
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structures and electronic states of trimer radical cations of coronene: DFT–ESR simulation study†
Hiroto
Tachikawa
 a and
Anders
Lund
*b
a and
Anders
Lund
*b
aDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Sapporo 060-8628, Japan
bDepartment of Physics, Chemistry and Biology, Linköping University, S-581 83 Linköping, Sweden. E-mail: anders.lund@liu.se
First published on 5th April 2022
Abstract
Coronene (C24H12), a charge transfer complex with low-cost and high-performance energy storage, has recently attracted attention as a model molecule of graphene nano-flakes (GNFs). The stacking structures of the trimer radical cation correlate strongly with the conduction states of the GNFs. In the present paper, the structures and electronic states of the monomer, dimer and trimer radical cations of coronene were investigated by means of density functional theory calculations. In particular, the proton hyperfine coupling constants of these species were determined. The radical cation of coronene+ (monomer) showed two structures corresponding to the 2Au and 2B3u states due to the Jahn–Teller effect. The 2Au state was more stable than the 2B3u state, although the energy difference between the two states was only 0.03 kcal mol−1. The dimer and trimer radical cations took stacking structures distorted from a full overlap structure. The intermolecular distances of the molecular planes were 3.602 Å (dimer) and 3.564 and 3.600 Å (trimer). The binding energies of the dimer and trimer were calculated to be 8.7 and 13.3 kcal mol−1, respectively. The spin density was equivalently distributed on both coronene planes in the dimer cation. In contrast, the central plane in the trimer cation had a larger spin density, ρ = 0.72, than the upper and lower planes, both with ρ = 0.14. The proton hyperfine coupling constants calculated from these structures and the electronic states of the monomer, dimer, and trimer radical cations of coronene were in excellent agreement with previous ESR spectra of coronene radical cations. The structures and electronic states of (coronene)n+ (n = 1–3) were discussed on the basis of the theoretical results.
1. Introduction
π-stacking is a non-covalent interaction caused by the overlapping of π-orbitals between π conjugated systems.1 This interaction plays an important role in structural arrangements and molecular assemblies in biology,2 chemistry,3 and materials science.4 The degree of π-stacking is an important factor in controlling the efficiency of photovoltaic power generation.5 Chou et al. investigated the effect of the molecular stacking orientation on the open-circuit voltage of pentacene-based organic solar cells.5 They demonstrated that a significant increase in the voltage from 0.28 to 0.83 eV occurred, owing to the π–π stacking of pentacene molecules.Coronene (C24H12, denoted as Coro) has recently attracted attention as a charge transfer complex6 with low-cost and high-performance energy storage.7–9 In particular, Coro is widely utilized as a model of graphene nano-flakes.10–13 As a piece of important information, it is known that Coro takes a stacking structure.14–16 The stacking and electronic structures in neutral and ionic states are strongly correlated with the conduction state of the graphene nano-flakes.17 However, the stacking structure of Coro was not accurately determined. In particular, the structures at the ion radical (cation) state, controlling the conduction state, are scarcely known, although several experiments were performed for the cation state.
The electron spin resonance (ESR) technique is one of the powerful tools to determine the structure and electronic states of ion radicals. The ESR measurements of the radical cation of Coro have a long history.18,19 In 1968, Willigen et al. reported the possibility of the formation of the trimer radical cation of Coro, (Coro)3+, in SO2 with BF3.20 However, the observed ESR spectrum was not sufficiently resolved to establish the existence of the trimer unambiguously. Ohya-Nishiguchi et al. measured the ESR spectra of the trimeric radical cation of Coro generated by the electrochemical oxidation technique.21 The proton-hyperfine coupling constants (hfcc's) in the observed spectrum were composed of 0.117 mT due to 12 equivalent protons (12H) and 0.020 mT due to 24 equivalent protons (24H), where mH denotes the number of equivalent protons. They proposed a full overlapped stacking structure of (Coro)3+. Komaguchi et al. measured highly resolved ESR spectra of the monomer, dimer and trimer radical cations of Coro in a solution of 1,1,1,3,3,3-hexafluoro-2-propan-2-ol (HFP) containing thallium(III) trifluoroacetate (oxidant) at room temperature.22 The spectra, consisting of multiple lines of 0.0766 mT (24H) and 0.013 mT (6H), were attributable to a mixture of the dimer with the trimer radical cations. For the dimer radical cation, the hfcc's were measured to be 0.077 mT. Based on an analysis of the previously published data,20 a staggered sandwich structure was proposed for the “global” minimum of the trimer cation with hfcc's of 0.122 (12H) and 0.015 (24H), respectively.22 Thus, the experimental data have been accumulated gradually. However, a structure that satisfies the previous experimental findings has not been determined yet.
In the present study, density functional theory (DFT) calculations were performed for the monomer, dimer, and trimer radical cations of Coro in order to elucidate their electronic states and stacking structure. Furthermore, simulations of ESR spectra based on the DFT calculations were carried out for the stacking structure. The stacking structures of radical cations of Coro were determined based on the ESR spectra and the DFT calculations.
