DOI:
10.1039/D2CE01416E
(Paper)
CrystEngComm, 2022,
24, 8390-8398
Co-crystals of zwitterionic GABA API's pregabalin and phenibut: properties and application†
Received
13th October 2022
, Accepted 17th November 2022
First published on 17th November 2022
Abstract
We present several multicomponent crystalline species formed by zwitterionic GABA analogues pregabalin and phenibut. These compounds are evaluated based on their crystal structure in congruence with properties such as melting behaviour, solubility, and lattice energies. Furthermore, it is discussed how major property distinctions between a homo- and heterochiral co-crystalline system enable enantiopurification of pregabalin.
1 Introduction
In crystal engineering of active pharmaceutical ingredients (APIs) the enhancement of attributes such as solubility or drug stability is an ongoing task. Various approaches are used to identify influences on crystal properties and increasingly more sensitive ways are developed to synthesize a desired target product.1–10 In many cases, thermodynamic factors such as lattice energies play an important role in the outcome of an attempted crystallization. Computational methods based on density functional theory (DFT) as well as force field applications have proven to be useful in determining energetic properties of single as well as multicomponent species.11–15 A common approach in using DFT-methods for lattice energy calculations is to compare energies of larger crystalline slabs to energy sums of occurring crystallographically independent molecules in the unit cell. However, differing molecular charges can make that task difficult. Aside from their effect on energy calculations, molecular charges build a basis for the classification of crystalline solids, for example as either a salt or a neutral co-crystal. Childs et al. describe the salt-cocrystal continuum in their 2007 contribution, evaluating molecular influences of the co-formers and their acidity as important factors for the charge status of the received product.16 In 2010 Braga et al. coined the term ionic co-crystal. After describing multicomponent crystalline entities composed of neutral barbituric acid and various alkali bromides or caesium iodide, their publication closes with the suggestion to further examine enthalpic and entropic contributions via theoretical evaluations.17 They brought up the topic again in 2018, focussing on inorganic and organic co-crystalline systems, highlighting pharmaceutical and agricultural applications and especially the enantiomeric resolution of racemates as interesting uses.18 Additional discussions on the influence of ionicity on structure, properties or formation of co-crystalline species containing various charged molecular species have been conducted over the years,19–21 also in conjunction with organic/organic multicomponent crystalline entities. Various definitions for such structures have been proposed.22–26 As the established terminology has found acceptance in the community,27–30 methods to explicitly distinguish between salts and ionic co-crystals were designed.31,32 Next to varying molecular charges and acidity, a further molecular influence which can impact crystalline structure and lattice makeup is molecular chirality.18,33,34 Mandelic acid and its derivatives have shown remarkable chirality-based influences on co-crystalline systems when used as co-formers.35–37 In the past, several processes were patented that amongst other compounds use mandelic acid to separate e.g. (S)-pregabalin ((S)-4-amino-3-isobutylbutanoic acid) from its (R)-enantiomer. (S)-Pregabalin, the eutomer, which is the enantiomer that shows the desired pharmaceutical properties, and (R)-pregabalin, its distomer, with no or undesired pharmaceutical effects, can be separated via co-crystal formation with mandelic acid and subsequent co-former removal.38–44 Pregabalin is a pharmaceutically active γ-amino butanoic acid (GABA) derivative with a considerable amount of uses as an API. Pregablin remained a staple treatment API for such diseases as epilepsy, neuropathic pain and anxiety disorders over the years since its release.45–54 It is chiral, shows zwitterionic charges on its GABA-subunit and being an amino acid is slightly acidic. A further related nootropic and anxiolytic drug is phenibut ((RS)-4-amino-3-phenylbutanoic acid) where the (R)-enantiomer is the eutomer,55–57 that has fallen out of favour for its abuse potential.58–62 As another GABA-derivative, it also contains a zwitterionic subunit, poses chirality in C3 and is also a weakly acidic amino acid. In the present work various co-crystalline embodiments of enantiomers of pregabalin (1) and racemic phenibut (2) with co-formers mandelic acid (3) and malic acid (4) are examined (Scheme 1). Pregabalin exhibits an unusual behaviour to form a zwitterionic/neutral co-crystal with mandelic acid as first described by Samas et al. in 2007,63 or a system composed solely of charged molecules depending on whether a homo- or heterochiral set of co-formers is co-crystallized. For comparison a set of multicomponent systems of pregabalin with malic acid and phenibut with mandelic acid is presented to show that the described behaviour is unique for pregabalin and mandelic acid only. Molecular influences such as zwitterionicity, chiral information and compound acidity in multicomponent entities are discussed in congruence with structural makeup and intermolecular interactions, especially hydrogen bond (HB) as well as properties like melting behaviour, solubility in aqueous medium and lattice energies. These findings are then applied to explain how the enantiopurification of pregabalin according to the established process with mandelic acid can be understood and improved on. It is shown how a specific set of molecular and structural influences enables a useful application.
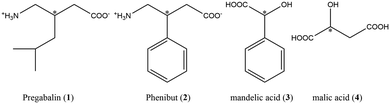 |
| | Scheme 1 Compounds used in this work. APIs pregabalin and phenibut as well as coformers mandelic- and malic acid. | |
2 Experimental and computational methods
Single-crystal X-ray diffraction
Suitable single crystals were selected from the sample and mounted on a cryo-loop under protective oil. Diffraction data were recorded with a Rigaku XtaLAB Synergy S diffractometer with Hybrid Pixel Arrow detector and a PhotonJet X-ray source using Cu-Kα radiation (λ = 1.54182 Å) at 100.0 ± 0.1 K with ω-scans. Data reduction and absorption correction were conducted on CrysAlisPRO v. 42 software, numerical absorption correction based on Gaussian integration over a multifaceted crystal model and empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm was used.64 The single crystal structures were analyzed and refined by using direct methods (SHELXT-2015), full-matrix least-squares refinements on F2 were performed using SHELXL2017/01 software package.65,66 Structure solution and refinements were conducted with OLEX2-1.5 software package. Hydrogen atoms were experimentally refined, all esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.67 Figures were prepared with Mercury software v. 2020.2.0.68
Differential scanning calorimetry
Measurements were performed on a Linkam DSC 600 with nitrogen cooling and heating range from −190–600 °C in alumina crucibles.
X-ray powder diffraction
Measurements were performed on a Rigaku Miniflex diffractometer in θ/2θ geometry at ambient temperature using Cu-Kα radiation (λ = 1.54182 Å).
Lattice energy calculations
Quantum Espresso (QE) PWSCF v. 6.6 was used to perform periodic calculations based on the Perdew–Burke–Ernzerhof (PBE) functional.69 The PBEsol basis set was used to describe pseudo potentials. Lattice energies were determined by geometric optimization energies of crystal structures as measured compared to ideal gas states of the participating molecules in a vacuum cluster, similar to the method described by Marchese Robinson et al. and Voronin and colleagues.12,14
Diffraction quality single crystals
Multicomponent species (S,S)-1:3, (R,R)-1:3, (S,R)-1:3, (R,S)-1:3, (S,S)-1:4, (S,R)-1:4 and (R,S)-2:3 were obtained by co-crystallization of equimolar amounts of co-formers from aqueous solution and subsequent evaporation of the solvent at room temperature. Single crystals of single component species 1 and 2 were obtained by dissolution of powdery substance in aqueous medium and subsequent evaporation of the solvent at room temperature.
