
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An in situ study of thermal crystallization of amorphous calcium phosphates†
Lorenzo
Degli Esposti
 a,
Marco
Fosca
b,
Aurélien
Canizares
c,
Leire
Del Campo
c,
Marco
Ortenzi
b,
Alessio
Adamiano
a,
Julietta V.
Rau
b and
Michele
Iafisco
*a
a,
Marco
Fosca
b,
Aurélien
Canizares
c,
Leire
Del Campo
c,
Marco
Ortenzi
b,
Alessio
Adamiano
a,
Julietta V.
Rau
b and
Michele
Iafisco
*a
aInstitute of Science and Technology for Ceramics (ISTEC), National Research Council (CNR), Via Granarolo 64, 48018 Faenza, Italy. E-mail: michele.iafisco@istec.cnr.it
bIstituto di Struttura della Materia, Consiglio Nazionale delle Ricerche (ISM-CNR), Via del Fosso del Cavaliere, 100-00133 Rome, Italy
cCNRS, CEMHTI UPR3079, Univ. Orléans, F-45071 Orléans, France
First published on 19th September 2022
Abstract
The heat-induced crystallization of amorphous calcium phosphate (ACP) is an intriguing process not yet well comprehended. This is because most of the works on this topic are based on ex situ studies where the materials are characterized after the heat and cooldown cycles, thus missing transient structural changes. Here, we used time-resolved energy dispersive X-ray diffraction and infrared spectroscopy to study, for the first time, the thermal crystallization of ACP in situ. The thermal crystallization of two kinds of citrate-stabilized carbonated ACP was studied, as they are promising materials for the preparation of advanced bioceramics. The behavior of these samples was compared to that of two citrate-free ACPs, either doped or non-doped with carbonate ions. Our results evinced that several phenomena occur during ACP thermal annealing. Before crystallization, all ACP samples undergo a decrease in the short-range order process, followed by several internal reorganizations. We have assessed that differently from carbonate-free ACP, carbonated ACPs with and without citrate directly crystallize into a biomimetic poorly crystalline carbonated hydroxyapatite. Citrate-stabilized ACPs in comparison to citrate-free ACPs have a faster hydroxyapatite formation kinetics, which is due to their higher specific surface area. This work reveals the necessity and the potentialities of using in situ techniques to effectively probe complex processes such as the heat-induced crystallization of ACPs.
Introduction
Calcium phosphates are excellent materials for application in medicine, agriculture, environmental remediation, cosmetics, catalysis, and many other sectors.1–3 Their most successful and widespread use is in the form of bioceramics, employed in dentistry and orthopedic surgery.4 Among calcium phosphate phases, there is renewed interest in the metastable amorphous calcium phosphate (ACP). ACP does not have a precise stoichiometry and is represented by the general formula Cax(PO4)y·nH2O, and it usually consists of rounded nanoparticles containing Ca9(PO4)6 clusters surrounded by 3–5 water molecules that “attach” the clusters together.5 The short-range (local) ordered ACP clusters, defined as Posner's clusters, have a structural similarity to some crystalline calcium phosphates, such as hydroxyapatite (HA, Ca5(PO4)3(OH), Ca/P molar ratio = 1.67) or tricalcium phosphate (TCP, Ca3(PO4)2, Ca/P molar ratio = 1.5).6 ACP has found successful application as a biomaterial for dental remineralization, as a component of self-setting injectable bone cements, and as a coating material of orthopedic prostheses.7–10 For these applications, the material is used in its amorphous phase, and then it crystallizes, forming the desired crystalline product in situ.Crystallization of ACP is an intriguing process not yet well comprehended and of great relevance for any application. ACP crystallizes both by reacting with water, as well as by heating.11–13 Thermal crystallization of ACP is a multistep process, which starts with a non-reversible dehydration of its structural water and concludes with the transformation into a crystalline phase, usually occurring in the temperature range between 500 and 800 °C. The thermal crystallization of ACP is thought to be controlled by a solid-state internal lattice reordering mechanism, and usually, the product is TCP, either in the α- or β-form.7 Previous reports have suggested that the ACP composition, and thus its Ca/P molar ratio, influences the type of crystalline product, usually leading to the formation of mixtures of TCP with other calcium phosphate phases. Recently, we have studied the thermal crystallization of carbonate-doped ACP nanoparticles stabilized by citrate ions (hereafter denoted Cit-ACP). We have demonstrated that the thermal crystallization of Cit-ACP uniquely leads to the formation of pure HA instead of TCP, as its Ca/P molar ratio (1.70) is closer to the HA's one (1.67).14 In addition, Cit-ACP was doped with fluoride and carbonate ions, and these ions were maintained in the HA structure formed by heating, allowing the production of a multi-ion doped bioceramic of medical interest.14
However, several phenomena may occur to ACP under heating. This is a relevant issue as to the best of our knowledge all the studies on this topic reported in the literature are “ex situ”, meaning that ACPs were characterized after being heated and subsequently cooled down or quenched to room temperature.11–13,15–21 Previous ex situ studies could have missed transient transformations (i.e. very fast-occurring phenomena) such as structural changes occurring during heating or cooling steps.11–21 So, there is a need for extensive and reliable in situ experimental data at high temperatures. For example, in situ high-temperature energy dispersive X-ray diffraction (HT-EDXRD) allows observing the processes of crystallization, crystal growth, and changes in crystal phases as a function of temperature.22–24 Another important technique in this field is the in situ temperature-dependent Fourier-transform IR spectroscopy (TD-FTIR) that provides information on the variations in local order that occur in functional groups. TD-FTIR is sensitive to short-range ordered (amorphous) phases and allows discriminating between amorphous and proto-crystalline phases25,26 as well as studying the thermal stability of volatile substances.27 Therefore, the aim of this work was to investigate in detail the transformations of Cit-ACP during heating by using complementary HT-EDXRD and TD-FTIR, as these techniques allow the investigation of the heat-induced transformations from the crystallographic point of view as well as at atomic and molecular levels.
