
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBandgaps of atomically precise graphene nanoribbons and Occam's razor†
Aristides D.
Zdetsis

Molecular Engineering Laboratory, Department of Physics, University of Patras, Patras 26500, GR, Greece. E-mail: zdetsis@upatras.gr
First published on 5th April 2022
Abstract
Rationalization of the “bulk” (ΔEac) or “zigzag-end” (ΔEzz) energy gaps of atomically precise armchair graphene nanoribbons (AGNRs), which are directly related to fundamental applications in nanoelectronics, could be challenging and largely controversial with respect to their magnitude, origin, substrate influence (ΔEsb), and spin-polarization, among others. Hereby a simple self-consistent and “economical” interpretation is presented, in full accordance with Occam's simplicity principle, which is highly successful (within less than 1%) in predicting all energy gaps of the 5-, 7-, and 9-AGNRs, in contrast to other complicated and/or contradicting prevailing views in the literature for ΔEac, ΔEzz, and ΔEsb. The present approach is based on “appropriate” DFT (TDDFT) calculations, general symmetry principles, and plausibility arguments. The excellent agreement with experiments and the new insight gained is achieved by invoking the approximate equivalence of Coulomb correlation energy with the staggered sublattice potential. Breaking established stereotypes, we suggest that the measured STS gaps are virtually independent of the substrate, essentially equal to their free-standing values, and that the “true” lowest energy state is a closed singlet with no conventional magnetism. The primary source of discrepancies is the finite length of AGNRs together with inversion/reflection symmetry conflict and the resulting topological end/edge states. Such states invariably mix with other “bulk” states making their unambiguous detection/distinction difficult. This can be further tested by eliminating end-states (and ΔEzz), by eliminating “empty” zigzag rings.
1. Introduction
Edge or end states in graphene nanoribbons (GNRs), and in particular armchair GNRs (AGNRs), have attracted much interest recently,1–8 due to their anticipated magnetic properties,9,10 although their presence in finite nanographenes (NGRs) was predicted a long time ago.11 However, the significance and importance of end states for AGNRs was recognized only recently,1–8 after the pioneering bottom-up synthesis of atomically precise AGNRs of finite lengths L with short zigzag ends, and their characterization by scanning tunnelling microscopy (STM) and spectroscopy (STS).3–16 Clearly no end states appear in the common infinite AGNRs fabricated via the usual top-bottom techniques, which are theoretically described by periodic boundary conditions at their two ends.1 The new developments have brought to the forefront new concepts and properties such as the “bulk band gaps” ΔEac (or Δac1,6) i.e., the energy gaps between delocalized states, and the energy separation of the zigzag-end-localized “end-states”, denoted here by (ΔEzz) (or Δzz6–8), thus increasing both the quantity and quality of the key properties to be rationalized, understood, or interrelated at the atomic scale. At the same time, despite the increased complexity, such advances have also allowed the study of the L-dependence of key-quantities such as the bandgaps4–7 (both, ΔEac and ΔEzz), conductivity, aromaticity,1,3,4 and even Raman spectra.17 The L-dependence studies4,5 revealed that the changes in such properties versus length are not always gradual (or smooth). The presence of a metal-insulator-like phase transition at a critical length Lc was advocated by two different recent studies, Lawrence et al.8 and Zdetsis et al.,4 almost simultaneously. However, these two studies have offered different assessments and interpretations on the nature of the transition and magnetism, as well as the value of Lc.4,8 This is not something new or unusual in a rapidly grown pioneering field like this,1 and this is not the only existing “discrepancy”. Other conflicting (or conflicting-looking) results (experimental and theoretical) include the magnitude and nature of the bandgaps,1,6,7,12,13 the existence and nature of magnetism in the edge states,1,3–7 and the magnitude of the substrate's influence on these properties.1,5–7 For example, the magnitude of the bandgap for the 5-AGNRs has been measured by (at least) three different groups8,12,13 to be 0.85 eV,8 2.8 eV,12 and 0.1 eV13 respectively, while the theoretical values vary from 0.1 eV1 to 1.7 eV.14 For the 7-AGNRs the measured values of ΔEzz vary between 1.9 eV6 and 2.5 eV,7 whereas the measured ΔEac values range from 2.3 eV to 3.2 eV,6,7,9,15 overlapping significantly with the range of ΔEzz. Thus, the unambiguous distinction between ΔEac and ΔEzz is another subtle point together with the bridging of the measured and calculated ΔEac values, which also vary widely from 2.3 eV to 3.7 eV.1,6,7,14 Some of the (different) measured or calculated values correspond to AGNRs of different lengths, but in the literature the quoted values are usually given without reference to the actual length which is, thus, treated as a hidden variable. However, the biggest problem seems to be the large difference between the measured values of the gap(s) in relation to the “official” theoretical values obtained by the GW method,14 which are widely recognized as an almost universal point of reference. For example, the measured STS gap value(s) for the 7-AGNRs is between 2.3 eV and 2.8 eV,6,7,9,15 whereas the theoretical GW value14 is 3.7 eV (i.e. about 1.5 eV higher). Thus, one possible interpretation is that the real STS gap is indeed 3.7 eV, and the difference of ∼1.5 eV is due to screening from the metallic substrate (usually Au), which remains to be seen. On the other hand, the simpler interpretation suggested here (in accordance with Occam's principle) consistent with our results, is that the real (theoretical and experimental) STS gap is in reality ∼2.5 ± 0.3 eV and the reference GW results14 are overestimated by about 1.5 eV. This claim (see Section 2.4) is supported by both theoretical6 and experimental6,7 evidence:(1) The theoretical reference GW results14 have been obtained for AGNRs of infinite length, not for finite atomically precise AGNRs. Recent GW results by Wang et al.6 for the finite length (7, 24) AGNRs showed a gap of 2.8 eV in full support of the present results. Moreover,
(2) experimental measurements6,7 for 7-AGNRs supported on non-metallic substrates, such as NaCl and MgO (without metallic screening) found an STS gap of ∼2.5 eV, in full agreement with the present interpretation. As a result, it appears that there are several conflicting results and interpretations (or highbrow “solutions”) about the STS gaps, although the real solution seems to be much simpler (but not always obvious), as could be possibly argued on the basis of Occam's principle. Thus, there seem to be several subtle points behind the controversy for the energy gaps of atomically precise AGNRs, which include:
(1) The finite length of the AGNRs.
(2) The overestimation of the reference theoretical values.
(3) The magnitude of the substrate interaction.
(4) The topological end-states.
(5) The inversion/reflection symmetry conflict.
(6) The “exact” exchange in the DFT functionals.
(7) The mixing of zigzag-end localized topological end-states with other delocalized “bulk” states, which leads to.
(8) Difficulties in distinguishing between ΔEac, ΔEzz gaps, and in part Δεζζ, which is the real energy separation between the end-states.