Enantiopurification of pregabalin
Two batches of racemic pregabalin hydrate (1.375 g, 7.756 mmol, 1 eq.) and enantiopure mandelic acid (1.180 g, 7.756 mmol, 1 eq.) were co-grinded with a Retsch MM400 ball mill for 40 min at 25 Hz using two 25 mL stainless containers fitted with one PTFE-ball (ø 2 cm) each. The received co-crystalline substance was subsequently washed with water and dried at 45 °C thrice, using 12 mL water in the first wash, 6 mL water in the second wash and 4 mL water in the third washing instance to remove the more soluble heterochiral species. The remaining residue was then stirred in a mixture of 75 mL acetone and 500 μL water for 40 h at room temperature. Filtration of the powdery residue led to enantiopurified pregabalin.
IR spectra
IR-spectra were recorded on the Bruker Tensor 27 Fourier transformed IR device in attenuated total reflectance mode in the range 4000 cm−1 to 400 cm−1. Spectra are shown in the ESI.†
1H-NMR spectra
1H-NMR spectra for solubility determination were recorded on a Bruker Avance III NMR-spectrometer at 600 MHz and are shown in the ESI.†
Chemicals
The following chemicals were purchased and used without further purification: (rac)-pregabalin hydrate from abcr, (rac)-phenibut from BLDpharm, (S)-mandelic acid from G&K, (R)-mandelic acid from TCI, (S)-malic acid from GLENTHAM LIFE SCIENCES and (R)-malic acid from BLDpharm. (S)-Pregabalin and (R)-pregabalin were produced by enantiomeric enrichment from (rac)-pregabalin hydrate according to the described procedure.
3 Results and discussion
3.1 Properties
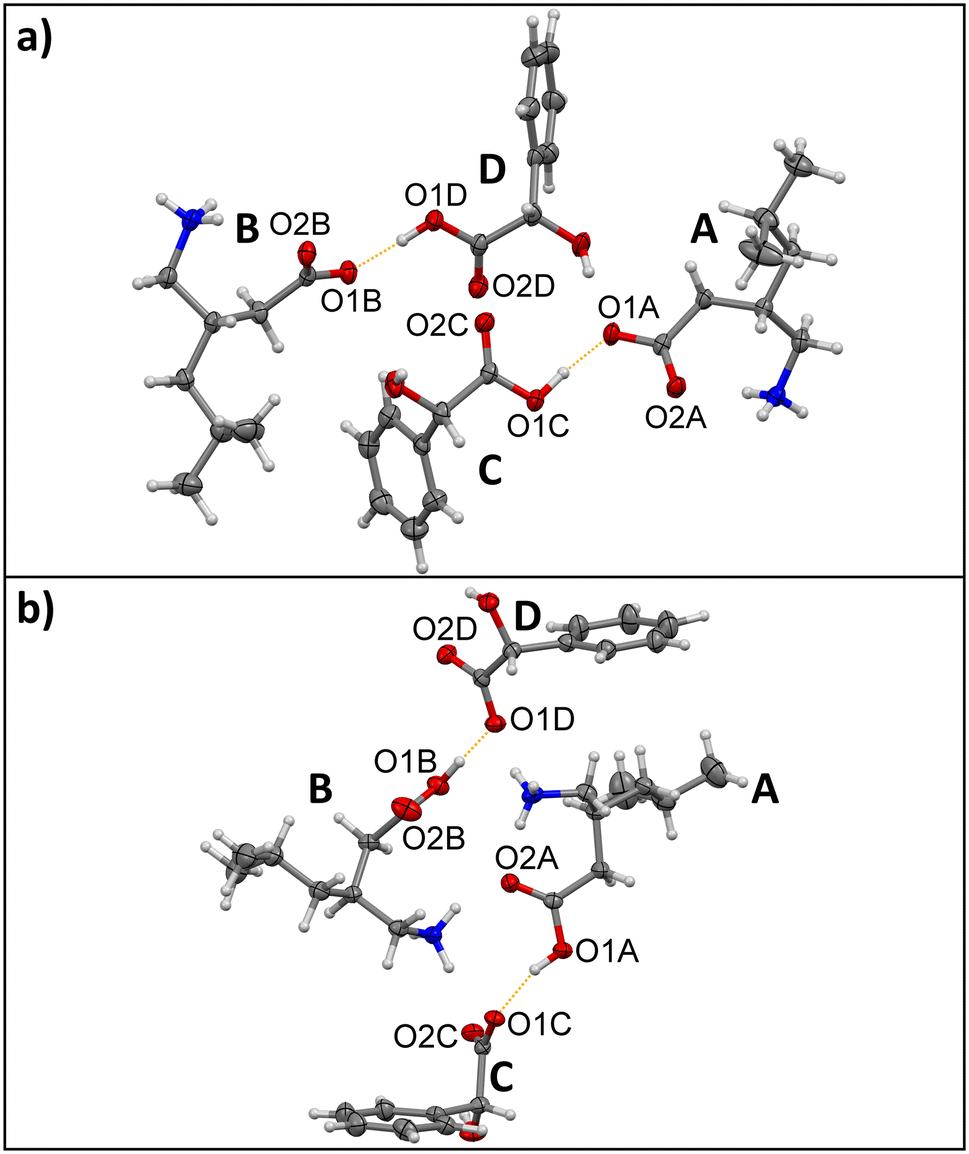
In 1:3-systems no protonic shift occurs between co-formers if a homochiral (S,S) or (R,R) chirality is present, while each molecule becomes formally charged in species of (S,R) or (R,S) chirality in pregabalin:mandelic acid co-crystalline entities (Fig. 1). Homochiral forms crystallize isostructurally in regard to each other as do the heterochiral species. Both homo- and heterochiral pairs can be synthesized from solution as well as through neat grinding. Considering 1 and 3 properties, the only significant difference is their molecular chirality. In terms of acidity, a pKa1 value of 4.2 is reported for 1 and 3.41 for 3,70,71 resulting in a ΔpKa value of 0.79. According to Childs et al. this matches a defined murky area of ΔpKa = 0–3 for the solid-state protonation where both a salt as well as a co-crystal formation can occur, and additional molecular influences play a role.16 Similar observations for small structural changes have been conducted in the past. Trifluoro acetic acid becomes an ionic solid at low temperatures when hydrogenated, but a molecular solid when deuterated.72 It appears likely that the slight change in molecular chirality has a similar energetic effect in 1:3-species, favouring ionization in heterochiral forms while keeping default charges in homochiral ones. To highlight the uniqueness of the described system, further similar compounds (S,S)-1:4, (S,R)-1:4 as well as (R,S)-2:3, were co-crystallized from aqueous solution. Structural characteristics as well as thermodynamic properties were compared between API's and the received multicomponent species. A detailed comparison of hydrogen bonds shows similar characteristics in all compounds regardless of their protonation status (Table 1). The pKa values should be considered to enhance the understanding of the present HB characteristics. The pKa1 value of 4 is 3.40,73 and that for 2 is 4.44,74 giving ΔpKa values of 0.8 for the 1:4-species and 1.03 for the 2:3 system. All systems are placed in the previously mentioned range of poor definability of the solid-state classification and all form strong – mid strength HBs considering their intermolecular distances. Small ΔpKa values also facilitate the formation of stronger hydrogen bonds.75 This shows why the investigated substances behave structurally similar. Even though some of them are composed of overall neutral molecules and some are charged, they all fall into a range of inconclusive pKa-influence on protonation behaviour, with the oddity present between homo- and heterochiral 1:3-systems to stay default or protonate/deprotonate apparently solely based on the molecular chirality. Closer examination of HB properties highlights the similarities even further. Except for single species 1 and 2, where the average HB distance is about 2.75–2.77 Å, the HB average is closer to 2.8 Å in all multicomponent compounds. Interestingly, the shortest interactions never occur between two charged subunits like ammonium and carboxylate. While the shortest HB lengths are observed in malic acid homo- and heterochiral forms (S,S)- and (S,R)-1:4 which are both composed of charged molecules with 2.446(4) Å and 2.475(2) Å, the mandelic acid overall neutral homochiral (S,S)-1:3 Å comes closely thereafter with 2.487(3) Å. The corresponding average HB for this system with 2.784 Å is the shortest among the HB averages in multicomponent entities suggesting the strongest overall HB-motif. Average HB distances in multicomponent systems show a rather large deviation from 0.043 Å up to 0.124 Å. On the other hand, single component species 1 and 2 show a more uniform dispersion of HB distances. Here, the average variation is only 0.016 Å, 0.004 Å in the shortest and 0.028 Å in the longest HB. Hydrogen bonds in single-component structures are not exceptionally short, but on average slightly shorter than in the multicomponent systems. This emphasizes that while molecular charges might shift in the described compounds, they all behave structurally similar regarding their HB characteristics, and multicomponent species stay similar to single component APIs. In general, in the most of these systems it is not possible to discern which structural features are influenced by pKa distinctions and which by molecular influences, but for the 1:3-species chirality inversion seems to be the most probable cause for the ionization behaviour. Furthermore, ionicity does not appear to have a significant impact on the structural makeup, especially regarding HB lengths and angles. While structurally similar compounds are formed in all investigated species, they do show some significant distinctions in their melting behaviour, solubility and lattice energies (Table 2). As was established in the past, higher melting points correlate with lower solubilities.76–78 The highest solubility is present in 1:4 that melt