
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyridine interaction with γ-CuI: synergy between molecular dynamics and molecular orbital approaches to molecule/surface interactions†
Titouan B.
Duston
 a,
Robert D.
Pike
a,
David A.
Welch
b and
Aaron D.
Nicholas
*c
a,
Robert D.
Pike
a,
David A.
Welch
b and
Aaron D.
Nicholas
*c
aDepartment of Chemistry, William & Mary, Williamsburg, VA 23187, USA. E-mail: tbdustondevill@wm.edu; rdpike@wm.edu
bChemistry Department, Farmingdale State College, Farmingdale, NY 11784, USA. E-mail: welchd@farmingdale.edu
cPacific Northwest National Laboratory, 902 Battelle Boulevard, Richland, WA 99354, USA. E-mail: aaron.nicholas@pnnl.gov
First published on 14th March 2022
Abstract
We have used a synergistic computational approach merging Molecular Dynamics (MD) simulations with density functional theory (DFT) to investigate the mechanistic aspects of chemisorption of pyridine (Py) molecules on copper iodide. The presence of both positive and negative ions at the metal halide surface presents a chemical environment in which pyridine molecules may act as charge donors and/or acceptors. Computational results reveal that Py molecules interact with the γ-CuI(111) surface owing to a combination of noncovalent Cu⋯N, Cu/I⋯π/π*, and hydrogen bonding interactions as determined via Natural Bonding Orbitals (NBO). Introduction of surface defect sites alters the interaction dynamics, resulting in a “localizing effect” in which the Py molecules clump together within the defect site. Significant enhancement of hydrogen bonding between C–H σ* and I 6p orbitals results in more tightly surface-bound Py molecules. Our findings provide a platform for understanding the interaction between Py and Py-derivative vapors and metal-based surfaces that contain both electron acceptor and donor atoms.
Introduction
Next-generation smart materials exhibit observable and rapid responses to target compounds or classes of compounds in the environment. The response of such sensor materials may arise due to either physical or chemical interaction, ranging from chemisorption, to solventochromism, to reversible or irreversible bonding.1–3 The response may be electrochemical, such as a change in voltage or conductivity, or it may be spectral, such as a visible color change, or either the enabling or quenching of luminescence behavior.Transition metal d10 centers offer a particularly wide range of chemical and photophysical behaviors that can potentially be leveraged for use as smart materials in chemical detection. Chemically speaking, d10 ions such as Cu(I), Ag(I), and Au(I) exhibit flexible coordination spheres ranging from 2- to 4-coordination with no crystal field preferences. They readily form metal–organic polymers and other networks with bridging ligands, leading to a diverse family of materials.4–8 They also exhibit strong chemical bonding preferences for soft ligands such as aromatic nitrogen, sulfur and phosphorus nucleophiles which constitute a wide range of industrial, military, and medically relevant compounds. In terms of their photophysical characteristics, d10 complexes tend to be photoemissive and are known to exhibit metal-centered (e.g. d → p), cluster-centered (e.g. in halometallates), metal-to-ligand charge transfer, and halide-to-ligand charge transfer transitions.9,10 In many instances, a single chromophore will show more than one of these behaviors, with observed intensity and/or wavelength depending upon solvent, temperature, or exposure to chemical agents resulting in a unique spectroscopic signature. The foregoing chemical and photophysical diversity and flexibility has spurred investigations of d10 systems as biosensors, chemical sensors and optical sensors.11–13
We have previously reported on the conversion of solid γ-CuI upon exposure to gaseous pyridine (Py) to emissive [CuI]4Py4 and are currently studying the applications of this reaction for chemical sensors.14 The solid transformation of zinc blende lattice γ-CuI into discrete [CuI]4Py4 clusters involves substantial chemical reorganization, as illustrated in Scheme 1; nevertheless, it appears to occur relatively quickly – on the minutes time scale. Surface bonding of Py is almost certainly involved in the process, as suggested in the scheme. Nevertheless, the nature of that bonding is unclear, and questions remain surrounding the involvement of the nitrogen lone pair as well as aromatic π-electron systems, especially given the soft character of both Cu(I) and iodide ions. Many reports have focused on nucleophilic molecule interactions with elemental Cu, Ag, and Au surfaces or nanoclusters.15–20 Consideration of metal halides, such as CuI, introduces the possibility of additional interactions associated with the potentially electron-accepting, yet relatively electron-rich, metal ions as well as electron-donating halide centers. For example, a previous density functional theory (DFT) study of Py (along with benzene and thiophene) adsorption to Cu(111) surface confirmed the experimental result that Py molecules lie flat along the metal surface,21,22 rather than upright as suggested in Scheme 1. Importantly, no experimental or computational studies have yet addressed the chemisorption behavior of Py on the more electronically complex CuI surfaces.
