
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetically induced ring currents in metallocenothiaporphyrins†
Rashid R.
Valiev
ab,
Theo
Kurten
b,
Lenara I.
Valiulina
a,
Sergey Yu.
Ketkov
 c,
Viktor N.
Cherepanov
a,
Maria
Dimitrova
b and
Dage
Sundholm
*b
c,
Viktor N.
Cherepanov
a,
Maria
Dimitrova
b and
Dage
Sundholm
*b
aDepartment of Optics and Spectroscopy, Tomsk State University, Tomsk, 634050, Russia
bDepartment of Chemistry, Faculty of Science, University of Helsinki, FIN-00014, Helsinki, Finland. E-mail: sundholm@chem.helsinki.fi
cLaboratory of Structures of Organometallic and Coordination Compounds, G. A. Razuvaev Institute of Organometallic Chemistry RAS, 49 Tropinin St., Nizhny Novgorod 603950, Russia
First published on 20th December 2021
Abstract
The magnetically induced current–density susceptibility tensor (CDT) of the lowest singlet and triplet states of the metallocenothiaporphyrins, where the metal is V, Cr, Mn, Fe, Co, Ni, Mo, Tc, Ru, or Rh, have been studied with the gauge-including magnetically induced currents (GIMIC) method. The compounds containing V, Mn, Co, Tc or Rh were studied as cations because the neutral molecules have an odd number of electrons. The calculations show that the aromatic nature of most of the studied molecules follows the Hückel and Baird rules of aromaticity. CDT calculations on the high-spin states of the neutral metallocenothiaporphyrins with V, Mn, Co, Tc or Rh also shows that these molecules follow a unified extended Hückel and Baird aromaticity orbital-count rule stating that molecules with an odd number of occupied conjugated valence orbitals are aromatic, whereas molecules with an even number of occupied conjugated orbitals are antiaromatic.
Introduction
Classical metallocenes are organic sandwich compounds formed by two cyclopentadienyl rings and a transition metal (M). The general formula of metallocene structures is (η5-C5H5)2M where the metal ion in the oxidation state 2+ is located between two parallel cyclopentadienyl anions at equal or nearly equal distances from all carbon atoms of the two cyclopentadienyl ((C5H5)2) rings. The metal in the 3d and 4d metallocenes is bound to the ligands via covalent chemical bonds except for manganocene exhibiting ionic bonding.1 Vanadocene and chromocene have partly ionic metal–ligand bonds.2 The covalent contribution to the M–C5H5 bonds is provided mainly by the overlap between the 3d orbitals of the metal and the π-orbitals of the C5H5 ring.3Ferrocene, which is the first discovered metallocene, was synthesized independently in 1951 by the groups led by Pauson, and Miller.4,5 The intriguing ferrocene sandwich structure was suggested by the groups of Woodward, Wilkinson6 and Fischer.7 The discovery of ferrocene inspired subsequent synthesis of new similar metal complexes. Already by 1954, metallocenes of most of the d elements had been synthesized and characterized. However, ferrocene is the most stable of all known metallocenes.
The stability of ferrocene is explained by the so-called 18-electron rule. According to this rule, the most stable structure is obtained when the sum of the valence electrons of a transition metal and the ligand electrons participating in bonding is 18. Only the metallocenes with a metal from group VIII can be neutral 18-electron complexes. Metals from other groups form less stable, reactive complexes, and do not always lead to the formation of an ideal sandwich structure.1 For first-row transition metals, the stability of the metallocenes decreases in the following order: Fe > Ni > Co > V ≫ Cr > Ti.2 Due to their unique chemical properties, metallocenes are widely used in various applications.8–15
The reactivity of ferrocene resembles that of benzene. It is, therefore, referred to as a non-benzoic cylindrically aromatic system with 4n conjugated electrons.16,17 Ferrocene has been the subject of many studies.18–21 The conjugation transmitted by the d electrons of ferrocene and ruthenocene has been studied by incorporating it into a porphyrinoid compound.22,23 Porphyrins and their derivatives are widely used in various fields due to their rigid molecular structure and strong light absorption in the visible region.24–30
The wide variety of porphyrin compounds and their unique electronic structure enable studies of the relationship between aromaticity and physicochemical properties. The aromatic character influences the optical properties of porphyrins such as light absorption, excitedstate lifetimes, fluorescence quantum yields, and nonlinear optical properties including two-photon absorption.31,32
Since metallocenes and porphyrins are important molecules in many contexts, the combination of the two compounds by incorporating various metallocenes into the porphyrin macrocycle may result in molecules with intriguing properties.22,23 The number of d electrons contributing to the electron delocalization determines the aromatic character and can be adjusted by choosing the metal and the number of the inner hydrogens connected to the pyrrole rings. The current–density flux and the ring current passing the metal of the ferrocene moiety could not be elucidated neither spectroscopically nor computationally22 because the current–density flux is very complicated with a helical flux at the ferrocene as shown in this study.
Aromaticity can be assessed using a range of criteria, among which is the ring-current criterion employed in this study. An external magnetic field induces a diatropic ring current in aromatic molecular rings, whereas in antiaromatic molecules the ring current flows in the non-classical, i.e., the paratropic direction. The degree of aromaticity can be judged by determining the magnitude of the induced ring-current strengths,33 which are obtained by integrating the current density flux using the GIMIC program. Detailed information about the GIMIC program and its capabilities can be found in recent reviews.34–36
The aim of this work is to investigate the aromaticity and electronic delocalization in porphyrinoid rings with incorporated metallocenes. We have studied how the current density is affected when reducing the molecule by adding two inner hydrogen atoms to the porphyrinoid. We have formulated a generalization of the Hückel and Baird aromaticity rules, which can be extended to systems with more than two open shells.
