
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStressing the differences in alizarin and purpurin dyes through UV-visible light absorption and 1H-NMR spectroscopies†
Roger-Charles
Tissier
a,
Baptiste
Rigaud
b,
Pierre
Thureau
 a,
Miquel
Huix-Rotllant
a,
Maguy
Jaber
b and
Nicolas
Ferré
*a
a,
Miquel
Huix-Rotllant
a,
Maguy
Jaber
b and
Nicolas
Ferré
*a
aAix-Marseille Univ, CNRS, Institut Chimie Radicalaire, Marseille, France. E-mail: nicolas.ferre@univ-amu.fr; Tel: +33(0)413945889
bSorbonne Université, CNRS, Laboratoire d'Archéologie Moléculaire et Structurale, Paris, France
First published on 25th July 2022
Abstract
Three anthraquinone-based chromophores (9,10-anthraquinone, alizarin, purpurin) are compared from the point of view of their experimental and computed NMR and UV-visible light absorption spectra. Using a hybrid (explicit/implicit) solvent model, each proton chemical shift can be reproduced with an error of less than 7%, even when such protons are engaged in inter-molecular hydrogen bonds with the solvent or when the analyzed sample contains a significant amount of impurities, for instance, 9,10-anthraquinone in purpurin. All the steady-state UV-visible absorption spectra feature a significant vibrational progression in the first absorption band. The shape of the corresponding computed spectra, including vibronic couplings obtained with the adiabatic Hessian approach and the Franck–Condon and Herzberg–Teller approximation of the transition dipole, are in excellent agreement with the experimental ones. The importance and the nature of the vibronic couplings are different for the three molecules, even if they only differ by the number of hydroxyl groups.
1 Introduction
Anthraquinone-based molecules and, especially, the hydroxy-anthraquinone (HAQ) ones (e.g. alizarin, purpurin, see Fig. 1, but also kermesic and carminic acids, …) are particularly important in our cultural heritage. Either in their pigment1–4 or dye forms,5 these chromophores absorb light in different spectral ranges, depending on different parameters such chemical substitutions, pH and chemical environment (solvent, metal, inorganic matrix, etc.).6 HAQs can be obtained through extraction of natural plants or insects (e.g. carminic acid from cochineal7 or plants (e.g. alizarin from roots8), or, for most of them, are commercially available. Despite purification processes that have been employed by artists throughout history, the naturally extracted dyes – for example, madder dyes extracted from Rubia tinctorum – are present as a mixture of different molecules. These molecules could be highlighted by different techniques such as HPLC followed by NMR or UV-vis absorption spectroscopy.5 Nevertheless, some of them are not sensitive to the techniques employed. For instance, anthraquinone could not be detected on LC-MS experiment – even on reference compound9 – and does not absorb light in the visible region. Therefore “ghost” molecules could be present and affect the physico-chemistry properties, such as the photo-stability of the dye of interest. Considering the variety of molecules involved in the biosynthesis of carminic acid,10 purpurin or alizarin, a closer investigation seems necessary.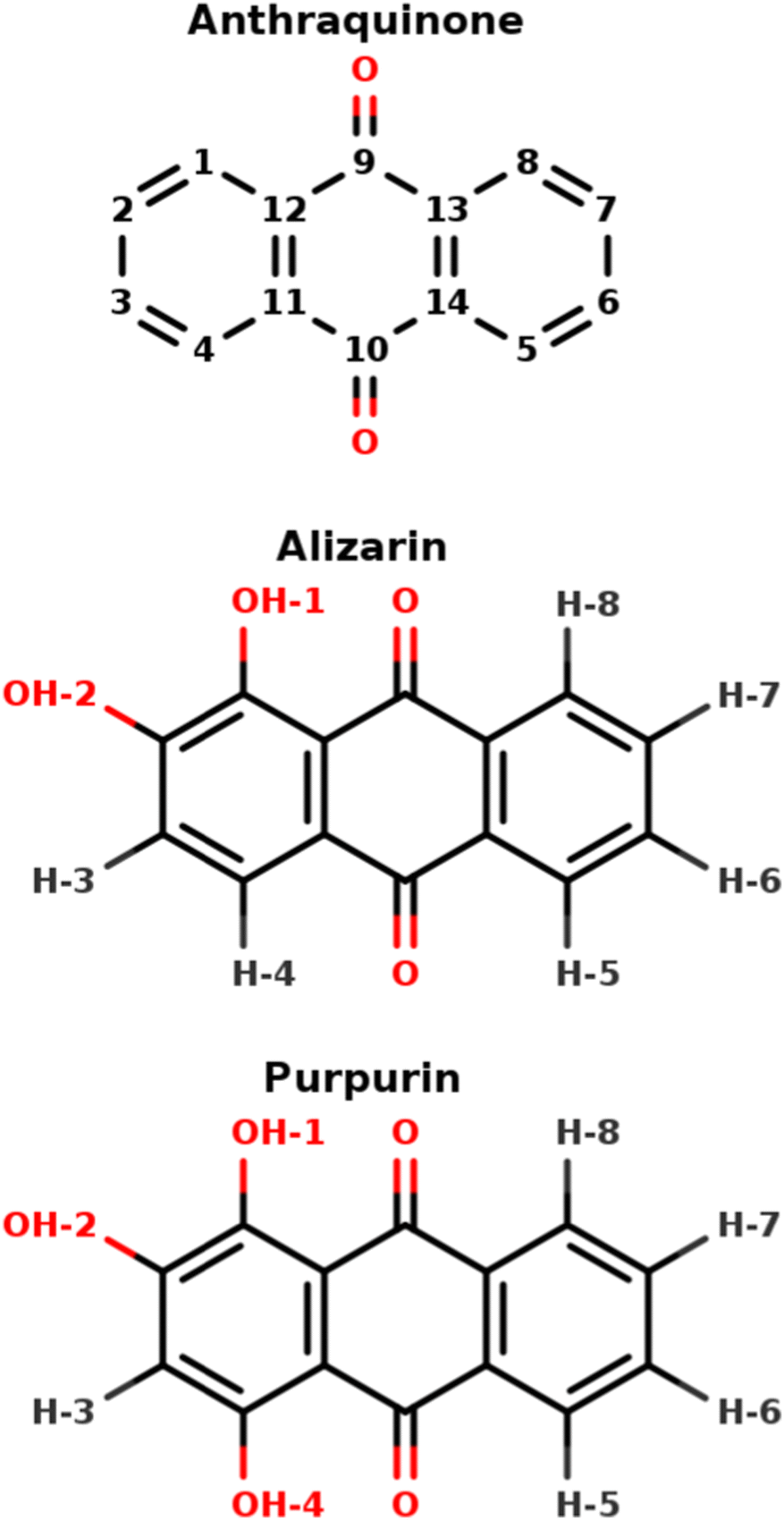 | ||
| Fig. 1 9,10-Anthraquinone, alizarin and purpurin structures, featuring 0, 2 and 3 hydroxyl groups, respectively. The indicated atom labeling is used throughout the present article. | ||
Despite the easy access to these molecules and their broad and various light absorption capabilities, HAQ-based paints suffer from a high sensibility to light (UV and/or visible), leading ultimately to a loss of color, called fading.4 Some of us studied the efficient singlet–triplet photoconversion in similar aromatic ketones, which could be involved in the degradation.11–15 The desire to identify and control the possible fading processes requires a better understanding of HAQ physico-chemical properties, especially when HAQ compounds are found in a complex chemical environment, as is the case in pigments and paintings. This can be achieved using various analytical techniques, among which we herein focus on Nuclear Magnetic Resonance (NMR) spectrometry and UV-visible absorption spectroscopy, as well as those quantum chemical studies aiming at rationalizing the experimental data. The presence of a large number of other chemical species in the studied medium may tremendously complicate the identification of HAQ spectroscopic signature. This would be the case when one can use portable techniques like UV-induced fluorescence16 or unilateral NMR,17 meant for in situ and non-destructive characterization of paintings. From a bottom-up perspective, the present work aims at comparing two commercially available, i.e. not pure, HAQ compounds, alizarin and purpurin. We demonstrate that the confrontation of experimental and computational data enables us to characterize the differences in alizarin and purpurin, but also to quickly identify an impurity present in a purchased sample.