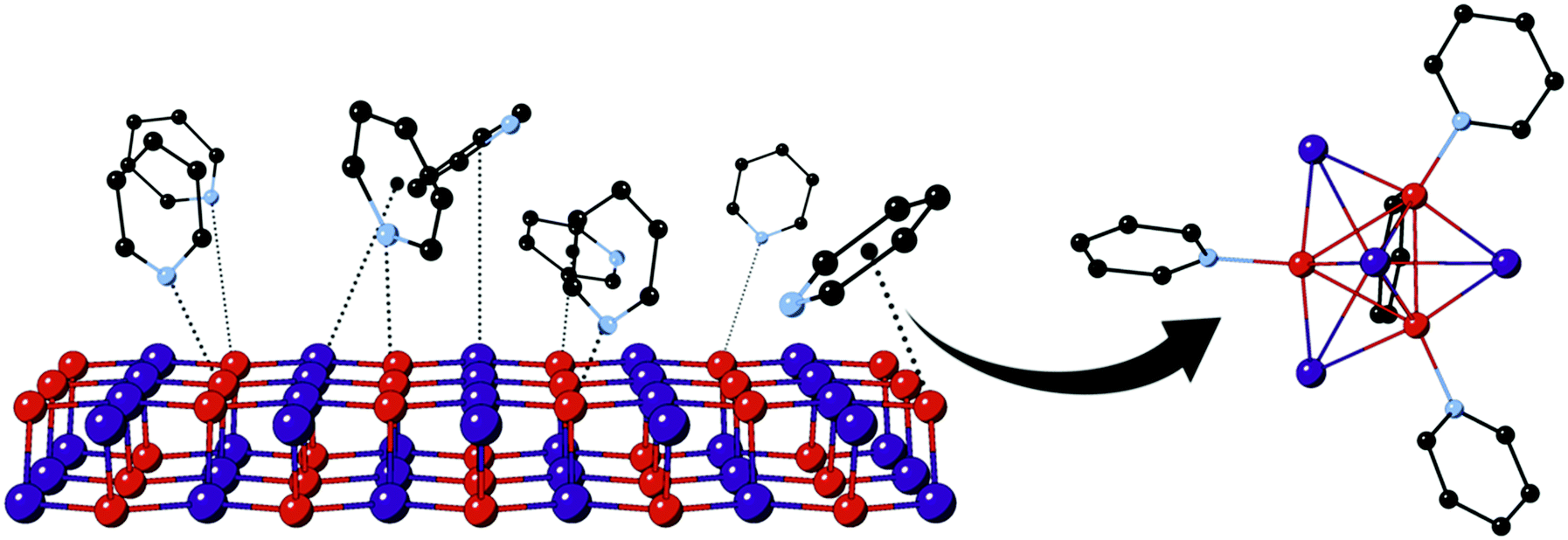 | ||
| Scheme 1 Envisioned reaction of Py (g) with γ-CuI (s) to form [CuI]4Py4 (s). | ||
Our recent computational study on the interaction of dimethyl sulfide (Me2S) vapor with γ-CuI demonstrated that this nucleophile was able not only to adsorb to the Ag(I)-doped CuI surface, but ultimately to embed itself into the solid.23 Nevertheless, the relatively simple Lewis base Me2S is distinctly different from Py, which offers additional reactivity through its π-electrons, including possible acceptor behavior. Interestingly, in the case of Me2S, Ag(I) doping was shown to open up the CuI lattice, making it more subject to nucleophile embedding. As part of that study, we highlighted the value of “atomistic” molecular dynamics studies to produce small molecule-surface ensemble models at relatively little computational expense. These rather large models can include hundreds of vapor molecules interacting on CuI slabs >5 nm in thickness. Specific interactions uncovered by MD may then be examined in detail by trimming the ensemble and then re-examining it in molecular orbital studies using DFT.
In this contribution we expand on the synergism between these complimentary computational techniques, this time studying a more complex system wherein the orientation of the nucleophile (Py) is chemically non-trivial. We show that the atomistic approach, which considers only dipole–dipole interactions, offers computationally inexpensive models that can be used in computationally demanding single-point DFT orbital calculations. DFT modeling allowed us to examine CuI–Py interactions according to electrostatic potentials (ESPs) between polarized species and charge-transfer perspectives via natural bond orbitals (NBO). The Py plane-to-surface bonding angle is investigated and optimized according to bonding strength and distance. Additionally, using a feed-back loop between atomistic and DFT models, we have constructed a comprehensive model for the behavior of Py at the γ-CuI (111) surface. Our findings show that, while the charge transfer from the N lone electron pair to the empty Cu 4s are the strongest interactions, they are not the sole driving force in the geometric orientation of the Py molecule with respect to the surface. Instead a combination of π/π* orbitals interacting with Cu 4s and I 6p atomic orbitals, as well as hydrogen bonding with I 6p orbitals, give rise to strong adsorption of Py molecules to the surface. Using Py as a model molecule, this study represents a milestone in terms of the comprehensiveness and rigor in the analysis of supramolecular assembly and tuning of CuI-based photoemissive materials.
Computational methods
Molecular dynamics simulations
Molecular Dynamics (MD) calculations were performed with the NANIM simulation program.24 MD simulations were performed in the NVT ensemble with a time step of 2 fs and an equilibration period of 100 ps. The temperature was set to 298 K via the velocity-rescaling thermostat (coefficient = 0.01 ps).25 Surface slab generation was performed with the zinc blende structure for copper iodide (assuming a cubic lattice parameter of 0.620 nm). The slab had two-dimensional periodicity with dimensions of 5.2609 × 5.3127 nm. The slab depth was ∼5.1 nm, with the top surface layer being a (111) face made up of cations and the bottom surface layer being a (111) face made up of anions. The surface described thus far was used in the simulations of a pyridine monolayer interacting with an ideal surface. Additionally, simulations were performed for a kinked surface, as well as a surface containing a variably sized “crater.” To create a kinked surface from an ideal surface, first a terrace was generated along the cation-terminated surface by removing one ∼2.3 nm wide layer of metal-iodide pairs (see Fig. 1). Second, half of the metal-iodide pairs located along the upper part of the surface steps were removed in order to create kink sites. To alternatively create a one-layer deep crater on an ideal surface, one, two, four, or eight metal-iodide pairs were removed from the center of both the top surface layer and the bottom surface layer. After surface construction was completed, seventy Py molecules were distributed along the top surface layer. Two stationary walls confine this system along the non-periodic dimension. One wall (wall A) was placed well above the Py monolayer, and a second wall (wall B) was placed well underneath the (111) anion-terminated surface of the slab. The atomic interaction model consists of Coulombic, Lennard-Jones, and Vashishta-Rahman interactions for the intermolecular potential and harmonic stretching/bending terms for the intramolecular potential.26–32 Details of the interaction potential were largely described in our previous work.23 Additionally, the effective atomic charges assigned to carbon, nitrogen, and hydrogen (which we derived from DFT calculations of APT charges for a Py molecule interacting with a CuI cluster, via the LANL2dz basis set) were +0.0129, −0.3166, and +0.0632, respectively. System walls were assigned the following Lennard-Jones potential parameters: A = 90.415 eV Å−9 and B = 0.39459 eV Å−3 for wall A; A = 90.415 eV Å−9 and B = 3.9459 eV Å−3 for wall B. | ||
| Fig. 1 Atomistic model for the kinked (111) copper iodide surface. This surface model is used to assess the interaction of the pyridine monolayer with copper(I) iodide substrate. Image made with VMD.33 | ||
