
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular synthon hierarchy in cyclopropyl-containing peptide-derived compounds†
Joanna
Bojarska
 *a,
Martin
Breza
b,
Milan
Remko
c,
Paweł
Borowiecki
d,
Andrzej
Fruziński
a,
Izabela D.
Madura
e,
Krzysztof
Kaczmarek
f,
Zbigniew
Leśnikowski
gh,
Agata
Kraj
h,
Piotr
Zielenkiewicz
ij and
Wojciech M.
Wolf
a
*a,
Martin
Breza
b,
Milan
Remko
c,
Paweł
Borowiecki
d,
Andrzej
Fruziński
a,
Izabela D.
Madura
e,
Krzysztof
Kaczmarek
f,
Zbigniew
Leśnikowski
gh,
Agata
Kraj
h,
Piotr
Zielenkiewicz
ij and
Wojciech M.
Wolf
a
aChemistry Department, Institute of Ecological and Inorganic Chemistry, Technical University of Lodz, 116 Zeromskiego St., 90-924 Lodz, Poland. E-mail: joanna.bojarska@p.lodz.pl
bDepartment of Physical Chemistry, Slovak Technical University, Radlinskeho 9, SK-81237 Bratislava, Slovakia
cRemedika, Luzna 9, SK-85104, Bratislava, Slovakia
dLaboratory of Biocatalysis and Biotransformation, Department of Drugs Technology and Biotechnology, Faculty of Chemistry, Warsaw University of Technology, 75 Koszykowa St., 00-662 Warsaw, Poland
eFaculty of Chemistry, Warsaw University of Technology, 3 Noakowskiego St., 00-664 Warsaw, Poland
fInstitute of Organic Chemistry, Faculty of Chemistry, Lodz University of Technology, 116 Zeromskiego St., 90-924 Lodz, Poland
gInstitute of Medical Biology, Polish Academy of Sciences, Laboratory of Medicinal Chemistry, 106 Lodowa St., 92-232 Lodz, Poland
hInstitute of Medical Biology, Polish Academy of Sciences, National Library of Chemical Compounds POL-OPENSCREEN, 106 Lodowa St., 92-232 Lodz, Poland
iInstitute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawinskiego 5a, 02-106 Warsaw, Poland
jDepartment of Systems Biology, Institute of Experimental Plant Biology and Biotechnology, University of Warsaw, Miecznikowa 1, 02-096 Warsaw, Poland
First published on 4th November 2022
Abstract
Considering the increasing importance of cyclopropyl-containing peptide-derived compounds in the pharmaceutical industry, herein, we report the crystal engineering of a series of novel derivatives, i.e., diethyl 2-acetamido-2-(cyclopropylmethyl)malonate (1), 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid (2) [Ac-b-cyclopropyl-(R,S)-Ala-OH], 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid hydrate (3), 2-acetamido-3-cyclopropylpropanoic acid (4), and (2S)-2-[cyclopropyl(9H-fluoren-9-ylmethoxycarbonyl)amino]propanoic acid (5) [Fmoc-b-cyclopropyl-(S)-Ala-OH]. Although several cyclopropyl-based peptide-derived structures have been reported in the literature, to the best of our knowledge, studies on the synthon hierarchy in this class of structures are limited. Thus, to address this gap, herein, we report a multidisciplinary study to shed light on the role of cyclopropyl synthons in (bio)supramolecular assemblies, opening a new vista for the further applications of this unique scaffold. The synthesis was achieved via a multi-step protocol in good yield and the structures were determined by single-crystal X-ray diffraction. The diverse supramolecular synthons responsible for the arrangement of molecules in the crystal lattice of either new compounds or all cyclopropyl-containing peptide-derived solids deposited in the CSD thus far were specified and summarized, building a library. The self-assembly is directed by the cooperative interplay of H-bonds and π-stacking interactions. Quantum-chemical computational studies revealed that the cyclopropyl ring is capable of  ,
,  ,
,  and
and  interactions. The molecular docking study delineated the C–H⋯C(cyclopropyl) and C–H(cyclopropyl)⋯π interactions of the cyclopropyl moiety with the essential amino acid residues inside the active pocket of the human androgen receptor, highlighting the vital role of cyclopropyl in the supramolecular landscape of the bio-complex. Indeed, 2 shows a significant docking score with effective binding affinity, and thus is a promising candidate for prostate cancer prevention or management.
interactions. The molecular docking study delineated the C–H⋯C(cyclopropyl) and C–H(cyclopropyl)⋯π interactions of the cyclopropyl moiety with the essential amino acid residues inside the active pocket of the human androgen receptor, highlighting the vital role of cyclopropyl in the supramolecular landscape of the bio-complex. Indeed, 2 shows a significant docking score with effective binding affinity, and thus is a promising candidate for prostate cancer prevention or management.
1. Introduction
Cyclopropyl is a unique motif attracting wide interests in synthetic, supramolecular, and medicinal chemistry, chemical biology, and molecular recognition processes due to its wide bio-applications and specific chemical nature, i.e., coplanarity of carbon atoms, short C–H and C–C bonds, and enhanced π-character.1–8 Interestingly, an extensive list of cyclopropyl-based drugs contains more than a hundred therapeutic agents related to cancers, infectious diseases (viral, bacterial, and fungal), cardiovascular, metabolic, neurological, skin, dental, and eye disorders, depression, and many other conditions.9–27 Remarkably, about 20 peptide-derived compounds with the cyclopropyl moiety have been approved by the Food and Drug Administration (FDA) in the last decade28 (Table S1†). Thus, cyclopropyl exists in ∼10% of all new therapeutics, which together with fluorinated methyl (CF3), is one of the most popular pharmaceutical scaffolds.29,30 However, it should be highlighted that fluorinated drugs are controversial due to the possible tissue-specific fluorination of proteins.31 Alternatively, the small cyclopropyl system possesses appealing features such as increased potency, metabolic stability, brain permeability, reduced off-target effects, and plasma clearance32 as well as enhanced cytotoxicity toward tumor cells.33 Cyclopropyl may contribute to entropically more favorable binding to biological targets due to its conformational rigidity.32 It is worth mentioning that cyclopropyl forms hydrophobic interactions with the protein target,34 which can be amplified by π-based inter-contacts. Also, π-sigma interaction can help intercalate the ligand (drug) in the binding site and enhance the affinity between the ligand and receptor.35 This can be one of the key issues in the explaining the differences in activity related to dynamic behavior.36 Currently, peptide-derived compounds have gained increasing attention as safe and effective next-generation therapeutic agents due to their high affinity and excellent specificity towards protein receptors with recent progress in biotechnology overcoming their shortcomings.37–39An understanding of the molecular topology and modification of the supramolecular framework is essential to design new drugs with the desired bio-pharmacokinetic properties due to their close relationship.40 The behavior of drug molecules in bio-systems depends on the combination of H-bond acceptors and donors (and their molecular shape). Hirshfeld surface (HS) studies provide a valuable source of information on the supramolecular properties of potential bioactive substances. Interestingly, the HS concept is also being used to profile the shape of the protein pocket and generate the HS around the ligand molecule. Consequently, the design of effective ligand molecules inside the protein pocket by using proper H-bonding synthons41 from the model library of interactions may be possible in the future.42 A crystal is a supermolecule,43 and a bio-complex can also be considered a supermolecule. In this context, the cyclopropyl-based synthon data are clearly needed, but to the best of our knowledge cyclopropyl-based supramolecular synthons have received much less attention to date.
The aim of this work was to hierarchize the supramolecular synthons responsible for the molecular arrangement in cyclopropyl-containing peptide-derived crystals. Specifically, a series of novel cyclopropyl-based modified amino acids and closely related compounds, i.e., diethyl 2-acetamido-2-(cyclopropylmethyl)malonate (1), 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid (2) [Ac-b-cyclopropyl-(R,S)-Ala-OH], 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid hydrate (3), 2-acetamido-3-cyclopropylpropanoic acid (4), and (2S)-2-[cyclopropyl(9H-fluoren-9-ylmethoxycarbonyl)amino]propanoic acid (5) [Fmoc-b-cyclopropyl-(S)-Ala-OH], were successfully synthesized, thoroughly investigated by solid-state structural analysis and theoretical calculations, and compared with structures deposited in the CSD, providing a library of H-bonding supramolecular motifs. Notably, we revealed the potential of the cyclopropyl ring to create π-based supramolecular synthons. In addition, we evaluated the bio-pharmaceutical profiles of the new molecules using computational methods, and their binding modes in the active site of human androgen receptor (hAR) were rationalized via molecular docking analysis.
These studies fit into the mainstream of our ongoing project that concentrates on supramolecular exploration and bioactivity of peptide-based molecules.37,38,44–82
2. Experimental
2.1. Synthesis and single-crystal X-ray diffraction (SCXRD)
The synthesis of the novel compounds 1–5 was based on the literature83 and our experience, which is discussed in the ESI† in detail.X-ray diffraction data for compounds 1–5 were collected at 100 K on an XtaLAB Synergy Dualflex Pilatus 300 K diffractometer (Rigaku Corporation, Tokyo, Japan) and processed using the Olex2 software.84 The structures were solved with SHELXT,85 using CrysAlis PRO for data reduction,86 and refined with SHELXL87 using least-squares minimization. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were positioned geometrically and refined with isotropic thermal displacement parameters [Uiso(H) = 1.2Ueq(CH, CH2) and Uiso(H) = 1.5Ueq(CH3)]. The details of the X-ray measurements and crystal data for all the compounds studied are presented in Table 1. The CCDC No. of the title compounds 1–5 are as follows: CCDC 2203890 (1), 2203891 (2), 2203892 (3), 2203893 (4) and 2203894 (5).