From these data, we have evaluated the lifetime of the amorphous phase as a function of temperature, the formation of transient phases, the reaction mechanisms, and the kinetics of HA crystallization, shedding more light on the ACP thermal crystallization process.
Experimental section
Materials
All reagents were purchased from Sigma Aldrich (St. Luis, MO, USA) and were used without purification. Reagents comprise calcium chloride dihydrate (CaCl2·2H2O, ≥99.0% pure), hydrochloric acid (HCl, ≥37.0% pure), sodium citrate tribasic dihydrate (Na3(C6H5O7)·2H2O, ≥99.0% pure), sodium phosphate dibasic dihydrate (Na2HPO4·2H2O, ≥99.0% pure), and sodium carbonate monohydrate (Na2CO3·2H2O, ≥99.0% pure). All solutions were prepared with ultrapure water (18.2 MΩ cm, 25 °C, Arium© pro, Sartorius).Sample preparation
Cit-ACP samples (hereafter denoted Cit-ACP-1 and Cit-ACP-4 as a function of the different nominal Cit/Ca ratio used) were synthesized as reported in the work of Iafisco et al.28 Cit-ACP was prepared by mixing two solutions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 200 mL total) of (A) 100 mM CaCl2 + X mM Na3(C6H5O7) (where X = 100 mM for Cit-ACP-1 and 400 mM for Cit-ACP-4) and (B) 120 mM Na2HPO4 + 200 mM Na2CO3 under vigorous stirring at room temperature. Before mixing solution (B), pH was adjusted to 9.5 with HCl. After 1 min of stirring, the particles were repeatedly washed with ultrapure water by centrifugation (7000 rpm, 5 min, 4 °C) and then freeze-dried overnight.
:1 v/v, 200 mL total) of (A) 100 mM CaCl2 + X mM Na3(C6H5O7) (where X = 100 mM for Cit-ACP-1 and 400 mM for Cit-ACP-4) and (B) 120 mM Na2HPO4 + 200 mM Na2CO3 under vigorous stirring at room temperature. Before mixing solution (B), pH was adjusted to 9.5 with HCl. After 1 min of stirring, the particles were repeatedly washed with ultrapure water by centrifugation (7000 rpm, 5 min, 4 °C) and then freeze-dried overnight.
Citrate-free ACP samples doped and non-doped with carbonate ions and hereafter denoted Ref-CO3-ACP and Ref-ACP, respectively, were synthesized by slightly modifying the chemical precipitation described above and used as reference samples. In detail, Ref-CO3-ACP was prepared by mixing two solutions (1:1 v/v, 200 mL total) of (A) 10 mM CaCl2 and (B) 12 mM Na2HPO4 + 20 mM Na2CO3 at 4 °C, and the precipitate was washed three times by centrifugation (12500 rpm, 4 min, 4 °C), the first two with water at 4 °C and the last with a solution of ethanol–water (70:30 v/v) at 4 °C, and then freeze-dried. Ref-ACP was prepared using a procedure similar to that used for Ref-CO3-ACP without using Na2CO3.
Chemical and crystallographic characterization of ACP samples
Powder X-ray diffraction (PXRD) patterns of the samples were recorded on a D8 Advance diffractometer (Bruker, Karlsruhe, Germany) using Cu Ka radiation, with source parameters set at 40 kV and 40 mA. The patterns were collected in the 2θ range from 10° to 60° with a step size of 0.02° and an acquisition time of 0.5 s.The Ca and P contents of the samples were measured by using an Agilent 5100 inductively-coupled plasma optical emission spectrometer (Agilent Technologies, Santa Clara, CA, USA). For the analysis, 10 mg of the sample was dissolved in 50 mL of a 1 wt% HNO3 aqueous solution, and a calibration curve was constructed by using certified standard solutions (Sigma Aldrich, St. Luis, MO, USA). Citrate and carbonate contents were measured through thermogravimetry analysis using an STA 449F3 apparatus (Netzsch GmbH, Selb, Germany). Ca. 10 mg of the sample was put in a platinum crucible and heated from room temperature to 1100 °C under air flow at a heating rate of 10 °C min−1; citrate was quantified with the weight loss from 350 to 700 °C and carbonate with the weight loss from 700 to 1000 °C.28
The specific surface area (SSABET) of the samples was measured using a Surfer instrument (Thermo Fisher Scientific, Waltham, MA, USA) through the Brunauer–Emmett–Teller approach for N2 gas adsorption.
In situ energy dispersive XRD with a controlled temperature program
In situ FT-IR spectroscopy with a controlled temperature program
In addition, the TD-FTIR spectra were further processed to highlight the crystallization of ACP into HA as a function of temperature. In detail, the second derivative of the spectra was calculated by applying the Savitzky–Golay algorithm combined with spectral smoothing to improve the signal-to-noise ratio. Afterward, the second derivative of the spectra was inverted to show the negative peaks.
