
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An experimental and computational study of the reaction between 2-methylallyl radicals and oxygen molecules: optimizing master equation parameters with trace fitting†
Timo T.
Pekkanen
a,
Raimo S.
Timonen
a,
Struan H.
Robertson
b,
György
Lendvay
 c,
Satya P.
Joshi
a,
Timo T.
Reijonen
a and
Arkke J.
Eskola
*a
c,
Satya P.
Joshi
a,
Timo T.
Reijonen
a and
Arkke J.
Eskola
*a
aDepartment of Chemistry, University of Helsinki, P.O. Box 55 (A.I. Virtasen aukio 1), 00014 Helsinki, Finland. E-mail: arkke.eskola@helsinki.fi
bDassault Systèmes, 334 Science Park, Cambridge CB4 0WN, UK
cInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Magyar Tudósok krt. 2., Budapest H-1117, Hungary
First published on 27th January 2022
Abstract
We have investigated the reaction between 2-methylallyl radicals and oxygen molecules with experimental and computational methods. Kinetic experiments were conducted in a tubular laminar flow reactor using laser photolysis for radical production and photoionization mass spectrometry for detection. The reaction was investigated as a function of temperature (203–730 K) and pressure (0.2–9 torr) in helium and nitrogen bath gases. At low temperatures (T < 410 K), the reaction proceeds by a barrierless reaction to form 2-methylallylperoxyl. Equilibration of the peroxyl adduct and the reactants was observed between 350–410 K. Measurements were extended to even higher temperatures, up to 730 K, but no reaction could be observed. Master equation simulations of the reaction system were performed with the MESMER program. Kinetic parameters in the master equation model were optimized by direct fitting to time-resolved experimental 2-methylallyl traces. Trace fitting is a recently implemented novel feature in MESMER. The trace approach was compared with the more traditional approach where one uses experimental rate coefficients for parameter optimization. The optimized parameters yielded by the two approaches are very similar and do an excellent job at reproducing the experimental data. The optimized master equation model was then used to simulate the reaction under study over a wide temperature and pressure range, from 200 K and 0.01 bar to 1500 K and 100 bar. The simulations predict a small phenomenological rate coefficient under autoignition conditions; about 1 × 10−18 cm3 s−1 at 400 K and 5 × 10−16 cm3 s−1 at 1000 K. We provide modified Arrhenius expressions in PLOG format for the most important product channels to facilitate the use of our results in combustion models.
1 Introduction
Master equation (ME) models of gas-phase reactions typically contain many parameters that are not known very accurately, so they often need to be adjusted to obtain quantitative agreement with experiment. Common adjustable parameters are well-depths, barrier heights, and the 〈ΔE〉down and Lennard-Jones parameter values of different species. To evaluate the goodness of a master equation model, one usually compares the phenomenological Bartis–Widom rate coefficients1 produced by the model to experimental rate coefficients. Parameter optimization can then be performed to minimize the difference between modeled and measured rate coefficients. This approach is satisfactory in many cases, but runs into trouble in at least two scenarios.1. When kinetic traces are multi-exponential. In this case, the functions that need to be fitted to experimental traces tend to be complicated, and it can be difficult, if not impossible, to obtain reliable values for the parameters in the fitting function. This uncertainty will be reflected in the rate coefficients because the rate coefficients are functions of these parameters.
2. When chemically significant eigenvalues (CSEs) mix with internal energy relaxation eigenvalues (IEREs). The Bartis–Widom technique that is used to obtain phenomenological rate coefficients from master equation simulations is only valid when CSEs and IEREs are well separated. When they are not, a rate coefficient description of the reaction system is difficult to define.
Both of these problems can be avoided if one compares experimental and modeled species traces, rather than rate coefficients, with each other. The traces produced by a master equation simulation are valid even in cases where a rate coefficient description does not exist. The trace approach is superior to the conventional rate coefficient approach also in the sense that a more direct comparison between experiments and simulations is made. After all, it is time-resolved kinetic traces that kineticists measure, not rate coefficients. Typically, of course, experiments are never simple and will contain secondary chemistry or diffusion phenomena that affect the time-resolved behaviour of the measured traces. This needs to be accounted for when performing trace fitting, just as it does for the conventional approach.
Trace fitting was implemented in MESMER version 6.0.2 The details of this implementation are presented by Medeiros et al.3 In version 6.1, this feature was extended to allow the user to specify first-order loss rates (diffusive losses) for all species present in the reaction system. The purpose of these loss rates is to account for processes other than the studied reaction which have an effect on the measured traces. For example, the loss rate could be the rate at which a species diffuses out of the monitored reaction zone. In other experiments, this loss rate could be the rate at which the studied species reacts with reactor surfaces. In this work, we use the trace fitting feature in MESMER for the reaction between 2-methylallyl radicals (2-methylprop-2-en-1-yl) and oxygen molecules. The reaction system under study is
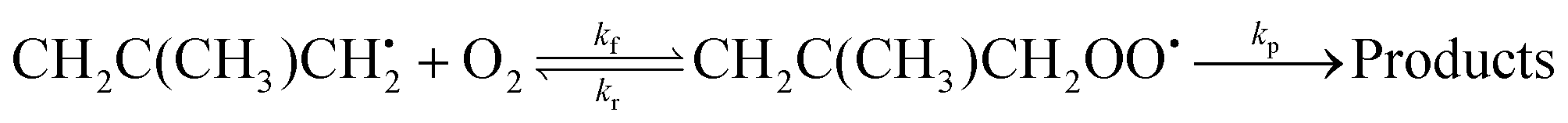 | (1) |
Here kf and kr are the forward and reverse rate coefficients of the initial association reaction, kp is the irreversible first-order loss rate of the peroxyl adduct (this includes its wall rate), and kw is the wall rate of 2-methylallyl. The wall rate kw is the loss rate of 2-methylallyl in the absence of added oxygen and is almost entirely due to the heterogeneous reaction of 2-methylallyl with the reactor wall. The reaction system is relatively simple and the rate coefficients can be obtained by performing single- or double-exponential fits to the measured traces. Thus, both the rate coefficient and trace approach can be used for parameter optimization and should yield similar results.
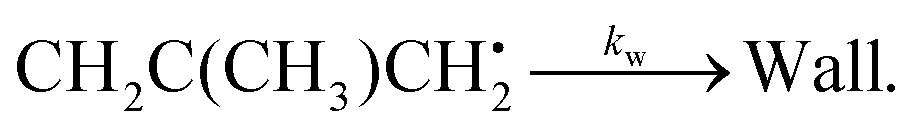
In combustion systems, 2-methylallyl radicals can be produced by abstracting hydrogen from isobutene (2-methylpropene), a major oxidation and pyrolysis product of iso-octane (2,2,4-trimethylpentane).4 Hydrogen abstraction from isobutene is far more likely to produce 2-methylallyl radicals than vinylic 2-methylprop-1-en-1-yl radicals because six of the eight hydrogens in isobutene are allylic, which are much easier to abstract than vinylic hydrogens. 2-Methylallyl is a resonance stabilized hydrocarbon radical (RSHR, see Fig. 1). Such radicals exhibit decreased reactivity towards O2 and increased thermal stability compared to similar-sized hydrocarbon radicals that are not resonance-stabilized.5 Therefore, RSHRs can accumulate in combustion environments to reach concentrations where their cross-reactions become relevant. Reactions between RSHRs are an important step in forming “the first aromatic ring”, a key precursor in soot formation.6 Since RSHR + O2 reactions compete with soot-initiating RSHR + RSHR reactions, quantitative data is needed for both categories of reactions to accurately model soot formation.
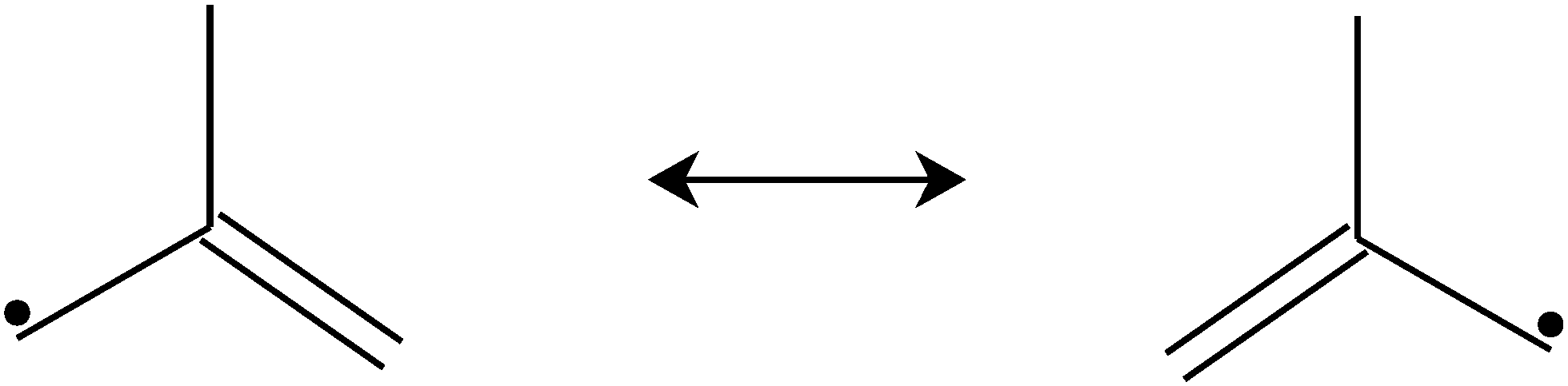 | ||
| Fig. 1 The equivalent resonance structures of 2-methylallyl. | ||
Because O2 addition causes RSHRs to lose their resonance stabilization, RSHR + O2 → RO2 reactions have shallow wells (zero-kelvin binding enthalpies) and, consequently, begin to equilibrate at relatively low temperatures. For allylic and propargylic radicals, the reverse reaction has been observed to become significant already between 300–400 K.7–14 RSHR + O2 reactions are therefore expected to be “dead-ends” in combustion systems unless new reaction channels become accessible at higher temperatures. Bimolecular product channels at high temperatures (T > 500 K) have been observed for propargylic RSHRs and for 1,1-dimethylallyl,7,11,14 but such reactions appear to be very slow for allyl and 1-methylallyl.12,15 The relatively high reactivity of 1,1-dimethylallyl with molecular oxygen at high temperatures is explained by the presence of a low-barrier, well-skipping channel that produces hydroperoxyl and 2-methylbuta-1,3-diene. This conjugate-alkene-forming channel is not available for allyl, which would explain its low reactivity with molecular oxygen at high temperatures. However, this channel is present for 1-methylallyl, but Knyazev and Slagle were not able to observe any reaction between 1-methylallyl and molecular oxygen even at 700 K.12 This suggests that the conjugate-alkene-forming channel has a noticeably higher barrier for 1-methylallyl than 1,1-dimethylallyl. Similarly to allyl, there is no conjugate-alkene-forming channel for 2-methylallyl, so one might expect it to exhibit low reactivity towards oxygen at high temperatures. However, there are three allylic hydrogens available for internal abstraction in 2-methylallylperoxyl, so it may have important QOOH chemistry.
