
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effects of glycine to alanine mutations on the structure of GPO collagen model peptides†
Konstantin
Röder

Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, Cambridge, UK. E-mail: kr366@cam.ac.uk
First published on 7th December 2021
Abstract
Collagen proteins are the main constituents of the extracellular matrix (ECM), and fulfil a number of wide-ranging functions, including contributions to the mechanical and biological behaviour of the ECM. Due to the heterogeneous nature of collagen in tissue samples it is difficult to fully explain the experimental observation, and hence the study of shorter model peptides is common place. Here, the computational energy landscape framework is employed to study Gly to Ala mutations in a GPO model peptide. The results show good agreement with the experimental observations for the GPO reference and a triply mutated peptide, demonstrating the validity of the approach. The modelling predicts that changes in structure are moderate and localised, with an increased dynamic in the backbone and alterations to the hydrogen bonding pattern. Two mechanisms for adjusting to the mutations are observed, with potential consequences regarding protein binding. Finally, in line with a hypothesis that proline puckering allows controlled flexibility (Chow et al., Sci. Rep., 2018, 8, 13809), alterations in the puckering preferences are observed in the strained residues surrounding the mutational sites.
1 Introduction
The extracellular matrix in multicellular organisms is a dynamic and heterogeneous biomaterial.1–4 It facilitates cell growth and differentiation, while detachment of cells from the extracellular matrix leads to cell death, a key feature of tumour metastasis.3,5 The main constituent of the extracellular matrix is collagen proteins, a family of proteins self-assembling into triple helices6 and characterised by regular repeats of three amino acids.These regular repeats are commonly referred to as triplets, with the sequence GXY. The regular repetition of glycine facilitates the formation of the characteristic triple helix.7 Due to the crowded centre, only glycine as the smallest amino acid can be accommodated without structural deformations.
The other two residues in the three residue repeats, X and Y, are more varied. The X position is enriched in proline, and in the Y position a high occupancy of (2S,4R)-hydroxyproline (Hyp or O) is observed. The vast majority of collagen proteins adopt trans conformations for these residues. While the abundance of Pro and Hyp in these repeats is higher than in other proteins, a significant proportion of repeats in the collagen samples contain neither Pro nor Hyp. Nonetheless, GPO and GPP repeats are commonly used as models to study collagen fibres in experiments and in silico.
The three strands are staggered by one residue, and as a result there is a leading, middle and trailing strand. Due to this stagger, each register has a Gly, an X and a Y position amino acid. This architecture is stabilised by interchain hydrogen bonds, formed between the carbonyl group on the amino acid in the X position and the amide hydrogen on glycine in the next register.8
While the canonical architecture of collagen is well described, the heterogeneity in its sequences and in the different types of collagen complicate experimental studies. In addition, while local interactions are key to the behaviour of collagen fibres, their macroscopic behaviour, interaction with other molecules, and response to external influences such as mechanical forces form the basis for its biological role.6 As a result, no experimental technique is available to study all relevant length scales at the required resolution.
Computational techniques can supplement experimental approaches, as they can model the dynamic nature of the collagen molecules at atomistic resolution. Such detailed descriptions can aid experimental work, as the computational models can be translated into observables, and may predict experimental signatures of distinct structural features.
GPP and GPO model collagen peptides are fairly rigid due to their regular structure, but as already indicated these sequences are somewhat simplistic. It is therefore of interest to understand in more detail how mutations and deletions affect the behaviour of collagen, where the altered local interactions and the resulting changed dynamics may influence properties on a larger scale. Unfortunately, researchers face two challenges in studying such distinct changes. Firstly, the experimental resolution may not allow for a differentiation between local structures if they are highly dynamic. Secondly, the synthesis of such model peptides generally leads to mutation in all three collagen strands,9 and therefore does not lead to a clear picture about the effect of a single mutation.
In this study, the energy landscapes are explored for such mutated sequences and compared to the ‘canonical’ GPO repeats. The exploration of energy landscapes allows us to model structural and dynamic properties, and the effect of mutations can be clearly analysed and linked to predicted, observable features, such as interatomic distances. We find that the modelling approach shows good agreement with previous experimental work on the GPO model peptide and with structures reported for collagen peptides with glycine to alanine (G to A) mutations in all strands. A mutation from Gly to Ala in a single strand has very similar effects, with the interruption in the hydrogen bonding observed for both being very similar. The structural ensembles for all mutated peptides show shifts in the endo/exo-populations around the mutated registers, allowing them to accommodate strain introduced by the larger required volume for Ala. A fine balance between backbone strain and the interruption of hydrogen-bonding is observed, where for a single mutation, the methyl group is pointing to the centre of the triple helix, while for multiple mutations this methyl orientation competes with rotated methyl groups that introduce strain, but allow for the formation of hydrogen bonds between hydroxyproline and proline in the same register. These distinct mechanisms likely leave identifiable signatures in experimental observations.
