
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCs sorption of Mn–Fe based Prussian blue analogs with periodic precipitation banding in agarose gel
Hisashi
Hayashi

Department of Chemical and Biological Sciences, Faculty of Science, Japan Women's University, 2-8-1 Mejirodai, Bunkyo-ku, Tokyo 112-8681, Japan. E-mail: hayashih@fc.jwu.ac.jp
First published on 30th March 2022
Abstract
We report the understanding of Cs sorption/desorption properties of Mn–Fe based Prussian blue analogs (Mn–Fe PBA) and Prussian blue (PB) in agarose gel via X-ray fluorescence spectroscopy and scanning electron microscopy. After contacting a 2 mass% agarose gel containing 100 mmol dm−3 [Fe(CN)6]3− ions (inner electrolyte gel) with a 1 mass% agarose gel containing 550 mmol dm−3 Mn2+ or Fe2+ ions (outer electrolyte gel) in a plastic straw for 11 days, the clipped 10 mm-long gel columns of uniformly formed precipitates (named “Mn–Fe PBA gel” and “PB gel”) were used to investigate their Cs sorption/desorption properties. The Mn–Fe PBA gel showed several interesting features that were not observed in the PB gel. The Cs sorption capacity of the Mn–Fe PBA gel increased over time, and after ∼1200 h reached a value comparable to the most optimal values previously reported. During Cs sorption, Mn2+ ions were constantly released from the Mn–Fe PBA gel, and several periodic precipitation bands were generated. The positions of the periodic bands agreed with those of the peak distributions of Cs, Mn, and Fe, suggesting that the concentration fluctuation of MnII–FeIII PBA in the gel resulted in the periodic band formation. In these bands, large crystallites (>10 μm) were dominant, suggesting the contribution of Ostwald ripening. During Cs desorption, while Mn2+ ions were released from the Mn–Fe PBA gel, the release of Cs+ ions was considerably suppressed (∼1/3 for the PB gel). Based on these results, a model for Cs sorption by the Mn–Fe PBA gel was proposed, and its potential applications were discussed.
Introduction
The Fukushima nuclear accident on March 11, 2011, triggered a massive release of radioactive elements into the environment and generated a large amount of contaminated water. Among others, the two radioactive cesium (Cs) isotopes, i.e., 137Cs and 135Cs, pose serious threats to the environment, particularly aquatic environments, because of their long half-lives (30.17 and 2.3 × 106 years, respectively), and high water solubility and diffusivity. Thus, highly selective, efficient, and inexpensive sorbents for Cs+ ions in solutions are highly desired for (1) their control and collection in the nuclear industry, (2) their removal from the environment, and (3) the alleviation of internal exposure.Currently, the most promising Cs sorbents are Prussian blue (PB) and their analogs (PBA), which are known to have a high affinity for Cs over a wide pH range.1–14 These compounds are generally represented by the formula AxMy[M′(CN)6]z□1−z·nH2O, where A is an alkali metal element (e.g., Na, K, and Cs), and M (e.g., Mn, Fe, Co, and Cu) and M′ (e.g., Cr, Fe, and Co) are mainly transition metal elements.14–17 Here, if M = M′ = Fe, the compounds are generally called “PB”; if not, they are called “M–M′ based PBA.” The symbol □ indicates M′(CN)6 vacancies, that is, defects resulting from missing M′(CN)6 moieties; for most PBA, the ranges are 0 ≤ x ≤ 2, y = 1, and 0 < z ≤ 1.
The preparation of PB/PBA and their application for the removal of radioactive Cs+ ions from the contaminated water have been studied over the past 65 years1,5 due to their relevance in the nuclear industry. Considerable effort has been devoted to the development and optimization of Cs decontamination procedures using PB/PBA, and because of such efforts, for instance, PB has been approved by the US Food and Drug Administration as a drug used in clinics for the treatment of radioactive exposure.8,14 However, many of the previous studies have been prompted by specific applications, which have different requirements. Even when the applications have many similarities, the small differences frequently result in different conclusions.5 Moreover, PB and PBA exhibit a wide variety of compositions, crystal structures, and Cs sorption abilities, and these properties strongly depend on the reaction conditions and preparation methods.5,14 Consequently, in many earlier methodological studies on PB/PBA preparation, reports with contradictory results are fairly common.5 Partly because of this, the Cs sorption mechanism by PB/PBA-based sorbent has not been elucidated yet.6,12,14,18–20 Additionally, in practical applications, it is also important to consider effective means of supporting PB/PBA because PB/PBA precipitations give slurries of fine powders otherwise, which can pose different technical challenges (e.g., separating the sorbent from the purified solutions).5 Thus, the preparation methods of PB/PBA with a higher Cs sorption capacity on suitable supporting materials5,11,13,21 (preferably inexpensive, easy-to-handle, and environmentally friendly) continue to attract significant scientific interest.
Most of the previous studies utilized ferrocyanides (M′ = FeII), such as CoII–FeII based PBA (CoII–FeII PBA)3–5,7 and CuII–FeII based PBA (CuII–FeII PBA),6,10,18 to remove Cs+ ions from solutions. Ferricyanides (M′![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) FeIII) have received significantly less attention because they are usually more soluble and tend to have lower Cs sorption capacities.5 Nevertheless, our recent study22 showed that the precipitation bands of ferricyanide, MnII–FeIII based PBA (MnII–FeIII PBA), which spontaneously formed in agarose gel,23 can trap Cs+ ions more effectively than the band of PB (if the Cs processing time is longer than 10 days), suggesting their potential use as a new Cs sorbent. To implement this application in practice, first and foremost, Cs removal from aqueous solutions by MnII–FeIII PBA precipitates must be demonstrated. Additionally, the morphological change in the crystallites formed in the precipitation band during Cs sorption, if any, should be carefully investigated because it can influence the performance of Cs sorption.
FeIII) have received significantly less attention because they are usually more soluble and tend to have lower Cs sorption capacities.5 Nevertheless, our recent study22 showed that the precipitation bands of ferricyanide, MnII–FeIII based PBA (MnII–FeIII PBA), which spontaneously formed in agarose gel,23 can trap Cs+ ions more effectively than the band of PB (if the Cs processing time is longer than 10 days), suggesting their potential use as a new Cs sorbent. To implement this application in practice, first and foremost, Cs removal from aqueous solutions by MnII–FeIII PBA precipitates must be demonstrated. Additionally, the morphological change in the crystallites formed in the precipitation band during Cs sorption, if any, should be carefully investigated because it can influence the performance of Cs sorption.
In the current study, we (1) prepared 10 mm-long agarose-gels in which MnII–FeIII PBA spontaneously precipitated, (2) monitored the Cs sorption/desorption from/into 50 mmol dm−3 Cs+-solution/deionized-water by the gels using X-ray fluorescence (XRF) spectroscopy, and (3) observed the morphological changes in the MnII–FeIII PBA crystallites during Cs sorption using scanning electron microscopy (SEM). For comparison, the corresponding gels containing PB precipitates were also investigated.