Along these lines the present work aims at deciphering all these subtle points, which are clearly interrelated. Thus, the present work can be considered as a touchstone or a positive synthesis of various conflicting views. Based on previous experience,1,3,19 it is expected that such synthesis should be proven successful and constructive, facilitating the successful and accurate functionalization of AGNRs for realistic applications. We can fully rationalize all known experimental data for the 5-, 7-, and 9-AGNRs within less than 1% accuracy, predict non-measured gaps, and pinpoint at the same time the sources of discrepancies.
2. Theoretical framework
For a consistent and transparent understanding and interpretation of the origin and magnitude of ΔEac, ΔEzz as well as the factors that influence their size, it is important to realize that practically all these quantities are dominated by the influence of the (“many-body”) Coulomb correlation energy combined with sublattice frustration, which gives rise to the staggered sublattice potential20 across the zigzag ends of finite length AGNRs. In fact, the sublattice frustration, which is equivalent to the inversion/reflection symmetry conflict, is the driving force for the generation of the end/edge states and, as we have illustrated earlier,2–4 constitutes the largest contribution to the Coulomb correlation energy. Such symmetry conflict is clearly illustrated in Fig. 1 and in particular in Fig. 1(a). As we can see in Fig. 1(a), the sublattice symmetry group, which is characterized by a horizontal C2 rotation symmetry axis and a vertical S2 rotation-reflection axis, lacks a center of inversion. As a result, end-atoms at the two zigzag ends belong to different sublattices. However, the molecular symmetry group, which reflects the symmetry of the Hamiltonian, is of D2h symmetry with a center of inversion and two (horizontal and vertical) C2 axes. Therefore, if we try to build the sublattice structure preserving the full D2h symmetry, we end up with a region in the middle, designated by a vertical ellipse in Fig. 1a, where there is a sublattice conflict with neighboring (pink in the figure) carbon atoms across the y-axis belonging to the same sublattice. This is actually the reason for the generation of topological end-states which do not penetrate in the symmetry-forbidden central region of the AGNRs.3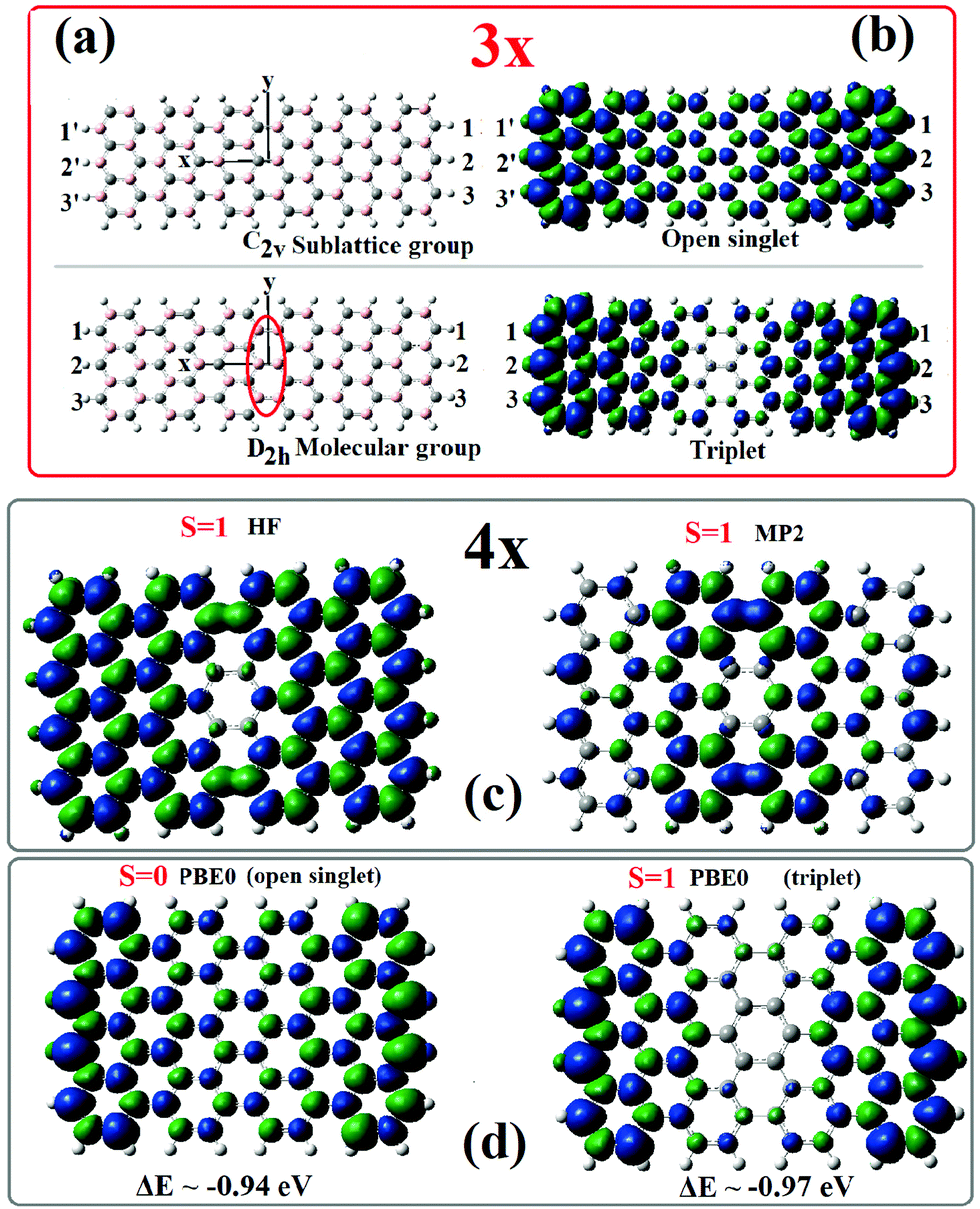 | ||
| Fig. 1 Molecular and sublattice symmetry of the 3 × 6 (7, 12) AGNR (a), reflected in the spin densities (b). Isovalue = 0.004. The vertical elliptic curve indicates the region of sublattice imbalance (and frustration). Comparison of the corresponding spin densities for the 4 × 4 (9, 8) AGNR at the HF, MP2 and DFT/PBE0 level is given in (c and d). | ||
Thus, the understanding that most (or all) of the Coulomb correlation energy is devoted to counterbalancing the topological frustration between sublattice and molecular symmetry-groups is the starting (and key) point of the present investigation. This principle together with the established2–5 (hidden) strong contributions of aromaticity and shell structure2–5 constitute the basis for a deeper understanding of all these quantities (ΔEac, ΔEzz, Δεζζ, and ΔEs). Hence, if we can properly alleviate the sublattice-molecular group symmetry frustration (which is equivalent to inversion/reflection symmetry conflict3–5), under the natural constrains of shell structure and aromaticity,2,5 we could effectively account for the (largest part of) Coulomb correlation energy. This would also explain why the open shell states (singlet or triplet) are not the real lower energy states, but rather “pseudo-states”.21