Results and discussion
Morphological, structural, and compositional characterization of Cit-ACP and Ref-ACP
Cit-ACP and citrate-free ACP samples are amorphous, as shown by their PXRD patterns (Fig. S1, ESI†). Cit-ACP samples were prepared at two different nominal Cit/Ca molar ratios, i.e. Cit/Ca = 4 or 1, as reported in our previous works we have found that the Cit/Ca ratio controls their specific surface area (SSABET) and this, in turn, influences their thermal crystallization as well as the crystallization in water.14,28 Citrate-free ACPs, used as reference samples, were prepared under similar reaction conditions to those used for preparing Cit-ACPs; however, it should be considered that they cannot be obtained using the same concentration of calcium and phosphate precursors employed for Cit-ACP, as the high supersaturation in the absence of citrate induces the immediate conversion of ACP into HA (data not shown). Therefore, Ref-CO3-ACP and Ref-ACP samples were prepared with the same precursor molar ratios of Cit-ACP, but the overall ion concentration was reduced by a factor of 10. Chemical composition and SSABET values of Cit-ACP samples in comparison with those of citrate-free ACP samples are reported in Table S1 (ESI†). Cit-ACP samples are similar to each other in terms of elemental composition and citrate/carbonate content. Ref-CO3-ACP has a carbonate content comparable to Cit-ACP samples, while for Ref-ACP, the carbonate content is negligible. The Ca/P molar ratio of Ref-ACP (1.35) matches with the standard value reported in the literature for one common type of ACP (called ACP2, Ca/P = 1.35–1.38).7 On the other hand, Cit-ACP samples and Ref-CO3-ACP have higher Ca/P values (1.70 and 1.66, respectively) due to the presence of carbonate ions substituting phosphate ions in the ACP structure, as previously demonstrated by other studies.11,29 The SSABET values of all materials are relatively high, as all samples are nanosized. The SSABET values of Ref-ACP and Ref-CO3-ACP samples are 50 and 130 m2 g−1, respectively, which are similar to other ACPs reported in the literature (50–160 m2 g−1).30,31 On the other hand, the presence of citrate leads to remarkably higher SSABET values for Cit-ACP samples, being ca. 200 and 300 m2 g−1 for Cit-ACP-4 and Cit-ACP-1, respectively. Our previous works have shown that the uncommonly high SSABET values of Cit-ACP are caused by Cit molecules, which adsorb on the surface of ACP nuclei, minimizing their size and increasing their specific surface area.14,28 In addition, the nominal Cit/Ca molar ratio influences the nucleation lag time, leading to different SSABET values. As a consequence, the different surface areas influence Cit-ACP crystallization kinetics and thermodynamics.14,28In situ HT-EDXRD analysis of Cit-ACP thermal crystallization
As the first step, the thermal crystallization of ACP samples was investigated in situ by using EDXRD coupled with a high-temperature cell.In Fig. 1, the HT-EDXRD patterns collected during heating of Cit-ACP samples (panels A and B) and citrate-free ACP samples (panels C and D) are presented as three-dimensional graphs together with the corresponding two-dimensional contour plots as well as stack plots in Fig. S2 (ESI†). HT-EDXRD patterns of Cit-ACP samples show that from 30 °C to ca. 550 °C both materials remain amorphous, as no diffraction peaks are present and only an ACP characteristic broad band centered around 30° 2θ was detected. In this temperature range, an increase in the intensity of the ACP band can be observed, which can be attributed to the thermal expansion of the sample holder, which is mainly constituted of metal, hence lifting the ACP powder. Consequently, a higher quantity of powder intercepts the incoming X-ray beam and contributes to the diffracted signal. Citrate-free ACP samples have also the same behavior, with a non-specific increase of ACP band intensity between room temperature and 550 °C. In the range between 550 and 700 °C, crystallization occurs in Cit-ACP samples, proved by the progressive formation of several diffraction peaks. This finding is in agreement with our previously reported differential thermal analysis (DTA) data.14 The crystallization product of Cit-ACP samples is HA as a single phase (PDF card file 00-009-0432) as some of its characteristic peaks can be observed: (002) at 25.6°, the triplets (211), (112), and (300) between 31–33°, respectively, and (310) at 40° (Fig. S3, ESI†). This finding is in agreement with previous ex situ analysis.14 A closer look at high-temperature patterns (Fig. 1 and Fig. S2, S3, ESI†) evidences the formation of another diffraction peak at 35.5° 2θ at temperature >680 °C for both Cit-ACP-1 and Cit-ACP-4, which is attributed to crystalline calcium oxide (CaO, PDF card file 00-037-1497). This peak was expected, as our previous ex situ study revealed the formation of a small amount of CaO (less than 1 wt%) due to the reaction between the excess calcium of Cit-ACP (their Ca/P ratio is above 1.67, the stoichiometric value for HA) and atmospheric oxygen during the thermal crystallization.14 Thanks to this in situ analysis, it has been clarified that CaO formation occurs after Cit-ACP crystallization into HA, and it is not concomitant to crystallization. Interestingly, Ref-CO3-ACP and Ref-ACP present a different behavior at high temperatures. Ref-CO3-ACP, similar to Cit-ACP samples, crystallizes into HA (together with traces of CaO), but at a higher temperature (at ca. 600 °C) due to its lower surface area. Ref-ACP remains amorphous up to 700 °C, as previously observed by ex situ DTA and XRD analyses. Ref-ACP crystallization into TCP occurs at more than 700 °C and could not be observed in our HT-EDXRD experiment.14 Therefore, these data prove that the chemical composition of ACP controls the nature of the final product obtained by heating.
 | ||
| Fig. 1 EDXRD patterns, collected as a function of heating temperature of (A) Cit-ACP-1, (B) Cit-ACP-4, (C) Ref-CO3-ACP, and (D) Ref-ACP. The two peaks at ca. 10° 2θ are due to instrumental contribution and correspond to the anisotropic Kα and Kβ fluorescence signals of the W anode tube. | ||
The HA formation and its crystalline domain dimension were assessed from HT-EDXRD patterns. HA formation was estimated by measuring the intensity of the HA (211) peak (Fig. 2A), as this parameter is proportional to the amount of the crystalline phase. This approach was chosen over other qualitative methods for the evaluation of the degree of crystallinity as the others can be used only for XRD patterns of crystalline materials characterized by several diffraction peaks. This study was carried out only for Cit-ACP samples and Ref-CO3-ACP, as no crystallization process occurred in Ref-ACP. The data show once again that Cit-ACP samples start to crystallize earlier than Ref-CO3-ACP (at 530 °C rather than at 570 °C), and the HA phase content increases with heating. The size of the HA crystalline domain along the (002) direction (D(002)) as a function of temperature was calculated using Scherrer's equation (Fig. 2B) to estimate the HA crystalline domain dimension along its major c-axis. D(002) values show that both Cit-ACP samples and Ref-CO3-ACP are nanocrystalline, with an average size of ca. 5 nm. Interestingly, the size of D(002) does not vary as a function of the temperature up to 700 °C, suggesting that with heating more and more crystalline HA nuclei form without domain size growth.