Experimental section
Chemicals
Analytical reagent grade MnSO4·H2O, FeCl2·4H2O, CsCl, and K3[Fe(CN)6] were obtained from Wako Pure Chemical Industries (Osaka, Japan). Agarose (gel strength: 1800–2300 g cm−3) was procured from Kanto Chemical (Tokyo, Japan). All chemicals were used as received without further purification. Aqueous solutions were prepared using deionized water obtained after purification of tap water using a cartridge water purifier (G-10, Organo, Tokyo, Japan).Preparation of gel samples
The agarose gel where the precipitation patterns (e.g., Liesegang bands,24,25cf.Fig. 1a) form and the electrolyte present therein are hereafter referred to as the “inner electrolyte gel” and “inner electrolyte”, respectively, while the agarose gel to be poured onto the inner electrolyte gel and the electrolyte present therein are referred to as the “outer electrolyte gel” and “outer electrolyte”, respectively. Here, the inner electrolyte is K3[Fe(CN)6], and the outer electrolyte is either MnSO4 or FeCl2. Three types of gel samples, namely “Blank gel,” “Mn–Fe PBA gel,” and “PB gel,” and three types of plastic straw setups containing these gel samples and solutions, namely “Blank-gel system,” “Mn–Fe PBA-gel system,” and “PB-gel system,” were prepared as follows (see Fig. 1). | ||
| Fig. 1 Schematic illustrations of the setups for the (a) preparation of Mn–Fe PBA/PB gel, (b) Cs sorption by the gels, and (c) Cs desorption of the gels. The setups shown in (b) and (c) are referred to as the “Mn–Fe PBA/PB-gel system.” The arrows in (b) and (c) indicate the diffusion direction of the Cs+ ions. | ||
To prepare the inner electrolyte gels, K3[Fe(CN)6] powder (0.988 g) was dissolved in deionized water at 25 °C to form a 100 mmol dm−3 [Fe(CN)6]3− solution (30 cm3). After adding 2 mass% agarose (0.63 g), the resultant mixture was stirred vigorously at 98 °C for 30 s to produce a uniform [Fe(CN)6]3− agarose sol. Using a Pasteur pipette, the prepared hot sol was immediately transferred to plastic straws (straw cells:26 4 mm diameter and 50 mm long) and allowed to cool to 25 °C, which formed solidified gels in the straw cells within 500 s. The height of the gel columns in the straw cells was approximately 30 mm. The obtained gels were used as inner electrolyte gels to prepare the Mn–Fe PBA and PB gels.
To prepare the outer electrolyte gels, an appropriate amount of MnSO4·H2O or FeCl2·4H2O powder was dissolved in deionized water at 25 °C to form a 550 mmol dm−3 outer electrolyte solution (30 cm3). After adding 1 mass% agarose, the resultant mixture was stirred vigorously at 98 °C for 30 s to produce a uniform MnSO4 or FeCl2 electrolyte sol, which was poured over the solid inner electrolyte ([Fe(CN)6]3−) gels in the straw cells using a Pasteur pipette within 200 s and allowed to cool to 25 °C, forming solidified outer electrolyte gels at the upper part of the straw cells within 1000 s. The height of each outer electrolyte gel in the straw cells was approximately 20 mm.
Ten gel samples were prepared for each outer electrolyte to conduct reproducibility tests for the Cs sorption/desorption experiments. The above electrolyte concentrations and agarose densities were selected because they were shown to generate the most evident Liesegang bands of MnII–FeIII PBA in agarose gel.23
The straw cells containing the gel samples were closed using styrene-resin stoppers wrapped with parafilm and were allowed to stand at 25 °C for 11 days for the precipitation bands to form in the inner electrolyte gels. After 11 days, the inner electrolyte gels were pulled out of the straw cells and immersed in deionized water (∼200 cm3) for 2 days to remove any unreacted ions. Subsequently, 10 mm-long gel columns were clipped from the inner electrolyte gels (3–13 mm region from the top, where precipitates of MnII–FeIII PBA/PB were uniformly formed) using a razor blade. These clipped 10 mm-long gels containing MnII–FeIII PBA and PB are the Mn–Fe PBA and PB gels, respectively. The Mn–Fe PBA and PB gels were placed at the bottom of the straw cells for Cs sorption (Fig. 1b) and desorption (Fig. 1c) experiments.
For comparison, Blank gels were prepared as follows. Agarose (0.63 g) was dissolved in 30 cm3 of deionized water at 98 °C for 30 s. This ∼2 mass% agarose sol was then transferred to the straw cells using a Pasteur pipette and allowed to cool to 25 °C, which resulted in the formation of solidified gel columns with a height of approximately 30 mm within 500 s. These straw cells were closed using styrene-resin stoppers wrapped with parafilm and were allowed to stand at 25 °C for 11 days. After 11 days, the gels were pulled out of the straw cells and immersed in deionized water (∼200 cm3) for 2 days, and subsequently, the 10 mm-long gel columns were clipped using a razor blade. These clipped gels (Blank gels) were also placed at the bottom of the straw cells for control experiments of Cs sorption and desorption.
Cs sorption and desorption experiments
For Cs sorption experiments, CsCl powder (0.842 g) was dissolved in deionized water (100 cm3) and vigorously stirred to prepare a 50 mmol dm−3 solution at 25 °C. This Cs+ solution was added to each gel in the straw cells using a Pasteur pipette to achieve a height of 40 mm for the solution (Fig. 1b). Depending on the gels present, the systems prepared constitute the Blank-gel, Mn–Fe PBA-gel, and PB-gel systems. These samples were mounted on handmade holders [acrylic resin, 80 (h) × 30 (w) × 10 mm3 (d)] and were allowed to stand at 25 °C for at least 10 days, intermittently measuring the XRF intensity of the solutions at 5, 10, and 15 mm above the top of the gels. After the Cs sorption within the gels, the supernatant Cs+ solutions were removed from the straw cells and deionized water was subsequently added onto each gel in the straw cells (40 mm height) using a Pasteur pipette for the Cs desorption experiments (Fig. 1c). In the desorption experiments, the gel systems were allowed to stand at 25 °C for at least 10 days, intermittently measuring their XRF intensity as described above.XRF intensity measurement
A homemade setup, as described previously,22 was used to measure the XRF intensity from the three types of systems. Briefly, the excitation source was Cu Kα1 X-rays from an 18 kW generator (RU-300, Rigaku, Tokyo, Japan) operating at 40 kV and 60 mA, and the beam was focused within 0.5 mm of the horizontal direction using a SiO2(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1) Johansson-type crystal monochromator. The sample mounted on the holder was placed on a computer-controlled X–Z stage.22 XRF signals from each measurement point of the gel system were detected using a silicon PIN detector (XR-100CR, Amptek, Bedford, MA, USA), and the data were collected for 90 s using a multichannel analyzer (MCA8000A, Amptek). After subtracting the constant background from the obtained XRF spectra, the integrated intensities of each XRF peak over the following energy ranges were deduced: Mn Kα (5691–6124 eV), Fe Kα (6214–6647 eV), and Cs L-lines including Cs Lα and Lβ (4124–5205 eV). The overlap of the Mn Kβ and Fe Kα lines in the region containing both these elements was estimated by taking the Mn Kβ (Kβ1 + Kβ2,5) components in the Fe Kα line to be 11.8% of the observed Mn Kα (Kα1 + Kα2) intensity, according to the literature.27
1) Johansson-type crystal monochromator. The sample mounted on the holder was placed on a computer-controlled X–Z stage.22 XRF signals from each measurement point of the gel system were detected using a silicon PIN detector (XR-100CR, Amptek, Bedford, MA, USA), and the data were collected for 90 s using a multichannel analyzer (MCA8000A, Amptek). After subtracting the constant background from the obtained XRF spectra, the integrated intensities of each XRF peak over the following energy ranges were deduced: Mn Kα (5691–6124 eV), Fe Kα (6214–6647 eV), and Cs L-lines including Cs Lα and Lβ (4124–5205 eV). The overlap of the Mn Kβ and Fe Kα lines in the region containing both these elements was estimated by taking the Mn Kβ (Kβ1 + Kβ2,5) components in the Fe Kα line to be 11.8% of the observed Mn Kα (Kα1 + Kα2) intensity, according to the literature.27
The Mn, Fe, and Cs concentrations in the solution part of the three types of systems were determined from the integrated intensities of the Mn Kα, Fe Kα, and Cs L lines obtained using calibration curves prepared experimentally. Here, 10, 20, 30, 40, and 50 mmol dm−3 aqueous solutions of MnSO4, K3[Fe(CN)6], and CsCl were used to prepare the calibration curves for Mn, Fe, and Cs, respectively. The obtained concentrations were averaged to monitor the concentration changes in Mn, Fe, and Cs during Cs sorption or desorption.