Density functional theory
Density functional theory (DFT) calculations were performed using the Gaussian16 software program.34 Method validation was performed utilizing the well-known CuI cubane compound [CuI]4Py4 and the M0635 or B3LYP36,37 functionals paired with the cep-121G(d),38–41 LANL2dz,42,43 def2tzvp/6-311+g(d,p),44–47 or aug-cc-pvdz48–51 basis sets (Fig. S1 and Table S1, ESI†). Validation results demonstrate the B3LYP and lanl2dz functional and basis set pairing as the best match with experimentally determined crystallographic parameters52 and were utilized for following calculations. The agreement of the B3LYP/lanl2dz computational results with experimental values show that the use of van der Waals corrections (i.e., Grimme style, D3 or D4) are unnecessary and as such were not utilized in this study. Atomic force tensors (Table S2, ESI†) were determined as part of the DFT calculations and used for Molecular Dynamics simulation input variables. DFT adsorption energies (Eads) were calculated by the equation:| Eads = EPy/CuI − (EPy + ECuI) |
Electrostatic potential surfaces
The donor/acceptor regions of Py molecules were investigated via electrostatic potential (ESP) analysis. Calculations were performed within the Gaussian16 program on an optimized Py molecule using the basis set described above. Electrostatic potentials were generated on an electron density surface at 0.002 ε bohr−1 where areas of positive/negative potential indicate electron-rich/deficient regions. Electrostatic potentials generated at this isodensity value have been determined by previous studies to be an accurate indicator for supramolecular assembly.56,57Natural bonding orbitals (NBO) and quantum theory of atoms in molecules (QTAIM)
NBO calculations were performed using the NBO7 software suite utilizing the individual components constructed from the Molecular Dynamic outputs. NBO second order perturbation theory was applied to parse out the donor–acceptor interaction between the Py electron donor and the Cu or I electron acceptor including deconvoluting the sub-noncovalent interaction types, i.e. halogen, hydrogen bonding, as well as identify the atomic or molecular orbitals involved. Orbitals were rendered using the Jmol (v.14.30.1) java applet.58,59 Models generated and used for NBO calculations can be found in Fig. S2 and S3 (ESI†).Results and discussion
Electrostatic potential surface of pyridine
Previous work on the interaction of nucleophilic volatile organic compound (VOC) vapors with metal-based surfaces have largely focused on elemental metallic systems, wherein noncovalent interactions between species give rise to regium bonding.15,18,19,21 These interactions are thus an important component in understanding adsorption and catalytic mechanisms of metallic compounds.16,17,20,60,61 Simply described, interactions occur between electron acceptor–donor pairs, and in metallic cases metal centers act as acceptors while the VOC donor may contribute through either a lone electron pair or π aromatic system.20,62–64 Pyridine, as a simple N-heteroaromatic, is capable of electron donation via either the N lone pair (n-interactions) or the π cloud (π-interactions), giving rise to a potential competition between σ- and π-interactions. This was noted by Atodiresei et al., who reported that Py is capable of adsorbing onto Cu(110) and Ag(110) surfaces in either a parallel (π-interaction) or perpendicular (n-interaction) geometry.22 In this example only the metal center, acting as an electron acceptor, is capable of interacting with the incoming electron-donating VOC molecule. This dynamic changes in metal halide systems, wherein the halide may act as an electron donor to empty (electron-accepting) antibonding molecular orbitals of the VOC. As such, a number of other noncovalent interactions are possible, including X⋯H and X⋯π* bonding.The interaction of vapor-phase molecules and CuI surfaces can first be partially described as a coulombic attraction between polarized regions along either CuI surface or the Py molecule. As such, electron density surfaces are useful in identifying regions of relative electron richness (Vs,min) and depletion (Vs,max) that may serve as electron donors or acceptors, respectively, in facilitating supramolecular assembly, catalytic reactions, and surface adsorption.16,65,66 For Py (Fig. 2) polarization about the molecule results in a region of electron richness adjacent the N atom owing to the presence of lone pair electrons. This region has an electrostatic potential value of −223 kJ mol−1. As the most negative region (Vs,min), this site is the strongest potential electron donor for formation of noncovalent interactions with the CuI surface, thus indicating a stronger propensity to interact via σ-interaction. Turning towards π-interactions, regions of electron richness are also located directly above and below the ring plane adjacent the C atoms. Here the electrostatic potential is slightly negative at −20 kJ mol−1 and indicates significantly poorer electron-donor behavior compared to the N lone pair. Less obviously, the Py molecule features a region of positive electrostatic potential, and thus may also act as an electron acceptor. This region of positive potential (Vs,max) is located about the terminal end of the para and meta C–H bond and has a potential value of +129 kJ mol−1.