HAQ is any derivative of anthraquinone in which one or several hydrogen atoms are replaced with hydroxyl groups (Fig. 1). From the 1H-NMR perspective, 3 main types of protons exist in HAQ, depending on the nuclei to which they are bound: (i) sp2 carbon participating in an extended conjugated π electronic system, (ii) hydroxy oxygen atoms involved in intra-molecular hydrogen bonds and (iii) hydroxy oxygen atoms involved in inter-molecular hydrogen bonds with the HAQ surroundings. It must be immediately noted that HAQ molecules feature acidic protons and tautomer forms, possibly complicating their chemical spaces, which in turn complicates the analysis and the interpretation of HAQ 1H-NMR spectra.18,19 From previous works in which experimental and theoretical NMR spectra of HAQ systems are confronted, it turns out that Density Functional Theory (DFT) is most of the time accurate enough for reaching a 5 to 10% error with respect to the experimental chemical shifts of protons bound to sp2 carbon atoms.20–22 Regarding protons involved in intra-molecular hydrogen bonds, a study by Siskos has demonstrated that a 0.1 Å variation of the O–H bond length can impact the proton chemical shift by more than 5 ppm.23 Consequently, the selection of the exchange–correlation functional in DFT, as well as the basis set on which the Kohn–Sham orbitals are expanded, is particularly important. The specific case of protons (partially or totally) shared with the solvent requires a much more involved molecular model: DFT needs to be applied to an extended explicit model featuring the HAQ molecule in interaction with the closest solvent molecules and surrounded by an implicit electrostatic solvent model. Such a complex model is required for reaching an almost quantitative agreement with the experimental spectrum.24 In 1H-NMR spectroscopy, each and every proton chemical shift is temperature-dependent. It is known for a long time that possible hydrogen-bonding and/or proton exchange with the solvent is witnessed by a much stronger variation of the proton chemical shift with the temperature, a few tenths of ppm each 10 K.25–27 Variable-temperature 1H-NMR is a valuable tool for further characterizing HAQ systems.28 The expansion (until second order) of the thermal average of the chemical shift constant reveals that the anharmonicity and centrifugation terms are widely responsible for this temperature dependence on the chemical shift in the gas phase.29 In a solvent, both the chemical shifts and the band full width at half maximum are affected.30 In HAQ-related systems involving the cathecol function (quercetin, caffeic acid,…) Vassiliki and coworkers have shown that even in organic solvent such as acetone or DMSO, some hydroxyl groups can be involved in a fast proton exchange with the residual water molecules.27 From the theoretical point of view, such a temperature-based effect has been linked to entropy and can be qualitatively introduced in an extended model built from the solute, the closest solvent molecules to which the solute is hydrogen-bonded and an electrostatic model for the rest of the solvent.31
UV-visible absorption spectroscopy is a complementary analytical method for investigating the HAQ photophysical properties.32 These molecules can potentially feature many n → π* and π → π* transitions resulting in spectrum bands whose positions and relative intensities depend on specific intra- and inter-molecular interactions and the solvent dielectric constant. For instance, the visible region of alizarin in acidic solution presents an absorption maximum at about 425 nm attributed to a π → π* transition.33 Successive studies confirm the existence of such a broad band around this maximum in various experimental conditions.34–39 Refined vibrationally-resolved UV-visible absorption spectra of several HAQ systems solvated in pentane32 and more recently for alizarin in n-heptane40 show a clear vibronic progression, calling for a deeper investigation of its origin. Purpurin vibrationally-resolved UV-visible absorption spectrum was obtained in water–dioxane mixture at acidic pH. It shows three maxima in the visible region at 455 nm, 480 nm and 510 nm.34 Purpurin has also been studied in DMSO by Machatová.38 Three maxima have been identified at 458 nm, 486 nm and 520 nm together with a much less intense absorption band at 600 nm attributed to a proton transfer to the solvent. HAQ UV-visible absorption has also been studied using different theoretical protocols. On one hand, several studies explored the photophysics of alizarin, purpurin and other HAQ molecules using the simplest vertical excitation method at the time-dependent density functional theory (TD-DFT) level.32,36,41 On the other hand, the UV-visible spectrum of alizarin in its protonated form has been obtained within the ensemble framework, taking into account the possible anisotropic solute–solvent interactions.42 Neither tautomerization nor deprotonation was considered in this study. To include vibronic effects, TD-DFT based approaches have been developed43–45 and successfully applied to molecular systems with similar HAQ skeleton, both in the time-independent46 and time-dependent6 frameworks. Jacquemin et al.46 have highlighted the influence of the basis set (converged spectra are achieved using the 6-311+G* basis set) and the stronger impact of the exchange–correlation functional. Range-separated functionals are necessary to improve the calculated spectra, eventually reaching a quantitative agreement with the experimental ones. As already mentioned, HAQ tautomeric forms could exist because of intra-molecular proton transfer between ketone and alcohol moieties. Alizarin tautomers have been studied using several quantum chemical approaches.6,36,41 In particular, Le Person et al.36 suggest the existence of a tautomeric form by comparing experimental and theoretical absorption spectra. Nevertheless, none of the above-mentioned studies succeeded in obtaining a free energy difference between tautomers small enough to account for more than one tautomer at room temperature. More recently, independent theoretical and experimental studies suggest that only a single tautomeric form is present in the alizarin ground state.39,47
The above-mentioned literature is abundant and manifold. Still, it often lacks a unified picture, in terms of protocols used to analyze and compare several HAQ compounds. With this idea in mind, together to compare real-life, i.e. not pure, HAQ solutions, we have combined theoretical quantum chemistry calculations with different experimental techniques, namely steady-state UV-visible absorption spectroscopy and NMR spectroscopies, for the investigation and comparison of two molecules representative of the HAQ family: alizarin and purpurin (Fig. 1), using 9,10-anthraquinone as a reference.
2 Methodologies
2.1 Calculations
UV-visible spectra have been computed using time-dependent density functional theory (TDDFT) using the ωB97xD exchange–correlation functional48 and the 6-31+G(d) basis set.49–51 This level of theory has been extensively tested by different authors in the specific case of vibronic resolved UV-visible spectra of similar compounds.46 NMR calculations have required another level of theory in order to accurately compute chemical shifts.27 Therefore the ωB97xD/6-311+G(2d,p) level of theory together with the Gauge-Independent Atomic Orbital approach52,53 have been used. The chemical shifts have been obtained by subtracting the tetramethyl silane anisotropic shielding tensor from the one of the molecule of interest, both calculated at the same level of theory. The ground state geometry in each system has been optimized using DFT and the same basis sets mentioned above. Potential energy surfaces (PES) of different tautomers and rotamers have been explored. The nature of each stationary point has been confirmed by means of vibrational frequency analysis. The very same workflow has been used in the case of excited state PES using TDDFT. The effect of solvent bulk has been taken into account using the polarizable continuum model (PCM).54 Even if some of our experiments have been performed in acetone (alizarin and purpurin) or in heptane (9,10-anthraquinone), the recorded NMR spectra have shown specific interactions with solvent molecule and/or water traces. Hence we have also considered the limiting case of water as the bulk. In the case of NMR chemical shift calculations, specific solute–solvent interactions have been taken into account. Therefore, a hybrid cluster + continuum approach using both PCM and explicit solvent molecules close to the solute hydroxyl positions was used. The vibronic resolved UV-Visible spectra have been obtained using the integrated protocols available in Gaussian16, based on the time-dependent approach to vibronic spectroscopy, including both the Franck–Condon (FC) and Herzberg–Teller (HT) terms (the zeroth and first order in the transition dipole moment expansion) in combination with the vertical gradient (VG) or the adiabatic Hessian (AH)45 approaches. A half-maximum (HWHM) equal to 235 cm−1 has been used in all spectra. The coupled normal modes have been identified using the time-independent calculation of the very same vibronic couplings.55 All calculations have been performed using the Gaussian16 code,56 while Natural Transition Orbitals (NTOs) have been produced using the Theodore package.572.2 1H-NMR experiments
The 1H NMR spectra were carried out on a Bruker Avance III 500 (11.7T) equipped with a wide band broad probe of 5 mm with a frequency of 500.07 MHz. The experiments concerning alizarin and purpurin were performed using either a 90° pulse (zg) sequences and 30° pulse (zg) sequences, respectively. Spectra were recorded using 512 scans for each sample and with a recycling delay of 2 s. Temperature-dependent NMR experiments have been performed at 298, 283, 273, 263, 253 or 243 K.2.3 UV-vis experiments
UV-visible experiments have been carried out using Ocean Optics Deuterium-Tungsten Halogen and Ocean Optics Flame detector (FLMS00699) with Ocean Optics QP400-1-UV-VIS glass fibers for acquisition from 200 to 850 nm. Data were collected with OceanView 1.5.0 software in absorption measurement mode, and an average of 100 scans of 4.1 ms integration time was performed for each measurement.3 Results
3.1 9,10-Anthraquinone
The 9,10-anthraquinone (Fig. 1 top), a highly symmetrical (![[scr D, script letter D]](https://www.rsc.org/images/entities/char_e523.gif) 2h) molecule in its equilibrium geometry, does not feature any proton which could exchange with the solvent. Accordingly, its spectroscopic properties can be computed straightforwardly. NMR 1H and 13C chemical shifts characterizing the 9,10-anthraquinone molecule have been reported experimentally by Jiang et al.58 We have used DFT calculations to compute these chemical shifts (Table 1). Overall, our results show a good agreement with the reported experimental ones, with an overestimation not larger than 7%. More importantly, the chemical shift differences between non-equivalent nuclei are almost quantitatively reproduced, hence validating the chosen level of theory.
2h) molecule in its equilibrium geometry, does not feature any proton which could exchange with the solvent. Accordingly, its spectroscopic properties can be computed straightforwardly. NMR 1H and 13C chemical shifts characterizing the 9,10-anthraquinone molecule have been reported experimentally by Jiang et al.58 We have used DFT calculations to compute these chemical shifts (Table 1). Overall, our results show a good agreement with the reported experimental ones, with an overestimation not larger than 7%. More importantly, the chemical shift differences between non-equivalent nuclei are almost quantitatively reproduced, hence validating the chosen level of theory.
The 9,10-anthraquinone experimental UV-visible absorption spectrum has been previously published by Khan and Khan32 in n-pentane. Two main bands were reported: the first one in the 275–340 nm region and the second (higher in energy) one in the 210–275 nm region. Herein, we are merely interested in the simulation of the first region which exhibits a small shoulder at 300 nm, as well as 2 clear maxima at 317 nm and 328 nm (Fig. 2). Since such features could be the signature of vibronic effect, we have decided to test different computational methodologies: (i) vertical gradient, (ii) adiabatic Hessian within the Franck–Condon approximation and (iii) within the Franck–Condon–Herzberg–Teller one, denoted VG|FC, AH|FC and AH|FCHT, respectively. Taking into account the 6 first singlet excited states, the resulting spectra are reported in Fig. 2. Overall, only the AH|FCHT-based spectrum satisfactorily fits the experimental one, when a 55 nm red-shift is applied to the computed raw spectrum.