2. Computational method
2.1. DFT calculations
The geometries of the neutral Coro, the monomer, dimer and trimer radical cations of coronene were fully optimized at the CAM-B3LYP/6-311G(d,p) level of theory.23,24 In the geometry optimizations, thresholds of maximum force, root-mean square (RMS) force, maximum displacement of the geometrical parameter and RMS of displacement of the geometrical parameter were set to 4.50 × 10−4 (a.u.), 3.00 × 10−4 (a.u.),1.80 × 10−3, and 1.20 × 10−3 (hartrees bohr−1 and hartrees radians−1), respectively. A quadratically convergent (QC) Hartree–Fock method was used in all self-consistent field (SCF) calculations.25 SCF convergence on energy was set to 1.00 × 10−5 (a.u.). Open-shell systems (i.e., radical cations) were calculated using the unrestricted method. The vibrational frequencies of the coronene radical cation (monomer) were calculated to verify the energy minima on the potential energy surface. Frequency calculations for the dimer and trimer radical cations were not possible with the available computer resources. The dimer and trimer radical cations observed by ESR20–22 had hfcc's in excellent agreement with the calculated structures, however, indicating that those structures are the stable ones observed experimentally. The standard Gaussian 09 program package was used for the DFT calculations.262.2. ESR simulation method
The isotropic ESR spectra of the trimer radical cation of coronene were simulated exactly using the EasySpin software available at https://www.easyspin.org/. The proton hyperfine coupling constants in Table 3 were used to simulate the spectra predicted by the DFT calculations in this work, by an experimental analysis,21 and by an interpretation22 of the spectrum obtained by Willigen et al.20| C–C bond | Neutral | State | |
|---|---|---|---|
| 2Au | 2B3u | ||
| R1 | 1.423 | 1.416 | 1.411 |
| R2 | 1.423 | 1.409 | 1.418 |
| R3 | 1.361 | 1.379 | 1.360 |
| R4 | 1.420 | 1.400 (shortened) | 1.432 (elongated) |
| R5 | 1.420 | 1.436 (elongated) | 1.398 (shortened) |
| R6 | 1.361 | 1.351 (shortened) | 1.388 (elongated) |
| Form | ΔE | E bind | d(A–B) | d(A–C) |
|---|---|---|---|---|
| ABA | 0.00 | −13.29 | 3.600 | 3.600 |
| ABC | 0.10 | −13.20 | 3.622 | 3.629 |
| Full | 2.44 | −10.85 | 3.901 | 3.901 |
| No. of Coro | Chemical | hfcc/mT | State or form | |
|---|---|---|---|---|
| n = 1 | (C24H12)+ | −0.1560(12H) | 2Au | Present calc. |
| −0.1556(12H) | 2B3u | Present calc. | ||
| −0.153(12H) | Ref. 18 (expl.) | |||
| −0.157(12H) | Ref. 20 (expl.) | |||
| −0.156(12H) | Ref. 22 (expl.) | |||
| n = 2 | (C24H12)2+ | −0.0771(24H) | Present calc. | |
| 0.0770(24H) | Ref. 20 (expl.) | |||
| 0.0766(24H) | Ref. 22 (expl.) | |||
| n = 3 | (C24H12)3+ | −0.1112(12H) | ABA | Present calc. |
| −0.0213(12H) | ||||
| −0.0213(12H) | ||||
| −0.1110(12H) | ABC | Present calc. | ||
| −0.0211(12H) | ||||
| −0.0212(12H) | ||||
| −0.1072(12H) | Full overlap | Present calc. | ||
| −0.0249(24H) | ||||
| 0.117(12H) | Ref. 21 (expl.) | |||
| 0.020(24H) | ||||
| 0.122(12H) | Ref. 20 (expl.) | |||
| 0.015(24H) |
3. Results
3.1. Radical cations of coronene (Coro+)
First, the structure of the neutral coronene (denotes as Coro) was optimized at the CAM-B3LYP/6-311G(d,p) level. The optimized structure and geometrical parameters are given in Fig. 1 and Table 1, respectively. The C–C bonds of the benzene ring in the central region of Coro were calculated to be R1 = R2 = 1.423 Å. In the edge region, the bond lengths for R3–R6 were 1.361, 1.420, 1.420, and 1.361 Å, respectively.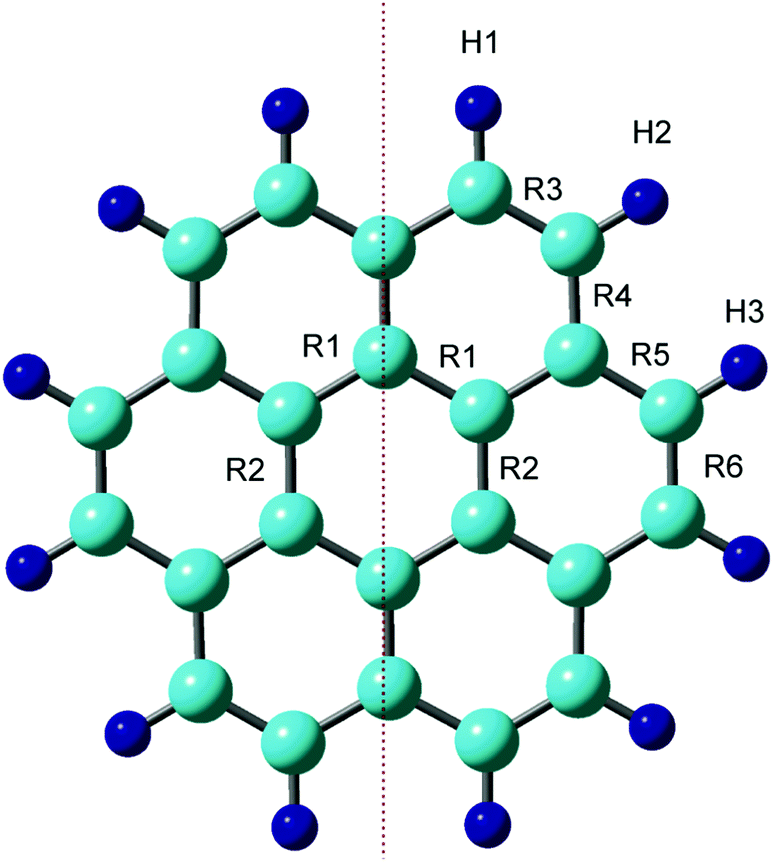 | ||
| Fig. 1 Optimized structure of coronene at the neutral state calculated at the CAM-B3LYP/6-311G(d,p) level. | ||
Next, the radical cation of coronene (denoted as Coro+) was optimized by considering the Jahn–Teller (JT) effects. The energy diagram of the coronene radical cation (Coro+) is given in Fig. 2. Two energy levels were degenerated under D6h symmetry (2E2u state). These energy levels were split into two states (2Au and 2B3u states) in D2h symmetry due to the Jahn–Teller effect.17,27,28 Therefore, Coro+ has two low-lying electronic states. In the present study, the structure and electronic state of Coro+ at the 2Au and 2B3u states were investigated.
