
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dehydrogenation of ammonia on free-standing and epitaxial hexagonal boron nitride†
Anthony J. R.
Payne
 a,
Neubi F.
Xavier
a,
Glauco F.
Bauerfeldt
b and
Marco
Sacchi
*a
a,
Neubi F.
Xavier
a,
Glauco F.
Bauerfeldt
b and
Marco
Sacchi
*a
aDepartment of Chemistry, University of Surrey, Guildford, GU2 7XH, UK. E-mail: m.sacchi@surrey.ac.uk
bInstituto de Química, Universidade Federal Rural do Rio de Janeiro, 23890-000 Seropédica-RJ, Brazil
First published on 11th August 2022
Abstract
We report a thermodynamically feasible mechanism for producing H2 from NH3 using hBN as a catalyst. 2D catalysts have exceptional surface areas with unique thermal and electronic properties suited for catalysis. Metal-free, 2D catalysts, are highly desirable materials that can be more sustainable than the ubiquitously employed precious and transition metal-based catalysts. Here, using density functional theory (DFT) calculations, we demonstrate that metal-free hexagonal boron nitride (hBN) is a valid alternative to precious metal catalysts for producing H2via reaction of ammonia with a boron and nitrogen divacancy (VBN). Our results show that the decomposition of ammonia proceeds on monolayer hBN with an activation energy barrier of 0.52 eV. Furthermore, the reaction of ammonia with epitaxially grown hBN on a Ru(0001) substrate was investigated, and we observed similar NH3 decomposition energy barriers (0.61 eV), but a much more facile H2 associative desorption barrier (0.69 eV vs 5.89 eV). H2 generation from the free-standing monolayer would instead occur through a diffusion process with an energy barrier of 3.36 eV. A detailed analysis of the electron density and charge distribution along the reaction pathways was carried out to rationalise the substrate effects on the catalytic reaction.
1 Introduction
Several green energy technologies employing hydrogen to decarbonise transport and industry are of considerable economic interest. However, H2 has a low energy density by volume (2.4 kWh l−1)1 and is difficult to store and transport, therefore more effective chemical storage methods are required. Ammonia, which can be stored as a liquid and contains 17.65% H by mass, can provide a convenient source of H2.2 Decomposition of ammonia will only produce H2 and N2, the latter a non-toxic, non-greenhouse gas. However, decomposing NH3 is difficult to achieve, often requiring rare transition metal catalysts such as Ru or Te.3 Furthermore, often further steps are required to purify H2 before use. Clearly, there is a need to design materials that can simultaneously decompose ammonia and produce pure H2.Hydrogenation reactions involve the addition of H2 to an element or molecule (generally an unsaturated compound) and are widely used in the food,4 petrochemical,5 and fine chemical industries.6 For enabling hydrogenation, a catalyst is employed to activate molecular hydrogen and allow the reaction to proceed. A range of metal-based catalysts make a variety of hydrogenation reactions possible, including: the conversion of alkenes to alkanes using Pt,7,8 carboxylic acids to alcohols or alkanes using Re/Pd,9 and phenols to cyclohexanone/cyclohexanol using Rh.10 Such catalysis enables new synthesis routes and the production of high-quality feedstocks for fine chemical industries and mass-produced products such as Nylon.
However, the decrease in the availability of metals such as Pt, Pd, and Re, combined with an increase in prices due to high energy demands,11 and local environmental damage,12 has reduced the appeal of these catalysts. Greener and more sustainable alternative materials with similar catalytic properties need to be found. Hexagonal boron nitride (hBN) nanosheets are of interest for applications including electrical,13 optoelectronic14 and spintronic15 devices. hBN consists of sp2 hybridised N and B atoms forming 2D sheets.16 Layers of hBN stack in an ABAB pattern held together by van der Waals forces.17 Charge localisation gives hBN a large experimental band gap of 6 eV.18 The fabrication techniques used to produce monolayer hBN include chemical vapour deposition,19 direct reaction,20 and mechanical methods.21 Additives can be added during sonication to aid the exfoliation of hBN to nanosheets. Ammonia facilitates hBN exfoliation via a Lewis base mechanism.22 The methods used to produce hBN can result in a range of thicknesses, with the fabrication of monolayer hBN possible.23
Despite the recent advances in the fabrication process of hBN,24 hBN sheets often contain a range of defects, including impurities, vacancies, edges and grain boundaries.25 These defects have been investigated as metal-free sites for a wide range of reactions, such as; the hydrogenation of alkenes,26 capture of CO2,27 conversion of methane to methanol,28 and N2 fixation to NH3.29 Ball milling has been established as a way of creating additional defect sites.30,31 Longer milling durations have shown increased activity and selectivity for the nitroaldol reaction at 100 °C.32 Clearly, reactions of hBN defects are possible under achievable conditions.
A range of vacancy defects including boron vacancies (VB) and nitrogen vacancies (VN) have been observed using high-resolution transmission electron microscopy.33,34VB and VN have been shown to react with atmospheric components to create catalytically active sites; for example, carbon sites can be introduced by the reaction of VB with CO2.35 These sites can then catalyse NO reduction by CO to form N2 and CO2.27 These promising results have sparked interest in hBN as a heterogeneous catalyst. Pristine hBN has several properties which are intrinsically suited for a catalyst, including high thermal stability,16,36 a low coefficient of thermal expansion,37 and high specific surface area.38 The range of active sites available and the physical stability make hBN an excellent candidate for industrial catalysis.
hBN with a boron antisite defect where a nitrogen is replaced with an additional boron atom has been explored as a potential site for hydrogen release from NH3 using density functional theory (DFT).39 NH3 is chemisorbed onto the antisite, and H2 can be released by overcoming a calculated energy barrier of 0.65 eV, leaving behind a NH species which requires an energy of 4.68 eV to remove and regenerate the original site. In this study, we focus on the application of vacancy defects as active sites for the dehydrogenation of ammonia. Each dangling bond on VB and VN defects can be hydrogenated to become more stable.40 Hydrogenated VB can behave as H donor sites, able to hydrogenate a range of alkanes at high yields as shown by experiment and supported with DFT.26 However, an efficient way of creating hydrogenated VB defects has yet to be proposed.
In addition to VN and VB defects, larger vacancies such as BN divacancies (VBN) and triangular (VN+3B) have been the subject of previous research, with the VBN defect shown to be more stable under neutral and nitrogen rich conditions.41 Calculations have shown that VB vacancies can migrate much more easily than VN.42 Therefore VB can become trapped when contacting a VN, forming VBN through a mutual annihilation process. This process of forming VBN from VB and VN has been used to justify the low concentration of paramagnetic centres measured in irradiated hBN.43VBN has been investigated for use in H2 activation and production, requiring an energy barrier of approximately 400 kJ mol−1 to be overcome to release H2 and regenerate VBN.44 Dehydration of alkanes to alkenes using VBN sites has also been investigated as a metal-free alternative to commercial production of alkenes.45 An activation energy barrier of 112.1 kJ mol−1 (1.16 eV) was found to convert ethane (C2H6) to ethene (C2H4), resulting in a hydrated VBN. Given the stability, properties and previous work on VBN, we expect VN vacancies to be a source of novel reactivity.