For the Mn–Fe PBA-gel systems, the XRF intensity distribution of the Mn–Fe PBA gels in the horizontal (X) direction (the X-dependence of the XRF intensities) was measured by moving the sample along the X direction in 0.5 mm increments. Before and after each XRF distribution measurement, the intensity of the incident X-rays was monitored using an ion chamber (the variations were within 2%).
SEM observation
The SU 8220 microscope (Hitachi, Japan) operated at 2 kV and at working distances of 14.3–15.3 mm was used to observe the SEM morphologies of the Mn–Fe PBA gels and PB gels. To prepare the samples for SEM, the gels were taken out of the straw cells and cut into ∼1 mm-thick pieces. Prior to the SEM observations, the gel pieces were stuck on double-sided adhesive carbon tape, mounted on an aluminum stub, and dried for ∼3 days in an ambient laboratory environment.Results and discussion
Preparation of inner electrolyte gels and their properties
In Fig. 2a and b are shown the typical, fully developed precipitation patterns formed at 25 °C in the inner electrolyte gels for the Mn–Fe PBA-gel and PB-gel systems, respectively, after 11 days of the addition of the outer electrolyte sols. The characteristic colors of MnII–FeIII PBA and PB, i.e., dark brown and deep blue, respectively, were clearly observed. The patterns were almost fully developed and exhibited negligible further changes over time. We observed the formation of periodic bands (Liesegang bands) followed by a continuous zone (occasionally called “turbulent zone”)28 in the MnII–FeIII PBA inner electrolyte gel, although the bands were relatively few in number and significantly broad in width as reported earlier.23 In contrast, the precipitation band that formed in the PB inner electrolyte gel was primarily continuous in accordance with previous observations.22,23 The essentially continuous precipitation zones for both the inner electrolyte gels, which are marked by a dotted red square (Fig. 2), were used for Cs sorption/desorption experiments. The dry mass of these sections (i.e., Mn–Fe PBA and PB gels) was 0.008 ± 0.001 g, whereas that of the Blank gel was 0.003 ± 0.001 g; hence, the MnII–FeIII PBA/PB mass in the Mn–Fe PBA/PB gels was estimated to be 0.005 ± 0.002 g. | ||
| Fig. 2 Typical precipitation patterns formed at 25 °C in the inner electrolyte gels for (a) MnII–FeIII PBA and (b) PB after 11 days of the addition of the outer electrolyte sols. In the MnII–FeIII PBA gel, Liesegang bands that were followed by a turbulent zone are observed. The regions which were used for Cs sorption/desorption experiments are indicated by a dotted red square. A scale bar is provided at the bottom of the images. | ||
Here, it should be noted that MnII–FeIII PBA are more soluble than PB, even in agarose gels. To illustrate this, Fig. 3 shows the side views of beakers in which the MnII–FeIII PBA and PB inner electrolyte gels were immersed in deionized water at 25 °C for 650 h. Whereas the PB inner electrolyte gel showed no significant change, the characteristic dark brown color of the MnII–FeIII PBA inner electrolyte gel considerably faded and the solution turned yellow, suggesting that MnII–FeIII PBA were decomposed and consequently, the generated [Fe(CN)6]3− ions escaped into the solution.
 | ||
| Fig. 3 Side views of beakers in which the MnII–FeIII PBA and PB inner electrolyte gels were immersed in deionized water (∼50 cm3) at 25 °C for 650 h. | ||
Despite the relatively high solubility, the color fading of the MnII–FeIII PBA inner electrolyte gel and associated solution yellowing were not observed even after ∼1500 h (not shown here) when this gel was in direct contact with aqueous solutions in a straw cell. This finding suggests that MnII–FeIII PBA are potentially applicable as a relatively robust Cs sorbent if they are packed in a gel column and used one-dimensionally, as illustrated in Fig. 1b and c. Despite the relatively high solubility of MnII–FeIII PBA being an apparent drawback, this results in several interesting and important properties in the Mn–Fe PBA gel as a Cs sorbent, vide infra.
Cs sorption of Mn–Fe PBA/PB gels
Fig. 4 shows the variation in the Cs concentration (initial concentration: 50 mmol dm−3) in the solution part (40 mm long) over time for the Mn–Fe PBA-, PB-, and Blank-gel systems. | ||
| Fig. 4 Time-dependence of Cs concentration of the solution part in Mn–Fe PBA- and PB-gel system after introducing 50 mmol dm−3 Cs+ solution. For comparison, the result of the Blank-gel system is also plotted. | ||
After the introduction of the Cs+ solution onto the gel samples, the Cs concentration rapidly decreased (from 50 to ca. 45 mmol dm−3) for all systems during the first 4 h. This rate similarity between the Mn–Fe PBA/PB-gel and Blank-gel systems during this period suggests that the observed concentration decrease is primarily due to the diffusion of Cs+ ions from the solution into the gel.
After approximately 20 h, the Cs concentrations of both the Blank-gel and PB-gel systems hardly changed within experimental uncertainty, indicating that these systems reached a steady state. The average Cs concentrations of the Blank-gel and PB-gel systems after ∼20 h were 42 ± 1 and 40 ± 1 mmol dm−3, respectively. This result suggests that approximately (42–40) mmol dm−3 × 5.0 × 10−4 dm3 = 1 × 10−3 mmol of Cs+ ions were adsorbed by PB in the PB-gel system because the volume of the introduced solutions were 3.14 × 0.202 cm2 × 4.0 cm ≈ 0.50 cm3 = 5.0 × 10−4 dm3. Thus, the Cs sorption capacity of PB in the PB gel was estimated to be 1 × 10−3 mmol/(5 × 10−3 g) = 0.2 mmol g−1, which is comparable to that of commercial PB nanoparticles (0.22 mmol g−1).9
Up to approximately 45 h, the time-dependence of the Cs concentration in the Mn–Fe PBA-gel system was similar to that in the PB- and Blank-gel systems. However, after approximately 50 h, the Cs concentration in the Mn–Fe PBA-gel system continued to decrease and did not reach a steady state even after 250 h (Fig. 4).