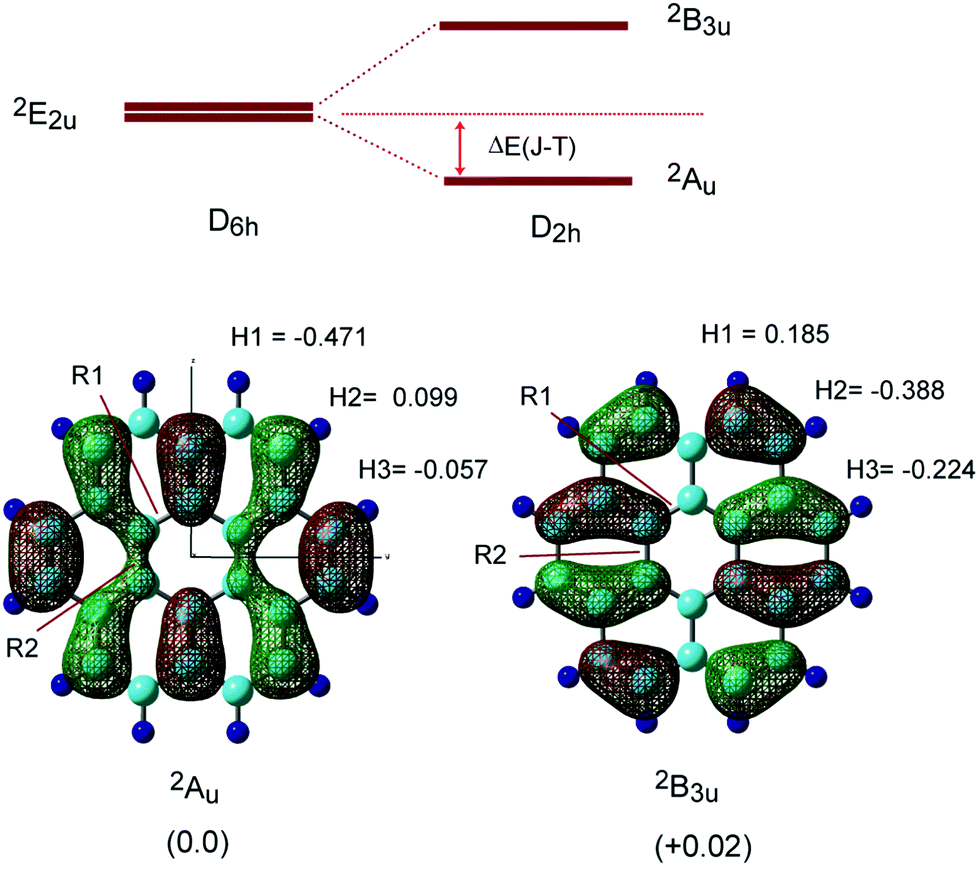 | ||
| Fig. 2 (upper) Schematic illustration of the energy levels of coronene radical cations under D6h and D2h symmetries. The energy levels of the coronene radical cation (2E2u) are split into 2Au and 2B3u states due to the Jahn–Teller effect. ΔE(J–T) means stabilization energy (in kcal mol−1). (lower) Optimized structures and SOMOs of the coronene radical cation, and proton hyperfine coupling constants of the coronene radical cation (in mT). Relative energies between the 2Au and 2B3u states are given in parentheses (in kcal mol−1). Calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. | ||
The optimized structures and singly occupied molecular orbitals (SOMOs) of Coro+ are illustrated in Fig. 2 (lower), and the geometrical parameters are given in Table 1. Typical differences of Coro+ from neutral state Coro clearly appeared in the C–C bond lengths of R4, R5 and R6. Compared with the neutral state (R4 = 1.420, R5 = 1.420, and R6 = 1.361 Å), R4 and R6 were significantly shortened at the 2Au state (R4 = 1.400 and R6 = 1.351 Å), whereas they were elongated at the 2B3u state (R4 = 1.432 and R6 = 1.388 Å). In addition, R5 was elongated at the 2Au state (R5 = 1.436 Å) and was shortened at the 2B3u state (R5 = 1.398 Å). These structural changes are easily interpreted in terms of the phase of the SOMO. If the orbital of the C–C bond is of a bonding or anti-bonding nature, the C–C bond is shortened or elongated by hole capture, respectively. The structural change of Coro+ occurred according to these rules. The stabilization energy due to Jahn–Teller effects was 2.29 kcal mol−1 (2Au state). These features are similar to the Jahn–Teller effect in the benzene radical cation: compressed and elongated structures are obtained after the ionization of benzene.29
In order to elucidate the stability of Coro+ on the potential energy surface (PES), harmonic vibrational frequencies were calculated for the two electronic states. Coro+ (2Au) possessed all positive frequencies, whereas Coro+ (2B3u) showed an imaginary frequency, suggesting that Coro+ (2B3u) is located at a saddle point on the PES. This tendency was not dependent on the calculation methods. The vibrational frequencies are given in the ESI.†
