
Multiple-cation wide-bandgap perovskite solar cells grown using cesium formate as the Cs precursor with high efficiency under sunlight and indoor illumination†
Qiang
Guo
 a,
Yuanjia
Ding
ab,
Zheng
Dai
ab,
Zongwei
Chen
a,
Mengzhen
Du
ab,
Zongtao
Wang
ab,
Lei
Gao
a,
Chen
Duan
a,
Qing
Guo
a and
Erjun
Zhou
*b
a,
Yuanjia
Ding
ab,
Zheng
Dai
ab,
Zongwei
Chen
a,
Mengzhen
Du
ab,
Zongtao
Wang
ab,
Lei
Gao
a,
Chen
Duan
a,
Qing
Guo
a and
Erjun
Zhou
*b
aHenan Institute of Advanced Technology, Zhengzhou University, Zhengzhou 450003, China
bCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing, 100190, China. E-mail: zhouej@nanoctr.cn
First published on 14th July 2022
Abstract
Owing to the advantages of adjustable bandgap, low-cost fabrication and superior photovoltaic performance, wide-bandgap (WBG) perovskite solar cells (PSCs) are considered as the promising top-cell for multi-junction solar cells. At the same time, WBG PSCs have also shown great potential for indoor photovoltaic applications. To further improve the performance of WBG PSCs, in this work, we fabricated efficient WBG PSCs via introducing cesium formate (CsFa) as the Cs precursor. Due to the HCOO·Pb+ and HCOOH·Cs+ complex formation and HCOOH volatilization accompanying the crystallization process, the crystallization of the perovskite using the CsFa precursor (CsFa-perovskite) is promoted. Compared to the perovskite prepared using the CsBr precursor (CsBr-perovskite), the WBG CsFa-perovskite shows better perovskite crystallization, reduced trap-state density, and better phase stability under light illumination. Finally, the 1.63 eV WBG PSCs based on the CsFa-perovskite achieve a significant PCE of 20.01% under one sun illumination (AM 1.5G, 100 mW cm−2), which is higher than that of PSCs based on the CsBr-perovskite (18.27%). Moreover, the PCE of CsFa-perovskite PSCs also under indoor warm-white 2700 K LED light illumination (1000 lux) is as high as 38.52%. Our results demonstrate that CsFa as the Cs precursor is a promising candidate to promote the device performance of WBG PSCs.
Introduction
Organic–inorganic hybrid perovskite solar cells (PSCs) have attracted tremendous research attention in the last decade and the highest certified power conversion efficiency (PCE) of single-junction devices has already reached 25.7%.1 However, it is still far away from the Shockley–Queisser (S–Q) thermodynamic limit of single-junction solar cells. To surpass the S–Q limit, it is a feasible way to prepare multi-junction solar cells. Since the bandgap of perovskite materials can be easily tuned by adjusting A-site cations, B-site metal ions and X-site halide ions in the perovskite crystal, wide-bandgap (WBG) perovskites have been widely employed as the top-cell for multi-junction solar cells, such as perovskite–silicon,2,3 perovskite–perovskite,4,5 perovskite–CIGS,6,7 and perovskite–organic8,9 tandem solar cells. In addition, owing to the unprecedented development of the Internet of Things (IoT), the application of solar cells for indoor dim-light energy harvesting has attracted much attention recently.10,11 Because the fluorescence spectrum of a fluorescent lamp or a light-emitting diode is only limited in the visible range, the absorption spectrum of the WBG perovskite can match the spectrum of indoor light sources well. At the same time, a wider bandgap makes WBG PSCs have the potential to obtain higher voltage, making them more suitable for indoor photovoltaic applications than low bandgap PSCs.12–14 Hence, the WBG perovskite also shows great potential for indoor photovoltaic applications.The mixed-A-site cation strategy containing formamidinium (FA), methylamine (MA) and cesium (Cs) was usually employed to improve the phase- and photo-stability of the perovskite layer. For this reason, the majority of studies on WBG PSCs have adopted multiple-cation-mixed perovskites.14–19 In this work, we deposited the WBG perovskite film by interdiffusion of a precursor stacking layer, which was proposed by Xiao et al.20 In this interdiffusion approach, the MAI/isopropanol solution was spin-coated onto a wet PbI2 layer and then the bilayer was diffused during the annealing process to form perovskite crystals. It is not necessary to require PbI2 to form a porous structure for the interdiffusion approach, which is different from the conventional two-step deposition method. Meanwhile, there is no need to use an anti-solvent, which makes it more suitable for large-scale production in the future.
However, since cesium halides are insoluble in isopropanol, cesium halides can only be dissolved in PbBr2 and PbI2 precursors in this interdiffusion approach. Cesium halides tend to aggregate with PbBr2 and PbI2 rapidly to form an inorganic perovskite core during the first spin-coating process, leading to rapid nucleation of the perovskite. Hence, further delaying the formation of perovskite nucleation and controlling the perovskite crystallization is the key for improving the device performance of WBG PSCs. It is recognized that, in the process of preparing perovskites by the solution method, forming an appropriate intermediate phase during the crystallization process can effectively improve the crystallization of the perovskite.21 The PbI2–DMSO and PbI2–NMP complexes were widely adopted as the intermediate phase to slow down the reaction of PbI2 and MAI or FAI, thus promoting the crystallization of the perovskite.22–24 Recently, formate (HCOO−) based salts and formic acid were introduced as precursors, additives, or solvents to form high-quality perovskite films.25–29 The strong interaction between HCOO− and PbI2 induces an intermediate phase formed and retards the crystal growth of the perovskite, resulting in a high-quality perovskite layer and excellent device performance. Inspired by this, we introduced CsFa as the Cs precursor using the sequential spin-coated interdiffusion method to prepare the WBG perovskite layer. At the same time, HPbBr3 with an equal molar ratio of CsFa was added to the first-step solution.
The WBG perovskite derived from the CsFa precursor (CsFa-perovskite) showed stronger crystallization, reduced trap-state density and better optoelectrical properties in comparison to the WBG perovskite derived from the CsBr precursor (CsBr-perovskite). The charge carrier dynamics of the CsFa-perovskite and CsBr-perovskite were analyzed by transient absorption (TA) measurements. Meanwhile, the TA measurement results also demonstrated that the CsFa-perovskite possesses better photo-stability than the CsBr-perovskite. Finally, the 1.63 eV WBG PSCs based on the CsFa-perovskite also achieve more superior photovoltaic performance (20.01% under 1 sun illumination and 38.52% under LED light illumination) and long-term stability.