below 100 °C with 85 °C for (S,S) and 95 °C for (S,R) respectively. In 1:3-systems the heterochiral forms melt 25 °C lower than their homochiral counterparts and their solubility is about nine times higher at 316 ± 18 g L−1 and 307 ± 6 g L−1 for (S,R) and (R,S) as compared to 37 ± 1 g L−1 and 40 ± 4 g L−1 for (S,S)- and (R,R)-species. All in all, multicomponent systems composed of charged molecules reach far higher solubilities than their zwitterionic or neutral counterparts. This is interesting especially in the case of 1:3 homo- and heterochiral forms. Even though solubility of 3 is about five times higher than that of 1 with 203 ± 3 g L−1 and 35 ± 0.4 g L−1 or 33 ± 0.3 g L−1 respectively, a substantial increase in solubility is not reached in the homochiral co-crystal forms. In co-crystalline entities the solubility is generally linked to several parameters such as co-former solubility, solvent as well as its pH value, co-former pKa values, co-former complexation by solvent, co-former ratio and ionicity.79–82 As the same pH-stable solvent conditions were used in all cases and neither the co-former, the co-former ratio nor the co-former pKa was changed, it is highlighted how the chirality induced change in ionicity possibly affects solvent complexation and thereby increases solubility in heterochiral 1:3-forms compared to the homochiral systems. It is furthermore surprising that while the melting point in homochiral 1:3-forms is decreased and 3 is substantially more soluble than 1, the solubility increase is practically negligible. This indicates that ionicity plays a key-role for the dissolution behaviour. In case of 1:4-forms, the mentioned criteria for the co-crystal solubility are fulfilled more uniformly. The solubility of 4 with 2061 ± 76 g L−1 is exceedingly high compared to that of pure 1, all components are ionized and a very high solubility of more than 800 g L−1 is reached in both cases. In 2:3 an increase of solubility up to 71 ± 3 g L−1 is also achieved, which is comparatively low. Here, the increase is most probably due to the large difference between the solubilities of 2 and 3 with 18 ± 1 g L−1 and 203 ± 3 g L−1 respectively. It is noteworthy that this system is heterochiral, yet here differing chirality does not cause a difference in ionicity as is the case in heterochiral 1:3-species. This further confirms that ionicity plays an important role in the solubility increase in these compounds, having a larger impact than the co-former solubility alone. Lattice energies do not appear to impact solubility in a significant manner, as all multicomponent species are in a similar range, the lowest values present in homochiral 1:3-forms with about −320 kJ mol−1 and the highest values in 1:4-systems with −356.66 kJ mol−1 in (S,S) and −363.05 kJ mol−1 in (S,R). As HB-properties are also similar in all investigated compounds, the influence of the charged species on the strength of intermolecular interactions seems to be questionable. While the highest values are present in 1:4-systems composed of charged molecules, the second most favourable energy is given for 2:3 with −343.23 kJ mol−1 composed of zwitterionic and neutral molecules. The calculated lattice energies for the discussed compounds are higher than those established by Marchese Robinson et al. for a large number of neutral single component API's,14 but lower compared to those for ionic liquids containing inorganic ions,13 and far lower than those for inorganic systems.83,84 However, the values are slightly higher than values established by Voronin et al. for carbendazim maleates.12 This suggests that (zwitter)ionicity does generally have an influence on lattice energies, even though the final molecular protonation status in multicomponent species of the investigated compounds does not impact structural features meaningfully. The unusually beneficial lattice energy of 2 with −367.68 kJ mol−1 might appear too high especially when compared to 1-forms with about −195 kJ mol−1. However, 2 behaves rather different than 1 in two key aspects: firstly, 2 does not form a stable hydrate as racemic 1 does.85,86 Contrary to the latter, 2-hydrate transitions to anhydrous 2 quickly, suggesting a preferable form in the anhydrous species. Furthermore, experimental observations conducted in the presented work suggest a higher stability of 2 as compared to 2:3. While co-crystalline systems of 1 with the used co-formers form readily and without evidence for impurities under the investigated conditions, the 2:3 system cannot be obtained as reliably. 2:3 single crystals could only be received once; neat co-milling did not work under the chosen conditions and co-crystallization from solution still contains impurities visible in the powder pattern (see ESI†). Therefore, a comparatively beneficial lattice energy seems likely. This might stem from an increased connectivity via π-interactions enabled by phenyl-subunits in 2. Further support for unusually high lattice energy in 2 is given through its melting behaviour. It is the only discussed compound that decomposes without melting at temperatures above 200 °C which indicates strong intermolecular connectivity. To conclude, the comparison of the presented compounds based on the discussed structural and thermodynamic properties uncover their commonalities. They all behave structurally similar as their co-formers are in a specific murky ΔpKa range that enables formation of salts as well as co-crystals. Some of the chosen systems retain zwitterionic/neutral molecular makeup and some obtain molecular charges. Energetically, they all are in a similar range, posing more beneficial lattice energies than neutral compounds but less favourable ones than organic/inorganic or purely inorganic compounds. However, this border-region in the salt-co-crystal continuum can have high impact on properties such as melting and solubility behaviours. The shown compounds that become charged upon co-crystallization melt at lower temperatures and are vastly more soluble than their neutral/zwitterionic counterparts. This specific behaviour seems to largely depend on the molecular chirality in case of homo- and heterochiral 1:3-forms and the comparatively low lattice energy in 1 enables easy co-crystallization. As 1 is a commercially viable API the presented results can be used to better understand, modify and improve an established crystallization-based enantiopurification processes.