The proton hyperfine coupling constants (H-hfcc's) were calculated to be a(H1) = −0.471 mT, a(H2) = 0.099 mT, and a(H3) = −0.057 mT at the 2Au state, while a(H1) = 0.185 mT, a(H2) = −0.388 mT, and a(H3) = −0.224 mT at the 2B3u state. The average hfcc values are 〈a(H)〉 = −0.143 mT at the 2Au state, and 〈a(H)〉 = −0.143 mT at the 2B3u state, suggesting that the two states cannot be distinguished at higher temperatures. The energy difference between the 2Au and 2B3u states was calculated to be 0.03 kcal mol−1, indicating that the structure at the 2Au state is slightly lower in energy than that of the 2B3u state, although the difference is quite small. The hfcc's of all protons are given in Table S1 (ESI†).
3.2. Dimer cations of coronene (Coro)2+
The optimized structure and the SOMO of the coronene dimer cation (Coro)2+ are illustrated in Fig. 3. The interplane distance was calculated to be 3.602 Å. The molecular planes of coro(A) and coro(B) were almost parallel to each other in the dimer cation. The structure was distorted from the full overlap structure by one-half of the C–C bond length. The symmetry of the structure was Ci. The binding energy of Coro+ to Coro was calculated to be 8.71 kcal mol−1. The binding energy is defined as follows:| −Ebind = E(Coro)2+ − [E(Coro+) + E(Coro)] | (1) |
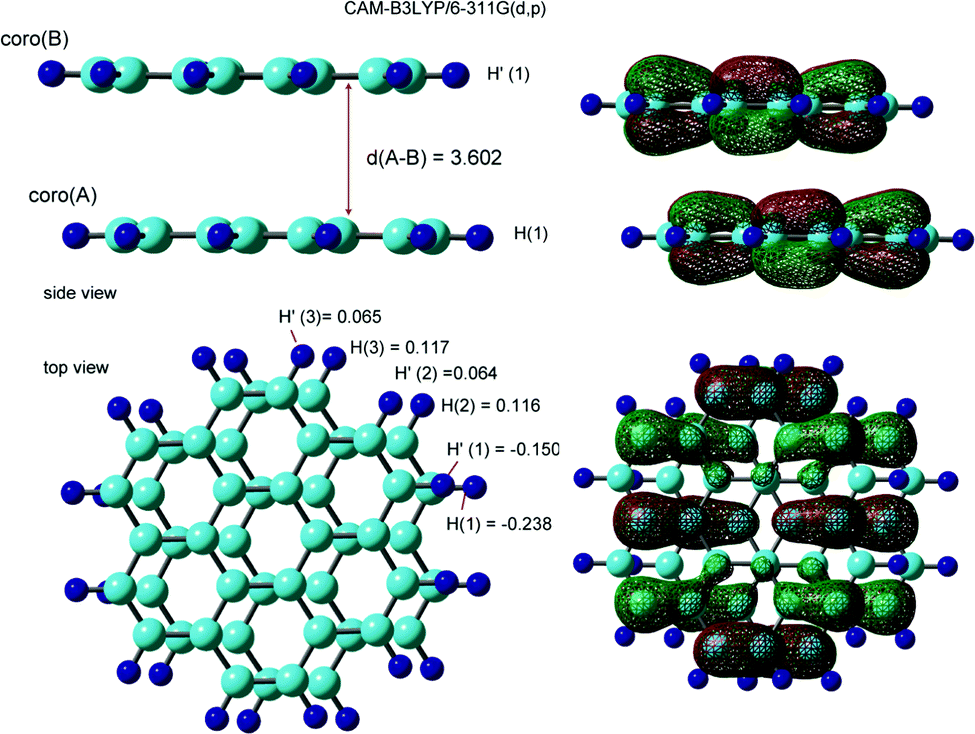 | ||
| Fig. 3 Optimized structures and SOMOs of the dimer cation of coronene, and proton hyperfine coupling constants (in mT). Interplane distance, d(A–B), is in Å, 1 Å = 0.1 nm. Calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. | ||
The hfcc's of (Coro)2+ were calculated to be a(H1) = −0.238 mT, a(H2) = 0.116 mT, and a(H3) = 0.117 mT in the lower coronene, coro(A), while a(H1′) = −0.150 mT, a(H2′) = 0.064 mT, and a(H3′) = 0.065 mT in the upper coronene, coro(B). The hfcc's of all protons are given in Table S2 (ESI†).
