
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of the hydrogen extraction reactions of isopentane molecules and ions
Yi
Gao
 a,
Bin
Yang
a and
Yang
Zhao
*b
a,
Bin
Yang
a and
Yang
Zhao
*b
aCenter for Combustion Energy and Key Laboratory for Thermal Science and Power Engineering of MOE, Tsinghua University, Beijing, 100084, China
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: e0185978@u.nus.edu
First published on 20th September 2022
Abstract
In plasma-assisted combustion, excited species, ions, radicals, and other active intermediates can be produced by the excitation, ionization, and dissociation processes, which are conducive to steady combustion under the conditions of low temperature and pressure. Positive ions are reported to play an important role in electron impact ionization of fuels, but the associated kinetic processes are rarely discussed owing to the lack of experimental data and uncertain reaction pathways in the model. As they are the smallest geometric structures with primary, secondary, and tertiary hydrogen atoms, a study is presented to discuss the hydrogen abstraction potential energy surfaces of isopentane molecules and positive ions by hydrogen radicals at the DLPNO-CCSD(T)/aug-cc-pVTZ//B3LYP-D3(BJ)/6-311G(2df,2p) and DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2x-D3/ma-def2-TZVP levels. The strengths of C–C and C–H bonds were characterized by bond order analyses to predict changes in the active sites of isopentane molecules and positive ions. Molecular orbitals were analyzed to identify simple feature patterns for establishing forming rules, showing consistency with the potential energy surface (PES) analysis. Structure selectivity of different reactions was discussed based on the predicted rate constant calculations in the temperature range of 300–2000 K, and the mechanisms were compared based on the isopentane pyrolysis model constructed with the combination of Reaction Mechanism Generator (RMG) and updated rate constants determined in this work.
1. Introduction
Ignition and combustion stability covering a wide range of operating conditions has remained a hotspot issue in modern energy systems. The optimization and improvement of energy-conversion combustion systems still present great challenges, especially under extreme conditions.1 The application of a plasma-assisted approach seems promising to overcome the limits of region of ultra-lean fuel–air mixtures in high speed flows, low temperatures, low pressure of high-altitude flights2–5 and so on.In a plasma-assisted system, high-energy electrons, ions, excited species, and radicals are generated through excitation, ionization, and dissociation processes.6 Therefore, it is a key challenge to quantitatively examine the contributions of different species to combustion enhancement at the fundamental response level.7 The crucial step is to detect the critical species during the plasma-assisted combustion (PAC) kinetic process. In the air, oxygen-containing species such as O, O3, and O(1D) and nitrogen-containing species, including N, NO, and NO2, as well as excited states, ions, etc. have been identified.8,9 Mintoussov et al.10 investigated the influence of a pulsed nanosecond atmospheric pressure barrier discharge based on the OH, CH, and C2 emission profiles by using emission spectroscopy. Ombrello et al. isolated ozone or trioxygen (O3) produced by dielectric barrier plasma discharge from C3H8/O2/N2 laminar lifted flames11 and O2(a1Δg) produced in a microwave discharge plasma from ethylene (C2H4)-lifted flames.12 Quantitative measurements were performed by using absorption spectroscopy, and the flame propagation speed was determined to be increased by several percentage points. Wang et al.13 reported a novel microwave PAC system with emission spectroscopy and pulsed cavity ring-down spectroscopy (CRDS) and hypothesized that OH(X) radicals play a more prominent role in flame stabilization, but OH(A) radicals play a more dominant role in the ignition enhancement. Although the dominant species can be identified in experiment, the role of species produced by a plasma discharge of the fuel has not been discussed in an elementary reaction.
A validated kinetic model is required to understand the PAC reaction at the microscopic level. However, even in homogeneous plasma reactors, current kinetic models are likely to introduce large errors to predict time-dependent primary species histories.14 Zhang et al.15 coupled a dielectric barrier discharge flow reactor with a molecular beam mass spectrometer for conducting detailed species diagnostics in a CH4/O2/Ar system and detected hydrocarbons ranging between C2 and C5. The uncertainty factor for quantification is estimated to be 20% for major species and 50% for intermediates. Sun et al.16 performed path flux analysis in the non-equilibrium plasma-assisted methane (CH4) oxidation mechanism and proposed that the fuel was dissociated primarily by H abstraction with OH, O, and H or via collisions with an electron, Ar+ and Ar* before further oxidation. They predicted the formation of O and CH2O reasonably, overestimated the concentrations of CO, H2O, and H2, and underestimated the concentration of CO2. In addition, several studies on PCA have focused on small molecule fuels, such as methane,17–20 which is the main component in natural gas and has a simple structure both numerically and experimentally. Compared with steady combustion, there has been relatively little discussion of C2–C5 fuels and their structure selectivity in PAC. On the other hand, many of the reactions are difficult to study experimentally due to the relative complexity of separating reactions and/or species and the sometimes very slow time scales involved.21 Therefore, theoretical calculations would provide the necessary useful tools for predicting reaction pathways and estimating rate parameters.
Herein, as isopentane (i-C5H12) being the smallest alkane with primary, secondary, and tertiary hydrogen atoms, i-C5H12 molecules (C5H12), ions (C5H12+) and their hydrogen extraction reactions by H atoms were evaluated and compared at DLPNO-CCSD(T)/aug-cc-pVTZ//B3LYP-D3(BJ)/6-311G(2df,2p) and DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2x-D3/ma-def2-TZVP levels. Then PESs were discussed at the same level with explanation by wave function analysis. Finally, the structure selectivity of hydrogen extraction reactions by H atoms was discussed depending on the combination of the isopentane pyrolysis model constructed using RMG and calculated rate constants to compare the kinetics and products between PAC and steady combustion.