2.1 A synopsis of the aromatic/topological shell model
The “shell model” was initially invoked by the present author22 as a molecular bridge from benzene to graphene through a sequence (termed the “main sequence”) of hexagonal (D6h symmetry) polycyclic hydrocarbons (PAHs) of the general form C6n2H6n, n = 1, 2,…, containing n hexagonal monocyclic rings surrounding each other. The integer n is also called the shell number since, if we consider the nth PAH of the sequence, we can uncover a shell structure with a benzene nucleus (n = 1) surrounded by n − 1 annulene layers or “shells”, as is shown in Fig. 2. This shell structure has been shown to be a realistic concept, with characteristic peaks and dips in the aromaticity indices, which is analogous to the electronic atomic shell model, responsible for the periodic table of the elements. In the current aromatic shell model, the shell number n corresponds to the principal quantum number n, and the number of layers (around the benzene nucleus) l = n − 1 is analogous to the angular momentum quantum number L. The role of the ±ML values is played by the number of the u and g parity MOs in the given shell.22 Moreover the shell model (shell structure) is not merely an abstract concept, but it has been used as a very useful and powerful tool in rationalizing the exotic properties of graphene at the molecular level,2 and for understanding the key properties of AGNRs (such as the triple periodicity in terms of width),3 as well as the aromaticity and stability of various NGRs.5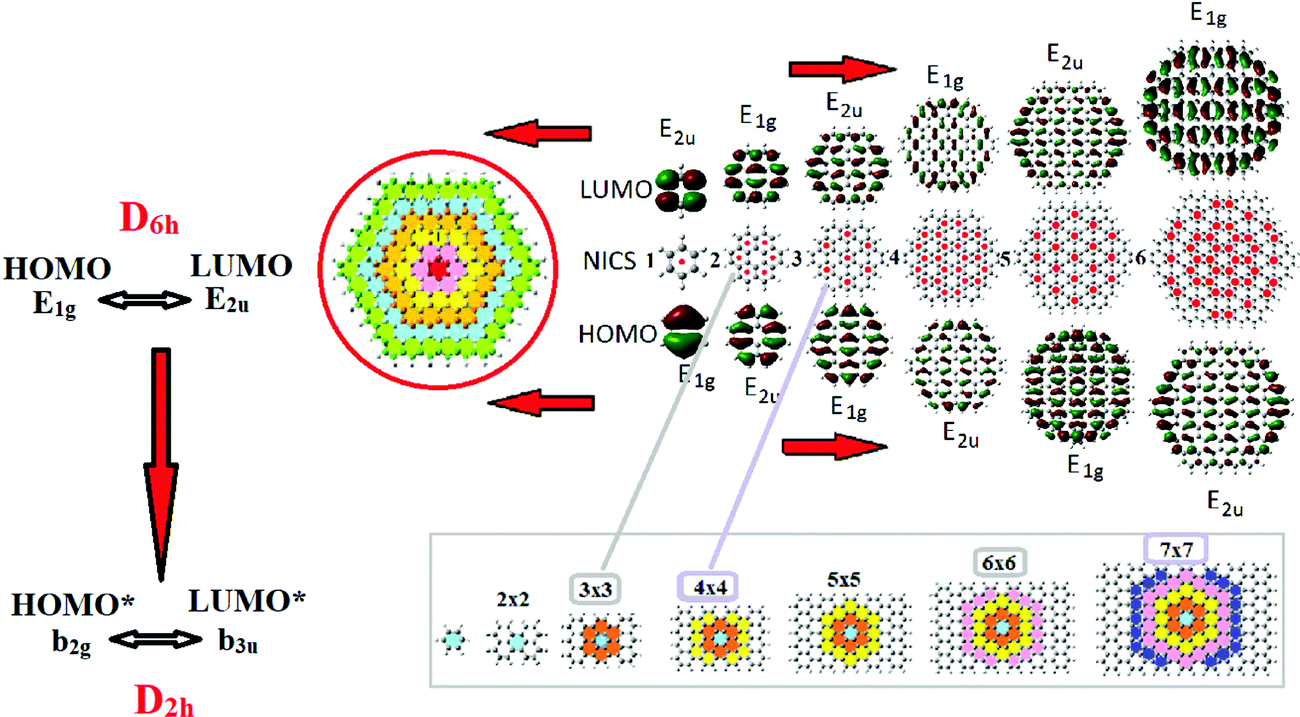 | ||
| Fig. 2 The shell model and the “main sequence of PAHs (shown up to n = 6), illustrating the aromaticity patterns (with red dots at the centers of the aromatic or “full” rings), and the structure and symmetry of the HOMO and LUMO orbitals (top). The bottom part illustrates the analogy with corresponding rectangular NGRs or AGNRs with an equal number of armchair and zigzag rings (see the text). | ||
The shell model is responsible2,22 for the double periodicity in the aromaticity patterns, and the structure/symmetry of the HOMOs and LUMOs (E1g and E2u) of hexagonal PAHs seen in Fig. 2. The even parity E1g HOMOs (and odd parity E2u LUMOs) are associated with a triangular Clar-type aromaticity pattern and odd shell number, like n = 3 circumcoronene (CIRCO), whereas E2u HOMOs (and E1g LUMOs) are connected with hexagonal non-Clar-type aromaticity and an even shell number like n = 2 coronene (CO).2,5,22 As we can see in the figure, the HOMO of the nth PAHs is similar to the LUMO of the (n − 1)th and vice versa. Likewise, for rectangular NGRs and AGNRs we have a three-member periodicity specified by their shorter dimension, i.e. the width defined by the number of zigzag rings, Z, which (due to the shell structure) has the form:3Z = 3n, 3n + 1, or Z = 3n − 1, n = 1, 2,…. This is the well-known 3n, 3n ± 1 rule of AGNRs. The first two types correspond to PAHs with CIRCO and CO aromaticity patterns with effective HOMOs* of odd (b3u) and even (b2g) parities respectively; where “effective” means effective “bulk” MOs, defined by neglecting the frontier end-state MOs.3 Obviously the LUMOs* follow the exactly opposite trend. Therefore, the shell structure (or shell model) is both a conceptual and efficient tool for graphene-based structures, such as AGNRs.