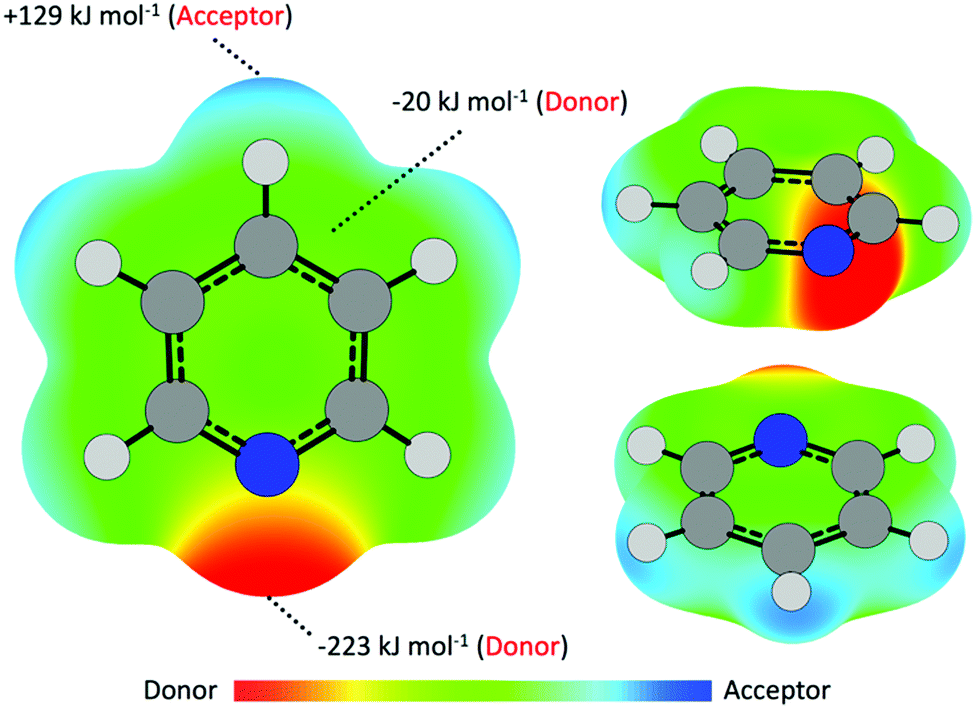 | ||
| Fig. 2 Electrostatic potentials of a pyridine molecule mapped onto a 0.002 ε bohr−1 isodensity surface highlighting regions which may act as electron donors (negative potentials) or acceptors (positive potentials). | ||
As previously discussed, the iodide is an electron donor within the CuI surface and thus may interact with this electron-acceptor region on the Py molecules. The large positive potential value (+129 kJ mol−1vs. −20 kJ mol−1) indicates that this region might potentially out-compete interactions with the π cloud. Given the dual electron-donating/-accepting capacity of the Py molecule, the question of how competition between these sites results in preferred bonding geometries about CuI remains unclear based on ESP surfaces alone and thus warrants detailed investigation.
Ideal copper(I) iodide surface
Surfaces that do not contain terminal atoms or defect sites, termed ‘ideal’, are historically used in DFT modelling to understand the chemisorption mechanism of small molecules to metal-based surfaces. These models typically are limited to elemental metals or metal oxides and exclude families of metal halides such as perovskites. While less realistic owing to the lack of defect sites or terminal atoms, these models are nevertheless important as a first step in understanding some basic principles as to how small molecules may interact with metal-based surfaces. As such, we have opted to start with an ideal CuI surface in which Py molecules are free to interact along the (111) plane in order to determine (i) the prediction of the Py geometry along the surface, (ii) the Py–CuI adsorption energy, and (iii) identify and quantify the atomic and molecular orbitals involved in the charge transfer process. | ||
| Fig. 3 Molecular Dynamics simulations (left) for the Py monolayer at an ideal surface and (right) angular distribution of Py molecules (i.e. flat orientation = 0°; upright orientation = 90°). Image made with VMD.33 | ||
The N atom resides almost directly above a positively charged Cu metal center (Fig. S4, ESI†). Fig. 4 shows the potential energies for a series of calculations wherein the Py molecule is removed from the CuI surface in a step-wise manner while being held at a plane-to-surface angle of 90°. The results of this study reveal a minimum energy value at a surface⋯N distance of 2.65 Å. Fitting a Morse potential, we are able to determine an adsorption energy of 0.528 eV, a value consistent with other studies on adsorption of heterocyclic aromatic molecules to metallic (Cu/Ag/Au) surfaces.19,21,22 This adsorption energy value is significantly lower than the calculated value of 0.714 eV for the removal of a formally coordinated Py from the known cubane [CuI]4Py4 complex. Absorption of a Py molecule on a CuI surface can conceivably produce an orientation of the molecular plane anywhere from perpendicular to parallel to the surface.
 | ||
| Fig. 4 Interaction energy between pyridine and a CuI surface as a function of (top) surface⋯N distance and (bottom) pyridine tilting angle. | ||
Starting at the optimized bond distance, the interaction energy was calculated as a function of tilt angle of the Py ring in relation to the CuI surface (Fig. 4). Tilting of the Py molecule reveals a ground state in which the molecular plane is canted at an angle of 90° with respect to the CuI surface. This position is lower in energy by 1.75 eV and 3.53 eV for the two parallel orientations 0° and 180°, respectively. Orientation of the Py molecule at angles other than 90°, however, undoubtedly facilitates interaction of the CuI surface with both the N lone pair and the aromatic π cloud on the face of the Py plane. Importantly, the ground state calculations reveal that the Py molecules preferentially adsorb perpendicular to the CuI surface, as we might expect if only the N lone pair were involved in the interaction.