 | ||
| Fig. 2 (A) Diffraction intensity of the (211) HA peak, and (B) HA D(002) size as a function of heating temperature. | ||
Overall, our data prove that HA is directly formed through heating of carbonated ACP without the formation of intermediate phases, and rules out the hypothesis that HA is formed by ACP recrystallization during the cooling process. Even if the heating temperature is relatively high and can induce a strong crystal growth, we show that the newly formed diffraction peaks are broad and poorly defined, indicating that HA has nanometric crystalline domains and low crystalline order and that any growth of crystalline domains along the investigated temperature range occurs. This means that the material obtained at high temperature is rather similar to biogenic apatite, which is ion substituted, contains carbonates, and is poorly crystalline.32 The implication of this finding is that a biomimetic HA ceramic material can be prepared by heating a Cit-ACP precursor.
In situ TD-FTIR spectroscopy of Cit-ACP thermal crystallization
Complementary to HT-EDXRD, TD-FTIR allowed us to investigate the compositional and structural changes that occurred to Cit-ACP samples during heating which are not detectable by XRD. In Fig. 3, the FTIR spectra as a function of temperature for Cit-ACP samples (panels A and B) and citrate-free ACP samples (panels C and D) are represented as three-dimensional graphs together with the corresponding two-dimensional contour plots as well as stack plots in Fig. S4 (ESI†). At room temperature, all samples display the broad absorption bands at ca. 560 and 1050 cm−1 that are associated, respectively, with the bending (ν4PO4) and stretching (ν3PO4) modes of phosphate groups in an amorphous environment. In addition, Cit-ACP samples and Ref-CO3-ACP present additional bands at ca. 870 cm−1 and in the range 1400–1500 cm−1 that were attributed to the carbonate substitution ions inside the ACP structure (ν2CO3 and ν3CO3 modes, respectively).33 Finally, Cit-ACP samples present a band at 1560 cm−1 that is attributed to the stretching of carboxylate groups of citrate (νCOO).34 For the sake of clarity, the spectra of the samples recorded before the thermal treatment are reported in Fig. S5 (ESI†). Enlargements of contour plots focusing on phosphate and carbonate bands are provided in Fig. S6–S8 (ESI†).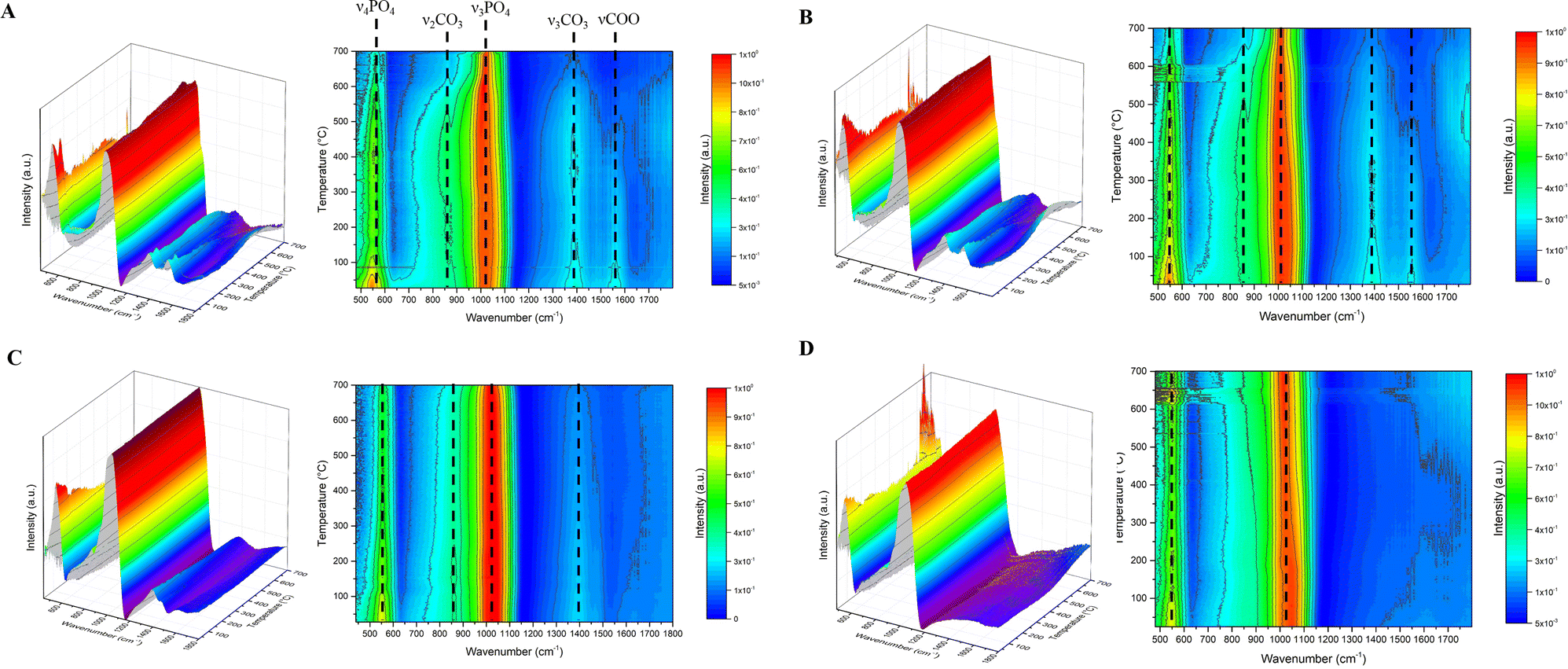 | ||
| Fig. 3 Normalized TD-FTIR spectra, collected as a function of heating temperature of (A) Cit-ACP-1, (B) Cit-ACP-4, (C) Ref-CO3-ACP, and (D) Ref-ACP. | ||
The TD-FTIR spectra of Cit-ACP-4 and Cit-ACP-1 show that several changes occur during the annealing as shown in Fig. 3 and 4 and Fig. S6–S8 (ESI†) and are summarized in Table 1.
