © the Owner Societies 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Jens
Beckmann
*a and
Jens
Beckmann


 ) to the 1 M standard state (
) to the 1 M standard state ( ).88 Comparable results were obtained in computational studies on C70 fullerenes49 as well as on star-shaped super-alkali clusters (C5Li7+ and O2Li5+)52 or for series of dianionic [NgBeB11(CN)11]2− and [NgB12(X)11]2− (X = F–I, CN) superelectrophiles.29,33 Whereas on the one hand fullerenes rely solely on the confinement approach, they have to overcome considerable Ng atom insertion barriers. The proposed super-alkali clusters and superelectrophiles on the other hand rely solely on direct bonding and can’t benefit from stabilizing confinement effects. Our conceptual design of an “open dynamic cage” with a direct bonding partner inside the void attempts to overcome both limitations. An estimate of the energy requirements for the distortion of the ligand system (which also might be considered as “lattice distortions”) necessary to accommodate for the size of the individual Ng atom was obtained by single-point calculations on the optimized coordinates of each adduct after removing the Ng atom (in other word the free ligand system), see lower left part of Table 1. For He, Ne, and Ar, the energy of the ligand system is within a ±0.3 kJ mol−1 range identical to the energy of the optimized structure of the active state (2c), for Kr only 3.2 kJ mol−1 had to be afforded, which confirms that the ligand system is clearly able to fully encapsulate all tested Ng atoms with little to no energy cost. However, this is only the steady-state (permanent) energetic cost after adduct formation, so in order to estimate the costs of the dynamic process of adduct formation, a potential energy scan was performed with fixed Si⋯Kr distances of 3, 4, 5, 7.5, and 10 Å, see Fig. 3. Kr (2g) was chosen, because it is the biggest Ng atom, providing the maximum cost estimate. Starting from 3 Å to larger Si⋯Kr distances, the ΔE curve rises steeply and levels off at about 85 kJ mol−1 for 7.5–10 Å. On the one hand, this means, it will not be easy to get the Ng atoms into the void experimentally against this kinetic barrier, on the other hand this means, once the Ng atom is in the void, losing the Ng atom again is strongly hindered, adding to the thermodynamic gain to the kinetic barrier. Dispersion plays a crucial role in molecules including large organic groups. Accordingly, large contributions of the Grimme dispersion correction of more than 1250 kJ mol−1 were obtained for all xan-models, see Table S4 and Fig. S1 (ESI†), and the values rise along the Ng series, as expected. The averaged value for all calculations of the active ligand (active- and “no Ng”-states) was subtracted from the respective Ng-adducts to get an estimate of the effect of dispersion to the ligand–Ng interaction, revealing it to be the dominant force of Ng binding (−6.3 to −84.3 kJ mol−1), compare italic-style values in Table 1 with Table S4 (ESI†). All in all, the energetic considerations indicate that adduct formation is potentially feasible also experimentally for most if not all of the four Ng atoms. [(5-Ph2B-xan-4-)3Si]+ (2c) can also trap gaseous N2 (ΔE = −82.1 kJ mol−1, see also Fig. S7, ESI†), so any experimental attempt to trap Ng atoms should include the use of pure Ng gases. With d(Si–N) = 1.988 Å, d(B⋯N) > 3.3 Å, and d(N
).88 Comparable results were obtained in computational studies on C70 fullerenes49 as well as on star-shaped super-alkali clusters (C5Li7+ and O2Li5+)52 or for series of dianionic [NgBeB11(CN)11]2− and [NgB12(X)11]2− (X = F–I, CN) superelectrophiles.29,33 Whereas on the one hand fullerenes rely solely on the confinement approach, they have to overcome considerable Ng atom insertion barriers. The proposed super-alkali clusters and superelectrophiles on the other hand rely solely on direct bonding and can’t benefit from stabilizing confinement effects. Our conceptual design of an “open dynamic cage” with a direct bonding partner inside the void attempts to overcome both limitations. An estimate of the energy requirements for the distortion of the ligand system (which also might be considered as “lattice distortions”) necessary to accommodate for the size of the individual Ng atom was obtained by single-point calculations on the optimized coordinates of each adduct after removing the Ng atom (in other word the free ligand system), see lower left part of Table 1. For He, Ne, and Ar, the energy of the ligand system is within a ±0.3 kJ mol−1 range identical to the energy of the optimized structure of the active state (2c), for Kr only 3.2 kJ mol−1 had to be afforded, which confirms that the ligand system is clearly able to fully encapsulate all tested Ng atoms with little to no energy cost. However, this is only the steady-state (permanent) energetic cost after adduct formation, so in order to estimate the costs of the dynamic process of adduct formation, a potential energy scan was performed with fixed Si⋯Kr distances of 3, 4, 5, 7.5, and 10 Å, see Fig. 3. Kr (2g) was chosen, because it is the biggest Ng atom, providing the maximum cost estimate. Starting from 3 Å to larger Si⋯Kr distances, the ΔE curve rises steeply and levels off at about 85 kJ mol−1 for 7.5–10 Å. On the one hand, this means, it will not be easy to get the Ng atoms into the void experimentally against this kinetic barrier, on the other hand this means, once the Ng atom is in the void, losing the Ng atom again is strongly hindered, adding to the thermodynamic gain to the kinetic barrier. Dispersion plays a crucial role in molecules including large organic groups. Accordingly, large contributions of the Grimme dispersion correction of more than 1250 kJ mol−1 were obtained for all xan-models, see Table S4 and Fig. S1 (ESI†), and the values rise along the Ng series, as expected. The averaged value for all calculations of the active ligand (active- and “no Ng”-states) was subtracted from the respective Ng-adducts to get an estimate of the effect of dispersion to the ligand–Ng interaction, revealing it to be the dominant force of Ng binding (−6.3 to −84.3 kJ mol−1), compare italic-style values in Table 1 with Table S4 (ESI†). All in all, the energetic considerations indicate that adduct formation is potentially feasible also experimentally for most if not all of the four Ng atoms. [(5-Ph2B-xan-4-)3Si]+ (2c) can also trap gaseous N2 (ΔE = −82.1 kJ mol−1, see also Fig. S7, ESI†), so any experimental attempt to trap Ng atoms should include the use of pure Ng gases. With d(Si–N) = 1.988 Å, d(B⋯N) > 3.3 Å, and d(N