2.2 Calculation of ΔEzz and ΔEac from the one-body DFT calculations
Within the 1-electron approximation underlining the DFT and Hartree–Fock (HF) self-consistent fields, the symmetry frustration between molecular (D2h) and sublattice (C2V) symmetry groups can be alleviated by effectively breaking (or redefining) the symmetry of the additional degrees of freedom (besides spatial coordinates) i.e., the spin and/or pseudospin (for real-space calculations). In the first case we can introduce non-zero spin values preserving the molecular symmetry,3 whereas in the second case we are forced to break molecular symmetry, by introducing open-shell singlet states, which when optimized geometrically converge normally to C2v symmetric geometries compatible with sublattice symmetry, thus breaking the inversion/reflection symmetry. This occurs because the HOMO (and LUMO) orbitals of the open singlet are obtained, by construction, by mixing the HOMO and LUMO orbitals of the closed singlet. These orbitals have opposite parities, u, or g, (and opposite behaviour under the σy or σxz reflection plane2,3). Thus, by losing the σy (or σxz) reflection plane of the D2h symmetry group, as is illustrated in Fig. 1(a), we can get sublattice distribution with opposite sublattice points at the two ends. This facilitates frontier molecular orbitals (HOMO, LUMO) localized only at one end (left or right) of the AGNR, producing an antisymmetric (pseudo)spin density, as is shown in Fig. 1(b), reflecting the sublattice symmetry and structure. Obviously, the reverse picture with the pseudospins interchanged is equally valid. On the other hand, the molecular D2h symmetry demands the same type (same sublattice) of atoms at the two ends, as shown in the lower part of Fig. 1(a), thus producing a symmetric, with respect to the y-axis, (pseudo)spin distribution (bottom of Fig. 1(b)). In both cases the (pseudo)spin distribution is almost zero at the middle part of the AGNR. This is reproduced in the corresponding “spin” densities (b). Such spin densities, in Fig. 1(b–d), which are generated self consistently (through the DFT convergence process) are in fact pseudospin densities. The open singlet reflects the sublattice symmetry (with different types of atoms at the two ends) with an equal number of up and down (pseudo) spins, or A and B sublattice points. This results in a balanced (nearly zero spin distribution in the middle). The triplet state on the other hand has also a region of zero spin in the middle, exactly where the sublattice imbalance occurs. In contrast to the band description (in k-space), in the real-space (“molecular”) calculations the sublattice degree of freedom does not enter in the spatial Hamiltonian and can only be introduced as (pseudo)spin. Then, due to the better account of Coulomb interaction, open shell states (triplet or singlet) appear energetically lower than the closed singlet state. This is because the additional degree of freedom of “pseudospin”, introduced to take care of the sublattice topology (and the staggered potential), facilitates the optimization of Coulomb interaction by keeping away each of the other electrons of different spin (for which Pauli repulsion is not operative), but of identical pseudospins. Moreover, as was illustrated in Section 2.1 (and Fig. 2), based on the shell model,2,3 the unoccupied states of the “previous” (shell number smaller by 1) PAH are the occupied ones of the current PAH. In AGNRs this is responsible for the interplay between odd and even parity HOMOs as the width of AGNRs is growing (3n AGNRs have an odd HOMO and even LUMO, whereas 3n + 1 AGNRs are characterized by an even HOMO and odd LUMO).2,3 This is also responsible for the well-known 3n, 3n ± 1 width rule for AGNRs.3 Note that the (pseudo)spin densities invariably reflect the sublattice (pseudospin) structure within the frustrated molecular (D2h) symmetry3 in the first case, or the sublattice symmetry (C2v) in the latter, where opposite end sites have opposite spins. It should be emphasized at this point that for wider AGNRs (where n > 1 in the above rule for width3), higher spin states are required3 to lower the total energy (within the molecular D2h symmetry group). Such larger (pseudo)spin-polarized states optimize better the sublattice distribution (within the D2h molecular group),3 whereas the open-shell singlets lie higher in energy and revert to the closed singlet state. This illustrates emphatically that the open-singlet state is not the true ground (lowest energy) state of AGNRs (and, consequently, no conventional magnetism is truly present). Nevertheless, the open singlet state is still a very useful and efficient concept for the description of end-states. It should be emphasized that in both cases of Fig. 1, when correlation is introduced even at the MP2 level, the energetical ordering is reversed and the lowest energy structure is a closed singlet.4 In addition, the MP2 correlated “spin” density of the triplet, as we can see in Fig. 1(c), is rather correcting the HF failure (having the opposite sign) than reflecting the full sublattice structure. Note also in Fig. 1(c and d) that the triplet state is slightly lower than the open singlet, and that the energy difference of the open shell singlet and triplet states (which are practically isoenergetic) from the closed singlet is about 0.95 eV. This should be a good estimate of the “missing” Coulomb energy in this case and based on the (approximate) electron–hole symmetry, the expected (HOMO–LUMO) separation of the open singlet (or the triplet) should be about twice as large (∼2 eV). Indeed, the calculated open-singlet HOMO–LUMO gap for the 9-AGNRS (or 4x) is 2.2 eV, and so is ΔEzz (vide infra). Even more important is the fact that the corresponding value for the 3 × 6 or (7, 12) AGNR is also about 0.93 eV, suggesting an open-singlet gap of about 1.9 which is in excellent agreement with both the measured value6 of ΔEzz (1.90 eV), and the calculated open singlet gap. It is important to observe also that the open-singlet value “ΔEzz” = 1.2 eV for the 5-AGNRs and the 1.9 eV open singlet gap for the 7-AGNRs are practically equal to the correlation improved GW-LDA bandgap differences,14 which is highly suggestive for the essential correctness of our claim. Thus, within the one-electron approximation we have established the correct basis for discussion and analysis of both ΔEzz and ΔEac. ΔEzz is identified as the open-singlet HOMO–LUMO gap, whereas ΔEac can be identified as the difference |(HOMO−1)-(LUMO+1)|, with the understanding that both HOMO and LUMO are end-states. Nevertheless, similar values of gaps and analogous estimates can be found in the triplet state as well. Furthermore, it should be emphasized that the central meaning of ΔEzz is only valid for lengths L longer than the critical length (L ≥ Lc), although the open-singlet HOMO–LUMO is defined for almost all lengths and is practically constant, as was also verified by Wang et al.62.3 Introducing many-body corrections to the gaps
For both gaps (ΔEac and ΔEzz) we can further correct if we wish their (one-body) values by considering additional many body contributions through time-dependent DFT (TDDFT), which has been shown1 to provide very good (“many-body”) estimates of the gaps, so that the STS spectrum overall looks very much like the (luminous) optical spectrum, because both are dominated by molecular overlaps between transition states. This is further illustrated and “verified” from the results below. Furthermore, the use of TDDFT allows the clear and unambiguous identification of the energy separation of the end/edge states, which according to the present investigation is not given by ΔEzz, as Wang et al.6 have suggested, but by another type of gap which here is denoted as Δεζζ. In the usual one-body approximation Δεζζ corresponds to the HOMO–LUMO separation of the closed singlet true ground state for L ≥ Lc, which is always only a few 0.1 eV (∼0.1 eV, for L → ∝) in accord with the association of the end states with the Dirac points3,4 (and charge neutrality points4) located “very close” to the Fermi level. TDDFT indeed verifies that in contrast to Δεζζ which involves transition from one purely end-localized HOMO state to an opposite-parity end-localized LUMO state, the ΔEzz gap always involves transitions from a mixture (∼60% to ∼40%) of “surface”-“bulk” states to another state of about equal amount of mixing. Thus, although ΔEzz involves a large number of localized end-states, it should not be associated with the energy separation of the end-states. Another way, besides TDDFT, to distinguish between “bulk” and “surface” energy gaps is by comparing to the corresponding “edge-modified” AGNRs,5 obtained by eliminating “empty” (i.e., non-aromatic) end-rings, which also eliminates topological end-states (and, therefore, ΔEzz and Δεζζ).2.4 The substrate influence on the measured STS gaps