The reaction investigated in this work, CH2C(CH3)CH2˙ + O2, has only been studied once experimentally. Schleier et al. measured the rate coefficient of the reaction at 298 K and 0.001–0.003 bar and found k = 8.5 ± 1.7 × 10−13 cm3 s−1,16 independent of pressure, from which they concluded that the reaction is already at its high-pressure limit. Computational studies on the reaction have been reported by Chen and Bozzelli as well as by Zheng et al.17,18 Chen and Bozzelli searched for the stationary points (local minima and transition structures connecting neighbouring minima) on the potential energy surface (PES) of reaction (1) using several levels of theory (CBS-q, MP2, and B3LYP) and combined their quantum chemical calculations with QRRK/master equation simulations. They found that the most probable reaction pathways for the initial association product are dissociation back to reactants as well as formation of the QOOH radical by an internal hydrogen abstraction reaction. The dominant bimolecular product channel was predicted to be the formation 2-methylidene-1,3-epoxypropane and hydroxyl radical. Zheng et al. computed the stationary points of reaction (1) at the CBS-QB3 level of theory. Except for a few transition structures, their relative energies were in good agreement with those of Chen and Bozzelli. Zheng et al. further investigated the subsequent QOOH + O2 reaction pathway and found the formation of 2-(hydroperoxymethyl)prop-2-enal, a ketohydroperoxide (KHP) species, and hydroxyl radical to be the most likely bimolecular product channel.
In this work, we have measured reaction (1) as a function of temperature (203–730 K) and pressure (0.2–9 torr) in helium and nitrogen bath gases using laser-photolysis photoionization mass spectrometry (LP-PIMS). Quantum chemical calculations and master equation simulations were performed to complement our experimental work. Kinetically important parameters in the master equation model were optimized using the trace fitting feature implemented in MESMER. The results of this optimization were compared with the more conventional approach where one uses experimental rate coefficient data in parameter optimization. The optimized master equation model was then used to simulate reaction (1) over a wide temperature and pressure range.
2 Experimental
We have measured the kinetics of reaction (1) in real time using a laminar flow reactor coupled to a photoionization mass spectrometer. Laser-photolysis was used for radical production. The measurements were performed at low pressures (p < 10 torr) using tubular stainless steel (halocarbon wax coating), Pyrex (polydimethylsiloxane coating), and quartz (boric oxide coating) reactors. The stainless steel reactors had inner diameters of 0.80 cm or 1.70 cm, the Pyrex reactor had an inner diameter of 1.65 cm, and the quartz reactors had inner diameters of 0.85 cm or 1.70 cm.A brominated precursor (3-bromo-2-methylpropene, purity ≥97%, Sigma-Aldrich) was used in all experiments. The liquid precursor was degassed with several freeze–pump–thaw cycles before its use. Gaseous precursor was introduced to the reactor by bubbling helium (or nitrogen) through temperature-controlled liquid precursor. Helium (purity 99.9996%), molecular nitrogen (purity 99.9996%), and molecular oxygen (purity 99.9995%) were used as supplied. The radical under study was homogeneously produced along the reactor by photolysis of the precursor with a pulsed KrF exciplex laser (λ = 248 nm). The laser fluences used were between 4–44 mJ cm−2. The major photolysis reaction observed is
| CH2C(CH3)CH2Br + hν → CH2C(CH3)CH2˙ + Br˙. | (2) |
We performed the experiments under pseudo-first-order conditions ([O2] ≫ [CH2C(CH3)CH2˙]) with helium bath gas being in large excess over molecular oxygen (pO2/ptot < 0.05). Nitrogen (N2) was used as bath gas in a few experiments. A small, known portion (≤20%) of the flowing gas mixture was sampled through a small hole on the side of the reactor into a vacuum chamber containing the photoionization mass spectrometer. Either a xenon lamp (E = 8.44 eV) with a sapphire window or a chlorine lamp (E = 8.9–9.1 eV) with a CaF2 or BaF2 window was used to ionize 2-methylallyl radicals. Products were sought using a hydrogen lamp (E = 10.2 eV) with a MgF2 window and a neon lamp (E = 16.7 eV). With the neon lamp we used a collimated hole structure (CHS) in place of a salt window. We observed product signals at m/z = 15 and m/z = 40 with H/MgF2 when no oxygen was added, which indicates that these products are produced directly by the photolysis event. The signals most likely correspond to methyl radical (m/z = 15) and allene (propadiene, m/z = 40).
In a typical bimolecular rate coefficient measurement at a fixed p and T, we perform the following steps:
1. The wall rate kw of the studied radical is measured at the beginning of the rate coefficient measurement. The wall rate is the decay rate of the radical in the absence of molecular oxygen (or some other reactant). The wall rate accounts for the reaction of the radicals with the reactor surface, the self-reaction of the radicals, and for the reaction between the radicals and the radical precursor molecules. Low radical and precursor concentrations are typically used to minimize the effect of self-reactions and radical-precursor reactions. The wall rate measurement is repeated at the end of a bimolecular rate coefficient measurement to ensure that it has remained approximately constant. A single-exponential function
| [R˙] = A + [R˙]0e−kwt | (3) |
2. After the wall rate measurement, a known concentration of oxygen is added into the reactor. The radical decay rate is then monitored and a single-exponential function
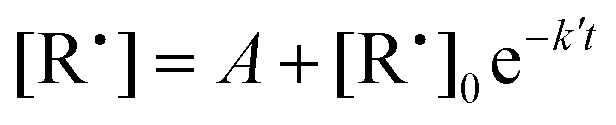 | (4) |
| k′ = k[O2] + kw, | (5) |
3. Finally, the obtained pseudo-first order rate coefficients are plotted as a function of [O2]. The slope of a linear fit made onto these data points gives the bimolecular rate coefficient k. The intercept of the fit gives kw, and the kw value obtained from the fit is compared with the measured kw value to assess the reliability of the fit. We typically report both values, and this is also done in this work (see Table S1, ESI†).
When redissociation of the association adduct (R˙ + O2 ← RO2˙) is significant, the procedure described above cannot be used, because the radical trace is not single- but double-exponential. For double-exponential decays, we use an alternative approach that permits us to determine both the forward and reverse rate coefficients.10 The procedure is as follows:
1. The wall rate is measured (same as previously).
2. A known concentration of oxygen is added to the reactor and the radical decay rate is monitored.
3. A double-exponential function
| [R˙] = A + Be−λ1t + Ce−λ2t | (6) |
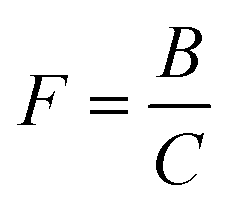 | (7) |
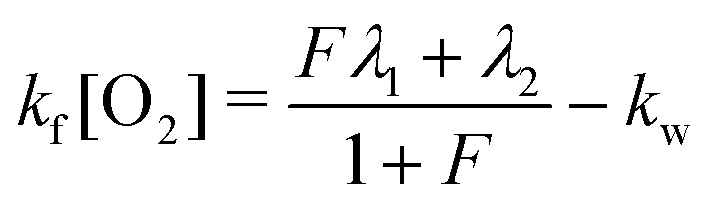 | (8) |
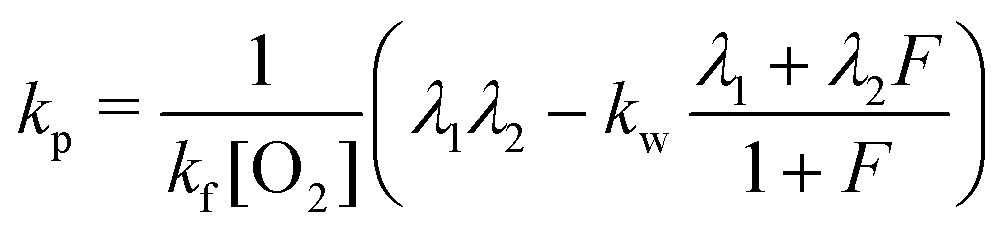 | (9) |
| kr = λ1 + λ2 − kf[O2] − kw − kp. | (10) |
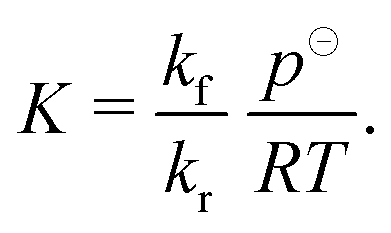 | (11) |
The experimental apparatus has been described in more detail elsewhere.19
3 Quantum chemistry
The geometries of the stationary points on the PES of reaction (1) were optimized at the MN15/Def2TZVP level of theory.20,21 We performed intrinsic reaction coordinate (IRC) calculations to ensure that each saddle point on the PES connects the right local minima. The same level of theory was used to compute harmonic frequencies and zero-point energies (ZPEs) and to perform relaxed PES scans (5° increments) to obtain one-dimensional hindered rotation potentials. The harmonic frequencies and zero-point energies were scaled by a factor of 0.979.22Single-point energies were calculated for the stationary points at the ROHF-CCSD(T),23 UHF-CCSD(T), ROHF-DLPNO-CCSD(T1),24,25 and CASPT2 levels of theory. The T1 in the parentheses means that an improved, iterative triples calculation is used in the DLPNO method; it should not be confused with the T1 diagnostic.26 The correlation consistent basis sets cc-pVDZ, cc-pVTZ, and cc-pVQZ were used in the single-point calculations.27 A three-parameter exponential function
| EHF(X) = EHF,∞ + BHFe−αHFX | (12) |
| Ecorr(X) = Ecorr,∞ + BcorrX−αcorr. | (13) |
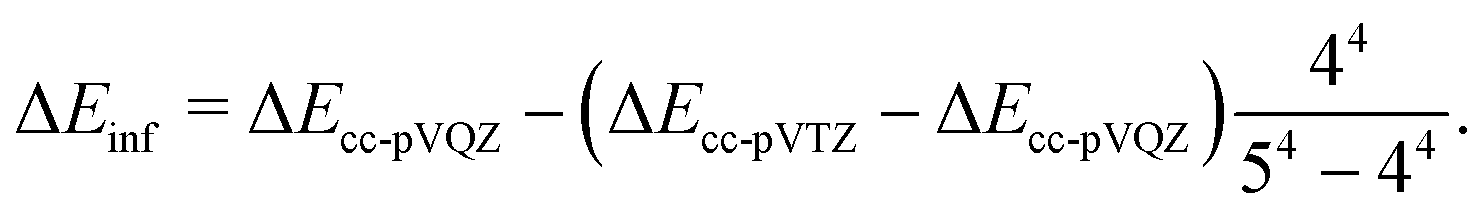 | (14) |
4 Master equation modeling
We used MESMER (version 6.1)2 in our master equation simulations. MESMER is a one-dimensional master equation code, meaning that angular momentum dependence is not considered (not even in an averaged manner). Neglecting to properly account for angular momentum effects introduces some error, but it is not believed to be the main source of uncertainty in master equation simulations of unimolecular reactions.2 RRKM theory was used to calculate microcanonical rate coefficients for reactions that have well-defined barriers. For barrierless reactions, we used the Inverse Laplace Transform (ILT) approach implemented in MESMER to obtain the number of states available for the loose transition state. The expression that is transformed is the modified Arrhenius equation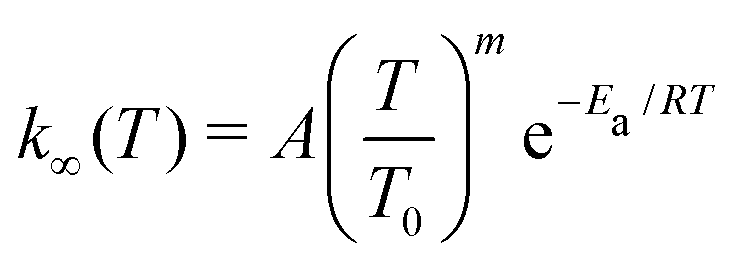 | (15) |
The exponential-down model
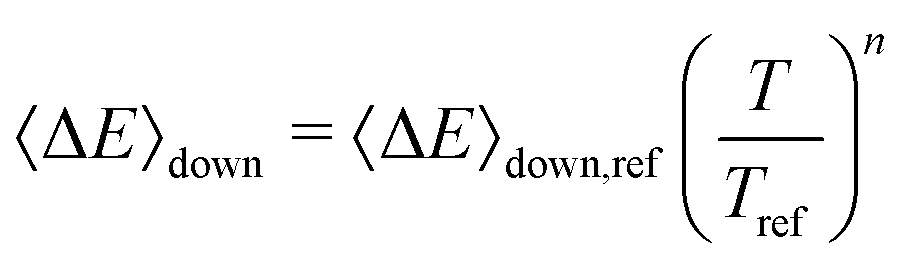 | (16) |
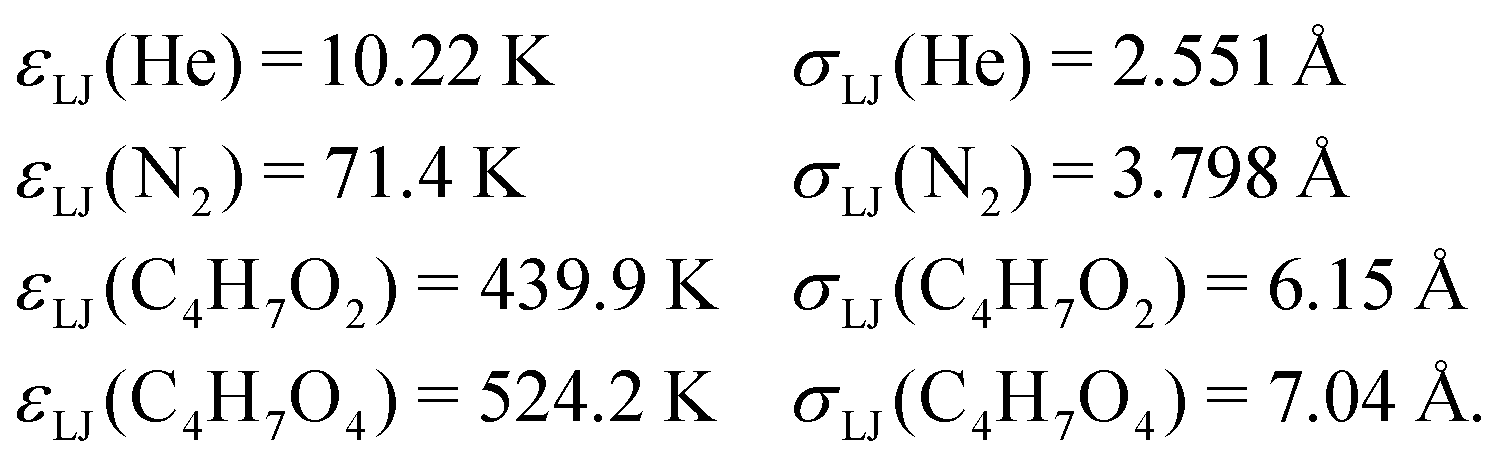
An energy grain size of 75 cm−1 was used in all simulations. The cut-off energy was set to 25kBT above the highest energy stationary point. The Eckart tunneling model was used to calculate tunneling corrections for hydrogen abstraction reactions.