Fig. 5 shows the variation in the concentrations of Cs, Mn, and Fe (with initial concentrations of 50, 0, and 0 mmol dm−3, respectively) in the solutions of the Mn–Fe PBA-gel system over 1200 h. The Cs concentration after 1200 h was 10 ± 1 mmol dm−3, which did not seem to reach a steady state yet. This result suggests that more than [(42–10) ± 2] mmol dm−3 × 5.0 × 10−4 dm3 = (1.6 ± 0.1) × 10−2 mmol of Cs+ ions can be adsorbed by MnII–FeIII PBA in the Mn–Fe PBA gel. The suggested Cs sorption capacity of MnII–FeIII PBA in the Mn–Fe PBA gel was estimated to be more than (1.6 ± 0.1) × 10−2 mmol/[(5 ± 2) × 10−3 g] = 3.9 ± 1.8 mmol g−1. This Cs sorption capacity value is comparable to or larger than the values recently reported for sophisticated PBA-related materials, such as hollow PB nanoparticles (1.97 mmol g−1),9 zinc-modulated Fe-Co PBA (1.92 mmol g−1),12 and PB@PAAm/PAMPS hydrogel composite (3.16 mmol g−1).21
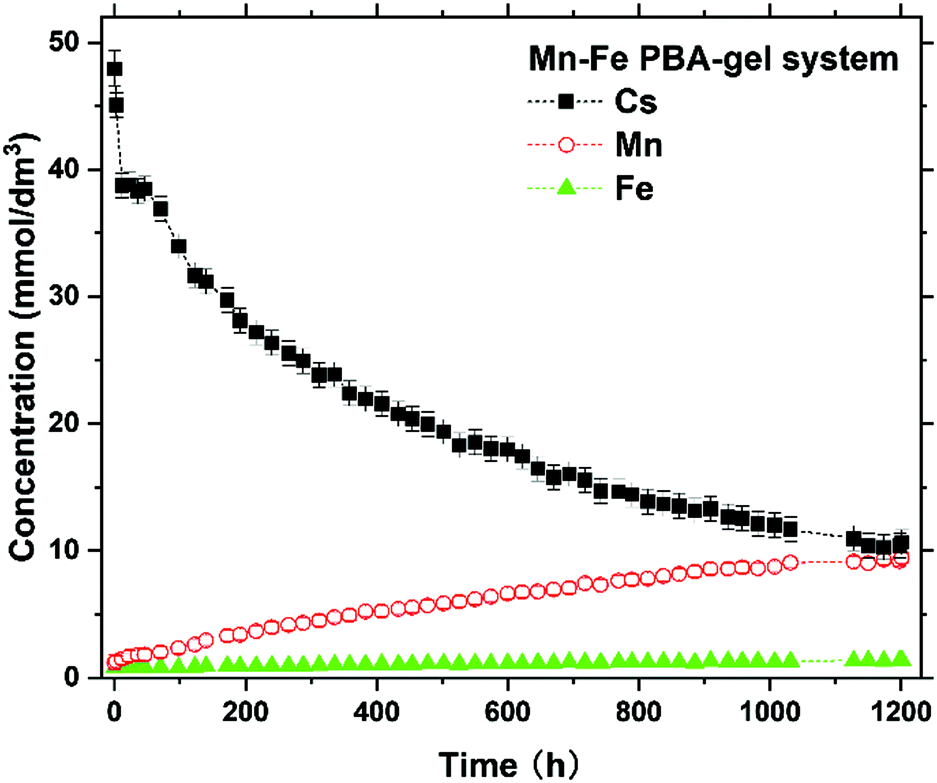 | ||
| Fig. 5 Time-dependence of the concentration of Cs, Mn, and Fe of the solution part in the Mn–Fe PBA-gel system over 1200 h after introducing 50 mmol dm−3 Cs+ solution. | ||
As can be seen in Fig. 5, the Mn concentration shows a correlation with the Cs concentration, suggesting that more Cs+ ions are adsorbed by the Mn–Fe PBA gel and more Mn2+ ions are released in the solution. In contrast, the Fe concentration was significantly low and largely independent of time, suggesting that the release of [Fe(CN)6]3− ions was well suppressed. In this situation, MnII–FeIII PBA crystallites in the Mn–Fe PBA gel become negatively charged, providing the driving force for the entry of Cs+ ions. Thus, the Mn2+ emission is a possible cause for the relatively large Cs capacity of the Mn–Fe PBA gel. Notably, similar phenomena were also reported for other PBA systems showing large Cs capacities, such as zinc-modulated Fe–Co PBA (showing Zn2+ emission)12 and Cu–Fe PBA (showing Cu2+ emission).6 The detailed Cs sorption mechanism with the release of Mn2+ ions is discussed later.
Cs desorption from Mn–Fe PBA/PB gels
Fig. 6 shows the time-dependence of the Cs concentration of the solution part in the Mn–Fe PBA- and PB-gel systems after the Cs+ desorption experiment started. The gel samples were in contact with deionized water in the straw cells (Fig. 1c). Before this desorption experiment, the gel samples adsorbed Cs+ ions from the initial 50 mmol dm−3 Cs solution in the straw cells for 270 h.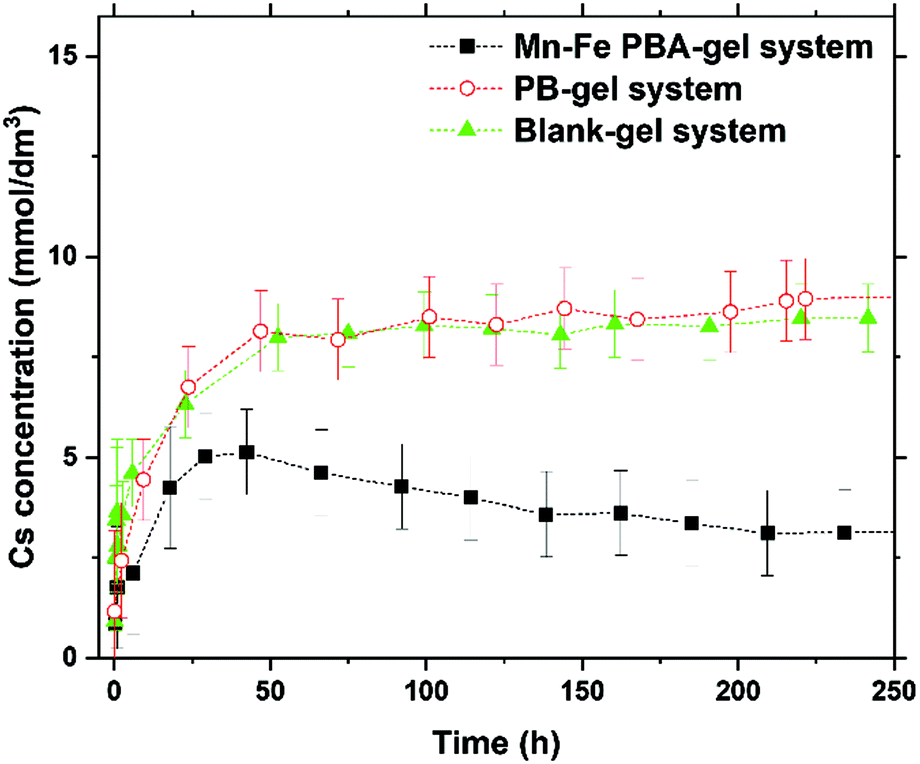 | ||
| Fig. 6 Time-dependence of Cs concentration of the solution part in Mn–Fe PBA- and PB-gel systems after the Cs+ desorption experiment started. For comparison, the result of the Blank-gel system is also plotted. | ||
After approximately 50 h from the introduction of deionized water, the Cs concentration of the Blank- and PB-gel systems did not change significantly within experimental uncertainty, indicating that these systems reached a steady state. The average Cs concentrations of the Blank-gel and PB-gel systems after ∼50 h were almost identical (8 ± 1 mmol dm−3). This result indicates that in these systems, approximately 8 mmol dm−3 × 5.0 × 10−4 dm3 = 4 × 10−3 mmol of Cs+ ions were released in the solution by diffusion. This suggests that the Blank gel and PB gel contained ∼4 μmol of Cs+ ions, which were not strongly trapped.