As we have mentioned above, the main crucial property under possible dispute is the magnitude of the substrate influence (screening) ΔEsb on the measured STS gap. According to our earlier estimates1 ΔEsb should be of the order of a few 0.1 eV. However, almost in all cases ΔEsb larger than 1 eV is needed to bridge the experimental STS measurements for AGNRs deposited on metal surfaces (usually Au) and the theoretical values for free standing AGNRs. The theoretical values widely recognized as an almost universal point of reference are the GW results of Yang et al.,14 which among the theoretical values reported earlier are clearly the largest (and many times, by far). As a result, ΔEsb which is defined as the difference of the STS measurements and the theoretical reference values are unrealistically large. For example, for the 7-AGNRs the theoretical GW gap14 is 3.7 eV, whereas the experimental STS gap value obtained by various groups6,7,9,15 is 2.5 ± 0.2 eV. Thus, ΔEsb should be at least 1.2 eV. However, the STS value of 2.5 eV was also obtained for 7-AGNRs deposited on non-metallic substrates, such as NaCl6 and MgO,7 for which such a large ΔEsb value is clearly unrealistic. On the basis of their STS measurements on samples grown on MgO, Kolmer et al.7 concluded that ΔEsb should be marginal, which is in full agreement with our present results. However, the general consensus, with few exceptions1,7,18 is largely different (up to now). In this work we are led to conclude that the GW results14 overestimate the bandgaps mainly due to the size effect, since the GW results of Yang et al.14 were obtained for infinite AGNRs, whereas the atomically precise AGNRs have finite length (and topological end-states). This could be sufficient to explain the resulting unrealistically large ΔEsb values. Yet, besides the infinite size (and the corresponding periodic boundary conditions) the lack of exact exchange in the LGA wavefunctions building the Green's function could also be important since exchange interaction is very sensitive to inversion symmetry frustration. Nevertheless, judging from our TDDFT results, it is more reasonable to attribute the gap difference between the infinite and the finite size AGNRs (size effect) to the mixing of edge/end states with the infinite “bulk” states (and the scattering at the zigzag edges) which can drastically reduce the gap. This is corroborated by the GW results6 of Wang et al.6 for the finite (7, 24) AGNR (and slightly longer), who obtained a gap of 2.8 eV clearly closer to the measured (by several groups) gap, and substantially smaller compared to the 3.7 eV (G0W0) value14 for the infinite 7-AGNR. Parenthetically, it should be mentioned at this point that even in the worst-case scenario where the substrate interaction is strong (especially when the distance of the STS tip from the surface is small), this leads to mixed substrate-AGNR states1 which can be easily recognized (and excluded) from the measurements by comparing the two separate STS spectra. Moreover, such states would be expected to have low overlap with the pure AGNR-excited-states, and consequently the corresponding transition(s) would have very low intensity and would be difficult to detect. Thus, we assert here that ΔEsb should indeed be marginal, in full agreement with the experimental results (for 7-AGNRs grown directly on the MgO substrate) and conclusions of Kolmer et al.72.5 Computational details
The theoretical and computational details of the present investigation are described in ref. 1–5. The computations, as before, have been performed with the Gaussian23 program package using DFT and TDDFT employing the PBE0 functional24 (which includes “exact” exchange, in contrast to PBE) and the 6-G31(d) basis set. The same computational package was used for the Møller–Plesset many-body perturbation theory of 2nd order (MP2) calculations, with the same basis set. The PBE0 functional and the 6-31G(d) basis set23 have been constantly, consistently, and successfully used over the last 7–8 years1–5,22,25 by the present author for all graphene-based structures (PAHs, NGRS, AGNRs), small, medium, and large (up to ∼1440 atoms, 8190 electrons).25 This functional (PBE0) and basis set have been consistently used for all types of calculations (geometry optimizations, energies, and/or vibrational frequencies) which is very important for the broad range of structures and comparisons used. For smaller structures the results have been successfully compared with the results obtained with larger basis sets. However, the best test for the PBE0/6-31g(d) approach is the excellent agreement obtained with measured quantities such as the STS gaps (e.g., the measured open singlet gap of 1.90 eV for the 3 × 6 AGNR,6 or the optical gap of perylene1) and other properties, including cohesive energies,25 even for non-graphene-like structures (finite or infinite). For example, the calculated band gap and cohesive energy of diamond was calculated using this PBE0/6-31g(d) theoretical level with almost chemical accuracy.25 Thus, this level of calculation is clearly adequate for our purposes. The convergence criterion for SCF was set to tight, which corresponds to 10−8 change in the RMS density matrix, whereas the RMS force criterion for the geometry optimization was set to 1 × 10−5 atomic units (tight) for the “key structures” and 3 × 10−4 for the rest of the calculations. Finally, the visualization of the results was accomplished using GaussView software.262.6 Synopsis of the theoretical approach
For the atomically precise AGNRs examined here it has been illustrated that although the closed singlet is the correct lowest energy state, due to (inversion) symmetry frustration (which is also a sublattice frustration), open shell states, such as open shell singlet and triplet appear energetically lower due to better account of the Coulomb correlation energy (within the 1-electron approximation).3 Thus, such open shell states should be considered as “pseudo-states”, and their resulting spin distribution within the one-electron DFT framework should be characterized as the “pseudospin” distribution. The full amount (or most of it) of the Coulomb correlation energy can be estimated from the total energy difference of the closed singlet and the open singlet or triplet states. Thus, using standard DFT calculations (with functionals including exact exchange, such as the PBE023) for the closed and open singlet, and/or triplet states we can have a very good estimate of all (“surface” and “bulk”) energy gaps, measured by STS as follows:(1) The HOMO–LUMO gap of the closed singlet corresponds to the real energy separation of the end-states localized at the zigzag ends, Δεζζ. This is true for long enough AGNRs for which both HOMO and LUMO are zigzag-end-localized.
(2) In this case the (HOMO−1)–(LUMO+1) difference corresponds to the “bulk” bandgap ΔEac between states delocalized over the entire AGNR.
(3) ΔEac can be further verified, if desired, by comparing to the HOMO–LUMO gap of the edge modified AGNRs,4,5 which contain no zigzag end bonds, and no end-states.
(4) On the other hand, the zigzag-end-localized HOMO–LUMO gaps of the open-shell singlet or the triplet states provide a very good estimate of the “mixed” ΔEzz gap.