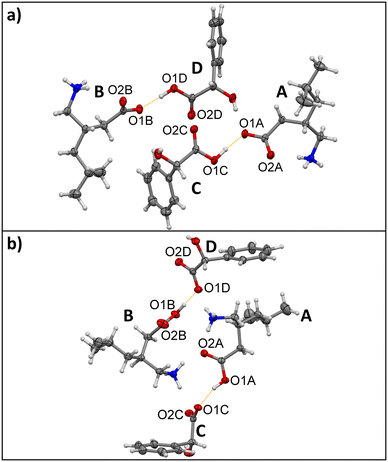 |
| | Fig. 1 Illustration of the protonic shift in the asymmetric units of a) homochiral (S,S)-1:3 and b) heterochiral (S,R)-1:3. Four crystallographically non-equivalent occurring molecules are marked as A, B, C and D respectively. Proton transfer occurs in (S,R)-1:3 between pregabalin A's O1A and mandelic acid C's O1C as well as pregabalin B's O1B and mandelic acid D's O1D. Carbon atoms are depicted in grey, hydrogen atoms in white, nitrogen atoms in blue and oxygen atoms in red. The protonic shift is highlighted in a dotted orange line. | |
Table 1 An overview of the overall number of hydrogen bonds in the systems, compared to those occurring between two charged subunits. Average, the shortest and the longest HB donor/acceptor distances and their corresponding angles are compared. The average values are calculated for each different occurring HB interaction in the unit cell. Samples composed solely of charged molecules and entries solely involving interactions of charged subunits are written in bold. Carboxylate or ammonium residues in zwitterionic forms are considered charged even though the molecules are overall formally neutral
| Sample |
Charged HB/all HB |
Ø D⋯A [Å]/D–H⋯A [°] |
Min. D⋯A [Å]/D–H⋯A [°] |
Max. D⋯A [Å]/D–H⋯A [°] |
| (S)-1 |
3/3 |
2.751/171
|
2.728(0)/171(2)
|
2.771(1)/172(2)
|
| (R)-1 |
3/3 |
2.752/172
|
2.731(2)/174(2)
|
2.767(2)/172(3)
|
| (rac)-2 |
3/3 |
2.767/168
|
2.732(3)/160(3)
|
2.795(3)/173(2)
|
| (S,S)-1:3 |
6/12 |
2.784/158 |
2.487(3)/174(2) |
3.036(4)/151(4)
|
|
(R,R)-1:3
|
6/14 |
2.818/152 |
2.489(7)/172(4) |
3.038(2)/148(3)
|
|
(S,R)-1:3
|
4/14 |
2.799/145 |
2.570(3)/179(6) |
3.012(3)/121(3) |
|
(R,S)-1:3
|
4/14 |
2.797/144 |
2.570(2)/174(4) |
3.006(2)/129(2) |
| (R,S)-2:3 |
3/7 |
2.821/151 |
2.504(2)/175(4) |
3.020(2)/117(2) |
|
(S,S)-1:4
|
2/8 |
2.827/153 |
2.446(4)/172(3) |
3.154(3)/112(2) |
|
(S,R)-1:4
|
2/8 |
2.798/154 |
2.475(2)/175(3) |
3.103(2)/127(3) |
Table 2 Overview on the determined melting points, solubilities in aqueous phosphate buffer at pH 6.8 and 37 °C as well as lattice energies calculated with quantum espresso. Bold written samples are composed solely of charged molecules. Mandelic and malic acid solubilities are given in ESI†
| Sample |
Melting point [°C] |
Solubility [g L−1] |
E
lat [kJ mol−1] |
|
(rac)-Phenibut decomposes prior to melting at about 200 °C, as such no melting point could be determined.
The maximum solubility could not be determined. No reliable results could be obtained from the highly viscous substance at higher concentrations.
|
| (S)-1 |
185 |
35 ± 0.4 |
−195.08 |
| (R)-1 |
187 |
33 ± 0.3 |
−195.17 |
| (rac)-2 |
—a |
18 ± 1 |
−367.68 |
| (S,S)-1:3 |
138 |
37 ± 1 |
−320.36 |
| (R,R)-1:3 |
132 |
40 ± 4 |
−320.99 |
|
(S,R)-1:3
|
111 |
316 ± 18 |
−304.66 |
|
(R,S)-1:3
|
105 |
307 ± 6 |
−307.77 |
| (R,S)-2:3 |
150 |
71 ± 3 |
−343.23 |
|
(S,S)-1:4
|
85 |
>800b |
−356.66 |
|
(S,R)-1:4
|
95 |
>800b |
−363.05 |
3.2 Enantiopurification of pregabalin
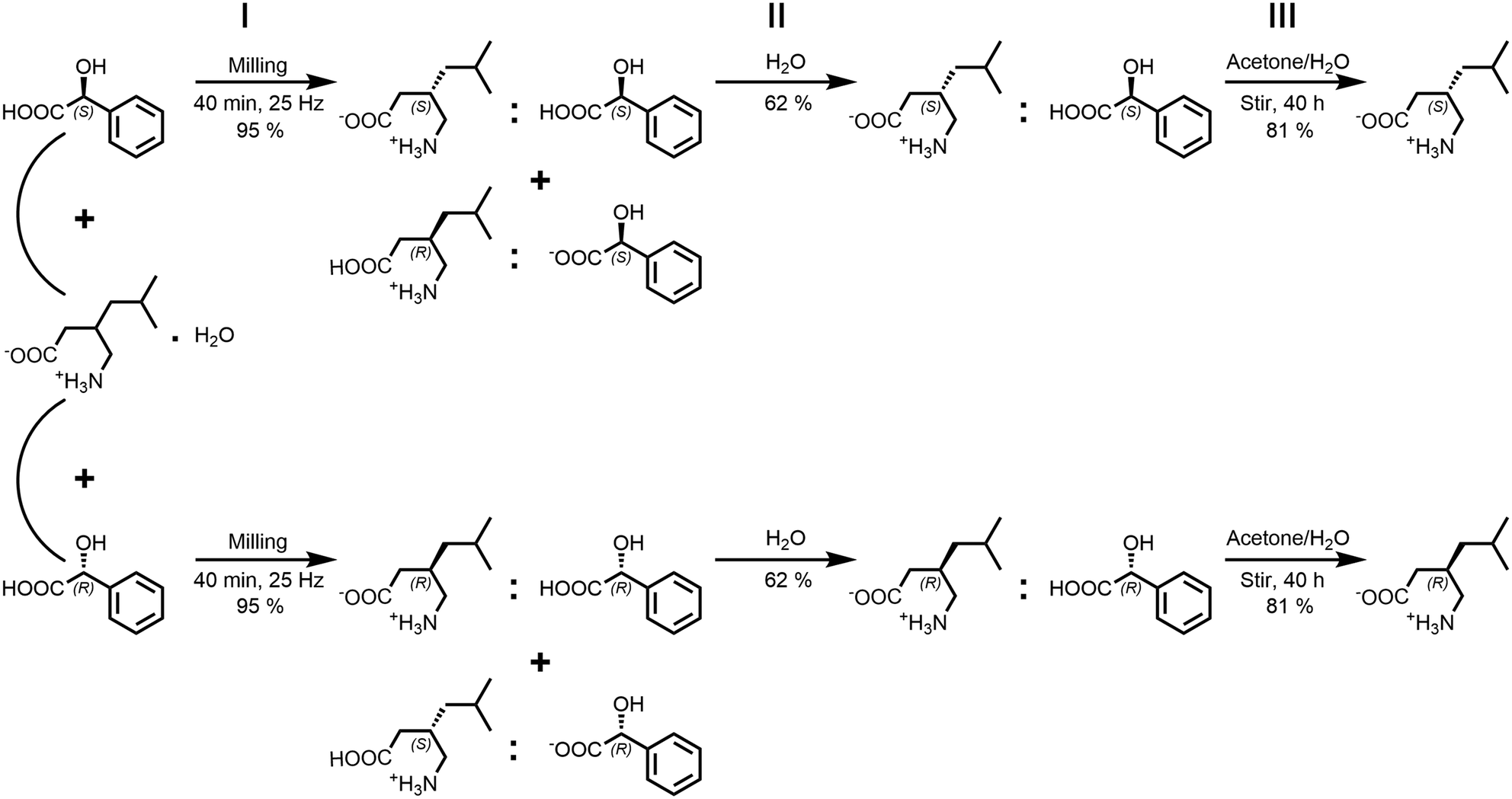
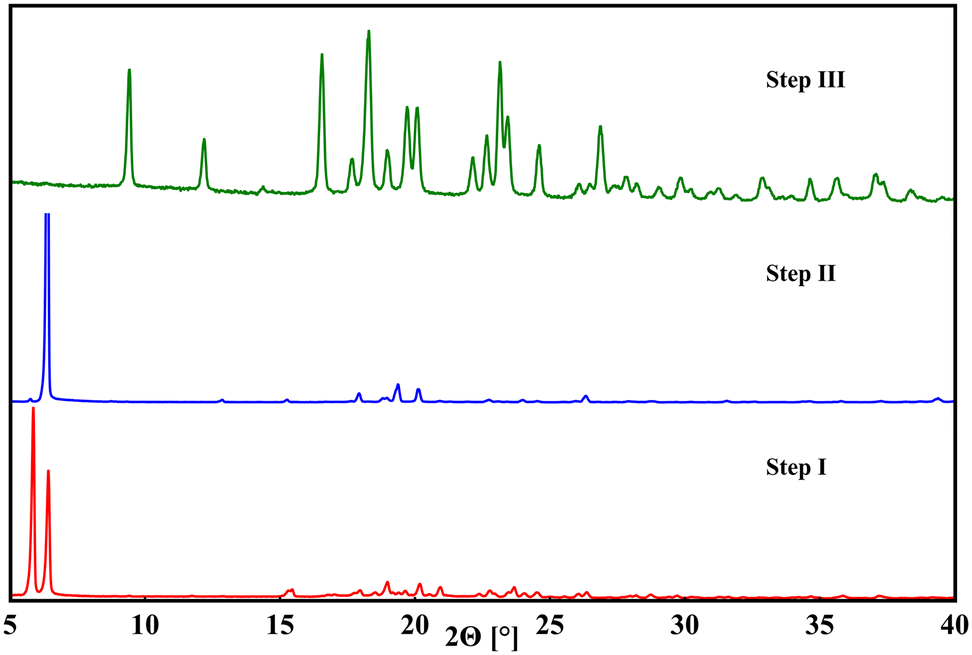
Based on the described findings, a process for the racemic separation of the racemic pregabalin hydrate ((rac)-1·H2O) could be improved on and simplified (Scheme 2). Similar enantiopurification methods were previously patented,38–44 that followed a bottom-up approach in their racemic separation processes. By dissolving varying quantities of (rac)-pregabalin hydrate with (S)-mandelic acid and subsequently cooling or crystallization via vaporization the (S,S)-species was formed. In the patents it is then removed from solution and further processed. As was shown in the present study the formation of 1:3 is energetically favourable compared to crystallization of 1. Furthermore, (S,R)- as well as (R,S)-1:3 