As can be seen in Fig. 6, the Cs concentrations of the Mn–Fe PBA-gel system (e.g., ∼3 mmol dm−3 at 234 h) are lower than those of the Blank- and PB-gel systems. This result is consistent with our previous conclusion22 that in agarose gel, the Cs+ ions are captured by MnII–FeIII PBA with a higher affinity in comparison to PB.
Interestingly, the Cs concentration of the Mn–Fe PBA-gel system showed a slight tendency to decrease over time after 50 h. This was supported by an additional measurement, which showed that the Cs concentration after 1340 h of the introduction of deionized water was 2 ± 1 mmol dm−3. These results suggest that the affinity of the Mn–Fe PBA gel for Cs+ ions increased over time, allowing the re-trapping of Cs+ ions that were released in the solution.
Fig. 7 shows the time-dependence of the concentration of Cs, Mn, and Fe in the solution part of the Mn–Fe PBA-gel system after the Cs+ desorption experiment started. Interestingly, in accordance with the results shown in Fig. 5, the Mn concentration increased over time, whereas the Fe concentration was significantly low and largely independent of time. This result demonstrates that the Mn release from the Mn–Fe PBA gel is independent of the Cs concentration of the solution (at least for <50 mmol dm−3), suggesting that the Mn2+ release provides the driving force for the re-entry of Cs+ ions and increasing affinity for Cs+ ions in this gel.
 | ||
| Fig. 7 Time-dependence of the concentration of Cs, Mn, and Fe of the solution part in the Mn–Fe PBA-gel system after the Cs+ desorption experiment started. | ||
Periodic banding in the Mn–Fe PBA gel
Several periodic bands gradually appeared in the Mn–Fe PBA gels and were clearly perceived approximately 300 h after the introduction of the 50 mmol dm−3 Cs+ solution to the straw cell. Even when the Cs+ solution was replaced with deionized water for the desorption experiment, the periodic bands were always perceived in the gel ∼300 h after its introduction, suggesting that the observed periodic banding is independent of the Cs concentration of the solution. As an example, Fig. 8a shows periodic bands formed in the Mn–Fe PBA gel 1200 h after the introduction of the 50 mmol dm−3 Cs+ solution. The spaces between the bands were ∼1.5 mm and their widths were in the range of 0.1–1.0 mm. | ||
| Fig. 8 (a) Periodic bands formed in Mn–Fe PBA gel after 1200 h of the introduction of the 50 mmol dm−3 Cs+ solution into the straw cell. For comparison, a 1 mm scale is shown. (b) XRF intensity distributions of the Mn–Fe PBA gel. An image of the gel is displayed at the top of the graph to allow a comparison of the XRF distributions with the positions of the periodic bands. | ||
In Fig. 8b are shown the XRF intensity distributions of the Mn–Fe PBA gel shown in Fig. 8a. Here, the horizontal axis indicates the distance from the gel boundary (X), and its value is positive at the bottom of the straw cell. The vertical axis represents the relative XRF intensity. These XRF distributions provide a good approximation of the Cs, Mn, and Fe distributions in the gel.22,29 An image of the measured Mn–Fe PBA gel is displayed at the top of this graph to allow a comparison of the XRF distribution with the positions of the periodic bands. The color of this image is somewhat different from that of Fig. 8a because the images in Fig. 8a and b were taken for the gel without and within the straw cell, respectively, under different lighting conditions.
As can be seen in Fig. 8b, the peak positions of the Cs, Mn, and Fe distributions coincided well with one another, strongly suggesting that the observed distributions could be attributed to the distribution of MnII–FeIII PBA containing Cs+ ions. The peak positions of the three XRF distributions also coincided well with the positions of the periodic bands. This finding suggests that periodic bands were generated owing to the concentration fluctuation of MnII–FeIII PBA in the gel. Interestingly, when aqueous solutions were not introduced at the top of the Mn–Fe PBA gel, no periodic band was formed even after 3000 h of the gel preparation. This suggests that the concentration fluctuation of MnII–FeIII PBA in the gel was triggered by the introduction of aqueous solutions on the top of the gels.
To the best of our knowledge, the periodic band formation in the turbulent zone after forming Liesegang bands (such as that shown in Fig. 8a) has not been reported yet, and the detailed mechanism is currently unclear. The models of conventional Liesegang bands may be useful as a starting point for constructing a theoretical framework. Modern theoretical models for Liesegang band formations can be approximately classified into pre- or post-nucleation models, depending on the sequence of elementary events.24,25,30–32 Pre-nucleation models assume that the precipitation bands are formed because of repeated cycling of supersaturation, nucleation, and depletion of the reaction products. In these models, the crystallites are formed by rapid nucleation and crystallization triggered by supersaturation, and hence they should be relatively small in size and irregular in shape.23 In contrast, post-nucleation models assume competitive growth of small particles of reaction products (including crystallites). According to these models, the crystallites in the Liesegang bands must be relatively large in size because most of them have undergone Ostwald ripening.23 Overall, the sizes and shapes of the crystallites in Liesegang bands can be used to identify the dominant formation processes: small and irregular crystallites are likely formed according to the pre-nucleation models, whereas relatively large crystallites indicate the dominance of the post-nucleation processes.