| Compound | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Empirical formula | C13H21NO5 | C9H13NO5 | C9H13NO5, H2O | C8H13NO3 | C21H21NO4 |
| Formula weight | 271.31 | 215.20 | 233.22 | 171.19 | 351.39 |
| Crystal system | Orthorhombic | Triclinic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | Pna21 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca | P 121/c1 | I 121 |
| a (Å) | 9.589(2) | 7.49868(19) | 13.72920(10) | 14.66220(10) | 24.5330(2) |
| b (Å) | 12.661(3) | 8.0934(2) | 10.90840(10) | 11.42930(10) | 5.04577(4) |
| c (Å) | 11.779(3) | 9.4854(2) | 14.82920(10) | 21.61680(10) | 30.1674(3) |
| α (°) | 90 | 83.844(2) | 90 | 90 | 90 |
| β (°) | 90 | 73.807(2) | 90 | 90.6010(10) | 109.5159(10) |
| γ (°) | 90 | 69.194(3) | 90 | 90 | 90 |
| Volume (Å3) | 1430.0(6) | 516.76(3) | 2220.87(3) | 3622.32(4) | 3519.81(6) |
| Z | 4 | 2 | 8 | 16 | 8 |
| d calc (mg m−3) | 1.260 | 1.383 | 1.395 | 1.256 | 1.326 |
| Wavelength | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| T (K) | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| No. of unique reflections | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 453 453 |
2043 | 2229 | 7387 | 6882 |
| Absorption correction | None | Multi-scan | None | Gaussian | Gaussian |
| No. of refined parameters | 257 | 140 | 152 | 442 | 518 |
| No. of restraints | 1 | 0 | 0 | 0 | 1 |
| GOF on F2 | 1.01317 | 1.05569 | 1.05542 | 1.04768 | 1.03688 |
| R 1 | 0.0533 | 0.0282 | 0.0294 | 0.0353 | 0.0254 |
| wR 2 | 0.1381 | 0.0719 | 0.0789 | 0.0930 | 0.0655 |
| Min/max residual density | 0.410/−0.442 | 0.360/−0.185 | 0.381/−0.247 | 0.362/−0.294 | 0.243/−0.151 |
Geometrical calculations were performed using the PLATON program,88 while the crystal structure diagrams and analysis of H-bond graph-sets performed using Mercury 4.0.89–91
2.2. Computational details
| βk = γk + ∑αk | (1) |
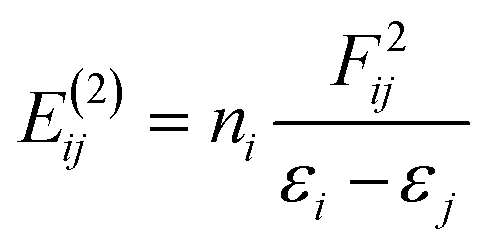 | (2) |
Using the AIM2000 software101 Quantum Theory of Atoms-in-Molecules (QTAIM),102 atomic volumes and charges were calculated by integration over their atomic basins up to the 0.001 a.u. level. The QTAIM bond characteristics in terms of electron density, ρ, and its Laplacian, ∇2ρ, given by eqn (3)
| ∇2ρ = λ1 + λ2 + λ3 | (3) |
| ε = λ1/λ2 − 1 | (4) |
The pKa and logPoct/w measurements114–117 for 5 are described in the ESI.†
2.3. CSD/PDB survey
A search of the CSD (ver. 5.43 updates March 2022)118 was carried out using a cyclopropyl scaffold related to modified amino acids and peptide-related compounds. 225 crystal structures containing cyclopropyl were found, which confirmed the novelty of the synthesized compounds 1–5. Next, we restricted the search in terms of the following filters: 3D coordinates determined, R factor ≤0.05, only non-disordered, no errors, not polymeric, only single crystal structures, only organics, and no repeated entries. Consequently, 33 crystal structures consisting of cyclopropyl were extracted. The CSD refcodes of these entries are as follows: ADELOM, ADELUS, ADEMAZ, ADEMED,119 APOFOD,120 BUTCIE,121 CARYAY,122 CATWEA,123 CEGVUH,124 CERQUM,125 EDIWIZ,126 FIJDIO,127 GENYUU,128 HILXIM,129 HORZEU,130 IHUFEY,131 JAWLAU,132 KEZNEJ,133 KUDZIS,134 LIKFIX,135 MENHIW,136 MOYSEB,137 PEDWOM,138 ROPQUL,139 VEHDIY,140 VETKEL,141 WICMIH,142 WOVXAG,143 XICRAC,144 ZAMJEF, ZAMJOP,145 ZEGGEZ,146 and ZUQBIY.147 The molecular views and basic crystallographic data for the literature ‘hits’ are summarized in Tables S2 and S3,† respectively. It is noteworthily that no structure is a close relative of the new compounds 1–5. Therefore, a detailed discussion of these structures, besides those of cyclopropyl-based synthons, is beyond the scope of this article.Furthermore, in the Brookhaven RCSB Protein Data Bank (PDB database, https://www.rcsb.org/pdb/), 814 structures with cyclopropyl scaffolds have been deposited so far. Interestingly, 59 entries (Table S2†) of them were deposited this year.
3. Results and discussion
3.1. Structural description and supramolecular analysis
The perspective views of the molecular structures of 1–5 with the thermal ellipsoidal plots and the non-hydrogen atom labelling schemes are presented in Fig. S1,† while Scheme S1† shows the superposition of all the structures, revealing the similarities of 1–3, and differences between 4 and 5. The molecule of 4 has R and S chiral centres at C3, C3A, C3B, and C3C, while in 5 has S at C3 and C3A. In 5, the fluorenyl methoxycarbonyl (Fmoc) group and the carbamate linkages [N–C9(O3)–O4–C10/NA–C9A(O3A)–O4A–C10A] are planar. In all the compounds, the amide bonds are in a trans conformation. The structures are stabilized by diverse intra-N–H⋯O, C–H⋯O and intermolecular O–H⋯O, N–H⋯O, C–H⋯O and H–bonding interactions, in the range of 2.5342(1) Å in 3 to 3.5379(1) Å in 4 due to the presence of rich H-bonding donors and acceptors, mainly in terms of amine, carbonyl, and carboxylic groups. H-bonds with a distance of D–A ≤2.55 Å for O–H⋯O (observed in 2–4) and ≤2.65 Å for N–H⋯O (in 1–3) are called short strong H-bonds. Interestingly, all the h-atoms of the NH and OH groups and O atoms of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in all the reported compounds are involved in the H-bonding as donors and acceptors, respectively (only in 2 a weak NH⋯C bond is observed). The geometric parameters of the H-bonding interactions are collected in Table 2, while the π-stacking interactions in 5 are shown in Table S4† (π⋯π) and Table S5† (C–H⋯π). Notably, the H⋯Cg distances are in the range of 2.53–2.83 Å, and according to Malone,148 the C–H⋯π hydrogen bonds are classified as type I.
O groups in all the reported compounds are involved in the H-bonding as donors and acceptors, respectively (only in 2 a weak NH⋯C bond is observed). The geometric parameters of the H-bonding interactions are collected in Table 2, while the π-stacking interactions in 5 are shown in Table S4† (π⋯π) and Table S5† (C–H⋯π). Notably, the H⋯Cg distances are in the range of 2.53–2.83 Å, and according to Malone,148 the C–H⋯π hydrogen bonds are classified as type I.
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | (DHA) |
|---|---|---|---|---|
| 1 | ||||
| N–H⋯O4a | 0.86 | 2.29 | 2.6579(7) | 106 |
| N–H⋯O1i | 0.86 | 2.12 | 2.9565(8) | 162 |
| C2–H2A⋯O1i | 0.96 | 2.39 | 3.2680(8) | 152 |
| C2–H2B⋯O4ii | 1.00 | 2.43 | 3.3481(9) | 151 |
| C4–H4B⋯O5a | 0.99 | 2.57 | 2.9123(7) | 100 |
| C10–H10C⋯O4iii | 0.96 | 2.54 | 3.1685(8) | 123 |
| (i) −1/2 + x, ½ − y, z; (ii) 1 − x, −y, −1/2 + z; (iii) ½ + x, ½ − y, z | ||||
| 2 | ||||
|---|---|---|---|---|
| N–H⋯O2a | 0.86 | 2.23 | 2.6273(1) | 108 |
| N–H⋯O2i | 0.86 | 2.41 | 3.0318(1) | 130 |
| O3–H3⋯O1ii | 0.82 | 1.78 | 2.5893(1) | 170 |
| O5–H5⋯O4iii | 0.82 | 1.84 | 2.6590(1) | 177 |
| C2–H2A⋯O2iv | 0.96 | 2.37 | 3.2729(1) | 156 |
| C5–H5A⋯O2i | 0.98 | 2.57 | 3.4524(1) | 150 |
| (i) 1 + x, y, z (ii) −1 + x, y, z; (iii) −x, 1 − y, 2 − z; (iv) 1 − x, −y, 1 − z | ||||
| 3 | ||||
|---|---|---|---|---|
| N–H⋯O4a | 0.86 | 2.20 | 2.5928(1) | 108 |
| O–HA⋯O4i | 0.85 | 1.89 | 2.7291(1) | 171 |
| O–HB⋯O2ii | 0.85 | 1.92 | 2.7646(1) | 175 |
| O3–H3⋯Oiii | 0.82 | 1.75 | 2.5659(1) | 171 |
| O5–H5⋯O1iv | 0.82 | 1.72 | 2.5342(1) | 175 |
| C2–H2A⋯O2v | 0.96 | 2.50 | 3.3219(1) | 144 |
| C2–H2C⋯O3vi | 0.96 | 2.51 | 3.1663(1) | 126 |
| (i) X, y, z; (ii) 1 − x, −1/2 + y, ½ − z; (iii) 1 − x, −y, 1 − z; (iv) ½ + x, y, ½ − z; (v) x, ½ − y, −1/2 + z; (vi) −1/2 + x, ½ − y, 1 − z | ||||
| 4 | ||||
|---|---|---|---|---|
| N–H⋯O2a | 0.88 | 2.35 | 2.6862(1) | 103 |
| N–H⋯O2Ai | 0.88 | 2.19 | 3.0055(1) | 153 |
| O3A–H3A⋯O1Bii | 0.84 | 1.72 | 2.5521(1) | 170 |
| NA–HA⋯O2i | 0.88 | 2.10 | 2.9542(1) | 164 |
| NA–HA⋯O2Aa | 0.88 | 2.34 | 2.6848(1) | 104 |
| O3–H3⋯O1Ciii | 0.84 | 1.72 | 2.5497(1) | 171 |
| NB–HB⋯O2Ba | 0.88 | 2.38 | 2.6972(1) | 102 |
| NB–HB⋯O2Cii | 0.88 | 2.08 | 2.9408(1) | 165 |
| O3B–H3B⋯O1Aiv | 0.84 | 1.72 | 2.5535(1) | 172 |
| O3C–H3C⋯O1i | 0.84 | 1.69 | 2.5268(1) | 172 |
| NC–HC⋯O2Ca | 0.88 | 2.35 | 2.6913(1) | 103 |
| NC–HC⋯O2Bv | 0.88 | 2.15 | 2.9815(1) | 158 |
| C2–H2B⋯O2Ai | 0.98 | 2.48 | 3.2904(1) | 140 |
| C2–H2C⋯O2Cii | 0.98 | 2.39 | 3.3114(1) | 157 |
| C4–H4A⋯O3a | 0.99 | 2.55 | 2.8941(1) | 100 |
| C2C–H2CA⋯O2v | 0.98 | 2.59 | 3.5379(1) | 163 |
| C2C–H2CB⋯O2Bv | 0.98 | 2.60 | 3.2454(1) | 124 |
| C2B–H2BB⋯O2Cii | 0.98 | 2.48 | 3.2948(1) | 140 |
| C2A–H2AB⋯O2i | 0.98 | 2.51 | 3.3590(1) | 144 |
| (i) x, y, z; (ii) 1 − x, −1/2 + y, ½ − z; (iii) 1 − x, ½ + y, ½ − z; (iv) −1 + x, y, z (v) −x, −1/2 + y, ½ − z | ||||
| 5 | ||||
|---|---|---|---|---|
| a Intramolecular interactions. | ||||
| N–H⋯O3i | 0.88 | 2.05 | 2.7972(1) | 142 |
| NA–HA⋯O3Ai | 0.88 | 2.18 | 2.9466(1) | 146 |
| C4A–H4AA⋯O1Ai | 0.99 | 2.44 | 3.3310(1) | 150 |
| O2–H2⋯O1Aii | 0.94 | 1.72 | 2.6573(1) | 175 |
| O2A–H2A⋯O1iii | 0.92 | 1.75 | 2.6583(1) | 166 |
| C4A–H4AA⋯O1Ai | 0.99 | 2.44 | 3.3297(1) | 150 |
| C3–H3⋯O3a | 1.00 | 2.49 | 2.8753(1) | 102 |
| C21A–H21A⋯O4iv | 0.95 | 2.48 | 3.4100(1) | 165 |
| (i) x, 1 + y, z; (ii) ½ − x, 1/2 + y, ½ − z; (iii) ½ − x, −1/2 + y, ½ − z; (iv) −1/2 + x, −1/2 + y, −1/2 + z | ||||
To gain deeper insight into the supramolecular preferences of the novel structures in the context of all the cyclopropyl-containing peptide-derived compounds known thus far, 33 entries from the CSD, described in the Experimental section and included in Tables S2 and S3,† were re-analyzed and considered. Consequently, with these additional crystal data, we identified the main basic synthons, highlighting the new H-bonding motifs realized for the first time in novel compounds 1–5. However, obtaining appropriate information was a challenge because of the complexity of the CSD structures with diverse functional groups with multiple H-bond donors and acceptors. All the identified types of both non-cyclopropyl- and cyclopropyl-based supramolecular interactions (synthons) are presented in Fig. 1 and S2,† respectively. Interestingly, the molecules show a greater preference for hetero- than homo-synthons. The cyclic motifs are built by carboxylO–H⋯Ocarbonyl/carboxyl, N–H⋯Ocarbonyl and arylC–H⋯Ocarbonyl/carboxyl interactions. Linear H-bonding patterns are created by hydroxyl/carboxylO–H⋯Ocarbonyl, N–H⋯Ocarbonyl, arylC–H⋯Ocarbonyl/carboxyl, arylC–H⋯Ohydroxyl, S–H⋯S–H, and arylC–H⋯S–H. The H-bonding synthon motifs called B3 and B5 are formed by N–H⋯Ocarbonyl and arylC–H⋯Ocarbonyl, respectively, as well as the bifurcated synthon C1 resulting from the most popular in this class of compounds. At this point, more attention can be paid to the occurrence of the bifurcated H-bonding topology in the studied crystals.