Dehydration of ammonia borane (H3N–BH3) by VN, VB and VN+3B has been investigated using DFT as a way of producing H2.46 The results showed low energy barriers to transfer 2H to defects in the hBN sheet followed by further reaction to release a single H2. However, only 2 of the 6 available hydrogen atoms are released, leaving behind a H2N–BH2 species. The incompleteness of the reaction and the limited uses of ammonia borane in industry hinder its practical and widespread adaption as a fuel source. Conversely, ammonia is widely used in industry, most notably in the production of fertilisers and has been shown to decompose completely.47 Literature has investigated the use of NO and NO2 to repair VN defects, ultimately ‘healing’ the vacancy and producing oxygen.48 This process is energetically facile with a rate-limiting barrier of 0.82 eV. Ammonia has the potential to behave similarly and produce hydrogen.
Our computational study shows that the VBN defects of hBN can provide an excellent site for the reaction of ammonia to form a hydrogenated VB site or H2 depending on surface temperature. The hydrogenated VB can then be used as a source of H2 for green energy and key hydrogenation reactions such as the hydrogenation of alkenes to alkanes.
2 Computational methodology
First-principles electronic structure calculations were carried out using CASTEP.49 The PBE functional50 was used to treat exchange and correlation in combination with the Tkatchenko and Scheffler dispersion correction method.51 The plane wave basis set was expanded to a cut-off energy of 400 eV using Vanderbilt ultrasoft pseudopotentials.52 All calculations used a Monkhorst–Pack k-point sampling of the first Brillouin zone53 with a uniform grid of (10 × 10 × 1) or (4 × 4 × 1) for modelling (1 × 1) and (5 × 5) unit cells, respectively. The surface structures were optimised until the maximum force on each atom was less than 0.025 eV Å−1 and the total energy changed by less than 1 × 10−6 eV. The transition states along the reaction pathways were calculated with the Linear and Quadratic Synchronous Transit (LST/QST) algorithm, with a force tolerance of 0.1 eV Å−1. A (5 × 5) supercell was employed for all defect calculations, with a vacuum region of at least 20 Å to separate the periodically repeated images and avoid spurious interactions.Adsorption energies EAds were calculated using the standard formula:
| EAds = EAB − EA − EB | (1) |
Formation energies Ef were calculated using the formula:
| Ef = EV − EhBN + nNEN + nBEB | (2) |
3 Results and discussion
3.1 Ammonia decomposition on hBN
Initially, a (1 × 1) cell consisting of a single boron and nitrogen atom was converged with respect to cell size and k-points. When optimised, a bond length of 1.433 Å and lattice parameter of 2.483 Å was achieved, in close agreement with experimental values.23 It is well established that GGA functionals such as PBE underestimate the band gap; our value of 4.5 eV is in close agreement with other DFT studies.55,56 Each vacancy was constructed by removing the corresponding atoms, and the energy converged with respect to cell size and k-points.The two possible mono-vacancies can be seen in Fig. 1. VN has D3h symmetry and B–B distances of 2.367 Å around the vacancy site and a magnetic moment of 1 ℏ/2. In contrast, VB has C2v symmetry, two N–N distances of 2.786 Å and one of 2.845 Å, with a magnetic moment of 1 ℏ/2, due to Jahn-Teller effects. The geometry and properties of these vacancies is consistent with the literature.55,57,58 The Ef of VN and VB are 8.63 eV and 9.75 eV, respectively. Therefore, VN is more stable, in agreement with previous works.59–62VBN can be formed by removing a pair of adjacent B and N atoms achieving a N–N distance of 2.379 Å, B–B distances of 2.052 Å, B–N distances of 3.099 Å, an overall magnetic moment of 0 ℏ/2, and C2v symmetry, consistent with previous calculations.63 The B–B distance is shorter due to a weak covalent bond, and there is no overall magnetic moment as the electrons are all able to pair. The Ef of the divacancy has been calculated to be 11.83 eV, approximately 2 eV higher than the mono vacancies consistent with literature.64,65 The combined formation energy of VB and VN is 18.38 eV, much higher than that for a VBN, thus the probability of forming a VBN defect is higher than forming separate VB and VN defects, as has been previously suggested.65
 | ||
| Fig. 1 (a) Pristine hBN sheet. (b) VBN (Ef = 11.83 eV, spin = 0 ℏ/2) defect. (c) and (d) are VB (Ef = 9.75 eV, spin = 1 ℏ/2), and VN (Ef = 8.63 eV, spin = 1 ℏ/2) respectively. Boron atoms are shown in green and nitrogen atoms shown in grey. | ||
Firstly, the adsorption of ammonia above a pristine hBN monolayer was considered. A range of sites and orientations were investigated. The most stable physisorption site can be seen in Fig. 2a with an EAds of −0.172 eV. As in previous work, the most stable site is above the B atom.56,66–68 However, we found the configuration with hydrogens orientated towards the hBN is preferred by 0.015 eV, with a N distance of 3.34 Å from the surface. In terms of NH3 orientation upon adsorption, literature results vary depending on the exchange and correlation functional, basis set, and tolerances used, but they generally agree with our results. The barrier to invert the H–H bonds (umbrella inversion) is small and was calculated to be 0.158 eV, similar to values calculated for an isolated ammonia molecule.69Fig. 2b and d show the charge accumulation (blue) and charge depletion (red) of ammonia above hBN, with respect to isolated ammonia in the gas phase. The difference in intensity and topology of the charge accumulation reveals a much stronger dispersion interaction when the hydrogen atoms are orientated towards the hBN. The ammonia has a partial charge of 0.13 e and 0.06 e when the hydrogens are orientated towards and away from the hBN, respectively. The greater partial charge is indicative of stronger dispersion interactions, resulting in increased stability when the hydrogen atoms are orientated towards the hBN monolayer.