Experimental
Materials
PbI2, PbBr2, HPbBr3, FAI, MABr, MACl, Spiro-OMeTAD, TBP, Li-TFSI and Co-TFSI were brought from Xi’an Polymer Light Technology Corp. Cesium formate was purchased from Macklin Biochemical Technology Corp. Extra dry isopropyl alcohol, chlorobenzene, acetonitrile, dimethyl sulfoxide (DMSO) and N,N-dimethylformamide (DMF) were purchased from Acros Organics. Tin(IV) oxide (15% in H2O colloidal dispersion) was purchased from Alfa Aesar.Precursor preparation
SnO2 precursor solution was prepared by diluting tin(IV) oxide (15% in H2O colloidal dispersion) for 2 times with ultra-pure water. For the 1.63 eV WBG perovskite fabricated using the CsBr precursor, the first-step precursor was prepared by dissolving 13.85 mg of CsBr, 479.44 mg of PbI2, and 95.42 mg of PbBr2 in 1 mL of DMF and DMSO mixed solution (DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO = 95:5). For the 1.63 eV WBG perovskite fabricated using the CsFa precursor, the first-step precursor was prepared by dissolving 11.57 mg of CsFa, 29.12 mg of HPbBr3, 479.44 mg of PbI2, and 71.57 mg of PbBr2 in 1 mL of DMF and DMSO mixed solution (DMF:DMSO = 95:5). For the 1.7 eV WBG perovskite fabricated using the CsFa precursor, the first-step precursor was prepared by dissolving 11.57 mg of CsFa, 29.12 mg of HPbBr3, 389.55 mg of PbI2, and 143.15 mg of PbBr2 in 1 mL of DMF and DMSO mixed solution (DMF:DMSO = 95:5). The first-step precursor solution was stirred at 60 °C for 2 h and then filtered before use. For all these WBG perovskites, the second-step precursors were obtained by dissolving 50 mg of FAI, 6.3 mg of MABr and 6.3 mg of MACl in isopropyl alcohol.
:DMSO = 95:5). For the 1.63 eV WBG perovskite fabricated using the CsFa precursor, the first-step precursor was prepared by dissolving 11.57 mg of CsFa, 29.12 mg of HPbBr3, 479.44 mg of PbI2, and 71.57 mg of PbBr2 in 1 mL of DMF and DMSO mixed solution (DMF:DMSO = 95:5). For the 1.7 eV WBG perovskite fabricated using the CsFa precursor, the first-step precursor was prepared by dissolving 11.57 mg of CsFa, 29.12 mg of HPbBr3, 389.55 mg of PbI2, and 143.15 mg of PbBr2 in 1 mL of DMF and DMSO mixed solution (DMF:DMSO = 95:5). The first-step precursor solution was stirred at 60 °C for 2 h and then filtered before use. For all these WBG perovskites, the second-step precursors were obtained by dissolving 50 mg of FAI, 6.3 mg of MABr and 6.3 mg of MACl in isopropyl alcohol.
Device fabrication
The pre-cleaned ITO was ultraviolet ozone-treated for 15 min. The SnO2 precursor was spin-coated onto the ITO substrate and then annealed at 150 °C for 30 min. Then, the obtained ITO/SnO2 was placed in an N2-filled glove box and the deposition of the perovskite layer was completed in the glove box. 40 μL of the first-step precursor solution was spin-coated onto the ITO/SnO2 substrate at 3000 rpm for 30 s. After spin-coating stops, 80 μL of the second-step precursor solution was added onto the substrate and then spin-coated at 3000 rpm for 30 s. The wet film was annealed at 150 °C for 10 min to form the WBG perovskite layer. Subsequently, the hole transport layer was deposited by spin-coating the spiro-OMeTAD solution (90 mg of spiro-OMeTAD, 35 μL of TBP, 23 μL of Li-TFSI (520 mg mL−1 in acetonitrile), and 10 μL of Co-TFSI (375 mg mL−1 in acetonitrile) dissolved in 1 mL of chlorobenzene) at 3000 rpm for 30 s. Finally, the 100 nm Ag electrode was thermally evaporated at a pressure of 3 × 10−4 Pa.Instrumentation and characterization
Ultraviolet-visible (UV-vis) absorption spectra were recorded using a Shimadzu UV-3600 spectrophotometer. Photoluminescence (PL) and time-resolved photoluminescence (TRPL) measurements were measured using an Edinburgh Fluorescence Spectrometer (FLS 1000). X-ray diffraction (XRD) was measured using a PANalytical Empyrean diffractometer. Scanning electron microscopy (SEM) images were obtained using a Zeiss Sigma 300 field emission scanning electron microscope at an acceleration voltage of 3 kV. Atom force microscopy (AFM) measurements were performed using a HORIBA AIST-NT scanning probe microscopy system. TA spectra were recorded using a regenerative amplified Ti:sapphire laser system (Coherent) as the laser source and EOS spectrometer (Ultrafast Systems LLC) as the spectrometer. The monochromatic high-energy pump pulse used to trigger the photoreaction of the sample was set at 380 nm. The current–voltage (J–V) curves were obtained using a Keithley 2400 digital source meter under standard one sun AM1.5G (100 mW cm−2) illumination. The irradiance and illumination intensity of LED light (IKEA of Sweden AB, LED 1919G8 E27) were measured using a Spectral Irradiance Colorimeter (EVERFINE Corporation, SPIC-300AW). The active area of devices for the J–V test is 4 mm2. The scan rate of the J–V test is 150 mV s−1. The J–V curves of the SCLC device were measured in a dark environment. The external quantum efficiency (EQE) was measured using a Zolix SCS10-X150-DSSC system. The 13C MAS NMR spectrum was tested using a Magnet System 400’89 Ascend.Results and discussion