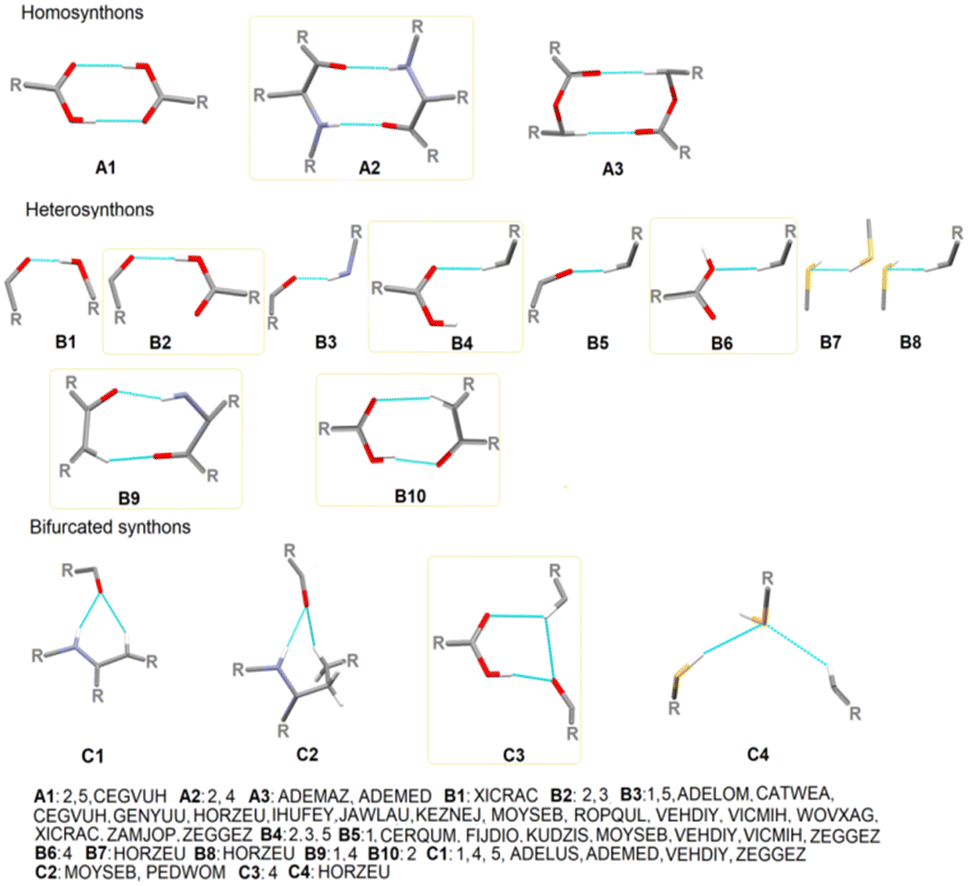 | ||
| Fig. 1 Particular basic supramolecular H-bonding patterns identified in compounds 1–5 and those derived in CSD (new motifs are indicated by yellow frames). | ||
The tendency of the oxygen atoms of the CO and S of SH groups to act as acceptors of two H-bond interactions related to the NH/CH and SH/CH groups, respectively, as well as the hydrogen atom of CH to act as a donor of two H-bond interactions related to the SH/CH groups, is a specific feature of the supramolecular organization of the novel crystals, introducing a stronger cooperativity effect. An additional feature that arises from this work is that a wide variety of H-bonding supramolecular patterns result from the cyclopropyl moiety (Fig. S3†). Specifically, cyclopropylC–H⋯Ocarbonyl, cyclopropylC–H⋯Ohydroxyl, cyclopropylC–H⋯OH2O, cyclopropylC–H⋯OC–O–C, cyclopropylC–H⋯Caryl, arylC–H⋯Ccyclopropyl and cyclopropylC–H⋯Ccyclopropyl, cyclopropylC–H⋯Narom, cyclopropylC–H⋯S, and cyclopropylC–H⋯F, interactions employed in the cyclopropyl-based synthons are specified (Table 3). The last three interactions, together with cyclopropylC–H⋯OC–O–C, are observed only in the literature structures. The synthon denoted as B4cyclopropyl, created by cyclopropylC–H⋯Ocarbonyl and C1cyclopropyl, formed by an additional NH⋯Ocarbonyl, is the most favorable cyclopropyl-based motif.
| Type of interaction | Occurrence | |
|---|---|---|
| cyclopropylC–H⋯Ocarbonyl | 2, 5 | ADELOM, ADELUS, ADEMAZ, ADEMED, BUTCIE, CEGVUH, EDIWIZ, GENYUU, HILXIM, IHUFEY, JAWLAU, ROPQUL, VEHDIY, VETKEL, ZAMJEE, ZAMJOP |
| cyclopropylC–H⋯Ohydroxyl | 2, 3, 5 | CATWEA |
| cyclopropylC–H⋯OH2O | 3 | |
| cyclopropylC–H⋯OC–O–C | ADEMAZ, PEDWOM | |
| cyclopropylC–H⋯Caryl | 1, 4 | |
| arylC–H⋯Ccyclopropyl | 1, 5 | |
| cyclopropylC–H⋯Ccyclopropyl | 2, 4 | |
| cyclopropylC–H⋯Narom | CERQUM, GENYUU, PEDWOM, ZUQBIY | |
| cyclopropylC–H⋯S | CERQUM | |
| cyclopropylC–H⋯F | PEDWOM, WICMIH | |
In summary, the heterosynthon called R12(6), via N–H⋯Ocarbonyl and aryl/cyclopropylC–H⋯Ocarbonyl interactions, designated as C1/C1cyclopropyl, is the most common cyclic motif in all the compounds derived from cyclopropyl-containing peptide-based compounds known thus far. All the identified graph-set notations (up to 20-membered motifs) are collected as a library in Table S6,† while all the cyclopropyl-based H-bonding patterns, at two graph-set theory levels, in 1–5, are shown in Fig. S4–S8,† respectively.
O group acts as a bifurcated acceptor to N–H and C–H donors (Fig. 2). In addition, N–H⋯O1carbonyl together with the arylC2–H2B⋯O4carbonyl interaction generates another simple dimer motif, designated as R22(9), (synthon B9cyclopropyl). The fused heteromeric R12(6) and R22(9) motifs propagate alternately along the crystallographic a-direction, forming a ribbon. Interestingly, R12(6) and R22(9) are arranged in the form of a triple H-bonding motif (the three-point synthon). In the perpendicular plane, the cyclopropyl moieties connect molecules together into larger fused cyclic patterns (Table S6†).