2 Methodology
2.1 System setup
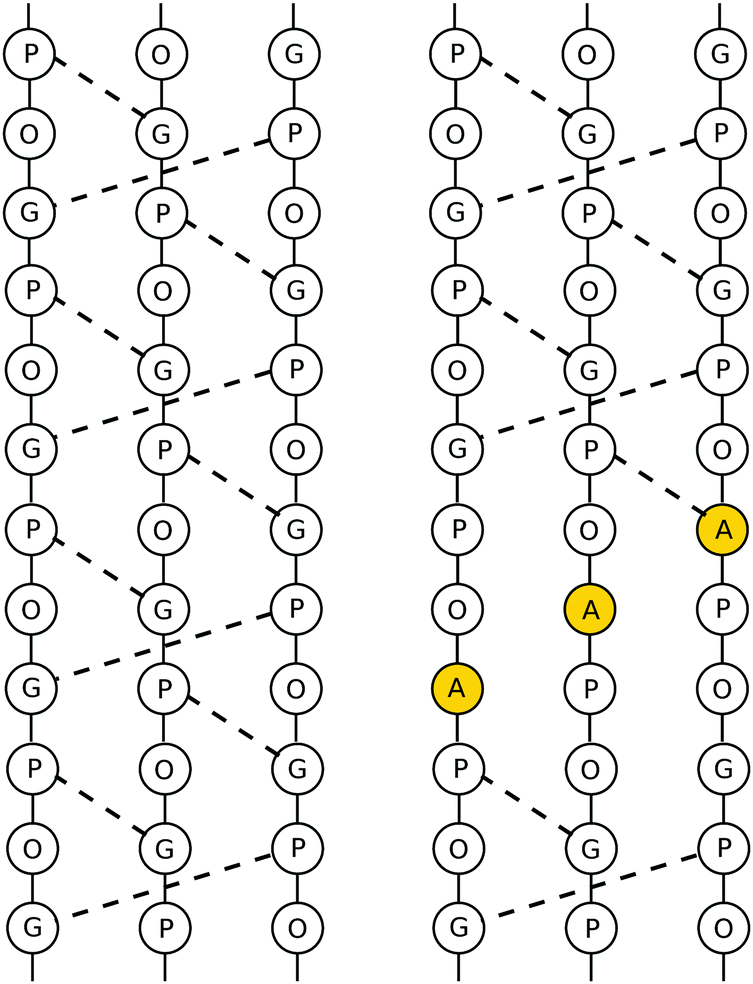
The mutated collagen systems under consideration are based on an experimentally reported GPO-based structure (PDB id 1V4F). The three chains consist of seven GPO repeats, and each chain fragment is capped with terminal methyl groups. The mutated glycine residues are in the fourth repeat in each chain, i.e. in the centre of the fragments. Hence, the three possible mutation sites are in close proximity, but staggered following the register shift between chains, as illustrated in Table 1. A number of different possible mutation patterns arise from this setup, and the four patterns studied here are given in Table 2. Alongside these mutated molecules, we further studied an unmutated version of this system as a reference. The reference and a mutated system are shown in Fig. 1.| α3 | α2 | α1 |
|---|---|---|
| Gly | Pro | Hyp |
| Pro | Hyp | Gly |
| Hyp | Gly | Pro |
| Gly | Pro | Hyp |
| Pro | Hyp | Gly |
| Mutation pattern 1 | Mut1 | α1 only |
| Mutation pattern 2 | Mut2 | α1 and α3 |
| Mutation pattern 3 | Mut3 | α1 and α2 |
| Mutation pattern 4 | Mut4 | All three chains |
 | ||
| Fig. 1 Depiction of a triply mutated collagen system (top, mutational pattern 4) and the unmutated reference (bottom). The alanine and glycine residues are highlighted in orange. | ||
The AMBER ff14SB10 force field was used, properly symmetrised,11,12 with an implicit Generalised Born solvation model (igb = 8)13 and an effective ion concentration of 0.1.
2.2 Computational methods
The potential energy landscapes for the five different collagen fragments were explored using the computational potential energy landscape framework.14,15 Searches for low energy structures to seed the sampling of the energy landscapes were undertaken using basin-hopping global optimisation.16–18 Discrete path sampling (DPS)19,20 was employed to obtain kinetic transition networks.21,22 Transition state candidates were located with the doubly-nudged elastic band (DNEB) algorithm,23–25 and refined with hybrid eigenvector-following (HEF).26 The sampling followed previously published protocols and more details can be found elsewhere.14,152.3 Analysis
A number of key characteristics were used to identify structural changes in the mutated systems. CPPTRAJ27 was used to calculate the distances between residues, the existence of hydrogen bonding, and dihedral angles. The first set of distances are between the Cα atoms in the glycine and proline residues involved in the interchain hydrogen bonds. A second set of distances measured contains the Cα distances between the neighbouring Gly, Hyp and Pro residues in the three chains, i.e. the distances between residues in a row in Table 1. χ1(N–Cα–Cβ–Cγ) and χ2(Cα–Cβ–Cγ–Cδ) were calculated to monitor the exo/endo configuration of Pro and Hyp residues. Furthermore, the LCPO28 algorithm in CPPTRAJ27 was used to calculate the surface area for the fragment containing the three registers before the mutational site in the α1 chain, the mutated registers, and the three registers after the mutational site in the α3 chain, i.e. a segment containing nine amino acids in each chain.The final metric used in this study is the orientation of the methyl groups on the alanine residues with respect to the other chains. The angle used to represent this orientation is the angle between the vector pointing from the Cα atom of Ala to the midpoint between the two Cα atoms of the Pro and Hyp residue in the other two chains in the same register, and the vector from Cα to Cβ in alanine, projected into the plane formed by the three Cα atoms. The setup is illustrated in Fig. 2.
 | ||
| Fig. 2 Scheme illustrating the setup for the calculation to determine the methyl group orientation in the mutated collagen chains relative to the other two chains. | ||
3 Results
In this section, we will report the structural changes observed in the mutated collagen fragments. The first two sections discuss the GPO model peptide and the system, in which each chain carries a mutation. As experimental results are available for these systems, they serve as a reference and show that the method and the chosen models are able to reproduce even nuanced changes between the GPO model and the mutants. These sections are followed by descriptions of the changes observed for a single or two mutations, and a section looking at the orientation of the methyl groups in all mutants. The sections contain a description of the most important changes, and all relevant data sets and analysis scripts are available online.The energy landscapes for the reference and collagens with Gly to Ala mutations exhibit a single shallow and broad funnel with little distinguishable substructure. This observation indicates a large number of relatively similar structures, without the existence of competing morphologies with different backbone arrangements.
3.1 The GPO model peptide
The GPO model peptide has been studied experimentally, and a number of structures have been published on the protein database. Therefore, comparison between these published structures and the average structures found in the computational exploration of the energy landscape serves as validation for the employed methodology, and at the same time provides a reference to compare the modelled structural features for the mutated peptides.Each register has three distinct distances that we can compare across registers, namely the distances between Pro and Gly, Gly and Hyp, and Hyp and Pro. As the protein adopts a regular structure these distances are conserved along the triple helix. We can therefore look at the average distances between these residues to compare the structures from the experiment with the modelling. For the structures found on the energy landscape, we can employ two different approaches: either we consider the lowest energy minimum as a representative sample, or we use a thermally weighted average. The thermal weights in this case are the Boltzmann factor based on the potential energy difference to the global minimum. The averaged distances between the Cα atoms for the four structures from the PDB and the thermal average and the lowest PE minimum are provided in Table 3 (top). The computed averages are within the standard deviation for all experimental structures, whether we look at the thermal average or the lowest potential energy minimum only. We can extend our comparison, and also compare it to experimental distances for the GPP model peptides, and we again observe a good agreement (see Table 3, bottom), indicating a good agreement between the computational work with the canonical triple helix structure.