To treat the coupling between internal and external rotations, we used the method of Gang et al. implemented in MESMER.35 This treatment is classical, so to ensure that the ZPEs of the internal rotations are not double-counted, we subtracted these ZPEs from the ZPE-corrected relative energy of each species. We calculated the ZPEs for internal rotations with the uncoupled quantum mechanical hindered rotor model. While the method of Gang et al. is general (within the classical approximation), the current implementation in MESMER 6.1 does not explicitly treat potential coupling between hindered rotors.
When using experimental rate coefficient data (ki,exp(p,T)) for parameter optimization in MESMER, the expression that is being minimized is
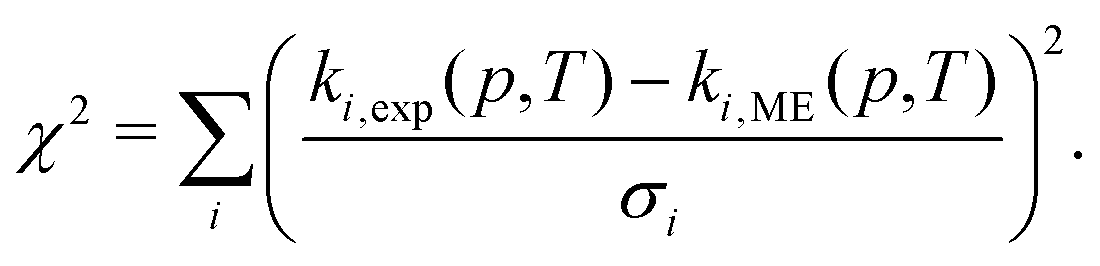 | (17) |
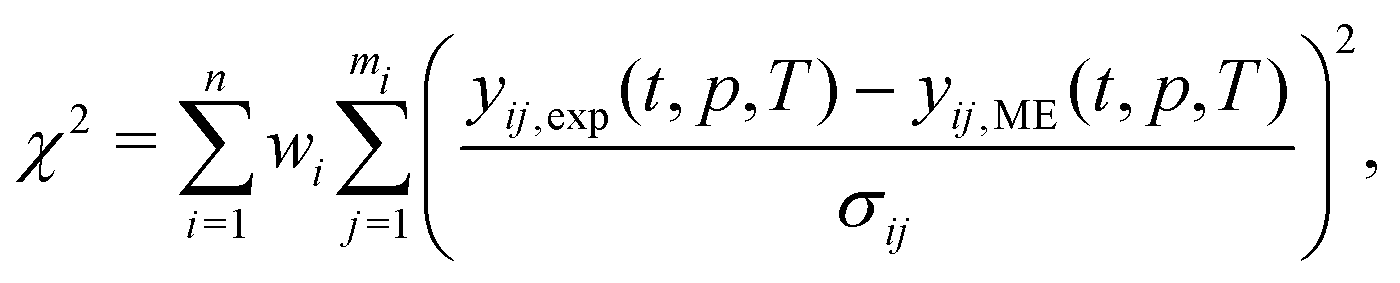 | (18) |
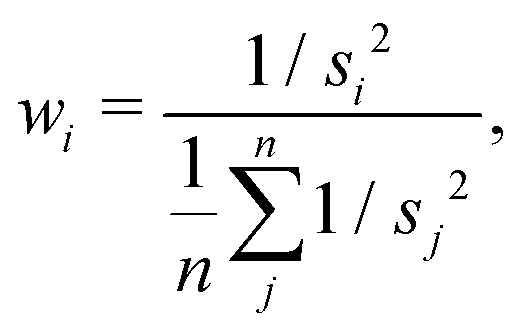 | (19) |
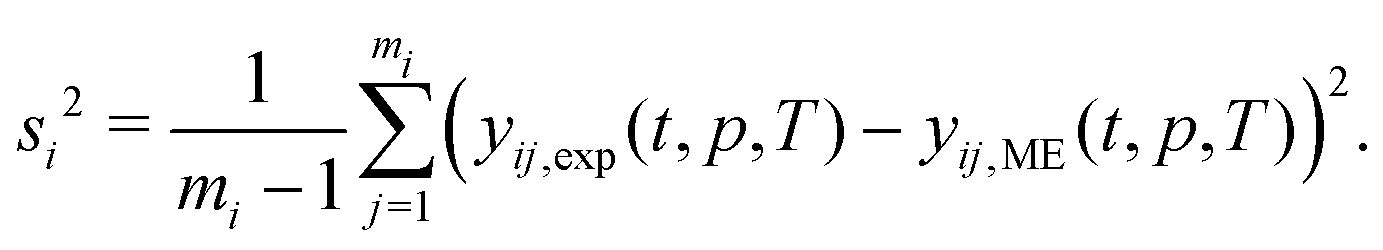 | (20) |
A practical problem with fitting traces is that the initial concentration of the deficient reactant is not known in the experiments. This issue has been addressed by Medeiros et al. using a least squares fit of the amplitude of the calculated trace to the measured trace. The details are presented in ref. 3.
5 Results and discussion
5.1 Experimental
In the experimental temperature range, the bimolecular rate coefficient kf exhibits negative temperature dependence and depends on bath gas density. At slightly elevated temperatures (347–410 K), the dissociation reaction back to reactants becomes significant and the observed radical traces were double-exponential. In this temperature range, both kf and kr were determined, which were then used to calculate the equilibrium constant of the association reaction. We further investigated reaction (1) at high temperatures, but no clear reaction between CH2C(CH3)CH2˙ and O2 could be observed even at T = 730 K and [O2] = 2.54 × 1016 cm−3. At this temperature, we determined an upper limit value of 2 × 10−16 cm3 s−1 for the rate coefficient. At temperatures above 730 K the radical precursor 3-bromo-2-methylpropene becomes thermally unstable and kinetic measurements were no longer possible. Examples of measured traces are shown in Fig. 2 and 3. An example of a bimolecular plot is given in Fig. 3. This figure also displays a modified van't Hoff plot of the measured equilibrium constants. We have included tables in the ESI,† that detail the experimental conditions and result of each measurement (Tables S1–S3, ESI†). No reaction products were observed at any temperature. We estimate the uncertainty of the bimolecular rate coefficient and equilibrium constant measurements to be ±15%. The main source of uncertainty is the uncertainty in the employed O2 concentration, which results from uncertainties in measured helium and oxygen flow rates. Uncertainties in the fitting parameters of the fitting functions are also a source of uncertainty–especially in the measurements where a four-parameter function was fitted to the traces.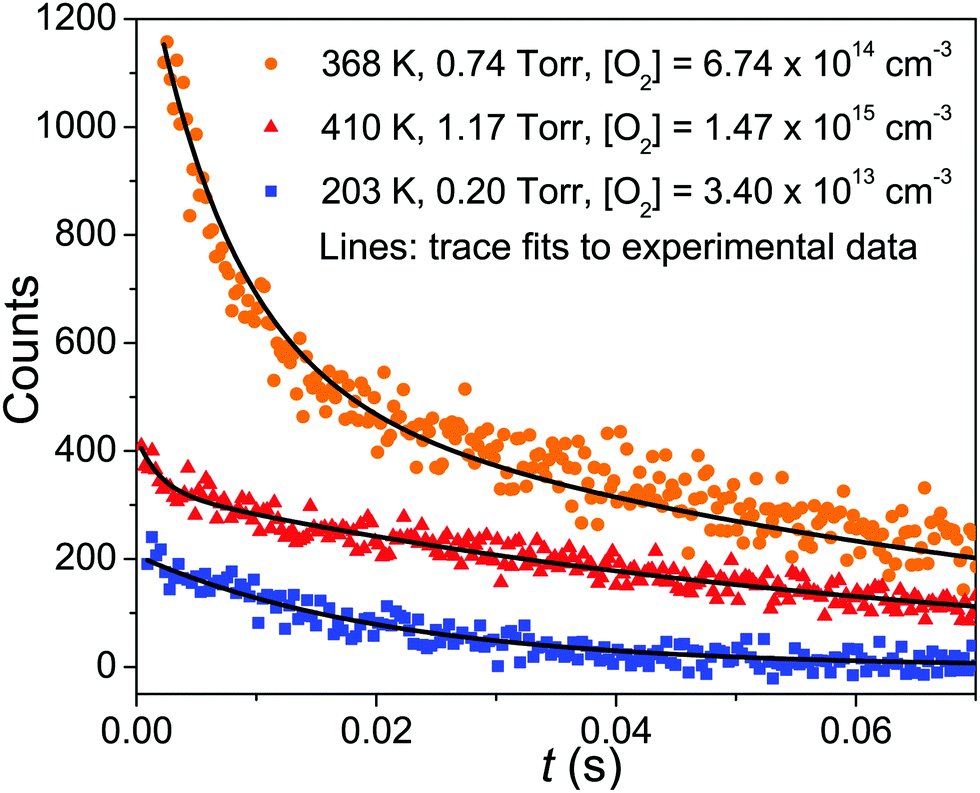 | ||
| Fig. 2 Examples of measured kinetic traces (symbols). The lines are simulated traces from our optimized master equation model. | ||
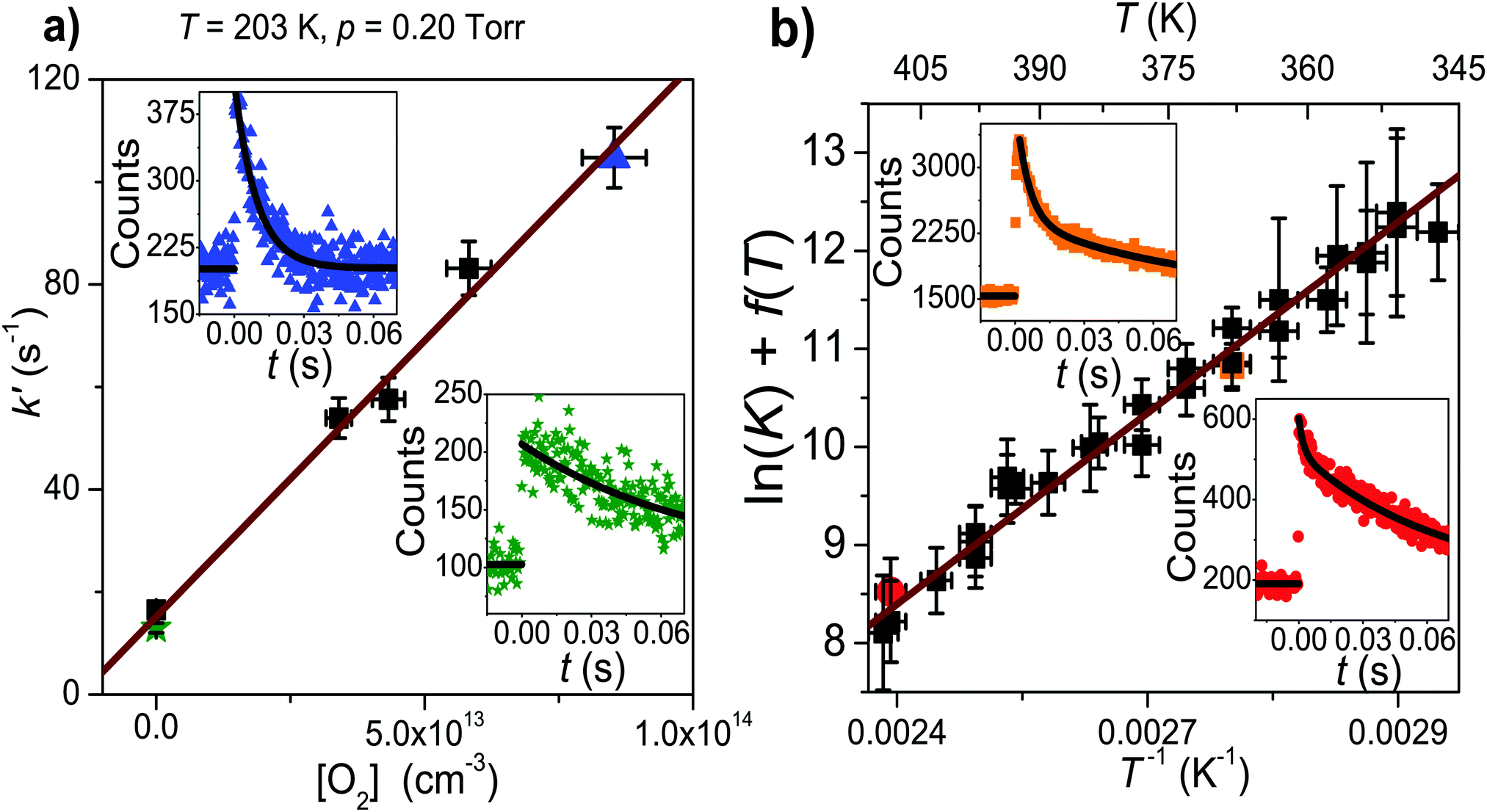 | ||
| Fig. 3 (a) A typical low-temperature bimolecular plot. A radical trace in the absence of O2 is shown in the bottom right corner. A radical trace with added oxygen is shown in the top left corner. (b) A modified van't Hoff plot of ln(K) + f(T) versus reciprocal temperature for the CH2C(CH3)CH2˙ + O2 ⇌ CH2C(CH3)CH2OO˙ reaction. Radical traces measured at 368 K (orange) and 410 K (red) are shown in the top left and bottom right corners, respectively. In both figures, the coloured symbols depict the measurements that correspond to the shown traces. | ||
5.2 Quantum chemistry and thermochemistry