 | ||
| Fig. 2 (a) The most favourable orientation of ammonia above a pristine hBN sheet has the three N–H bonds pointing towards the surface and an adsorption energy of −0.172 eV at a distance of 3.68 Å from the surface. (c) Inverted ammonia adsorption, with an adsorption energy of −0.157 eV at a distance of 2.92 Å from the surface. These orientations can be inter-converted by overcoming an energy barrier of 0.158 eV. (b) and (d) show charge accumulation (blue) and charge depletion (red) with respect to isolated ammonia (isosurface cutoff 0.001 e Å−3). | ||
Several sites and orientations of ammonia above VBN were considered including on top of B, on top of N, the centre of the vacancy and between N–N, B–B and B–N atoms. Optimised structures gave an EAds of −0.25 eV for the most stable adsorption geometry. The nitrogen atom is positioned almost directly above the vacant nitrogen position of the VBN at a height of 2.76 Å. Contrary to what was observed for NH3 physisorption on pristine hBN, ammonia adsorbs on top of a divacancy in a slightly tilted configuration to enable the lone pair on the nitrogen to interact with the electron-deficient B–B bond, forming a weak intermolecular bond. Each of the three N–H bonds are consequently pointing away for the vacancy. This geometry is the first step of the deportation reaction which mechanism is illustrated in Fig. 6 and in Fig. S1 of the ESI.†
Next, the reaction of NH3 and VBN was investigated. A number of pathways, products and intermediates were considered in order to find the minimum energy pathway and the most plausible reaction mechanism. The first intermediate is produced by breaking the strong N–H bond to produce a chemisorbed NH2 and H species. The surface boron atom plays an active role in stabilising the transition state (TS1) by forming a bond to nitrogen and being ready to accept the deprotonated hydrogen atom as seen in Fig. 3. The resulting energy barrier is 0.52 eV and is the highest barrier to NH3 dissociation.
 | ||
| Fig. 3 (a) the geometry and (b) the electron density isosurface (0.5 e Å−3) of the transition state TS1 for the dissociative chemisorption of ammonia over a hBN VBN. The energy barrier is 0.52 eV and the limiting step for ammonia disassociation. The geometry of this intermediate shows puckering of the anchoring boron atom 0.4 Å above the plane as the N–B bonds are stretched away from their equilibrium positions. The electron density isosurface shows a weak orbital overlap between the ammonia nitrogen and a hydrogen atom indicative of the first deprotonation step. Once this transition is overcome, the structure collapses to create an adsorbed NH2 and H. | ||
As the barriers to NH3 desorption and dissociation are 0.25 eV and 0.52 eV respectively both of these processes are competitive. Therefore the kinetics of the aforementioned reactions we considered to suggest the formation rate of the NH2 and H species on the surface. Rate coefficients were obtained by means of the Transition State Theory, at the temperatures of 600 K, 800 K, 1000 K and 1200 K which were possible operation temperature values of catalytic H2 production by methods as described in the ESI.† It was possible to suggest that, at 600 K, the complete convergence of the adsorbed NH3 into NH2 and H, was obtained, at roughly 400 seconds (6.6 minutes), whereas, complete convergence was achieved in 0.4 seconds at 1200 K as illustrated in Fig. S2 of the ESI.† Therefore, NH3 is expected to dissociate into NH2 and H in a reasonable amount of time.
The second barrier of 0.15 eV over TS2 reflects the migration of a hydrogen atom to satisfy a nitrogen dangling bond. The third transition state (TS3) reflects the migration of two hydrogen atoms to satisfy all the dangling bonds of VB and form a stable hydrogenated boron vacancy after overcoming a small energy barrier of 0.04 eV. TS3 involves the migration of an H breaking one H–N bond and forming a second N–H bond on the other side of the vacancy with the migration of a second H breaking a B–H bond and forming an H–N bond in place of the original. Both migrations in TS3 are expected to take place in a single step as no relaxed geometry of a single migration was observed. The reaction pathway leads to the formation of a hydrogenated VB, with a ΔE reaction of −8.11 eV and can be seen in Fig. 6.
Following the creation of a hydrated VB vacancy, the three remaining hydrogen atoms could diffuse away sequentially; the possible paths are shown in Fig. 4. Considering the first diffusion (Fig. 4a) the hydrogen could either move on top of a nitrogen atom that is adjacent to the vacancy (HNA) with energy barriers of 3.44 eV (TS5) or move directly away from the vacancy onto a nitrogen that is towards the pristine sheet (HNP) with energy barriers of 3.36 eV (TS4). No relaxed structures where the hydrogen atom had migrated to the boron atom were observed in geometry optimisations. The boron atom refuses to accept the hydrogen as its pz orbital is empty. The average bond population of the B–N bond increases from 0.90 e where a N–H bond is present to 1.15 e; this compensates for the loss of connectivity and creation of a dangling bond. The nitrogen atoms surrounding the vacancy have available and occupied pz orbitals, these bind to the diffusing hydrogen. Therefore, the hydrogen is not accepted by the boron, and moves onto a surrounding nitrogen instead. We found that the HNP pathway has a lower energy barrier by 0.07 eV (2.10%) and is the dominant route. This slight difference in transition state stability is attributed to a very weak stabilising B–H bond with a population of 0.11 e that is present in TS4 but not TS5. The trend of hydrogen diffusion following the HNP pathway continues as hydrogen's are removed from the vacancy as TS6 is higher in energy than TS7 by 2.29% and TS8 is higher in energy than TS9 by 1.45%.
 | ||
| Fig. 4 Sequential diffusion of the first (a), second (b) and third (c) hydrogen atoms away from the hydrogenated VB vacancy to HNA or HNP sites. Transition states are identified by ‡. Diffusion to a HNP site is preferred in each case with an increasing energy barrier increasing with each diffusion event (3.40 eV 3.63 eV and 3.88 eV). | ||
The average energy barrier to diffuse increases as the number of hydrogen atoms increases from 3.40 eV (TS4-5), 3.63 eV (TS6-7) and 3.88 (TS8-9). Hydrogen diffusion creates nitrogen dangling bonds the loss of connectivity destabilises the vacancy resulting in higher barriers hindering further diffusion steps. Diffused hydrogens could then migrate across the surface, where they may find another vacancy or hydrogen. In the latter case, it has been found that two surface hydrogens can desorb from a pristine hBN surface with an energy barrier of 0.41 eV and form H2 as shown in Fig. 5. This barrier reflects the energy required to break B–H and N–H bonds. The ΔE of the reaction is −2.04 eV; hence it is thermodynamically favourable for surface hydrogen to desorb as H2. Fig. 6 shows the reaction scheme from ammonia adsorption to diffusion of the first hydrogen via a hydrogenated VB vacancy.
 | ||
| Fig. 5 Combination of two Hads to create H2 and desorb from a pristine hBN surface following diffusion from a hydrogenated VB vacancies. Transition states are identified by ‡. The ΔE of the reaction is −2.04 eV. | ||
 | ||
| Fig. 6 The reaction pathway of ammonia with a VBN to produce a hydrogenated VB defect in monolayer hBN. Transition states are identified by ‡ and labelled TS1-4. The activation energy for the reaction is 0.52 eV reflecting the energy required to deprotonate a hydrogen atom from ammonia. Subsequent steps occur with a negligible energy barrier to produce a hydrogenated boron vacancy with a ΔE reaction of −8.11 eV. A hydrogen atom can then diffuse away from the vacancy after overcoming an energy barrier of 3.36 eV. | ||
Our results show that dehydration of ammonia followed by the insertion of a nitrogen atom into the VBN to form a hydrogenated VB is energetically facile as N becomes part of the lattice. Subsequent removal of H2 from VB is more challenging to achieve and requires higher temperatures.