In the current case, the periodic band formation occurred in a continuous precipitation band (turbulent zone) after the precipitation pattern was fully developed and most unreacted Mn2+ and [Fe(CN)6]3− ions were removed. Hence, pre-nucleation models (where supersaturation of unreacted ions are fundamentally important) may not be able to explain the band formation mechanism. On the other hand, post-nucleation models seem to be a better fit because Ostwald ripening is possible after the formation of precipitation band(s).33 Partial dissociation of MnII–FeIII PBA accompanied by Mn2+ emission (as indicated by Fig. 5 and 7) may support Ostwald ripening because such emission of constituent ions more strongly promotes dissolution of smaller crystallites via the Gibbs–Thomson effect. To get a better insight, the sizes of the crystallites formed in the Mn–Fe PBA gel are surveyed in the next section.
SEM observation
Fig. 9a and b show SEM images (×10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) of crystallites in the PB gel before and after the Cs sorption experiment for 250 h, respectively. In the PB gel, the observed crystallites were primarily cubic, which is characteristic of PB and PBA.16,17 These crystallites are relatively small (<1 μm lateral size), which means they should have a large surface area that is expected to facilitate both the adsorption and desorption of Cs+ ions by allowing more frequent contact with Cs+ ions (for adsorption) and solvent molecules (for desorption). Thus, in the PB-gel system, Cs+ ions would be rapidly adsorbed and easily desorbed, and consequently, the Cs concentration of the solution part would become constant within a relatively short period. Moreover, a comparison between Fig. 9a and b shows that the sizes of the crystallites did not significantly change with the addition of the 50 mmol dm−3 Cs+ solution, suggesting that they are considerably stable for the Cs+ solutions. This robustness of the crystallites explains the observation of steady states for the Cs concentration as shown in Fig. 4 and 6.
000) of crystallites in the PB gel before and after the Cs sorption experiment for 250 h, respectively. In the PB gel, the observed crystallites were primarily cubic, which is characteristic of PB and PBA.16,17 These crystallites are relatively small (<1 μm lateral size), which means they should have a large surface area that is expected to facilitate both the adsorption and desorption of Cs+ ions by allowing more frequent contact with Cs+ ions (for adsorption) and solvent molecules (for desorption). Thus, in the PB-gel system, Cs+ ions would be rapidly adsorbed and easily desorbed, and consequently, the Cs concentration of the solution part would become constant within a relatively short period. Moreover, a comparison between Fig. 9a and b shows that the sizes of the crystallites did not significantly change with the addition of the 50 mmol dm−3 Cs+ solution, suggesting that they are considerably stable for the Cs+ solutions. This robustness of the crystallites explains the observation of steady states for the Cs concentration as shown in Fig. 4 and 6.
 | ||
| Fig. 9 SEM images (×10000) of crystallites in the PB gel (a) before and (b) after the Cs sorption experiment for 250 h and SEM images (×1000) of crystallites in the Mn–Fe PBA gel (c) before and (d) after Cs sorption experiment for 1200 h. These images were acquired at X ≈ 4 mm, and the scale bars are shown in yellow. | ||
Fig. 9c and d show SEM images (×1000) of crystallites in the Mn–Fe PBA gel before and after the Cs sorption experiment for 1200 h, respectively. In the Mn–Fe PBA gel, the observed crystallites were primarily cubic with sizes approximately 10-fold larger than those observed in the PB gel, suggesting that crystal ripening occurs more easily in the Mn–Fe PBA-gel system than in the PB-gel system. A comparison between Fig. 9c and d shows that by addition of the 50 mmol dm−3 Cs+ solution, smaller crystallites (<5 μm) mostly decreased and larger crystallites (>10 μm) significantly increased; in other words, Ostwald ripening of the crystallites occurred in the Mn–Fe PBA gel. Thus, Fig. 9c and d strongly support the important role of Ostwald ripening of MnII–FeIII PBA crystallites in periodic banding (Fig. 8). The occurrence of Ostwald ripening implies that the Mn–Fe PBA-gel system is unstable (unlike the PB-gel system), which is consistent with the Cs concentration of its solution part not reaching a steady state even after 1200 h following the addition of the Cs+ solution.
If MnII–FeIII PBA crystallites undergo Ostwald ripening, the Cs+ ions that were originally adsorbed in smaller crystallites would be, at least partially, incorporated inside the formed larger crystallites. The Cs+ ions trapped inside the crystallites are expected to be less desorbed into the solution than those adsorbed on the surface. This inference is consistent with the observed high affinity for Cs+ ions in the desorption experiments (Fig. 6).
Cs sorption mechanism of MnII–FeIII PBA in the gel
Based on the results of Fig. 4–9, we propose a qualitative model of Cs sorption mechanism by MnII–FeIII PBA in the Mn–Fe PBA-gel system, a schematic of which is shown in Fig. 10. During the sorption process, Mn2+ ions are released into the solution, followed by the one-dimensional entry of Cs+ ions. The MnII–FeIII PBA crystallites temporarily carry negative charges after the release of Mn2+ ions, providing the driving force for the entry of Cs+ ions. Furthermore, the release of Mn2+ ions generates relatively large defect sites, which have been suggested to be the primary trapping sites for Cs+ ions in PB19 and PBA,20 and thus, the increase in the defects is also expected to contribute to an increase in the Cs sorption capacity over time. These MnII–FeIII PBA crystallites undergo Ostwald ripening over time to form periodic bands in the gel. The introduction of Cs+ ions inside the crystallites by the ripening procedure improves the Cs affinity, which slows the desorption process. | ||
| Fig. 10 Sorption mechanism of Cs+ ions by MnII–FeIII PBA crystallites in agarose gel. | ||
This model presupposes that the sorption of Cs+ ions occurs far from equilibrium and is basically a dynamic (i.e., unstable) process. Ion exchange mechanisms over a long time have been proposed for Zn-modulated Fe–Co PBA12 and Cu–Fe PBA systems.6 However, to the best of our knowledge, a dynamic Cs sorption mechanism accompanied by Ostwald ripening-driven pattern formation, such as the current model, has never been presented. It is noteworthy that in this model, spontaneous precipitation patterning is fundamentally related to the improvement in performance for Cs sorption, suggesting a new possibility for the self-organization of precipitates.34
Possible application
MnII–FeIII PBA form hardly soluble precipitates in aqueous solution like other PB/PBA; even so, as shown in Fig. 3, MnII–FeIII PBA are more soluble than PB, even in agarose gels. Hence, the use of MnII–FeIII PBA or gels containing them as fully immersed Cs sorbents (e.g., in waste storage tanks) is challenging; during the long-term procedure of Cs removal, MnII–FeIII PBA may be, at least partially, decomposed. Nevertheless, as suggested in this study, if MnII–FeIII PBA are used in a gel column where Cs+ solutions can make contact only with the top surface of the column, the dissolution/decomposition of MnII–FeIII PBA is largely prevented (increasing their Cs sorption capacity), and their precipitates could be maintained in a gel column for >1500 h.One disadvantage of the MnII–FeIII PBA sorbent is the escape of Mn2+ ions into the solution. Hence, complementary packaging strategies are required. A possible solution is that a gel containing Mn2+ ions is set at the opposite side of the Cs+ solution (cf.Fig. 11). Interestingly, the region forming Liesegang bands in the well-developed inner electrolyte gel for MnII–FeIII PBA (Fig. 2a) is expected to contain a significant amount of unreacted Mn2+ ions if the gel was not immersed in deionized water to remove them. This is because the initial concentration of Mn2+ ions in the outer electrolyte gel should be sufficiently higher than that of [Fe(CN)6]3− ions in the inner electrolyte gel to form precipitation bands therein23 (e.g., the initial concentration ratio, [Mn2+]/[[Fe(CN)6]3−], is 5.5 in the current study). In fact, the persistence of Mn2+ ions in the region forming Liesegang bands was suggested in a previous study (Fig. 2a in ref. 22). Thus, conveniently, the well-developed inner electrolyte gel for MnII–FeIII PBA (without the removal of unreacted ions and subsequent clipping) may be used as a composite gel containing both the Cs sorption part (turbulent zone) and Mn2+ supply part (Liesegang bands zone).