2. Method
The conformational space of isopentane (i-C5H12) molecules and ions was studied with the MM+ force field in the HyperChem22 package. The Gaussian 16 program23 was used to perform ab initio calculations. The equilibrium geometries of the i-C5H12 local minima were optimized at superfine grids for the dispersion-corrected density functionals24–26 B3LYP-D3(BJ) with Becke–Johnson damping27 with the triple-ζ basis set 6-311G(2df,2p) and M06-2x-D3 with the minimally augmented polarized triple zeta basis set ma-def2-TZVP.28,29 This methodology helped to suppress the basis set superposition error (BSSE) to afford a better description for weakly bonded complexes.30,31 The potential energy surfaces of the i-C5H12 global minimum were also explored. Vibrational characteristics with hindered internal rotor correction of stationary points (isomers, reactants, and product complexes, and transition states (TSs)) were determined at B3LYP-D3(BJ)/6-311G(2df,2p) and M06-2x-D3/ma-def2-TZVP levels. The single point energies of PESs were refined using the DLPNO-CCSD(T) method (default setting, TightPNO, TightSCF and Grid = 6) with the aug-cc-pVTZ basis sets and ORCA32 software which enabled this to approach the results of the coupled-cluster CCSD(T) method within 1 kJ mol−1.33,34The rate constants were calculated based on the transition-state theory (TST) for the abstraction processes in the temperature range of 300–2000 K with the Mesmer program.35 The Eckart,36,37 Wigner38 and zero-curvature tunneling (ZCT)39 approximations were applied with the Polyrate program.40 Wave function analyses were performed using the Multiwfn program.41 The chemical kinetics mechanisms of i-C5H12 were constructed through the combination of automated methods Reaction Mechanism Generator (RMG)42 and updated rate constants determined in this work for analyses using CHEMKIN-PRO software.43
3. Results and discussion
3.1 Geometric parameters of i-C5H12 and i-C5H12+
As shown by the geometries and Fischer formula shown in Fig. 1(a) and (b), two local minima with C1 and Cs symmetry were optimized at B3LYP-D3(BJ)/6-311G(2df,2p) and M06-2x-D3/ma-def2-TZVP levels after relaxed PES scan of the C1–C2–C3–C4 dihedral angles by 360 degrees in steps of 10 degrees at the same level. C1 symmetry was found to be a global minimum (at both CCSD(T)/cc-pVTZ//M06-2x-D3/ma-def2-TZVP and CCSD(T)/cc-pVTZ//B3LYP-D3(BJ)/6-311G(2df,2p) levels for C5H12 and C5H12+) because the steric hindrance effect caused by three-branched methyl groups in Cs symmetry led to a larger dipole moment, as marked by yellow arrows in the given figure. Ionization results in a significant contraction in the electron cloud density of C1, C4, and C5, as the lighter color of C2 and C3 indicates the loss of an electron of pentane (C5H12) and C5H12+ determined by the ADCH44 population. A compliance matrix45 predicted that, when compared with C5H12, the strength of the C–C ([2]–[3]) bond in C5H12+ decreased distinctly, while the strength of other C–C ([1]–[2], [3]–[4] and [4]–[5]) bonds slightly increased in lower compliance constants. Fig. 1(c) shows that the energies of the highest occupied molecular orbital (EHOMO) of the Cs symmetry in both C5H12 and C5H12+ were higher than those of C1 symmetry, while the energies of the lowest unoccupied molecular orbital (ELUMO) were lower in C5H12 and C5H12+. Compared with C5H12, C5H12+ with larger dipole moments had more discrete but lower energy levels. Populations of two conformations46 at B3LYP-D3(BJ)/6-311G(2df,2p) and M06-2X-D3/ma-def2-TZVP Gibbs free energies (ΔG) were calculated using eqn (1) and (2). | (1) |
 | (2) |
 | ||
| Fig. 1 Comparison of two conformers of isopentane molecules and ions. (a1) C1 conformer of C5H12; (a2) Cs conformer of C5H1; (b1) C1 conformer of C5H12+; (b2) Cs conformer of C5H12+; (c) molecular orbital diagram of C5H12 and C5H12+; (d) conformation populations with temperature of C5H12 and C5H12+. | ||
The strength of bonds was reported to be closely related to the activity and selectivity.47 Laplacian bond order (LBO),48 fuzzy bond order (FBO),49 and Wiberg bond order (WBO),50 which revealed a direct correlation with the bond dissociation energy (BDE) of C5H12 in Fig. 2(b), were listed to characterize C–C and C–H bonds as shown in Fig. 2(a). Both bond order analysis and BDE results showed that the C–C bond sequence of C5H12 was: secondary–tertiary ([2]–[3]) < branched primary–tertiary ([3]–[4] and [3]–[5]) < primary–secondary ([2]–[3]). The sequence of C–H bond strength in C5H12 was: secondary ([2]) < tertiary ([3]) < primary ([1]) ≈ branched primary ([4]). The green rectangles in Fig. 2(b) show that the differences between CBS-QB3 and CBS-APNO were within 1 kcal mol−1 and could become larger with lower bond dissociation energies (BDEs). Furthermore, FBO and WBO displayed better differentiation between secondary and tertiary hydrogen atoms than LBO, while CBS-APNO performed better than the CBS-QB3 method. Bonds of C5H12+ were predicted by the bond order analysis using the CBS-APNO method. A significant decline in secondary–tertiary ([2]–[3]) and a slight increase in primary–tertiary ([3]–[4] and [3]–[5]) and primary–secondary ([2]–[3]) could be noticed. Smaller differences could be observed in the C–H bonds of C5H12+ than those of C5H12; these differences are caused by atomic composition and electron density, as shown in Fig. 1(a). An increase of C and H atom composition in primary ([1]) and secondary ([2]) environments, and a decrease of C and H in tertiary ([3]) and primary ([4] and [5]) environments tended to balance the effect of ionization on carbon atoms, so hydrogen bonds became weaker but more average.