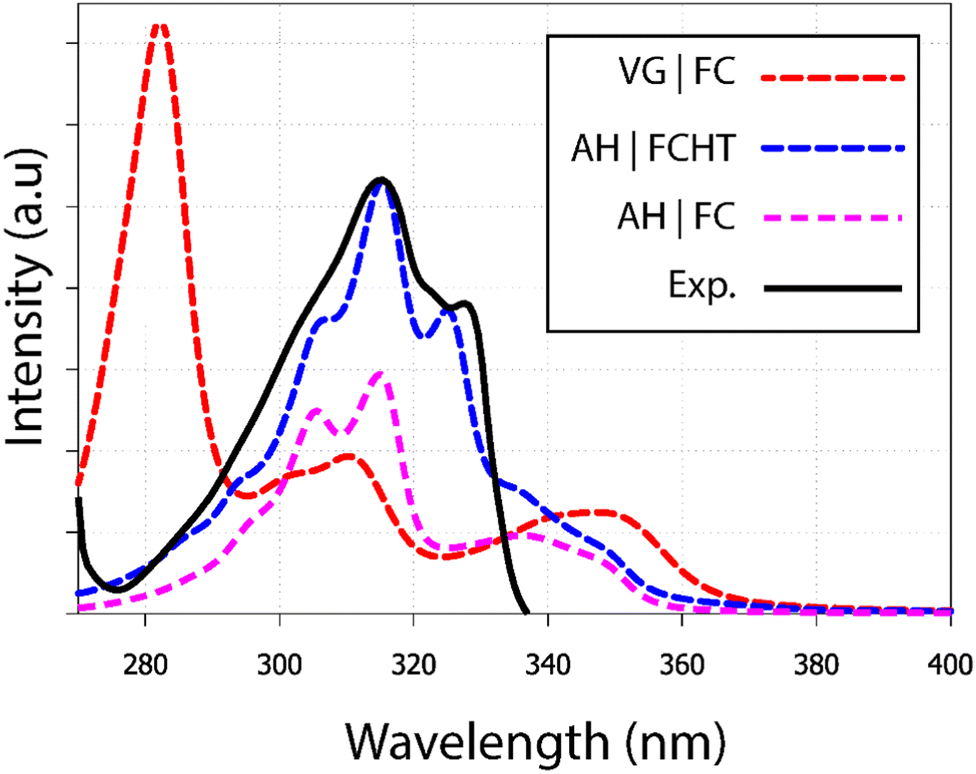 | ||
| Fig. 2 UV-visible absorption spectra of 9,10-anthraquinone in n-pentane. Using different methodologies (i) VG|FC; (ii) AH|FCHT and (iii) AH|FC, each calculated spectrum includes at least the six first electronic transitions (see Fig. S1, ESI†). A shift of 55 nm has been applied on all calculated spectra. The experimental spectrum has been extracted from the publication of Khan.32 | ||
The analysis of the spectrum starts with the vertical transitions in the Franck–Condon region. While the first two lowest transitions are dark n → π* ones, the four next ones can be characterized as π → π* ones (ESI† Table T1). In the spectral region of interest, the vertical excitation calculation indicates that two π → π* electronic transitions, S0 → S4 and S0 → S6, feature a significant oscillator strength, 0.23 and 0.32 respectively, while the two other ones show identically null oscillator strengths. The latter result can be easily understood by looking at the symmetry of each hole and particle orbital in subsequent NTO analysis (Fig. 3 and ESI† Table S1).
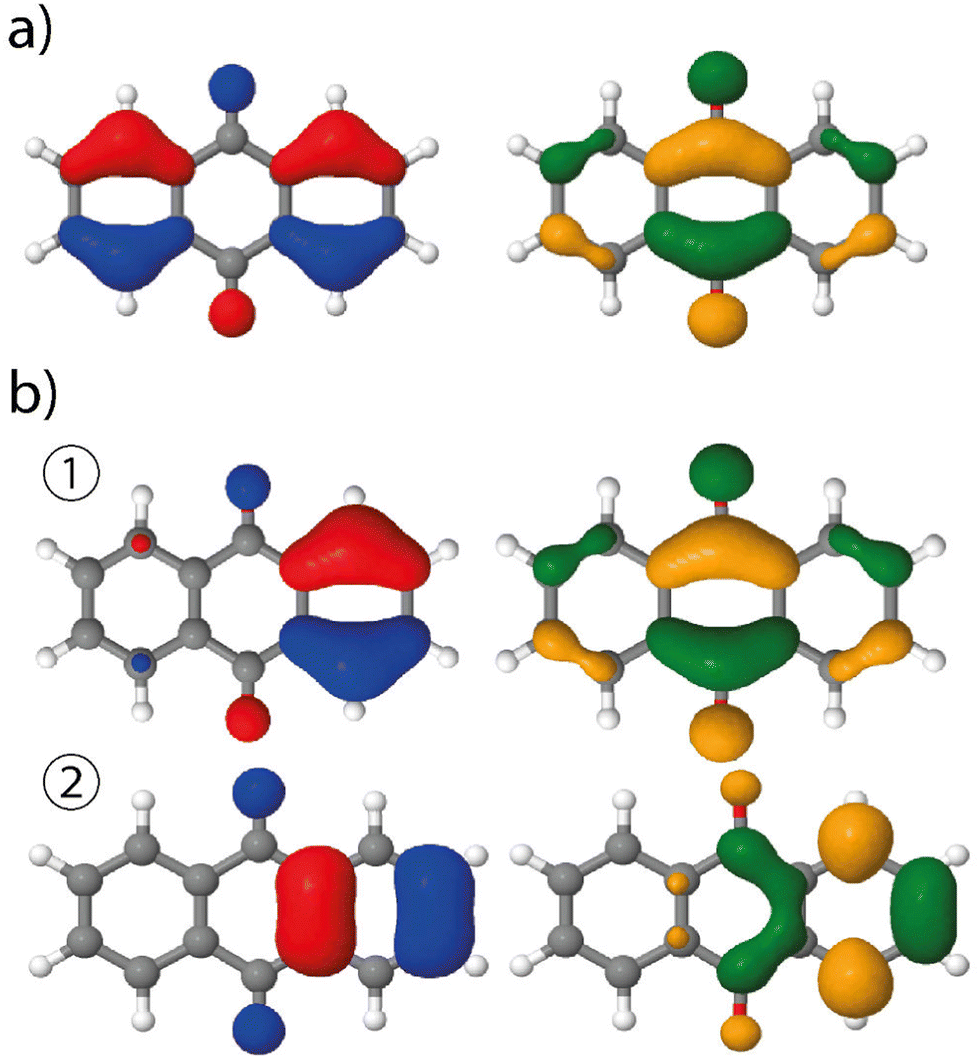 | ||
| Fig. 3 NTOs of 9,10-anthraquinone (left: hole orbital; right: particle) involved in the S0 → S5 transitions, using (a) the ground state minimum energy geometry or (b) a modified geometry based on the displacement along the 58th normal mode in S5. This vertical transition features one NTO pair at 9,10-anthraquinone equilibrium geometry whereas two pairs, ① and ②, are involved at the distorted geometry, the associated coefficients being 0.94 and 0.05, respectively. | ||
Vibrationally-resolved spectra obtained from VG|FC or AH|FC vibronic couplings do not compare well with the experimental spectrum. However, using the AH|FCHT approach, the S0 → S5 transition does now contribute significantly to the spectrum (see ESI† Fig. S1 for a decomposition of the total spectrum into individual contributions). Actually, a single vibronic transition (|0,0〉 → |5,58〉) corresponding to an in-plane motion (Fig. 4), associated with b2u irreducible representation, is responsible for the largest peak. Further evidence of the major role played by this vibration is obtained by manually distorting the 9,10-anthraquinone geometry along the corresponding normal mode. Using a 5% geometry modification, 2 effects are evidenced: (i) a large increase in the oscillator strength characterizing the S0 → S5 vertical transition (f = 0.000 up to f = 0.062) and (ii) the localization of the NTO hole in one of the two phenyl moieties (Fig. 3). The 9,10-anthraquinone minimum-energy structure keeps the 2h symmetry in S5, i.e. identical to the one in S0. However, the 58th normal mode induces a 2h to ![[capital script C]](https://www.rsc.org/images/entities/char_e522.gif) 2v symmetry lowering which impacts the NTOs and their symmetries (in terms of irreducible representation). While NTOs are delocalized over all the molecule in 2h symmetry, they localize in some regions of the molecule in 2v symmetry. Note also that the electronic transition S0 → S5 is characterized by a second pair of NTOs in this distorted geometry. Similar to the 2h symmetry-based analysis given above, we can explain the finite value of the S0 → S5 oscillator strength using 2v symmetry elements (ESI,† Table S1).
2v symmetry lowering which impacts the NTOs and their symmetries (in terms of irreducible representation). While NTOs are delocalized over all the molecule in 2h symmetry, they localize in some regions of the molecule in 2v symmetry. Note also that the electronic transition S0 → S5 is characterized by a second pair of NTOs in this distorted geometry. Similar to the 2h symmetry-based analysis given above, we can explain the finite value of the S0 → S5 oscillator strength using 2v symmetry elements (ESI,† Table S1).