Molecular structures
We denote the metallocenothiaporphyrin and the dihydrometallocenothiaporphyrin compounds (C5H5)2MP and H2-(C5H5)2MP, respectively. The molecular structures of (C5H5)2MP and H2-(C5H5)2MP are shown in Fig. 1. We have studied (C5H5)2MP and H2-(C5H5)2MP compounds, where M is a 3d transition metal from Sc to Zn or a 4d transition metal from Y to Cd. However, many of the studied molecules were unstable at the employed level of theory and were not studied further. We have investigated the lowest singlet and triplet states of the neutral M = Cr, Fe, Ni, Mo, and Ru compounds as well as the lowest singlet and triplet states of the M = Co, Mn, V, Tc, and Rh containing (C5H5)2MP+ and H2-(C5H5)2MP+ cations. We have also studied the sextet ground state of the neutral Mn and Tc compounds as well as the quartet ground state of the neutral vanadium compound. | ||
| Fig. 1 The molecular structures of the studied metallocenothiaporphyrins, where M = V, Cr, Mn, Fe, Co, Ni, Mo, Tc, Ru and Rh. | ||
The porphyrin macrocycle consists of two pyrrole rings and one thiophene ring which are connected at their α-carbon atoms via methine bridges. The metallocene is linked via methine bridges to the two pyrrole rings of the thiaporphyrin. The pyrrole rings of H2-(C5H5)2MP have inner hydrogen atoms increasing the formal number of π electrons by two compared to (C5H5)2MP.
Computational methods
The molecular structures were optimized at the density functional theory (DFT) level using the Becke, Lee, Yang, Parr (B3LYP) functional37,38 and the Karlsruhe def2-TZVP39 basis set. The Cartesian coordinates of the molecular structures are given in the ESI.† The average bond lengths (in Å) between the metal and the carbon atoms of the cyclopentadienyl rings are also reported in the ESI.† Calculations of the vibrational frequencies showed that the optimized molecular structures are minima on the potential energy surface.The magnetically induced current density susceptibility tensors (CDT) were calculated using the GIMIC method,33 which uses the Cartesian coordinates of the atoms, basis-set information, and density matrices as input data. The first-order magnetically perturbed density matrices are obtained by performing nuclear magnetic resonance (NMR) shielding calculations, while the unperturbed density matrix is obtained in self-consistent field (SCF) DFT calculations. The density matrices were computed at the B3LYP/def2-TZVP level using Gaussian 16.40 A tool that converts the Gaussian output into appropriate input format for GIMIC was employed.33,41
The ring-current strengths (I in nA T−1) were obtained by integrating the current–density flux passing through a plane placed perpendicularly to the molecular structure crossing a given chemical bond.35 The sign and magnitude of the ring-current strengths are used for determining the character and degree of aromaticity. Negative ring-current strengths indicate antiaromatic character, that is, the paratropic ring current dominates, while positive ring-current strengths suggest that the molecule is aromatic with a dominating diatropic ring current.33–35 Previous current–density studies showed that DFT calculations using the B3LYP functional42–44 yield accurate ring-current strengths and degree of aromaticity for aromatic and weakly antiaromatic molecules. However, B3LYP calculations overestimate the strength of the paratropic ring current of strongly antiaromatic molecules.45 We have studied the CDT, ring-current strengths, and aromatic pathways of the lowest singlet and triplet states of (C5H5)2VP+, (C5H5)2CrP, (C5H5)2MnP+, (C5H5)2FeP, (C5H5)2CoP+, (C5H5)2NiP, (C5H5)2MoP, (C5H5)2TcP+, (C5H5)2RuP, and (C5H5)2RhP+ as well as for the quartet state of the neutral (C5H5)2VP and the sextet state of the neutral (C5H5)2MnP and (C5H5)2TcP.
Results and discussion
Aromaticity rules
The total number of electrons participating in the bond conjugation can be determined by accounting for the number of electrons in the porphyrinoid part of the conjugation pathway, the valence electrons of the metal and the π electrons of the C5H5 rings. The cyclopentadienyl anions have 12π electrons of which two are energetically low-lying. The number of electrons contributing to the conjugation pathway of the other metallocenes originating from the metal is equal to the number of valence d electrons of the metal. Fe2+ has 6 d electrons. Thus, the ferrocene unit provides 16 electrons to the bond conjugation. For (C5H5)2MP, the porphyrinoid part contributes with 20π electrons. Combined with the 16 valence electrons in the ferrocene moiety, this yields 36 electrons in the conjugation pathway of (C5H5)2FeP. Given that the number of electrons fulfills Hückel's rule for antiaromaticity, the singlet state of (C5H5)2FeP is expected to be antiaromatic. H2-(C5H5)2FeP has 38 conjugated electrons since the porphyrinoid part contributes with 22π electrons implying that the singlet state of H2-(C5H5)2FeP is expected to be aromatic.46–48 Similarily, the triplet states of (C5H5)2FeP and H2-(C5H5)2FeP are expected to be aromatic and antiaromatic, respectively, according to Baird's aromaticity rule.48–50The same aromaticity rules can be applied to the isoelectronic (C5H5)2CoP+ and H2-(C5H5)2CoP+ cations as well as to the corresponding compounds containing the 4d metals: (C5H5)2RuP, (C5H5)2RhP+, H2-(C5H5)2RuP and H2-(C5H5)2RhP+. The vanadium complexes (C5H5)2VP+ and H2-(C5H5)2VP+ have four electrons less than the [Fe]-cenothiaporphyrins and are expected to have the same aromatic character as the [Fe]-cenothiaporphyrins.