| A | |||
|---|---|---|---|
| (GPO)x structure | d(O–G)/Å | d(P–G)/Å | d(O–P)/Å |
| a Seven registers starting with Gly in the leading chain. b Two GPO repeats starting with Gly in the leading chain. c Eleven GPO repeats with distances averaged for each structure, and then weighted using Boltzmann weights to obtain a weighted average. d Eleven GPO repeats averaged for the lowest potential energy minimum. e Two GPP repeats starting with the Y-position proline (ProY) in the leading chain. f Seven registers starting with the X-position proline (ProX) in the leading chain. | |||
PDB 1V4Fa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 29 |
5.033 ± 0.167 | 4.757 ± 0.130 | 4.818 ± 0.143 |
| PDB 1V6Qb29 |
4.966 ± 0.118 | 4.840 ± 0.150 | 4.863 ± 0.058 |
| PDB 1V7Hb29 |
4.956 ± 0.200 | 4.788 ± 0.150 | 4.786 ± 0.166 |
| PDB 3B0Sb30 |
4.946 ± 0.190 | 4.763 ± 0.100 | 4.799 ± 0.204 |
| Thermal averagec | 5.050 ± 0.008 | 4.830 ± 0.006 | 4.824 ± 0.031 |
| Lowest PE minimumd | 5.035 ± 0.066 | 4.824 ± 0.048 | 4.869 ± 0.156 |
The largest error we observe in the set of distances is in the distance between Hyp and Pro. However, this error is not simply due to more flexibility in these two residues, but is directly related to the endo/exo configurations of the proline in the X position. While the hydroxyproline, in agreement with the experiment, is in an exo configuration,7,33,34 the configuration of the proline is more flexible.35,36 The thermal averaged distance for an endo-configuration is 4.97 Å, while the distance is reduced to 4.65 Å if the proline is in an exo-configuration. Based on the thermal average for all structures, and using the fact that we do not observe any planar configurations in the low energy structures that have a significant population,‡ on average 55% of all prolines are in an endo-configuration at 25 °C. At lower temperatures, i.e. when we only see contributions from the lowest energy minima, the endo-configuration is somewhat more common with 59% of all prolines adapting it.
The exploration of the energy landscape gives us additional insight to this predicted structural behaviour. The lowest PE minimum in the database has four prolines in an exo- and seven prolines in an endo-configuration. It is linked by a single transition state to a minimum, where one additional proline is switched from endo to exo. The energy difference between the two minima is 0.14 kcal mol−1, so significantly smaller than kT at room temperature, and the potential energy barrier is also low at 2.92 kcal mol−1 relative to the lowest PE minimum. This barrier corresponds to a mean-first encounter time for this transition of a few nanoseconds, in agreement with previous experimental work.37–44
Importantly, while this analysis seems to indicate that it is roughly equally likely to observe both configurations, the low energy structures show additional interesting features. In Fig. 4, the fraction of endo- and exo-configurations is shown. While the data towards either end of the molecules might be affected by finite size effects, we observe an alteration over two or three registers from mainly endo to exo and vice versa.
 | ||
| Fig. 3 The distances between Cα atoms in the three chains for two registers in the middle of the modelled sequences. Each point corresponds to an individual minimum in the database with the respective distance and potential energy. The dashed lines indicate the energy of the lowest potential energy minimum and the thermal weighted average for the inter-residue distances for the entire database. These are the same values that are provided in Table 3. While the Hyp–Gly and Gly–Pro distances are clustered around the thermal average, the Pro–Hyp distances form two distinct clusters, related to the endo/exo configuration of the proline in the X-position. | ||
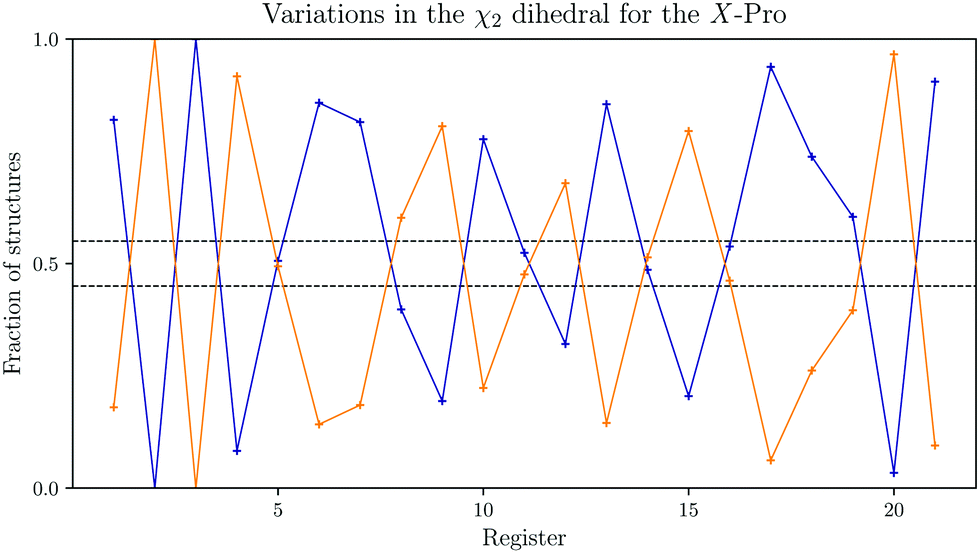 | ||
| Fig. 4 Variation in the χ2 dihedral angle Boltzmann-weighted average for the X-position proline in all registers of the reference GPO model peptide. The fraction of the exo-configuration is shown in orange, and the endo-configuration in blue. The dashed lines indicate the average number of configurations. | ||
The regular structure is stabilised by hydrogen bonds, as introduced above. Similar to the interchain distances, we observe a fixed distance for the residues involved in the hydrogen bonds as well, with a Cα–Cα distance of 6.234 ± 0.007 Å. The regular pattern formed by the hydrogen bonds is shown in Fig. 5 on the left hand side.
 | ||
| Fig. 5 Hydrogen bonding patterns from the structures located on the energy landscape for the reference GPO model peptide (left) and for the peptide with Gly to Ala mutations in every chain (right). The mutations lead to local interruptions in the hydrogen bonds without any effects on the global structure. | ||
3.2 Alanine mutations in all chains
Aside from the GPO model peptides, experimental structures are available for structures, which carry a mutation in every chain. A comparison between the computational work and the experimental structures is therefore again possible, however, in this case with some caveats. Firstly, the number of structures available is more limited, namely PDB entries 5Y45 and 5Y46.9 These structures furthermore contain an additional mutation as the sequence of interest is GPO–ALO–GPO.In Fig. 6, the relative change in interchain distances between the three chains are plotted for four models contained in PDB 5Y45.9 The first noticeable property is the localisation of the changes, i.e. the interchain distances are only perturbed within the immediate vicinity of the mutations. The maximum change in distance is around 2 Å, with the middle chain being displaced away from the helix, while the leading and trailing chains stay close.