3.3. Trimer cations of coronene (Coro)3+
We examined three stacking forms for the structure of the trimer radical cation, which are schematically illustrated in Fig. 4: ABA, ABC and full overlap forms. The ABA and ABC forms correspond to distorted structures from the full overlapped form. These three structures were fully optimized without symmetry restriction. The initial geometries of the ABA and ABC forms were started from slightly distorted structures of the full overlap form. The optimized geometrical parameters and relative energies are given in Table 2. The ABA stacking form was the most stable in energy. Although the ABC form was slightly less stable than the ABA form, the energy difference was quite small. Therefore, both forms are possible to exist by energy considerations. The full overlap form was 2.34 and 2.07 kcal mol−1 higher in energy than the ABA and ABC forms, respectively.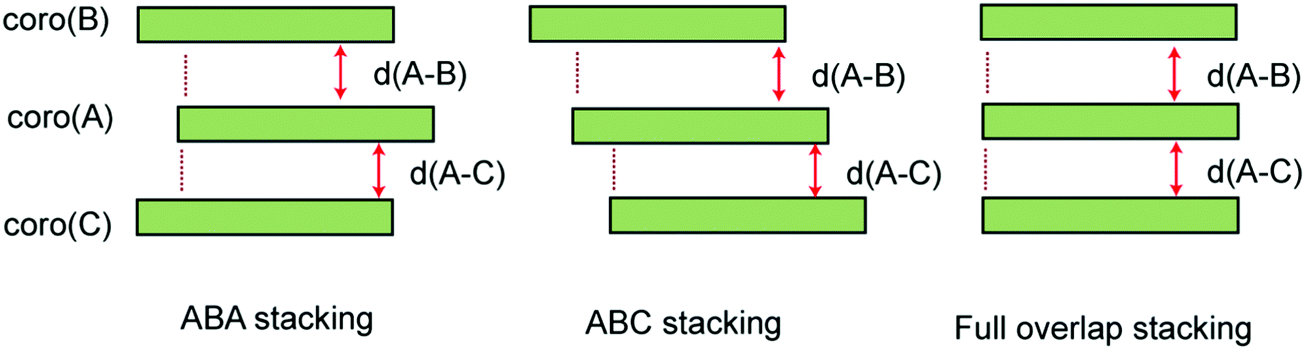 | ||
| Fig. 4 Schematic illustration of three types of π-stacking forms in the coronene trimer cation. Notations d(A–B) and d(A–C) mean interplane distances between coro(A)–coro(B) and coro(A)–coro(C), respectively. | ||
The fully optimized structure and spin density distribution of the trimer cation (Coro)3+ in the ABA stacking form are illustrated in Fig. 5. The interplane distances of coro(B) and coro(C) from coro(A) were 3.600 and 3.600 Å, respectively, which are slightly shorter than for the dimer cation (3.602 Å). (Coro)3+ had no symmetry. The binding energy of the three Coro units, Coro+ + Coro + Coro → (Coro)3+, was calculated to be 13.29 kcal mol−1. The binding energy is defined as follows:
| −Ebind = E(Coro)3+ − [E(Coro+) + 2E(Coro)] | (2) |
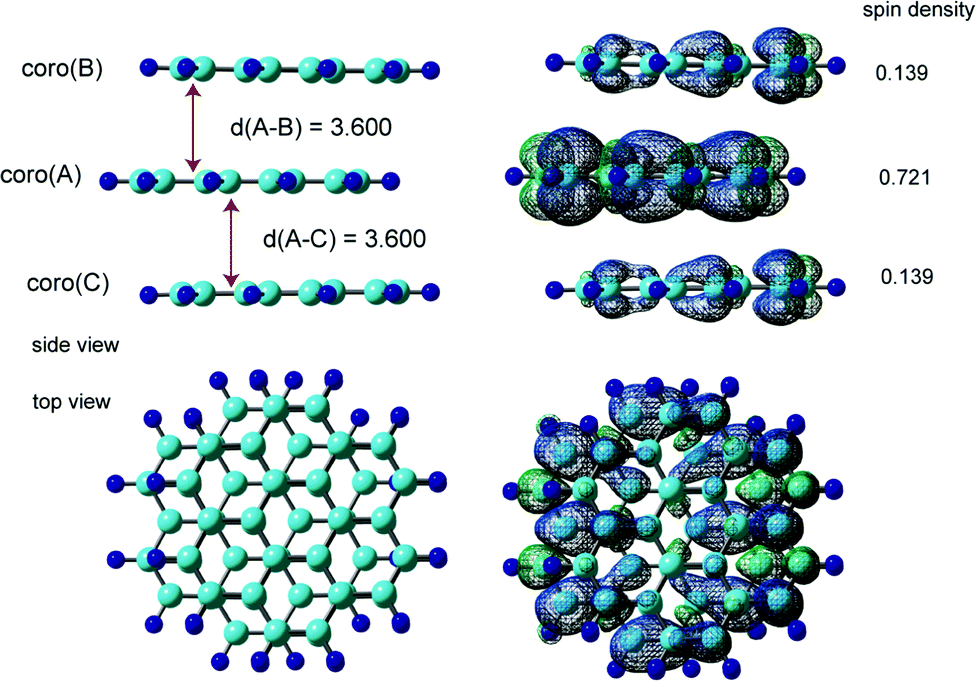 | ||
| Fig. 5 Optimized structures and spin density distributions of the trimer cation of coronene (ABA stacking form). Interplane distances, d(A–B) and d(A–C), are in Å. The spin density values on the coronene molecules are indicated. Calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. | ||
The spin densities were 0.721 in coro(A), 0.139 in coro(B), and 0.139 in coro(C), indicating that the unpaired electron is mainly distributed on coro(A) in the centre plane (72.1%). The average hfcc's for coro(A), coro(B), and coro(C) were −0.1019, −0.0195, and −0.0195 mT, respectively. The central coro(A) has a larger average hfcc.