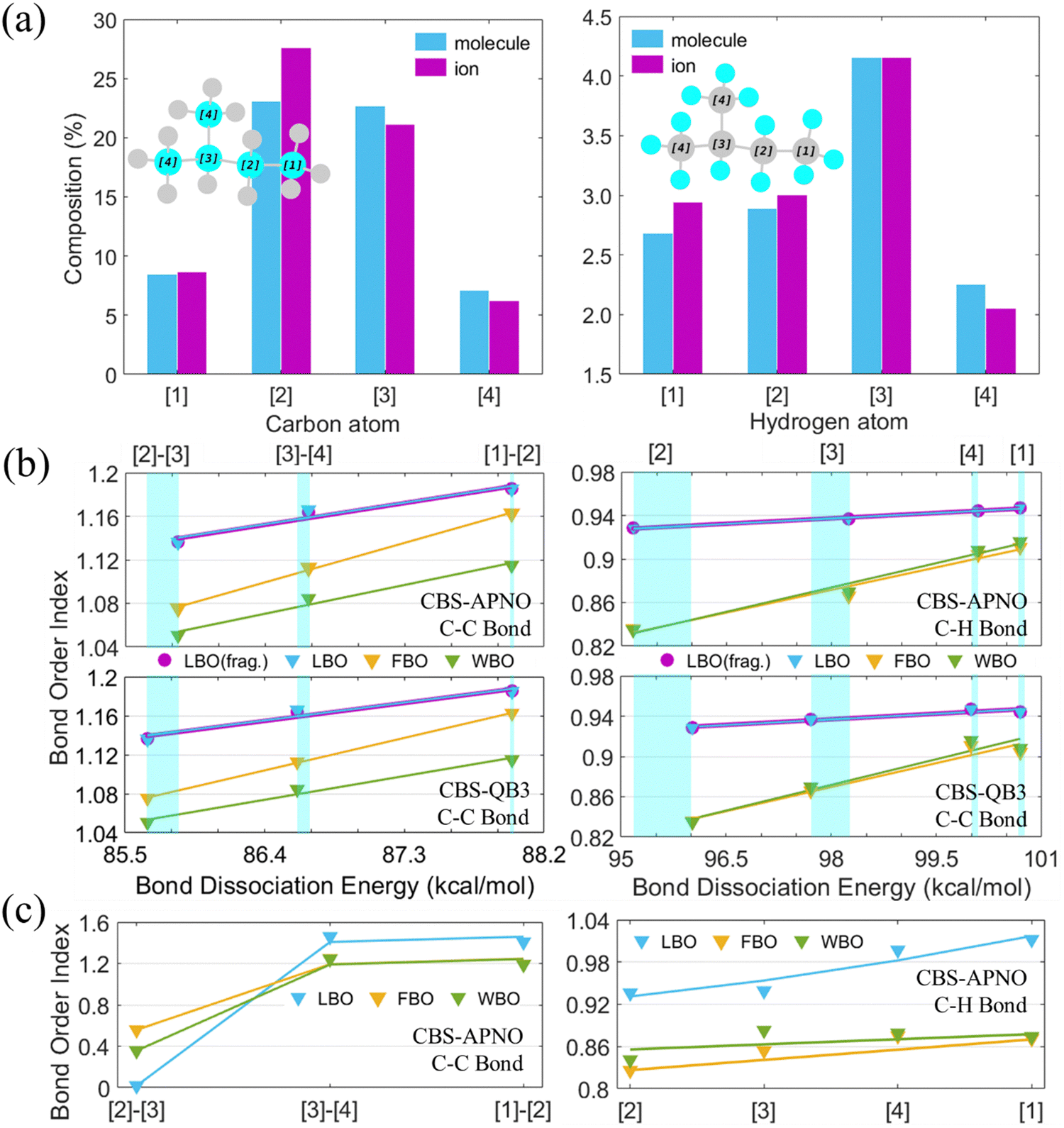 | ||
| Fig. 2 Bond comparison of C5H12 and C5H12+C1 conformers. (a) Composition of carbon and hydrogen atoms to valence electron density. (b) LBO, FBO, WBO, and BDE of the C–C and C–H bonds of C5H12 showed a direct correlation at CBS-QB3 and CBS-APNO. (c) C–C and C–H bonds of C5H12+ characterized by LBO, FBO, and WBO at CBS-APNO. | ||
3.2 Mechanism of the abstraction reaction by the H atoms of isopentane molecules and ions
In this study, four abstraction reaction paths for the H atoms of C5H12 and eight C5H12+ were obtained. Fig. 3 shows the transition states (TS) and the corresponding products (PR). Each geometry in the same reaction path was marked with the same double-digit, such as 01_ts. The first digit 0 represents the reactant isomer C5H12 and 1 represents C5H12+. The second digit corresponds to the reactive sites marked in pink, as shown in Fig. 2(a) (CH3)2CHCH2CH2 + H2 (01_PR), (CH3)2CHCH˙CH3 + H2 (02_PR), CH3(CH2˙)CHCH2CH3 + H2 (03_PR) and CH3(CH3)C˙CH2CH3 + H2 (04_PR) could be obtained when the H atom reacts with C5H12. In addition to the product H2 by a H atom reacting with another H atom at the reactive site of the reactant isomer C5H12, the reaction paths of C5H12+ showed more abundant products. The abstraction reactions of the H atom in the C5H12+ ion can also be combined with the cleavage of the C–C bond to perform alkyl extraction, so small molecular structures of CH4, C2, and C3 may be found in the products.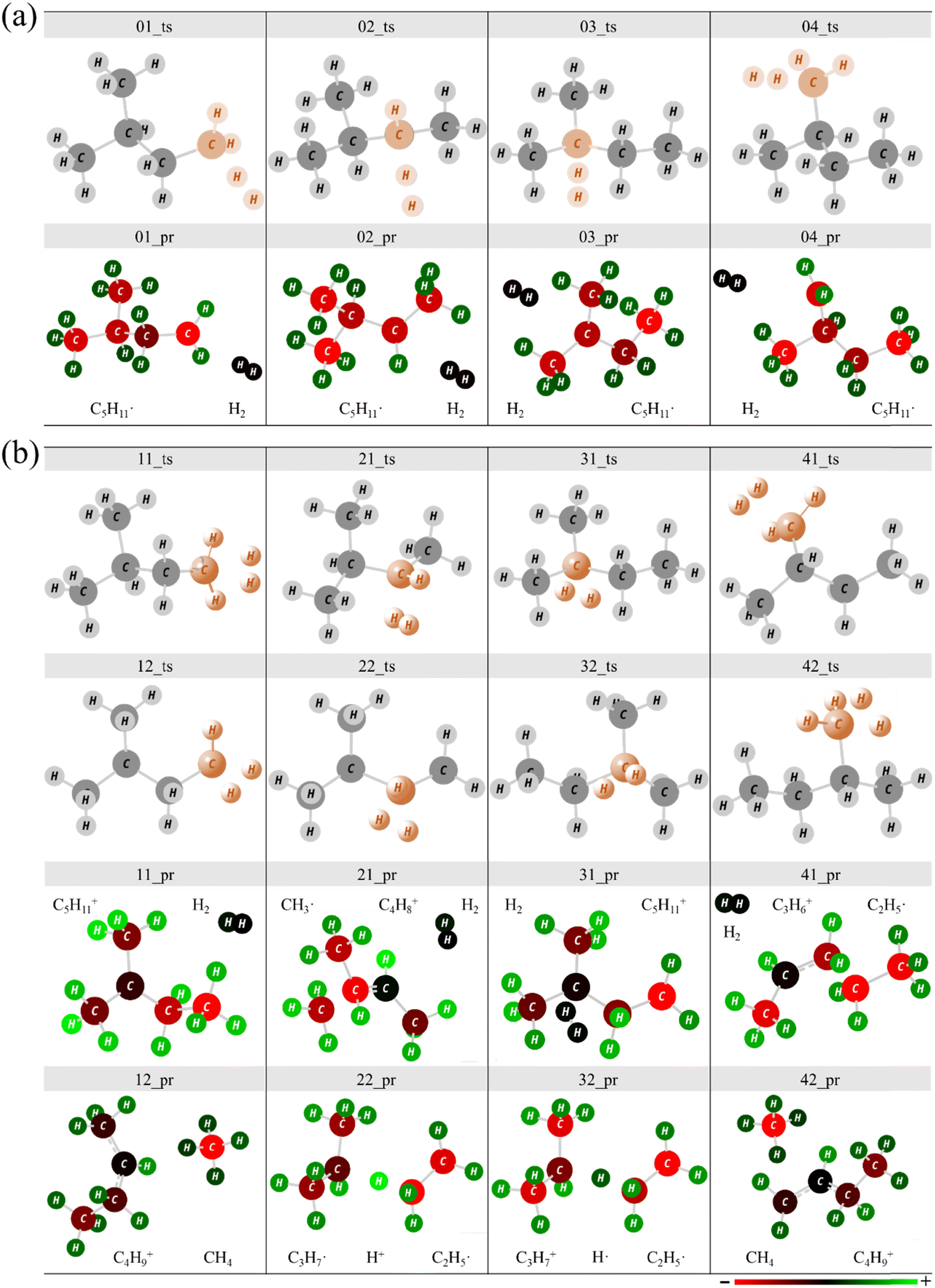 | ||
| Fig. 3 Optimized geometries of transition states and the corresponding products (marked with the same number): (a) C5H12 and (b) C5H12+. | ||
Fig. 4 shows a pictorial representation of the calculated potential energy surface at the DLPNO-CCSD(T)/aug-cc-pVTZ//B3LYP-D3(BJ)/6-311G(2df,2p) (in parentheses) and DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2X-D3/ma-def2-TZVP levels, for some possible abstraction reactions of C5H12 and C5H12+ by H atoms. Fig. 4(a) shows reaction paths 01 and 04, abstraction of primary H by a H atom, showing higher strength of the C–H bond. The energy barrier of the C–C bond in Fig. 2 is the highest, about 10 kcal mol−1. Followed by reaction paths 02 and 03, the abstraction of secondary H and tertiary H in C5H12 is about 7 kcal mol−1 higher than the reactant isomer. So the sequence of reactivity may be: 03 > 02 > 01 > 04. For the products of C5H12 + H, the sequence of stability was 04 > 03 ≈ 02 > 01.