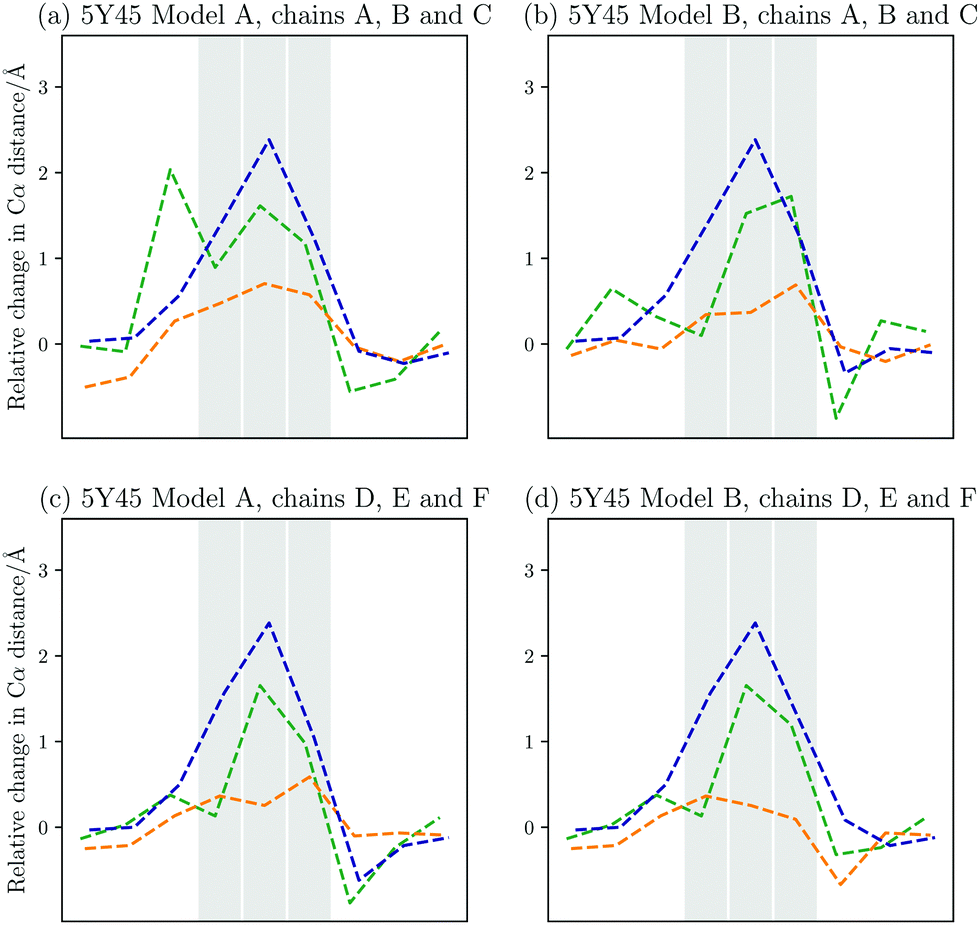 | ||
| Fig. 6 Changes in the interchain Cα–Cα distances compared to the GPO model peptide between the leading and middle chain (green), the leading and the trailing chain, (orange), and the middle and trailing chain (blue) for the different models from PDB 5Y45.9 The registers that contain the Ala to Gly mutations are highlighted. The middle chain moves away from the other two chains up to 2 Å relative to the GPO model. The distance between the leading and trailing chain is fairly similar to the model, though some variation is observed. Overall, the changes are very local to the mutation sites. | ||
In Fig. 7(d), the Boltzmann weighted distances from the landscape exploration are shown. As in the case of the GPO model peptide, the computational work agrees well with the available experimental data. In both sets of data, the middle chain is moved away from the other two chains, where the distance between the middle and trailing chain increased most by up to 2 Å. The changes are localised, and the computational work shows that only the neighbouring registers to the three mutated registers are affected. This limitation in the structural changes on the immediate vicinity of the mutations is also evident from the predicted changes in the hydrogen bonding. The hydrogen bonding is interrupted around the mutations, but away from the mutational sites no changes are observed (see Fig. 5 on the right).
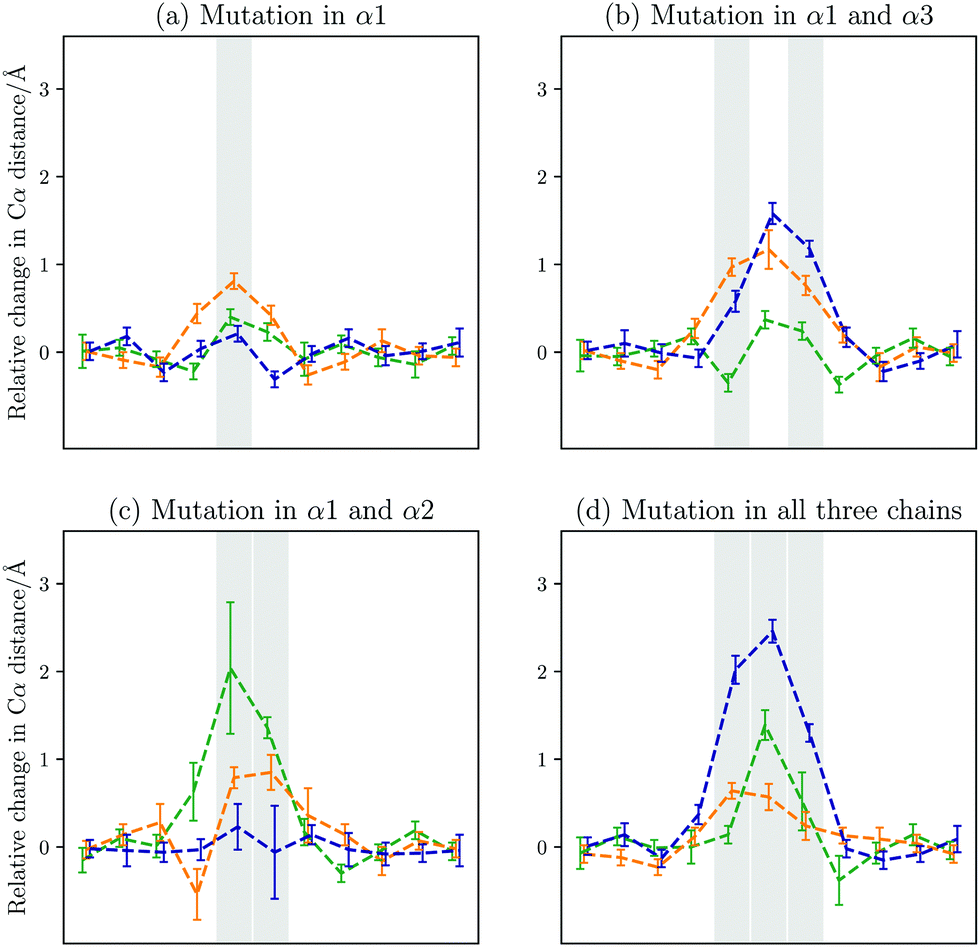 | ||
| Fig. 7 Changes in the interchain Cα–Cα distances compared to the GPO model peptide between the leading and middle chain (green), the leading and the trailing chain, (orange), and the middle and trailing chain (blue) for the different mutational patterns. The registers that contain the Ala to Gly mutations are highlighted. The contributions from the individual minima are Boltzmann weighted. | ||
Another structural effect of the mutations is a change in the distribution of endo/exo-configurations for the X-position prolines around the mutated register. In Table 4, the percentage of residues in an exo-configuration are given, again Boltzmann-weighted to yield a realistic picture of the observable distributions. While our findings indicate an oscillation in the distribution for the GPO model peptide as well (see Fig. 4), the introduction of the alanine residues results in structures exclusively adopting exo- or endo-configurations.