The hfcc's of all protons are given in Tables S3 and S4 (ESI†). The hfcc's in six of the protons were close to zero (less than 0.001 mT). These protons are located on coro(B) and coro(C). The average hfcc was calculated to be 〈aH〉 = −0.0513 mT. All data are given in the ESI.†
The fully optimized structure and spin density distribution of (Coro)3+ with the ABC form are illustrated in Fig. S2 (ESI†). The interplane distances were d(A–B) = 3.622 and d(A–C) = 3.623 Å, which are comparable to those of the ABA form. In the case of full overlap form, the interplane distance was significantly longer (d = 3.901 Å). In the neutral state of (Coro)3, the interplane distance was calculated to be 4.308 Å (see, Fig. S3 in the ESI†), meaning that π-stacking takes place after hole capture of (Coro)3.
3.4. Hfcc's of (Coro)n+ (n = 1–3)
In the present study, the DFT method was applied to obtain the hfcc's of (Coro)n+ (n = 1–3) in order to determine the structures and electronic states. For n = 1 (monomer radical cation), the average hfcc was calculated to be −0.143 mT (12H). This value is in good agreement with the ESR experiments (0.153–0.156 mT), although the theoretical value is slightly underestimated (the factor is f = 1.092, indicating that the hfcc's were estimated to be within 10% by the used DFT method). The hfcc's of (Coro)n+ (n = 1–3) adjusted by this factor are given in Table 3 together with previous ESR data.In the dimer radical cation, the theoretical value was −0.0771 mT (24H), which is in excellent agreement with the experiments: 0.077 mT (24H) and 0.0766 mT (24H). The hfcc's of the trimer radical cation with the ABA stacking form [−0.1112 mT (12H), −0.0213 mT (12H), and −0.0213 mT (12H)] agreed excellently with previous experiments. The hfcc's of the ABC form [−0.1110 mT (12H), −0.0211 mT (12H) and −0.0212 mT (12H)] agreed well with previous experiments, indicating that the Coro trimer radical cation has structures distorted from the full overlap form. The hfcc values at the centre (12H) and outer (24H) Coro units, [−0.1072 mT (12H) and −0.0249 mT (24H)] of the full stacking form differed more from the experiment, however, and were considered inconsistent with the experimental data.
Based on the DFT data, the ESR spectrum of the trimer radical cation (ABA form) was simulated and is plotted in Fig. 6. The ESR spectra measured by van-Willigen and Ohya-Nishiguchi are also given for comparison. As clearly seen in these spectra, the simulated line shape of (Coro)3+ with the stacking structure (ABA form) reproduces excellently the experimental ESR spectra. The ESR spectra of ABC and full stacking forms were also simulated, and the spectra are given in Fig. S4 (ESI†). Similar shapes of the spectra were obtained for both forms. However, the values of the hfcc's of the full overlap form were inconsistent with the experiments. The full overlap form is also less stable with respect to energy than the ABA and ABC forms. The present DFT-based ESR simulation demonstrates that the Coro trimer radical cation is composed of stacking structures (ABA and ABC forms).