We prepared WBG perovskite thin films using CsBr and CsFa as Cs precursors. The proportion of Cs was controlled at 5% and the bandgap of the perovskite was adjusted by changing the ratio of I:Br. We choose WBG perovskites with band gaps of 1.63 and 1.70 eV for experiments in that previous works have studied that 1.63–1.70 eV WBG perovskites are ideal top cells for perovskite–silicon monolithically tandem solar cells.2,30–32 In order to simplify the analysis, the following analysis is based on the 1.63 eV perovskite. Firstly, to investigate the morphology of the perovskite film derived from different precursors, SEM and AFM measurements were performed. As shown in Fig. 1(a) and (b), the CsFa-perovskite shows larger crystal grain size and more compact crystal grains compared to the CsBr-perovskite. The increased crystal size of the CsFa-perovskite can be ascribed to that of the complex HCOOH·Cs+ was formed during the first-step spin-coating,25 which can impede the Cs+ coordinate with Pb2+, I− and Br− to form the perovskite core. Besides, HCOO− can also coordinate with Pb2+ to form the complex HCOO·Pb+.28 The same information about the change of the film morphology can be verified from AFM images (Fig. 1(c) and (d)). Although the CsFa-perovskite exhibits a large grain size than the CsBr-perovskite, the root-mean-square roughness (RMS) of the CsFa-perovskite (18.10 nm) is little smaller than that of the CsBr-perovskite (18.77 nm), meaning the more smoother surface of the CsFa-perovskite. Then, the crystallization of the perovskite was studied by XRD. Fig. 1(e) illustrates the X-ray diffraction spectra of the CsFa-perovskite and the CsBr-perovskite. There are obvious diffraction peaks at 14°, 28.3°, 31.7° and 40.5° in the two XRD spectra, which are related to the (110), (220), (312) and (224) lattice planes of the perovskite. However, the intensity of the XRD peaks of the CsFa-perovskite is stronger than that of the CsBr-perovskite, demonstrating stronger crystallization of the CsFa-perovskite. Fig. 2 illustrates the schematics of CsBr-perovskite and CsFa-perovskite crystallization processes. For the CsFa-perovskite, during the annealing process, the HCOO·Pb+ and HCOOH·Cs+ complexes need to be de-coordinated before they crystallize with FA+, MA+ and halogen to form the perovskite. Then, HCOO− is replaced by I− (Br−); HCOO− is coordinated with H+ to form HCOOH and volatilizes during the annealing process. Both processes are favorable to slow down the crystallization rate of the perovskite and enlarge the crystal grain size. The first-step precursor film was prepared by annealing the first-step precursor solution at 150 °C for 10 min. Because we added HPbBr3 to the first precursor solution, some studies suggest that HPbBr3 may be DMAPbBr3, thereby introducing DMA ions during the preparation of the perovskite.33 Therefore, we annealed the first step solution at 150 °C and dried it for solid-state nuclear magnetic resonance (NMR) analysis. The 13C MAS NMR spectrum of the first-step precursor film with CsFa and HPbBr3 shows that DMA is not present in the product (Fig. S1, ESI†). | ||
| Fig. 1 SEM images of the (a) CsFa-perovskite and (b) CsBr-perovskite. AFM topography images of the (c) CsFa-perovskite and (d) CsBr-perovskite. (e) XRD spectra of the CsFa-perovskite and CsBr-perovskite. | ||
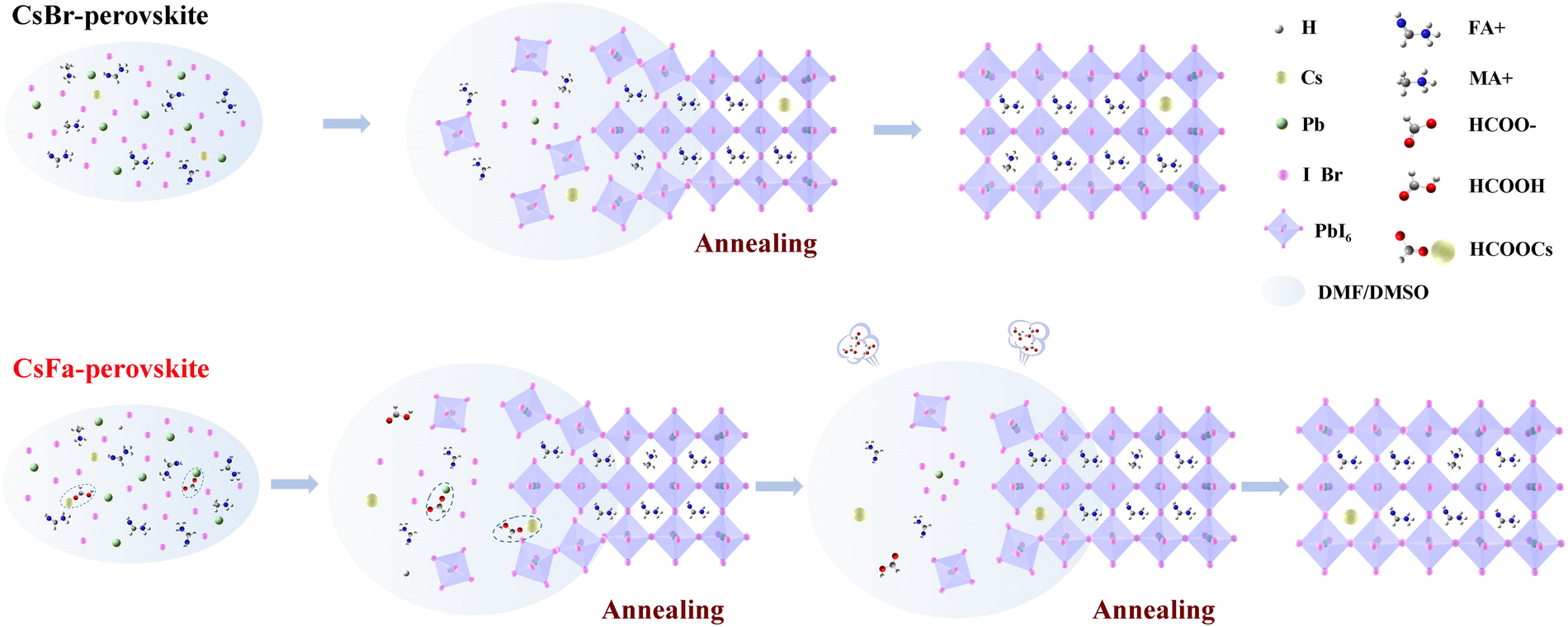 | ||
| Fig. 2 Schematics of the CsBr-perovskite and CsFa-perovskite crystallization processes. | ||
Besides the morphology and crystallization, the photophysical properties of the active layer are the other important factors affecting the final device performance.34,35 The bandgap of the perovskite was detected using UV-vis absorption spectra measurements. Fig. 3(a) and (b) show the UV-vis absorption spectra and Tauc plot bandgap of the CsBr-perovskite and the CsFa-perovskite. The UV-vis absorption spectra of the CsBr-perovskite and the CsFa-perovskite exhibit the same absorption edge. According to the Tauc plot, the bandgap of the CsBr-perovskite and the CsFa-perovskite is about 1.63 eV. Then, to study the changes in the photophysical properties of the CsFa-perovskite and the CsBr-perovskite, steady-state PL and TRPL were conducted. As shown in Fig. 3(c), the CsFa-perovskite and the CsBr-perovskite show strong PL centers at 754 and 752 nm, respectively. In contrast, the