3.2 Ammonia decomposition on hBN/Ru(0001)
So far, the hBN system studied is that of an isolated “freestanding” monolayer. However, this does not represent the only or the most probable case for technological applications in catalysis. In practice, epitaxial single-layer hBN is typically grown and employed as a catalytic material on a metal substrate. Currently, high-quality hBN surfaces can be epitaxially grown by CVD on a Ru(0001) surface.70 Therefore we expanded our investigations to include vacancies over a Ru(0001) substrate.Ru(0001) has a hexagonal-close-packed hcp crystal structure. It has been found that the most stable hBN over Ru(0001) configuration occurs when boron atoms continue the hcp stacking arrangement and nitrogen atoms sit on the adjacent top site directly above the Ru in the layer below.71–73 Charge transfer between hBN and Ru(0001) leads to repulsive/attractive forces acting on the nitrogen/boron of the hBN overlayer.73 Consequently, the boron atoms are marginally closer to the surface than the nitrogen. It is important to note that larger cells of hBN/Ru(0001) lead to the formation of a 12 × 12 nanomesh, however, experimentally the nanomesh consists of large pores 2.2 Å above the Ru(0001) surface,71 a similar distance to our calculated value of 2.17 Å and where vacancies would have adequate space to form.
The Ru(0001) lattice is 8.5% larger than that of hBN. This lattice mismatch stretches the hBN N–B bonds to match those of the Ru(0001) surface below in order to create N–Ru bonds. The Mulliken atomic charge distributions in boron and nitrogen atoms change from 2.18 to 2.63 e (+20%) and from 5.82 to 5.64 e (−3%) respectively, when comparing pristine hBN to pristine hBN/Ru(0001), showing that the B–N bond becomes more polarised as a consequence of the substrate interaction. The difference in populations reflects the reduction in B–N bond strength when above a Ru(0001) surface due to the additional strain caused by the substrate. The Mulliken charges on the boron and nitrogen atoms change dramatically (from 0.82 to 0.37 e for boron and from −0.82 to −0.64 for nitrogen). The atomic partial charges indicate that the nature of hBN interlayer bonding has changed due to the Ru(0001) layer below. As with the isolated monolayer, vacancy defects can be introduced when above a Ru(0001) surface with Ef of 2.37 eV, 3.45 eV and 5.38 eV for VB, VN, and VBN defects, respectively. These energies are lower than for a monolayer (−63% on average), owing to the added stability provided by the Ru layer below.
The calculated pathway for the reaction of NH3 with a hBN VBN on a Ru(0001) surface can be seen in Fig. 7 showing intermediates and transition states.
 | ||
| Fig. 7 The reaction pathway of ammonia with a VBN hBN defect above a Ru(0001) substrate. Transition states are identified by ‡ and labelled TS10-14. The ammonia is readily chemisorbed onto the surface after overcoming a low energy barrier of 0.02 eV. A hydrogenated VB defect is produced by sequential deprotonation steps. The highest energy barrier is 0.61 eV and reflects the energy to deprotonate the final hydrogen atom. The ΔE of the reaction is −3.21 eV. H2 can then associativity desorb by overcoming an energy barrier of 0.69 eV. | ||
As with the monolayer, several adsorption sites and orientations of ammonia on a hBN/Ru(0001) surface were considered. Optimised structures gave an EAds of −0.52 eV for the most stable adsorption geometry. This is indicative of strong physisorption with NH3 being adsorbed more strongly onto hBN/Ru(0001) than a hBN monolayer with an EAds of −0.25 eV. This difference in adsorption energy is attributed to the increased polarisation of the B–N bonds; thus, the interaction between the ammonia lone pair and the electron-deficient boron atoms of the vacancy is stronger.74 Similar to the monolayer, ammonia is physisorbed onto the hBN/Ru(0001) surface with a slightly tilted orientation allowing the nitrogen lone pair to point towards the B–B bond at a height of 1.90 Å above the surface, as can be seen in Fig. 8.
 | ||
| Fig. 8 NH3 over a hBN/Ru(0001) surface. (a) and (b) physisorption and chemisorption of NH3 respectively. (c) A hydrogenated VB hBN monolayer above a Ru(0001) substrate. The reaction proceeds via physisorption and chemisorption followed by a series of deprotonations to (c). | ||
Unlike adsorption onto an isolated monolayer, NH3 chemisorbed state with an adsorption energy of −1.57 eV was also observed as shown in Fig. 8b. The chemisorbed state has the ammonia's nitrogen bonded with one of the vacancy's boron atoms at a height of 1.07 Å and an average H–N–B bond angle of 110.3°. This chemisorption step is easily achieved by overcoming a very low energy barrier of 0.02 eV as only inter-molecular repulsion needs to be overcome in TS10. The existence of a chemisorbed NH3 species can be explained by the overall weaker N–B bonding in the hBN lattice due to stretching to fit the Ru(0001) surface unit cell, resulting in average N–B distances of 1.57 Å (9.8% longer compared to the optimum bond length of 1.43 Å, which makes the B atoms more electrophilic, binding to the ammonia more readily. Additionally, the N–Ru bond reduces the electron density available to the nitrogen for binding with boron atoms. Subsequent deprotonation of a H atom to a chemisorbed NH2 species occurs with a barrier of 0.21 eV (TS11) to saturate a dangling bond on the nitrogen occurs. This results in a B–NH2 and a N–H satisfying half of the dangling bonds in the vacancy. Next, the NH2 rotates around the B–N bond and binds to the baron atom on the other side of the vacancy after overcoming an energy barrier of 0.36 eV (TS12). The next step involves breaking a second N–H bond to satisfy the final dangling bond and hydrogenate the vacancy in TS13. For the decomposition of NH3 on hBN/Ru(0001) TS13 has the highest barrier of 0.61 eV due to the stability of the final intermediate and change from sp3 to sp2. The product of the reaction is similar to that of the previous mechanism investigated for the monolayer (Fig. 6), with hydrogen atoms satisfying the dangling bonds of the VB pointing away from the Ru as shown in Fig. 8c. The final structure has an EAds of −3.73 eV. The difference in pathway and energy barriers reflect the impact of the substrate. The small lattice mismatch between hBN and Ru(0001) allows the hBN monolayer to map onto the Ru(0001) lattice. Consequently, the B–N bonds stretch away from the ideal free-standing hBN lattice vectors causing, an overall weakening of the B–N bonds, affecting the band structure and electron density in the epitaxial layer in agreement with previously reported by experimental measurement.75 The change in electronic structure stabilises key intermediates for the reaction making a chemisorbed state possible which was not observed for the isolated monolayer. Despite these differences in mechanism, the product of NH3 decomposition on suspended hBN and hBN/Ru(0001) is similar and correspond to the creation of a hydrogenated VB vacancy.