show ionicity-based increased solubilities compared to the (S,S)- and (R,R)-species. In opposition, 1:4-compounds are prone to form viscous residue instead of crystalline materials which is one of the reasons those are not suitable for the separation process. Due to the energetic favourability as well as mechanic and thermal stability of 1:3-species they can be prepared in a ball mill via mechanochemical synthesis. This enables a top-down approach where the scale depends solely on the largest available milling vessel. By milling of (rac)-1·H2O with either mandelic acid enantiomer (S)-3 or (R)-3 in equimolar amounts a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the (S,S)/(R,S) or (R,R)/(S,R) multicomponent systems is formed in good yields (step I in Scheme 2). Missed yield at this point stems from losing some material on the milling vessel walls. As the (S,R)- and (R,S)-systems pose an about nine times higher solubility in water compared to their homochiral counterparts they can then be removed by subsequent washing and drying steps with increasingly smaller amounts of water. This approach therefore offers the advantage of quickly producing co-crystalline product and necessitates only low amounts of water to remove the unwanted heterochiral species. To give an example, 4.580 g of co-crystal mixture was produced by milling in step I. After three washing and drying steps with 12 mL, 6 mL and 4 mL water, 1.442 g (62%) of enantiopurified homochiral co-crystal were received in step II. Low amounts of washing water further simplify regaining the missed yield: the lost product can be recrystallized by water vaporization. The washing process can then be repeated with even smaller amounts of water. Our approach does not require further additives like organic solvents, additional acidic or caustic compounds nor is heating necessary which is an advantage compared to previously reported procedures. This also allows for an environmentally friendly, low effort process. To separate 3 from 1 in step III a slight variation to the patented process by Pradhan and colleagues,40 who proposed a number of different separation methods, is introduced. Washed homochiral co-crystalline product is stirred for 40 h in acetone with catalytic amounts of water added. To follow up on the previous example, 1.442 g of powdery co-crystal were put in a glass vessel with 75 mL of acetone and 500 μL of water and stirred for 40 h. The powdery substance was then filtered, washed with additional 20 mL of acetone and 0.612 g (81%) of enantiopurified 1 were received. In contrast to the previously proposed method the stirring process is much longer but does not require heating. Acetone was chosen as it is cheap, easily available and poses less environmental risks than other organic solvents that could be suitable for this process. The proposed enantiopurification can be controlled through powder X-ray diffraction during each step (Fig. 2). The described method relies on property differences unique to the 1:3-systems. Inversion of molecular chirality leads to related multicomponent species that still differ in their key attributes. Higher solubility in one of the received multicomponent species in congruence with the mechanical stability enables the described process for 1. The previously discussed properties in malic acid systems deem them unusable for this in contrast to 1:3-forms, as 1:4-species preferably form viscous liquids with similar solubilities. On the other end, phenibut co-crystallization with mandelic acid is energetically unfavourable compared to pure 2-formation. Mechanochemical synthesis under the same herein discussed conditions did not lead to 2:3. Furthermore, 2 does not show chirality dependent varying crystallization products, a trait inherent only to 1.