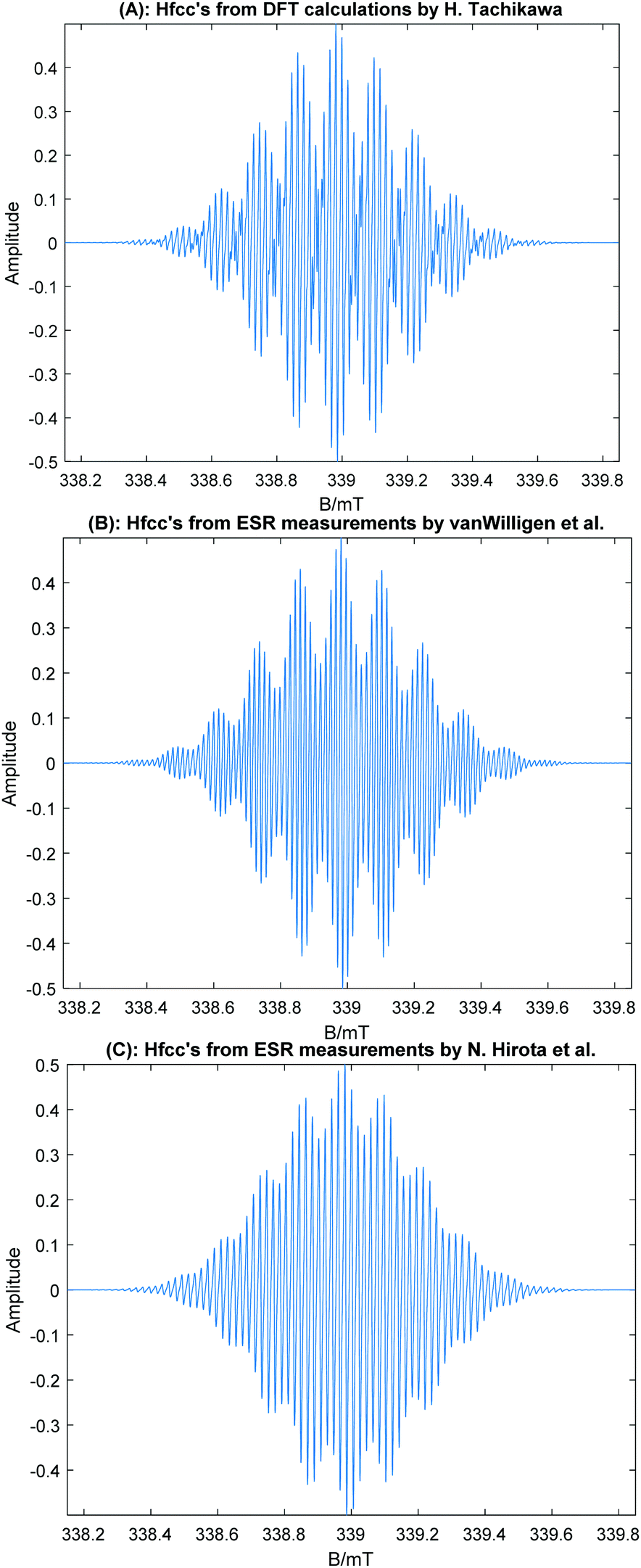 | ||
| Fig. 6 Simulated ESR spectra of the coronene trimer radical cation using the hfcc's in Table 3. (A) Based on the hfcc's calculated at the CAM-B3LYP/6-311G(d,p) level multiplied by f = 1.092. (B) Based on the hfcc's deduced from the experimental spectrum by H. Van Willigen et al.20,22 (C) Based on the experimental hfcc's obtained by Ohya-Nishiguchi et al.21 The peak–peak line width was 0.005 mT (Lorentzian shape) in all cases. | ||
3.5. Effects of functionals
The effects of functionals in DFT on the structure, binding energy and electronic states were examined, and the corresponding results are shown in the ESI,† Tables S5–S7. First, the structure of the benzene dimer cation was calculated using CAM-B3LYP, xB97XD, LC-wB97XD, M062X, wB97X, and B3LYP functionals30–33 in order to test the effects of functionals. The interplane distances were calculated to be 3.077–3.133 Å, except for B3LYP (Table S5, ESI†), which are in good agreement with that of CAM-B3LYP (3.194 Å). The B3LYP functional gave a poor result for the benzene dimer cation: the interplane distance (3.354 Å) was significantly longer than with the other functionals. These results indicate that long-range dispersion interactions play an important role in the stacking of radical cations of polycyclic aromatic hydrocarbons.Next, the Jahn–Teller stabilization energy (ΔE(J–T)) was calculated (Table S6, ESI†): 2.29 (CAM-B3LYP), 2.27 (xB97XD), and 2.11 kcal mol−1 (M062X), which also agreed with each other. Lastly, the relative energies of ABA, ABC, and full overlap forms of (Coro)3+ were calculated (Table S7, ESI†). Single point energy calculations with the functionals were performed using the optimized structure obtained at the CAM-B3LYP/6-311G(d,p) level. The calculations with all functionals except for B3LYP gave that the ABA and ABC forms were energetically more stable than the full overlap form. Slightly distorted structures of the full-overlap form, namely the ABA and ABC stacking forms, are candidates for the coronene trimer cation observed in the ESR measurements.
4. Conclusion
The π-stacking of graphene nano-flakes (GNFs) plays an important role in hole and electron transfer along the layer structure. Coro is widely utilized as a model molecule of GNFs. However, information on the stacking structure of Coro has been limited. In particular, the structure and electronic states of the Coro trimer radical cation, which is strongly correlated with the conduction state, were not determined yet, although the experimental data have been gradually accumulated. In the present study, DFT calculations were carried out for the Coro monomer, dimer, and trimer radical cations (Coro)n+ (n = 1–3). The calculated hfcc's of (Coro)n+ were in good agreement with previous ESR data. As for the trimer radical cation, structures distorted slightly from the full-overlap form were favoured in energy as the stacking form (i.e., ABA and ABC forms). ESR simulations based on the hfcc's predicted using the DFT method were carried out for the trimer radical cation. The simulated spectra of (Coro)3+ (ABA and ABC forms) were in excellent agreement with previous ESR spectra. It is therefore concluded that the Coro trimer radical cation has layer structures (ABA and ABC) slightly distorted from the full overlap stacking form.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The author (HT) acknowledges partial support from JSPS KAKENHI (Grant Numbers 21K04973 and 21H05415).References
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900, 10.1039/c1cp20404a.
- C. Lin, R. Chinnappan, K. Acharya, J.-L. Pellequer and R. Jankowiak, Biophys. Chem., 2011, 154, 35–40, DOI:10.1016/j.bpc.2010.12.005.