Reversed aromaticity rules are expected for the corresponding Ni compounds with two electrons more than the Fe containing compounds. The Cr and Mo containing compounds with two valence electrons less than [Fe]-cenothiaporphyrin and the valence isoelectronic Mn+ and Tc+ cations are also expected to have reverse aromaticity compared to the corresponding [Fe]-cenothiaporphyrin.
(C5H5)2FeP and H2-(C5H5)2FeP and isoelectronic compounds of group VIII
Calculations of the magnetically induced ring current (MIRC) of the singlet state of (C5H5)2FeP yield a ring-current strength of −10.4 nA T−1 when the external magnetic field is nearly perpendicular to the porphyrinoid ring, whereas H2-(C5H5)2FeP with two inner hydrogens is aromatic sustaining a MIRC of 13.8 nA T−1, which is comparable to the strength of the MIRC of benzene of 12 nA T−1.51 The triplet state of (C5H5)2FeP is calculated to be 12.0 kcal mol−1 above the singlet state and the triplet state of H2-(C5H5)2FeP is analogously 17.8 kcal mol−1 above the singlet state. The calculated ring-current strength of the triplet state of (C5H5)2FeP is 6.2 nA T−1 and the triplet state of H2-(C5H5)2FeP sustains a ring current of −9.0 nA T−1. Thus, the aromatic character of the studied states of the [Fe]-cenothiaporphyrins follows the Hückel rule of aromaticity, since the singlet states have 4n + 2 electrons in the conjugated bonds, while 4n electrons lead to antiaromaticity. The antiaromatic singlet states become an aromatic triplet state and vice versa, following the Baird rule for triplet-state aromaticity.49,50,52 The MIRC strengths are summarized in Table 1.
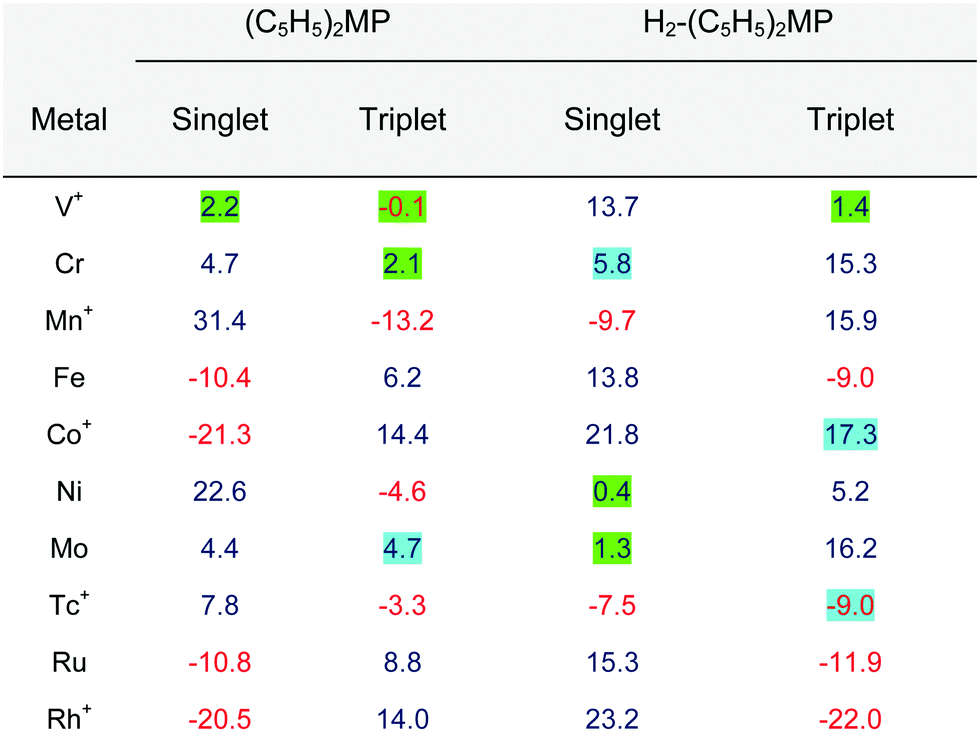
|
Previously calculated nucleus-independent magnetic shielding functions and experimental 1H NMR chemical shifts also suggested that the singlet state of (C5H5)2FeP is antiaromatic and the singlet state of H2-(C5H5)2FeP is aromatic.22
The calculations showed that the ring current of the porphyrinoid part splits into an inner and an outer route at each heterocyclic ring. At the pyrrole rings of the singlet (C5H5)2FeP it mainly follows the innermost pathway, whereas at the thiophene ring, the ring-current strength of the inner pathway is about a factor of two stronger than that of the outer one.
The calculations showed that the ring current of (C5H5)2FeP in the porphyrinoid part splits into an inner and outer route at each heterocyclic ring. At the pyrrole rings of the singlet (C5H5)2FeP it mainly follows the innermost pathway, whereas at the thiophene ring, the ring-current strength of the inner pathway is about a factor of two stronger than that of the outer one. The pyrrole rings sustain a weak local diatropic ring current of 1.6 nA T−1. The strengths of the MIRC obtained by integrating the current density passing along selected chemical bonds are shown in Fig. 2. The main ring-current pathways of the singlet and triplet states of (C5H5)2FeP and H2-(C5H5)2FeP are shown in Fig. 3.