:1 mixture of the (S,S)/(R,S) or (R,R)/(S,R) multicomponent systems is formed in good yields (step I in Scheme 2). Missed yield at this point stems from losing some material on the milling vessel walls. As the (S,R)- and (R,S)-systems pose an about nine times higher solubility in water compared to their homochiral counterparts they can then be removed by subsequent washing and drying steps with increasingly smaller amounts of water. This approach therefore offers the advantage of quickly producing co-crystalline product and necessitates only low amounts of water to remove the unwanted heterochiral species. To give an example, 4.580 g of co-crystal mixture was produced by milling in step I. After three washing and drying steps with 12 mL, 6 mL and 4 mL water, 1.442 g (62%) of enantiopurified homochiral co-crystal were received in step II. Low amounts of washing water further simplify regaining the missed yield: the lost product can be recrystallized by water vaporization. The washing process can then be repeated with even smaller amounts of water. Our approach does not require further additives like organic solvents, additional acidic or caustic compounds nor is heating necessary which is an advantage compared to previously reported procedures. This also allows for an environmentally friendly, low effort process. To separate 3 from 1 in step III a slight variation to the patented process by Pradhan and colleagues,40 who proposed a number of different separation methods, is introduced. Washed homochiral co-crystalline product is stirred for 40 h in acetone with catalytic amounts of water added. To follow up on the previous example, 1.442 g of powdery co-crystal were put in a glass vessel with 75 mL of acetone and 500 μL of water and stirred for 40 h. The powdery substance was then filtered, washed with additional 20 mL of acetone and 0.612 g (81%) of enantiopurified 1 were received. In contrast to the previously proposed method the stirring process is much longer but does not require heating. Acetone was chosen as it is cheap, easily available and poses less environmental risks than other organic solvents that could be suitable for this process. The proposed enantiopurification can be controlled through powder X-ray diffraction during each step (Fig. 2). The described method relies on property differences unique to the 1:3-systems. Inversion of molecular chirality leads to related multicomponent species that still differ in their key attributes. Higher solubility in one of the received multicomponent species in congruence with the mechanical stability enables the described process for 1. The previously discussed properties in malic acid systems deem them unusable for this in contrast to 1:3-forms, as 1:4-species preferably form viscous liquids with similar solubilities. On the other end, phenibut co-crystallization with mandelic acid is energetically unfavourable compared to pure 2-formation. Mechanochemical synthesis under the same herein discussed conditions did not lead to 2:3. Furthermore, 2 does not show chirality dependent varying crystallization products, a trait inherent only to 1.
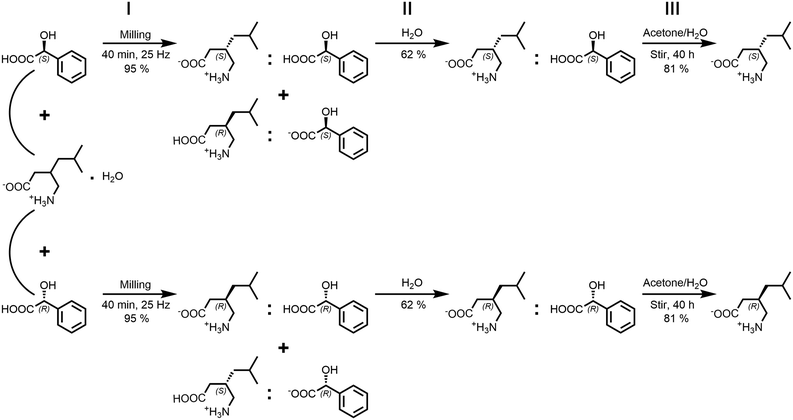 |
| | Scheme 2 Steps for the racemic separation of (rac)-1·H2O with mandelic acid: step I - mechanochemical co-crystallization, step II - washing powdery product with water and drying the residue, step III - extract mandelic acid by stirring the residue in acetone/water mixture for 40 h. | |
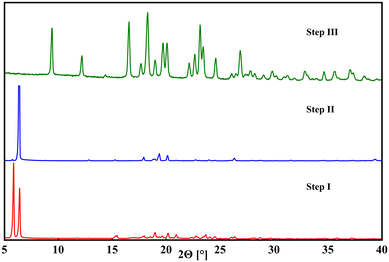 |
| | Fig. 2 Powder patterns of products received after enantiopurification process step I (red), step II (blue) and step III (green). In step I, both homo- and heterochiral multicomponent species are present, visible by the two strong diffraction reflexions at 5.9° and 6.4° 2Θ. In step II the heterochiral compound was removed by washing, diminishing the 5.9° signal. In step III, mandelic acid was removed, solely leaving enantiopure pregabalin. | |
4 Conclusions
In this work we presented an atypical case concerning co-crystallization of zwitterionic APIs of the GABA-family. Multicomponent crystalline species of related compounds pregabalin and phenibut were characterized regarding their structural and thermodynamic properties. Homo- and heterochiral pregabalin:mandelic acid species exhibit a remarkably different solubility and melting behaviour based on molecular charge differences, even though they are structurally very similar. It was shown that this hardly predictable behaviour occurs in a vague range of the salt-cocrystal continuum and can, in this specific case, be attributed to the co-former chirality inversion. It seems likely that this small change in the molecular makeup crosses an energetic barrier needed for ionization when heterochiral co-formers pregabalin and mandelic acid are used. For the given set of compounds, the results indicate that complex multicomponent crystalline species exist on a spectrum and their properties are influenced more impactfully by molecular makeup rather than their crystal structure. It was further presented how investigations of crystal properties in the borderline regions of the spectrum can offer interesting and unexpected results. An optimized and simplified top-down process of (rac)-pregabalin hydrate enantiopurification was developed and its functionality was explained by the previously stated properties. This work shows that careful examination of multicomponent systems composed of similar co-formers in areas where multiple molecular and thermodynamic influences compete to determine product properties can lead to fruitful and surprising results.