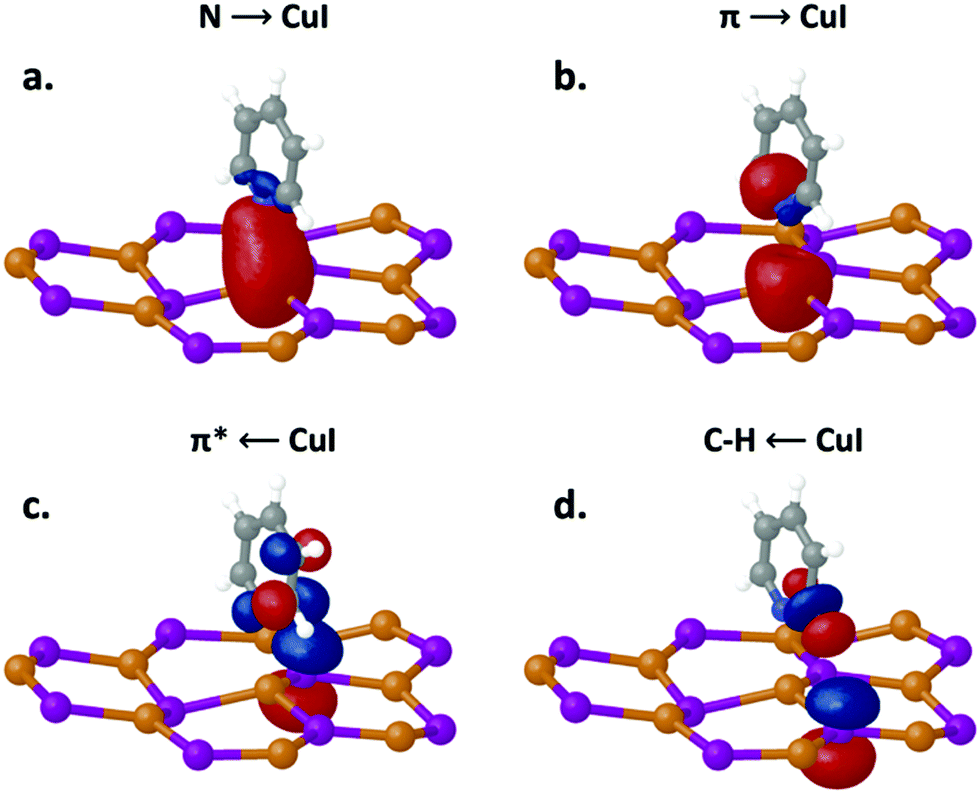 | ||
| Fig. 5 Orbitals involved in the noncovalent interaction between Py and the CuI surface in the ground state. The Py tilt angle is 90° in relation to the CuI surface. Arrows denote the direction of the electron charge transfer. | ||
To better understand their directing role and to relate the strength of these interactions to one another, we have calculated their respective stabilization energies (Table 1). The total stabilization energy at a tilt angle of 90° is 12.64 kcal mol−1. As expected, when Py is oriented perpendicular to the CuI surface (90°–120°), the N lone pair interaction with Cu is strongest, contributing 37% (4.64 kcal mol−1) of the total stabilization energy. Interestingly, despite the geometry, interaction via the π and π* is nearly as strong, contributing 35% toward the total stabilization energy. Of this amount, the major portion is due to Py charge donor behavior involving the Py π orbitals at 2.89 kcal mol−1, while the remainder (1.48 kcal mol−1) is associated with charge acceptor behavior involving the Py π*. In its interaction via the C–H orbitals the Py molecule acts as either a charge donor or acceptor. As described above, in cases where the Py acts as a charge donor the C–H (σ) orbitals interact with the Cu 4s orbitals, contributing 1.29 kcal mol−1, while in cases where the Py molecule acts as a charge acceptor the C–H (σ*) orbitals interact with the I 6p orbitals, contributing 2.34 kcal mol−1. The sum of the stabilization energies for the C–H interactions is 3.63 kcal mol−1, representing a 29% contribution. These findings highlight three important factors with regard to the interactions of Py with CuI: (i) interaction with the N lone pair, while the strongest, still represents a minority of the overall interaction strength, (ii) the interaction energies are stronger for the Py π and C–H σ orbitals than for the Py π* and C–H σ* orbitals, indicating that Py charge donor behavior is more important than its acceptor behavior, and (iii) hydrogen bonding interactions are non-negligible and contribute significantly to the overall interaction.
| Interaction type | Pyridine role | ||
|---|---|---|---|
| Donor (kcal mol−1) | Acceptor (kcal mol−1) | Donor + acceptor (kcal mol−1) | |
| N–CuI | 4.64 | 0.00 | 4.64 |
| π/π*-CuI | 2.89 | 1.48 | 4.37 |
| C–H (σ/σ*)-CuI | 1.29 | 2.34 | 3.63 |
| Total stabilization energy | 12.64 | ||
We note that there is a divergence of MD- and DFT-determined geometric orientation preference of the Py molecule with respect to the CuI surface for the ground state. As we have previously noted, MD simulations consider only dipole–dipole interactions between molecules, making them computationally inexpensive even for large models. This is evident in our results which show maximization of N⋯N distances between Py molecules. Furthermore, MD simulations contain multiple Py molecules which invariably influence the overall statistical Py geometric orientation as they interact with one another. DFT calculations, on the other hand, consider many more properties that influence the Py–CuI geometry including polarization and orbital interactions. These additional considerations provide a much more detailed understanding of the Py–CuI ensemble. However, DFT calculations require sizeable computational resources and thus can only be performed on models containing a fraction of the atoms in MD simulations. Our findings expose a critically important flaw in current modelling techniques that may rely on solely ideal surfaces or a lone computational methodology. While further exploration of the divergence of MD- and DFT-determined geometric orientation preference is outside the scope of this study, we bring attention to it as a point for future research.