 | ||
| Fig. 4 Schematic diagrams of the potential energy surfaces refined at the DLPNO-CCSD(T)/aug-cc-pVTZ level and optimized at B3LYP-D3(BJ)/6-311G(2df,2p) (in parentheses) and M06-2X-D3/ma-def2-TZVP levels: (a) C5H12 and (b) C5H12+. | ||
Because bond strength dropped with the decrease of electron cloud density after ionization, the abstraction reaction paths of C5H12+ + H in Fig. 4(b) could be presented in two ways: the first way was formed by breaking the C–H bond similar to that of C5H12 + H as shown in Fig. 4(a); the second was formed by C–C bond cleavage. For the first way (abstraction products), although the overall energy barrier is lowered compared to C5H12 + H, the sequence of reactivity seems to remain as 13 > 12 > 11 > 14. As illustrated in Fig. 2(a), the apparent increase in the activity of 12 and 13 (abstraction products) might be due to the rise of carbon and hydrogen atom composition to valence electron density in secondary and tertiary carbon. For the products of C5H12+ + H (abstraction products), the sequence of stability was 11 > 13 > 14 > 12. The improved product stability of the primary reaction path (11 and 14) may be caused by the trend of the electron density average of the three primary groups (CH3–). For the second way of C5H12+ + H (cleavage products), the sequence of reactivity was 14 ≈ 11 > 12 > 13, which seems to be opposite in polarity to the first way (abstraction products), depending on the strength of the nearby C–C bonds being abstracted. For the products (cleavage products), the sequence of stability was 14 > 11 > 12 > 13. The cleavage of the primary groups (CH3) to form CH4 seemed to possess lower energy barriers and more stable products. Because the cleavage products, H2 and CH4, were neutral molecules without charge, the C4 ions had a unit positive charge. The charged C2 and C3 were complex and may be verified experimentally.