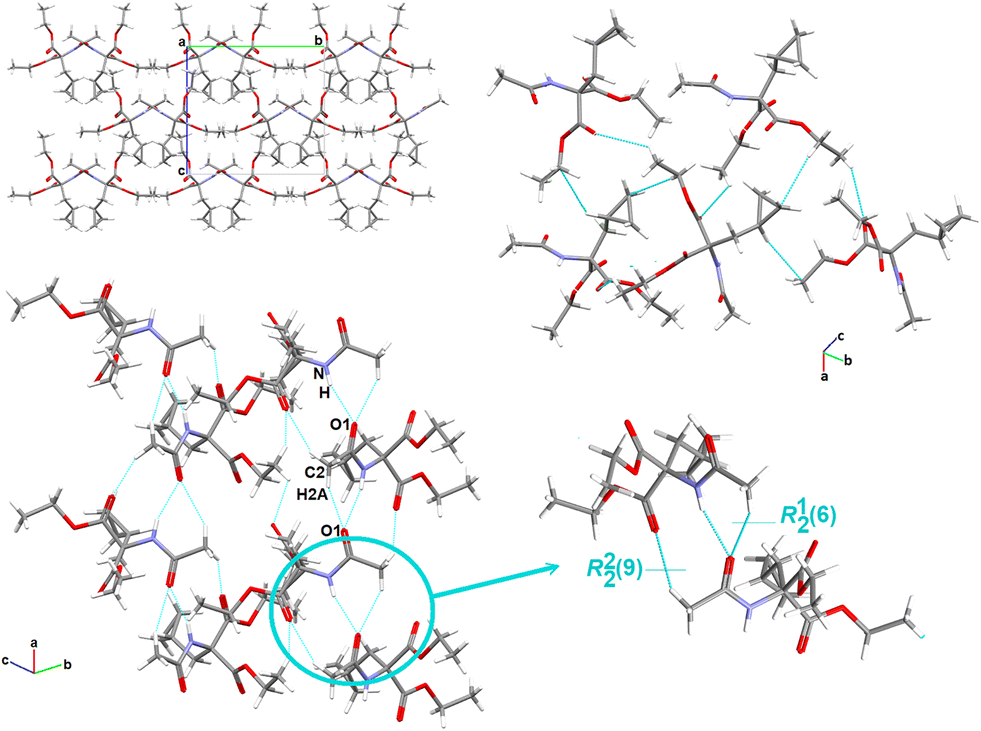 | ||
| Fig. 2 Crystal packing in 1, showing the intermolecular interactions and the supramolecular H-bonding synthons. | ||
Another feature worth considering is the long-range synthon Aufbau module (LSAM), a concept close to the cooperative effect, which is related to a stable packing motif constructed from different synthons,149 and based on Kitaigorodskii's ideas on the Aufbau principle.150 The LSAMs, so-called large synthons, consist of more than one type of inter-contact to summarize all the discovered H-bonding motifs controlling the packing of the molecules. In this context, in 1, the lack of COOH groups in the molecule leads to the activation of peptide N–H⋯O bonds, which join molecules into a 1D structure along the a-direction and symmetry of the pc rod group (blue lines in Fig. 3). The chains are packed almost hexagonally, and the distortions are caused by secondary C–H⋯O interactions, which are marked as thin green lines.
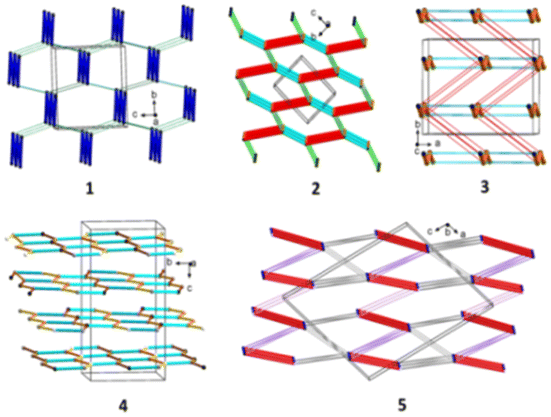 | ||
| Fig. 3 Long-range synthon Aufbau modules, showing the H-bonding interactions in 1–5 (figure was prepared using Diamond v.4.6.6).118 | ||
Crystal packing of compound 2. The crystal of 2 belongs to the centrosymmetric triclinic P
space group. The molecules are arranged in the ‘head-to-tail’ type (Fig. 4). In the crystal lattice, the basic linear motifs, denoted as C(7), formed by hydroxylO3–H3⋯O1carbonyl (synthon B2, see Fig. 1), arylC2–H2A⋯O2carbonyl (synthon B4) and cyclopropylC7–H7A⋯O3hydroxyl (synthon B1cyclopropyl, see Fig. S2†), respectively, at the first level of the graph-set theory, are found. The first two chains are parallel to the a-axis, while the chain generated by the cyclopropyl-based interaction is prolonged along the b-direction. Remarkably, the combination of these H-bonds closes the rings, resulting in a basic cyclic homosynthon, featuring the graph-set notation as R22(8), built by hydroxylO5–H5⋯O4carbonyl H-bonds (synthon A1, Fig. 1), leading to the supramolecular linear chain propagated along the cb-plane. Perpendicularly, the heterosynthonic basic structural unit, represented by the same graph-set notation R22(8), is formed through the hydroxylO3–H3⋯O1carbonyl and arylC2–H2A⋯O2carbonyl interactions (synthon B10, Fig. 1), at the second graph set level of theory, propagated into another supramolecular linear chain extending along the crystallographic a-axis. Consequently, these tapes are interrelated into a 2-D planar sheet in the ac-plane, unveiling primary tetramer synthons, denoted as R44(26) and R34(24), between tapes. In another plane, cyclopropyl participates in the building of secondary H-bonding patterns. Further insight shows that the molecules are linked by 3-centre, bifurcated N–H⋯O and C–H⋯O interactions, leading to the very important R12(7) synthon (C4cyclopropyl, presented in Fig. S2†).
 | ||
| Fig. 4 Crystal packing in 2, showing the intermolecular interactions and the H-bonding supramolecular synthons. H-atoms that are not engaged in the interactions are omitted for clarity. | ||
Considering LSAMs, a supramolecular layer parallel to the crystallographic bc-plane is observed it is consistent with the P space group symmetry, while its topology can be described as a 4-connected simple uninodal network, with a node being the centre of gravity of the molecule (black dot in Fig. 3), and edges in line with the observed H-bonds. Hence, the red lines represent a carboxylic dimer, cyan for the connections between the NH donor and the COOH acceptor, and the orange is for the COOH donor and peptide carbonyl atom. The network may be regarded as square planar with a {4·4} Schläfli symbol if the edges are indistinguishable. The thinner pink and green lines represent secondary C–H⋯O interactions, which lead to a 3D structure.
Crystal packing of compound 3. The crystal structure of 3 was determined in the orthorhombic, centrosymmetric Pbca space group, with one symmetry-independent 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid molecule and one crystallographically unique water molecule in the asymmetric unit. Herein, the adjacent molecules in the crystal lattice establish the wave-like architecture in the ab-plane (Fig. 5). The water molecule, as a bifurcated donor or bifurcated acceptor, acts as a bridge between the two main molecules that form the C22(8) motif viaarylC4–H4A⋯O4carbonyl and cyclopropylC6–H6A⋯O5hydroxyl interactions. Consequently, a supramolecular zigzag chain is created. Moving forward, a similar supramolecular chain, denoted as C22(9), is built by the H2OO–HA⋯O4carbonyl and cyclopropylC6–H6B⋯OH2O interactions, in which the cyclopropyl ring is employed. A perpendicular chain C(7) is formed through the cyclopropylC6–H6A⋯O5carboxyl interaction (synthon B1cyclopropyl, Fig. S2†). By deeper exploration of the crystal lattice, diverse cyclic H-bonding patterns were also found, resulting in a 3D network. After further LSAMs analysis, the pc rod group symmetry describes the symmetry of the supramolecular chain structure, along the c-direction, formed by the H-bonds between the COOH group and the peptide carbonyl oxygen atom (thick orange lines in Fig. 3). The rectangular alignment of the main chains as viewed along the c-axis is more noteworthy rather than approximately hexagonal packing of rods. The presence of crystalline water molecules is responsible for this. In the figure, the water bridges are represented by thin red and cyan lines.
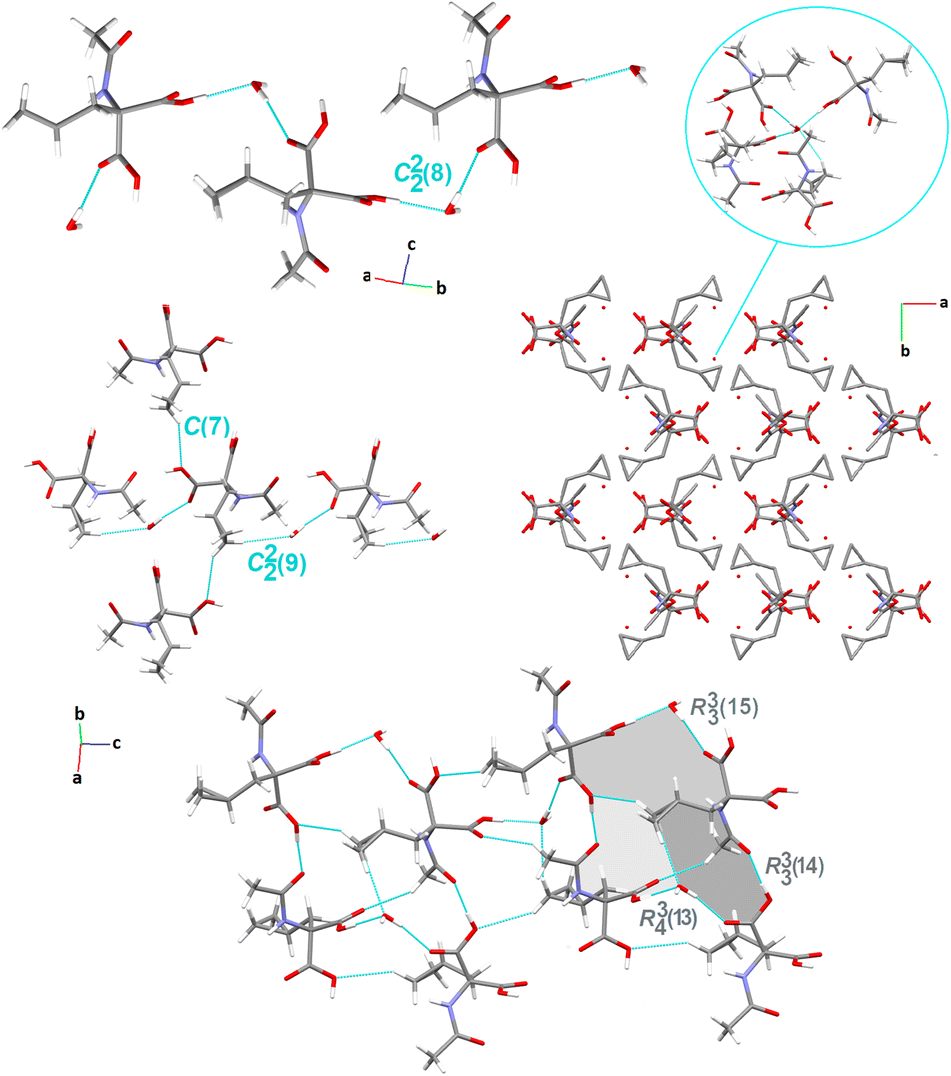 | ||
| Fig. 5 Crystal packing in 3, showing the intermolecular interactions and the H-bonding supramolecular synthons. H-atoms that are not engaged in the interactions are omitted for clarity. | ||
Crystal packing of compound 4. Compound 4 crystallizes in the monoclinic system, centrosymmetric P121/c1 space group, with a surprisingly high number of crystallographically independent racemic Ac-b-cyclopropyl-(R,S)-Ala-OH moieties, Z′ = 16. In this context, 4 provides exclusivity in comparison with the cyclopropyl-containing peptide-based structures deposited in the CSD. Notably, these compounds attract special attention for pharmaceutical applications.151 Among the studied crystals, the supramolecular assembly of 4 (Fig. 6) is constructed and stabilized through the most diverse inter-contacts, i.e., O⋯H/H⋯O, O⋯C/C⋯O, C⋯H/H⋯C, N⋯H/H⋯N, O⋯N/N⋯O, and O⋯O (see section below). Specifically, the arylC2A–H2C⋯O3Ahydroxyl interaction links adjacent molecules into the sinusoidal supramolecular chain, denoted as C(7) (synthon B6, Fig. 1), which is parallel to the a-axis. O–H⋯O, N–H⋯O and C–H⋯O H-bonds participate in the construction of an interesting sheet consisting of fused cyclic two heterosynthons and one homosynthon, with the graph-set notation of R12(6), (synthon C1), R22(6) (synthon C3), and R22(10) (synthon A2). The stacking of the sheets is observed, providing an extra advantage for this structure. Alternatively, these tapes are interconnected by cyclopropyl-based interactions. Within the tapes, the H-bonding interactions are built in the mode of the repeated patterns of fused cyclic supramolecular H-bonding motifs.