 | ||
| Fig. 4 Results of peak integration analysis. (A) FWHM of the ν3PO4 peak, (B) area of νCOO and ν3CO3 peaks. | ||
| Sample | Temperature range (°C) | Event | Interpretation |
|---|---|---|---|
| Cit-ACP-1 | Ca. 25–150 | Broadening of ν3PO4 | Decrease of local short-range order36 |
| Ca. 270–400 | Decrease of νCOO intensity | Decrease of citrate content | |
| Ca. 300–500 | Redshift of ν3PO4 (from ca. 1015 to 1005 cm−1) | Variation of the local short-range environment (shrinking of volume or change in geometry)36 | |
| Ca. 450–550 | Deconvolution of ν2CO3 from ν3PO4 (ca. 865 cm−1) | Increase of local short-range order | |
| Ca. 500–600 | Narrowing of ν4PO4 | Increase of long-range order (i.e., crystallization)36 | |
| Ca. 530–700 | Decrease of ν2CO3 and ν3CO3 intensity | Decrease of carbonate content | |
| >570 | Splitting of ν4PO4 in two bands (560 and 600 cm−1) | Formation of an ordered apatitic environment38 | |
| Ca. 500–620 | Narrowing of ν3PO4 | Increase of long-range order (i.e., crystallization)36 | |
| Ca. 580–620 | Blueshift of ν3PO4 (from ca. 1005 to 1025 cm−1) | Variation of the long-range environment13,36 | |
| Cit-ACP-4 | Ca. 25–150 | Broadening of ν3PO4 | Decrease of local short-range order36 |
| Ca. 270–400 | Decrease of νCOO intensity | Decrease of citrate content | |
| Ca. 300–500 | Redshift of ν3PO4 (from ca. 1015 to 1005 cm−1) | Variation of the local short-range environment (shrinking of volume or change in geometry)36 | |
| Ca. 450–550 | Deconvolution of ν2CO3 from ν3PO4 (ca. 860 cm−1) | Increase of local short-range order | |
| Ca. 520–650 | Narrowing of ν4PO4 | Increase of long-range order (i.e., crystallization)36 | |
| Ca. 530–700 | Decrease of ν2CO3 and ν3CO3 intensity | Decrease of carbonate content | |
| >620 | Splitting of ν4PO4 in two bands (560 and 600 cm−1) | Formation of an ordered apatitic environment38 | |
| Ca. 600–650 | Narrowing of ν3PO4 | Increase of long-range order (i.e., crystallization)36 | |
| Ca. 600–650 | Blueshift of ν3PO4 (from ca. 1005 to 1010 cm−1) | Variation of the long-range environment13,36 | |
| Ref-CO3-ACP | Ca. 25–150 | Broadening of ν3PO4 | Decrease of local short-range order36 |
| Ca. 200–400 | Redshift of ν3PO4 (from ca. 1040 to 1020 cm−1) | Variation of the local short-range environment (shrinking of volume or change in geometry)36 | |
| Ca. 500–700 | Decrease of ν2CO3 and ν3CO3 intensity | Decrease of carbonate content | |
| >650 | Narrowing of ν3PO4 and ν4PO4 | Increase of long-range order (i.e., crystallization)36 | |
| Ref-ACP | Ca. 25–200 | Broadening of ν3PO4 | Decrease of local short-range order36 |
| Ca. 200–400 | Redshift of ν3PO4 (from ca. 1030 to 990 cm−1) | Variation of the local short-range environment (shrinking of volume or change in geometry)36 |
First, from room temperature to ca. 120 °C, there is an increase in the ν3PO4 band broadness (Fig. S6A and B, ESI†). The evolution of band broadness was measured by peak integration analysis, calculating the FWHM of ν3PO4 as a function of temperature (Fig. S6A, ESI†). Calcium phosphate bands are constituted by the superposition of several sub-bands associated with P–O vibrations in different local environments.35 In the case of ACP, the non-ordered structure causes the formation of many different local ionic geometries around phosphate anions, and therefore, their vibrational bands appear broad and without distinctive peaks.7 Therefore, an increase of FWHM indicates an increment of such distribution (i.e. a decrease of short-range order) which can be associated with ACP dehydration, as previously described.36 No significant differences in ν3PO4 FWHM between Cit-ACP-1 and Cit-ACP-4 were observed, suggesting that this event is not influenced by SSABET of the samples. The increase of FWHM occurs also for Ref-CO3-ACP and Ref-ACP (Fig. 4A and Fig. S6C and D, ESI†).
Second, from ca. 270 to 400 °C, the νCOO band of citrate in Cit-ACP samples decreases in relative intensity in comparison to the ν3PO4 band and completely disappears at ca. 400 °C (Fig. 4B and Fig. S7, ESI†). This finding confirms that citrate undergoes a thermal decomposition to carbon dioxide before crystallization, as previously suggested by DTA analysis.14 Also, in this case, the different SSABET values of Cit-ACP-1 and Cit-ACP-4 do not influence the degradation rate of citrate, as their νCOO area curves are superimposed.