The results of our quantum mechanical calculations are given in the ESI† (Table S4). We found that the energy difference between UHF-CCSD(T)/CBS and ROHF-CCSD(T)/CBS energies were generally less than two kJ mol−1. However, some transition structures had huge spin contaminations and in these cases the energy differences between UHF-CCSD(T) and ROHF-CCSD(T) could be quite large. For this reason, ROHF-CCSD(T) calculations were preferred. Fig. S1 in the ESI† shows the optimized geometries of all stationary points. Preliminary master equation simulations were run with the ROHF-CCSD(T)/CBS energies to find the reaction channels that had non-negligible branching ratios at high temperatures and pressures. Reaction channels with negligible branching ratios were removed from the model to reduce the computational cost of the simulations. The reaction enthalpy profile shown in Fig. 4 shows the kinetically relevant reaction pathways and this is the profile that was used in master equation simulations (except for the preliminary tests). In Table 1 we provide the computational details for the species that were included in the final model. An optical symmetry number of two was specified for all species except for 2-methylallyl, O2, Int1, Int2, and Int7. 2-Methylallyl and O2 do not have optical isomers and in Int1, Int2, and Int7 internal rotations permit interconversion between mirror images.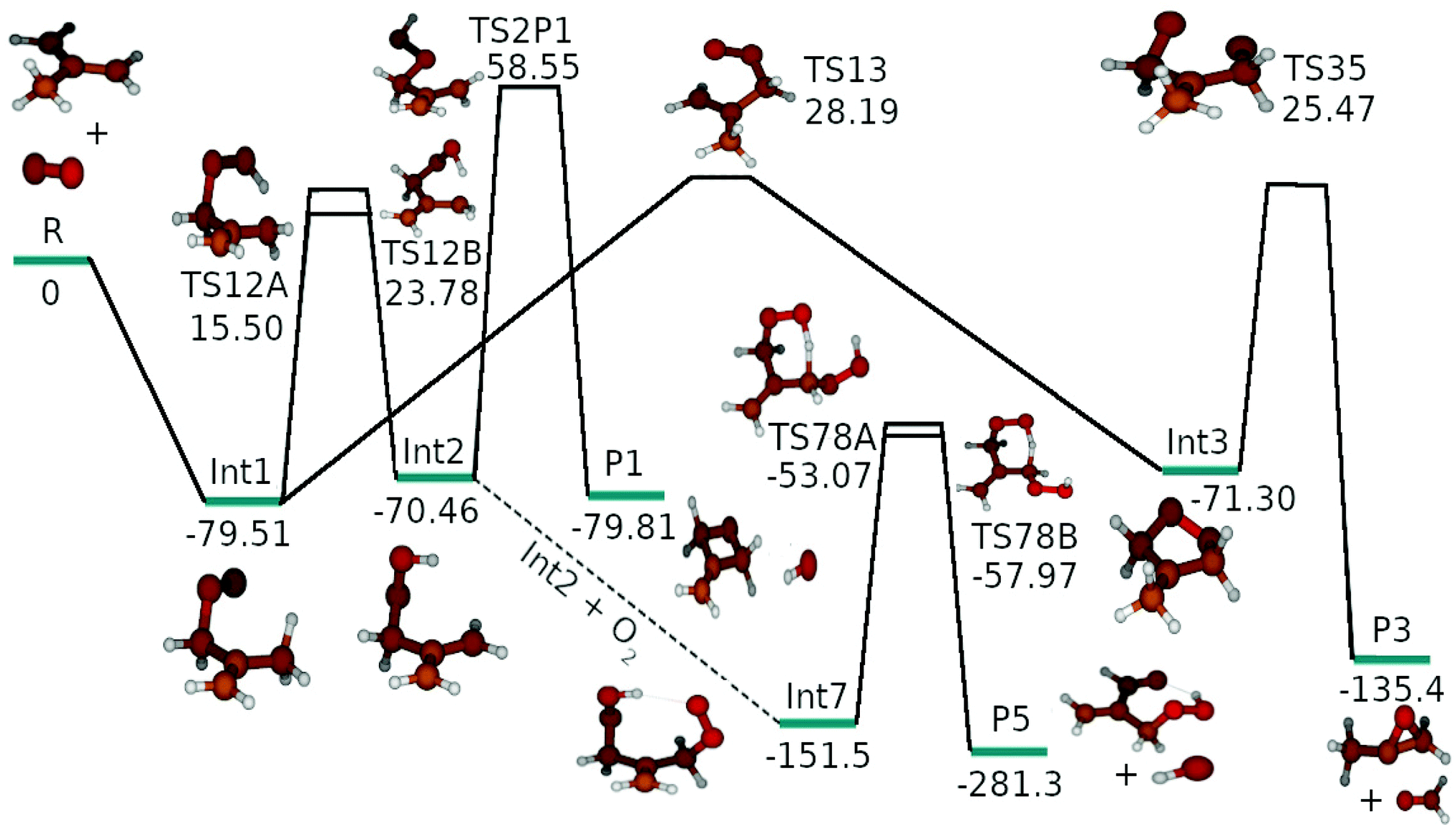 | ||
| Fig. 4 The zero-kelvin reaction enthalpy profile used in the master equation simulations. The energies are in kJ mol−1. | ||
| Species | (σextσint)/mopta | ROHF-CCSD(T)b | DLPNO-ROHF-CCSD(T1)c | CASPT2de |
|---|---|---|---|---|
| kJ mol−1 | kJ mol−1 | kJ mol−1 | ||
| a Here σext and σint are the external and internal symmetry numbers, respectively, and mopt is the optical symmetry number. b The value in the parentheses is the T1 diagnostic. c The value in the parentheses is the T1 diagnostic. The tight PNO setting was specified in the DLPNO calculations. d The value in the parentheses is the reference weight. e The CASPT2 energies here are reported relative to Int1. The reported Int1 value is the optimized well depth from the weighted trace fit. | ||||
| R | (2·3)/1, (2·1)/1 | 0 (0.029, 0.017) | 0 (0.028, 0.017) | |
| Int1 | (1·3)/1 | −81.12 (0.025) | −80.57 (0.023) | −79.51 (0.78)e |
| Int2 | (1·2)/1 | −70.46 (0.023) | −72.51 (0.022) | −73.52 (0.78) |
| Int3 | (1·3)/2 | −71.30 (0.015) | −73.73 (0.014) | −69.49 (0.78) |
| TS12A | (1·1)/2 | 21.52 (0.027) | 23.30 (0.024) | 15.50 (0.77) |
| TS12B | (1·1)/2 | 27.81 (0.024) | 23.78 (0.77) | |
| TS2P1 | (1·1)/2 | 66.69 (0.026) | 68.31 (0.025) | 58.55 (0.77) |
| TS13 | (1·3)/2 | 32.69 (0.026) | 35.07 (0.030) | 28.19 (0.78) |
| TS35 | (1·3)/2 | 13.43 (0.070) | 39.24 (0.046) | 25.47 (0.78) |
| P1 | −79.81 (0.011, 0.013) | −86.18 (0.011, 0.016) | ||
| P3 | −135.4 (0.018, 0.015) | −138.4 (0.017, 0.015) | ||
| Int7 | (1·1)/1 | −151.5 (0.022) | ||
| TS78A | (1·1)/2 | −53.07 (0.021) | ||
| TS78B | (1·1)/2 | −57.97 (0.021) | ||
| P5 | −281.3 (0.013, 0.016) | |||
For the initial association reaction, R → Int1, a transition structure was found at the MN15/Def2TZVP level of theory. At this level of theory the relative energy of this structure was slightly negative, −0.5054 kJ mol−1. A ROHF-CCSD(T)/CBS single-point calculation at this geometry reduces the energy of the submerged barrier to −5.588 kJ mol−1, but this energy is not reliable because the T1 diagnostic is quite high, 0.047. Nevertheless, the experimental findings are consistent with a barrierless association reaction. We did not investigate the association potential in more detail because this information is not needed if one uses the Inverse Laplace transform approach to compute microcanonical rate coefficients for the association reaction.