| System | Register −2 (α1) | Register −1 (α2) | Register 0 (α3) | Register 1 (α1) | Register 2 (α2) | Register 3 (α3) | Register 4 (α1) |
|---|---|---|---|---|---|---|---|
| Reference | 80.6% | 22.3% | 47.6% | 67.9% | 14.5% | 51.4% | 79.5% |
| Mut1 | 97.9% | 99.9% | 0.0% | 87.7% | 51.4% | 1.5% | 8.5% |
| Mut2 | 98.8% | 0.0% | 99.9% | 75.9% | 5.4% | 98.4% | 79.6% |
| Mut3 | 13.6% | 98.8% | 1.9% | 87.1% | 90.8% | 56.5% | 85.7% |
| Mut4 | 99.6% | 44.3% | 1.4% | 7.5% | 99.4% | 98.7% | 9.9% |
3.3 The effect of Gly to Ala mutations in a single or two chains
When analysing the effects of a single and two mutations, a key point of interest is whether the changes observed in the triply mutated system are very similar to those observed with fewer mutations, or whether these changes are much smaller. These options will lead to distinct behaviour in real systems, and alter potential binding interactions. As an experiment cannot probe single and double mutations, computational modelling is the best approach to answer this question.The alterations in the interchain distances shown in Fig. 3 show much smaller changes for a single mutation. The main change occurs between the leading chain and the trailing chain. The increased distances lead to interruption of the hydrogen bonding pattern. The changes are localised to the mutated register and the two registers on either side. The canonical hydrogen bond between the alanine in the leading chain and the proline in the middle chain in the preceding register is only found in around 2% of structures. Similarly, the hydrogen bonds between the proline two registers before and after the alanine in the leading chain to glycines in the trailing chain are only found in 4 and 10% of structures, respectively. This prediction means that of the three hydrogen bonds in the vicinity of the mutation that involve residues in the mutated strand, none survive. The only other hydrogen bond in this region between the middle and trailing chain is preserved in about a third of structures. A small number of structures, around 5 to 10%, form hydrogen bonds between the carbonyl oxygen atoms in the affected proline residues in the leading strand and the hydroxyl group in the hydroxyprolines in the same register, which are located in the α3 chain. The pattern is illustrated on the left in Fig. 8.
 | ||
| Fig. 8 Hydrogen bonding patterns from the structures located on the energy landscape for the Mut1 (left) and Mut2 (right) mutational patterns. The mutations lead to local interruptions in the hydrogen bonds without any effects on the global structure. The partial preservation of a hydrogen bond in Mut1 and the formation of an alternative hydrogen bond in Mut2 are predicted. | ||
Introducing two mutations in the collagen peptide can happen in multiple ways. Here, two distinct mutational patterns were studied: Mut2, where there is a register between the two mutation sites, and Mut3, where the mutated registers are directly next to each other.
In Mut2, the mutations are in the leading and trailing chain. This pattern leads to the largest changes in distance between the trailing chain and the other two chains, i.e. the trailing chain is pushed away from the core of the triple helix. While these changes in distance are larger than for the single mutation, the change in hydrogen bonding is reduced. The hydrogen bonds preceding the first mutated register are preserved, including the hydrogen bond formed between the alanine in the leading chain and the proline in the preceding register in the trailing chain. Only two hydrogen bonds are lost, as can be seen in Fig. 8 (right). In addition, a new hydrogen bond is predicted to form between the proline carbonyl oxygen and the hydroxyprolone OH group in the register containing Ala in the α1 chain.
In Mut3, a large interchain distance is observed, which is at the same time associated with the largest standard deviation across all structures (see Fig. 3). As the mutations occur in neighbouring registers in the leading and middle chain, it is potentially not surprising that the distance between these chains increases the most. Above the mutation sites the leading and trailing chain are closer, while below the mutational sites they are further apart, showing a buckling in the backbone. The large standard deviation is a result of two competing structural families, which becomes evident when considering the distribution of interchain distances. The two structures are defined by two competing hydrogen bonding patterns. These patterns are shown in Fig. 9. The two structural families arise from a competition between the two mutational sites – either the hydrogen bond involving the alanine in the trailing chain or in the middle chain is conserved, but never both. The second case allows the formation of additional hydrogen bonds between proline and hydroxyproline in the same register, as observed for Mut2 as well. It should be noted here that these structures show a high degree of variation, and these features are the unifying themes that seem to separate the structures. Taken together they only account for roughly 80% of structures and other additional hydrogen bonds, such as between the alanine in the trailing chain and the glycine in the register above. The key change in all these structures is the loss of hydrogen bonds in the vicinity of the mutations, and a prediction of the appearance of new hydrogen bonding patterns formed within registers between proline and hydroxyproline.
 | ||
| Fig. 9 Competing hydrogen bonding patterns from the structures located on the energy landscape for the Mut3 mutational pattern. Either the hydrogen bond involving the alanine in the trailing chain is conserved (left), or the hydrogen bond formed by the alanine in the middle chain is present (right). The second case leads to the formation of additional hydrogen bonds, leading to a lower energy. | ||
In summary, the single and double mutations have a smaller effect on the relative displacement of the chains, and it is tempting to therefore suggest the changes are smaller than in the system with three mutations. However, the interruption of hydrogen-bonding is very similar in all mutated systems, with the possibility of alternative hydrogen bonding being formed. This observation suggests that the key features, i.e. the possibility to bind other molecules due to the higher backbone flexibility and the existence of alternative hydrogen bonds, exist for all mutants, independent of the number of mutations introduced.