Third, from ca. 300 to 500 °C, a redshift of the main phosphate peak ν3PO4, with the maximum shifting from ca. 1015 to ca. 1005 cm−1 (Fig. S6A and B, ESI†), was found. This modification can also be observed in Ref-CO3-ACP and Ref-ACP (Fig. 6C and D, ESI†), but for these samples, it occurs at a lower temperature (between ca. 200 and 400 °C) and at different wavenumbers (from 1025 to 1020 cm−1 for Ref-CO3-ACP and from 1030 to 990 cm−1 for Ref-ACP). A similar redshift was reported in the literature when other ACPs are heated, and it was tentatively attributed to phenomena associated with dehydration, and to the elimination of voids and the shrinking of the ACP volume.36 However, this shift can be either a variation of the P–O bond length or a change of phosphate vibrational geometry.37 Although the interpretation of this redshift is still open, all the hypotheses suggest that with heating, there is a change in the local environment around phosphate groups in all samples before crystallization, and this change occurs at higher temperatures for Cit-ACP samples in comparison to citrate-free ACP ones, independently of the presence of carbonate ions.
Fourth, at around 500 °C, a small peak centered at ca. 865 cm−1 (for Cit-ACP-1) or 860 cm−1 (for Cit-ACP-4) was revealed (Fig. 3 and Fig. S6A, B, ESI†). This peak is the ν2CO3 band, and its appearance due to the narrowing of the ν3PO4 band indicates a progressive increase of the local order around phosphate and carbonate ions. This suggests that Cit-ACP crystallization occurs through an internal reorganization of ions into a short-range ordered structure; afterward, the apatitic crystallites grow and can be detected by HD-EDXRD. Regarding carbonate bands, there is a slight decrease in the relative intensity and peak area of both ν2CO3 and ν3CO3 bands between ca. 530 and 700 °C (Fig. 4B and Fig. S6, S7, ESI†) that is due to the thermal decomposition of carbonates into CO2 occurring in parallel with Cit-ACP crystallization. In agreement with our previous DTA data, the amount of this loss is comparable between Cit-ACP-1 and Cit-ACP-4. It is worthy of note that the carbonate bands are still present at 700 °C after the complete crystallization event, confirming that the thermal treatment of Cit-ACP leads to the formation of carbonate-doped HA. The evolution of carbonate bands of Ref-CO3-ACP as a function of temperature is rather similar to the ones of Cit-ACP samples (Fig. 4B and Fig. S6, ESI†).
Finally, between 500 and 650 °C, the crystallization of Cit-ACP into HA is observed, which is evinced by three main changes to the TD-FTIR spectra. The first is the narrowing of ν3PO4 and ν4PO4 bands (Fig. 4A and Fig. S6A, B and S8, ESI†). As stated above, for calcium phosphate materials, the FWHM of a peak is inversely proportional to local order, and the narrowing of both phosphate bands indicates the rearrangement of ions in a more ordered (i.e., crystalline) structure as the number of possible geometries around phosphate anions decreases. Interestingly, at 700 °C, the phosphate bands are narrower than the pristine ones but are still relatively broad, suggesting that the final material is poorly crystalline. The second is the splitting of the ν4PO4 band into two sub-bands at ca. 560 and 600 cm−1 (Fig. S8, ESI†). This band deconvolution and structuring into sub-bands indicates that all phosphate ions acquire a precise local environment. In particular, the appearance of two bands at 560 and 600 cm−1 for ν4PO4 demonstrates that an apatitic crystal structure is obtained, as those are the characteristic ν4PO4 frequencies of HA.38 The last is the blue-shift of the ν3PO4 peak from ca. 1005 to ca. 1025 or 1010 cm−1 for Cit-ACP-1 and Cit-ACP-4, respectively (Fig. S6A and B, ESI†). The relationship between ACP crystallization and the blue-shift of the ν3PO4 peak to 1010–1020 cm−1 is well reported in the literature and described as the formation of an apatitic long-range ordered structure.13,36
The crystallization of the samples was also followed by calculating the second-order derivatives of the FTIR spectra, as this mathematical expression allows the magnification of the changes in the ν3PO4 position and shape.39,40 Querido et al. have validated the robustness of this data analysis for the study of ACP-to-HA crystallization in synthetic calcium phosphates, in complex materials such as bones, and in minerals formed in osteogenic cell cultures.39 In detail, the second-order derivative spectrum of ACP presents a characteristic negative peak at ca. 990–996 cm−1 while HA or TCP has a corresponding negative peak at 1015–1020 cm−1.39,40 Second-order derivatives of ACP ν3PO4 band are reported in Fig. 5 as three-dimensional graphs together with corresponding two-dimensional contour plots; an enlargement of the 950–1050 cm−1 range is provided in Fig. S9 (ESI†). The second-order derivative of Cit-ACP (Fig. 5A, B and Fig. S9A, B, ESI†) clearly shows a negative peak at ca. 985 cm−1 at room temperature that between 450 and 600 °C shifts to ca. 1010–1015 cm−1 due to the progressive decrease of ACP and increase of HA.39 This indicates that in Cit-ACP samples, the phosphate groups initially are in a non-ordered environment, and their transition to an apatitic environment occurs in the 450–500 °C range. Differently, Ref-ACP presents a negative peak at ca. 985–970 cm−1 which between 400 and 700 °C decreases in intensity without disappearing, while in parallel, a new negative peak at 1000–1020 cm−1 appears above 200 °C (Fig. 5C, D and Fig. S9C, D, ESI†). This result suggests that from 200 to 600 °C, a concomitant presence of phosphate ions in a non-ordered local environment and in an ordered environment exists. With heating, a progressive transformation from the former to the latter occurs, and only above 650–700 °C, the ordered environment is predominant. This finding was not observed in previous ex situ studies.40 It is important to mention that for Ref-ACP other crystalline phases could be present, making the second-order derivative analysis difficult to interpret. The second-derivative spectra of Ref-CO3-ACP (Fig. 5C and Fig. S9C, ESI†) have the features both of the “single peak shift” observed for Cit-ACP samples (i.e., a shift of the maximum between 500 and 650 °C) as well as the “double peak” observed for Ref-ACP (i.e., the permanence of the 985–970 cm−1 peak up to 700 °C), suggesting that carbonate ions prevent the early formation of the locally ordered phosphates observed in Ref-ACP.