Defect copper(I) iodide surfaces
While commonly used and well accepted in MD-based calculations, the use of defect surfaces is rarer for DFT-based modelling owing to the necessary increase in computational expense. Defective surfaces, however, represent a more realistic approximation of actual surfaces, which invariably suffer from defect or kink sites, and thus computations involving defect sites must lead to more meaningful predictions of chemical behavior. These sites, additionally, are critically important in the prediction and understanding of chemical activity. For example, in catalysis a defective region is more apt to give rise to a ‘localizing’ effect and may help hold molecules in close proximity to one another or increase their exposure to surface atoms. As such, we have included MD and DFT calculations using a simple defect CuI surface in which we have systematically removed atoms while maintaining a neutral charge balance. Our aims are to determine: (i) the change in preferred Py orientation, (ii) the difference in adsorption energy compared to the ideal surface model, and (iii) shifts in charge transfer dynamics between donor and acceptor orbitals. | ||
| Fig. 6 Molecular Dynamics simulations for the Py monolayer at a cratered surface (circled in red). The crater site ranges in size from (left to right) four to eight to sixteen Cu–I pairs removed from the top layer of the ideal surface. In general, the crater causes Py molecules to group inside the defect and exhibit higher tilting angles. Image made with VMD.33 | ||
 | ||
| Fig. 7 Interaction energy between pyridine and a defective CuI surface as a function of pyridine tilting angle. | ||
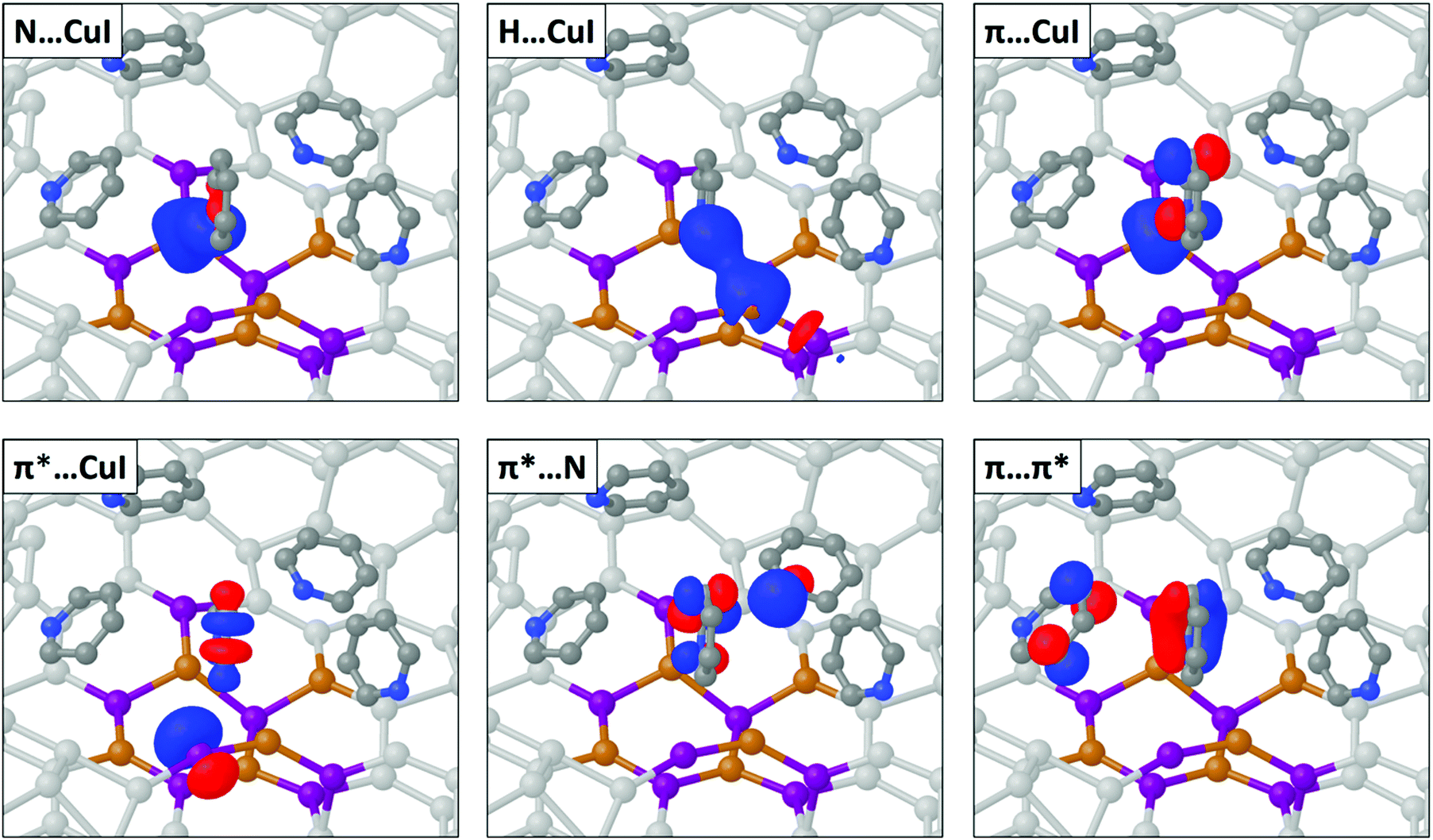 | ||
| Fig. 8 Orbitals involved in the noncovalent interaction between pyridine and the defect CuI surface in the ground state. The pyridine tilt angle is 90° in relation to the CuI surface. Noninteracting CuI atoms are colored light gray for clarity. | ||
To compare the relative strengths of the noncovalent interactions and reconcile the lower adsorption energy of the defect surface to the ideal surface, we have tallied the NBO determined stabilization energies for each donor/acceptor pairing in Table 2. At this optimized angle the N–CuI orbital pairs (N lone pair donor and Cu acceptor) dominate, contributing a total of 6.45 kcal mol−1 (44%). Notably, significant hydrogen bonding between Py and the CuI surface contribute the second largest stabilization energy of 4.86 kcal mol−1. Of this stabilization, 69% arises from the C–H σ* orbitals acting as charge acceptors from I donors, while the remaining 31% results from the C–H σ orbitals acting as charge donors. In comparison, π interactions with the CuI surface account for only 2.20 kcal mol−1 of the total stabilization energy, of which 73% is from Py π orbitals acting as charge donors. The Py⋯Py interactions are weakest, including both π–π and n–π interactions, and contribute only ∼8% to the total stabilization.
| Interaction type | Molecular pair | Pyridine role | ||
|---|---|---|---|---|
| Donor (kcal mol−1) | Acceptor (kcal mol−1) | Donor + acceptor (kcal mol−1) | ||
| N–CuI | Py/CuI | 6.45 | 0.00 | 6.45 |
| H–CuI | Py/CuI | 1.52 | 3.34 | 4.86 |
| π/π*-CuI | Py/CuI | 1.61 | 0.59 | 2.20 |
| π–π* | Py/Py | 0.53 | 0.09 | 0.62 |
| π*-N | Py/Py | 0.00 | 0.63 | 0.63 |
| Total Stabilization energy | 14.76 | |||
The stabilization energies highlight two points: (i) Py⋯Py interactions do not significantly stabilize Py molecules within the defect site, and (ii) the inclusion of I atoms as charge donors significantly enhances the overall stabilization energy. The latter finding demonstrates the importance of having an electron donor embedded within the surface for capture and subsequent bonding with Py vapor molecules, since only the I atoms are able to participate meaningfully with the C–H σ/σ* bonds. In the absence of these atoms the C–H orbitals would otherwise go unutilized or potentially lead to competitive Py⋯Py interactions, leading to weaker noncovalent interactions with the metallic surface. With respect to the weak Py⋯Py interactions, it becomes clear that the perpendicular geometric orientation is most likely the result of destabilizing steric repulsion rather than positive charge transfer attraction and that these interactions do not contribute significantly to the Py adsorption process. Repulsion is also credited with the lower DFT-determined adsorption energy which is excluded from the NBO determined stabilization energies. Interestingly, the results indicate that while the Py molecule interacts more strongly with the defective CuI surface, the repulsion amongst adjacent Py molecules results in an overall less stable configuration within the well. This sets up an intriguing competition wherein Py molecules are more readily able to interact with the surface but must balance this with the available space within the defect site. This simple, yet critical, point provides a rationalization to the observed localizing effect whereby the Py molecules almost exclusively prefer an upright orientation within the crater site. This geometry is best suited to maximize the number of Py molecules within the available space, while simultaneously minimizing Py–Py contacts.