Although T1 diagnostics were generally acceptable, below 0.04 (TS35 being an exception with a high T1 diagnostic of 0.070), we chose to calculate CASPT2 energies for the kinetically important structures. This was motivated by our recent work14 on the kinetics of the reaction between 1,1-dimethylallyl and O2 where we discovered that ROHF-CCSD(T) energies were unable to explain the observed kinetics at high temperatures. Similarly to the current system, the T1 diagnostics appeared acceptable. CASPT2 yielded lower barrier heights than ROHF-CCSD(T) and these barriers were more consistent with the experimental observations. In the present study, CASPT2 also predicts lower barriers, and the relative energies of transition structures are roughly 5–10 kJ mol−1 lower with CASPT2 than with ROHF-CCSD(T) (see Table S4, ESI†). Accordingly, we have chosen to use CASPT2 energies for all transition structures in this work. For stable intermediates, we decided to use the ROHF-CCSD(T) energies because they were very similar to the CASPT2 energies and, therefore, presumably reliable. We note, however, that in this work no reaction is observed at high temperatures, and there is no obvious way of telling whether the CASPT2 energies of the transition structures are more accurate than the ROHF-CCSD(T) energies.
An (11,11) active space was used in the CASPT(2) calculations. For the reaction channel Int1 → Int2 → P1, this active space consists of the bonding and anti-bonding O–O σ-orbitals (2,2), the bonding and anti-bonding C–O σ-orbitals (2,2), the bonding and anti-bonding C–C π-orbitals (2,2), the bonding and anti-bonding C–H σ-orbitals of the hydrogen that is abstracted (2,2), the non-bonding and anti-bonding orbitals of the lone pair of the non-terminal oxygen (2,2), and the radical orbital (1,1) of the terminal oxygen. For the five-membered ring channel, Int1 → Int3 → P3, the active space was the same except that the bonding and anti-bonding C–H σ-orbitals were replaced by the non-bonding and anti-bonding orbitals of the lone pair of the terminal oxygen.
For the QOOH + O2 (Int2 + O2) channel, DLPNO-ROHF-CCSD(T1) energies were used. This is for the simple reason that ROHF-CCSD(T) and CASPT2 calculations are prohibitively expensive for species of this size. One can see from Table 1 and Table S4 (ESI†) that the difference between the DLPNO-ROHF-CCSD(T1)/CBS and the ROHF-CCSD(T)/CBS energies is generally less than four kJ mol−1, so we do not believe there are gross errors in the energies in the QOOH + O2 channel.
We used the third-law method to evaluate the well-depth of the initial association reaction. To do this, a correction function
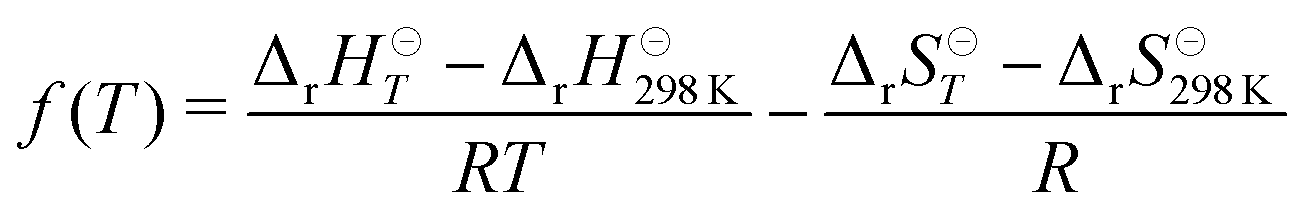 | (21) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K is fixed with a computational value and ΔrH⊖298K is obtained from the slope of a linear fit to ln(K) + f(T) data. This procedure yielded the following values:
K is fixed with a computational value and ΔrH⊖298K is obtained from the slope of a linear fit to ln(K) + f(T) data. This procedure yielded the following values:| ΔrS⊖298K = –128.7 J mol−1 K−1 (computational) |
| ΔrH⊖298K = –82.40 kJ mol−1 (computational) |
| ΔrH⊖298K = –81.03 ± 0.11 kJ mol−1 (third-law). |
K is provided for comparison, and as can be seen, the agreement between the fitted and computed value is very good. The difference between the two enthalpies results mainly from the difference between the actual and computed Int1 well depth. Thus, the computed well depth can be adjusted with the difference between the room-temperature enthalpies to obtain an “experimental” well depth ΔrH⊖0 = –79.75 kJ mol−1.
5.3 Master equation modeling
In the fitting simulations, only the initial association/dissociation reaction was considered (CH2C(CH3)CH2˙ + O2 ⇌ CH2C(CH3)CH2OO˙). This was done to reduce the computational cost of the simulations. Under the conditions of our experiments, only the initial association/dissociation reaction is significant. As mentioned before, we were unable to see any further reaction even at 730 K, indicating that any reaction over TS12A and TS12B is very slow. Furthermore, the values of the irreversible first order loss rate kp (see eqn (9)) remain approximately constant across the temperature range 347–410 K (see Table S2, ESI†). This very strongly suggests that this value is in fact just the wall rate of the peroxyl adduct and that unimolecular isomerization reactions do not contribute to kp. Thus, the kp values were used as the peroxyl adduct wall rates in the trace fit simulations. Fits were first performed using our helium bath gas data. In the nitrogen bath gas fits, all the parameters were fixed to their optimized helium bath gas values except 〈ΔE〉down,ref. The Arrhenius parameters and the well depth of Int1 do not depend on the bath gas. The parameter n does depend on the bath gas, but we have insufficient temperature-dependent data in nitrogen bath gas to fit this parameter. Altogether 173 experimental traces were simultaneously fitted in the helium bath gas trace fits. The corresponding number of traces in the nitrogen bath gas trace fits was 21.
The results of rate coefficient, unweighted trace, and weighted trace fits are tabulated in Table 2. We used the three sets of optimized parameters to plot fall-off curves for reaction (1) in helium bath gas and this is depicted in Fig. 5. The same figure shows the effect the well depth of Int1 has on the equilibrium constant. The simulation results are shown together with experimental results. As can be seen, all three fits give very similar values for the optimized parameters and the quality of the three fits is difficult to distinguish. In fact, the optimized parameters of the three sets coincide within or almost within fitting uncertainties (1σ). Based on a visual inspection of the fall-off curves, one can speculate that in the rate coefficient fit the fall-off curves begin to bend toward the high-pressure limit too early.
| Parameter | Rate coefficient fit | Unweighted trace fit | Weighted trace fit |
|---|---|---|---|
| a Fixed to the third-law analysis value. | |||
| A (10−12 cm3 s−1) | 1.95 ± 0.06 | 2.46 ± 0.44 | 2.42 ± 0.35 |
| m | −0.727 ± 0.086 | −0.351 ± 0.387 | −0.387 ± 0.318 |
| 〈ΔE〉(He)down,ref (cm−1) | 146 ± 3 | 139 ± 10 | 144 ± 11 |
| n | 0.0746 ± 0.1131 | 0.100 ± 0.216 | 0.0235 ± 0.363 |
| ΔrH⊖0 (kJ mol−1) | −79.75a ± 0.11 | −79.44 ± 0.05 | −79.51 ± 0.09 |
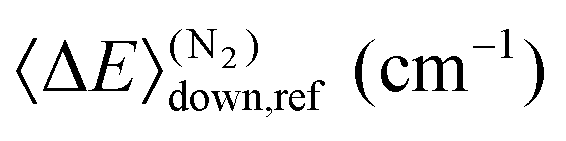
|
353 ± 7 | 339 ± 45 | 336 ± 57 |
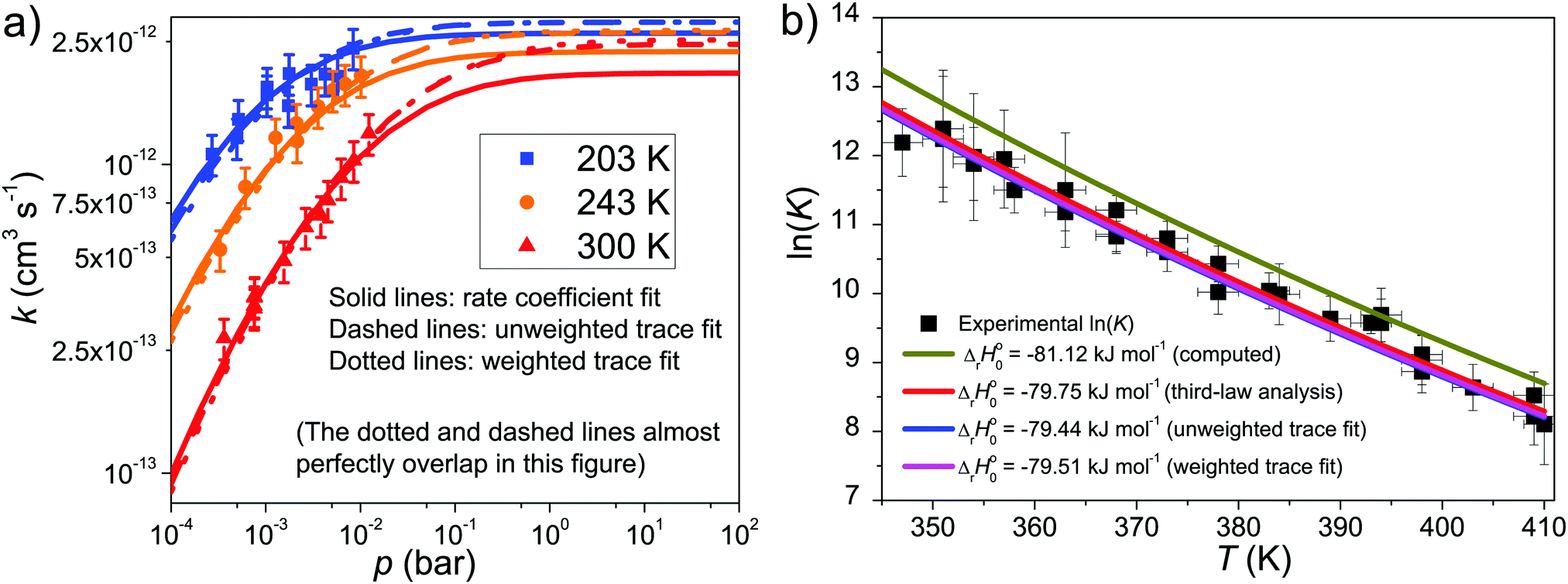 | ||
| Fig. 5 (a) Fall-off curves for reaction (1) calculated using three different sets of optimized parameters obtained from rate coefficient fitting or trace fitting with or without weighting. The results are shown alongside the experimental results. (b) The equilibrium constant of the CH2C(CH3)CH2˙ + O2 ⇌ CH2C(CH3)CH2OO˙ reaction plotted as a function of temperature with four different ΔrH⊖0 values. The results are shown alongside the experimental results. The bath gas is helium in all experiments and simulations. | ||
From this point onward, all simulations were run using the reaction enthalpy profile shown in Fig. 4 and the parameters obtained from the weighted trace fit. Unless otherwise stated, nitrogen bath gas used. The QOOH + O2 reaction (Int2 + O2 → Int7) is included in the ME model because we expected it to be the main sink of 2-methylallyl at low temperatures. We assumed this reaction to be barrierless and assigned it the optimized ILT Arrhenius parameters that were obtained for the CH2C(CH3)CH2˙ + O2 → CH2C(CH3)CH2OO˙ reaction. This is a very crude approximation, and the results obtained for the QOOH + O2 channel are semi-quantitative at best.