 | ||
| Fig. 2 The strength of the MIRC (in nA T−1) along selected bonds in the singlet and triplet states of (C5H5)2FeP and H2-(C5H5)2FeP. | ||
 | ||
| Fig. 3 (a) The dominating antiaromatic pathway (in red) of the singlet state of (C5H5)2FeP, (b) the dominating aromatic pathway (in blue) of the singlet state of H2-(C5H5)2FeP, (c) the dominating aromatic pathway (in blue) of the triplet state of (C5H5)2FeP, and (d) the dominating antiaromatic pathway (in red) of the triplet state of H2-(C5H5)2FeP. | ||
For the aromatic singlet state of H2-(C5H5)2FeP, the outer pathways of the diatropic MIRC dominate. The MIRC splits at the pyrrole rings, whereas the thiophene ring sustains a local ring current of −3.3 nA T−1. The diatropic MIRC of the aromatic triplet state (C5H5)2FeP takes the outer route at the pyrrole and thiophene rings. The thiophene ring sustains a local ring current of −5.3 nA T−1, whereas the current density does not take the inner route at the pyrrole rings.
The paratropic MIRC of the triplet state of H2-(C5H5)2FeP takes the inner pathway at the pyrrole rings, whereas it splits into an inner and outer pathway at the thiophene ring. The current density along the inner pathway is almost a factor of two stronger than the one along the outer route. The strength of the MIRC pathways is shown in Fig. 2 and the dominating ring-current pathways are shown in Fig. 3, where diatropic ring currents are marked in blue and the paratropic ones are red. The current densities of the singlet state of (C5H5)2FeP and H2-(C5H5)2FeP are shown in Fig. 4.
 | ||
| Fig. 4 The current density of (a) the singlet state of (C5H5)2FeP whose paratropic MIRC flows mainly on the inside; (b) the singlet state of H2-(C5H5)2FeP with diatropic MIRC flowing along the outer pathway. The paratropic MIRC of H2-(C5H5)2FeP on the inside of it is also seen. The helical current density at the ferrocene has the same chirality in both molecules. The current–density plots have been visualized using Paraview.53 The color scale represents the strength of the current density such that white is the strongest and black is the weakest one. | ||
An unusual current–density topology was observed in the ferrocene unit. In the aromatic singlet H2-(C5H5)2FeP, the diatropic MIRC flows on the outside of the C5H5 ring when it arrives to the ferrocene moiety. It forms a helical flow in the ferrocene making an extra lap around the other C5H5 ring before it leaves the ferrocene moiety on the other side. The paratropic MIRC flows on the inside of the porphyrinoid part, taking a shortcut at the ferrocene from one C5H5 ring to the other on inside of the ferrocene moiety.
In the antiaromatic (C5H5)2FeP, the paratropic MIRC flows on the inside of the porphyrinoid ring. When it approaches the ferrocene moiety, it makes a lap around the C5H5 ring before it continues with a helical flow to the other C5H5 ring where it leaves the ferrocene moiety on the inside of the C5H5 ring. The paratropic MIRC also takes a shortcut on the inside from one C5H5 ring to the other as in H2-(C5H5)2FeP. The helical current density at the ferrocene moiety of the two molecules has the same chirality. The MIRC fluxes of (C5H5)2FeP and H2-(C5H5)2FeP are shown in Fig. 5(a) and (b), respectively. More streamline representations of the MIRC are shown in the ESI.†
 | ||
| Fig. 5 (a) The paratropic MIRC of (C5H5) 2FeP taking the inner pathway and the helical flux at the ferrocene moiety. (b) The diatropic MIRC of H2-(C5H5)2FeP and the helical flux at the ferrocene. More pictures of the current densities and the MIRCs are shown in the ESI.† The current–density flow has been obtained using the Runge–Kutta algorithm in Paraview.53 | ||
(C5H5)2RuP, H2-(C5H5)2RuP, (C5H5)2RhP+ and H2-(C5H5)2RhP+ are isoelectronic with the corresponding Fe compounds. Their aromatic character is the same as for the valence isoelectronic [Fe]-cenothiaporphyrins. The MIRC of (C5H5)2RuP and H2-(C5H5)2RuP are 0.4–2.9 nA T−1 stronger than for the corresponding [Fe]-cenothiaporphyrins. The singlet ground state of (C5H5)2RuP is antiaromatic sustaining a MIRC of −10.8 nA T−1 and the singlet ground state of H2-(C5H5)2RuP is aromatic with a MIRC of 15.3 nA T−1. The triplet state of (C5H5)2RuP and H2-(C5H5)2RuP lie 13.2 kcal mol−1 and 20.2 kcal mol−1 above the singlet ground state, respectively. The triplet state of (C5H5)2RuP and H2-(C5H5)2RuP sustains MIRC of 8.8 nA T−1 and −11.9 nA T−1, respectively. The aromatic character of (C5H5)2RuP and H2-(C5H5)2RuP follows the Hückel and Baird aromaticity rules.
The triplet state of (C5H5)2RhP+ is 10.0 kcal mol−1 and the triplet state of H2-(C5H5)2RhP+ is 22.8 kcal mol−1 above the corresponding singlet ground states. The MIRC of the singlet ground state of (C5H5)2RhP+ is −20.5 nA T−1 and in the MIRC of singlet ground state of H2-(C5H5)2RhP+ is 23.2 nA T−1. Since the triplet states of (C5H5)2RhP+ and H2-(C5H5)2RhP+ also have reverse aromatic character as compared to the singlet ground states, their aromatic character follows the Hückel and Baird aromaticity rules.