(5) Further refinement of all these three gaps (“bulk”, “surface”) can be obtained, if needed, by the analysis of the orbital composition of the main peaks of the excitation spectrum obtained by TDDFT. This can also help the correct identification (and nature) of the gaps.
3. Results and discussion
3.1 5-AGNRs
Fig. 3 summarizes the present results for the 5-AGNRs (or 2 × AGNRs) which have also been studied by several groups.1,4,8,12,13,16 | ||
| Fig. 3 (a) Variation of the open singlet, ΔEzz, and the “bulk” |(HOMO−1)-(LUMO+1)| = ΔEac gaps (in eV) in terms of length L (in Å) for the 5-AGNRs (2×). (b) Variation in the ΔEac gap (in eV) as a function of length for the edge modified 5-AGNRs together with the usual polynomial fit (see text). (c) Excitation spectrum of the 2 × 22 and 2 × 23 AGNRs. Intensity(I) is in arbitrary units and excitation energy (ΔE) in eV. (d) Variation with length of the HOMO–LUMO, and ΔEzz, Δεζζ gaps, calculated by TDDFT as “first” and “second” optical gaps respectively, including the corresponding experimental values from ref. 8 and 13 (see text). | ||
3.2 7-AGNRs
Fig. 4 summarizes the results for the 7-AGNRs, represented by the 3 × 6 or (7, 12) AGNR, for which there are detailed experimental STS data.6 Results for all three spin states (closed singlet, open singlet, and triplet) are shown.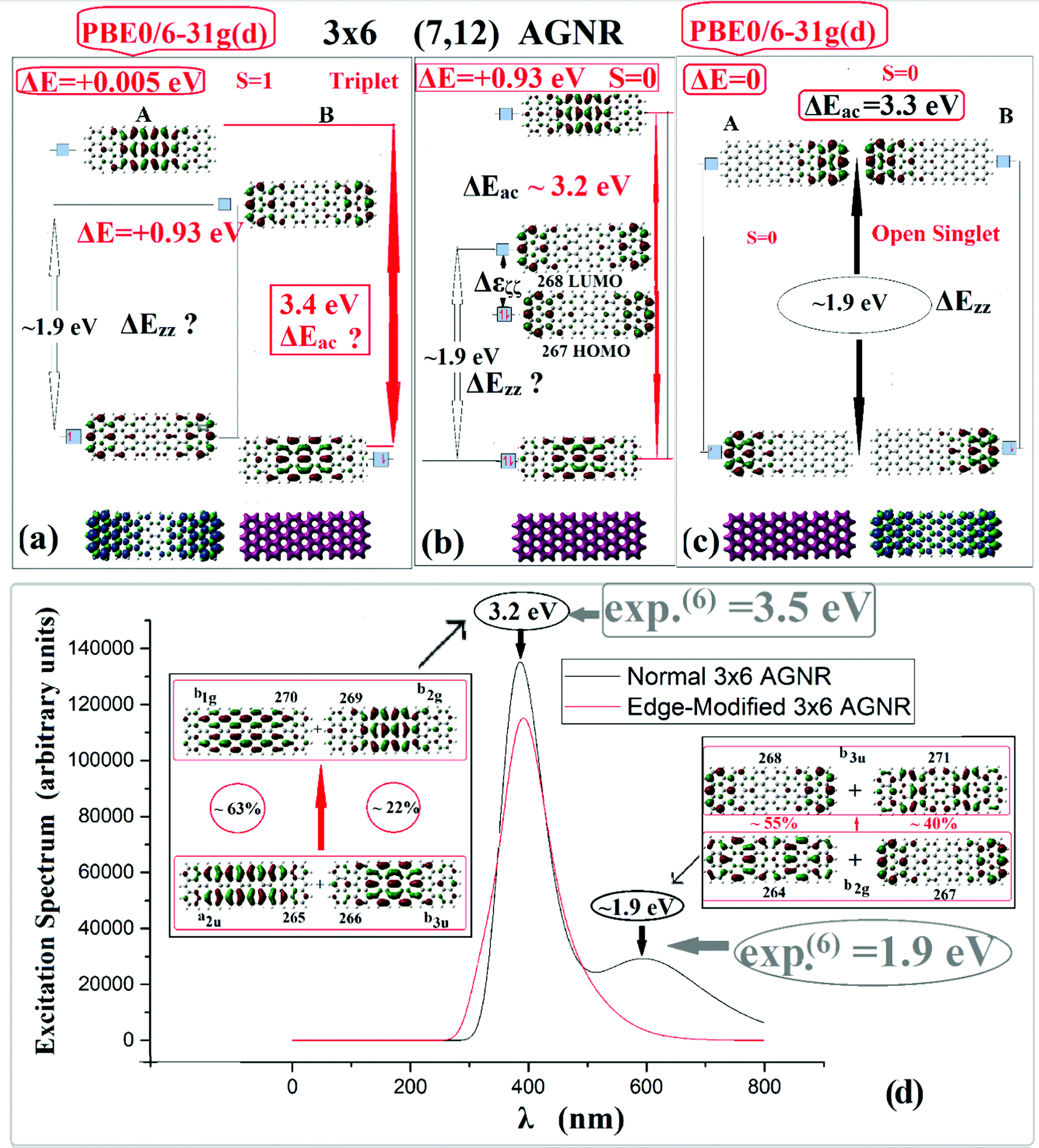 | ||
| Fig. 4 Spin states of the 3 × 6 (7, 12) AGNR: (a) triplet, (b) closed singlet, and (c) open singlet states, showing frontier MOs, and gaps, together with charge density and spin density (see text). (d) Excitation spectrum for the standard (black line) and edge-modified (red line on line) AGNRs. Intensity is given in arbitrary units, and excitation energy in eV. Boxes (rectangular and elliptical) emphasize the calculated values in agreement with the experimental measurements. Question marks indicate alternative possibilities and/or identifications of the corresponding gaps (see text). | ||
The end states, which are localized at the two zigzag ends, can be clearly seen in Fig. 4(b) illustrating the frontier MOs of the closed singlet. These end states, which comprise the HOMO and LUMO orbitals (labeled 267 and 268 respectively) have clearly zero density in the interior of the AGNR, in contrast to the MOs above and below them, which are (de)localized in the interior “bulk” region. However, although the end states in the closed singlet description appear in both ends, in the open shell case of Fig. 4(c) the end states appear only on one zigzag end at a time. Based on the closed singlet ground state, it becomes clear that the “real” energy separation of the end states is the HOMO–LUMO gap of the closed singlet state which is (almost always) about 0.1–0.3 eV (depending on the length). This is corroborated by the TDDFT results, giving rise to the Δεζζ gap, which was discussed earlier for the 5-AGNRs, and is consistent with the appearance of Dirac points close and around the Fermi level, whereas ΔEzz is due to mixed transitions involving both “end” and “bulk” states.