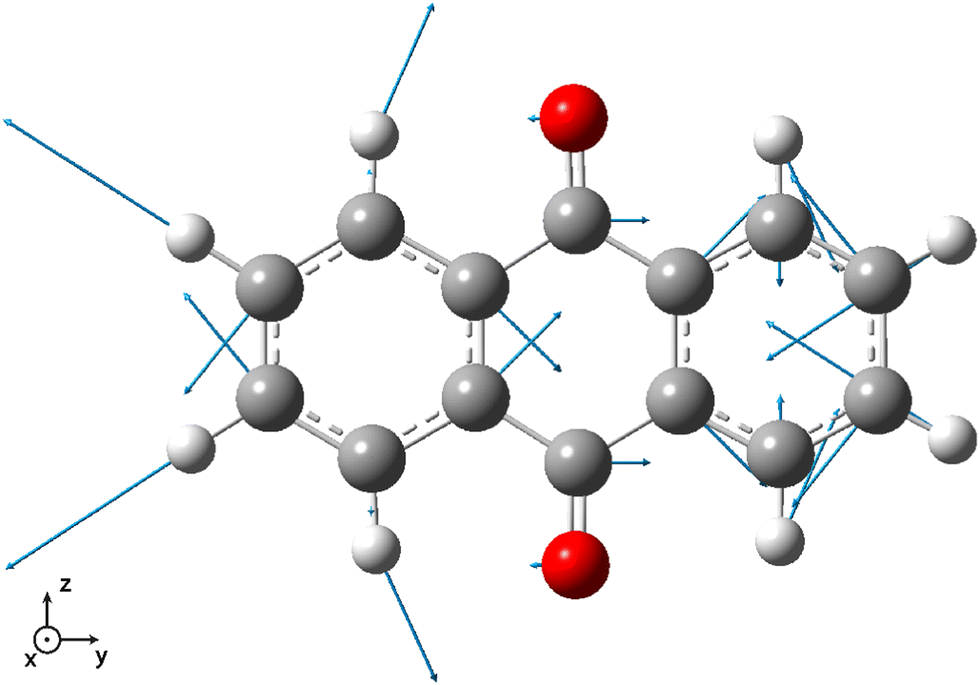 | ||
| Fig. 4 Vector displacement associated to the 58th normal mode (2501.6 cm−1) of 9,10-anthraquinone in its S5 electronic excited state. All the displayed vectors lie in the molecular plane. | ||
Finally, we should note the presence of a shoulder between 340 and 360 nm, already present when the HT correction is not taken into account. As a matter of fact, it is due to the bright S0 → S4 transition. Such a feature actually appears in the experimental spectrum (see Fig. 2 in ref. 32), however with a much lower intensity.
3.2 Alizarin
Compared to 9,10-anthraquinone, alizarin features two extra and vicinal enol groups (Fig. 1 middle). Because of these symmetry-lowering moieties, all the alizarin protons have to be considered as non-equivalent from the NMR point of view (Fig. 5 inset). As a matter of fact, the alizarin NMR spectrum recorded in acetone-d6 features 8 different peaks (Fig. 5). Several regions can be distinguished in these spectra: (i) 7 to 8.5 ppm assigned to hydrogen atoms bound to carbon atoms, (ii) 9.5 to 10.5 ppm attributed to the hydroxyl group involved in inter-molecular hydrogen bonds and (iii) 12.75 to 13 ppm corresponding to the hydroxyl group involved in intra-molecular hydrogen bonds. The attribution of the different protons peaks faces several potential issues that could be solved thanks to quantum chemical computations. First, some of the enol hydrogen atoms can participate in hydrogen bonds, either intermolecular (H-1) or intramolecular (H-2), whose positions can change if alizarin can exist in different tautomer forms resulting from intramolecular proton transfer. Computed free energy differences (ESI,† Fig. S2) strongly suggest that only the most stable tautomer form is present in solution at the temperatures considered in our study, in agreement with a similar finding.41 When the calculation is repeated in acetone instead of water, the free energy differences do not significantly change. Accordingly, we have considered only the most stable tautomer in the following.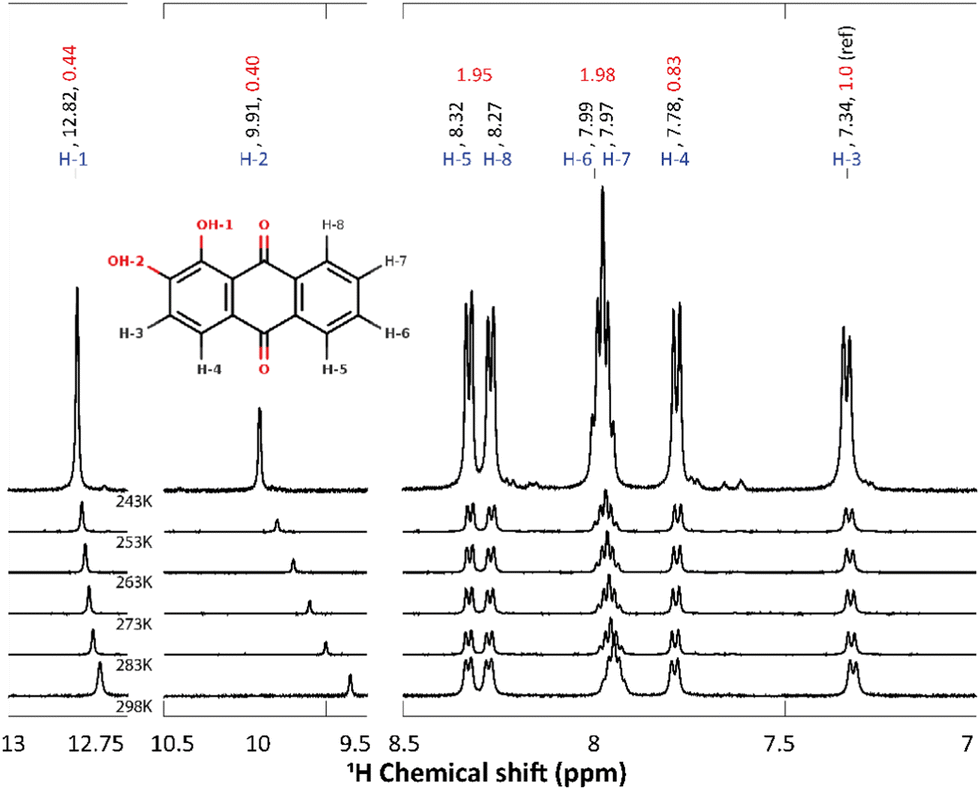 | ||
| Fig. 5 1H chemical shift of alizarin in acetone-d6 (50/50 ratio H2 O/DHO) at different temperatures (243, 253, 263, 273, 283 and 298 K). Atom labeling, chemical shifts values (in ppm) and integrated intensities are written in blue, black and red, respectively. The inset represents alizarin and its atom labeling. H-1 is involved in a strong intra-molecular H-bond, whereas H-2 is interacting with the alizarin surrounding. | ||
Second, OH proton exchange with the solvent may also be possible, even in acetone-d6 when some water traces are present. Temperature-dependent NMR spectra are usually helpful in identifying which protons can strongly interact with their surroundings. Accordingly, we have repeated 1H NMR experiments at different temperatures (298, 283, 273, 263, 253 and 243 K, Fig. 5) in order to: (i) highlight the bonding mode of each OH proton, i.e. intra- or inter-molecular; (ii) identify possible chemical exchange events; (iii) allow a better comparison with the computations (performed at 0 K). The most striking result is the strong temperature dependence of the H-2 signal (−0.01 ppm K−1). This behavior can simply be explained by the temperature-induced solvent reorganization around H-2, which is the most accessible proton to the solvent.27
Turning back to the measured NMR spectrum at 243 K (Fig. 5, the integrated peak areas corresponding to OH protons (i.e. located between 9 and 14 ppm) are 2 times smaller than the CH ones (region 7–9 ppm). This result can be explained assuming that alizarin co-exists in 2 different forms: (i) the fully protonated form exhibits 8 proton peaks, (ii) the deuterated form resulting from proton exchange with the solvent deuterium can lack one or two protons, i.e. the enol ones. This explanation has been confirmed by repeating the NMR experiment in non-deuterated acetone.
The experimental and the calculated 1H chemical shifts are summarized in Table 2. Overall, for all alizarin protons, we find chemical shift errors of less than 7%, similar to the ones obtained in the case of 9,10-anthraquinone (Table 1). Note that a cluster approach (in addition of a continuum representation of the solvent) has been employed to improve the computed chemical shift of the proton involved in the inter-molecular hydrogen-bond with the solvent. Therefore, an explicit acetone molecule has been placed close to H-2. Compared to the experimental H-2 chemical shift (9.91 ppm), this procedure demonstrated a noticeable improvement (10.44 ppm) with respect to the unrealistic value (6.08 ppm) obtained without this solvent molecule.
The experimental UV-visible absorption spectra of alizarin in polar solvents (acetone, DMSO etc.)40 do not show any vibrational structure. However, the same authors report a vibrational progression for alizarin in n-heptane (Fig. 6 as the black full line). Two maxima at 405 and 424 nm are evidenced (not visible in other solvents, see the inset of Fig. 6), as well as shoulders at 474, 450 and 388 nm.
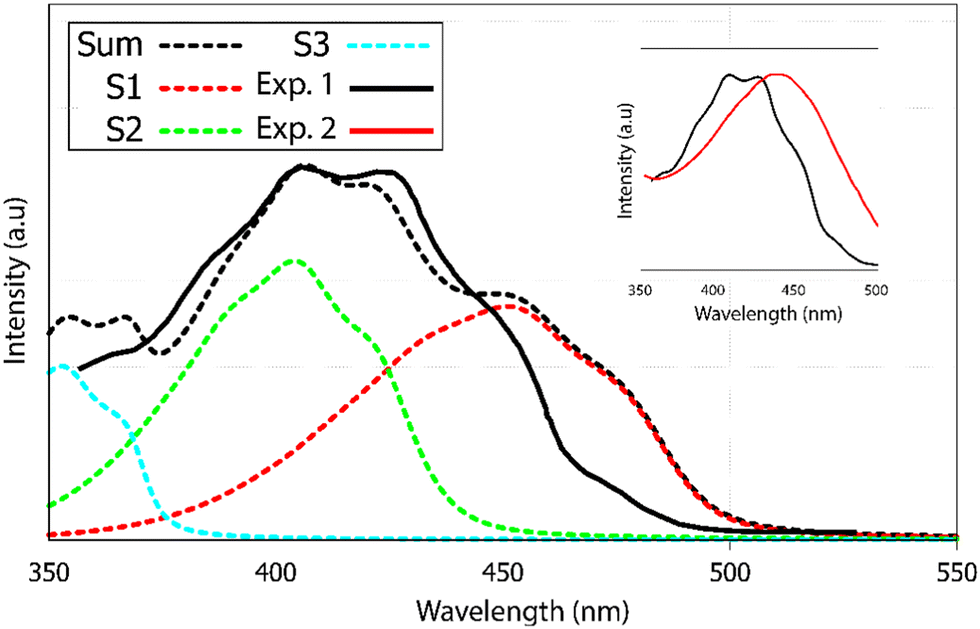 | ||
| Fig. 6 UV-visible absorption spectrum of alizarin. Experimental results are denoted Exp. 1 and Exp. 2, in heptane (black full line) and DMSO (red full line), respectively. Calculated (black dotted line) absorption spectra of alizarin in water. The calculated spectra is the sum of the first three electronic transitions to S1, S2 and S3 in red, green and blue dashed lines. The calculated spectra have been red-shifted by 55 nm. Two experimental spectra are extracted from Jen et al.40 and included in the inset. Note the blue-shift and the appearance of the vibrational progression when going from DMSO to heptane. | ||
Our absorption spectrum computation is performed using an implicit solvent model. Because of convergence issues, we could not use heptane or DMSO as an implicit solvent. Rather, we had to go for a very polar solvent like water in order to ensure correct computational behavior. Therefore the direct comparison between computed and experimental spectra has to be considered as qualitative. According to the TDDFT vertical excitation energies, the first 2 transitions, S0 → S1 and S0 → S2, exhibit similar wavelengths, 363 and 356 nm, respectively. While the first transition has a π → π* character with an oscillator strength equal to 0.210, the second transition is a forbidden one (n → π* with a null oscillator strength), see ESI† Fig. S4. Transitions to higher excited states are too energetic to contribute to the first absorption band observed experimentally.