(C5H5)2CoP+ and H2-(C5H5)2CoP+ are isoelectronic with the corresponding [Fe]-cenothiaporphyrins. When oxidizing metallocenes, the electronic charge is mainly removed from the C5H5 rings implying that the electron configuration of Co+ remains largely as 4s03d8. The same holds for the other cations of this study.54,55 However, the triplet state of H2-(C5H5)2CoP+ has a d occupation of 7.7 as compared to 8.2 for the other Co compounds. Thus, the exciton of the triplet state is partly localized to the metal leading to aromaticity of the triplet state of H2-(C5H5)2CoP+ instead of antiaromaticity as estimated from the Baird rule. The singlet ground state of (C5H5)2CoP+ and H2-(C5H5)2CoP+ are 7.6 kcal mol−1 and 9.1 kcal mol−1, respectively below the triplet state. The aromatic character of (C5H5)2CoP+ follows the Hückel and Baird aromaticity rules. The singlet ground state is antiaromatic sustaining a MIRC of −21.3 nA T−1 and the triplet state is aromatic sustaining an MIRC of 14.4 nA T−1. The singlet ground state of H2-(C5H5)2CoP+ is aromatic as expected from Hückel's aromaticity rule. It sustains an MIRC of 21.8 nA T−1, whereas the triplet state of H2-(C5H5)2CoP+ is unexpectedly also aromatic sustaining a strong diatropic MIRC of 17.3 nA T−1 implying that it does not follow Baird's aromaticity rule.
Group VI and isoelectronic compounds
The (C5H5)2MnP+ and H2-(C5H5)2MnP+ cations have an even number of electrons and two electrons less than the corresponding [Fe]-cenothiaporphyrins. The aromatic character and the aromatic pathways are therefore reversed as compared to the Fe containing compounds. The dominating pathways of the MIRC of the singlet and triplet states of (C5H5)2MnP+ and H2-(C5H5)2MnP+ are shown in Fig. 6. However, (C5H5)2MnP+ and H2-(C5H5)2MnP+ have a triplet ground state with the singlet state lying 24.1 kcal mol−1 and 18.0 kcal mol−1 higher in energy, respectively. Calculations of the current density for the singlet state of (C5H5)2MnP+ show that it is aromatic sustaining a MIRC of 31.4 nA T−1. The triplet state is antiaromatic with a MIRC of −13.2 nA T−1. The aromatic character of (C5H5)2MnP+ follows the Hückel and Baird rules, because it has 4n + 2 electrons in the bond conjugation. The aromatic character of H2-(C5H5)2MnP+ also follows the Hückel and Baird rules, since its singlet state is antiaromatic sustaining a MIRC of −9.7 nA T−1 and the triplet state is aromatic with a MIRC of 15.9 nA T−1. | ||
| Fig. 6 (a) The dominating aromatic pathway (in blue) of the singlet state of (C5H5)2MnP+, (b) the dominating antiaromatic pathway (in red) of the singlet state of H2-(C5H5)2MnP+, (c) the dominating antiaromatic pathway (in red) of the triplet state of (C5H5)2MnP+, and (d) the dominating aromatic pathway (in blue) of the triplet state of H2-(C5H5)2MnP+. | ||
The strengths of the MIRC along selected bonds are shown in Fig. 7. The ring current of the singlet state of (C5H5)2MnP+ splits at the pyrrole and thiophene rings. The current–density flux is about three times stronger along the outer route. The ring current of the triplet state follows mainly along the innermost pathway. It splits into an inner and an outer route at the thiophene ring. However, the inner one is twice as strong as the outer branch. The ring-current pathway of the singlet state of H2-(C5H5)2MnP+ is similar to the one for the triplet state of (C5H5)2MnP+. The diatropic ring current of the triplet state of H2-(C5H5)2MnP+ splits into an outer and inner branch at the pyrrole rings, whereas the thiophene ring sustains a weak local diatropic ring current. The outer pathway at the pyrrole rings is about three times stronger than the inner one.
 | ||
| Fig. 7 The strength of the MIRC (in nA T−1) along selected bonds in (a) the singlet state of (C5H5)2MnP+, (b) the singlet state of H2-(C5H5)2MnP+, (c) the triplet state of (C5H5)2MnP+ and (d) the triplet state of H2-(C5H5)2MnP+. | ||
The ground state of the neutral (C5H5)2MnP and H2-(C5H5)2MnP molecules is a sextet state. Calculations of the current density of the neutral (C5H5)2MnP and H2-(C5H5)2MnP show that they are non-aromatic sustaining a very weak MIRC of 1.5 nA T−1 and 0.9 nA T−1, respectively. The pyrrole and thiophene rings of (C5H5)2MnP sustain a local diatropic MIRC of 3.4 nA T−1. The local ring current of the thiophene ring of H2-(C5H5)2MnP is 6.9 nA T−1, whereas the pyrrole rings sustain a weak local MIRC of 2.6 nA T−1. The long distance between Mn and the C5H5 rings (see Table S1 in the ESI†) suggests that the sextet ground state of the neutral (C5H5)2MnP and H2-(C5H5)2MnP is weakly bound. The distance between Mn and the C5H5 rings is much shorter for the cations.