3.4 Methyl group orientations in the different mutational patterns
The structural changes predicted in the mutated sequences stem from the additional space required by the methyl groups in alanine. The space between the three chains cannot accommodate the methyl group, and therefore only two options remain. Either the chains are separated or the methyl group needs to point outwards leading to a twist of the backbone. The former is likely leading to the absence of some hydrogen bonds, while the latter leads to strain in the backbone, and also changes in the hydrogen bonding pattern.For the methyl groups, we observe three possible orientations: (a) pointing into the centre between the two other chains, (b) pointing towards one other chain, and (c) pointing slightly outwards. For case (a), the chain containing the alanine residue will be pushed away from the other two, but those chains remain close, and hence can preserve the hydrogen bonding. In case (b), the distance is only increased between two chains, namely the one containing the mutation and the one it points towards. This orientation preserves most hydrogen bonding, and is also the most compact. Finally, in the case of orientation (c), the backbone rotates allowing for additional hydrogen bonding involving the hydroxyproline on either side of the mutation, but it removes any possibility for hydrogen bonding involving the alanine, and it further introduces strain into the backbone. The orientation in cases (b) and (c) is as follows, the methyl group of Ala in α1 points at α3, the one in α3 points at α2 and the one in α2 points at α1.
If one mutation is introduced (Mut1), we see exclusively orientation (b). The orientation allows the preservation of more hydrogen-bonding, and the single mutation can still be accommodated this way. If a second mutation is introduced, the methyl orientation depends on the distance between them. When there is an offset, as in Mut2, both mutations, in the leading and trailing chain, have enough space in orientation (b). However, if the mutations are moved closer together, i.e. in Mut3, this is no longer possible. We observe two sets of orientations. Preserving the hydrogen bond between the alanine in the α1 chain and the proline in the α2 chain, the methyl in the leading group points towards the centre pushing the chains further apart, but allowing for the alanine in the α2 chain to still point at the α1 chain. Interestingly, in the second subset this orientation is unchanged, but rotation of the methyl group in the leading chain outwards allows the formation of alternative hydrogen bonds. If all three mutations are introduced, in most cases the leading alanine points to the centre, with the other two pointing more towards the chain, but due to the extended interchain distances and the loss of hydrogen bonds, there is a lot of flexibility, with the methyl groups taking up the space inside the pushed apart chains.
3.5 Changes in the surface area of the mutational sites
Not surprisingly, the changes in distance between the chains affect the solvent-accessible surface area of the molecule. The general trend is that the mutations increase the surface area, with a maximum 6% increase for three mutations compared to the GPO model reference. Interestingly, the only mutation pattern going against this trend is subset b for Mut3. The additional hydrogen bonds formed pull the structure together and the surface area is comparable to the reference. The thermally weighted values are provided in Table 5.| System | Solvent-accessible surface area/Å2 |
|---|---|
| Reference | 1362.1 ± 4.9 |
| Mut1 | 1390.5 ± 4.0 |
| Mut2 | 1398.5 ± 8.7 |
| Mut3 a | 1403.6 ± 5.2 |
| Mut3 b | 1366.3 ± 3.4 |
| Mut4 | 1446.9 ± 8.2 |
4 Discussion
4.1 Model validity and agreement with experiment
The first point to notice is the good agreement between the previously reported experimental structures and the predicted structural ensembles from our computational model. Two sets of comparison may be used in this context. Firstly, we can look at a direct comparison of the reported experimental structures and the structural ensembles found in this study. The structures are in good agreement, both for the lowest PE minimum and the thermally-weighted ensemble average. The canonical hydrogen bonding pattern8 is present in the structures located on the energy landscape. This agreement extends not only to good agreement in the GPO model peptide used as the reference, but also to the triple mutated system, for which experimental data is available in the protein data bank.The second comparison for which extensive experimental data exists, is the dynamic nature of the ring puckering of the proline and hydroxyproline residues. The Y-position hydroxyproline adopts an exo-configuration,7,34 which stabilises the collagen structure.45–48 In contrast, the proline in the X position is more dynamic, and shows transitions between endo- and exo-configurations on a nanosecond time scale.37–44 The mean first passage time for this transition from our modelling is in the nanosecond range as well. Furthermore, recent work using density functional approximations48 gave the difference between endo and exo as roughly 0.6 kcal mol−1, which we calculated for a specific example as around 0.2 to 0.3 kcal mol−1. Previous work combing experimental techniques with global optimisation techniques35 shows a range of energy differences between the conformations from close to 0.0 to 0.5 kcal mol−1. Importantly, this work on model peptides is in agreement with work on intact bone ECM samples.36 Furthermore, Chow et al.35 give a ratio of 60:40 between endo and exo for the X-position proline rings. From this more extensive exploration of the energy landscape this ratio is 55:45, but closer to 59:41 at lower temperatures when higher energy structures do not contribute to the structural ensembles.
The good agreement between these various reported experimental results and our computational exploration demonstrates the validity of the modelling approach. One factor that plays a role in this case is the rigidity of the collagen model peptides. The structures are fairly stable, and the chain length was chosen in this study to stop chain dissociation, as we are interested in the increased flexibility and the structures arising from it. This rigidity makes it more likely that we match experimental observables. However, this rigidity is not at the centre of some observables being well matched. First, the proline ring puckering is a dynamic process in nature, and the match between experiment and this study is due to an appropriate model choice. Secondly, the temperature variations, as reported through the ensemble averages, predicts small, but noticeable temperature effects on both interchain distances and ring puckering. Finally, the matching of interchain distance variations for the triply mutated system, including the correct relative variations between the chains is another observation not based on structural rigidity.
4.2 The impact of mutations is local, but significant
When we consider the impact of mutations on a biomolecule one of the key questions is how large the resulting structural and therefore functional changes are. In agreement with published experimental structures, the changes observed are restrained to the mutated registers, and those registers in the vicinity of the mutations. Consequently, the global structure of collagen is likely to be unaffected by the mutations, although the mutations still result in significant changes around the mutation sites.Two points merit further consideration in this context. The first is the mechanism of how the mutational effects are restrained locally. As the interchain distances increase and in some cases a change in backbone orientation is observed due to the orientation of the introduced methyl groups, strain is introduced into the affected backbones. Previously, it was suggested that the flexibility in the proline pucker provides controlled flexibility allowing mechanical deformations of collagen fibres.36,49 Consequently, following this hypothesis, one mechanism to absorb this strain is a change in puckering preferences around the mutational sites. Indeed, the puckering preferences for the X-position are changed around the mutations. This process allows for a local adjustment of structure without disturbing the hydrogen bonding pattern and structure further away.