 | ||
| Fig. 9 NH3 over a hBN/Ru(0001) surface. (a) and (b) physisorption and chemisorption of NH3 respectively. (c) A hydrogenated VB hBN monolayer above a Ru(0001) substrate. The reaction proceeds via physisorption and chemisorption followed by a series of deprotonations to (c). | ||
Hydrogen desorption from a hydrogenated VBN over a Ru(0001) surface was considered. The three surface hydrogen atoms are orientated away from the Ru(0001) substrate below (Fig. 8c). Two of the hydrogens were then found to desorb from the surface as H2 after overcoming a low energy barrier of 0.69 eV in TS14, producing H2. As with the monolayer, the remaining hydrogen atom could migrate across the surface and combine with a second hydrogen adatom, form H2, and desorb. The H2 desorption barrier from a pristine hBN/Ru(0001) surface was calculated to be 0.91 eV as shown in Fig. 6. The barrier for hydrogen to combine and desorb from the surface is higher than that for hBN/Ru(0001) than for the monolayer due to greater filling of the B pz orbital.
Alternatively, sequential diffusion of the final three hydrogen atoms was also considered. Unlike the monolayer where only HNA and HNP arrangements were found to be stable, the Boron atom adjacent to the vacancy has been found to accept a hydrogen atom HBA. Both of these routes are shown in Fig. 10 with the transition state of the HBA route lower in energy by 0.31 (6.5%) eV. This change in boron activity is due to B–Ru bonding which populates the B pz orbital. As this orbital is now populated, it can be used to bond with a diffusing hydrogen. After leaving the vacancy, the hydrogen atom would then diffuse over hBN and combine with another diffusing hydrogen after overcoming an energy barrier of 0.91 eV as shown in Fig. 9. Here unlike the monolayer, direct desorption of H2 is preferred with a low energy barrier of 0.69 eV compared to the diffusion process with barriers of 2.28 eV and 0.91 eV.
 | ||
| Fig. 10 Diffusion of hydrogen from a hydrated VB vacancy via HBA and HNA pathways. Transition states are identified by ‡. The HNA pathway has an activation energy of 2.59 eV and a ΔE reaction of 0.99 eV. The HBA pathway proceeds with an activation energy of 2.28 eV and a ΔE reaction of 0.37 eV. | ||
Attempting the associative desorption of H2 from the hydrogenated VB hBN monolayer results in a very high energy barrier of 5.98 eV. In this case, the adsorbed hydrogens rearrange to bring two adjacent hydrogens within 1.45 Å of each other, overcoming minimal activation energy of 0.07 eV as seen in Fig. S3 of the ESI.† The two hydrogen atoms associatively desorb by overcoming an energy barrier of 5.98 eV, leaving behind a boron vacancy containing a single hydrogen atom. The transition state barrier is much higher for associative desorption (5.98 eV) than diffusion (3.36 eV). Therefore, hydrogens are more likely released from the vacancy by diffusion from the isolated monolayer, in contrast to hBN/Ru(0001), where H2 associatively desorbs.
In order to explain the different reactivity of hBN/Ru(0001) compared to the free-standing hBN, we analysed the electron density of the pristine vacancy for the two systems. Fig. 11 shows the electron density slice of VBN. By comparing the two isosurfaces, we can see that when over Ru(0001), the hBN monolayer has a much lower electron density between localised the boron atoms and can therefore bind more readily with the incoming NH3. Additionally, the approaching ammonia experiences less repulsion from the reduced electron density in the vacancy. The lattice mismatch and consequent changes in B–N bonding have a profound effect on the reaction pathway. The reduced B–B bonding character and larger space provided by the lattice miss-match allows for the ammonia to chemisorb onto one of the boron atoms as seen in Fig. 8. The chemisorbed NH3 species is not stable for the isolated monolayer, instead, direct dissociative chemisorption occurs after the ammonia physisorption step. After the initial deprotonation, the effects of the substrate are evident for all remaining steps, with drastically different barrier heights and intermediates. The reduced electron density on the boron atoms caused by Ru(0001) increases the strength of the dipole–dipole bond between the surface and ammonia. Correspondingly the EAds for NH3 are −0.25 and −0.52 eV for the isolated monolayer and the monolayer above a Ru(0001) substrate, respectively.
 | ||
| Fig. 11 An electron density contour map from 0 to 4 e Å−3 of the hBN VBN monolayer (a) and the VBN monolayer over Ru(0001) (b). We can see that the electron density of the B–B bond in the vacancy (circled in red) is much higher for the isolated hBN monolayer than with a Ru(0001) substrate below. B–B distances are 2.05 Å and 2.98 Å for isolated hBN and hBN/Ru(0001). | ||
Fig. 12 shows the Highest Occupied Molecular Orbital (HOMO) for the physisorbed ammonia above the VBN and VBN over Ru(0001). Both plots show the electron density accumulation stretching from the physisorbed ammonia's nitrogen towards the surface boron atoms. The HOMO comprises of contributions from both the NH3 and the B–B atoms of the vacancy immediately underneath the adsorbate. The HOMO of the hBN/Ru(0001) system includes mixing of the boron sp2 orbitals with those of the Ru, which shifts the HOMO towards the centre of the vacancy. It follows that the reaction happens at the B–B site in order to minimise the energy of the HOMO orbital.