The molecular orbital (MO) of TSs could be seen as a mixture of the MO of potential products. The orbital interaction diagram analyses shown in Fig. 5 were used to understand the frontier MOs of TSs in C5H12 + H and C5H12+ + H reaction paths. Similarly, each geometry was marked with a double-digit, with 0 indicating C5H12 + H and 1 representing C5H12+ + H. The second digit of C5H12+ + H was used to classify reaction types: 1 represents reaction paths that possessed H2 as the abstraction product and 2 represents reaction paths to obtain the cleavage products. Fig. 6 shows that, for the reaction path with the abstraction product H2, the HOMOs of TSs were all mixed with those of two H atom (H atom as the reactant isomer and H atom at the reactive site) fragments and hydrocarbon fragments. The LUMOs of TSs in the C5H12+ + H reaction path were mainly composed of LUMOs of two H atom fragments, while those in the C5H12 + H reaction path were almost entirely LUMOs of hydrocarbon fragments. This probably makes the C5H12+ + H reaction path much easier to generate H2. For the C5H12+ + H reaction path with cleavage products, the LUMOs of TSs were mixed by the HOMOs of two H atom fragments and hydrocarbon fragments, which might predict the cleavage of C–C and C–H bonds to form smaller fragments.
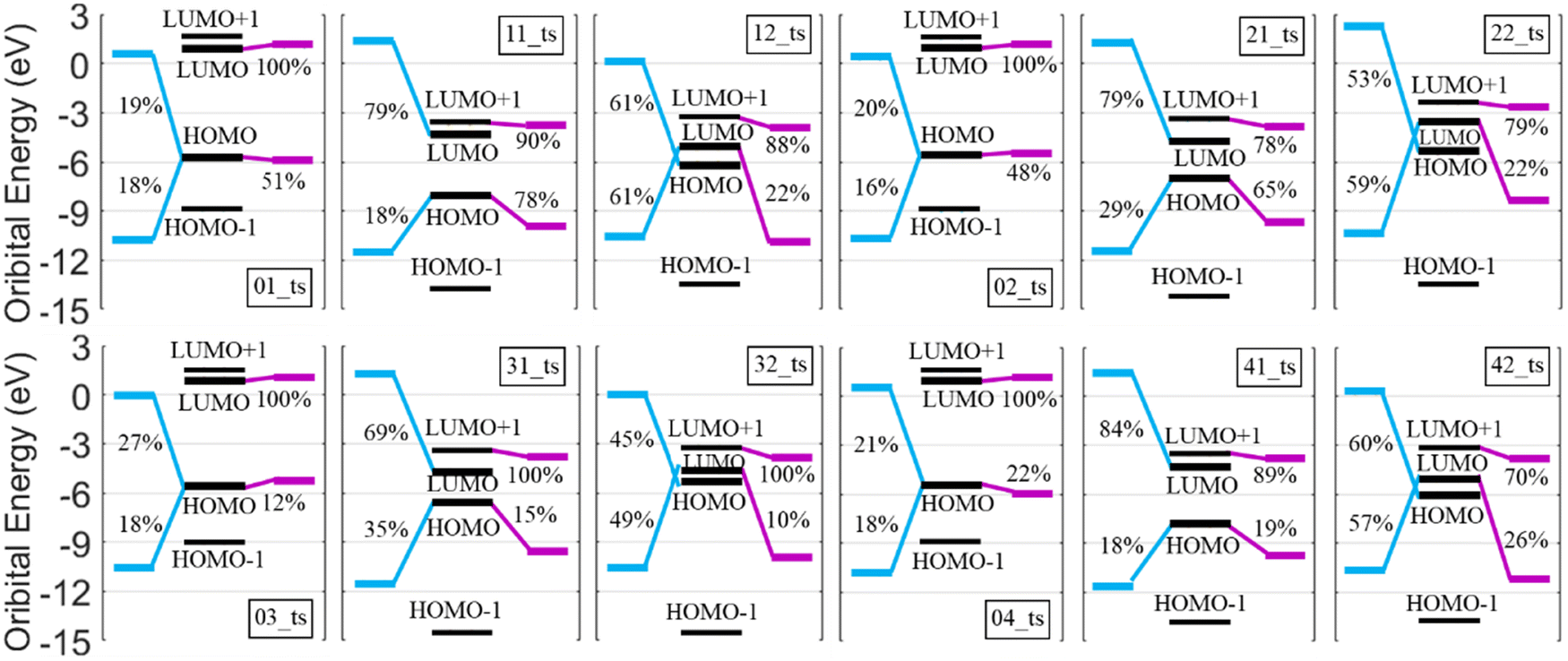 | ||
| Fig. 5 Orbital interaction diagram of TSs (marked in black lines) in C5H12 + H and C5H12+ + H abstraction reaction paths. The blue lines represent the HOMO and LUMO of the fragments formed by the H atom as the reactant isomer and the H atom at the reactive site. The HOMO and LUMO composited with other atoms in the system are marked with purple lines. The numbers marked beside the lines indicate the contribution (those smaller than 10% are not highlighted) from the fragmental MO to the corresponding TSs. | ||
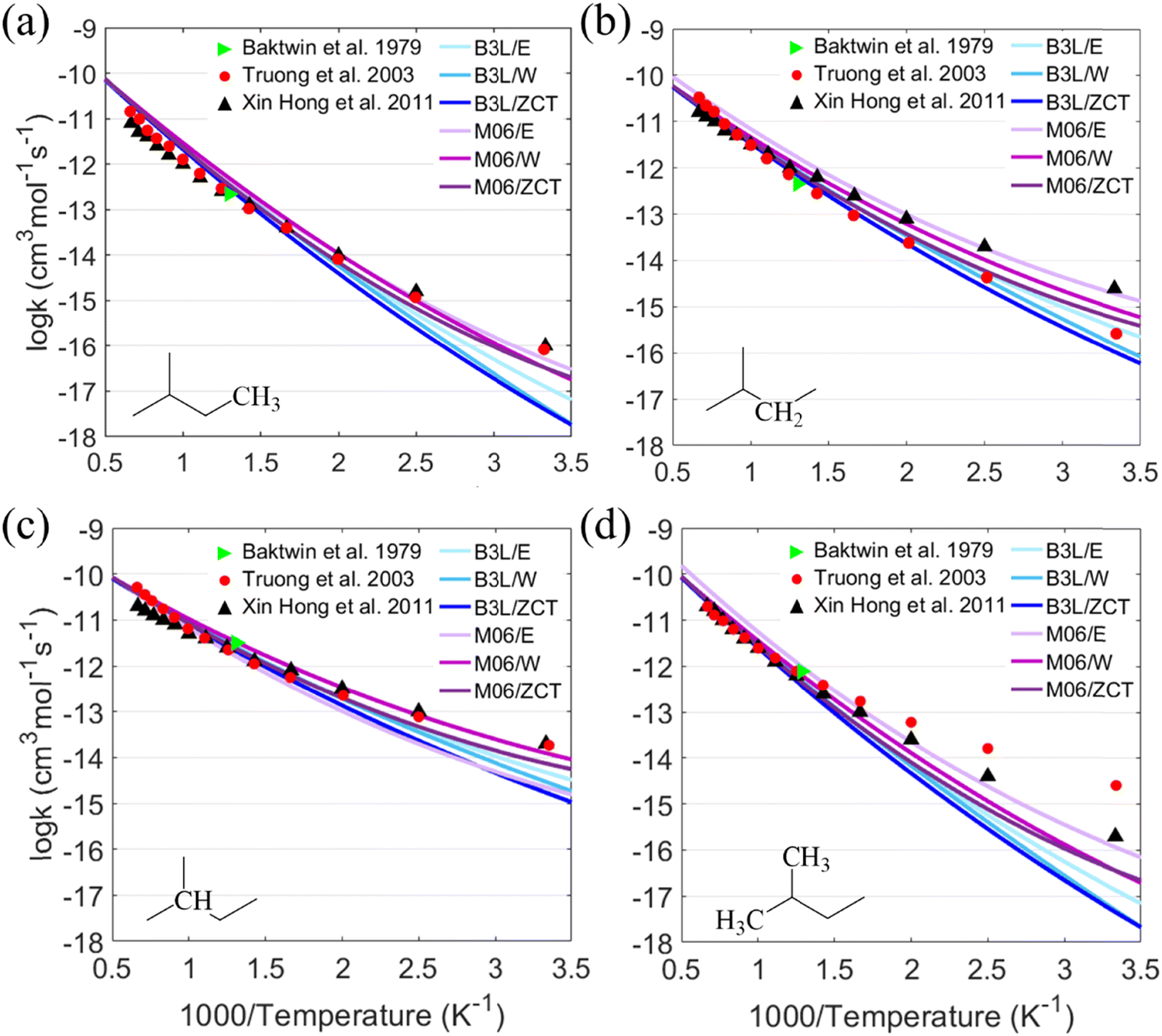 | ||
Fig. 6 The results of the calculated rate constants of C5H12 + H in this study (full lines in blue and purple) were compared to previous experimental data ( -Baktwin et al.51) and theoretical calculations ( -Baktwin et al.51) and theoretical calculations ( -Truong et al.;52 ▲-Xin Hong et al.7). -Truong et al.;52 ▲-Xin Hong et al.7). | ||