PL intensity of the CsFa-perovskite is stronger than that of the CsBr-perovskite. The enhanced PL intensity can be interpreted as the larger crystal size of the CsFa-perovskite reducing the generation of grain boundaries and more grain boundaries are often accompanied by more defects and increased non-radiative recombination. The TRPL decay curves in Fig. 3(d) also reflect the same trend. By fitting the decay curves with a single-exponential function, the PL lifetimes of the CsBr-perovskite and the CsFa-perovskite are 952 and 557 ns, respectively. In order to compare the trap-state densities of the CsBr-perovskite and the CsFa-perovskite further intuitively, the hole-only and electron-only space-charge-limited current (SCLC) devices were fabricated with the structure of ITO/PEDOT: PSS/perovskite/Spiro-OMeTAD/Ag and ITO/SnO2/perovskite/PCBM/Ag, respectively. The trap-state density (nt) can be calculated according to the following equation:36
| nt = 2(VTFLεε0)/(eL2) |
 | ||
| Fig. 3 (a) UV-vis absorption spectra and (b) Tauc plot bandgap of the CsBr-perovskite and C the sFa-perovskite. (c) PL and (d) TRPL curves of the CsBr-perovskite and the CsFa-perovskite. J–V curves of the (e) hole-only and (f) electron-only devices. | ||
Then, to understand the excited state dynamics of charge carriers, the femtosecond transient absorption (fs-TA) spectroscopy of the CsBr-perovskite and the CsFa-perovskite was studied. The CsBr-perovskite and CsFa-perovskite films for the fs-TA test were deposited on the quartz substrate. As shown in Fig. 4(a) and (b), both the CsBr-perovskite and the CsFa-perovskite show photobleaching at 460–480 and 740–750 nm, which correspond to the absorption of PbI2 and the perovskite. In comparison to the CsBr-perovskite, the CsFa-perovskite exhibit a weaker photobleaching peak at 470–480 nm and a stronger photobleaching peak at 740–750 nm, which verified the crystallization information indicated in the XRD measurement. As the perovskite film for the fs-TA measurement was illuminated using a 375 nm pump for about 30 min in ambient air, the fs-TA measurement is also an effective tool to check the photostability of the perovskite.17Fig. 4(c) and (d) display the transient absorption spectra of the CsBr-perovskite and the CsFa-perovskite after excitation at 5 ps, 50 ps and 1 ns. We found that, within a delay time of 10 ps, the CsBr-perovskite film shows the wider the bleaching peak, which can be divided into two peaks at 742 nm and 750 nm. Then, with a delay time increasing to 100 ps, the 740 nm peak disappears and only the 750 nm peak remains. However, the CsFa-perovskite film shows a bleaching peak that remains consistent at 742 nm. These results reveal that the CsFa-perovskite film possesses better phase stability under light illumination. According to the bleaching peak changes, we recorded the transient absorption decay kinetics of the CsFa-perovskite at 742 nm and the CsBr-perovskite at 742 and 750 nm and displayed in Fig. 4(e). For the CsBr-perovskite, whether it is at 742 nm or 750 nm, its ground state bleaching peak decays faster than the CsFa-perovskite. The faster decay of the ground state bleaching peak can be attributed to that more trap-assisted recombination occurs in the CsBr-perovskite, which is contented with the PL and TRPL results. Then, to study the relaxation dynamics, the decay kinetics of the CsFa-perovskite and the CsBr-perovskite (Fig. 4(e)) were fitted based on the equation y = y0 + A1exp(−x/τ1) + A2exp(−x/τ2) and the kinetic fit parameters are summarized in Table S1 (ESI†). The time constants, τ1 and τ2, of the CsBr-perovskite are much smaller than those of the CsFa-perovskite. The fast component can be attributed to charge carrier trapping at perovskite grain boundaries and the longer time constants (τ1) mean longer recombination times.37–39 Hence, longer time constants (τ1) of the CsFa-perovskite confirm that the probability of carrier trapping and recombination at the perovskite grain boundary is lower within the CsFa-perovskite.
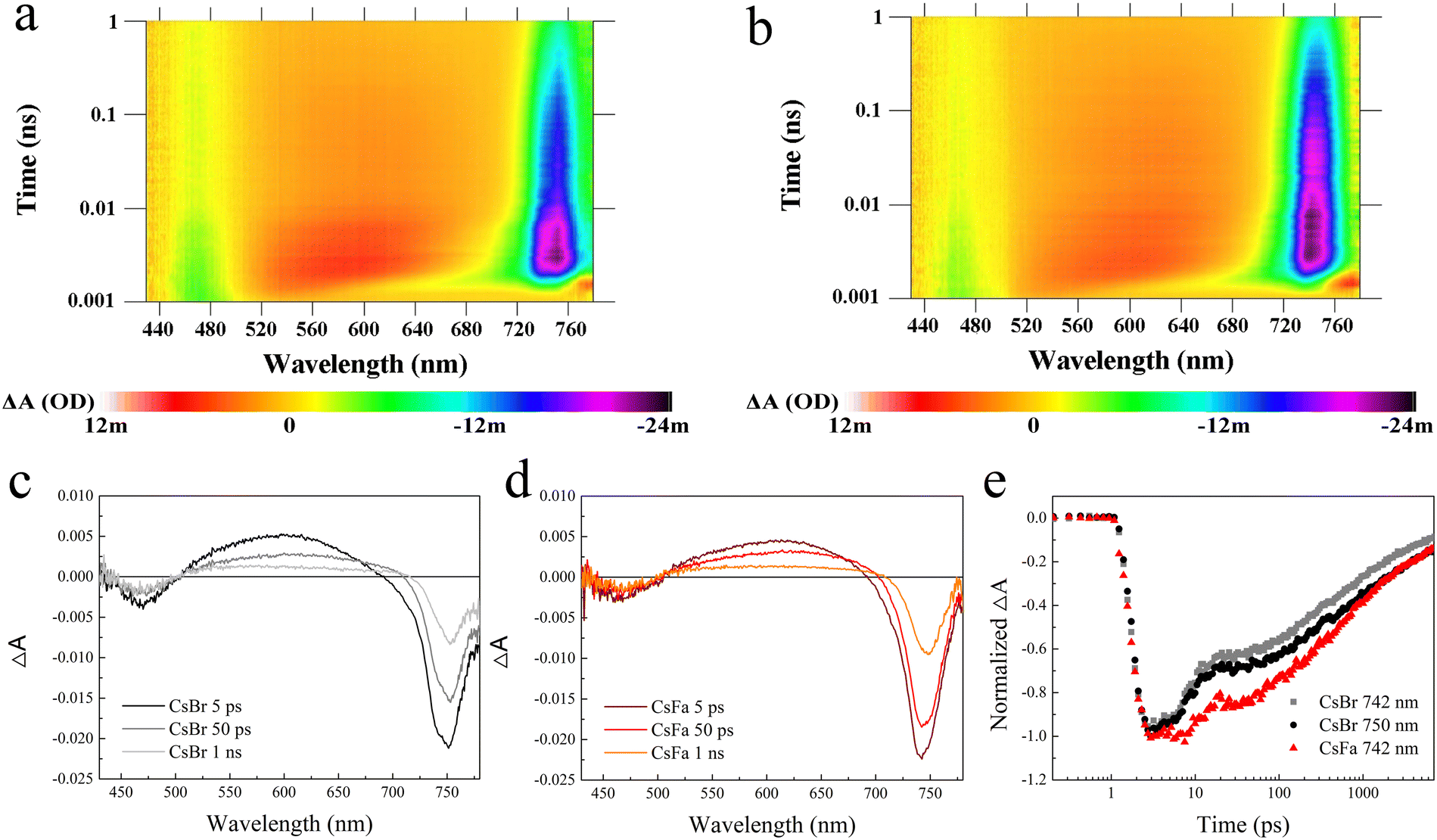 | ||
| Fig. 4 fs-TA studies of the (a) CsBr-perovskite and (b) CsFa-perovskite on quartz substrate. Transient absorption spectra of the (c) CsBr-perovskite and (d) CsFa-perovskite after excitation at 5 ps, 50 ps and 1 ns. (e) Transient absorption decay kinetics of the CsFa-perovskite and CsBr-perovskite. | ||