A caveat with this analysis is the χ2 variations reported in Fig. 4 for the model peptide. It appears that there is some deviation away from the 55:45 split for each individual residue, in a fairly regular manner. Especially towards the ends of the model molecules these deviations are extreme. A question therefore arises whether this feature is a finite size effect and disappears in longer segments, or whether the alterations are still observed, albeit in more moderate ways, as is predicted for the central residues here. In either case, the changes computed for the mutated peptides are significantly larger than the variation in the reference for the central residues, where the mutations are. The predicted puckering changes are therefore a feature of these mutated structures, albeit that the actual changes in preference may be slightly smaller.
The second point in this context relates to the behaviour of the energy landscapes when mutated. The stable sequences, i.e. sequences that form functional, stable biomolecules, are marginally stable,50 and single point mutations on the peptide level may or may not lead to more drastic changes in the energy landscape.51,52 Given that there seems to be a mechanism to constrain the changes to a small region within collagen, and the fact that the introduced changes are small, it is therefore not surprising that no larger changes are observed on the energy landscapes. All landscapes consist of a single broad funnel with a variety of structures, all of comparable energy. This availability of a large number of similar structures contributes to the dynamic character of collagen, which has been described based on experimental findings before.36,49 Based on statistical mechanics, it has been demonstrated that a high molecular symmetry generally leads to either very high or low energies,53 and that high symmetry in biomolecules is favourable.54 These observations are seen in three distinct ways in this study. The high symmetry of the reference collagen is the obvious first one. Secondly, we see it in the high symmetry adopted by the triple mutation. Finally, the retention of the canonical collagen structure on either site of the mutations may also be interpreted in this context. Importantly, this interpretation implies that the maximal changes should be observed in the mutants with lower symmetry. Indeed the most drastically changed ensemble is observed for the doubly-mutated collagen, where the mutations are in neighbouring registers. This proximity introduces overall larger alterations as the mutations will show physical interactions, and hence lead to larger changes. As a result we observe two competing structural ensembles, showing how even such modest changes such as from Gly to Ala can significantly alter the underlying energy landscape and hence the associated structural ensembles.
4.3 Two mechanisms for adapting to a Gly to Ala mutation exist
The methyl groups on the alanine have an increased volume and therefore cannot be accommodated in the centre of the triple helix. For a single mutation or mutations that are spaced out the lowest energy configuration adopted is for the methyl group to point towards the centre of another chain pushing out one of the chains. The closer the mutations are the more disruptive this mechanism becomes as the methyl groups not only clash with the other chains, but also with each other. Firstly, this clashing leads to an increase in the distance between chains. Secondly, it leads to a change in methyl orientation, where the methyl group twists outwards. Consequently the backbone also needs to change, resulting in hydrogen bonding changes. Importantly, it leads to intra-register hydrogen bonding involving the hydroxyl group of hydroxyproline. From this prediction, it is expected that we see changes in the backbone atoms, for example in the NMR signals, and also in the hydration shell around the mutational sites.4.4 The dynamic behaviour of mutated collagen chains
The dynamics of the mutated collagen fibres are affected in two ways. The dynamics of the ring puckering are affected as discussed before due to the changes in the backbone as the mutations push the collagen chains apart, which introduces strain. The increased distance between the chains and the absence of a number of hydrogen bonds changes the backbone dynamics. The predicted structural ensembles show an increased contribution of the backbone atoms in the vicinity of the mutations to the normal modes of the molecules compared to the GPO reference. Interestingly, the changes do not exhibit a clear pattern relating to the number of mutations introduced, but are similar across all mutational patterns probed. While the changes between the mutational patterns differ, in particular with respect to the distance between chains, the interruption of the hydrogen bonding patterns is very similar across the different patterns. As a result, a single Ala to Gly mutation is predicted to lead to a significant change in the dynamical behaviour around the mutational site.5 Conclusions
In this contribution, we employed the potential energy landscape framework to study the effect of Gly to Ala mutations in the GPO model peptides. The findings agree well with the available experimental data. The main contributions of this study are the modelling of experimentally inaccessible single and double mutations. While we predict that all changes due to the mutations are local, they have significant effects on the structure in the vicinity of the mutations. Most importantly, we observe that changes to the hydrogen bonding pattern in a peptide with a single mutation are very similar to a peptide with a mutation in every chain. This observation shows that even a single Gly to Ala mutation significantly alters the flexibility of the collagen chain in the mutated region, allowing for potential binding interactions with other molecules.These changes are a result of the additional space requirement of the alanines' methyl groups. Through exploration of the energy landscape, we obtained a detailed overview of the possible orientations of the methyl groups. Their presence either leads to straining through stretching or to straining through twisting, depending on the methyl group orientation. The distinct mechanisms of how the methyl groups are accommodated have been detailed here. Importantly, the number of mutations and their proximity has a decisive impact on the structure adopted, and we predict a different orientation for single or spaced-out double mutations than has been reported for the triple mutation.
Moreover, the strain from the structural alterations leads to changes in the puckering observed, which can be linked to a hypothesis by Chow et al.36 that the puckering dynamics allows for controlled flexibility in collagen changes. These changes in the puckering behaviour cannot be predicted from single experimental structures, as an ensemble view is necessary to clearly identify them. Given the locality of these changes, which nonetheless absorb significant elongation of the backbone, it can be seen how important this mechanism might be in the elasticity of collagen.
Given the wealth of information that can be extracted from this approach, future applications of the modelling towards binding domains, interrupting deletions of residues and the impact of mechanical forces on collagen fibres are desirable.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks Prof. Melinda Duer for her suggestion to study this system and the helpful discussions along the way. KR is funded by the Cambridge Philosophical Society.Notes and references
- M. Aumailley and B. Gayraud, J. Mol. Med., 1998, 76, 253–265 CrossRef CAS PubMed.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- C. Bonnans, J. Chou and Z. Werb, Nat. Rev. Mol. Cell Biol., 2014, 15, 786–801 CrossRef CAS PubMed.
- A. D. Theocharis, S. S. Skandalis, C. Gialeli and N. K. Karamanos, Adv. Drug Delivery Rev., 2016, 97, 4–27 CrossRef CAS PubMed.
- P. Lu, K. Takai, V. M. Weaver and Z. Werb, Cold Spring Harbor Perspect. Biol., 2011, 3, a005058 Search PubMed.
- S. Ricard-Blum, Cold Spring Harbor Perspect. Biol., 2011, 3, a004978 Search PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- A. Rich and F. H. Crick, J. Mol. Biol., 1961, 3, 483–506 CrossRef CAS PubMed.