 | ||
| Fig. 5 Second-order derivatives of the TD-FTIR spectra collected as a function of heating temperature of (A) Cit-ACP-1, (B) Cit-ACP-4, (C) Ref-CO3-ACP, and (D) Ref-ACP. | ||
Associating the crystallization event with the disappearance of the 985–970 cm−1 negative peak, the second-order derivative spectra confirm that the crystallization of Cit-ACP samples initiates at lower temperatures (450–500 °C) compared to citrate-free ACP samples (more than 650 °C for Ref-CO3-ACP, and more than 700 °C for Ref-ACP). The slight discrepancy in the crystallization temperature between HT-EDXRD (from 540–700 °C), TD-FTIR (from 450–500 to 650–700 °C), and our previous DTA analysis (from 500 to 700–800 °C) is due to the experimental setups, as the three setups have different sample holders, sample sizes, mass, volumes, heating systems, and temperature feedback mechanisms, and thus a different heat transfer to the sample.
Overall, the results of TD-FTIR experiments are in good agreement with those of HT-EDXRD and give several additional information on the events that occur in the materials before the crystallization, showing the strength of the technique for this kind of study. In particular, we have discovered that the thermal crystallization of Cit-ACP involves first a loss of order while the material is being dehydrated. This transformation is followed by a rearrangement of phosphate and carbonate ions into a pre-apatitic structure paralleled by citrate decomposition.
By comparing Cit-ACP with its reference samples, we have found that the role of citrate is to delay the first internal rearrangement of phosphate ions and to accelerate the onset of crystallization. It is likely that both effects are caused by the high surface area of Cit-ACP samples. We have also found out that carbonate ions influence the chemical composition (i.e., Ca/P ratio) of ACP, and this in turn leads to the selective formation of HA instead of TCP. In addition, the use of the second-order derivative on the FTIR spectra suggests that in Cit-ACP samples the transition from the non-ordered local environment to the ordered apatitic environment is discrete, while in citrate-free ACPs, the two local environments (ordered and disordered) coexist for some time. Finally, it must be noted that the TD-FTIR experiment not only shed more light on the thermal crystallization of ACP, but confirmed that the changes in the FTIR spectra observed by ex situ works were correctly attributed to the heating process and are not due to the subsequent cooling process.
Conclusions
Our work represents the first in situ, real-time study of ACP thermal crystallization using HT-EDXRD and TD-FTIR. We have discovered that several phenomena occur during the heating of ACP, and the crystallization is just the last step of several structural reorganizations. We also found out that citrate and carbonate ions play a critical role in the crystallization kinetics as well as have an effect on the nature of the crystalline product.This comprehensive investigation of the complex thermal crystallization of ACP paves the way for new research directions in the field. For example, thanks to the knowledge gained from this work, we envision that by using controlled heating of ACP, it will be possible to produce bioactive HA ceramics for medical applications. Indeed, the thermal treatment allows the consolidation of the material, and the use of Cit-ACP as a precursor would allow the improvement of the biomimetism of the ceramic by controlling crystallinity and ion doping. Finally, our work shows the high potentialities of in situ techniques that could be used to investigate in depth the crystallization mechanism of other kinds of amorphous materials.
Author contributions
L. D. E.: conceptualization, investigation, visualization, writing – original draft, and writing – review & editing; M. F.: investigation, writing – review & editing; A. C.: investigation, writing – review & editing; L. D. C.: writing – review & editing; M. O.: investigation; A. A.: writing – review & editing; J. V. R.: writing – review & editing; and M. I.: conceptualization, supervision, writing – original draft, and writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Italian Ministry of Health (Bando Ricerca Finalizzata 2016, no. GR-2016-02364704).References
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 CrossRef CAS PubMed.
- F. Carella, L. Degli Esposti, A. Adamiano and M. Iafisco, Materials, 2021, 14, 6398 CrossRef CAS PubMed.
- G. Fellet, L. Pilotto, L. Marchiol and E. Braidot, Agronomy, 2021, 11, 1239 CrossRef CAS.
- N. Eliaz and N. Metoki, Materials, 2017, 10, 334 CrossRef PubMed.
- E. Eanes, Calcif. Tissue Res., 1970, 5, 133–145 CrossRef CAS.
- E. Eanes, I. Gillessen and A. Posner, Nature, 1965, 208, 365–367 CrossRef CAS.
- C. Combes and C. Rey, Acta Biomater., 2010, 6, 3362–3378 CrossRef CAS PubMed.
- A. Tofighi, P. Chakravarthy, D. Lee, S. Mounic and C. Rey, Key Eng. Mater., 2001, 192, 769–772 Search PubMed.
- J. Zhao, Y. Liu, W.-B. Sun and H. Zhang, Chem. Cent. J., 2011, 5, 1–7 CrossRef PubMed.
- L. Degli Esposti and M. Iafisco, Biomater. Biosyst., 2022, 5, 100037 CrossRef.