Based on the 1.63 eV CsBr-perovskite and CsFa-perovskite, the PSCs with the structure of ITO/SnO2/perovskite/Spiro-OMeTAD/Ag were fabricated. The J–V curves of the PSCs are shown in Fig. 5(a) and the corresponding photovoltaic parameters are listed in Table 1. The PSCs based on the 1.63 eV CsBr-perovskite achieve a PCE of 17.43% for a forward scan with a short-circuit current density (Jsc) of 20.73 mA cm−2, an open-circuit voltage (Voc) of 1.165 V, a fill factor (FF) of 72.18% and a PCE of 18.27% for a reverse scan with a Jsc of 20.73 mA cm−2, a Voc of 1.175 V and an FF of 74.99%. In comparison, the optimized PSCs based on the 1.63 eV CsFa-perovskite achieved the highest PCE of 20.01% (Jsc = 21.58 mA cm−2, Voc = 1.204 V, FF = 77.01%) for a reverse scan and that of 19.47% (Jsc = 21.43 mA cm−2, Voc = 1.205 V, FF = 75.41%) for a forward scan. We also attempted to fabricate 1.63 eV PSCs with a large active area of 1 cm2, and the final devices achieved PCEs of 17.26% (CsFa-perovskite) and 15.26% (CsBr-perovskite) (Fig. S2, ESI†). Fig. S3 (ESI†) shows the distribution of the photovoltaic parameters of the 20 devices. From the statistical results, the Jsc and FF of CsFa-perovskite devices are generally higher than those of CsBr-perovskite devices. The promoted Jsc and FF can be ascribed to the improved crystallization and reduced trap-state density of the CsFa-perovskite film, thus reducing the Jsc and FF loss caused by carrier recombination. To analyze the charge recombination mechanism, the light-intensity (Plight) dependence of Jsc and Voc is introduced. The data of Jscversus Plight are plotted following the formula Jsc∝(Plight)α (Fig. S4a, ESI†), the recombination parameters α of CsBr-perovskite and CsFa-perovskite PSCs are 0.947 and 0.961, respectively, suggesting lower bimolecular recombination in CsFa-perovskite PSCs and thus resulting in higher FF.40,41 The data of Vocversus Plight are linearly fitted according to the formula Voc∝nkBT/qln(Plight) (Fig. S4b, ESI†), where q, T and kB represent the elementary charge, the temperature in kelvin and the Boltzmann constant, respectively. The slopes of the CsBr-perovskite and CsFa-perovskite PSCs are 1.32kBT and 1.27kBT, respectively, indicating higher monomolecular or trap-assisted recombination in the CsBr-perovskite.42,43Fig. 5(b) shows the corresponding external quantum efficiency (EQE) and integrated current density (Jint) curves and the Jint values calculated from the EQE test are 19.56 and 20.13 mA cm−2. The time dependence of the stabilized power output of these two PSCs under maximum power-point conditions was also investigated. As shown in Fig. 5(c), the PSCs with the CsBr-perovskite film show more serious PCE loss after 1000 s. The PCE loss is due to the photoinduced phase separation of the perovskite under continuous illuminations,44 which is consistent with the fs-TA test results discussed above. The obvious morphological differences are also detected by the AFM measurement. As the AFM topography images shown in Fig. S5 (ESI†), the morphology of the CsFa-perovskite and the CsBr-perovskite is quite different after continuous illumination under AM 1.5G sunlight for 7 hours. The two samples were placed in a nitrogen-filled glove box at room temperature and continuously illuminated for 7 h under AM1.5G illumination (100 mW cm−2). The temperature of the samples increased during the continuous illumination and we did not perform additional temperature control. The roughness of both perovskite films is increased after illumination. The change of the CsBr-perovskite is greater and there are obvious convex points on the CsBr-perovskite film (Fig. S5b, ESI†). Besides, we also fabricated 1.70 eV WBG PSCs using the CsFa-perovskite. The J–V curves of 1.70 eV PSCs are shown in Fig. 5(d), which achieve PCEs of 17.22% for the reverse scan and 16.67% for the forward scan. Furthermore, we also characterized the indoor photovoltaic performance of 1.63 eV PSCs. Fig. 5(e) and (f) show the J–V curves of the CsBr-perovskite and the CsFa-perovskite based 1.63 eV PSCs under 2700 K LED light illumination with light intensities of 300 lux (92.25 μW cm−2), 500 lux (153.51 μW cm−2) and 1000 lux (307.30 μW cm−2) and the emission power spectrum of the LED bulb is shown in Fig. S6 (ESI†). The detailed parameters are listed in Table 2. It is clearly showing that the CsFa-perovskite PSCs exhibit better performance than CsBr-perovskite PSCs at different light intensities. The highest PCE of 38.52% for CsFa-perovskite PSCs is achieved. Meanwhile, the best-performing CsFa-perovskite PSCs can also achieve a PCE of 35.41% under cold light illumination (6500 K, 1000 lux) (Fig. S7 and Table S2, ESI†). We compared our device performance with some recently reported photovoltaic performance of the state-of-the-art WBG PSCs (bandgap around ∼1.63 eV) (Table S3, ESI†) and efficient indoor-applied PSCs (Table S4, ESI†), respectively. The PCEs of our devices are among the best performances, especially the PCE under indoor light illumination.