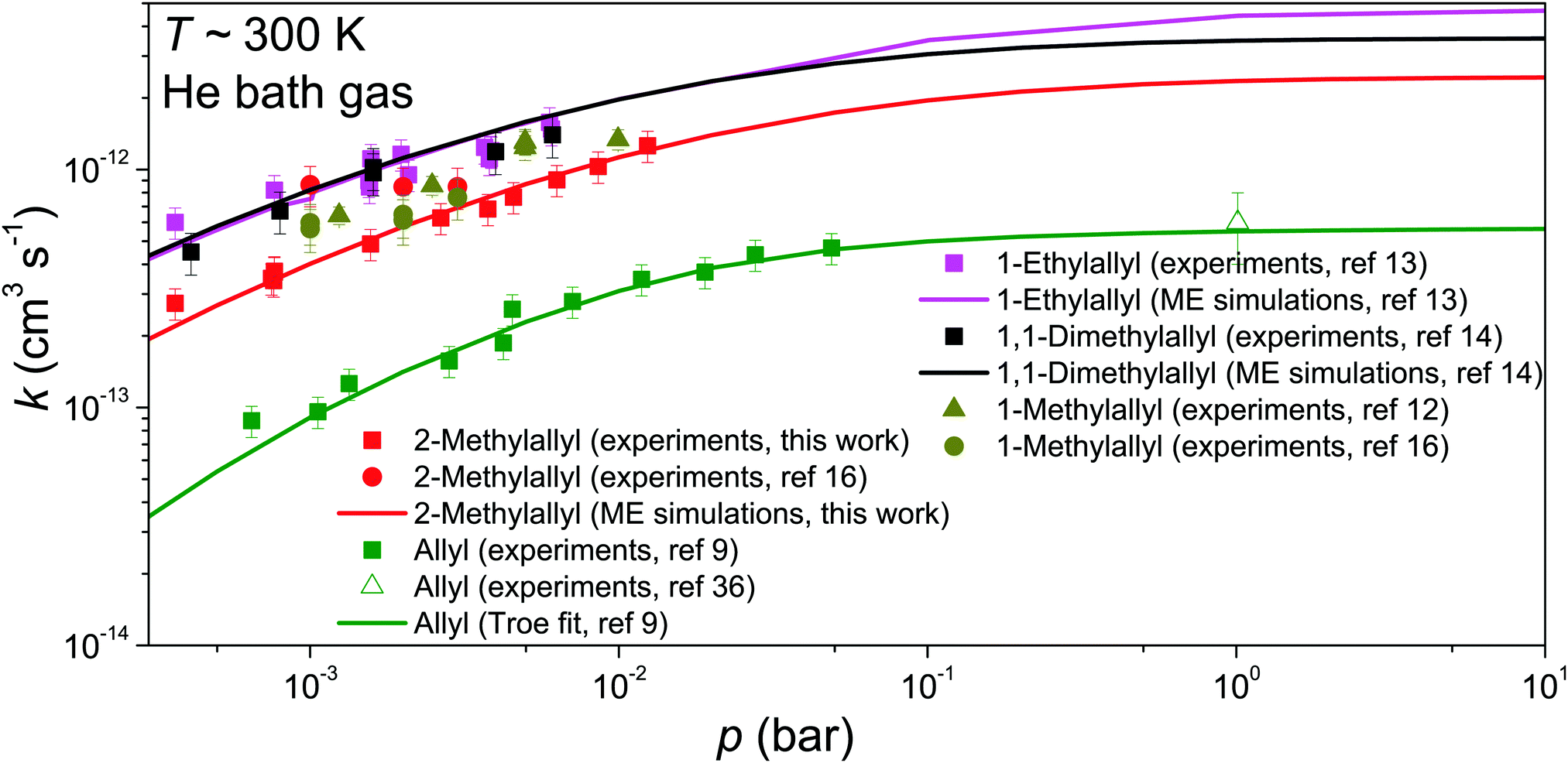 | ||
| Fig. 6 Fall-off curves for allylic radical + O2 reactions at around 300 K in helium bath gas. | ||
The comparison of our experimental and modeling results for 2-methylallyl with the experimental results of Schleier et al.16 shows that the measured rate coefficients are in pretty good agreement; at 0.003 bar the measurements agree within experimental uncertainty and at 0.001 bar there is only a factor of two difference. There is, however, a discrepancy. Schleier et al. observe no pressure dependence and conclude that the reaction is at its high-pressure limit already at 0.001–0.003 bar, whilst our results clearly show pressure dependence in this pressure range. In our view, our results are more in line with what is known about the pressure dependence of hydrocarbon radical + O2 reactions. Furthermore, we have consistently observed with our apparatus that allylic radical + O2 reactions are in the fall-off region between 0.001–0.01 bar. The same has been observed by Slagle et al. and Knyazev et al.,12,38 though it should be noted that their experimental technique is very similar to ours. We also believe that the features of the potential energy surface of reaction (1), namely the shallow well, support the finding that the reaction is still in the fall-off region between 0.001–0.003 bar. Pressure independence can be observed for a radical–molecule reaction if there is a well-skipping reaction pathway that leads directly to bimolecular products. In the present system, all bimolecular product channels have barriers with energies above that of the reactants, which means that well-skipping will not be significant at low temperatures.
We did not include in the comparison the computational results of Chen and Bozzelli and by Zheng et al.17,18 in Fig. 6 because their kinetic predictions are orders of magnitude slower than our experimental and modeling results. They found a small positive barrier for the association reaction between 2-methylallyl and O2 and this has a huge impact on the kinetic predictions. The positive barrier almost certainly results from the use of low-level single-reference methods. In general, multi-reference methods are needed to probe the association potentials in radical-molecule reactions, in particular those involving molecular oxygen.
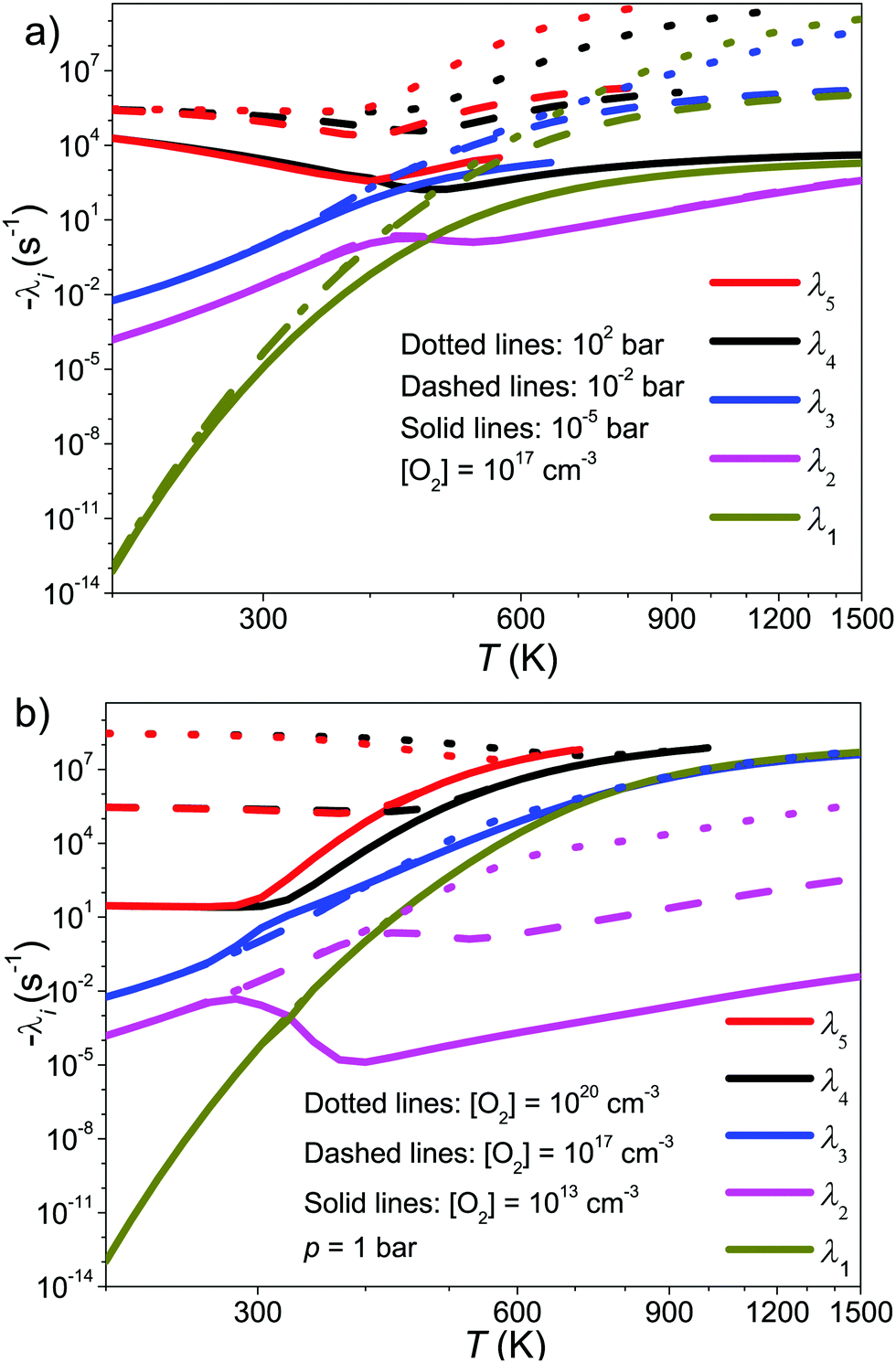 | ||
| Fig. 7 Chemically significant eigenvalues of the CH2C(CH3)CH2˙ + O2 reaction plotted as a function of temperature, pressure, and O2 concentration. | ||
In Fig. 7a we show the pressure and temperature dependence of the CSEs at constant [O2]. Some of the CSE curves terminate abruptly and this is because at the termination point they merge with the “sea of IEREs”. Several observations can be made from the subfigure:
1. Eigenvalues λ5 and λ4 correspond mainly to the reactions R ⇌ Int1 and Int2 ⇌ Int7, respectively, and are connected to their association and dissociation rate coefficients approximately by −λ4/5 ≈ kf[O2] + kr. At high temperatures, λ2 is the loss rate of 2-methylallyl and is connected to the phenomenological loss rate coefficient by kph[O2] ≈ −λ2. Eigenvalues λ1 and λ3 cannot be easily equated with a single reaction step, but it is clear from their pressure dependence that these describe the unimolecular isomerization reactions in the system.
2. There is a transition zone between 350–500 K where the behavior of some of the CSE curves change. Inspection of simulated traces shows that in this region 2-methylallyl decays are not single-exponential and there is equilibration between the reactants and the initial peroxyl adduct. Below 350 K, the decays are single-exponential and the eigenvalue that describes these decays is −λ5 ≈ kf[O2]. Above 500 K, single-exponential decays are again observed and the relevant eigenvalue is kph[O2] ≈ −λ2.