3.3 Kinetics of the abstraction reaction by the H atoms of isopentane molecules and ions
For the PESs optimized at B3LYP-D3(BJ)/6-311G(2df,2p) (marked as B3L) and M06-2X-D3/ma-def-TZVP (marked as M06) levels and refined at the DLPNO-CCSD(T)/aug-cc-pVTZ level, the calculated values of rate constants with the tunneling effect corrected by the Eckart (E), Wigner (W) and zero-curvature tunneling (ZCT) methods for the hydrogen abstraction of C5H12 by the H atom were compared with the literature values.9–11Fig. 6 shows that the rate constants calculated using the PESs optimized with the M06-2X functional were generally higher than those calculated using the B3LYP functional. Rate constants corrected by the Wigner and the ZCT tunneling revealed a better agreement than those corrected using the Eckart model. The results of M06/E were relatively close to those obtained by Hong et al. at a temperature above 500 K compared to the other methods in this study. Three-parameter Arrhenius expressions for the high-pressure-limit rate constants (cm3 mol−1 s−1) from 500 K to 2000 K at the DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2X-D3/ma-def2-TZVP level were fitted as follows:The sequence of the calculated rate constants was k03 > k02 > k04 > k01. In other words, the hydrogen abstraction reactivity of different reactive sites was tertiary > secondary > branched primary > primary, which was consistent with the abovementioned results of energy barriers and bond strengths.

The rate constants of C5H12+ + H were calculated at B3L and M06 levels and corrected using the Wigner and ZCT tunneling models, which showed good agreement with each other in the above discussion. The results of two reaction paths (abstraction products labeled 1 in blue lines and cleavage products labeled 2 in purple lines) are presented in Fig. 7. Tunneling appeared to have a smaller effect on primary and branched primary sites than on secondary and tertiary sites, and it deserved more consideration when the M06 method was used below 1000 K.