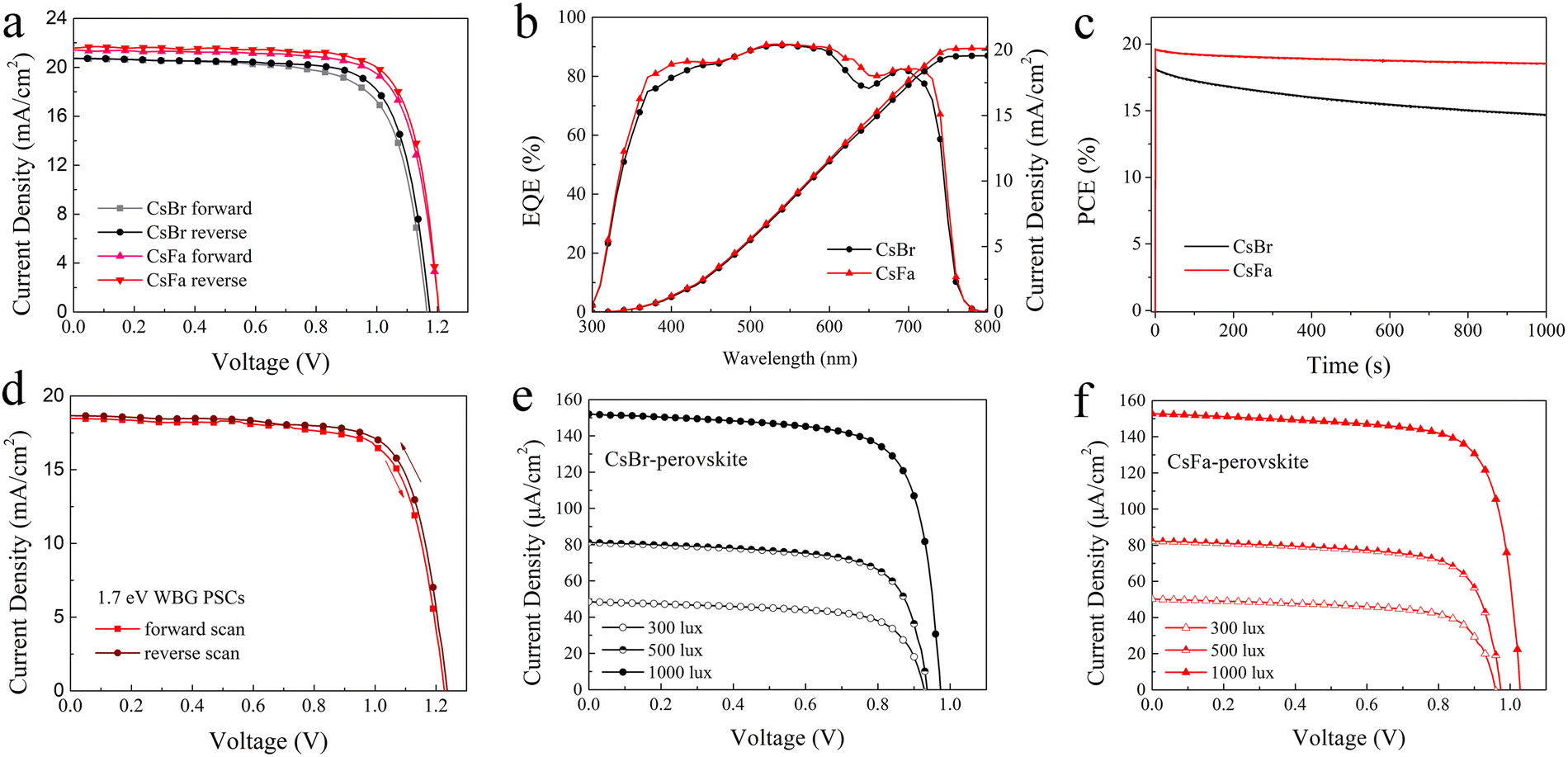 | ||
| Fig. 5 (a) J–V curves (AM 1.5G, 100 mW cm−2) and (b) EQE of 1.63 eV PSCs. (c) Stabilized power output of 1.63 eV PSCs under maximum power-point conditions. (d) J–V curves (AM 1.5G, 100 mW cm−2) of 1.7 eV PSCs based on the CsFa-perovskite. J–V curves of the (e) CsBr-perovskite and (f) CsFa-perovskite based 1.63 eV PSCs under a 2700 K LED light illumination condition. | ||
| J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | ||
|---|---|---|---|---|---|
| CsBr-perovskite | Forward | 20.73 | 1.165 | 72.18 | 17.43 |
| Reverse | 20.73 | 1.175 | 74.99 | 18.27 | |
| CsFa-perovskite | Forward | 21.43 | 1.205 | 75.41 | 19.47 |
| Reverse | 21.58 | 1.204 | 77.01 | 20.01 |
| Light intensity (lux) | Power density (μW cm−2) | J sc (μA cm−2) | V oc (V) | FF (%) | PCE (%) | |
|---|---|---|---|---|---|---|
| CsBr-perovskite | 300 | 92.25 | 48.44 | 0.928 | 68.40 | 33.33 |
| 500 | 153.51 | 81.17 | 0.936 | 69.90 | 34.59 | |
| 1000 | 307.30 | 152.02 | 0.973 | 73.45 | 35.35 | |
| CsFa-perovskite | 300 | 92.25 | 50.14 | 0.959 | 69.74 | 36.35 |
| 500 | 153.51 | 82.25 | 0.974 | 71.88 | 37.51 | |
| 1000 | 307.30 | 152.79 | 1.028 | 75.37 | 38.52 |
Finally, the long-term stability of 1.63 eV WBG PSCs was investigated. The PCE decay of samples stored in a N2-filled glove box and under an ambient atmosphere with a relative humidity (RH) of <10% was recorded and is shown in Fig. S8 (ESI†). All the samples for the stability test were stored in a dark environment at a temperature of 20–25 °C. After aging in the glove box for 1080 hours, the PCE of the WBG PSCs based on the CsFa-perovskite and the CsBr-perovskite retains 98.2% and 89.7% of the original value. For the WBG PSCs based on the CsFa-perovskite and CsBr-perovskite aging ambient atmosphere with an RH of <10%, their PCE retains the 84.2% and 69.0% of the initial PCE, respectively. Because the degradation of the PSCs is assisted by increased trap states,45 the improved stability of PSCs based on the CsFa-perovskite can be ascribed to the decreased trap-states of the CsFa-perovskite compared with those of the CsBr-perovskite.