3. Eigenvalue λ2 is pressure-independent for all practical purposes, meaning that the phenomenological loss of 2-methylallyl will be pressure-independent at elevated temperatures.
Fig. 7b illustrates the [O2]-dependence of the CSE curves and this subfigure confirms many of the conclusions that were made based on subfigure (a). Eigenvalues λ5 and λ4 depend linearly on [O2] at low temperatures, as they should, and λ2 depends linearly on [O2] at high temperatures, as it should. The eigenvalues λ1 and λ3 have very weak [O2]-dependence, which makes sense if they mainly describe unimolecular isomerization reactions. Furthermore, it can be seen that increasing O2 concentration pushes the transition zone (or equilibration zone) to higher temperatures, which is as one would expect.
Because λ2 is pressure-independent, it is tempting to assume that the branching ratios will also be pressure-independent. In Fig. 8a we display the branching ratios as a function of temperature and pressure, which indeed demonstrates that they have very weak pressure dependence. Fig. 8b shows the [O2]-dependence of the branching ratios. One can see the employed O2 concentration has a huge effect on the branching ratios and this is because the QOOH + O2 channel is included in the model. Under realistic autoignition conditions, (hydroperoxymethyl)prop-2-enal and hydroxyl radical (product channel P5) will be the dominant product channel. As temperature is increased, formation of product P3 (1,2-epoxypropan-2-yl + methanal) becomes the dominant product channel. At above 1000 K, P1 (2-methylidene-1,3-epoxypropane + hydroxyl) replaces P3 as the most important product channel.
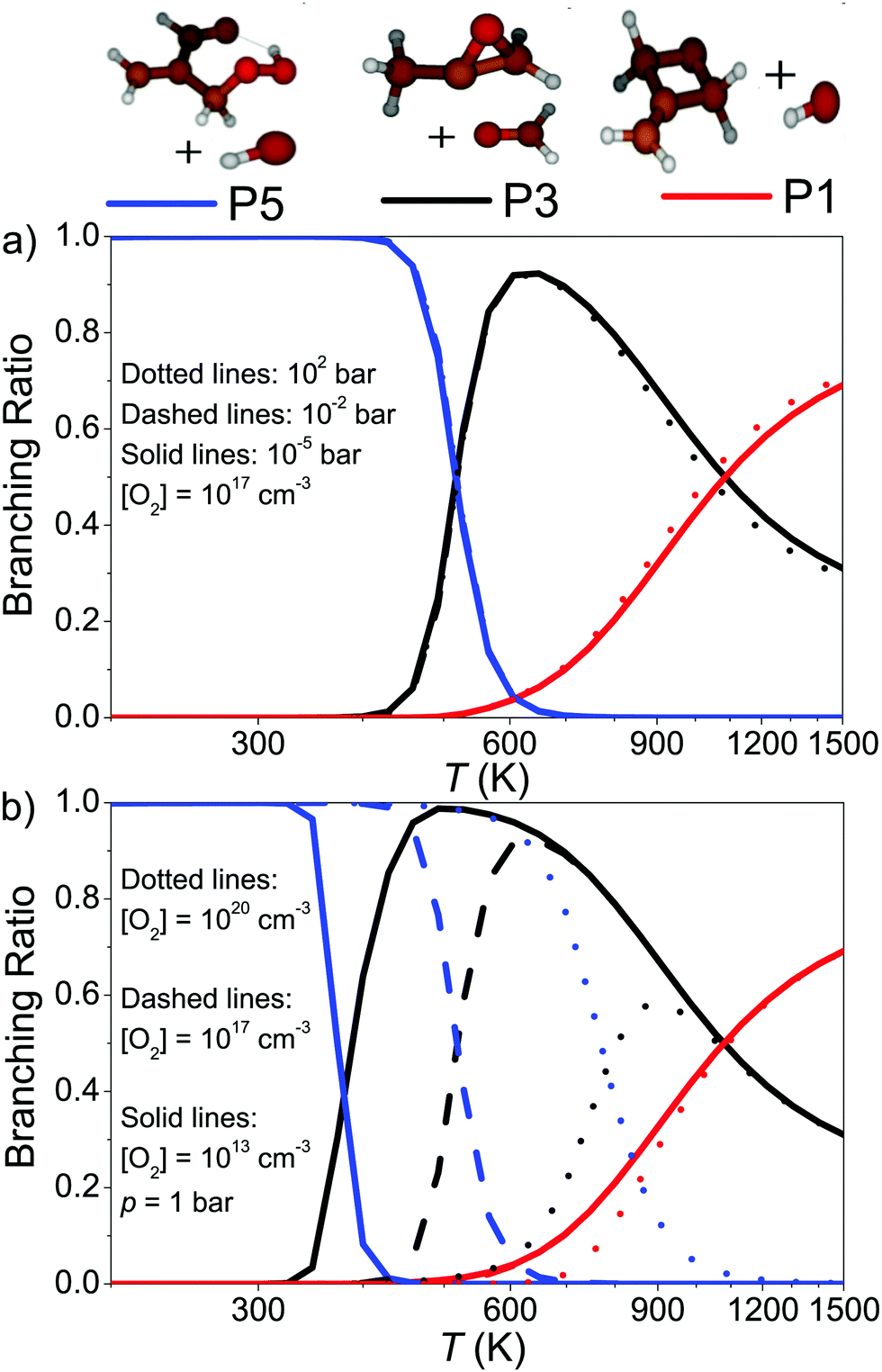 | ||
| Fig. 8 Branching ratios of the products of the CH2C(CH3)CH2˙ + O2 reaction plotted as a function of temperature, pressure, and O2 concentration. | ||
To provide a simple way of incorporating the results of our simulations in combustion models, we ran simulations between 0.01–100 bar and 400–1500 K, setting the O2 mole fraction to 0.21. To obtain channel-specific rate coefficients, we divided the −λ2 values shown as solid-lines in Fig. 7b with [O2] and multiplied this with the branching ratios obtained from the aforementioned simulations. The results of this procedure at 0.01, 1, and 100 bar are displayed in Fig. 9. This figure also shows why no reaction was observed at 730 K. At that temperature, the phenomenological bimolecular rate coefficient is around 5 × 10−17 cm3 s−1, so the reaction is too slow to observe with our experimental apparatus. Fits to the channel-specific rate coefficients with the simple modified Arrhenius expression showed that this formula is not flexible enough to capture the temperature dependence of the rate coefficients and there can be up to a factor of five difference between the Arrhenius and the modeled channel-specific rate coefficient. However, we believe the fits are good enough to give a rough description of chemistry of the CH2C(CH3)CH2˙ + O2 reaction at high temperatures. It should also be noted at very high O2 concentrations the equilibration zone can extend up to 800–900 K and under these conditions it is not entirely accurate to model the phenomenological loss of 2-methylallyl as a single-step reaction. We provide Arrhenius representations of the channel-specific rate coefficients in ChemKin compatible PLOG-format in the ESI† (PLOG.txt). The input file of our master equation model is also provided (C4H7+O2.xml).
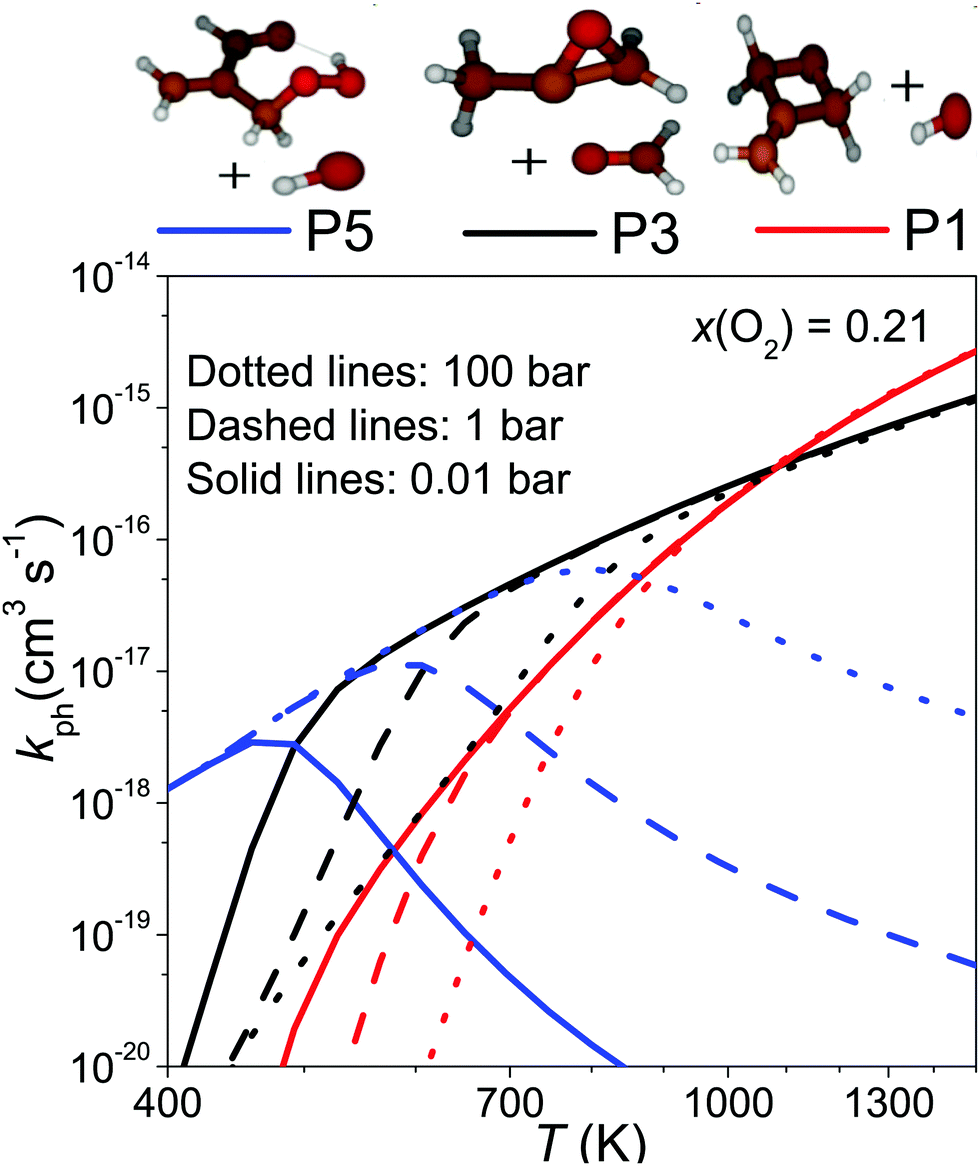 | ||
| Fig. 9 The channel-specific bimolecular rate coefficients plotted as a function of temperature and pressure at x(O2) = 0.21. | ||
| CH2C(CH3)CH2˙ + CH2C(CH3)CH2˙ → CH2C(CH3)CH2CH2C(CH3)CH2 | (22) |
| CH2C(CH3)CH2˙ → CH3˙ + CH2CCH2 | (23) |
| CH2CHCH2˙ + HO2˙ → Products. | (24) |
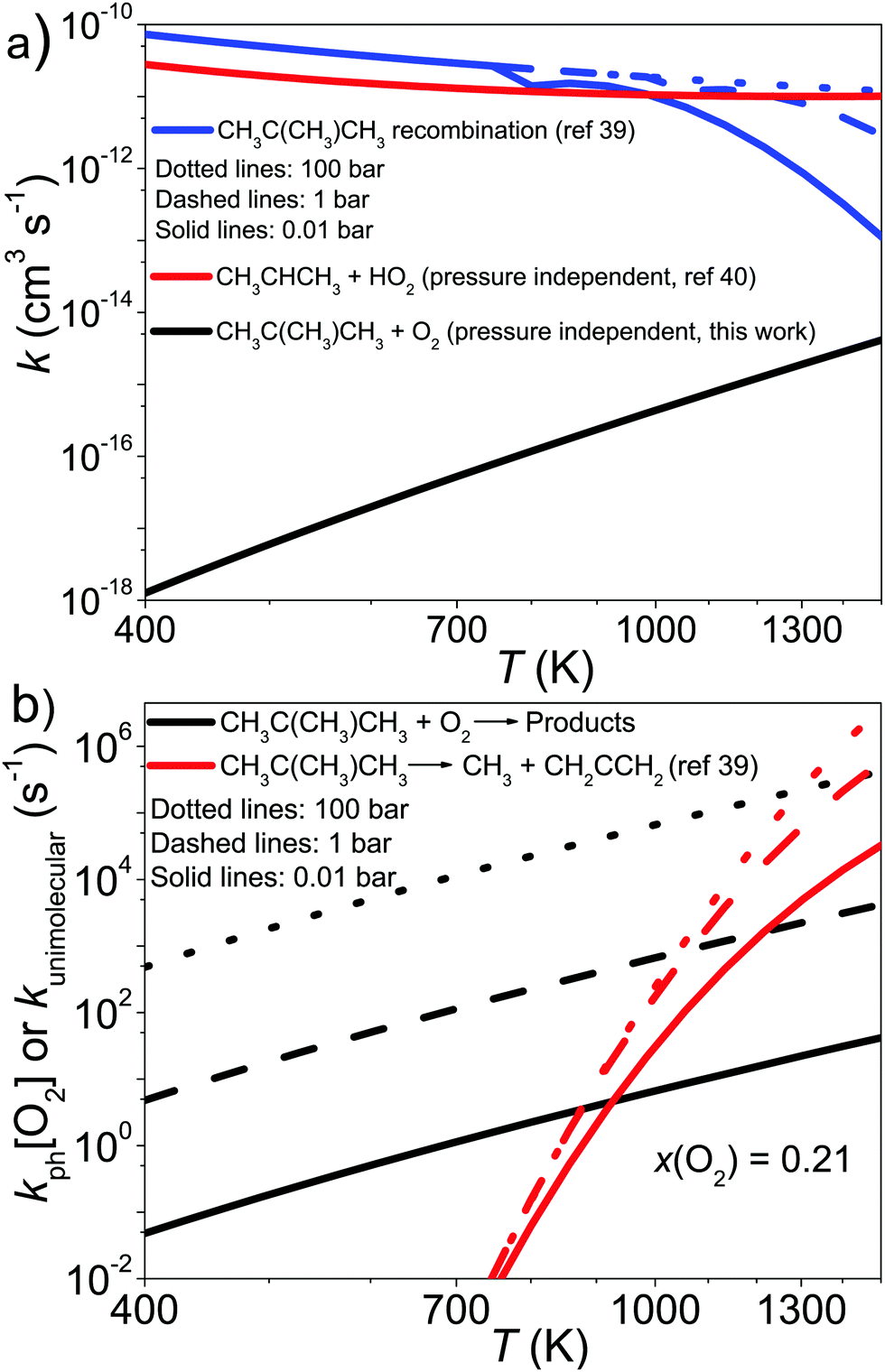 | ||
| Fig. 10 The high-temperature kinetics of the CH2C(CH3)CH2˙ + O2 reaction compared with the kinetics of the reactions (a) CH2C(CH3)CH2˙ + CH2C(CH3)CH2˙ → CH2C(CH3)CH2CH2C(CH3)CH2 and CH2CHCH2˙ + HO2˙ → Products and (b) CH2C(CH3)CH2˙ → CH3˙ + CH2CCH2. | ||
6 Conclusions
We have investigated the kinetics of the reaction between 2-methylallyl radicals (CH2C(CH3)CH2˙) and O2 with both experimental and computational methods. CH2C(CH3)CH2˙ traces were measured in real time under pseudo-first-order ([CH2C(CH3)CH2˙] ≪ [O2]) conditions using laser-photolysis photoionization mass spectrometry. At low temperatures (T ≤ 410 K), the reaction proceeds by a barrierless association to form 2-methylallylperoxyl. Between 347–410 K the reactants and products are found to equilibrate. No further reaction is observed even at 730 K. Quantum chemical calculations were performed to probe the possible reaction pathways. A master equation model was constructed that includes all the kinetically relevant reaction pathways. We used our experimental data to optimize various parameters in the model. This optimization was done both by comparing experimental and modeled rate coefficients with each other and by directly comparing experimental species traces with modeled ones. For the studied system, both approaches yielded very similar results and were able to reproduce the experimental data. The optimized model was used to simulate the reaction over the range 0.01–100 bar and 200–1500 K. According to the simulations, the CH2C(CH3)CH2˙ + O2 reaction is slow under autoignition conditions and combustion models are not expected to be sensitive to it. We provide modified Arrhenius representations for the high-temperature bimolecular product channels.Author contributions