The AH|FCHT scheme can be applied when the displacement of the nuclear positions between the ground and excited state is small enough, otherwise the expansion of the electronic dipole moment with respect to normal modes on an exited state will be incorrectly projected, leading to an irrelevant spectrum.59 Actually, alizarin features an intra-molecular proton transfer (ESIPT) in the S1 excited state when a triple-ζ basis set is used.46 Sticking to a double-ζ basis set avoids any ESIPT and allows us to apply the AH|FCHT approach. The vibronic structure of the first transition, S0 → S1, (Fig. 6, red dashed line) is mainly dominated by in-plane motions involving rocking of C–OH bonds with respect to the aromatic ring. While the S0 → S2 vertical transition is forbidden, applying the AH|FCHT scheme reveals several active vibronic couplings (|0,0〉 → |2,16〉, |0,0〉 → |2,21〉, |0,0〉 → |2,26〉, |0,0〉 → |2,33〉, |0,0〉 → |2,35〉 and |0,0〉 → |2,36〉 associated to A” irreducible representation of s symmetry, represented in ESI† Fig. S3). These couplings contribute to increase the intensity of this transition, eventually turning to be as intense as the S0 → S1 one (Fig. 6, green dashed line). All the above-mentioned normal modes involve out-of-plane motions, breaking the alizarin s symmetry. The analysis of the alizarin S0 → S2 natural transition orbitals is particularly insightful (Fig. 7). Comparing NTOs for the S0 optimized geometry and for a displaced one (along the 33rd normal mode, i.e. the one corresponding to the largest vibronic coupling), it is apparent that the n-like hole orbital gains some π character when alizarin motion is taken into account, explaining why the S0 → S2 transition is no longer dark. The consideration of the other coupling modes confirms this analysis.
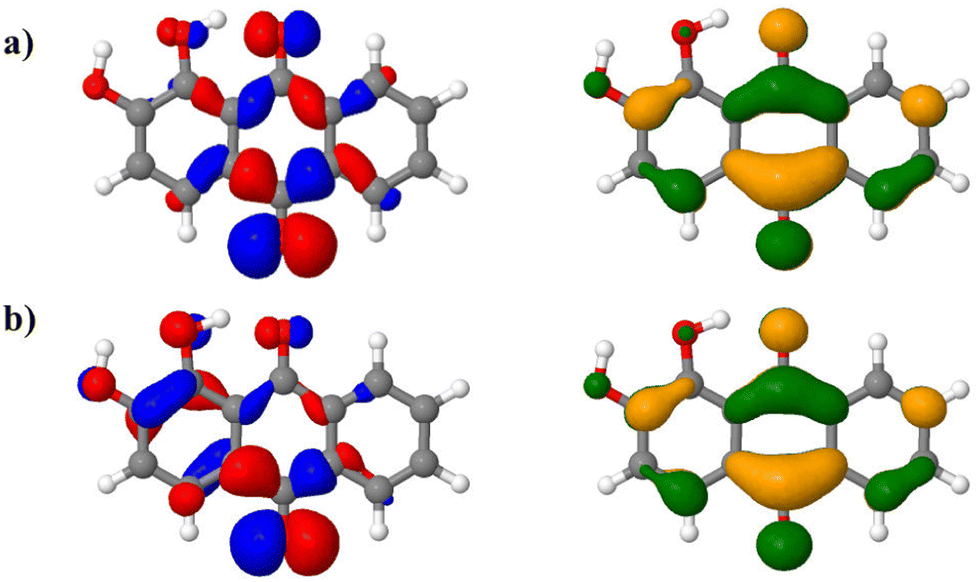 | ||
| Fig. 7 S0 → S2 natural transition orbitals (NTOs, left: hole, right: particle) of alizarin in (a) in its ground state optimized geometry and (b) in a displaced geometry using 15% of the displacement corresponding to the 33rd normal mode. | ||
The final alizarin absorption spectrum is presented in Fig. 6 as black dashed line. The calculated line shape matches well with the reported experimental one,40 highlighting again the importance of using first order terms in the electronic transition dipole moment. Note that the computed-to-experimental 55 nm shift is consistent with the one obtained on similar molecular systems by Jacquemin and coworkers.46 This shift is obtained by matching the experimental and computed maximum intensity wavelengths. However, this shift is probably too large for S0 → S1, as it appears as a too pronounced shoulder in the computed spectrum. Improved agreement could have been reached if explicit solvation would have been included in our models. However, vibronic couplings are much more difficult to converge when such specific solute–solvent interactions are considered.
3.3 Purpurin
Compared to alizarin, purpurin features an extra enol group, in para position with respect to alizarin intramolecular hydrogen-bonded enol group (Fig. 1 bottom). Therefore, purpurin can also feature several tautomeric forms as well as several intra- or inter-molecular hydrogen bonds. According to free energy calculations of each tautomer (ESI† Fig. S5 and Table S4), in the gas phase and the condensed phase using a continuum model for the solvent, only one stable form prevails at room temperature, represented in Fig. 8. Experimental NMR 1H spectrum of purpurin in acetone-d6 is reported in Fig. 8. Purpurin, like alizarin and 9,10-anthraquinone, features different proton families. Firstly, H-1, H-2 and H-4 present a much larger de-shielding (chemical shifts comprised between 10.0 and 13.5 ppm) compared to other protons. We can easily attribute them to the three enol moieties. Because only one of them features a temperature-dependent chemical shift, we conclude that there is only one proton (H-2) engaged in an intermolecular hydrogen bond with the solvent. Second, H-5 to H-8 are more deshielded than H-3 and show chemical shifts typical of the benzene molecule (8.5 to 7.75 ppm). Finally, H-3 features a chemical shift (comprised between 6.6 and 6.7 ppm, depending on the temperature) more shielded (compared to benzene), probably due to the enol groups bound to the same aromatic ring and perturbing the electron density current. These experimental observations are supported by our quantum chemistry calculations. Considering a cluster composed of the neutral form of purpurin represented in Fig. 8 with an explicit acetone molecule interacting with H-2, we obtain the calculated chemical shifts reported in Table 3. All the 3 families can be clearly identified and their chemical shift differences are correctly reproduced, with an excellent 4% average error. While the low-temperature NMR peak attribution is successful, their relative intensities are troublesome. Using H-3 as the reference, the accumulated intensities of the H-5/H-8 or H-6/H-7 pairs appear too large: about 3.5 instead of 2. Increasing the temperature up to 298 K, the H-5/H-8 peak is split into two different signals. However, this is not the case for the H-6/H-7 pair.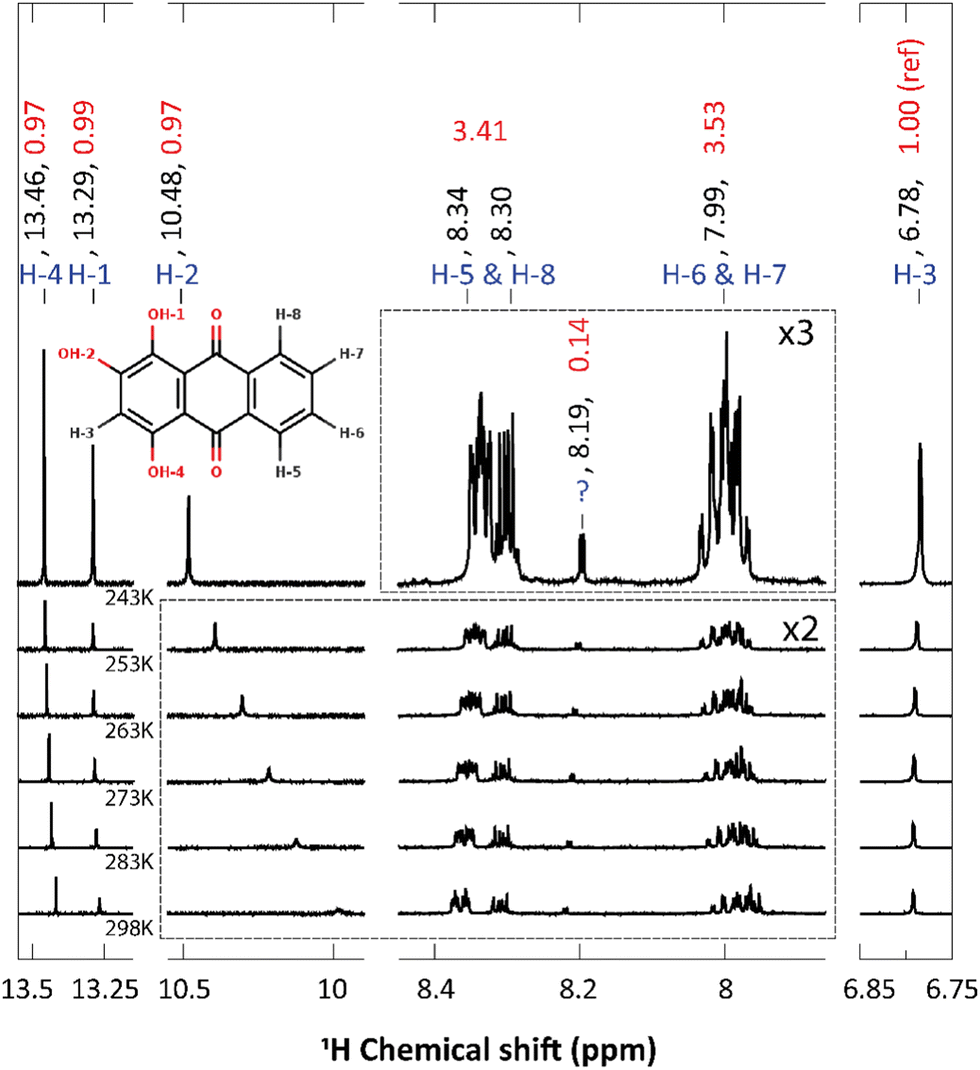 | ||
| Fig. 8 1H chemical shift of purpurin in acetone-d6 at different temperatures (243 K, 253, 263, 273, 283 and 298 K). Atom labeling, chemical shifts values (in ppm) and integrated intensities are written in blue, black and red, respectively. The inset represents purpurin and its atom labeling. H-1 and H-4 are involved in strong intra-molecular H-bonds, whereas H-2 is interacting with the solvent. The peak marked with '?' at 8.19 ppm is attributed to an impurity. Some parts of the spectra, delimited with dashed lines, have been enlarged with the mentioned zoom ratio. | ||
| Proton | Exp. | Calc. | Error (%) |
|---|---|---|---|
| H-1 | 13.29 | 13.18 | 1 |
| H-2 | 10.48 | 11.66 | 10 |
| H-3 | 6.78 | 6.97 | 3 |
| H-4 | 13.46 | 13.16 | 2 |
| H-5 | 8.34 | 8.92 | 7 |
| H-6 | 7.99 | 8.36 | 4 |
| H-7 | 7.99 | 8.30 | 4 |
| H-8 | 8.3 | 8.86 | 6 |