 | ||
| Fig. 6 Crystal packing in 4, showing the intermolecular interactions and the H-bonding supramolecular synthons. H-atoms that are not engaged in the interactions are omitted for clarity. | ||
From the LSAMs point of view, pseudo-centrosymmetric R22(10) dimers (light blue and cyan lines in Fig. 3) align alternately in the ab-plane and are further connected via topologically similar O–H⋯O bonds between COOH donors and peptide carbonyls (i.e., each dimer is both a donor and an acceptor). Together, the above-mentioned interactions establish a honeycomb-like layer perpendicular to the c-direction. The development of a layer occurs by translational vectors only. Hence, the layer symmetry is given by the p1 layer group. Assuming that all the vertices and edges are unified, the Schläfli symbol of the 2D supramolecular unit is {6·3·3}. There are four layers per unit cell related by symmetry elements of the P21/c space symmetry group. The secondary weak C–H⋯O interactions are visible between the layers.
Crystal packing of compound 5. Compound 5 crystallizes in a monoclinic system, in the I121 space group, and its asymmetric unit is comprised of two independent Fmoc-b-cyclopropyl-(S)-Ala-OH molecules. They are connected through hydroxylO2A–H2A⋯O1carbonyl H-bonds into a centrosymmetric dimer. Consequently, the cyclic array R22(8), a robust and very popular synthon (synthon A1, Fig. 1), is formatted. This pattern is propagated into columns along the b-direction, linking the molecules into tetramers, which are denoted as R44(38) motifs (Fig. 7). Through a deeper exploration of the crystal structure, the R23(7) trimer, built by O–H⋯O and C–H⋯O H-bonds (synthon C8cyclopropyl, Fig. S2†), unit is identified. This trimer is further fused to the mentioned earlier dimers, connecting tetramers and other cyclic motifs and creating a 2D layer. The parallel layers are connected by cyclopropylC6A–H6AB⋯O1Acarbonyl interactions, creating a chain called C(7) (synthon B2cyclopropyl, Fig. S2†). Interestingly, R23(7) and R22(8) are arranged in the four-point synthon. It should be noted that in contrast to all the other structures considered herein, 5 possesses the most intriguing feature, given that the electron-rich planar Fmoc ring system displays π-stacking behavior. Thus, H-bonding interactions are assisted by C–H⋯π and π⋯π interactions. These inter-contacts complete the final topology and stabilize the supramolecular assembly.
 | ||
| Fig. 7 Crystal packing in 5, showing the intermolecular interactions and the H-bonding supramolecular synthons. H-atoms that are not engaged in the interactions are omitted for clarity. | ||
At the LSAMs level, the earlier described basic synthons, pseudo-centrosymmetric dimers R22(8) (red lines in Fig. 3, and black and white nodes signify two crystallographically independent molecules), are joined into a 1D ladder structure through the peptide N–H⋯O hydrogen bond along the b-direction. The motif has only translational symmetry. The numerous C–H⋯O (pink lines) and C–H⋯π (grey lines) interactions combine these ladders into a 3D structure.
3.2. Hirshfeld surface analysis
To fully elucidate the supramolecular preferences among the new crystals, the Hirshfeld surface (HS) methodology103–107 was applied. The 3D HS maps with different properties (dnorm, de, di, electrostatic potential, shape index, curvedness, and fragment patch) of all the studied crystals are presented in Fig. S9.† The red-white-blue color scheme on the dnorm-mapped HSs is related to the inter-contacts shorter than, equal to, and longer than the sum of the vdW radii, respectively. The existence of bright red spots denotes the dominant close interactions and identifies the strongest H-bonds, such as O⋯H and N⋯H, which are the O-atoms of the carboxy groups, N-atoms of the amino groups, and H-atoms of the hydroxy/amino groups. The light blue smaller spots on the surface signify the weaker C⋯H and H⋯H inter-contacts. In 5, the shape index and the curvedness properties are helpful in the recognition of π-stacking arrangement via the presence of adjacent blue and red triangles (bow-tie pattern) and large green flat areas (brown circle in Fig. 8), respectively. The blue triangles correspond to the convex regions (the atoms of the molecule inside the surface) and the π-hole (the π-stacked molecule above it). This is consistent with the FP, which presents the contribution of the C⋯C interactions (the triangle near de = di ≈ 1.8 Å refers to the characteristic of π–π interactions). In the other compounds, π-stacking contacts are not observed. The circle in pink represents π-stacking from an aromatic proton outside the surface (red bulging area) around the CH2 moiety and the complementary proton inside the surface (blue hollow spot), indicating C–H⋯π interaction. As noted in Table S4,† the shortest face-to-face π⋯π contact was Cg(2)[C11–C12–C17–C18–C23]⋯Cg(3)[C12–C13–C14–C15–C16–C17] with an interaction distance of 3.9579(1) Å, while the T-shaped C–H⋯π was C10A H10B⋯Cg(9)[C11A–C12A–C17A–C18A–C23A] with 2.4488(1) Å (Table S5†). The fragment patch on the HSs characterize the coordination environment of the molecule in the crystal. The coordination number is determined by the curvedness of the HS. The extensive green flat areas of the HS have low values, whereas the sharp areas have high values of curvedness, resulting in the division of the surface into colour patches related to interactions among the nearest-neighbouring molecules. The collection of the color patches specifies the different independent species in the title structure.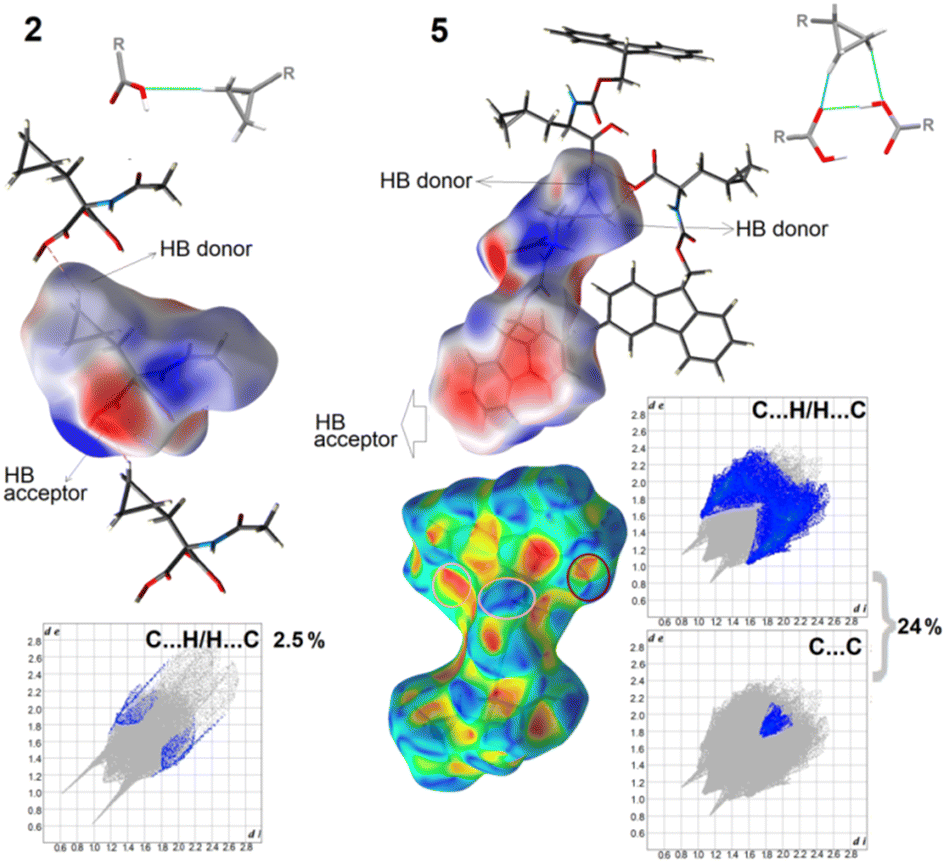 | ||
| Fig. 8 Electrostatic potential maps, showing selected supramolecular synthons, and delineated fingerprint plots into weak interactions in 2 and 5. | ||
The formation of H-bonds in the crystal molecules is elegantly driven by electrostatic interactions. The electrostatic potential (EP), mapped on the HS, reveals negative (red area) and positive (blue region) potentials, indicating the H-bond acceptor and donor sites, respectively. Thus, the EP maps provide direct insight into the sensitivity of a molecule toward chemical reactivity, which can be helpful in the identification of ‘3D pharmacophore points’, visualizing the donor and acceptor sites.42 To compare the total share of strong and weak interactions in the formation of synthons, we present the differences between the most extreme 2 and 5 crystals using fingerprint plots and EP maps (Fig. 8). In the latter, 24% inter-contacts is the sum of contributions of C⋯H/H⋯C (22.5%) and C⋯C (1.5%), while in 1, only 2.5% of all the interactions, derived from the C⋯H/H⋯C, participate in a set of H-bonding donors and acceptors. Nevertheless, in both crystals, weak cyclopropyl-based interactions are employed in the formation of the main synthons that govern the self-assemblies, such as C(7), denoted as B1cyclopropyl synthon, in 2 and R23(7), called C8cyclopropyl, in 5, visible in the EP mapped on the HSs. It should be noted that the electron-rich sites are located in the semi-transparent EP, mapped on the HSs, around the oxygen atoms (and the π-system of the Fmoc group). The electrostatic complementarities of the touching surface patches in the neighbouring molecules for all the studied crystals are presented in Fig. S9.†
Precisely, an analysis of all the fingerprints (FPs), which is useful in the comparison of the supramolecular features of all novel solids in terms of clear quantitative identification of each type of inter-contact, is necessary. The overall FPs are presented in Fig. 9, which are delineated into the corresponding types of interactions, such as H⋯H, O⋯H/H⋯O and C⋯H/H⋯C for compounds 1–3, H⋯H, O⋯H/H⋯O, C⋯H/H⋯C, C⋯O/O⋯C, N⋯H/H⋯N, O⋯N/N⋯O and O⋯O for compound 4 and H⋯H, O⋯H/H⋯O, C⋯H/H⋯C, C⋯O/O⋯C and C⋯C for compound 5 in Fig. S10.† The relative percentage contributions of the various close inter-contacts, both external and internal in the terms of diverse atoms, to the HS area of the compounds are demonstrated in Fig. 9 (Table S7†), where subtle differences are clearly highlighted. In particular, subtle H⋯H (∼50% in 2 and 3, 60% in 5, and 70% in 1 and 4) are the most significant interactions in all the analysed crystals due to the numerous H-atoms on the molecular surface where diverse vdW contacts are formed. Strong O⋯H/H⋯O H-bonds are the other major contributors to the HS, especially in the case of 2 and 3, where they account for ∼50%, due to the presence of additional carboxylic groups in the structure, while in other crystals are at the level of ∼20–25% of all the interactions. Alternatively, in 5, weak C⋯H/H⋯C inter-contacts have a considerable coverage (∼23%) of the total surface, while in the other crystals, their contribution is less than 3%. In contrast, the share of O⋯H/H⋯O H-bonds is the lowest among the crystal structures. Moreover, O⋯C/C⋯O contacts are observed in 4 (3.3%) and 5 (∼1%), while C⋯C only in 5 (1.5%) due to the presence of the fluorenyl group in this structure. Furthermore, N⋯H/H⋯N, O⋯N/N⋯O and O⋯O inter-contacts are present in 4, but the factors are only ∼1% (Table S7†). Thus, in 4, the most complex molecular surface with the greatest diversity of intercontacts is observed, following the order of H⋯H > O⋯H/H⋯O > O⋯C/C⋯O > C⋯H/H⋯C > N⋯H/H⋯N > O⋯N/N⋯O and O⋯O.