The agreement of MD and DFT modeling results highlights two important points: (i) ideal surfaces and those with defects produce entirely different results with regard to the geometric orientation and (ii) defect sites enhance the ability for electron donor I atoms to interact with electron acceptor C–H sites. With respect to the first point, we highlight this observation to draw attention to the need of DFT modelling to include defect models in addition to ideal surfaces. It is clear from our findings that the behavior of small molecules along a surface varies significantly when a defect site is present. Herein, we show that defect sites are important in enhancing adsorption and expanding the modes of interaction between the Py molecule and the CuI surface. These effects arise primarily from an enhancement of hydrogen bonding with terminal I atoms which are only present within the defect site.
Conclusion
We have investigated the interaction of nucleophilic pyridine molecules with copper(I) iodine surfaces utilizing a synergistic computational approach with Molecular Dynamic (MD) and density functional theory (DFT) modelling. Our findings feature the rarely reported CuI surface, which unlike historically investigated metallic surfaces, contains both cationic metal centers as well as anionic halides. Molecular Dynamic calculations were performed over several nanoseconds using a single layer of pyridine (Py) molecules to initially provide geometric information on the Py–CuI interaction. Electrostatic potentials along an isodensity surface of pyridine were used to achieve a molecular-level understanding of adsorption and to characterize the donor and acceptor sites of the neutral Py molecule in which polarization gives rise to regions of variable acceptor/donor strengths. The ESP values demonstrate the ability of Py to act as either an electron donor or acceptor, resulting in interactions with both Cu and I atoms. Natural bonding orbital (NBO) calculations show that the charge transfer from the N lone electron pair to the empty Cu 4s are the strongest interactions, but do not dominate as the most significant driving force in the geometric orientation of the Py molecule along the surface. Instead, a combination of π/π* orbitals interacting with Cu 4s and I 6p atomic orbitals alongside hydrogen bonding with I 6p orbitals give rise to strong adsorption of Py molecules to the surface. Introduction of defect sites along the CuI surface not only introduces Py⋯Py interactions via π stacking, but also significantly enhances the ability to form hydrogen bonds with terminal I atoms within the defect site. The result is a localizing effect within the defect site whereby Py molecules are tightly held close to the surface yet are oriented in such a way to minimize Py–Py contacts due to steric repulsion. Our findings provide a first-principle understanding on how orbital pair interactions and changes in metal halide surface leads to shifts in adsorption and noncovalent interactions utilizing a hybrid Molecular Dynamic-density functional theory methodology.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Prof. Christopher L. Cahill at The George Washington University for access and allocation of computational resources. The authors acknowledge William & Mary Research Computing for providing computational resources and/or technical support that have contributed to the results reported within this paper. URL: https://www.wm.edu/it/rc.References
- K. Dashtian, S. Shahbazi, M. Tayebi and Z. Masoumi, Coord. Chem. Rev., 2021, 445, 214097 CrossRef CAS.
- G. A. Leith, C. R. Martin, A. Mathur, P. Kittikhunnatham, K. C. Park and N. B. Shustova, Adv. Energy Mater., 2021, 2100441 Search PubMed.
- W. Luo and G. Wang, Adv. Opt. Mater., 2020, 8, 2001362 CrossRef CAS.
- A. Schlachter and P. D. Harvey, J. Mater. Chem. C, 2021, 9, 6648–6685 RSC.
- O. Veselska and A. Demessence, Coord. Chem. Rev., 2018, 355, 240–270 CrossRef CAS.
- S.-Y. Liu, J.-P. Zhang and X.-M. Chen, Cryst. Growth Des., 2017, 17, 1441–1449 CrossRef CAS.
- M. Shao, M.-X. Li, Z.-X. Wang, X. He and H.-H. Zhang, Cryst. Growth Des., 2017, 17, 6281–6290 CrossRef CAS.
- H. Fei, D. L. Rogow and S. R. J. Oliver, J. Am. Chem. Soc., 2010, 132, 7202–7209 CrossRef CAS PubMed.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2–18 CrossRef CAS.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Photochemistry and Photophysics of Coordination Compounds I, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 69–115 Search PubMed.
- Y. C. López, H. Viltres, N. K. Gupta, P. Acevedo-Peña, C. Leyva, Y. Ghaffari, A. Gupta, S. Kim, J. Bae and K. S. Kim, Environ. Chem. Lett., 2021, 19, 1295–1334 CrossRef.
- G. Evtugyn, S. Belyakova, A. Porfireva and T. Hianik, Sensors, 2020, 20, 6963 CrossRef CAS PubMed.
- J. Dong, D. Zhao, Y. Lu and W.-Y. Sun, J. Mater. Chem. A, 2019, 7, 22744–22767 RSC.
- J. P. Killarney, M. McKinnon, C. Murphy, K. M. Henline, C. Wang, R. D. Pike and H. H. Patterson, Inorg. Chem. Commun., 2014, 40, 18–21 CrossRef CAS.