We have recorded the steady-state UV-visible absorption spectrum of purpurin in acetone-d6. Its visible contribution (first band) is reported in Fig. 9 as black full line. This spectrum shows a clear vibrational structure, a maximum intensity at 482 nm and two clear shoulders at 516 and 456 nm. According to our TDDFT calculations, the first 2 raw vertical transitions, S0 → S1 and S0 → S2 are 402 and 339 nm, respectively. While the first transition has a π → π* character with an oscillator strength equal to 0.27, the second transition is a forbidden one (n → π* with a null oscillator strength). The corresponding NTOs are presented in ESI† Fig. S7. Transitions to higher excited states are too energetic to contribute to the first absorption band observed experimentally.
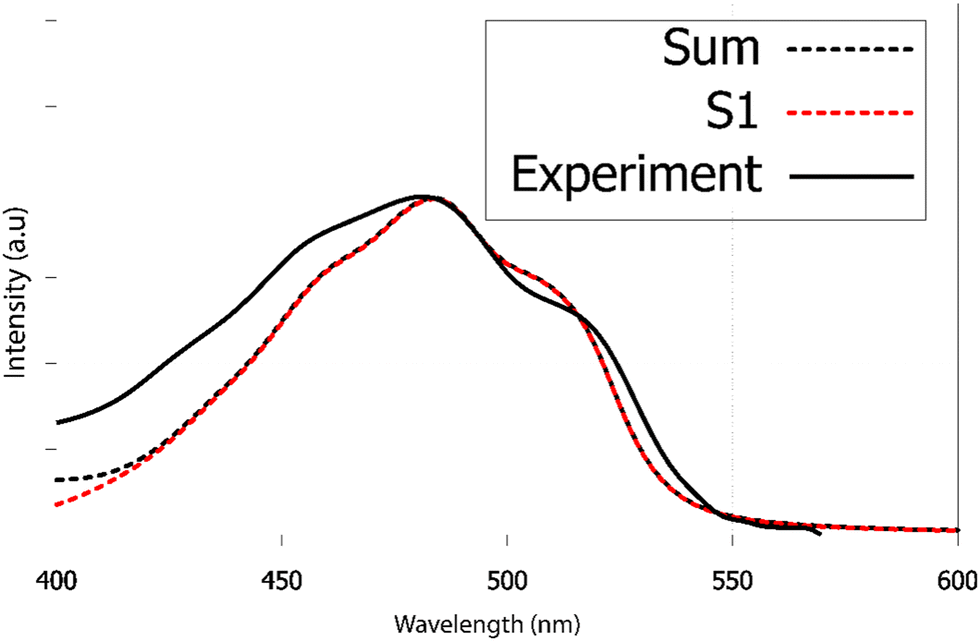 | ||
| Fig. 9 Experimental and calculated electronic absorption spectra of purpurin in acetone-d6. Excited states above S1 only contribute at high excitation wavelengths (shorter than 420 nm). The TDDFT spectrum has been red-shifted by 40 nm to match the experimental maximum wavelength. | ||
As already found in the case of alizarin, the AH|FCHT vibronic structure of the S0 → S1 first transition is dominated by in-plane motions involving rocking of C–OH bonds with respect to the aromatic ring scissoring (ESI,† Fig. S6). Considering the S0 → S2 transition, the HT term does not bring up sizeable couplings, hence this n → π* transition remains forbidden, independently of the solvation model (implicit with different solvents, water or acetone, or with a cluster approach). When red-shifted by 40 nm, the calculated vibrationally resolved absorption spectrum depicted in Fig. 9 as red dashed line is in excellent agreement with the experimental one, confirming that only S0 → S1 contributes to the purpurin first absorption band.
4 Discussions
Three molecules of the anthraquinone family, namely 9,10-anthraquinone, alizarin and purpurin have been studied by means of their respective 1H NMR and UV-visible absorption spectra. As long as a suitable model, including explicit solvent molecules and/or a continuum description of the solvent bulk is included, a good agreement between experiment and computation has been achieved. Since these three molecules only differ by the presence of 0, 2 and 3 hydroxyl moieties, their comparison is of interest.4.1 UV-visible absorption spectra
While the three UV-vis absorption spectra share the same general shape, it turns out that adding electron donating hydroxyl groups is shifting the spectrum from near UV (9,10-anthraquinone) to visible (alizarin: violet, purpurin: blue). Moreover, their first absorption band can be characterized by an intense π → π* vertical transition and several n → π* ones, as demonstrated by their respective NTOs (reported in ESI†). However, the calculation of vibronic couplings reveals several important differences we need to discuss further.In the Franck–Condon approximation,60 two main physical effects can be responsible for a vertical forbidden transition: (i) initial and final vibrational states do not overlap, (ii) the transition dipole moment is null because of symmetry reasons: the direct product of the initial state, final state, position operator irreducible representations does not belong to the fully symmetric one. For instance, 9,10-anthraquinone S0 → S5 vertical transition is forbidden because of reason (ii). However, the vibronic coupling with a single normal mode, whose motion lowers the molecule symmetry from 2h to 2v, is enough to trigger the transition, which eventually becomes the most important one in 9,10-anthraquinone vibrationally-resolved first absorption band. On the other hand, the energetic proximity of 4 excited states (S3 to S6) sharing the same π → π* character may suggest61,62 strong non-adiabatic coupling effects inducing intensity borrowing mechanism, in which the dark states S3, S5 could borrow intensity of the bright ones, S4, S6, which become less intense. Such an effect cannot be captured by the Herzberg–Teller perturbative treatment of the transition dipole moment. To analyze this mechanism, we have computed approximate interstate couplings from one dimensional potential energy surfaces cuts along the ground-state normal modes that couple the dark and bright states, and we performed quantum dynamics simulations15,63 for the modes that induce such coupling (see further details in ESI†). It turns out that such linear coupling effects are tiny and, thus, the intensity borrowing mechanism can be excluded. The vibrationally-induced large modifications of the 9,10-anthraquinone absorption spectrum can be therefore attributed to the dynamical lowering of the 9,10-anthraquinone molecular symmetry, resulting in a S0 → S5 dipole-allowed transition, hence highlighting the importance of including the first order term in the electronic transition dipole moment.
At variance with 9,10-anthraquinone, alizarin first absorption band comprises two vertical transitions, energetically very close, among which one is of n → π* character, with a zero oscillator strength. Vibronic couplings are responsible for π/σ mixing in alizarin electronic structure, as evidenced in Fig. 7, resulting in a significant increase in its contribution. Eventually, the alizarin first absorption band is made of two contributions with similar weights. Because of the near-degeneracy between the S1 and S2 states, intensity borrowing might also occur in this case, via the a′′ molecular motions that would trigger interstate coupling. Energy profile cuts along the relevant normal modes (ESI†) show that such couplings are also negligible in this case, thus excluding the intensity borrowing mechanism. While purpurin only contains one more –OH group than alizarin, the origin of its visible light absorption is significantly simpler: a single π → π* contributes. Moreover, its vibronic couplings are solely responsible for the spectrum vibrational progression. As a matter of fact, the extra intramolecular hydrogen bond brought by the third hydroxyl group prevents any structural change which could help π/σ mixing as it was found in the case of alizarin.