- Y. Li, F. Kong and W. Weng, J. Biomed. Mater. Res. B, 2009, 89, 508–517 CrossRef PubMed.
- L. Sinusaite, A. Kareiva and A. Zarkov, Cryst. Growth Des., 2021, 21, 1242–1248 CrossRef CAS.
- V. Uskoković, S. Marković, L. Veselinović, S. Škapin, N. Ignjatović and D. P. Uskoković, Phys. Chem. Chem. Phys., 2018, 20, 29221–29235 RSC.
- L. Degli Esposti, S. Markovic, N. Ignjatovic, S. Panseri, M. Montesi, A. Adamiano, M. Fosca, J. V. Rau, V. Uskoković and M. Iafisco, J. Mater. Chem. B, 2021, 4832–4845 RSC.
- S. Liu, W. Weng, Z. Li, L. Pan, K. Cheng, C. Song, P. Du, G. Shen and G. Han, J. Mater. Sci.: Mater. Med., 2009, 20, 359 CrossRef CAS.
- N. Döbelin, T. J. Brunner, W. J. Stark, M. Eggimann, M. Fisch and M. Bohner, Key Eng. Mater., 2009, 396–398, 595–598 Search PubMed.
- R. Kumar, P. Cheang and K. Khor, Acta Mater., 2004, 52, 1171–1181 CrossRef CAS.
- Y. Li, W. Weng and K. C. Tam, Acta Biomater., 2007, 3, 251–254 CrossRef CAS PubMed.
- M. Maciejewski, T. J. Brunner, S. F. Loher, W. J. Stark and A. Baiker, Thermochim. Acta, 2008, 468, 75–80 CrossRef CAS.
- S. Somrani, C. Rey and M. Jemal, J. Mater. Chem., 2003, 13, 888–892 RSC.
- C. Feng, K. Khor, S. Kweh and P. Cheang, Mater. Lett., 2000, 46, 229–233 CrossRef CAS.
- J. V. Rau, M. Fosca, V. S. Komlev, I. V. Fadeeva, V. Rossi Albertini and S. M. Barinov, Cryst. Growth Des., 2010, 10, 3824–3834 CrossRef CAS.
- J. V. Rau, A. Generosi, V. S. Komlev, M. Fosca, S. M. Barinov and V. R. Albertini, Dalton Trans., 2010, 39, 11412–11423 RSC.
- M. Fosca, V. S. Komlev, A. Y. Fedotov, R. Caminiti and J. V. Rau, ACS Appl. Mater. Interfaces, 2012, 4, 6202–6210 CrossRef CAS PubMed.
- D. D. S. Meneses, M. Eckes, L. Del Campo, C. N. Santos, Y. Vaills and P. Echegut, Vib. Spectrosc., 2013, 65, 50–57 CrossRef.
- Y. Wu, J. Ordonez-Miranda, S. Gluchko, R. Anufriev, D. D. S. Meneses, L. Del Campo, S. Volz and M. Nomura, Sci. Adv., 2020, 6, eabb4461 CrossRef CAS PubMed.
- J. Zawadzki and M. Wiśniewski, Carbon, 2003, 41, 2257–2267 CrossRef CAS.
- M. Iafisco, L. Degli Esposti, G. B. Ramírez-Rodríguez, F. Carella, J. Gómez-Morales, A. C. Ionescu, E. Brambilla, A. Tampieri and J. M. Delgado-López, Sci. Rep., 2018, 8, 17016 CrossRef PubMed.
- Y. Li, D. Li and W. Weng, Int. J. Appl. Ceram. Technol., 2008, 5, 442–448 CrossRef CAS.
- J. Vecstaudza and J. Locs, J. Alloys Compd., 2017, 700, 215–222 CrossRef CAS.
- R. Sun, M. Åhlén, C.-W. Tai, É. G. Bajnóczi, F. de Kleijne, N. Ferraz, I. Persson, M. Strømme and O. Cheung, Nanomaterials, 2020, 10, 20 CrossRef CAS.
- J. Gómez-Morales, M. Iafisco, J. M. Delgado-López, S. Sarda and C. Drouet, Prog. Cryst. Growth Charact. Mater., 2013, 59, 1–46 CrossRef.
- J. Termine and D. Lundy, Calcif. Tissue Res., 1974, 15, 55–70 CrossRef CAS PubMed.
- P. Ivanchenko, J. M. Delgado-López, M. Iafisco, J. Gómez-Morales, A. Tampieri, G. Martra and Y. Sakhno, Sci. Rep., 2017, 7, 8901 CrossRef PubMed.
- C. Rey, O. Marsan, C. Combes, C. Drouet, D. Grossin and S. Sarda, Advances in calcium phosphate biomaterials, 2014, 229–266 CAS.
- V. Uskokovic, Cryst. Growth Des., 2019, 19, 4340–4357 CrossRef CAS.
- S. R. Ryu, I. Noda and Y. M. Jung, Appl. Spectrosc., 2010, 64, 1017–1021 CrossRef CAS PubMed.
- A. Antonakos, E. Liarokapis and T. Leventouri, Biomaterials, 2007, 28, 3043–3054 CrossRef CAS PubMed.
- W. Querido, S. Bookbinder, M. C. Oliveira-Nunes, B. Krynska and N. Pleshko, Analyst, 2020, 145, 764–776 RSC.
- V. Uskoković, Vib. Spectrosc., 2020, 108, 103045 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical composition and SSABET analysis of ACP samples, the HT-EDXRD patterns of ACP samples at 25 °C and 700 °C, the TD-FTIR spectra of ACP samples at 27 °C, and enlargement of the TD-FTIR spectra in the ranges 800–1200 cm−1, 1200–1700 cm−1, and 480–650 cm−1. See DOI: https://doi.org/10.1039/d2cp02352k |
| This journal is © the Owner Societies 2022 |