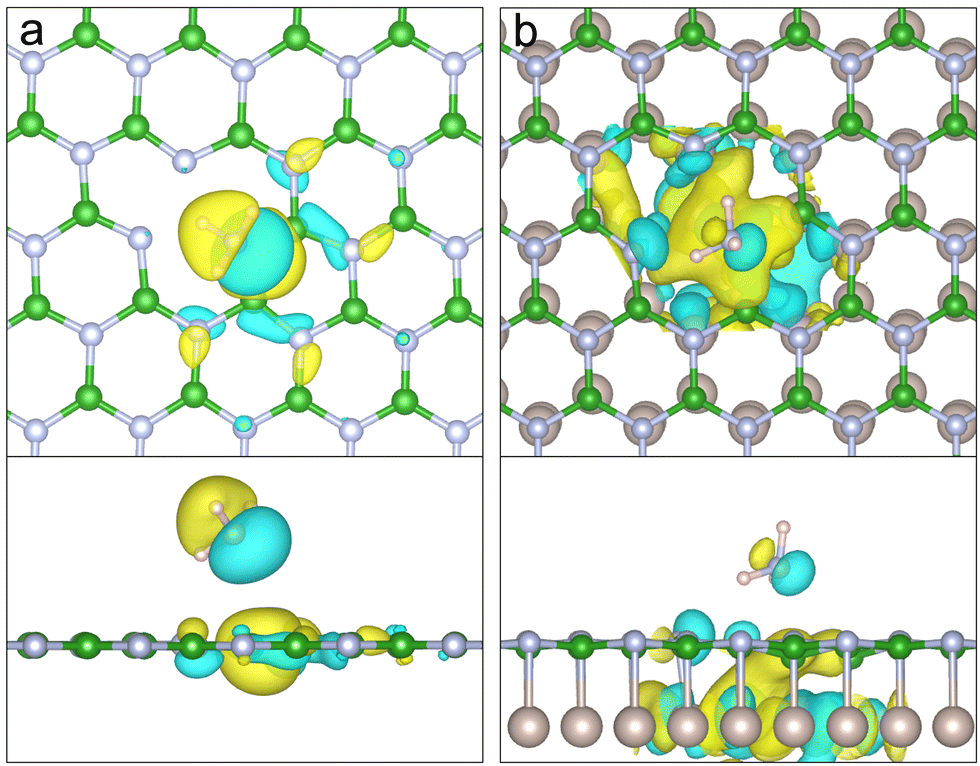 | ||
| Fig. 12 The HOMO of physisorbed NH3 over (a) VBN and (b) VBN over Ru(0001) with an isosurface value of 0.02 e Å−3. In both cases, the HOMO consists of ammonia and boron orbitals, however in (b) the HOMO is situated towards the centre of the vacancy. Showing where the ammonia reacts to lower the energy of these orbitals. Orbitals far from the vacancy have been omitted for clarity. | ||
Fig. 13a shows the electron density isosurface of NH3 chemisorbed onto a hBN VBN defect with a Ru(0001) substrate. Here we can see bond formation between the NH3 nitrogen and a surface boron atom in the vacancy. The tetragonally bonded nitrogen has a Mulliken charge of −0.85 e, and the B–N bond has a population of 0.62 e, showing more ionic character than a B–N hBN bond (typically 0.87 e). The result of electron donation from the substrate is that the B–B bond of the divacantcy is incredibly weak having been stretched to a length of 3.12 Å.
 | ||
| Fig. 13 (a) the electron density isosurface (0.5 e Å−3) and (b) the HOMO (0.02 e Å−3) for chemisorbed ammonia above a hBN/Ru(0001) VBN defect. The isosurface shows N–Ru bonding and the ammonia bonded to the surface boron atom, indicative of a chemisorbed state. The HOMO still contains significant B and Ru character, the energy of which can be minimalised by subsequent deprotonation of ammonia hydrogens. Orbitals far from the vacancy have been omitted for clarity. | ||
Observing ammonia in a similar position above an isolated monolayer, the nitrogen has a charge of −1.00 e and a reduced bond population of 0.39 e in the B–N bond. Yet, the B–B bond is strongly covalent with a population of 0.67 e. The existing B–B bond is therefore favoured over forming the new B–NH3. Furthermore, the ammonia's hydrogens are closer to the nitrogen dangling bonds on the other side of the vacancy. For the reaction to proceed, a hydrogen must deprotonate from the ammonia to form a stable surface intermediate as seen in Fig. 6. Fig. 13b shows the HOMO of chemisorbed NH3 above hBN/Ru(0001). The HOMO consists of mixed Ru and B atomic orbital characters, similar to that of the physisorbed of NH3Fig. 12b.
The second key difference between the monolayer hBN and hBN/Ru(0001) is the height of energy barrier for associative desorption of H2 from the hydrogenated VB. The latter system only requires an energy of 0.69 eV, while the former requires an energy of 5.98 eV. Clearly, the Ru(0001) has a significant role to play in aiding the desorption of H2. The N–Ru bonding stabilises the VB defect by partially satisfying the nitrogen dangling bonds as evidenced by the average Mulliken charge on the N–H bonded nitrogen staying around −0.74 e as the desorption proceeds. In contrast, the free-standing hBN monolayer values vary from −0.88 to −0.90 e. The differences in bonding contributes to dramatically reduce the H2 desorption energy barrier between the two hBN systems. Fig. 14 shows the change in energy of a VBN as a function of the number of hydrogens adsorbed on the divacancy. As the number of hydrogen atoms increases, so does the stability of the VBN. The defect stabilisation effect is much more pronounced for the monolayer than hBN/Ru(0001).
 | ||
| Fig. 14 The change in energy with the number of hydrogen atoms chemisorbed in a VB for both a hBN monolayer and hBN/Ru(0001). There is a trend where with decreasing number of hydrogen atoms, the energy of the site increases. The effect is more pronounced for the monolayer than for hBN/Ru(0001) due to stabilisation from the Ru and steric effects of neighbouring hydrogen atoms. | ||
To reduce steric clashes, the hydrogenated monovacancy positions hydrogen atoms above and below the hBN honeycomb maintaining an average H–H distance of 1.88 Å. When the Ru(0001) substrate is included in the calculations, the hydrogen atoms can only adsorb above the hBN plane (see Fig. 8), resulting in an average H–H distance of 1.74 Å. The increased H–H distance reduces the dipole–dipole interaction of the hydrogen atoms and the overall energy of the monolayer structure. In contrast, despite the larger hBN/Ru(0001) unit cell, the H–H distance is shorter, resulting in greater steric hindrance and reducing the change in binding energy. Overall, the energy of the system decreases by a smaller amount as hydrogens are added for VB hBN/Ru(0001) compared to the VB monolayer. The combination of electronic and steric effects allows H2 to desorb from a hydrogenated VB with a lower energy barrier in the hBN/Ru(0001) system than for the monolayer.
To determine if stretching of hBN affects the reactivity and lowers the H2 desorption energy of the hBN/Ru(0001) system a cell with the Ru(0001) removed but maintaining the B–N bond length of 1.57 Å was used. Geometry optimisations with a fixed cell size showed a difference in energy between the hydrated vacancy and the H2 desorbed state of 4.91 eV similar to that of the monolayer (4.77 eV). Furthermore, a hBN bilayer and a hBN monolayer with a graphene monolayer substrate was considered to compare the effect of different substrates. These showed a difference in energy between the hydrated vacancy and the H2 desorbed state of 4.65 eV and 4.60 eV respectively, similar to the monolayer. As the difference between the hydrated VB and desorbed H2 states are similar to that of the monolayer, it is presumed that the transition state energies would also be similar. Given the similarity of the results when considering stretching and different substrates it is clear that the Ru(0001) has a significant impact on the reactivity of hBN.
As NH3 can readily decompose into a VBN defect overcoming an achievable barrier of 0.52 eV and 0.61 eV, for the monolayer and hBN/Ru(0001) respectively. Both materials are excellent precursors for forming hydrogenated VB sites of catalytic interest. The high hydrogen diffusion energy of 3.36 eV limits the use of the monolayer as a source of H2. However, when a Ru(0001) substrate is placed below the monolayer H2 desorption energy reduces to 0.69 eV, demonstrating that substrate effects can change the catalytic activity of hBN/Ru(0001) vacancies, making hBN a promising candidate for NH3 decomposition to H2. In fact, the energy barrier of dehydrogenation on hBN is lower than that which has been reported for TiO276 and Molybdenum Nitrides77 with the H2, and is similar to that of metal complexes.78,79 Furthermore, as the nitrogen is incorporated into the hBN a pure product is expected.