To conclude this discussion regarding the modeling of UV-visible light absorption, vibronic couplings are mandatory if ones want to reproduce the spectrum of 9,10-anthraquinone, alizarin, purpurin, and probably other molecules sharing the same molecular architecture. Their spectrum vibrational progression is due to triggered forbidden transitions (9,10-anthraquinone: increased transition dipole; alizarin: π/σ mixing) and/or regular vibronic couplings of π → π* transitions (purpurin).
4.2 NMR spectra
Regarding the 1H NMR spectra, we generally obtain a good agreement between the experimental and computed proton chemical shifts when the model takes into account explicit interactions between the solute and the solvent molecules. However, it turns out that the purpurin spectrum (Fig. 8), when compared to 9,10-anthraquinone and alizarin ones, present unexplained features. Equivalent H-5 and H-8 peaks, as well as equivalent H-6 and H-7 peaks, integrate to a value that is about three times the one obtained for H-3, whereas only two times is expected. This issue is partially solved when increasing the temperature (Fig. 10): the H-5/H-8 signal splits into two peaks which can be integrated separately. At room temperature, we can observe one signal centered at 8.31 ppm and another one at 8.365 ppm with intensities equal to 1 and 2 respectively. Unfortunately, this is not the case for the H-6/H-7 pair. Nevertheless, the comparison of the purpurin chemical shifts (Table 3) with the alizarin spectrum (Fig. 5 and Table 2) suggests that signal at 8.365 ppm can be attributed to H-5, the one at 8.31 ppm to H-8 and the one between 7.95 and 8.01 ppm to the pair H-6/H-7. Given their relative intensities (Fig. 10), two extra protons need to be introduced: one at 8.365 ppm and one at 7.98 ppm. The identification of the origin of these two protons is not an easy task.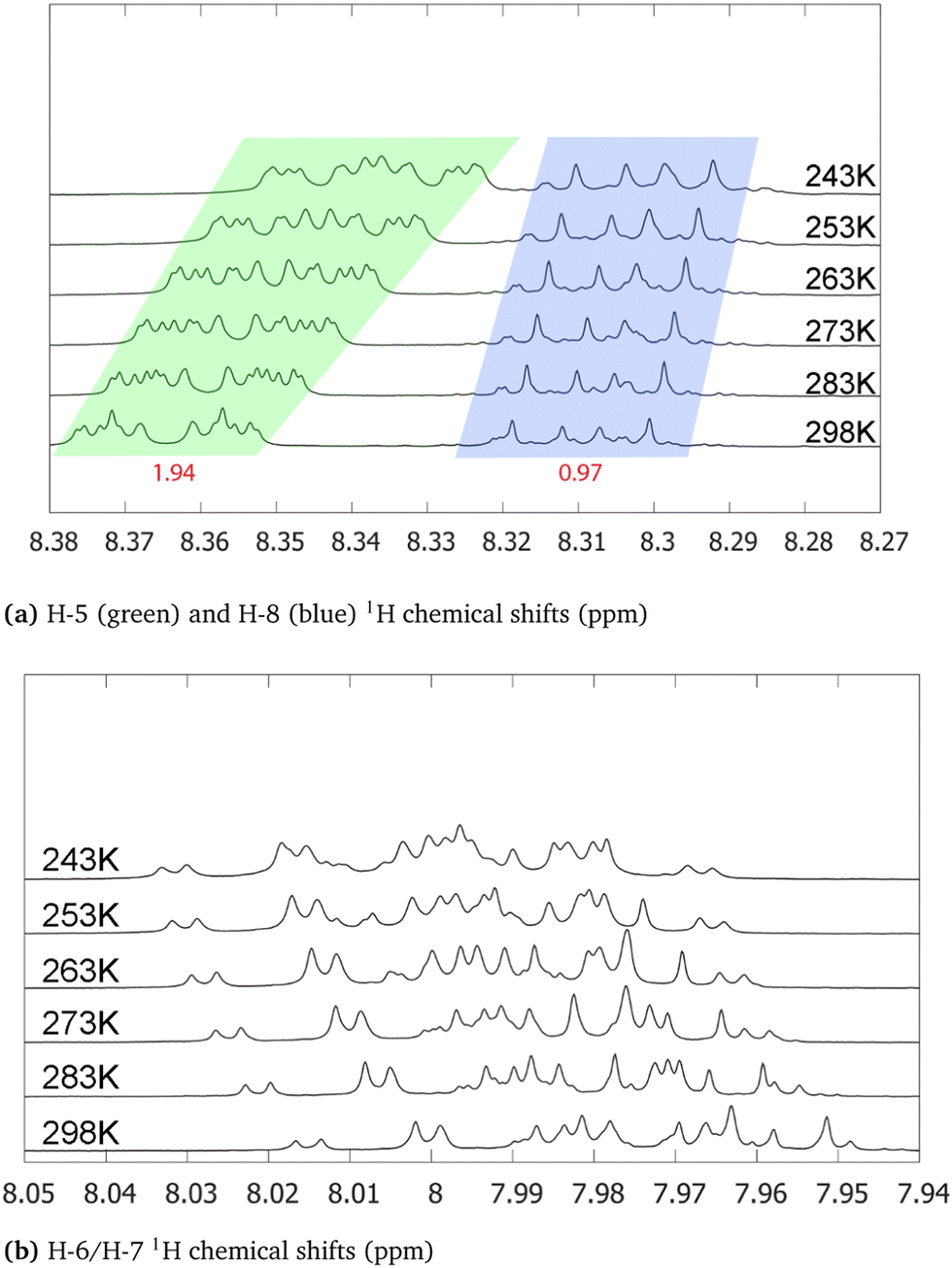 | ||
| Fig. 10 H-5/H-8 (a) and H-6/H-7 (b) 1H chemical shifts for purpurin in acetone-d6 at various temperatures (243 (top), 253, 263, 273, 283 and 298 K(bottom)). Integrated intensities (H-3 being the reference) are written in red. The individual integration of H-5 and H-8 areas can been performed in the spectrum at 298 K. However, their signal overlap is too important at 243 K to integrate their areas separately. | ||
Our first supposition is that the main compound in our sample is not purpurin or corresponds to a degraded form of purpurin which is known to exhibit a weak photoresistance, for instance.64,65 Repeating the NMR analysis on fresh samples results in identical spectra. Applying electrospray ionization mass spectrometry (in negative mode) to purpurin in acetone, the presence of purpurin was confirmed at m/z = 255, and supplemented by two additional signals at m/z = 227 and m/z = 183, assigned to purpurin–CO and purpurin–CO–CO2 (ESI,† Fig. S15).
Hence, we next consider the possibility of the presence of molecules in the sample featuring only two proton chemical shifts at about 8 and 8.35 ppm. The most probable candidate turns out to be 9,10-anthraquinone. As a matter of fact, its proton chemical shifts are 7.81 and 8.32 ppm in DMSO-d6 (Table 1). Since the purpurin sample has been solvated in acetone-d6 in our experiment, the small differences in the reported chemical shifts can be attributed to the different solvents. Furthermore, the 13C NMR spectrum of purpurin in acetone-d6 exhibits additional signals (for a total of 18) whereas 14 peaks are expected (ESI,† Fig. S16). These 4 extra peaks (one at 183.1 ppm, two toward 130 ppm and one at 127 ppm) support our hypothesis of the presence of 9,10-anthraquinone. Indeed, the chemical shifts are quite in agreement with the ones reported by Jiang et al. in DMSO–d6 (Table 1). Unfortunately, 9,10-anthraquinone is not soluble in acetone-d6, preventing us to record the corresponding NMR spectrum. Finally, notice that if 9,10-anthraquinone features two proton NMR signals, each of them corresponds to 4 equivalent protons. Accordingly, the presence of few amounts of 9,10-anthraquinone in the purpurin sample is expected to significantly contribute to the overall NMR spectrum.
5 Conclusions
In the present work, three anthraquinone-based chromophores have been compared: 9,10-anthraquinone, alizarin and purpurin. While they only differ by the number of hydroxyl substituents (respectively 0, 2 and 3), their corresponding 1H NMR and UV-visible absorption spectra show interesting differences we have investigated.From the NMR point of view, the comparison of spectra obtained at different temperatures with theoretical spectra computed at 0 K was enough to unambiguously attribute each signal, even in the case of purpurin whose sample presented non-negligible traces of 9,10-anthraquinone. Because of the presence of intermolecular hydrogen bonds between alizarin or purpurin with their solvents, the corresponding theoretical models need to include a proper description of the solvent effects using both short-range microsolvation and long-range electrostatic couplings with a polarizable continuum.
In the case of the UV-visible absorption spectra, all the three molecules exhibit a first absorption band featuring an important vibrational progression. The latter property has been successfully reproduced with models including vibronic couplings using the adiabatic Hessian approach and the Franck–Condon + Herzberg–Teller approximation of the transition dipole moment. Interestingly, these vibronic couplings impact each molecule differently. In 9,10-anthraquinone, the electronic coupling with symmetry lowering in-plane nuclear motions activates a π → π* transition, which was found dark when the corresponding vertical transition is considered only. In alizarin, vibronic couplings with out-of-plane nuclear motions induce π/σ mixing which is large enough to trigger the corresponding n → π* transition. In purpurin, vibronic couplings are much simpler: they shape the band of the single π → π* transition.
Finally, even if the three systems studied in this work look apparently chemically similar, we have found that their NMR and UV-visible absorption signatures originate from different microscopic behaviors. This exciting finding prompts us to expand this complementary experimental and computational approach to more involved HAQ systems like carminic acid.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Centre de Calcul Intensif dAix-Marseille is acknowledged for granting access to its high performance computing resources.Notes and references
- D. Guillermin, T. Debroise, P. Trigueiro, L. de Viguerie, B. Rigaud, F. Morlet-Savary, S. Balme, J.-M. Janot, F. Tielens, L. Michot, J. Lalevee, P. Walter and M. Jaber, Dyes Pigm., 2019, 160, 971–982 CrossRef CAS.