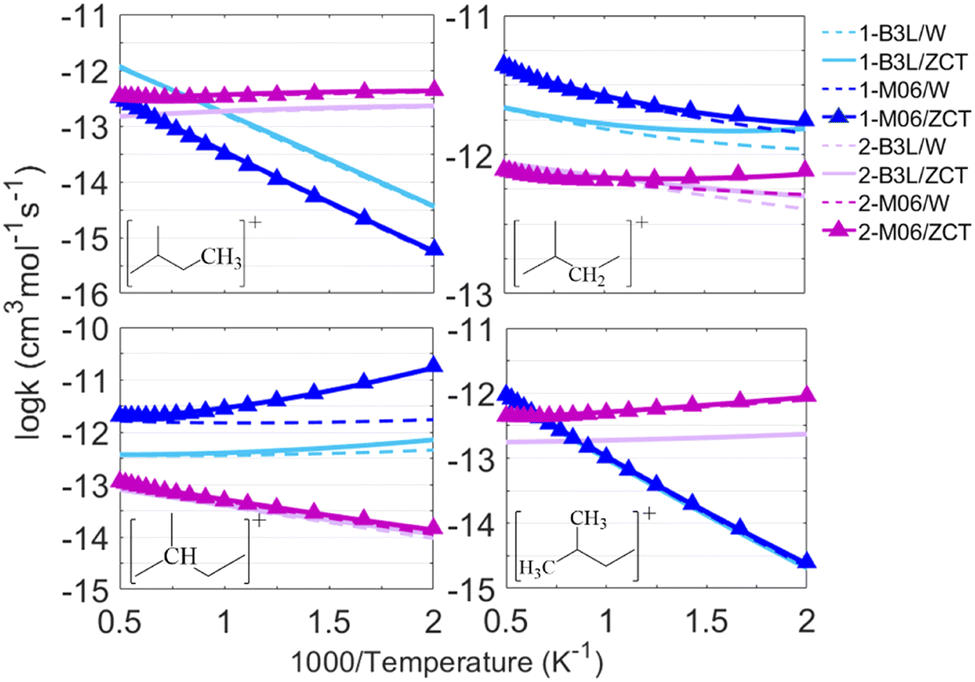 | ||
| Fig. 7 The results of the calculated rate constants of C5H12+ + H in this work. Those marked with blue lines stand for reaction paths with abstraction products. | ||
The primary and branched primary sites are more prone to abstraction products than to cleavage products, especially at low temperatures <1000 K, because higher C–C bond strengths make C–C bonds harder to be broken, and lower abstraction energy barriers facilitate abstraction as discussed above. Secondary and tertiary sites with weaker C–C bonds had advantages in formalizing cleavage products. High-pressure-limit rate constants (cm3 mol−1 s−1) at the DLPNO-CCSD(T)/aug-cc-pVTZ level//M06-2X-D3/ma-def2-TZVP level, as the highest accuracy achieved in this study, were fitted to a three-parameter Arrhenius expression over the temperature range from 500 K to 2000 K as follows:

To further discuss the potential influence of fuel ions on the reaction system, a basic model containing 204 species and 4682 reactions was built using RMG for the steam cracking of C5H12. Then the 12 calculated reaction rate constants were incorporated into the basic model. The updated model includes 210 species and 4690 reactions. Fig. 8 demonstrates the reaction path for the dissociation of C5H12 and C5H12+ at different temperatures. The normalized contributions to destruction were determined by rate-of-production (ROP) analysis with CHEMKIN-PRO, marked along with the reaction path arrows in Fig. 8. The final product showed a significant difference at different temperatures. Both C5H12 and C5H12+ tend to form methane (CH4) at low temperature (∼1000 K) and H2 at high temperature (∼2000 K). The result indicates that the C–H bond may dissociate at a higher temperature than the C–C bond, which is in good agreement with the dissociation energy of the C–C and C–H bonds shown in Fig. 2. To compare the reaction path of C5H12 and C5H12+, the dissociation intermediates differ in the number of carbon atoms. C5H12 tends to dissociate into large molecules, and C5H12+ dissociates more into small molecules, indicating that ionization improves the bond dissociation and makes the reaction easier.
 | ||
| Fig. 8 Reaction path of (a) C5H12 at 1000 K; (b) C5H12+ at 1000 K; (c) C5H12 at 2000 K; and (d) C5H12+ at 2000 K. | ||
Fig. 9 shows the sensitivity of CH4 and H2 in the top 15 related reactions. The reaction type means the role of the reaction in the reaction path, including direct reactions, i.e., abstraction, and indirect reactions, such as activation, addition, or isomerization of other reactants involved in abstraction reactions. The same reactants followed by different numbers indicate different activation sites or conformations. The same reaction shows a greater sensitivity of H2 at 2000 K and a higher sensitivity of CH4 at 1000 K, which matches the trend of final product formation in the reaction path.
 | ||
| Fig. 9 Sensitivity of (a) CH4 at 1000 K; (b) H2 at 1000 K; (c) CH4 at 2000 K; and (d) H2 at 2000 K in top 15 most related reactions. | ||
4. Conclusions
In conclusion, the minimum energy paths on the PESs for hydrogen abstraction of i-C5H12 molecules and positive ions (C5H12 and C5H12+) were optimized using DFT methods (B3LYP-D3(BJ)/6-311G(2df,2p) and M06-2X-D3/ma-def2-TZVP) and refined at the DLPNO-CCSD(T)/aug-cc-pVTZ level. Bond order analysis showed good agreement with the BDE results of C5H12 at CBS-QB3 and CBS-APNO levels and revealed that the bond strength sequence did not change after ionization. The C–C bond sequence was secondary–tertiary < branched primary–tertiary < primary–secondary, and the C–H bond strength was secondary < tertiary < primary ≈ branched primary. The sequence of structure selectivity of H abstraction of both C5H12 + H and C5H12+ + H to generate the abstraction product H2 was tertiary > secondary > branched primary > primary. For the decline in bond strengths caused by the decrease of electron density after ionization, C5H12+ + H possessed another way of abstraction reaction by cleavage of C–C bonds to generate small molecules such as CH4, C2, and C3 products, which showed activity towards the formation of final products, such as H2 at high temperature and CH4 at low temperature. The dominant cleavage products of C5H12 + H and C5H12+ + H reactions were obtained based on the pyrolysis model constructed using RMG and explained by the different components of their TS molecular orbitals using frontier orbital theory.Author contributions
Yi Gao: methodology, investigation, writing – original draft. Bin Yang: writing – review & editing. Yang Zhao: investigation, writing – original draft. All authors jointly analyzed and interpreted the data.Conflicts of interest
The authors declare no conflict of interest.References
- Y. Ju and W. Sun, Prog. Energy Combust. Sci., 2015, 48, 21–83 CrossRef.
- J. A. T. Gray and D. Lacoste, Combust. Flame, 2018, 199, 258–266 CrossRef.
- Y. Ju, J. K. Lefkowitz, C. B. Reuter, S. H. Won, X. Yang, S. Yang, W. Sun, Z. Jiang and Q. Chen, Plasma Chem. Plasma Process., 2016, 36, 85–105 CrossRef CAS.
- T. Shiraishi, SAE Int. J. Engines, 2019, 12, 101–113 Search PubMed.