4 Conclusions
In conclusion, using DFT calculations, we have investigated the reaction of ammonia with a VBN vacancy in both a “free-standing” hBN monolayer and a hBN monolayer above a Ru(0001) substrate. A stable hydrogenated VB is created in each case before desorption of H2. The atomistic mechanism describing NH3 dehydrogenation and H2 desorption has been described in detail before discussing the key differences due to substrate-effects, hBN bonding and geometry which result in differing reaction pathways. The activation energy barrier for the dehydrogenation of ammonia was found to be 0.52 eV for the free-standing monolayer and 0.61 eV for hBN/Ru(0001). Subsequent associative desorption of H2 is facile for hBN/Ru(0001) compared to the monolayer with barriers of 0.69 eV and 5.89 eV, respectively. H2 generation from the free-standing monolayer would instead occur through a diffusion process with an energy barrier of 3.36 eV. This huge difference in the recombinative desorption barrier between the system can be rationalised by analysing the widely differing electronic structure and bonding of the hydrogenated VB structures. This work shows that hBN could be an excellent candidate for generating H2 from NH3 for fuel as part of the emerging hydrogen economy and as a catalyst for hydrogenation reactions.Author contributions
A. P. conceived the presented idea, performed the DFT simulations and wrote the manuscript under the guidance of M. S. N. X. and G. B. performed kinetics calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work made use of ARCHER2, the UK's national high performance computing service, via the UK's HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/R029431). A. P. acknowledges the University of Surrey for access to the Eureka High Performance Computing facility and funding in the form of a PhD studentship. M. S. acknowledge the Royal Society for personal funding through a Royal Society University Research Fellowship URF\R\191029. Thanks to Robert Lawrence and Louie Slocombe for their help and conversation. Supplemental data for this article can be found at https://doi.org/10.15126/900374.Notes and references
- A. F. D. Center, US Department of Energy, 2014.
- G. Thomas, P. George, Potential Roles of Ammonia in a Hydrogen Economy, U.S. Department of Energy, 2006, 1–23 Search PubMed.
- K. E. Lamb, M. D. Dolan and D. F. Kennedy, Int. J. Hydrogen Energy, 2019, 44, 3580–3593 CrossRef CAS.
- A. J. Wright, A. Wong and L. L. Diosady, Food Res. Int., 2003, 36, 1069–1072 CrossRef CAS.
- N. López, B. Bridier and J. Pérez-Ramírez, J. Phys. Chem. C, 2008, 112, 9346–9350 CrossRef.
- Á. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185–221 CrossRef.
- O. Guselnikova, A. Olshtrem, Y. Kalachyova, I. Panov, P. Postnikov, V. Svorcik and O. Lyutakov, J. Phys. Chem. C, 2018, 122, 26613–26622 CrossRef CAS.
- M. Peer, A. Qajar, R. Rajagopalan and H. C. Foley, Carbon, 2014, 66, 459–466 CrossRef CAS.
- J. Ullrich and B. Breit, ., ACS Catal., 2018, 8, 785–789 CrossRef CAS.
- S. I. Fujita, T. Yamada, Y. Akiyama, H. Cheng, F. Zhao and M. Arai, J. Supercrit. Fluids, 2010, 54, 190–201 CrossRef CAS.
- T. Bossi and J. Gediga, Johnson Matthey Technol. Rev., 2017, 61, 111–121 CrossRef CAS.
- D. M. Díaz-Morales, J. H. Erasmus, S. Bosch, M. Nachev, N. J. Smit, S. Zimmermann, V. Wepener and B. Sures, Environ. Pollut., 2021, 286, 117284 CrossRef PubMed.
- S. A. Tawfik, S. Ali, M. Fronzi, M. Kianinia, T. T. Tran, C. Stampfl, I. Aharonovich, M. Toth and M. J. Ford, Nanoscale, 2017, 9, 13575–13582 RSC.
- X. Lü, S. He, H. Lian, S. Lv, Y. Shen, W. Dong and Q. Duan, Appl. Surf. Sci., 2020, 531, 147262 CrossRef.
- F. Zheng, G. Zhou, Z. Liu, J. Wu, W. Duan, B. L. Gu and S. B. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 1–5 Search PubMed.
- S. N. Perevislov, Refract. Ind. Ceram., 2019, 60, 291–295 CrossRef CAS.
- C. R. Hsing, C. Cheng, J. P. Chou, C. M. Chang and C. M. Wei, New J. Phys., 2014, 16, 113015 CrossRef.
- G. Cassabois, P. Valvin and B. Gil, Nat. Photonics, 2016, 10, 262–266 CrossRef CAS.
- X. Song, Q. Li, J. Ji, Z. Yan, Y. Gu, C. Huo and Y. Zou, 2D Mater., 2016, 3, 035007 CrossRef.
- R. T. Paine and C. K. Narulat, Chem. Rev., 1990, 90, 73–91 CrossRef CAS.
- K. Zhang, Y. Feng, F. Wang, Z. Yang and J. Wang, J. Mater. Chem. C, 2017, 5, 11992–12022 RSC.
- L. Cao, S. Emami and K. Lafdi, Mater. Express, 2014, 4, 165–171 CrossRef CAS.
- C. Jin, F. Lin, K. Suenaga and S. Iijima, Phys. Rev. Lett., 2009, 102, 1–4 Search PubMed.
- C. Maestre, B. Toury, P. Steyer, V. Garnier and C. Journet, J. Phys. Mater., 2021, 4, 044018 CrossRef CAS.
- M. Jana and R. N. Singh, Int. Mater. Rev., 2018, 63, 162–203 CrossRef CAS.
- D. J. Nash, D. T. Restrepo, N. S. Parra, K. E. Giesler, R. A. Penabade, M. Aminpour, D. Le, Z. Li, O. K. Farha, J. K. Harper, T. S. Rahman and R. G. Blair, ACS Omega, 2016, 1, 1343–1354 CrossRef CAS PubMed.
- M. D. Esrafili, S. Heydari and L. Dinparast, Struct. Chem., 2019, 30, 1647–1657 CrossRef CAS.
- A. Akça, Comput. Theor. Chem., 2021, 1202, 113291 CrossRef.
- Y. Zhang, H. Du, Y. Ma, L. Ji, H. Guo, Z. Tian, H. Chen, H. Huang, G. Cui, A. M. Asiri, F. Qu, L. Chen and X. Sun, Nano Res., 2019, 12(4), 919–924 CrossRef CAS.