- C. Deng, J. Chen and Q. Tang, J. Phys. Chem. C, 2021, 125, 4489–4497 CrossRef CAS.
- G. Li, J. H. Stenlid, M. S. G. Ahlquist and T. Brinck, J. Phys. Chem. C, 2020, 124, 14696–14705 CrossRef CAS.
- M. de las, N. Piña, A. Frontera and A. Bauzá, J. Phys. Chem. Lett., 2020, 11, 8259–8263 CrossRef PubMed.
- R. L. H. Freire, D. Guedes-Sobrinho, A. Kiejna and J. L. F. Da Silva, J. Phys. Chem. C, 2018, 122, 1577–1588 CrossRef CAS.
- X. Jia and W. An, J. Phys. Chem. C, 2018, 122, 21897–21909 CrossRef CAS.
- J. Halldin Stenlid, A. J. Johansson and T. Brinck, Phys. Chem. Chem. Phys., 2018, 20, 2676–2692 RSC.
- K. Tonigold and A. Groß, J. Chem. Phys., 2010, 132, 224701 CrossRef PubMed.
- N. Atodiresei, V. Caciuc, J.-H. Franke and S. Blügel, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 045411 CrossRef.
- A. D. Nicholas, F. H. Barnes, D. R. Adams, M. S. Webber, M. A. Sturner, M. D. Kessler, D. A. Welch, R. D. Pike and H. H. Patterson, Phys. Chem. Chem. Phys., 2020, 22, 11296–11306 RSC.
- D. A. Welch, T. J. Woehl, C. Park, R. Faller, J. E. Evans and N. D. Browning, ACS Nano, 2016, 10, 181–187 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- P. Vashishta and A. Rahman, United States, 1979.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- H. Heinz, R. A. Vaia, B. L. Farmer and R. R. Naik, J. Phys. Chem. C, 2008, 112, 17281–17290 CrossRef CAS.
- I. Y. Gotlib, A. K. Ivanov-Schitz, I. V. Murin, A. V. Petrov and R. M. Zakalyukin, Solid State Ionics, 2011, 188, 6–14 CrossRef CAS.
- S. Toxvaerd and J. C. Dyre, J. Chem. Phys., 2011, 134, 081102 CrossRef PubMed.
- C. J. Fennell and J. D. Gezelter, J. Chem. Phys., 2006, 124, 234104 CrossRef PubMed.
- P. Kumar, F. W. Starr, S. V. Buldyrev and H. E. Stanley, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 75, 011202 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Mont-gomery, J. E. Peralta Jr., F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- W. J. Stevens, H. Basch and M. Krauss, J. Chem. Phys., 1984, 81, 6026–6033 CrossRef.
- W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Can. J. Chem., 1992, 70, 612–630 CrossRef CAS.
- T. R. Cundari and W. J. Stevens, J. Chem. Phys., 1993, 98, 5555–5565 CrossRef CAS.
- S. Huzinaga, J. Andzelm, E. Radzio-Andzelm, Y. Sakai, H. Tatewaki and M. Klobukowski, Gaussian Basis Sets for Molecular Calculations, Volume 16, 1st Edition, Elsevier Science, 1984 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877–13883 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- A. W. Kelly, J. V. Handy, A. D. Nicholas, F. H. Barnes, H. H. Patterson, L. Wojtas and R. D. Pike, J. Inorg. Organomet. Polym. Mater., 2017, 27, 90–100 CrossRef CAS.
- K. Duanmu and D. G. Truhlar, J. Chem. Theory Comput., 2017, 13, 835–842 CrossRef CAS PubMed.
- R. G. Surbella, L. C. Ducati, K. L. Pellegrini, B. K. McNamara, J. Autschbach, J. M. Schwantes and C. L. Cahill, J. Am. Chem. Soc., 2017, 139, 10843–10855 CrossRef CAS PubMed.
- A. D. Nicholas, B. W. Walusiak, L. C. Garman, M. N. Huda and C. L. Cahill, J. Mater. Chem. C 10.1039/D0TC06000C.
- M. Borovina, I. Kodrin and M. Đaković, Cryst. Growth Des., 2019, 19, 1985–1995 CrossRef CAS.
- M. Borovina, I. Kodrin and M. Đaković, CrystEngComm, 2018, 20, 539–549 RSC.
- A. Herráez, Biochem. Mol. Biol. Educ., 2006, 34, 255–261 CrossRef PubMed.
- Jmol: an open-source Java viewer for chemical structures in 3D, http://www.jmol.org/, (accessed 12 April 2021).
- J. H. Stenlid and T. Brinck, J. Am. Chem. Soc., 2017, 139, 11012–11015 CrossRef CAS PubMed.
- F. Zhou, Y. Liu, Z. Wang, Q. Yang and B. Zheng, Comput. Theor. Chem., 2020, 1179, 112800 CrossRef CAS.
- A. Frontera and A. Bauzá, Chem. – Eur. J., 2018, 24, 7228–7234 CrossRef CAS PubMed.
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS.
- A. Bauzá and A. Frontera, Inorganics, 2018, 6, 64 CrossRef.
- J. Halldin Stenlid, A. J. Johansson and T. Brinck, Phys. Chem. Chem. Phys., 2019, 21, 17001–17009 RSC.
- C.-Y. Chiu, H. Wu, Z. Yao, F. Zhou, H. Zhang, V. Ozolins and Y. Huang, J. Am. Chem. Soc., 2013, 135, 15489–15500 CrossRef CAS PubMed.
- R. Thakuria, N. K. Nath and B. K. Saha, Cryst. Growth Des., 2019, 19, 523–528 CrossRef CAS.
- Z.-F. Yao, J.-Y. Wang and J. Pei, Cryst. Growth Des., 2018, 18, 7–15 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- E. M. Cabaleiro-Lago and J. Rodríguez-Otero, ACS Omega, 2018, 3, 9348–9359 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Description of DFT derived models, orbital interaction renderings, and additional NBO/NLMO calculations (PDF). See DOI: 10.1039/d1cp05888f |
| This journal is © the Owner Societies 2022 |