Conclusion
In summary, we developed efficient WBG PSCs via interdiffusion of the sequential spin-coated precursor stacking layer. To promote the crystallization of the perovskite, CsFa is introduced as the Cs precursor in the first-step. Since the HCOO·Pb+ and HCOOH·Cs+ complex formation and HCOOH volatilization occurs during the crystallization process, the perovskite nucleation formation rate is retarded and the crystal growth of the perovskite is enhanced. The CsFa-perovskite shows stronger crystallization, larger grain size and reduced trap-state density compared to the CsBr-perovskite. Moreover, fs-TA reveals that the CsFa-perovskite film also possesses better phase stability under light illumination. Finally, the 1.63 eV WBG PSCs based on the CsFa-perovskite achieve a significant PCE of 20.01%, which is obviously higher than that of PSCs based on the CsBr-perovskite (18.27%). The performance of CsFa-perovskite PSCs is also superior to that of CsBr-perovskite PSCs under warm-light LED illumination and the highest PCE of 38.52% (1000 lux) is achieved for CsFa-perovskite based PSCs. In addition, the WBG PSCs based on the CsFa-perovskite also exhibit better long-term stability in an N2-filled glove box and under an ambient atmosphere with a relative humidity (RH) of <10%. Our results indicate that CsFa as the Cs precursor is beneficial to improve the quality of the perovskite film and device performance.Author contributions
All authors gave approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank support from the National Natural Science Foundation of China (NSFC, No. 22109142), the Natural Science Foundation of Henan Province (202300410429), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB36000000), and the Outstanding Talent Research Fund of Zhengzhou University (32340035 and 32340100).References
- Best research-cell efficiencies NREL (2022); (http://www.nrel.gov/pv/assets/images/efficiency_chart.jpg).
- K. A. Bush, A. F. Palmstrom, Z. J. Yu, M. Boccard, R. Cheacharoen, J. P. Mailoa, D. P. McMeekin, R. L. Z. Hoye, C. D. Bailie, T. Leijtens, I. M. Peters, M. C. Minichetti, N. Rolston, R. Prasanna, S. Sofia, D. Harwood, W. Ma, F. Moghadam, H. J. Snaith, T. Buonassisi, Z. C. Holman, S. F. Bent and M. D. McGehee, Nat. Energy, 2017, 2, 17009 CrossRef CAS.
- A. Al-Ashouri, E. Köhnen, B. Li, A. Magomedov, H. Hempel, P. Caprioglio, J. A. Márquez, A. B. Morales Vilches, E. Kasparavicius, J. A. Smith, N. Phung, D. Menzel, M. Grischek, L. Kegelmann, D. Skroblin, C. Gollwitzer, T. Malinauskas, M. Jošt, G. Matič, B. Rech, R. Schlatmann, M. Topič, L. Korte, A. Abate, B. Stannowski, D. Neher, M. Stolterfoht, T. Unold, V. Getautis and S. Albrecht, Science, 2020, 370, 1300–1309 CrossRef CAS PubMed.
- R. Lin, K. Xiao, Z. Qin, Q. Han, C. Zhang, M. Wei, M. I. Saidaminov, Y. Gao, J. Xu, M. Xiao, A. Li, J. Zhu, E. H. Sargent and H. Tan, Nat. Energy, 2019, 4, 864–873 CrossRef CAS.
- P. McMeekin David, G. Sadoughi, W. Rehman, E. Eperon Giles, M. Saliba, T. Hörantner Maximilian, A. Haghighirad, N. Sakai, L. Korte, B. Rech, B. Johnston Michael, M. Herz Laura and J. Snaith Henry, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- H. Shen, T. Duong, J. Peng, D. Jacobs, N. Wu, J. Gong, Y. Wu, S. K. Karuturi, X. Fu, K. Weber, X. Xiao, T. P. White and K. Catchpole, Energy Environ. Sci., 2018, 11, 394–406 RSC.
- Q. Han, Y. T. Hsieh, L. Meng, J. L. Wu, P. Sun, E. P. Yao, S. Y. Chang, S. H. Bae, T. Kato, V. Bermudez and Y. Yang, Science, 2018, 361, 904–908 CrossRef CAS PubMed.
- X. Chen, Z. Jia, Z. Chen, T. Jiang, L. Bai, F. Tao, J. Chen, X. Chen, T. Liu, X. Xu, C. Yang, W. Shen, W. E. I. Sha, H. Zhu and Y. Yang, Joule, 2020, 4, 1594–1606 CrossRef CAS.
- S. Qin, C. Lu, Z. Jia, Y. Wang, S. Li, W. Lai, P. Shi, R. Wang, C. Zhu, J. Du, J. Zhang, L. Meng and Y. Li, Adv. Mater., 2022, 34, 2108829 CrossRef CAS PubMed.
- C. Y. Chen, J. H. Chang, K. M. Chiang, H. L. Lin, S. Y. Hsiao and H. W. Lin, Adv. Funct. Mater., 2015, 25, 7064–7070 CrossRef CAS.
- M. Li, C. Zhao, Z.-K. Wang, C.-C. Zhang, H. K. H. Lee, A. Pockett, J. Barbé, W. C. Tsoi, Y.-G. Yang, M. J. Carnie, X.-Y. Gao, W.-X. Yang, J. R. Durrant, L.-S. Liao and S. M. Jain, Adv. Energy Mater., 2018, 8, 1801509 CrossRef.
- M.-J. Wu, C.-C. Kuo, L.-S. Jhuang, P.-H. Chen, Y.-F. Lai and F.-C. Chen, Adv. Energy Mater., 2019, 9, 1901863 CrossRef CAS.
- R. Cheng, C.-C. Chung, H. Zhang, F. Liu, W.-T. Wang, Z. Zhou, S. Wang, A. B. Djurišić and S.-P. Feng, Adv. Energy Mater., 2019, 9, 1901980 CrossRef CAS.
- Z. Li, J. Zhang, S. Wu, X. Deng, F. Li, D. Liu, C. C. Lee, F. Lin, D. Lei, C.-C. Chueh, Z. Zhu and A. K. Y. Jen, Nano Energy, 2020, 78, 105377 CrossRef CAS.
- F. Peña-Camargo, P. Caprioglio, F. Zu, E. Gutierrez-Partida, C. M. Wolff, K. Brinkmann, S. Albrecht, T. Riedl, N. Koch, D. Neher and M. Stolterfoht, ACS Energy Lett., 2020, 5, 2728–2736 CrossRef.
- Y. Lin, B. Chen, F. Zhao, X. Zheng, Y. Deng, Y. Shao, Y. Fang, Y. Bai, C. Wang and J. Huang, Adv. Mater., 2017, 29, 1700607 CrossRef PubMed.
- H. Tan, F. Che, M. Wei, Y. Zhao, M. I. Saidaminov, P. Todorović, D. Broberg, G. Walters, F. Tan, T. Zhuang, B. Sun, Z. Liang, H. Yuan, E. Fron, J. Kim, Z. Yang, O. Voznyy, M. Asta and E. H. Sargent, Nat. Commun., 2018, 9, 3100 CrossRef PubMed.
- G. Kim, C. S. Moon, T.-Y. Yang, Y. Y. Kim, J. Chung, E. H. Jung, T. J. Shin, N. J. Jeon, H. H. Park and J. Seo, Sol. RRL, 2020, 4, 2000033 CrossRef CAS.