- Y. Ding, F. Torres-Davila, A. Khater, D. Nash, R. Blair and L. Tetard, MRS Commun., 2018, 8(3), 1236–1243 CrossRef CAS.
- L. H. Li, Y. Chen, B. Behan, H. Zhang, M. Petravic and A. M. Glushenkov, J. Mater. Chem., 2011, 21(32), 11862–11866 RSC.
- S. Torii, K. Jimura, S. Hayashi, R. Kikuchi and A. Takagaki, J. Catal., 2017, 355, 176–184 CrossRef CAS.
- C. Jin, F. Lin, K. Suenaga and S. Iijima, Phys. Rev. Lett., 2009, 102, 3–6 Search PubMed.
- J. C. Meyer, A. Chuvilin, G. Algara-Siller, J. Biskupek and U. Kaiser, Nano Lett., 2009, 9, 2683–2689 CrossRef CAS PubMed.
- Y. Jiao, A. Du, Z. Zhu, V. Rudolph, G. Q. Lu and S. C. Smith, Catal. Today, 2011, 175, 271–275 CrossRef CAS.
- D. R. Lide and G. Baysinger, CRC Handbook of Chemistry and Physics, 2019 Search PubMed.
- Q. Cai, D. Scullion, W. Gan, A. Falin, S. Zhang, K. Watanabe, T. Taniguchi, Y. Chen, E. J. Santos and L. H. Li, arXiv, 2019, 1–9.
- L. Xue, B. Lu, Z. S. Wu, C. Ge, P. Wang, R. Zhang and X. D. Zhang, Chem. Eng. J., 2014, 243, 494–499 CrossRef CAS.
- A. A. Peyghan, J. Mex. Chem. Soc., 2015, 59(1), 67–73 CAS.
- N. L. McDougall, J. G. Partridge, R. J. Nicholls, S. P. Russo and D. G. McCulloch, Phys. Rev. B, 2017, 96, 1–9 CrossRef.
- S. Okada, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 161404 CrossRef.
- A. Zobelli, C. P. Ewels, A. Gloter and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 75(9), 1–7 Search PubMed.
- M. Fanciulli, Philos. Mag. B, 1997, 76(3), 363–381 CrossRef CAS.
- R. Cortese and D. Campisi, Mol. Catal., 2020, 493, 110891 CrossRef CAS.
- R. Cortese, D. Campisi, A. Prestianni and D. Duca, Mol. Catal., 2020, 493, 110891 CrossRef CAS.
- A. Kuang, T. Zhou, G. Wang, Y. Li, G. Wu, H. Yuan, H. Chen and X. Yang, Appl. Surf. Sci., 2016, 362, 562–571 CrossRef CAS.
- I. Lucentini, X. Garcia, X. Vendrell and J. Llorca, Ind. Eng. Chem. Res., 2021, 60, 18560–18611 CrossRef CAS.
- B. Xiao, X. F. Yu and Y. H. Ding, RSC Adv., 2014, 4, 22688–22696 RSC.
- M. K. Agusta and I. Prasetiyo, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 6–9 CrossRef PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef PubMed.
- J. D. Monkhorst, J. Hendrik and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- K. Shirai, H. Dekura, Y. Mori, Y. Fujii, H. Hyodo and K. Kimura, J. Phys. Soc. Jpn., 2011, 80, 084601 CrossRef.
- B. Huang and H. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 1–8 Search PubMed.
- Y. J. Liu, B. Gao, D. Xu, H. M. Wang and J. X. Zhao, Phys. Lett. A, 2014, 378, 2989 CrossRef CAS.
- J. Strand, L. Larcher and A. L. Shluger, J. Phys.: Condens. Matter, 2020, 32, 055706 CrossRef CAS PubMed.
- K. Watanabe and T. Taniguchi, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 1–4 Search PubMed.
- M. S. Si and D. S. Xue, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 1–4 CrossRef.
- M. D. Esrafili and P. Mousavian, Appl. Surf. Sci., 2018, 440, 580–585 CrossRef CAS.
- O. L. Malkina, Phys. Chem. Chem. Phys., 2002, 4, 5467–5474 RSC.
- W. Amalia, P. Nurwantoro and Sholihun, Comput. Condens. Matter, 2019, 18, e00354 CrossRef.
- D.-H. Kim, H.-S. Kim, M. W. Song, S. Lee and S. Y. Lee, Nano Convergence, 2017, 4, 1–8 CrossRef PubMed.
- T. B. Ngwenya, A. M. Ukpong and N. Chetty, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 245425 CrossRef.
- M. S. Si, J. Y. Li, H. G. Shi, X. N. Niu and D. S. Xue, EPL, 2009, 86, 46002 CrossRef.
- A. A. Peyghan, H. Soleymanabadi and Z. Bagheri, J. Mex. Chem. Soc., 2015, 59, 67–73 CAS.
- H. Li, Z. Chen, X. Fang and D. Tie, Superlattices Microstruct., 2015, 88, 371–376 CrossRef CAS.
- M. Pashangpour, Int. Proc. Chem., Biol. Environ. Eng., 2011, 2, 113–116 Search PubMed.
- D. C. Ghosh, J. Jana and R. Biswas, Int. J. Quantum Chem., 2000, 80, 1–26 CrossRef CAS.
- Q. Zhang, Y. Chen, C. Zhang, C. R. Pan, M. Y. Chou, C. Zeng and C. K. Shih, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- T. Brugger, S. Günther, B. Wang, J. H. Dil, M. L. Bocquet, J. Osterwalder, J. Wintterlin and T. Greber, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 1–6 CrossRef.
- A. Kutana, A. Goriachko, Z. Hu, H. Sachdev, H. Over and B. I. Yakobson, Adv. Mater. Interfaces, 2015, 2, 1–8 Search PubMed.
- H. Li, H. Zhu and R. Zhao, New J. Chem., 2022, 46, 11035–11043 RSC.
- Á. Logadóttir and J. K. Nørskov, J. Catal., 2003, 220, 273–279 CrossRef.
- A. B. Preobrajenski, M. A. Nesterov, M. L. Ng, A. S. Vinogradov and N. Mårtensson, Chem. Phys. Lett., 2007, 446, 119–123 CrossRef CAS.
- D. Cheng, J. Lan, D. Cao and W. Wang, Appl. Catal., B, 2011, 106, 510–519 CrossRef CAS.
- J. Zhao, C. Cui, H. Wang, J. Han, X. Zhu and Q. Ge, J. Phys. Chem. C, 2019, 123, 554–564 CrossRef CAS.
- C. H. Christensen, T. Johannessen, R. Z. Sørensen and J. K. Nørskov, Catal. Today, 2006, 111, 140–144 CrossRef CAS.
- I. Lucentini, X. Garcia, X. Vendrell and J. Llorca, Ind. Eng. Chem. Res., 2021, 60, 18560–18611 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01392d |
| This journal is © the Owner Societies 2022 |