The experiments were conceived by Arkke Eskola and Raimo Timonen. The kinetic measurements were performed primarily by Timo Pekkanen, but he was significantly assisted by Satya Joshi, Timo Reijonen, and Raimo Timonen. The quantum chemical calculations and master equation simulations were performed by Timo Pekkanen under the supervision of György Lendvay. Struan Robertson assisted with the master equation simulations and modified the MESMER program to allow the user to specify first-order loss rates for all species present in a model. This modification enabled us to use our experimental data in trace fitting. Timo Pekkanen primarily wrote the manuscript. The manuscript was substantially amended by György Lendvay. The other authors reviewed and commented the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. T. P. acknowledges support from the Doctoral Programme in Chemistry and Molecular Sciences of the University of Helsinki and the Magnus Ehrnrooth foundation for funding. Project no. K129140 for G. L. has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the OTKA funding scheme. S. P. J., T. T. R., and A. J. E. acknowledge support from the Academy of Finland, grant numbers 294042, 319353, 325250, and 288377. The authors also acknowledge COST Action CM1404 (SMARTCATS) for support as well as CSC IT Center for Science in Finland for computational resources.Notes and references
- J. T. Bartis and B. Widom, J. Chem. Phys., 1974, 60, 3474–3482 CrossRef CAS.
- D. R. Glowacki, C.-H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- D. J. Medeiros, S. H. Robertson, M. A. Blitz and P. W. Seakins, J. Phys. Chem. A, 2020, 124, 4015–4024 CrossRef CAS PubMed.
- F. L. Dryer and K. Brezinsky, Combust. Sci. Technol., 1986, 45, 199–212 CrossRef CAS.
- J. Zádor, C. A. Taatjes and R. X. Fernandes, Prog. Energy Combust. Sci., 2011, 37, 371–421 CrossRef.
- J. A. Miller, M. J. Pilling and J. Troe, Proc. Combust. Inst., 2005, 30, 43–88 CrossRef.
- I. R. Slagle and D. Gutman, Proc. Combust. Inst., 1988, 21, 875–883 CrossRef.
- D. K. Hahn, S. J. Klippenstein and J. A. Miller, Faraday Discuss., 2002, 119, 79–100 RSC.
- M. P. Rissanen, D. Amedro, A. J. Eskola, T. Kurten and R. S. Timonen, J. Phys. Chem. A, 2012, 116, 3969–3978 CrossRef CAS PubMed.
- V. D. Knyazev and I. R. Slagle, J. Phys. Chem. A, 1998, 102, 1770–1778 CrossRef CAS.
- T. T. Pekkanen, R. S. Timonen, G. Lendvay, M. P. Rissanen and A. J. Eskola, Proc. Combust. Inst., 2018, 299–306 Search PubMed.
- V. D. Knyazev and I. R. Slagle, J. Phys. Chem. A, 1998, 102, 8932–8940 CrossRef CAS.
- M. Döntgen, T. T. Pekkanen, S. P. Joshi, R. S. Timonen and A. J. Eskola, J. Phys. Chem. A, 2019, 123, 7897–7910 CrossRef PubMed.
- S. P. Joshi, T. T. Pekkanen, P. Seal, R. S. Timonen and A. J. Eskola, Phys. Chem. Chem. Phys., 2021, 23, 20419–20433 RSC.
- Z. H. Lodhi and R. W. Walker, J. Chem. Soc., Faraday Trans., 1991, 87, 2361–2365 RSC.
- D. Schleier, E. Reusch, M. Gerlach, T. Preitschopf, D. P. Mukhopadhyay, N. Faßheber, G. Friedrichs, P. Hemberger and I. Fischer, Phys. Chem. Chem. Phys., 2021, 23, 1539–1549 RSC.
- C.-J. Chen and J. W. Bozzelli, J. Phys. Chem. A, 2000, 104, 9715–9732 CrossRef CAS.
- X. L. Zheng, H. Y. Sun and C. K. Law, J. Phys. Chem. A, 2005, 109, 9044–9053 CrossRef CAS PubMed.
- A. J. Eskola and R. S. Timonen, Phys. Chem. Chem. Phys., 2003, 5, 2557–2561 RSC.
- H. Yu, X. He, S. Louis Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. L. Bao, J. Zheng, I. M. Alecu, B. J. Lynch, Y. Zhao and D. G. Truhlar, 2017, https://comp.chem.umn.edu/freqscale/version3b2.htm, Accessed: 11.1.2022.
- J. D. Watts, J. Gauss and R. J. Bartlett, J. Chem. Phys., 1993, 98, 8718–8733 CrossRef CAS.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef PubMed.
- M. Saitow, U. Becker, C. Riplinger, E. F. Valeev and F. Neese, J. Chem. Phys., 2017, 146, 164105 CrossRef PubMed.
- Y. Guo, C. Riplinger, U. Becker, D. G. Liakos, Y. Minenkov, L. Cavallo and F. Neese, J. Chem. Phys., 2018, 148, 011101 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper and J. Olsen, Chem. Phys. Lett., 1999, 302, 437–446 CrossRef CAS.
- T. Helgaker, W. Klopper, H. Koch and J. Noga, J. Chem. Phys., 1997, 106, 9639–9646 CrossRef CAS.
- M. J. Frisch et al. , Gaussian-16 Revision B.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243–252 CrossRef CAS.
- B. E. Poling, J. M. Prausnitz and J. P. O'Connell, The Properties of Gases and Liquids, McGraw-Hill, 5th edn, 2001 Search PubMed.
- C. W. Gao, J. W. Allen, W. H. Green and R. H. West, Comput. Phys. Commun., 2016, 203, 212–225 CrossRef CAS.
- J. Gang, M. J. Pilling and S. H. Robertson, Chem. Phys., 1998, 231, 183–192 CrossRef CAS.
- M. E. Jenkin, T. P. Murrells, S. J. Shalliker and G. D. Hayman, J. Chem. Soc., Faraday Trans., 1993, 89, 433–446 RSC.
- G. Lendvay, in Unimolecular Kinetics, ed. S. H. Robertson, Elsevier, 2019, ch. 3, vol. 43, pp. 109–272 Search PubMed.
- I. R. Slagle, E. Ratajczak, M. C. Heaven, D. Gutman and A. F. Wagner, J. Am. Chem. Soc., 1985, 107, 1838–1845 CrossRef CAS.
- R. S. Tranter, A. W. Jasper, J. B. Randazzo, J. P. Lockhart and J. P. Porterfield, Proc. Combust. Inst., 2017, 36, 211–218 CrossRef CAS.
- C. F. Goldsmith, S. J. Klippenstein and W. H. Green, Proc. Combust. Inst., 2011, 33, 273–282 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp05591g |
| This journal is © the Owner Societies 2022 |