The aromatic character of the [Cr]- and [Mo]-cenothiaporphyrins are similar. The triplet ground state of (C5H5)2CrP and (C5H5)2MoP sustain a weak diatropic MIRC of 2.1 nA T−1 and 4.7 nA T−1, respectively. However, according to the aromaticity rules, one would expect them to sustain a paratropic MIRC. The singlet states of (C5H5)2CrP and (C5H5)2MoP that lie 29.5 kcal mol−1 and 2.1 kcal mol−1 higher in energy are weakly aromatic following Hückel's rule with MIRC strengths of 4.7 nA T−1 and 4.4 nA T−1, respectively. The triplet ground state of H2-(C5H5)2CrP and H2-(C5H5)2MoP is strongly aromatic sustaining an MIRC of 15.3 nA T−1 and 16.2 nA T−1, respectively, which is also predicted from Hückel's and Baird's aromaticity rules. However, their singlet states lying 23.6 kcal mol−1 and 10.2 kcal mol−1 above the triplet state also sustain a diatropic MIRC. The ring-current strength of H2-(C5H5)2CrP is 5.8 nA T−1 indicating weak aromaticity, whereas the strength of the MIRC of H2-(C5H5)2MoP is only 1.3 nA T−1 suggesting that it is nonaromatic. The partially ionic nature of the chemical bond between the metal and the C5H5 might be the reason for the unexpected aromatic properties of the [Cr]- and [Mo]-cenothiaporphyrins.2
Group VI and isoelectronic compounds
The singlet and triplet states of the (C5H5)2TcP+ and H2-(C5H5)2TcP+ cations are almost degenerate. The singlet state of H2-(C5H5)2TcP+ is 2.2 kcal mol−1 below the triplet, whereas the triplet state of (C5H5)2TcP+ is energetically 2.1 kcal mol−1 lower than the singlet. The aromatic character of the [Tc]-cenothiaporphyrin cations follows the Hückel and Baird aromaticity rules, except for the triplet state of H2-(C5H5)2TcP+, which is unexpectedly antiaromatic, sustaining a paratropic MIRC of −9.0 nA T−1. The singlet state of (C5H5)2TcP+ is aromatic with a MIRC of 7.8 nA T−1 and the singlet state of H2-(C5H5)2TcP+ is antiaromatic with a paratropic MIRC of −7.5 nA T−1. The triplet state of (C5H5)2TcP+ has a weak paratropic MIRC of −3.3 nA T−1. The electron configuration of the singlet state of H2-(C5H5)2TcP+ is 5s04d6.7 and the charge of the metal is 0.1e, whereas the triplet state of H2-(C5H5)2TcP+ has 6.3 electrons in the d shell and a charge of 0.4e on the metal. The exciton of the triplet state of H2-(C5H5)2TcP+ is partly located to the atom, implying that it does not follow the Baird rule.The sextet ground state of the neutral (C5H5)2TcP is antiaromatic sustaining a MIRC of −10.3 nA T−1. The MIRC of the sextet ground state of the neutral H2-(C5H5)2TcP is 16.3 nA T−1 showing that it is aromatic. The singlet and triplet states are 90.0 kcal mol−1 and 87.9 kcal mol−1 higher in energy.
Group X compounds
The [Ni]-cenothiaporphyrins have two electrons more than the [Fe]-cenothiaporphyrins. Thus, one expects a reverse aromatic nature as compared to the Fe compounds according to the Hückel and Baird aromaticity rules. The singlet state of (C5H5)2NiP is indeed strongly aromatic with a MIRC of 22.6 nA T−1, but the singlet state of H2-(C5H5)2NiP is nonaromatic with a weak MIRC of 0.4 nA T−1. However, (C5H5)2NiP and H2-(C5H5)2NiP have triplet ground states that are 4.4 kcal mol−1 and 8.1 kcal mol−1 below the singlet state. The triplet state of (C5H5)2NiP is weakly antiaromatic with a MIRC of −4.6 nA T−1 and the triplet state of H2-(C5H5)2NiP is weakly aromatic sustaining a diatropic MIRC of 5.2 nA T−1.Group IV compounds
The vanadium-containing (C5H5)2VP+ and H2-(C5H5)2VP+ are isoelectronic to group IV elements. They have formally only two valence electrons on the metal atom that can contribute to the bond conjugation and the aromatic pathway, which may explain why three of the four [V]-cenothiaporphyrins sustain very weak ring currents. The strengths of the MIRC of the ground triplet states of (C5H5)2VP+ and H2-(C5H5)2VP+ are −0.1 nA T−1 and 1.4 nA T−1, respectively. The singlet state of (C5H5)2VP+ that lies 21.7 kcal mol−1 above the triplet ground state sustains a weak diatropic MIRC of 2.2 nA T−1. The singlet state of H2-(C5H5)2VP+ lying 30.5 kcal mol−1 above the triplet is aromatic sustaining a MIRC of 13.7 nA T−1, which is also expected based on the aromatic nature of H2-(C5H5)2FeP when using Hückel's aromaticity rule.The quartet state of the neutral (C5H5)2VP is antiaromatic sustaining a MIRC of −6.0 nA T−1, which can indeed be deduced from the antiaromatic character of (C5H5)2FeP. In (C5H5)2FeP, the three valence orbitals of the metal are doubly occupied, whereas in the quartet state of the neutral (C5H5)2VP they are singly occupied. The aromatic character of the quartet state of H2-(C5H5)2VP can similarly be obtained from the aromatic nature of H2-(C5H5)2FeP, since the same number of orbitals of the metal are occupied in both cases even though it has three electrons less. H2-(C5H5)2VP is indeed aromatic with a MIRC of 8.6 nA T−1.