 | ||
| Fig. 9 2D fingerprint plots and percentage contributions of inter-contacts to the Hirshfeld surface in structures 1–5 (regions of particular interactions are indicated by the ovals in the FPs). | ||
To better understanding the propensity for the privileged and disfavoured interactions between a pair of chemical species in a crystal structure, the enrichment ratios (ER),152 based on the HS analysis, were determined for all the studied compounds and presented in Table S8.† In this regard, the inter-contacts with high propensity to construct stable supramolecular architecture are as follows: O⋯H/H⋯O and C⋯H/H⋯C in 1, 2, 3, 5; O⋯H/H⋯O, O⋯C/C⋯O, and O⋯N/N⋯O in 4. The thorough inspection revealed that H⋯H contacts are more favoured in 4, 1, and 5 (ER = 1.01, 0.97, and 0.96, respectively) than in others (ER = 0.88 in 2 and 0.89 in 3), which is consistent with the HS calculations. The opposite trend was observed for the O⋯H/H⋯O and C⋯H/H⋯C inter-contacts, where in 2 and 3, the ER is 1.35, while it is 1.25 for O⋯H/H⋯O and 1.12 for C⋯H/H⋯C in 5, 1.17 in 1, 1.02 for O⋯H/H⋯O and only 0.53 for C⋯H/H⋯C in 4. It is also important to note that in 4, O⋯C/C⋯O and O⋯N/N⋯O are greatly enriched with ER = 3.75 and 2.92, respectively, due to the relatively high value of their proportion on the molecular surface. In 5, C⋯C are slightly favoured (ER = 0.87).
Finally, to gain a complete view on the landscape of interactions, their relative strength and topologies in the studied crystals, we systematically compared the energy frameworks (EFs). In this regard, the crystal structures were studied by comparing their pairwise interaction energies. The molecular pairs are uniquely color-coded, as shown in Fig. S11,† while the energies of the inter-contacts are summarized in Table S9.† The 3D topology of the crystal packing is visualized via EFs for 1–5 in Fig. S12† to better understand the supramolecular rearrangement in all the crystal lattice directions. At first glance, some differences are noticeable. Crystal 5 possesses a unique architecture of molecular interaction topology. Specifically, the herringbone energy framework propagates along the a-direction, comprising molecules linked by weak interactions. The width of the cylinders correlates with the relative strength of the energy between the molecules (a greater radius indicates more substantial and prominent interactions). However, the diversity in the dimensions of these cylinders, i.e., pillars, columns, and crossbars, in the supramolecular architecture of the energy frameworks cannot be overlooked. The nature of the specific interactions is denoted by a red–green–blue colour scheme, signifying the electrostatic, dispersive, and total energy, respectively. A significant contribution of the electrostatic energy term, related to the strong classical interactions, is noticeable in 2 (the total electrostatic energy is −252.6 kJ mol−1), while in 1 (the total dispersive energy is −147.3 kJ mol−1), 4 (−93.1 kJ mol−1) and 5 (−322.9 kJ mol−1), where the dispersion terms responsible for weak interactions are favorable in terms of stabilization of the crystal lattice. In 3, the difference between the electrostatic (−148.1 kJ mol−1) and dispersion (−163.1 kJ mol−1) terms is not considerable. Indeed, a delicate balance is observed between the strong and weak interactions, with a slightly dominant dispersion component. In comparison, the polarization energy contributed slightly to the total energy of 1 (−21.2 kJ mol−1) and 4 (−14 kJ mol−1). The data in Table S9† indicate that the total energies follow the order of 5 (−327.3 kcal mol−1) > 2 (−265.6 kcal mol−1) > 3 (−175 kcal mol−1) > 1 (−140.7 kcal mol−1) > 4 (−94.3 kcal mol−1).
3.3. Quantum-chemical calculations: cyclopropyl
 | ||
| Fig. 10 On the left: M06/6-311++G(d,p) optimized structure of 4 dimer (C – black, H – white, O – red, and N – blue). On the right: M06/6-311++G(d,p) optimized structure of the 1 dimer (C – black, H – white, O – red, N – blue). | ||
 | ||
| Fig. 11 On the left: M06/6-311++G(d,p) optimized structure of compound 5 (C – black, H – white, O – red, and N – blue). On the right: molecular graph of compound 5 (C – black, H – grey, O – red, N – blue, bond critical points – small red, and ring critical points – yellow). | ||
| Bond | C3H6 | 1 | 2 | 4 | 5 |
|---|---|---|---|---|---|
| C6–C5 | 1.496 | 1.494 | 1.502 | 1.494 | 1.496 |
| C7–C5 | 1.496 | 1.502 | 1.495 | 1.502 | 1.497 |
| C6–C7 | 1.496 | 1.498 | 1.498 | 1.499 | 1.499 |
| C5–C4 | — | 1.502 | 1.502 | 1.503 | 1.502 |
| C4–C3 | — | 1.536 | 1.536 | 1.535 | 1.527 |
| C6–H6 | 1.085(2×) | 1.086 | 1.085 | 1.084 | 1.084 |
| 1.084 | 1.086 | 1.086 | 1.086 | ||
| C7–H7 | 1.085(2×) | 1.087 | 1.084 | 1.086 | 1.087 |
| 1.085 | 1.086 | 1.085 | 1.084 | ||
| C5–H5 | 1.085(2×) | 1.089 | 1.088 | 1.088 | 1.088 |
| C4–H4 | — | 1.096(2×) | 1.096(2×) | 1.098 | 1.087 |
| 1.096 | 1.084 |
| Bond | Atom | C3H6 | 1 | 2 | 4 | 5 |
|---|---|---|---|---|---|---|
| C6–C5 | C6 | 3.42 | 3.33 | 3.35 | 3.33 | 3.34 |
| C5 | 3.42 | 3.58 | 3.63 | 3.58 | 3.56 | |
| C7–C5 | C7 | 3.42 | 3.34 | 3.34 | 3.33 | 3.36 |
| C5 | 3.42 | 3.65 | 3.59 | 3.62 | 3.64 | |
| C6–C7 | C6 | 3.42 | 3.52 | 3.52 | 3.51 | 3.51 |
| C7 | 3.42 | 3.53 | 3.51 | 3.54 | 3.53 | |
| C5–C4 | C5 | — | 2.20 | 2.21 | 2.21 | 2.24 |
| C4 | — | 2.44 | 2.43 | 2.42 | 2.38 | |
| C4–C3 | C4 | — | 2.76 | 2.78 | 2.77 | 2.72 |
| C3 | — | 2.48 | 2.45 | 2.41 | 2.50 | |
| C6–H6 | C6 | 2.65(2×) | 2.66 | 2.63 | 2.67 | 2.64 |
| 2.64 | 2.66 | 2.64 | 2.66 | |||
| C7–H7 | C7 | 2.65(2×) | 2.66 | 2.64 | 2.66 | 2.65 |
| 2.64 | 2.66 | 2.63 | 2.62 | |||
| C5–H5 | C5 | 2.65(2×) | 2.94 | 2.93 | 2.93 | 2.89 |
| C4–H4 | C4 | — | 3.49 | 3.51 | 3.54 | 3.61 |
| 3.51 | 3.49 | 3.48 | 3.55 |
The σ–π* and π–σ* interactions between the C–C bonds in the cyclopropyl ring and π-type orbitals in the rest of the molecule were found in compound 5 only (Table 6). The stabilization energies related to the carbonyl or aromatic π orbitals and aromatic π* orbitals are relatively low. Interactions were also found between the aromatic π* orbitals and the cyclopropyl σ* orbitals. The QTAIM molecular graphs of the systems under study are presented in Fig. 11 and S18–S21.† The additional molecular paths between the cyclopropyl hydrogens and carboxyl oxygen atoms in compounds 2 and 1 increase the differences in the cyclopropyl group (see below). The QTAIM charges of all the relevant H and C atoms are nearly equal to zero, unlike the NBO treatment (Table S11†), except for the positive C3 atomic charges, which depend on the Ri substituents (Table S15†). The atomic volumes (Table S16†) of the secondary C6 and C7 atoms are significantly higher than that of the tertiary C5 atoms. The aliphatic secondary C4 atoms have even slightly lower volumes. The significantly higher volumes of C6 and C7 atoms with nearly equal charges with C5 atoms reflect the higher chemical reactivity102 of these cyclopropyl carbons. The BCP electron densities (Table S17†) indicate that the C–C bonds in cyclopropyl are comparable with the aliphatic C4–C3 bonds, whereas the C5–C4 bonds are stronger. The BCP Laplacians of the electron density (Table S18†) of the cyclopropyl C–C bonds are significantly less negative than the aliphatic ones. The BCP Laplacians of the electron density of all the C–H bonds are nearly equal and are significantly more negative than that of the C–C bonds. The extra high BCP ellipticities of the cyclopropyl C–C bonds (Table S19†) indicate a very high mechanical strain, which is slightly increased for weaker bonds (comparison shown in Table S17†). The aliphatic C5–C4 and C4–C3 bonds exhibit approximately one-order lower BCP ellipticities, i.e., pure σ bonding with only slight mechanical strain. The C–C-BCP angles in cyclopropyl (Table S20†) are nearly halved compared to the natural hybrid orbital deviations from the C–C lines (Table 4). This can be explained by the curvatures of the ‘banana’ bonds given that the hybrid orbital deviations are evaluated directly at the carbon atoms and the corresponding BCPs are in the middle of these bonds. Consequently, the BCP-C-BCP angles (Table S20†) are lower than the bond bending at the cyclopropyl carbons (Table S14†).