 | ||
| Fig. 11 Schematic illustration of packaging of a Mn–Fe PBA gel for potential applications in pipes. | ||
Even if Mn2+ ions are properly supplied to the MnII–FeIII PBA sorbent, the escape of Mn2+ ions from the sorbent might be unfavorable for application in the environment (as well as a drug for internal exposure), because of the potential risk of excessive Mn exposure, which can induce neurotoxicity in humans.35 Additionally, because the concentrations of radioactive Cs isotopes are relatively low in the environment (e.g., 10–200 Bq m−3 at the site of Fukushima rivers in FY2016–FY2017),36 the large Cs capacity of the MnII–FeIII PBA sorbent may not be entirely necessary.
Instead, the MnII–FeIII PBA sorbent is expected to be useful for long-term Cs removal in the nuclear industry, where high- concentration radioactive Cs+ solutions are routinely handled and the slight outflow of Mn2+ ions is not a significantly worrying problem. Particularly, its application for Cs removal in pipes (e.g., connecting pipes in nuclear waste disposal/treatment systems) seems to conform with the abovementioned features of MnII–FeIII PBA. Fig. 11 schematically illustrates a conceptual design for a device that can be used in such applications.
Note also that Cs+ ions captured by the Mn–Fe PBA gel are easily retrieved only by immersing the gel in water (where MnII–FeIII PBA may decompose to release Cs+ ions). This property can facilitate the reusability of radioactive Cs+ ions in diverse scientific fields, including radiochemistry and analytical chemistry.
Future issues for application
For practical applications of MnII–FeIII PBA as a Cs sorbent, several scientific and/or technological challenges remain.First, (1) the composition of the MnII–FeIII PBA formed in gel (in other words, the values of x, y, z, and n in (K/Cs)xMny[Fe(CN)6]z□1−z·nH2O), (2) its changes during Cs sorption/desorption, and (3) the Cs position(s) in the PBA crystallites should be investigated. The composition of PBA strongly influences the performance of Cs sorption and determines the upper limit of the capacity for Cs uptake.5 For instance, if the system contains k mol of MnII–FeIII PBA and their y value is 1, the maximum amount of Cs+ ions to be sorbed (by exchange of K+ ions under x = z = 1) must be k mol according to charge conservation. If the y value can decrease to y′ (<1), as is the present case, the maximum amount of the sorbed Cs+ ions can increase from k to k × (3 − 2y′) mol. Nevertheless, if y′ values are less than 1/2, the cubic structure of PBA (suggested in Fig. 9) is not expected to be maintained; thus k × (3 − 2 × 1/2) = 2k mol may be the Cs sorption limit. The Cs capacity per mass depends also on the n value. For instance, assuming x = y = z = 1 and n = 0, the maximum Cs capacity is 1/306.01 g mol−1 ≈ 3.3 mmol g−1 (interestingly, this value is approximately the same as the current experimental value: 3.9 ± 1.8 mmol g−1). Accurate x, y, z, and n values and their changes during Cs sorption/desorption are required for further understanding of the Cs sorption mechanism [including the suppression of Fe (possibly [Fe(CN)6]3− ions) emission] to further improve the performance of the MnII–FeIII PBA sorbent.
It is also important to understand the crystal structure, particularly the position(s) of the sorbed Cs+ ions in the MnII–FeIII PBA crystallites. Notably, Cs+ ions cannot be placed at the site of Mn2+ ions in MnII–FeIII PBA crystallites because both the charge and ionic radius (Cs+ = 0.173 nm and Mn2+ = 0.080 nm)37 differ. Indeed, an X-ray absorption fine structure study suggests that the Cs+ ions are localized at large defect sites close to the sub-cube faces in the jungle-gym-like structure of MnII–FeIII PBA.20 If such defect sites show some ordered alignments, the Cs sorption process should be a kind of phase transition. Here, note that in PBA structural phase transitions depending on the number of alkali metal element (x) are possible; e.g., Na1.40Mn[Fe(CN)6] (x = 1.40) and Na1.72Mn[Fe(CN)6] (x = 1.72) exhibit cubic and monoclinic phases, respectively.17 X-ray diffraction measurements,6,11–13,17–19,21 micro-XRF mapping,13,14,17,19 and inductively coupled plasma optical emission spectrometry12 of the MnII–FeIII PBA crystallites formed in the gel before and after the Cs sorption/desorption will be useful to assess these issues.
Second, quantitative models to describe Ostwald ripening-driven periodic pattern formation of MnII–FeIII PBA precipitates should be developed. Because the characteristic Cs sorption (as shown in Fig. 4 and 5) is suggested to be fundamentally related to the periodic banding (Fig. 10), such models are expected to be helpful for (1) quantitatively interpreting the kinetics of Cs uptake of MnII–FeIII PBA and resultantly, (2) properly analyzing the amount of sorbed Cs+ ions.
Finally, the prototype for the device schematically illustrated in Fig. 11 should be readily fabricated to demonstrate its effectiveness for removing Cs+ ions in pipes.
Thus, further research across several scientific fields, including physical chemistry, analytical chemistry, material sciences, and material engineering, is required.
Conclusions
The reaction–diffusion process of Mn2+ and [Fe(CN)6]3− ions in agarose gel spontaneously generates a continuous precipitation band of MnII–FeIII PBA, followed by relatively wide Liesegang bands at 25 °C. The 10 mm-long parts of the continuous band (Mn–Fe PBA gel) were used for Cs sorption/desorption experiments, and the associated processes were investigated via XRF spectroscopy and SEM observation. The Mn–Fe PBA gel showed several interesting features that were not observed in the PB gel. The Cs sorption capacity of the Mn–Fe PBA gel increased over time and reached a value comparable to the previously reported most optimal values after ∼1200 h. During Cs sorption, Mn2+ ions were constantly released from the gel, and periodic bands were generated in the continuous precipitation band in which cubic crystallites grew significantly, strongly suggesting a contribution of Ostwald ripening. During Cs desorption, while Mn2+ ions were released from the gel, the release of Cs+ ions was considerably suppressed (∼1/3 for the PB gel). Based on these results, a model for Cs sorption by the Mn–Fe PBA gel was proposed and its potential applications were discussed. Although ferricyanides, including MnII–FeIII PBA, have received relatively less attention than ferrocyanides, their usage involving one-dimensional packing in gel offers new insights into pattern-forming phenomena and heretofore unachieved applicability as a Cs sorbent. This scientifically interesting and practically important system is worthy of further investigation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded by JSPS KAKENHI (grant number JP19K05409). The author is grateful to Ms N. Taniguchi and Dr T. Takagi of Japan Women's University for their help with the preparation of the gel samples and their SEM observations, respectively.References
- G. B. Barton, J. L. Hepworth, E. D. McClanahan Jr, R. L. Moore and H. H. Van Tuyl, Ind. Eng. Chem., 1958, 50, 212 CrossRef CAS.