3.3 9-AGNRs
We can observe in Fig. S2(c) (ESI†) that the overall spectrum of the 4 × 6 AGNR which has the same length with the 3 × 6 AGNR, except for a suppression of the Δεζζ peak, looks at first sight very much like the one for the 3 × 6 AGNR. Clearly a (deep) “bulk” gap could be expected not to vary very much or be so sensitive to the exact AGNR's width; but for the peak around 2.0 eV, which up to now was associated with the ΔEzz gap of the 3×- AGNRs, further investigation is needed, which is described in Fig. 5. Fig. 5(a–c) are the corresponding analogues of Fig. 4(a–c) respectively. However, contrary to the 7-AGNRs, the experimental data for the 9-AGNRs are very limited.16 Therefore most of the results shown in Fig. 5 should be considered as predictions of the present work. As we can see in Fig. 5(c) for the open singlet the two fundamental gaps ΔEzz and ΔEac are very close together (2.2 eV and 2.4 eV respectively) and not exactly equal to the corresponding 3 × 6 gaps This is also true for the almost equal values of ΔEzz and ΔEac obtained from triplet (and closed singlet). Thus, the peak around 2.0 eV in Fig. 5(d) is the result of the overlap of the ΔEzz and ΔEac gaps, whereas the peak around 3.2 eV in the same figure, Fig. 5(d), although of “bulk” type (similarly to the 3 × 6 AGNR) is not the smallest “bulk” gap, and the real ΔEac for the 4 × 6 (9, 12) AGNR should be around 2.0 eV. This is verified in Fig. 5(d), which shows that the 2.1 eV “bulk” peak (together with the “deeper” 3.2 eV “bulk” peak) survives the elimination (total and partial) of the empty (non-aromatic) end-rings which generates the edge modified AGNRs (without end states, and ΔEzz). As is well known, this “bulk” peak value decreases as the length of the AGNR increases. For the 4 × 13 AGNR we find ΔEac = 1.6 eV, but for the longer 4 × 18 (9, 36) and 4 × 24 (9, 48) AGNRs of lengths L ≈ 78 Å and L ≈ 104 Å respectively, we obtain (by TDDFT) for both of them ΔEac = 1.45 eV. This value is in very good agreement with the recently measured gap of 1.4 eV by Talirz et al.,16 as is illustrated in Fig. S3 (ESI†). We can also clearly see in Fig. S3(b and c) (ESI†) the “surface” ΔEzz gap at about 2.1–2.2 eV. Thus, for the 9-AGNRs the predicted values for the gaps are ΔEac = 1.45 ± 0.1 eV, and ΔEzz = 2.1 ± 0.1 eV. | ||
| Fig. 5 Spin states of the 4 × 6 (9, 12) AGNR: (a) triplet, (b) closed singlet, and (c) open singlet, showing frontier MOs, gaps, and spin densities. (d) Excitation spectrum for the standard (black line), partially edge-modified (blue line on line), and fully edge modified AGNRs (red line on line). Intensity is given in arbitrary units, and excitation energy in eV. The frontier orbitals and aromaticity patterns of partially and fully edge modified AGNRs are shown in the right and left portions of the figure. The peak at 1.45 eV is further verified by Fig. S4 (ESI†) which shows the spectrum of the edge-modified 4 × 24 AGNR. | ||
4. Conclusions
We have achieved excellent agreement (within 1% or less) with the measured STS gaps (“bulk” and “surface”) for the known 5-, 7- and 9-AGNRs, although with the “surface” gaps, as illustrated in Table 1, namely:| AGNR | Δεζζ Calculated | Δεζζ Measured | ΔEzz Calculated | ΔEzz Measured | ΔEac Calculated | ΔEac Measured |
|---|---|---|---|---|---|---|
| a Values obtained in the present work. | ||||||
| 5- | 0.1(1) | 0.1(13) | 0.85 | 0.85(8) | 1.1a, ![[1 with combining low line]](https://www.rsc.org/images/entities/i_char_0031_0332.gif) . .![[7 with combining low line]](https://www.rsc.org/images/entities/i_char_0037_0332.gif) (14) (14) |
— |
| 7- | 0.1(1)a | — | 1.9a | 1.9(6), 2.5(7) |
2.5
, 2.8a, 2.8(6), ![[3 with combining low line]](https://www.rsc.org/images/entities/i_char_0033_0332.gif) .(14) .(14) |
2.8 (6), 2.5 ± 0.2(6,7,9,15) |
| 9- | 0.1(1)a | — | 2.2a | — |
1.45
, 1.6(1), ![[2 with combining low line]](https://www.rsc.org/images/entities/i_char_0032_0332.gif) . .![[0 with combining low line]](https://www.rsc.org/images/entities/i_char_0030_0332.gif) (14) (14) |
1.4 (16) |
(a) For the 5-AGNRs the measured8 gap value is 0.85 eV. The gap calculated here with DFT/PBE0 is 1.07, whereas the TDDFT/PBE0 value is exactly 0.85 eV, also indicating that this is a ΔEzz gap.
(b) Moreover, the measured13 0.1 eV gap is recognized to fully coincide with the Δεζζ gap calculated here (by both DFT-TDDFT/PBE0).
(c) For the 7-AGNRs the measured6,7,9,10,15 ΔEac gap of 2.3 ± 0.2 eV coincides with the ΔEac gap calculated here (with both DFT-TDDFT/PBE).
(d) Furthermore, for the (7, 28) AGNR the measured6 and GW-calculated6 2.8 eV gap fully coincides with the ΔEac gap calculated here (with both DFT-TDDFT/PBE0), whereas for the (7, 12) AGNR the measured6 and calculated6 ΔEac gap is ∼3.2 eV.
(e) The measured6 ΔEzz gap of 1.9 eV for the 7-AGNRs, (7, 12), and longer, is clearly identical to the ΔEzz gap calculated here of 1.9 eV (with both DFT-TDDFT/PBE0).
(f) For the 9-AGNRs the only known (to the present author) measurement16 for the gap is 1.4 eV. The present calculations (TDDFT/PBE0) yield a ΔEac value of 1.45 eV, and also predict ΔEzz = 2.1 ± 01 eV, quite close to the corresponding gap for the 7-AGNRs.
Note that for the 5- and 9-AGNRs the ΔEzz gaps are theoretical predictions of the present work, due to the lack of analogous experimental data.