- T. Okamoto, K. Nakahara, A. Saeki, S. Seki, J. H. Oh, H. B. Akkerman, Z. Bao and Y. Matsuo, Chem. Mater., 2011, 23, 1646–1649, DOI:10.1021/cm200356y.
- Y. Gao, G. Hu, W. Zhang, D. Ma and X. Bao, Dalton Trans., 2011, 40, 4542–4547, 10.1039/C0DT01392G.
- C.-T. Chou, C.-H. Lin, T. Yian, C.-H. J. Liu, L.-C. Chen and K.-H. Chen, J. Phys. Chem. Lett., 2012, 3, 1079–1083, DOI:10.1021/jz201615v.
- Y. Yoshida, K. Isomura, Y. Kumagai, M. Maesato, H. Kishida, M. Mizuno and G. Saito, J. Phys.: Condens. Matter, 2016, 28, 304001, DOI:10.1088/0953-8984/28/30/304001.
- K. Minami, T. Masese and K. Yoshii, New J. Chem., 2021, 45, 4921–4924, 10.1039/D1NJ00387A.
- M. L. P. Júnior, B. G. E. Neto, W. F. Giozza, R. T. S. Júnior, G. M. e Silva and L. A. R. Júnior, J. Mater. Chem. C, 2020, 8, 12100–12107, 10.1039/d0tc01319f.
- B. Mao, F. Cortezon-Tamarit, H. Ge, N. Kuganathan, V. Mirabello, F. J. Palomares, G. Kociok-Kohn, S. W. Botchway, D. G. Galatayud and S. I. Pascu, ChemistryOpen, 2019, 8, 1383–1398, DOI:10.1002/open.201900310.
- S. Prodhan, S. Mazumdar and S. Ramasesha, Molecules, 2019, 14, 730, DOI:10.3390/molecules24040730.
- S. D. Chakarova-Käck, A. Vojvodic, J. Kleis, P. Hyldgaard and E. Schröder, New J. Phys., 2010, 12, 013017, DOI:10.1088/1367-2630/12/1/013017.
- H. Tachikawa, Y. Izumi, T. Iyama and K. Azumi, ACS Omega, 2021, 6, 7778–7785, DOI:10.1021/acsomega.1c00243.
- H. Tachikawa and T. Iyama, J. Phys. Chem. C, 2019, 123, 8709–8716, DOI:10.1021/acs.jpcc.9b01152.
- T. Okazaki, Y. Iizumi, S. Okubo, H. Kataura, Z. Liu, K. Suenaga, Y. Tahara, M. Yudasaka, S. Okada and S. Iijima, Angew. Chem., Int. Ed., 2011, 50, 4853–4857, DOI:10.1002/anie.201007832.
- S. Bag and P. K. Maiti, Phys. Rev. B, 2017, 96, 245401, DOI:10.1103/PhysRevB.96.245401.
- J. Martinez-Blanco, A. Mascaraque, Y. S. Dedkov and K. Hom, Langmuir, 2012, 28, 3840–3844, DOI:10.1021/la205166m.
- Y. Yoshida, K. Isomura, M. Maesato, T. Koretsune, Y. Nakano, H. Yamochi and G. Saito, Cryst. Growth Des., 2016, 16, 5994–6000, DOI:10.1021/acs.cgd.6b01039.
- J. R. Bolton and A. Carrington, Mol. Phys., 1961, 4, 271–272, DOI:10.1080/00268976100100401.
- T. Sato, H. Tanaka, A. Yamamoto, Y. Kuzumoto and K. Tokunaga, Chem. Phys., 2003, 287, 91–102, DOI:10.1016/S0301-0104(02)00981-3.
- H. van Willigen and E. de Boer, J. Chem. Phys., 1968, 49, 1190–1192, DOI:10.1063/1.1670208.
- H. Ohya-Nishiguchi, H. Ide and N. Hirota, Chem. Phys. Lett., 1979, 66, 581–583, DOI:10.1016/0009-2614(79)80344.
- K. Komaguchi, K. Nomura, M. Shiotani, A. Lund, M. Jansson and S. Lunell, Spectrochim. Acta, Part A, 2006, 63, 76–84, DOI:10.1016/j.saa.2005.04.039.
- T. Yanai, D. Tew and N. A. Handy, Chem. Phys. Lett., 2004, 393, 51–57, DOI:10.1016/j.cplett.2004.06.011.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648, DOI:10.1063/1.438980.
- G. B. Bacskay, Chem. Phys., 1981, 61, 385–404, DOI:10.1016/0301-0104(81)85156-7.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- Y. Yoshida, M. Maesato, G. Saito and H. Kitagawa, Inorg. Chem., 2019, 58, 14068–14074, DOI:10.1021/acs.inorgchem.9b02080.
- T. Kato, K. Yoshizawa and T. Yamabe, J. Chem. Phys., 1999, 110, 249–255, DOI:10.1063/1.478100.
- H. Tachikawa, J. Phys. Chem. A, 2018, 122, 4121–4129, DOI:10.1021/acs.jpca.8b00292.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620, 10.1039/B810189B.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544, DOI:10.1063/1.1383587.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem., 2006, 110, 5121–5129, DOI:10.1021/jp060231d.
- D. Becke, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1cp04638a |
| This journal is © the Owner Societies 2022 |