| Type of interaction | Donor orbital population | Stabilization energy [kcal mol−1] |
|---|---|---|
| π(CO)R3 → σ*(C6–C7) |
1.989 | 13.90 |
| π(CO)R2 → σ*(C6–C7) |
1.995 | 0.58 |
| π(C–C)arom-sideR3 → σ*(C6–C7) | 1.662 | 9.00 |
| σ(C7–C5) → π*(C–C)arom-nearR3 | 1.952 | 0.59 |
| σ(C7–C5) → π*(C–C)arom-sideR3 | 1.952 | 3.51 |
| σ(C6–C7) → π*(C–C)arom-distR3 | 1.958 | 1.63 |
| σ(C6–C7) → π*(C–C)arom-nearR3 | 1.958 | 0.56 |
| σ(C6–C7) → π*(C–C)arom-distR3 | 1.958 | 2.60 |
| n π(O)esterR3 – σ*(C7–C5) | 1.820 | 78.25 |
|
n
π(O)COR3 → σ*(C7–C5) |
1.830 | 198.74 |
| n π(O)OHR2 → σ*(C7–C5) | 1.822 | 1292.47 |
| n π(N)R3 → σ*(C7–C5) | 1.770 | 77.38 |
| π*(C–C)arom-distR3 → σ*(C6–C7) | 0.370 | 22.19 |
| π*(C–C)arom-sideR3 → σ*(C6–C7) | 0.343 | 1.63 |
3.4. Biological screening
D is a distribution coefficient widely used to measure the lipophilicity of ionizable compounds, where the partition is a function of the pH. For non-ionizable compounds, logP = logD in the full pH range, whereas for ionizable compounds, logD takes into account the partition of both ionized and non-ionized forms. LogD is convenient for practical measurements, given that it takes into account solution pH, which is important for the analysis of the drug candidate properties in various biologic media with different pH values. In this context and at this juncture, we describe the results of the detailed experimental study of 5, the most controversial based on the in silico parameters among novel compounds. In particular, the logPoct/w calculated using the SwissADME software varies in the range of 2.76–5.99 with an average value of 3.84 (Fig. S25†). In this computational approach, the lipophilicity was calculated for the neutral form of 5 only, while the methodology used does not consider the fact that compound 5 is an ionizable molecule and the parameter defining the lipophilicity, more precisely, id logD. The experimental pKa for 5 is 3.60 and logP measured as a function of pH is 3.47 at pH 1 (stomach pH) when the compound is not ionized, whereas at higher pH such as 7.4 (blood pH), the lipophilicity is much lower and amounts to logD = −0.33. This may obviously affect the bioavailability of the compound, depending on the route of administration. Therefore, in future considerations regarding the lipophilicity and bioavailability of compounds 1–5 in the context of a possible route of administration, the instability of the cyclopropyl ring at very low pH should also be considered.
Furthermore, we checked via the SwissTargetPrediction program,155 that compounds 2–4 are potentially active toward protease inhibitor, enzyme, nuclear receptor, kinase, and membrane receptor, compound 1 shows activity toward protease, enzyme, family A G protein-coupled receptor, nuclear receptor, isomerase, kinase, writer and voltage-gated ion channel, while 5 shows activity toward protease, enzyme, family A G protein-coupled receptor, cytochrome P450, phosphatase and oxidoreductase (Fig. S26†). The bioactivity scores, ‘a measure of a molecule's ability to interact with receptors’, were predicted using the Molinspiration software.156 In addition, the prediction of tumor (and non-tumor) cell line cytotoxicity was calculated using the CLC-pred website tool, which is based on the relationship between structure-cell line cytotoxicity via the PASS procedure.113 Compound 2 (and 3) exhibited cytotoxicity against prostate carcinoma tumor cell lines, with a Pa value of greater than 0.5, indicating a high probability of action. This is a promising finding in the face of the increasing heterogeneity of prostate cancer and the constant need for new-generation safe and effective therapeutic agents and on-going research in this field. Notably, this fatal disease (the second-leading cause of cancer death for men) remains incurable despite the many recent advances in therapies.157
 | ||
| Fig. 12 In silico study of 2 binding to the human androgen receptor (hAR, PDB code: 1E3G). (A and B) Interactions between 2 and hAR in the substrate-binding pocket. (C and D) Surface model of the docking site showing 2 fits the pocket of hAR. The most significant amino acid residues contributing to the stabilization of the ligand molecules are shown in the stick (A) or line (B) representations, respectively. (B) Electron density map of ligand 2 corresponds to the Fo − Fc and is shown in the meshing representation. The carbon atoms are colored gray, the nitrogen atoms with blue color, the oxygen atoms red, and the polar hydrogen atoms white. The formation of potential intermolecular hydrogen bonds is represented by dashed magenta lines. The mutual distances between the amino acid residues and the respective ligand atoms are given in Å (see C and D). | ||
Overall, the results of the in silico studies showed that 2 is a promising candidate for further evaluation for prostate cancer prevention or management.
4. Conclusions
Herein, we reported the synthesis of novel cyclopropyl-containing compounds, namely, diethyl 2-acetamido-2-(cyclopropylmethyl)malonate (1), 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid (2) [Ac-b-cyclopropyl-(R,S)-Ala-OH], 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid hydrate (3), 2-acetamido-3-cyclopropylpropanoic acid (4), and (2S)-2-[cyclopropyl(9H-fluoren-9-ylmethoxycarbonyl)amino]propanoic acid (5) [Fmoc-b-cyclopropyl-(S)-Ala-OH]. Based on the in-depth supramolecular analysis, the influence of cyclopropyl in the construction of impressive structural landscapes at different levels of supramolecular architecture was unveiled. New H-bonding cyclopropyl synthons were identified and systematized in terms of all the cyclopropyl-containing peptide-derived compounds known thus far, building a library, which can be used for planning future studies on more effective, next-generation drugs of diverse categories due to the versatility of the analysed synthon functionalities. The Hirshfeld surface analysis revealed the intricacies of the H⋯H, H⋯O/H⋯O, and C⋯H/H⋯C interactions in 1–5, H⋯N/N⋯H, O⋯N/N⋯O, and O⋯O in 4, O⋯C/C⋯O in 4 and C⋯C related to π⋯π stacking forces and C–H⋯π inter-contact that steer the self-assembly of 5, and provided a deeper understanding of how cyclopropyl stabilizes the crystal packing. The relative contributions of the inter-contacts and enrichment ratios establish the cyclopropyl synthon as an important contributor, acting both as an H-bonding donor and acceptor, mainly via C–H⋯O interactions. Although weak cyclopropyl-based interactions play a secondary role, they introduce a stronger cooperativity effect. Electrostatic potential maps helped us to correlate the weak interactions with the electrostatic complementarity between them. The energy frameworks demarcated the dominant contribution of the electrostatic energy term (related to the strong classical interactions) in 2, while that in 1, 4, and 5 was the dispersion terms (weak interactions). In 3, the difference between both terms is not considerable. NBO and QTAIM treatments explained the relation between the p electron contributions in the spn hybrid orbitals in the carbon atoms and the mechanical strain in the corresponding bonds. The σ/σ* natural C–C bonds in the cyclopropyl rings require the presence of an aromatic substituent, although very distant, to allow their low-energy stabilizing interaction with π*/π bonds not only within the aromatic rings but also at the CO groups outside the aromatic part. Extra-high stabilizing energies correspond to the interaction of non-bonding π-type lone pairs at the O or N atoms with σ* natural C–C bonds in the cyclopropyl rings. In addition, the novel compounds have attractive in silico pharmacokinetic parameters. The molecular docking study revealed that cyclopropyl is engaged in the stabilization of the bio-complex via C–H⋯Ccyclopropyl and C–Hcyclopropyl⋯π interactions with the key amino acid residues inside the active pocket of the human androgen receptor. Cyclopropyl acts as a ‘hydrophobic anchor’, which allows the ligand molecule to be accommodated in the binding pocket of the enzyme. Notably, compound 2 (and 3) shows a significant docking score with effective binding affinity, and thus is a promising candidate for prostate cancer prevention or management.
Finally, we demonstrated that cyclopropyl plays an essential role in the (bio)supramolecular architecture. We hope that the findings reported herein will stimulate further investigations on new types of cyclopropyl synthons and will be useful for researchers working not only in the field of crystal engineering and supramolecular chemistry but also in peptide(ligand)–protein(target) interactions.
Author contributions
J. B.: conceptualization, investigation & writing – review & editing original draft; formal structural & supramolecular analysis; bioactivity & ADMET; CSD/PDB survey; interaction energy calculations, energy frameworks, visualization, enrichment ratio, full interaction maps, project administration, supervision, corrections; P. B.: molecular docking study & writing, software; M. B. & M. R.: quantum-chemical calculations & writing, software, corrections; A. F.: X-ray experiments, data curation; I. D. M.: LSAMs analysis; K. K.: synthesis & writing; Z. L. & A. K.: pharmacokinetic study & writing, software; P. Z. & W. M. W.: supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Slovak Scientific Grant Agency VEGA (contract no. 1/0139/20) and by the Slovak Science and Technology Assistance Agency (contract no. APVV-20-0213). We thank the HPC center at the Slovak University of Technology in Bratislava, which is a part of the Slovak Infrastructure of High Performance Computing (SIVVP Project No. 26230120002, funded by the European Region Development Funds), for computing facilities. The pKa and logPoct/w measurements for 5 were performed within the project POL-OPENSCREEN, financed by the Ministry of Science and Higher Education (Poland), decision no. DIR/WK/2018/06 of 24.10.2018.
This work is a part of project developed in the research laboratory of Professor Piotr Zielenkiewicz.
References
- K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312–322 CrossRef.
- E. Kochanski and J. M. Lehn, Theor. Chim. Acta, 1969, 14, 281–304 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- C. A. Coulson and T. H. Googwin, J. Chem. Soc., 1963, 3161 CAS.
- A. Galano, J. R. Alvarez-Idaboy and A. Vivier-Bunge, Theor. Chem. Acc., 2007, 118, 597–606 Search PubMed.
- M. E. Jason and J. A. Ibers, J. Am. Chem. Soc., 1977, 99, 6012–6021 CrossRef CAS.
- T. S. Cameron, A. Linden and K. Jochem, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 2110–2115 CrossRef.
- A. de Meijere, Angew. Chem., Int. Ed. Engl., 1979, 18, 809–826 CrossRef.
- M. R. Sun, H. L. Li, M. Y. Ba, W. Cheng, H. L. Zhu and Y. T. Duan, Mini-Rev. Med. Chem., 2021, 21(2), 150–170 CrossRef CAS PubMed.