- T. Xu, C.-Z. Zhou, J. Xiao and J. Liu, Biochemistry, 2018, 57, 1087–1095 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- E. Małolepsza, B. Strodel, M. Khalili, S. Trygubenko, S. N. Fejer and D. J. Wales, J. Comput. Chem., 2010, 31, 1402–1409 Search PubMed.
- E. Małolepsza, B. Strodel, M. Khalili, S. Trygubenko, S. N. Fejer and D. J. Wales, J. Comput. Chem., 2012, 33, 2209 CrossRef.
- H. Nguyen, D. R. Roe and C. Simmerling, J. Chem. Theory Comput., 2013, 9, 2020–2034 CrossRef CAS PubMed.
- J. A. Joseph, K. Röder, D. Chakraborty, R. G. Mantell and D. J. Wales, Chem. Commun., 2017, 53, 6974–6988 RSC.
- K. Röder, J. A. Joseph, B. E. Husic and D. J. Wales, Adv. Theory Simul., 2019, 2, 1800175 CrossRef.
- Z. Li and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 6611–6615 CrossRef CAS PubMed.
- Z. Li and H. A. Scheraga, J. Mol. Struct., 1988, 48, 333–352 CrossRef CAS.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111–5116 CrossRef CAS.
- D. J. Wales, Mol. Phys., 2002, 100, 3285–3305 CrossRef CAS.
- D. J. Wales, Mol. Phys., 2004, 102, 891–908 CrossRef CAS.
- F. Noé and S. Fischer, Curr. Opin. Struct. Biol., 2008, 18, 154–162 CrossRef PubMed.
- D. J. Wales, Curr. Opin. Struct. Biol., 2010, 20, 3–10 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. A. Trygubenko and D. J. Wales, J. Chem. Phys., 2004, 120, 2082–2094 CrossRef CAS PubMed.
- L. J. Munro and D. J. Wales, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 3969–3980 CrossRef CAS.
- D. Roe and T. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- J. Weiser, P. Shenkin and W. Still, J. Comput. Chem., 1999, 20, 217–223 CrossRef CAS.
- K. Okuyama, C. Hongo, R. Fukushima, G. Wu, H. Narita, K. Noguchi, Y. Tanaka and N. Nishino, Pept. Sci., 2004, 76, 367–377 CrossRef CAS PubMed.
- K. Okuyama, K. Miyama, K. Mizuno and H. P. Bächinger, Biopolymers, 2012, 97, 607–616 CrossRef CAS PubMed.
- R. Z. Kramer, L. Vitagliano, J. Bella, R. Berisio, L. Mazzarella, B. Brodsky, A. Zagari and H. M. Berman, J. Mol. Biol., 1998, 280, 623–638 CrossRef CAS PubMed.
- R. Berisio, L. Vitagliano, L. Mazzarella and A. Zagari, Protein Sci., 2002, 11, 262–270 CrossRef CAS PubMed.
- M. D. Shoulders, J. A. Hodges and R. T. Raines, J. Am. Chem. Soc., 2006, 128, 8112–8113 CrossRef CAS PubMed.
- M. D. Shoulders, K. A. Satyshur, K. T. Forest and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 559–564 CrossRef CAS PubMed.
- W. Y. Chow, D. Bihan, C. J. Forman, D. A. Slatter, D. G. Reid, D. J. Wales, R. W. Farndale and M. J. Duer, Sci. Rep., 2015, 5, 12556 CrossRef CAS PubMed.
- W. Y. Chow, C. J. Forman, D. Bihan, A. M. Puszkarska, R. Rajan, D. G. Reid, D. A. Slatter, L. J. Colwell, D. J. Wales, R. W. Farndale and M. J. Duer, Sci. Rep., 2018, 8, 13809 CrossRef PubMed.
- L. S. Batchelder, C. E. Sullivan, L. W. Jelinski and D. A. Torchia, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 386–389 CrossRef CAS PubMed.
- L. W. Jelinski, C. E. Sullivan, L. S. Batchelder and D. A. Torchia, Biophys. J., 1980, 32, 515–529 CrossRef CAS PubMed.
- H. Saitô, R. Tabeta, A. Shoji, T. Ozaki, I. Ando and T. Miyata, Biopolymers, 1984, 23, 2279–2297 CrossRef PubMed.
- H. Saitô and M. Yokoi, J. Biochem., 1992, 111, 376–382 CrossRef PubMed.
- D. Reichert, O. Pascui, E. R. de Azevedo, T. J. Bonagamba, K. Arnold and D. Huster, Magn. Reson. Chem., 2004, 42, 276–284 CrossRef CAS PubMed.
- D. Huster, J. Schiller and K. Arnold, Magn. Reson. Med., 2002, 48, 624–632 CrossRef CAS PubMed.
- S. K. Sarkar, C. E. Sullivan and D. A. Torchia, J. Biol. Chem., 1983, 258, 9762–9767 CrossRef CAS.
- S. K. Sarkar, C. E. Sullivan and D. A. Torchia, Biochemistry, 1985, 24, 2348–2354 CrossRef CAS PubMed.
- T. V. Burjanadze, Biopolymers, 1979, 18, 931–938 CrossRef CAS PubMed.
- J. Myllyharju, Top. Curr. Chem., 2005, 247, 115–147 CrossRef CAS.
- S. M. Krane, Amino Acids, 2008, 35, 703–710 CrossRef CAS PubMed.
- M. Cutini, M. Bocus and P. Ugliengo, J. Phys. Chem. B, 2019, 123, 7354–7364 CrossRef CAS PubMed.
- I. Goldberga, R. Li and M. J. Duer, Acc. Chem. Res., 2018, 51, 1621–1629 CrossRef CAS PubMed.
- E. D. Nelson and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10682–10686 CrossRef CAS PubMed.
- K. Röder and D. J. Wales, J. Chem. Theory Comput., 2017, 13, 1468–1477 CrossRef PubMed.
- K. Röder and D. J. Wales, J. Phys. Chem. B, 2018, 122, 10989–10995 CrossRef PubMed.
- D. J. Wales, Chem. Phys. Lett., 1998, 285, 330–336 CrossRef CAS.
- P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14249–14255 CrossRef CAS PubMed.
Footnotes |
| † All data available online. See DOI:10.5281/zenodo.5578060 |
| ‡ The endo/exo-configurations are determined based on the χ2 torsional angle in proline, where planar configurations are assumed for an absolute dihedral angle of 10° or less. Only 21 of the nearly 220000 χ2 torsional angles calculated fall into this category. |
| This journal is © the Owner Societies 2022 |