Author contributions
Conceptualization: Daniel Komisarek & Vera Vasylyeva. Data curation: Daniel Komisarek. Formal analysis: Daniel Komisarek. Funding acquisition: Vera Vasylyeva. Investigation: Daniel Komisarek. Methodology: Daniel Komisarek, Vera Vasylyeva, Takin Haj Hassani Sohi. Project administration: Vera Vasylyeva. Resources: Vera Vasylyeva. Supervision: Vera Vasylyeva. Validation: Daniel Komisarek, Vera Vasylyeva, Takin Haj Hassani Sohi. Visualization: Daniel Komisarek. Writing: Daniel Komisarek. Review & editing: Vera Vasylyeva.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank PD Dr. Oliver Weingart and Prof. Dr. Rochus Schmid for valuable discussions on theoretical aspects. Computational support and infrastructure was provided by the “Centre for Information and Media Technology” (ZIM) at the University of Duesseldorf (Germany). Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 440366605. Thanks to the CeMSA@HHU (Center for Molecular and Structural Analytics@Heinrich Heine University) for recording the NMR-spectroscopic data.
References
- V. Todaro and A. M. Healy, Drug Dev. Ind. Pharm., 2021, 47, 292–301 CrossRef CAS PubMed.
- M. Trampuž, D. Teslić and B. Likozar, Chem. Eng. Res. Des., 2021, 165, 254–269 CrossRef.
- A. Ainurofiq, K. E. Dinda, M. W. Pangestika, U. Himawati, W. D. Wardhani and Y. T. Sipahutar, Int. J. Res. Pharm. Sci., 2020, 11, 1621–1630 CrossRef.
- A. J. Al-Ani, C. Herdes, C. C. Wilson and B. Castro-Dominguez, Cryst. Growth Des., 2020, 20, 1451–1457 CrossRef CAS.
- D. Chen, Q. Sun, W. Huang and B.-S. Yang, Cryst. Growth Des., 2020, 20, 2251–2265 CrossRef CAS.
- L. Schenck, D. Erdemir, L. Saunders Gorka, J. M. Merritt, I. Marziano, R. Ho, M. Lee, J. Bullard, M. Boukerche and S. Ferguson,
et al.
, Mol. Pharmaceutics, 2020, 17, 2232–2244 CrossRef CAS.
- A. F. Shunnar, B. Dhokale, D. P. Karothu, D. H. Bowskill, I. J. Sugden, H. H. Hernandez, P. Naumov and S. Mohamed, Chemistry, 2020, 26, 4752–4765 CrossRef CAS.
- H.-L. Cao, J.-R. Zhou, F.-Y. Cai, J. Lü and R. Cao, Cryst. Growth Des., 2019, 19, 3–16 CrossRef CAS.
- S. Chattoraj and C. C. Sun, J. Pharm. Sci., 2018, 107, 968–974 CrossRef CAS.
- E. Hadjittofis, M. A. Isbell, V. Karde, S. Varghese, C. Ghoroi and J. Y. Y. Heng, Pharm. Res., 2018, 35, 100 CrossRef.
- F. Yang, C.-X. Yan, X. Yang, D.-G. Zhou and P.-P. Zhou, CrystEngComm, 2017, 19, 1762–1770 RSC.
- A. P. Voronin, A. O. Surov, A. V. Churakov, O. D. Parashchuk, A. A. Rykounov and M. V. Vener, Molecules, 2020, 25, 2386 CrossRef CAS.
- U. P. Preiss, D. H. Zaitsau, W. Beichel, D. Himmel, A. Higelin, K. Merz, N. Caesar and S. P. Verevkin, ChemPhysChem, 2015, 16, 2890–2898 CrossRef CAS PubMed.
- R. L. Marchese Robinson, D. Geatches, C. Morris, R. Mackenzie, A. G. P. Maloney, K. J. Roberts, A. Moldovan, E. Chow, K. Pencheva and D. R. M. Vatvani, J. Chem. Inf. Model., 2019, 59, 4778–4792 CrossRef CAS.
- H. K. Buchholz and M. Stein, J. Comput. Chem., 2018, 39, 1335–1343 CrossRef CAS PubMed.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS.
- D. Braga, F. Grepioni, L. Maini, S. Prosperi, R. Gobetto and M. R. Chierotti, Chem. Commun., 2010, 46, 7715–7717 RSC.
- D. Braga, F. Grepioni and O. Shemchuk, CrystEngComm, 2018, 20, 2212–2220 RSC.
- S. P. Kelley, A. Narita, J. D. Holbrey, K. D. Green, W. M. Reichert and R. D. Rogers, Cryst. Growth Des., 2013, 13, 965–975 CrossRef CAS.
- D. Yang, H. Wang, Q. Liu, P. Yuan, T. Chen, L. Zhang, S. Yang, Z. Zhou, Y. Lu and G. Du, Chin. Chem. Lett., 2021, 33, 3207–3211 CrossRef.
- T. Alkhidir, Z. M. Saeed, A. F. Shunnar, E. Abujami, R. M. Nyadzayo, B. Dhokale and S. Mohamed, Cryst. Growth Des., 2022, 22, 485–496 CrossRef CAS.
- M. Gryl, M. Kozieł and K. M. Stadnicka, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 53–58 CrossRef CAS.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. K. Thaper, S. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS.
- D. J. Berry and J. W. Steed, Adv. Drug Delivery Rev., 2017, 117, 3–24 CrossRef CAS PubMed.
- E. Grothe, H. Meekes, E. Vlieg, J. H. ter Horst and R. de Gelder, Cryst. Growth Des., 2016, 16, 3237–3243 CrossRef CAS.
- S. Guerin, S. Khorasani, M. Gleeson, J. O'Donnell, R. Sanii, R. Zwane, A. M. Reilly, C. Silien, S. A. M. Tofail, N. Liu, M. Zaworotko and D. Thompson, Cryst. Growth Des., 2021, 21, 5818–5827 CrossRef CAS.
- J. Nath and J. B. Baruah, Cryst. Growth Des., 2021, 21, 5325–5341 CrossRef CAS.
- W. Gong, P. K. Mondal, S. Ahmadi, Y. Wu and S. Rohani, Int. J. Pharm., 2021, 608, 121063 CrossRef CAS PubMed.
- T. A. Hegde, A. Dutta, T. C. Sabari Girisun and G. Vinitha, Chem. Phys. Lett., 2021, 781, 138971 CrossRef CAS.
- A. Kiguchiya, R. Teraoka, T. Sakane and E. Yonemochi, Chem. Pharm. Bull., 2019, 67, 945–952 CrossRef CAS PubMed.
- L. Zhao, M. P. Hanrahan, P. Chakravarty, A. G. DiPasquale, L. E. Sirois, K. Nagapudi, J. W. Lubach and A. J. Rossini, Cryst. Growth Des., 2018, 18, 2588–2601 CrossRef CAS.
- W. Ji, B. Xue, S. Bera, S. Guerin, L. J. Shimon, Q. Ma, S. A. Tofail, D. Thompson, Y. Cao, W. Wang and E. Gazit, Mater. Today, 2021, 42, 29–40 CrossRef CAS.
- P. Vaňkátová, A. Kubíčková and K. Kalíková, J. Chromatogr. A, 2022, 1673, 463074 CrossRef.
- C. C. Da Silva and F. T. Martins, RSC Adv., 2015, 5, 20486–20490 RSC.
- P. Rajasekar, C. Jose, M. Sarkar and R. Boomishankar, Angew. Chem., Int. Ed., 2021, 60, 4023–4027 CrossRef CAS.
- L. Zeng, Q. Yi, Q. Liu, K. Tang and B. van der Bruggen, Sep. Purif. Technol., 2021, 257, 117884 CrossRef CAS.