Conclusions
The magnetically induced current–density susceptibility tensor (CDT) has been studied for the lowest singlet and triplet states of the metallocenothiaporphyrins, where the metal M is V, Cr, Mn, Fe, Co, Ni, Mo, Tc, Ru, or Rh. The cations of the V, Mn, Co, Tc or Rh compounds were studied because the neutral molecules have an odd number of electrons. The relative energy of the singlet and triplet states are reported in Table S2 of the ESI.† The ground states of the metallocenothiaporphyrins have largely the same spin state as the corresponding metallocenes.56–58 See Table S3 of the ESI.† The wave functions of the studied molecules are expected to be dominated by a single Slater determinant as for the metallocenes.1 The CDT was calculated using the gauge-including magnetically induced currents (GIMIC) method. Strengths of the magnetically induced ring currents (MIRC) were obtained by integrating the CDT passing through integration planes across specific chemical bonds. The strength and direction of the MIRC yield the aromatic character, the degree of aromaticity, and the ring-current pathway. The calculations show that the aromatic nature of most of the studied molecules can be estimated from the Hückel and Baird aromaticity rules. For the singlet and triplet states of the four compounds that are isoelectronic to the Fe compounds, only the triplet state of H2-(C5H5)2CoP+ has incorrect tropicity judged from the Baird aromaticity rule. The triplet state of H2-(C5H5)2TcP+ is antiaromatic, while the triplet state of (C5H5)2MoP and the singlet state of H2-(C5H5)2CrP are aromatic, although the Hückel and Baird aromaticity rules suggest the opposite behaviour. The same holds for six other states. However, their MIRC are very weak (smaller than 3 nA T−1). Hence, they should be considered nonaromatic. Population analysis suggests that triplet states do not follow the Baird aromaticity rule when their exciton is partly located to the metal. Three of the four [V]-cenothiaporphyrins are nonaromatic probably because the ligand-metal bonding in vanadocene and chromocene is partly ionic. They have an electron configuration of 4s03d4 but formally only two valence electrons to transport the ring current across the metallocene.GIMIC calculations on the high-spin states of the neutral metallocenothiaporphyrins with M = V, Mn, Co, Tc or Rh show that the Hückel and Baird aromaticity rules can be unified. The combined Hückel and Baird aromaticity rules that molecules with an odd number (2n + 1) of occupied orbitals in the bond conjugation are aromatic, whereas molecules with an even number (2n) of occupied conjugation orbitals are antiaromatic. The sextet ground state of (C5H5)2TcP has 15 spin-down electrons and 20 spin-up valence electrons. The electrons occupy 20 conjugated orbitals of which 5 are singly occupied. Since the number of occupied orbitals is even, the molecule is antiaromatic according to the present orbital-count rule of aromaticity. Our calculations showed that the sextet state of the (C5H5)2TcP is indeed antiaromatic sustaining a MIRC of −10.3 nA T−1. H2-(C5H5)2TcP with a MIRC of 16.3 nA T−1 has 21 conjugated valence orbitals. The sextet state of H2-(C5H5)2TcP is expected to be aromatic because it has an odd number of conjugated orbitals. The Hückel rule of aromaticity is covered by the combined aromaticity orbital-count rule, since in closed-shell molecules, all orbitals are doubly occupied, yielding the well-known 4n + 2 electron-count rule. The Baird aromaticity rule for triplet states is also part of the combined aromaticity rule that can be employed for high-spin states such as those studied in this work.
The calculations show that the singlet ground state of [Fe]-cenothiaporphyrin ((C5H5)2FeP) is antiaromatic sustaining a paratropic MIRC of −10.8 nA T−1 and that the singlet ground state of dihydro[Fe]-cenothiaporphyrin (H2-(C5H5)2FeP) is aromatic sustaining a diatropic MIRC of 13.8 nA T−1. The lowest triplet states of (C5H5)2FeP and H2-(C5H5)2FeP are aromatic and antiaromatic, respectively. Calculations of the current density reveal a helical flow at the ferrocene moiety, whose chirality is the same for both the aromatic and antiaromatic molecules. The aromatic character of the studied metallocenothiaporphyrins with a noticeable MIRC (I > 3 nA T−1) can with a few exceptions, be obtained by applying the combined Hückel–Baird aromaticity rule. The obtained ring-current strengths and aromatic character of the studied molecules are summarized in Table 1, where the MIRC strength is colour-coded according to its tropicity. When the molecule follows the Hückel and Baird rules, the table has two red numbers and two blue numbers on the corresponding row. The green background of the MIRC strength indicates that the molecule can be considered nonaromatic, whereas incorrect tropicity is marked with a turquoise background.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Academy of Finland through project number 314821, by the Magnus Ehrnrooth Foundation, the Finnish Cultural Foundation, and the Swedish Cultural Foundation in Finland. V. N. Cherepanov thanks the Russian Scientific Foundation (18-19-00268-II). We acknowledge computational resources from the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533) and the CSC-IT Center for Science, Finland.Notes and references
- Z.-F. Xu, Y. Xie, W.-L. Feng and H. F. Schaefer, J. Phys. Chem. A, 2003, 107, 2716–2729 CrossRef CAS.
- F. A. Cotton and G. Wilksinson, Advanced Inorganic Chemistry: A Comprehensive Text, John Wiley & Sons, 2nd edn, 1967 Search PubMed.
- C. Elschenbroich, Organometallics, Wiley-VCH, 3rd edn, 2006 Search PubMed.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 114, 632–635 RSC.
- G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef CAS.
- E. O. Fischer and W. Pfab, Z. Naturforsch. B, 1952, 7, 377–379 CrossRef.
- K. P. Schug, H.-J. Guttmann, A. W. Preuss and K. Schädlich, SAE Tech. Pap., 1990, 900154 CrossRef.
- K. Park, D. Yeo, S. Oh, H. Kim, M. Hoon Ha, H. Oh, M. Park, J. Park, D. Park, D. Jung, H. Chae, H. Kim and J.-H. Boo, Adv. Mater. Res., 2011, 415–417, 1360–1363 Search PubMed.