At the same time the present work has provided a simple physical understanding/rationalization of the origin and properties of these gaps. We have shown that such excellent agreement can be obtained using a transparent approach, using a minimum of computational resources, avoiding high level many-body methods, such as the advanced GW approach.14 This is accomplished by recognizing (and suitably exploiting) the fact that most (or all) of the Coulomb correlation energy is devoted to offset the (inversion) symmetry conflict. This is an added insight. Thus, simple DFT calculations with or without “fictitious” open shell states (such as open shell singlets or triplets) can give accurate results, especially when augmented by TDDFT calculations which can further refine the results, provided that the chosen DFT functional includes the “exact” exchange (such as the PBE0 functional23 used here, proven to provide excellent results1–5,24), and the finite length of the AGNRs is taken into account (recall the synopsis of the theoretical approach in Section 2.5). Under the same provisions (a finite size of AGNRs and “exact exchange”) the GW approach would also give the correct results, as is illustrated in ref. 6, where taking into account the finite size of the 7-AGNRs has lowered the GW gap by about 1 eV, in very good agreement with the measured STS value. As a result, a similarly large overestimation of the expected substrate screening would be avoided, since the GW results14 are widely used as reference values for the free standing AGNRs. This is corroborated by STS measurements of AGNRs on non-metallic substrates.6,7 Thus, the measured STS gaps are practically independent of the substrate and virtually equal to the free-standing values, obtained by any of the three computational methods: DFT, TDDFT, and GW (from the simplest to the more complex), provided the finite size and the “exact” exchange are taken into account. Obviously, the simplest (and computationally most economical) approach should be normally preferred, in accordance also with Occam's principle. A combination of DFT and TDDFT, as is used here, should be considered ideal.
Additional supplementary material with more details and comparisons for the spectra of 5-, 7-, and 9-AGNRs are given in the ESI.†
Conflicts of interest
There are no conflicts to declare.References
- A. D. Zdetsis and E. N. Economou, Rationalizing and Reconciling Energy Gaps and Quantum Confinement in Narrow Atomically Precise Armchair Graphene Nanoribbons, Carbon, 2017, 116, 422–434 CrossRef CAS.
- A. D. Zdetsis, Bridging the Physics and Chemistry of Graphene(s): From Hückel's Aromaticity to Dirac's Cones and Topological Insulators, J. Phys. Chem. A, 2020, 124, 976–986 CrossRef CAS PubMed.
- A. D. Zdetsis, Do We Really Understand Graphene Nanoribbons? A New Understanding of the 3n, 3n ± 1 Rule, Edge “Magnetism” and Much More, J. Phys. Chem. C, 2020, 124, 7578–7584 CrossRef CAS.
- A. D. Zdetsis and E. N. Economou, Topological Metal-Insulator Transition in Narrow Graphene Nanoribbons?, Carbon, 2021, 176, 548–557 CrossRef CAS.
- A. D. Zdetsis, 4n + 2 = 6n? A Geometrical Approach to Aromaticity?, J. Phys. Chem. A, 2021, 125, 6064–6074 CrossRef CAS PubMed.
- S. Wang, et al., Giant edge state splitting at atomically precise graphene zigzag edges, Nat. Commun., 2016, 7, 1150, DOI:10.1038/ncomms115077.
- M. Kolmer, et al., Rational synthesis of atomically precise graphene nanoribbons directly on metal oxide surfaces, Science, 2020, 369, 571–575, DOI:10.1126/science.abb8880.
- J. Lawrence, et al., Probing the Magnetism of Topological End States in 5-Armchair Graphene Nanoribbons, ACS Nano, 2020, 14, 4499–4508 CrossRef CAS PubMed.
- Y.-C. Chen, D. G. Oteyza, Z. Pedramrazi, C. Chen, F. R. Fischer and M. F. Crommie, Tuning the Band Gap of Graphene Nanoribbons Synthesized from Molecular Precursors, ACS Nano, 2013, 6123–6128 CrossRef CAS PubMed.
- S. Song, et al., On-surface synthesis of graphene nanostructures with π-magnetism, Chem. Soc. Rev., 2021, 50, 3238–3262 RSC.
- K. Nakada, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Edge state in graphene ribbons: Nanometer size effect and edge shape dependence, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954 CrossRef CAS PubMed.
- H. Zhang, H. Lin and K. Sun, et al., On-surface synthesis of rylene-type graphene nanoribbons, J. Am. Chem. Soc., 2015, 137, 4022–4025 CrossRef CAS PubMed.
- A. Kimouche, M. M. Ervasti, R. Drost, S. Halonen, A. Harju, P. M. Joensuu, J. Sainio and P. Liljeroth, Ultra-narrow metallic armchair graphene nanoribbons, Nat. Commun., 2015, 6(10177), 1–6 Search PubMed.
- L. Yang, C.-H. Park, Y.-W. Son, M. L. Cohen and S. G. Louie, Quasiparticle energies and band gaps in graphene nanoribbons, Phys. Rev. Lett., 2007, 99, 186801 CrossRef PubMed.
- P. Ruffieux, J. Cai, N. C. Plumb, L. Patthey, D. Prezzi, A. Ferretti, E. Molinari, X. Feng, K. Müllen and C. A. Pignedoli, et al., Electronic Structure of Atomically Precise Graphene Nanoribbons, ACS Nano, 2012, 6, 6930–6935 CrossRef CAS PubMed.
- L. Talirz, H. Söde and T. Dumslaff, et al., On-Surface Synthesis and Characterization of 9-Atom Wide Armchair Graphene Nanoribbons, ACS Nano, 2017, 11, 1380–1388 CrossRef CAS PubMed.
- J. Overbeck, et al., A Universal Length-Dependent Vibrational Mode in Graphene Nanoribbons, ACS Nano, 2019, 13, 13083–13091 CrossRef CAS PubMed.
- A. Varykhalov, J. Sánchez-Barriga, A. M. Shikin, C. Biswas, E. Vescovo, A. Rybkin, D. Marchenko and O. Rader, Electronic and Magnetic Properties of Quasifreestanding Graphene on Ni, Phys. Rev. Lett., 2008, 101, 157601 CrossRef CAS PubMed.
- A. D. Zdetsis, The real structure of the Si6 cluster, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 64, 23202 CrossRef.
- Y.-W. Son, M. L. Cohen and S. G. Louie, Energy Gaps in Graphene Nanoribbons, Phys. Rev. Lett., 2006, 97, 216803 CrossRef PubMed.
- M. Huzak, M. S. Deleuze and B. Hajgató, Half-metallicity and spin-contamination of the electronic ground state of graphene nanoribbons and related systems: An impossible compromise?, J. Chem. Phys., 2011, 135, 104704 CrossRef CAS PubMed.
- A. D. Zdetsis, Classics Illustrated: Clar's Sextet and Hückel's (4n + 2) π Electron Rules, J. Phys. Chem. C, 2018, 122, 17526–17536 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods Without Adjustable Parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- R. Dennington; T. Keith and J. Millam, GaussView, version 5.09, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- A. D. Zdetsis and S. Niaz, From Zero to Infinity: Customized Atomistic Calculations for Crystalline Solids —Applications to Graphene and Diamond, Adv. Mater. Lett., 2021, 12(9), 21091659, DOI:10.5185/amlett.2021.091659.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp00650b |
| This journal is © the Owner Societies 2022 |