-
V. Gore, D. Datta, M. Gadakar, K. Pokharkar, V. Mankar and S. Wavhal, WO Pat., WO2009122215, 2009 Search PubMed.
-
V. Gore, D. Datta, M. Gadakar, K. Pokharkar, V. Mankar and S. Wavhal, US Pat., US20110124909, 2011 Search PubMed.
-
B. S. Pradhan, IN Pat., IN2010CH01584, 2010 Search PubMed.
-
A. Khaja, V. S. R. Potla, S. Govind, B. R. Konudula, Y. K. Chauhan and D. Datta, WO Pat., WO2009125427, 2009 Search PubMed.
-
K. B. Mafatlal, K. N. Kagathara, K. Sivaprasad, P. C. Rajendra, V. P. Bhikhalal, B. U. Rajaram and M. I. Ambalal, IN Pat., IN2009MU01587, 2009 Search PubMed.
-
S. R. D. Reddy, S. R. Velivela and R. R. V. Reddy, IN Pat., IN2010CH00299, 2010 Search PubMed.
-
R. K. Thaper, M. D. Prabhavat, S. K. Arora, Y. D. Pawar, D. K. P. Varma, V. S. Kamble and V. S. Shinde, IN Pat., IN2008KO00929, 2008 Search PubMed.
- D. K. Baidya, A. Agarwal, P. Khanna and M. K. Arora, J. Anaesthesiol., Clin. Pharmacol., 2011, 27, 307–314 CrossRef CAS.
- M. J. Brodie, Epilepsia, 2004, 45(Suppl 6), 19 CrossRef CAS.
- C. A. Federico, J. S. Mogil, T. Ramsay, D. A. Fergusson and J. Kimmelman, Pain, 2020, 161, 684–693 CrossRef PubMed.
- N. M. Gajraj, Anesth. Analg., 2007, 105, 1805–1815 CrossRef CAS.
- D. R. Guay, Am. J. Geriatr. Pharmacother., 2005, 3, 274–287 CrossRef CAS PubMed.
- R. Kavoussi, Eur. Neuropsychopharmacol., 2006, 16(Suppl 2), S128–S133 CrossRef CAS.
- N. Kumar, A. Laferriere, J. S. C. Yu, A. Leavitt and T. J. Coderre, J. Neurochem., 2010, 113, 552–561 CrossRef CAS.
- B. A. Lauria-Horner and R. B. Pohl, Expert Opin. Invest. Drugs, 2003, 12, 663–672 CrossRef CAS PubMed.
- R. A. Moore, S. Straube, P. J. Wiffen, S. Derry and H. J. McQuay, Cochrane Database Syst. Rev., 2009, 3, CD007076 Search PubMed.
- P. Ryvlin, E. Perucca and S. Rheims, Neuropsychiatr. Dis. Treat., 2008, 4, 1211–1224 CrossRef CAS.
- L. Zvejniece, E. Vavers, B. Svalbe, G. Veinberg, K. Rizhanova, V. Liepins, I. Kalvinsh and M. Dambrova, Pharmacol., Biochem. Behav., 2015, 137, 23–29 CrossRef CAS PubMed.
- I. Lapin, CNS Drug Rev., 2001, 7, 471–481 CrossRef CAS PubMed.
- M. Dambrova, L. Zvejniece, E. Liepinsh, H. Cirule, O. Zharkova, G. Veinberg and I. Kalvinsh, Eur. J. Pharmacol., 2008, 583, 128–134 CrossRef CAS PubMed.
- M. A. Downes, I. L. Berling, A. Mostafa, J. Grice, M. S. Roberts and G. K. Isbister, Clin. Toxicol., 2015, 53, 636–638 CrossRef PubMed.
- Y. B. Joshi, S. F. Friend, B. Jimenez and L. R. Steiger, J. Clin. Psychopharmacol., 2017, 37, 478–480 CrossRef.
- W. Li and B. Madhira, Am. J. Ther., 2017, 24, e639–e640 CrossRef.
- D. J. McCabe, S. A. Bangh, A. M. Arens and J. B. Cole, Am. J. Emerg. Med., 2019, 37, 2066–2071 CrossRef PubMed.
- D. R. Owen, D. M. Wood, J. R. H. Archer and P. I. Dargan, Drug Alcohol Rev., 2016, 35, 591–596 CrossRef PubMed.
- B. Samas, W. Wang and D. B. Godrej, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3938–o3938 CrossRef CAS.
-
CrysAlisPRO, Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS.
- P. Giannozzi, O. Baseggio, P. Bonfà, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. de Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru and S. Baroni, Chem. Phys., 2020, 152, 154105 CAS.
-
Physicians' desk reference 2007, ed. Thomson P. D. R., Thomson PDR, Mondvale, NJ, 2007 Search PubMed.
- G. Kortüm, W. Vogel and K. Andrussow, Pure Appl. Chem., 1960, 1, 187–536 CrossRef.
- D. Mootz and M. Schilling, J. Am. Chem. Soc., 1992, 114, 7435–7439 CrossRef CAS.
- R. M. Zelle, E. de Hulster, W. A. van Winden, P. de Waard, C. Dijkema, A. A. Winkler, J.-M. A. Geertman, J. P. van Dijken, J. T. Pronk and A. J. A. van Maris, Appl. Environ. Microbiol., 2008, 74, 2766–2777 CrossRef CAS.
- Chemicalize was used for prediction of pKa properties, 09/2022, https://chemicalize.com/ developed by ChemAxon (http://www.chemaxon.com).
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- E. Batisai, A. Ayamine, O. E. Y. Kilinkissa and N. B. Báthori, CrystEngComm, 2014, 16, 9992–9998 RSC.
- K. A. Chu and S. H. Yalkowsky, Int. J. Pharm., 2009, 373, 24–40 CrossRef CAS PubMed.
- G. L. Perlovich, Cryst. Growth Des., 2021, 21, 5058–5071 CrossRef CAS.
- M. Banik, S. P. Gopi, S. Ganguly and G. R. Desiraju, Cryst. Growth Des., 2016, 16, 5418–5428 CrossRef CAS.
- S. J. Bethune, N. Huang, A. Jayasankar and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9, 3976–3988 CrossRef CAS.
- D. J. Good and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9, 2252–2264 CrossRef CAS.
- D. J. Good and N. Rodríguez-Hornedo, Cryst. Growth Des., 2010, 10, 1028–1032 CrossRef CAS.
- S. Kaya and C. Kaya, Inorg. Chem., 2015, 54, 8207–8213 CrossRef CAS.
- S. Kaya, A. Robles-Navarro, E. Mejía, T. Gómez and C. Cardenas, J. Phys. Chem. A, 2022, 126, 4507–4516 CrossRef CAS PubMed.
- D. Komisarek, M. Pallaske and V. Vasylyeva, Z. Anorg. Allg. Chem., 2021, 647, 984–991 CrossRef CAS.
- M. Herbst, D. Komisarek, T. Strothmann and V. Vasylyeva, Crystals, 2022, 12, 1393 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Takin
Haj Hassani Sohi
,
Takin
Haj Hassani Sohi