- A. Xia, J. E. Knox, M. J. Heeg, H. B. Schlegel and Ch. H. Winter, Organometallics, 2003, 22, 4060–4069 CrossRef CAS.
- R. A. Collins, A. F. Russell and P. Mountford, Appl. Petrochem. Res., 2015, 5, 153–171 CrossRef CAS.
- H. Köpf and P. Köpf-Maier, Angew. Chem., Int. Ed. Engl., 1979, 18, 477–478 CrossRef.
- J. B. Waern, C. T. Dillon and M. M. Harding, J. Med. Chem., 2005, 48, 2093–2099 CrossRef CAS.
- S. Gómez-Ruiz, A. García-Peñas, S. Prashar, A. Rodríguez-Diéguez and E. Fischer-Fodor, Materials, 2018, 11, 224 CrossRef.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5986 CrossRef CAS.
- D. E. Bean, P. W. Fowler and M. J. Morris, J. Organomet. Chem., 2011, 696, 2093–2100 CrossRef CAS.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- M. D. Rausch, Can. J. Chem., 1963, 41, 1289–1314 CrossRef CAS.
- K. Heinze and H. Lang, Organometallics, 2013, 32, 5623–5625 CrossRef CAS.
- D. Astruc, Eur. J. Inorg. Chem., 2017, 6–29 CrossRef CAS.
- A. Pal, S. Ranjan Bhatta and A. Thakur, Coord. Chem. Rev., 2021, 431, 213685 CrossRef CAS.
- I. Simkowa, L. Latos-Grażyński and M. Stępień, Angew. Chem., Int. Ed., 2010, 49, 7665–7669 CrossRef CAS.
- I. Simkowa, L. Latos-Grażyński and M. Stępień, Angew. Chem., Int. Ed., 2013, 52, 1044–1048 CrossRef.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2004, 1, 279–293 CrossRef CAS.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Draina, Coord. Chem. Rev., 2010, 254, 2297–2310 CrossRef CAS.
- J. C. Barona-Castaño, C. C. Carmona-Vargas, T. J. Brocksom and K. T. de Oliveira, Molecules, 2016, 21, 310 CrossRef.
- G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Angew. Chem., Int. Ed., 2017, 57, 1251–1255 CrossRef.
- T. Woller, P. Geerlings, F. D. Proft, B. Champagne and M. Alonso, J. Phys. Chem., 2019, 123, 7318–7335 CAS.
- Z. S. Yoon, D.-G. Cho, K. S. Kim, J. L. Sessler and D. Kim, J. Am. Chem. Soc., 2008, 130, 6930–6931 CrossRef CAS.
- J. M. Lim, Z. S. Yoon, K. S. Kim, M.-C. Yoon and D. Kim, Chem. Commun., 2009, 261–273 CAS.
- D. Sundholm, H. Fliegl and R. Berger, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef.
- S. Taubert, D. Sundholm and J. Jusélius, J. Chem. Phys., 2011, 134(054123), 1–12 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 16, Revision A.02, Gaussian, Inc., Pittsburgh, PA, 2016 Search PubMed.
- M. Rauhalahti, S. Taubert, D. Sundholm and V. Liegeois, Phys. Chem. Chem. Phys., 2017, 19, 7124–7131 RSC.
- R. R. Valiev, H. Fliegl and D. Sundholm, Chem. Commun., 2017, 53, 9866–9869 RSC.
- R. R. Valiev, H. Fliegl and D. Sundholm, Phys. Chem. Chem. Phys., 2017, 19, 25979–25988 RSC.
- R. R. Valiev, G. V. Baryshnikov and D. Sundholm, Phys. Chem. Chem. Phys., 2018, 20, 30239–30246 RSC.
- R. R. Valiev, I. Benkyi, Y. V. Konyshev, H. Fliegl and D. Sundholm, J. Phys. Chem. A, 2018, 122, 4756–4767 CrossRef CAS.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- E. Hückel, Z. Phys., 1931, 72, 310–337 CrossRef.
- R. Breslow, Acc. Chem. Res., 1973, 6, 393–398 CrossRef CAS.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- K. Jorner, B. O. Jahn, P. Bultinck and H. Ottosson, Chem. Sci., 2018, 9, 3165–3176 RSC.
- H. Fliegl, D. Sundholm, S. Taubert, J. Jusélius and W. Klopper, J. Phys. Chem. A, 2009, 113, 8668–8676 CrossRef CAS.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS.
- J. Ahrens, B. Geveci and C. Law, Visualization Handbook, Elsevier, 2005, ISBN-13: 978-0123875822, see also http://www.paraview.org Search PubMed.
- J. Weber, A. Goursot, E. Penigault, J. H. Ammeter and J. Bachmann, J. Am. Chem. Soc., 1982, 104, 1491–1506 CrossRef CAS.
- S. Y. Ketkov, S.-Y. Tzeng, E. A. Rychagova, G. Markin, S. Makarov and W.-B. Tzeng, Dalton Trans., 2021, 50, 10729–10736 RSC.
- B. Rudshteyn, J. Weber, D. Coskun, P. A. Devlaminck, S. Zhang, D. R. Reichman, J. Shee and R. Friesner, ChemRxiv, 2021 DOI:10.26434/chemrxiv.14751684.v1.
- C. Cauletti, J. C. Green, M. R. Kelly, P. Powell, J. van Tilborg, J. Robbins and J. Smart, J. Electron Spectrosc. Relat. Phenom., 1980, 19, 327–353 CrossRef CAS.
- J. C. Green, Struct. Bonding, 1986, 43, 37–112 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp04779e |
| This journal is © the Owner Societies 2022 |