- S. J. Barkley, K. Zhu, J. E. Lynch, J. Michael and T. Sippel, Combust. Flame, 2019, 199, 14–23 CrossRef CAS.
- C. Gong, L. Yi, K. Wang, K. Huang and F. Liu, Energy, 2020, 192, 116598 CrossRef CAS.
- X. Hong, H. Sun and C. K. Law, Comput. Theor. Chem., 2011, 963, 357–364 CrossRef CAS.
- A. Starikovskiy and N. Aleksandrov, Prog. Energy Combust. Sci., 2013, 39, 61–110 CrossRef CAS.
- Z. H. Wang, L. Yang, B. Li, Z. S. Li, Z. W. Sun, M. Aldén, K. F. Cen and A. A. Konnov, Combust. Flame, 2012, 159, 120–129 CrossRef CAS.
- E. Mintoussov, S. Pancheshnyi and A. Starikovskii, 42nd AIAA Aerospace Sciences Meeting and Exhibit, 2004 DOI:10.2514/6.2004-1013.
- T. Ombrello, S. H. Won, Y. Ju and S. Williams, Combust. Flame, 2010, 157, 1906–1915 CrossRef CAS.
- T. Ombrello, S. H. Won, Y. Ju and S. Williams, Combust. Flame, 2010, 157, 1916–1928 CrossRef CAS.
- C. Wang and W. Wu, Combust. Flame, 2014, 161, 2073–2084 CrossRef CAS.
- A. Rousso, S. Yang, J. Lefkowitz, W. Sun and Y. Ju, Proc. Combust. Inst., 2017, 36, 4105–4112 CrossRef CAS.
- R. Zhang, H. Liao, J. Yang, X. Fan and B. Yang, Proc. Combust. Inst., 2019, 37, 5577–5586 CrossRef CAS.
- W. Sun, M. Uddi, S. H. Won, T. Ombrello, C. Carter and Y. Ju, Combust. Flame, 2012, 159, 221–229 CrossRef CAS.
- N. L. Aleksandrov, S. V. Kindysheva, E. N. Kukaev, S. M. Starikovskaya and A. Y. Starikovskii, Plasma Phys. Rep., 2009, 35, 867 CrossRef CAS.
- W. Wang, R. Snoeckx, X. Zhang, M. S. Cha and A. Bogaerts, J. Phys. Chem. C, 2018, 122, 8704–8723 CrossRef CAS.
- X. Mao, A. Rousso, Q. Chen and Y. Ju, Proc. Combust. Inst., 2019, 37, 5545–5552 CrossRef CAS.
- T. Y. Chen, A. C. Rousso, S. Wu, B. M. Goldberg, H. van der Meiden, Y. Ju and E. Kolemen, J. Phys. D: Appl. Phys., 2019, 52, 18LT02 CrossRef CAS.
- R. Gilbert and S. Smith, Theory of Unimolecular Recombination Reactions, 1990 Search PubMed.
- HyperChem(TM) Professional 7.51, Hypercube, Inc.: 1115 NW 4th Street, Gainesville, Florida 32601, USA, 2011.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Rev. A.03), Wallingford CT, 2016.
- A. R. Hajipour, M. Karimzadeh Younjali, S. Jalilvand, H. Farrokhpour and A. Chermahini, Comput. Theor. Chem., 2014, 1045, 10 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- E. Papajak and D. Truhlar, J. Chem. Theory Comput., 2010, 7, 10–18 CrossRef PubMed.
- Y. Gao, Y. Zhao, Q. Guan and F. Wang, Chem. Phys. Lett., 2020, 759, 138035 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- D. Liakos and F. Neese, J. Chem. Theory Comput., 2015, 11, 4054–4063 CrossRef CAS PubMed.
- J. Martin, F. Neese, D. Liakos, M. Kesharwani and M. Sparta, J. Chem. Theory Comput., 2015, 11, 1525–1539 CrossRef PubMed.
- D. Glowacki, C.-H. Liang, C. Morley, M. Pilling and S. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- S. H. Lipoff and D. R. Herschbach, Mol. Phys., 2010, 108, 1133–1143 CrossRef CAS.
- C. A. Gonzalez, T. C. Allison and F. Louis, J. Phys. Chem. A, 2001, 105, 11034–11040 CrossRef CAS.
- Y. Wang and J. M. Bowman, J. Chem. Phys., 2008, 129, 121103 CrossRef PubMed.
- R. Steckler, W.-P. Hu, Y.-P. Liu, G. C. Lynch, B. C. Garrett, A. D. Isaacson, V. S. Melissas, D.-h Lu, T. N. Truong, S. N. Rai, G. C. Hancock, J. G. Lauderdale, T. Joseph and D. G. Truhlar, Comput. Phys. Commun., 1995, 88, 341–343 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- C. W. Gao, J. W. Allen, W. H. Green and R. H. West, Comput. Phys. Commun., 2016, 203, 212–225 CrossRef CAS.
- S. An and J. C. Jung, Case Stud. Therm. Eng., 2020, 21, 100694 CrossRef.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163–183 CrossRef CAS.
- G. von Frantzius, R. Streubel, K. Brandhorst and J. Grunenberg, Organometallics, 2006, 25, 118–121 CrossRef CAS.
- T. Lu and S. Manzetti, Struct. Chem., 2014, 25, 1521–1533 CrossRef CAS.
- V. Gomonaj and H. Toulhoat, ACS Catal., 2018, 8, 8263–8272 CrossRef CAS.
- T. Lu and F. Chen, J. Phys. Chem. A, 2013, 117, 3100–3108 CrossRef CAS PubMed.
- I. Mayer and P. Salvador, Chem. Phys. Lett., 2004, 383, 368–375 CrossRef CAS.
- A. Michalak, R. L. DeKock and T. Ziegler, J. Phys. Chem. A, 2008, 112, 7256–7263 CrossRef CAS PubMed.
- R. R. Baldwin and R. W. Walker, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 140–154 RSC.
- S. Zhang and T. N. Truong, J. Phys. Chem. A, 2003, 107, 1138–1147 CrossRef CAS.
| This journal is © the Owner Societies 2022 |