- F. Fournier, L. de Viguerie, S. Balme, J.-M. Janot, P. Walter and M. Jaber, Appl. Clay Sci., 2016, 130, 12–17 CrossRef CAS.
- P. Trigueiro, F. A. Pereira, D. Guillermin, B. Rigaud, S. Balme, J.-M. Janot, I. M. dos Santos, M. G. Fonseca, P. Walter and M. Jaber, Dyes Pigm., 2018, 159, 384–394 CrossRef CAS.
- C. Ahn and S. K. Obendorf, Text. Res. J., 2004, 74, 949–954 CrossRef CAS.
- Z. Amar, H. Gottlieb, L. Varshavsky and D. Iluz, BioScience, 2005, 55, 1080 CrossRef.
- L. Carta, M. Biczysko, J. Bloino, D. Licari and V. Barone, Phys. Chem. Chem. Phys., 2013, 16, 2897–2911 RSC.
- M. Borges, R. Tejera, L. Díaz, P. Esparza and E. Ibáñez, Food Chem., 2012, 132, 1855–1860 CrossRef CAS.
- D. De Santis and M. Moresi, Ind. Crop Prod., 2007, 26, 151–162 CrossRef CAS.
- G. C. H. Derksen, H. A. G. Niederländer and T. A. van Beek, J. Chromatogr. A, 2002, 978, 119–127 CrossRef CAS PubMed.
- S. Rasmussen, K. Kongstad, P. Khorsand-Jamal, R. Kannangara, M. Nafisi, A. Van Dam, M. Bennedsen, B. Madsen, F. Okkels, C. Gotfredsen, D. Staerk, U. Thrane, U. Mortensen, T. Larsen and R. Frandsen, Insect Biochem. Mol. Biol., 2018, 96, 51–61 CrossRef CAS PubMed.
- M. Huix-Rotllant, D. Siri and N. Ferré, Phys. Chem. Chem. Phys., 2013, 15, 19293–19300 RSC.
- M. Huix-Rotllant and N. Ferré, J. Chem. Phys., 2014, 140, 134305 CrossRef PubMed.
- M. Huix-Rotllant, E. Dumont, N. Ferré and A. Monari, Photochem. Photobiol., 2015, 91, 323–330 CrossRef CAS PubMed.
- M. Huix-Rotllant, I. Burghardt and N. Ferré, Cr. Chim., 2016, 19, 50–56 CrossRef CAS.
- M. Alías-Rodríguez, C. de Graaf and M. Huix-Rotllant, J. Am. Chem. Soc., 2021, 143, 21474–21477 CrossRef PubMed.
- G. Conejo-Barboza, E. Libby, C. Marín and O. A. Herrera-Sancho, Heritage Sci., 2020, 8, 125 CrossRef.
- N. Marchettini, A. Atrei, F. Benetti, N. Proietti, V. D. Tullio, M. Mascalchi, I. Osticioli, S. Siano and I. T. Memmi, Surf. Eng., 2013, 29, 153–158 CrossRef CAS.
- L. Zhu, W. Wang, J. Miao, X. Yin, X. Hu and Y. Yuan, J. Mol. Struct., 2017, 1141, 462–468 CrossRef CAS.
- L. Kučera, O. Kurka, M. Golec and P. Bednář, Molecules, 2021, 26, 1–12 Search PubMed.
- D. Flaig, M. Maurer, M. Hanni, K. Braunger, L. Kick, M. Thubauville and C. Ochsenfeld, J. Chem. Theory Comput., 2014, 10, 572–578 CrossRef CAS PubMed.
- E. Toomsalu and P. Burk, J. Mol. Model., 2015, 21, 244 CrossRef PubMed.
- C. Benzi, O. Crescenzi, M. Pavone and V. Barone, Magn. Reson. Chem., 2004, 42, S57–S67 CrossRef CAS PubMed.
- M. Siskos, M. Choudhary and I. Gerothanassis, Molecules, 2017, 22, 415 CrossRef PubMed.
- M. Doskocz, K. Kubas, A. Frackowiak and R. Gancarz, Polyhedron, 2009, 28, 2201–2205 CrossRef CAS.
- J. T. Arnold and M. E. Packard, J. Chem. Phys., 1951, 19, 1608–1609 CrossRef CAS.
- N. Muller and R. C. Reiter, J. Chem. Phys., 1965, 42, 3265–3269 CrossRef CAS.
- P. Charisiadis, V. Kontogianni, C. Tsiafoulis, A. Tzakos, M. Siskos and I. Gerothanassis, Molecules, 2014, 19, 13643–13682 CrossRef PubMed.
- C. Shao, Z. She, Z. Guo, H. Peng, X. Cai, S. Zhou, Y. Gu and Y. Lin, Magn. Reson. Chem., 2007, 45, 434–438 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist, American Chemical Society (ACS), 2010, vol. 29, pp. 2176–2179 Search PubMed.
- P. Hore, Nuclear Magnetic Resonance, Oxford University Press, 2015 Search PubMed.
- V. Balevicius and K. Aidas, Appl. Magn. Reson., 2007, 32, 363–376 CrossRef CAS.
- M. S. Khan and Z. H. Khan, Can. J. Anal. Sci. Spectrosc., 2002, 47, 12 Search PubMed.
- A. Navas Diaz, J. Photochem. Photobiol. A, 1990, 53, 141–167 CrossRef CAS.
- C. Miliani, A. Romani and G. Favaro, J. Phys. Org. Chem., 2000, 13, 10 CrossRef.
- A. Niazi, A. A. Rezaei and F. Shahhosseini, Ann. Chim., 2007, 97, 199–211 CrossRef CAS PubMed.
- A. Le Person, J.-P. Cornard and S. Say-Liang-Fat, Chem. Phys. Lett., 2011, 517, 41–45 CrossRef CAS.
- P. Cysewski, T. Jeliński, M. Przybyłek and A. Shyichuk, New J. Chem., 2012, 36, 1836 RSC.
- Z. Machatová, Z. Barbieriková, P. Poliak, V. Jančovičová, V. Lukeš and V. Brezová, Dyes Pigm., 2016, 132, 79–93 CrossRef.
- M. A. P. Turner, M. D. Horbury, V. G. Stavros and N. D. M. Hine, J. Phys. Chem. A, 2019, 123, 873–880 CrossRef CAS PubMed.
- M. Jen, S. Lee, K. Jeon, S. Hussain and Y. Pang, J. Phys. Chem. B, 2017, 121, 4129–4136 CrossRef CAS PubMed.
- J. Preat, A. D. Laurent, C. Michaux, E. A. Perpète and D. Jacquemin, THEOCHEM, 2009, 901, 24–30 CrossRef CAS.
- T. J. Zuehlsdorff, P. D. Haynes, F. Hanke, M. C. Payne and N. D. M. Hine, J. Chem. Theory Comput., 2016, 12, 1853–1861 CrossRef CAS PubMed.
- F. Santoro, R. Improta, A. Lami, J. Bloino and V. Barone, J. Chem. Phys., 2007, 126, 084509 CrossRef PubMed.
- F. Santoro, A. Lami, R. Improta, J. Bloino and V. Barone, J. Chem. Phys., 2008, 128, 224311 CrossRef PubMed.
- A. Baiardi, J. Bloino and V. Barone, J. Chem. Theory Comput., 2013, 9, 4097–4115 CrossRef CAS PubMed.
- D. Jacquemin, E. Brémond, A. Planchat, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2011, 7, 1882–1892 CrossRef CAS PubMed.
- A. Amat, C. Miliani, A. Romani and S. Fantacci, Phys. Chem. Chem. Phys., 2015, 17, 6374–6382 RSC.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2009, 131, 174105 CrossRef PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- J. Gauss, Ber. Bunsenges. Phys. Chem., 1995, 99, 1001–1008 CrossRef CAS.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. Biczysko, J. Bloino, F. Santoro and V. Barone, Time-Independent Approaches to Simulate Electronic Spectra Lineshapes: From Small Molecules to Macrosystems, John Wiley & Sons, Ltd, 2011, ch. 8, pp. 361–443 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- H. Jiang, H. Sun, S. Zhang, R. Hua, Y. Xu, S. Jin, H. Gong and L. Li, J. Incl. Phenom. Macro., 2007, 58, 133–138 CrossRef CAS.
- J. Bloino, A. Baiardi and M. Biczysko, Int. J. Quantum Chem., 2016, 116, 1543–1574 CrossRef CAS.
- P. W. Atkins and R. S. Friedman, Molecular Quantum Mechanics, Oxford University Press, Oxford, New York, 5th edn, 2010 Search PubMed.
- Y. Liu, D. Aranda and F. Santoro, Phys. Chem. Chem. Phys., 2021, 23, 16551–16563 RSC.
- D. Aranda and F. Santoro, J. Chem. Theory Comput., 2021, 17, 1691–1700 CrossRef CAS PubMed.
- K. Falahati, H. Tamura, I. Burghardt and M. Huix-Rotllant, Nat. Commun., 2018, 9, 4502 CrossRef PubMed.
- S. Ahmadi, G. Absalan, D. Craig and D. Goltz, Dyes Pigm., 2014, 105, 57–62 CrossRef CAS.
- C. Clementi, W. Nowik, A. Romani, F. Cibin and G. Favaro, Anal. Chim. Acta, 2007, 596, 46–54 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 9,10-Anthraquinone, alizarin, purpurin tautomers and their free energy differences; natural transition orbital analysis; normal mode analysis; linear vibronic coupling models for 9,10-anthraquinone and alizarin, purpurin electrospray mass and 13C NMR spectra. See DOI: https://doi.org/10.1039/d2cp00520d |
| This journal is © the Owner Societies 2022 |