- L. Wang, G. Wang, Z. Yan, J. Qiu, C. Jia, W. Zhang, C. Zhen, C. Xu, K. Tai, X. Jiang and S. Yang, Sol. RRL, 2020, 4, 2000098 CrossRef CAS.
- Z. Xiao, C. Bi, Y. Shao, Q. Dong, Q. Wang, Y. Yuan, C. Wang, Y. Gao and J. Huang, Energy Environ. Sci., 2014, 7, 2619–2623 RSC.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- W. Fu, J. Yan, Z. Zhang, T. Ye, Y. Liu, J. Wu, J. Yao, C.-Z. Li, H. Li and H. Chen, Sol. Energy Mater. Sol. Cells, 2016, 155, 331–340 CrossRef CAS.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- Y. Jo, K. S. Oh, M. Kim, K.-H. Kim, H. Lee, C.-W. Lee and D. S. Kim, Adv. Mater. Interfaces, 2016, 3, 1500768 CrossRef.
- C. Y. Duan, J. Cui, M. M. Zhang, Y. Han, S. M. Yang, H. Zhao, H. T. Bian, J. X. Yao, K. Zhao, Z. K. Liu and S. Z. Liu, Adv. Energy Mater., 2020, 10, 2000691 CrossRef CAS.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- L. Chao, Y. Xia, B. Li, G. Xing, Y. Chen and W. Huang, Chem, 2019, 5, 995–1006 CAS.
- J. Y. Seo, T. Matsui, J. S. Luo, J. P. Correa-Baena, F. Giordano, M. Saliba, K. Schenk, A. Ummadisingu, K. Domanski, M. Hadadian, A. Hagfeldt, S. M. Zakeeruddin, U. Steiner, M. Gratzel and A. Abate, Adv. Energy Mater., 2016, 6, 1600767 CrossRef.
- L. Meng, Q. Wei, Z. Yang, D. Yang, J. Feng, X. Ren, Y. Liu and S. Liu, J. Energy Chem., 2020, 41, 43–51 CrossRef.
- B. Chen, S.-W. Baek, Y. Hou, E. Aydin, M. De Bastiani, B. Scheffel, A. Proppe, Z. Huang, M. Wei, Y.-K. Wang, E.-H. Jung, T. G. Allen, E. Van Kerschaver, F. P. García de Arquer, M. I. Saidaminov, S. Hoogland, S. De Wolf and E. H. Sargent, Nat. Commun., 2020, 11, 1257 CrossRef CAS PubMed.
- L. Mazzarella, Y. H. Lin, S. Kirner, A. B. Morales-Vilches, L. Korte, S. Albrecht, E. Crossland, B. Stannowski, C. Case, H. J. Snaith and R. Schlatmann, Adv. Energy Mater., 2019, 9, 1803241 CrossRef.
- B. Chen, Z. Yu, K. Liu, X. Zheng, Y. Liu, J. Shi, D. Spronk, P. N. Rudd, Z. Holman and J. Huang, Joule, 2019, 3, 177–190 CrossRef CAS.
- W. Ke, I. Spanopoulos, C. C. Stoumpos and M. G. Kanatzidis, Nat. Commun., 2018, 9, 4785 CrossRef PubMed.
- Z. Wang, K. Gao, Y. Kan, M. Zhang, C. Qiu, L. Zhu, Z. Zhao, X. Peng, W. Feng, Z. Qian, X. Gu, A. K. Y. Jen, B. Z. Tang, Y. Cao, Y. Zhang and F. Liu, Nat. Commun., 2021, 12, 332 CrossRef CAS PubMed.
- Y. Sun, T. Liu, Y. Kan, K. Gao, B. Tang and Y. Li, Small Sci., 2021, 1, 2100001 CrossRef.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- L. Wang, C. McCleese, A. Kovalsky, Y. Zhao and C. Burda, J. Am. Chem. Soc., 2014, 136, 12205–12208 CrossRef CAS PubMed.
- E. Serpetzoglou, I. Konidakis, G. Kakavelakis, T. Maksudov, E. Kymakis and E. Stratakis, ACS Appl. Mater. Interfaces, 2017, 9, 43910–43919 CrossRef CAS PubMed.
- N. Jiang, Z. Wang, Y. Zheng, Q. Guo, W. Niu, R. Zhang, F. Huang and D. Chen, Nano Energy, 2022, 97, 107181 CrossRef CAS.
- K. Gao, Y. Kan, X. Chen, F. Liu, B. Kan, L. Nian, X. Wan, Y. Chen, X. Peng, T. P. Russell, Y. Cao and A. K. Y. Jen, Adv. Mater., 2020, 32, 1906129 CrossRef CAS PubMed.
- K. Gao, J. Miao, L. Xiao, W. Deng, Y. Kan, T. Liang, C. Wang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, H. Wu and X. Peng, Adv. Mater., 2016, 28, 4727–4733 CrossRef CAS PubMed.
- A. Tang, W. Song, B. Xiao, J. Guo, J. Min, Z. Ge, J. Zhang, Z. Wei and E. Zhou, Chem. Mater., 2019, 31, 3941–3947 CrossRef CAS.
- R. Ma, K. Zhou, Y. Sun, T. Liu, Y. Kan, Y. Xiao, T. A. Dela Peña, Y. Li, X. Zou, Z. Xing, Z. Luo, K. S. Wong, X. Lu, L. Ye, H. Yan and K. Gao, Matter, 2022, 5, 725–734 CrossRef CAS.
- M. C. Brennan, S. Draguta, P. V. Kamat and M. Kuno, ACS Energy Lett., 2018, 3, 204–213 CrossRef CAS.
- D. Song, J. Ji, Y. Li, G. Li, M. Li, T. Wang, D. Wei, P. Cui, Y. He and J. M. Mbengue, Appl. Phys. Lett., 2016, 108, 093901 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 13C MAS NMR spectrum of the first-step precursor film with CsFa; J–V curves of 1.63 eV PSCs with large active area of 1 cm2; photovoltaic parameters (reverse scan) distribution of 20 individual 1.63 eV WBG PSCs based on CsBr-perovskite and CsFa-perovskite; Jsc and Vocversus light intensity of the 1.63 eV WBG PSCs; AFM topography images of CsFa-perovskite and CsBr-perovskite after continuous illumination under AM 1.5G sunlight for 7 hours; emission power spectrum of the LED bulb; J–V curves of 1.63 eV PSCs under a 6500K LED light illumination condition; normalized PCEs of unencapsulated 1.63 eV WBG PSCs based on CsBr-perovskite and CsFa-perovskite after aging in an N2-filled glovebox for 1080 hours and in an ambient environment (relative humidity <10%) for 1032 hours. See DOI: https://doi.org/10.1039/d2cp02358j |
| This journal is © the Owner Societies 2022 |