- P. Nielsen, B. Dresow and H. C. Heinrich, Z. Naturforsch., 1987, 42B, 1451 CrossRef.
- J. Lehto, R. Harjula and J. Wallace, J. Radioanal. Nucl. Chem., 1987, 111, 297 CrossRef CAS.
- J. Lehto, A. Paajanen and R. Harjula, J. Radioanal. Nucl. Chem., 1992, 164, 39 CrossRef CAS.
- P. A. Haas, Sep. Sci. Technol., 1993, 28, 2479 CrossRef CAS.
- S. Ayrault, B. Jimenez, E. Garnier, M. Fedoroff, D. J. Jones and C. Loos-Neskovic, J. Solid State Chem., 1998, 141, 475 CrossRef CAS.
- D. V. Ca and J. A. Cox, Microchim. Acta, 2004, 147, 31 CAS.
- M. Altagracia-Martínez, J. Kravzov-Jinich, J. M. Martínez-Núňez, C. Ríos-Castaňeda and F. López-Naranjo, Orphan Drugs: Res. Rev., 2012, 2, 13 CrossRef.
- N. L. Torad, M. Hu, M. Imura, M. Naito and Y. Yamauchi, J. Mater. Chem., 2012, 22, 18261 RSC.
- D. Parajuli, A. Takahashi, H. Noguchi, A. Kitajima, H. Tanaka, M. Takasaki, K. Yoshino and T. Kawamoto, Chem. Eng. J., 2016, 283, 1322 CrossRef CAS.
- H. Yang, L. Sun, J. Zhai, H. Li, Y. Zhao and H. Yu, J. Mater. Chem. A, 2014, 2, 326 RSC.
- J. Liu, X. Li, A. I. Rykov, Q. Fan, W. Xu, W. Cong, C. Jin, H. Tang, K. Zhu, A. S. Ganeshraja, R. Ge, X. Wang and J. Wang, J. Mater. Chem. A, 2017, 5, 3284 RSC.
- I. Lee, S.-H. Kim, M. Rethinasabapathy, Y. Haldorai, G.-W. Lee, S. R. Choe, S.-C. Jang, S.-M. Kang, Y.-K. Han, C. Roh, W.-S. Cho and Y. S. Huh, Sci. Rep., 2018, 8, 4540 CrossRef PubMed.
- J. Estelrich and M. A. Busquets, Int. J. Mol. Sci., 2021, 22, 780 CrossRef CAS PubMed.
- M. Verdaguer and G. Girolami, Magnetic Prussian Blue Analogs, in Magnetism: Molecules to Materials V, ed. J. S. Miller and M. Drillon, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2004 Search PubMed.
- F. Ma, Q. Li, T. Wang, H. Zhang and G. Wu, Sci. Bull., 2017, 62, 358 CrossRef CAS.
- W.-J. Li, C. Han, G. Cheng, S.-L. Chou, H.-K. Liu and S.-X. Dou, Small, 2019, 1900470 CrossRef PubMed.
- C. Loos-Neskovic, S. Ayrault, V. Badillo, B. Jimenez, E. Garnier, M. Fedoroff, D. J. Jones and B. Merinov, J. Solid State Chem., 2004, 177, 1817 CrossRef CAS.
- M. Ishizaki, S. Akiba, A. Ohtani, Y. Hoshi, K. Ono, M. Matsuba, T. Togashi, K. Kananizuka, M. Sakamoto, A. Takahashi, T. Kawamoto, H. Tanaka, M. Watanabe, M. Arisaka, T. Nankawad and M. Kurihara, Dalton Trans., 2013, 42, 16049 RSC.
- H. Hayashi, S. Aoki, M. Takaishi, Y. Sato and H. Abe, Phys. Chem. Chem. Phys., 2019, 21, 22553 RSC.
- P. Zhao, W. Zhang, Y. V. Kaneti, A. Azhar, A. A. Alshehri, Y. Yamauchi and M. Hu, Bull. Chem. Soc. Jpn., 2018, 91, 1357 CrossRef CAS.
- H. Hayashi, Y. Sato, S. Aoki and M. Takaishi, J. Anal. At. Spectrom., 2019, 34, 979 RSC.
- H. Hayashi, S. Aoki and T. Suzuki, RSC Adv., 2019, 9, 36240 RSC.
- H. Nabika, Curr. Phys. Chem., 2015, 5, 5 CrossRef CAS.
- H. Nabika, M. Itatani and I. Lagzi, Langmuir, 2020, 36, 481 CrossRef CAS PubMed.
- H. Hayashi and M. Takaishi, Anal. Sci., 2019, 35, 651 CrossRef CAS PubMed.
- G. Zschornack, Handbook of X-Ray Data, Springer-Verlag, Berlin, 2007 Search PubMed.
- B. A. Grzybowski, Chemistry in Motion: Reaction−Diffusion Systems for Micro- and Nanotechnology, John Wiley & Sons, Chichester, UK, 2009 Search PubMed.
- H. Hayashi and H. Abe, J. Anal. At. Spectrom., 2016, 31, 1658 RSC.
- F. Izsák and I. Lagzi, Models of Liesegang pattern formation, in Precipitation Patterns in Reaction–Diffusion Systems, ed. I. Lagzi, Research Signpost, Kerala, India, 2010 Search PubMed.
- M. Chacron and I. L’Heureux, Phys. Lett. A, 1999, 263, 70 CrossRef CAS.
- I. L’Heureux and R. Bektursunova, Modeling Liesegang periodic precipitation patterns in geochemical systems, in Precipitation Patterns in Reaction−Diffusion Systems, ed. I. Lagzi, Research Signpost, Kerala, India, 2010 Search PubMed.
- H. Hayashi, Y. Sato and H. Abe, J. Anal. At. Spectrom., 2018, 33, 957 RSC.
- E. Nakouzi and O. Steinbock, Sci. Adv., 2016, 2, e1601144 CrossRef PubMed.
- P. Chen, S. Chakraborty, T. V. Peres, A. B. Bowman and M. Aschner, Toxicol. Res., 2015, 4, 191 CrossRef CAS PubMed.
- T. Nakanishi and K. Sakuma, Chemosphere, 2019, 215, 272 CrossRef CAS PubMed.
- Y. Marcus, Chem. Rev., 1988, 88, 1475 CrossRef CAS.
| This journal is © the Owner Societies 2022 |