- Z. Casar, Synthesis, 2020, 52, 1315–1345 CrossRef CAS.
- M. Yoshida, M. Ezaki, M. Hashimoto, M. Yamashita, N. Shigematsu, M. Okuhara, M. Kohsaka and K. Horikoshi, J. Antibiot., 1990, 43(7), 748–754 CrossRef CAS PubMed.
- M. Ansseau, Inpharma Wkly., 1989, 698, 4–5 Search PubMed.
- M. T. Smith, L. E. J. Evans, M. J. Eadie and J. H. Tyrer, Eur. J. Clin. Pharmacol., 1979, 16, 141–147 CrossRef CAS PubMed.
- D. Chessman, L. Kostenko and T. Lethborg, et al. , Immunity, 2008, 28, 822–832 CrossRef CAS PubMed.
- A. J. Rapkin and S. A. Winer, Expert Opin. Pharmacother., 2007, 8, 989–999 CrossRef CAS PubMed.
- P. Kawalec, J. Kryst, A. Mikrut and A. Pilc, PLoS One, 2013, 8, e76587–e76598 CrossRef CAS PubMed.
- C. D. Bernstein, K. L. Albrecht and D. A. Marcus, Expert Opin. Pharmacother., 2013, 14, 905–916 CrossRef CAS PubMed.
- O. Burkhardt and T. Welte, Expert Rev. Anti-infect. Ther., 2009, 645–668 CrossRef CAS PubMed.
- S. Amlani, T. Nadarajah and R. A. McIvor, Expert Opin. Pharmacother., 2011, 12, 2119–2128 CrossRef CAS PubMed.
- M. M. P. Bastos, C. C. Costa, T. C. Bezerra, F. C. Da Silva and N. Boechat, Eur. J. Med. Chem., 2016, 108, 455–465 CrossRef CAS PubMed.
- A. Corsini and R. Ceska, Curr. Med. Res. Opin., 2011, 27, 1551–1562 CrossRef CAS PubMed.
- S. Ahmad and R. F. Storey, Curr. Pharm. Des., 2012, 18, 5240–5260 CrossRef CAS PubMed.
- F. S. Mah and C. M. Sanfilippo, Ophthalmol. Ther., 2016, 5, 1–20 CrossRef PubMed.
- S. Dhillon, Drugs, 2015, 75, 1783–1796 CrossRef CAS PubMed.
- Z. T. Al-Salama, G. M. Keating and S. J. Keam, Drugs, 2017, 77, 2025–2036 CrossRef CAS PubMed.
- K. P. Garnock-Jones, Drugs, 2012, 72, 2431–2456 CrossRef CAS PubMed.
- C. M. Perry, Drugs, 2012, 72, 619–641 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2019, 18, 85–89 CrossRef PubMed.
- N. A. J. Meanwell, Med. Chem., 2018, 61, 582 Search PubMed.
- Y. Zhou, J. Wang, J. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- A. Gawor, Z. Gajewski, L. Paczek, B. Czarkowska-Paczek, A. Konopka, G. Wryk and E. Bulska, Int. J. Mol. Sci., 2022, 23, 4202–4213 CrossRef CAS PubMed.
- T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed.
- W. Wang, H. Lu, M. Zhang, H. Ma, X. Cheng, Y. Ding and A. Hu, J. Mater. Chem. B, 2021, 9, 4502–4509 RSC.
- H. Lu, Q. Zhou, J. He, Z. Jiang, C. Peng, R. Tong and J. Shi, Signal Transduct. Target. Ther., 2020, 5, 213–236 CrossRef PubMed.
- D. E. Arthur and J. Uzairu, J. King Saud Univ., Sci., 2019, 31, 1151–1166 CrossRef.
- Y. Lee, S. Kim, J. Y. Kim, M. Arooj and S. Kim, et al. , PLoS One, 2014, 9(1), e85827 CrossRef PubMed.
- V. Apostolopoulos, J. Bojarska, T. T. Chai, S. Elnagdy, K. Kaczmarek and J. Matsoukas, et al. , Molecules, 2021, 26, 430–475 CrossRef CAS PubMed.
- V. Apostolopoulos, J. Bojarska, J. Feehan, J. Matsoukas and W. M. Wolf, Front. Pharmacol., 2022, 13, 914467–914475 CrossRef CAS PubMed.
- C. H. Stammer, WO1985000809A1, 1985.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd., UK, 2nd edn, 2009 Search PubMed.
- G. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- P. R. Spackman, L. J. Yu, C. J. Morton, M. W. Parker, C. S. Bond and M. A. Spackman, et al. , Angew. Chem., 2019, 131, 16936–16940 CrossRef.
- J. D. Dunitz, Pure Appl. Chem., 1991, 63, 177–185 CrossRef CAS.
- J. Bojarska, M. Breza, M. Remko, M. Czyz, A. Gajos-Michniewicz, M. Zimecki, K. Kaczmarek, I. D. Madura, J. M. Wojciechowski and W. M. Wolf, Int. J. Mol. Sci., 2022, 23, 7173–7205 CrossRef CAS PubMed.
- V. P. Chavda, J. Ajabiya, D. Teli, J. Bojarska and V. Apostolopoulos, Molecules, 2022, 27, 4315–4325 CrossRef CAS PubMed.
- V. Apostolopoulos, J. Bojarska, T. T. Chai, J. Feehan, K. Kaczmarek, J. M. Matsoukas, O. Paredes Lopez, M. Saviano, M. Skwarczynski and J. Smith-Carpenter, et al. , Molecules, 2022, 27, 3635–3639 CrossRef CAS PubMed.
- J. Bojarska, A. Mieczkowski, Z. M. Ziora, M. Skwarczynski, I. Toth, A. O. Shalash, K. Parang, S. A. El-Mowafi, E. H. M. Mohammed and S. Elnagdy, et al. , Biomolecules, 2021, 11, 1515–1579 CrossRef CAS PubMed.
- J. Bojarska and W. M. Wolf, Proceedings, 2021, 79(1), 10–18 Search PubMed.
- J. Bojarska, V. Apostolopouos, J. Matsoukas, J. Feehan, H. Ridgway, P. Zielenkiewicz and W. M. Wolf, Acta Crystallogr., Sect. A: Found. Adv., 2021, 77, c794–c795 Search PubMed.
- J. Bojarska, Acta Crystallogr., Sect. A: Found. Adv., 2021, 77, c1070 Search PubMed.
- J. Bojarska, Acta Crystallogr., Sect. A: Found. Adv., 2021, 77, c1210 Search PubMed.
- J. Bojarska, R. New, P. Borowiecki, M. Remko, M. Breza, I. D. Madura, A. Fruzinski, A. Pietrzak and W. M. Wolf, Front. Chem., 2021, 9, 679776–679800 CrossRef CAS PubMed.
- J. Bojarska, M. Remko, M. Breza, I. D. Madura, A. Fruzinski and W. M. Wolf, Pharmaceuticals, 2020, 13, 338–380 CrossRef CAS PubMed.
- J. Bojarska, Int. J. Nutr. Sci., 2021, 6(1), 104–108 Search PubMed.
- J. Bojarska, Int. J. Nutr. Sci., 2020, 5(1), 1039–1044 Search PubMed.
- J. Bojarska, M. Remko, I. D. Madura, K. Kaczmarek, J. Zabrocki and W. M. Wolf, Acta Crystallogr., Sect. C: Struct. Chem., 2020, 76, 328–345 CrossRef CAS PubMed.
- J. Bojarska, M. Remko, M. Breza, I. D. Madura, K. Kaczmarek and J. Zabrocki, et al. , Molecules, 2020, 25, 1135–1162 CrossRef CAS PubMed.
- J. Bojarska, M. Remko, I. D. Madura, J. M. Wojciechowski, A. Olczak and K. Kaczmarek, et al. , J. Mol. Struct., 2019, 1190, 11–22 CrossRef CAS.
- J. Bojarska, K. Kaczmarek, J. Zabrocki and W. M. Wolf, Int. J. Nutr. Sci., 2019, 4, 1035–1037 Search PubMed.
- J. Bojarska, A. Fruzinski, L. Sieron and W. Maniukiewicz, J. Mol. Struct., 2019, 1179, 411–420 CrossRef CAS.
- J. Bojarska, K. Kaczmarek, J. Zabrocki and W. M. Wolf, Novel Approaches in Drug Designing and Development, 2019, 129, 1–27 Search PubMed.
- J. Bojarska, J. Zabrocki, K. Kaczmarek, M. Remko and W. M. Wolf, Acta Crystallogr., Sect. A: Found. Adv., 2019, 75, e588 Search PubMed.
- J. Bojarska, K. Kaczmarek, J. Zabrocki and W. M. Wolf, Supramolecular Chemistry of Modified Amino Acids and Short Peptides, in Advances in Organic Synthesis, ed. A. Rahman, Bentham Science Publishers Ltd., Sharjah, UAE, 2018, vol. 11, pp. 43–107 Search PubMed.
- J. Bojarska, M. Remko, A. Fruzinski and W. Maniukiewicz, J. Mol. Struct., 2018, 1154, 290–300 CrossRef CAS.
- J. Bojarska, A. Fruzinski and W. Maniukiewicz, J. Mol. Struct., 2016, 1116, 22–29 CrossRef CAS.
- J. Bojarska and W. Maniukiewicz, J. Mol. Struct., 2015, 1099, 419–426 CrossRef CAS.
- J. Bojarska, W. Maniukiewicz, A. Fruzinski, L. Sieron and M. Remko, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 199–203 CrossRef CAS PubMed.
- J. Bojarska, W. Maniukiewicz, A. Fruzinski, M. Jedrzejczyk, J. Wojciechowski and H. Krawczyk, J. Mol. Struct., 2014, 1076, 126–135 CrossRef CAS.
- J. Bojarska, W. Maniukiewicz, M. Glówka, L. Sierón and M. Remko, J. Chil. Chem. Soc., 2013, 58, 1530–1533 CrossRef CAS.
- J. Bojarska, W. Maniukiewicz, L. Sieron and M. Remko, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 630–633 CrossRef CAS PubMed.
- J. Bojarska, W. Maniukiewicz and L. Sieron, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 781–786 CrossRef CAS PubMed.
- J. Bojarska, W. Maniukiewicz, L. Sieron, A. Fruzinski, P. Kopczacki and K. Walczynski, et al. , Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, o341–o343 CrossRef CAS PubMed.
- J. Bojarska, W. Maniukiewicz, L. Sieron, P. Kopczacki, K. Walczynski and M. Remko, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, o443–o446 CrossRef CAS PubMed.
- A. Olczak, M. Główka, M. Szczesio, J. Bojarska, W. L. Duax and B. M. Burkhart, et al. , Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 319–327 CrossRef CAS PubMed.
- A. Olczak, M. L. Główka, M. Szczesio, J. Bojarska, Z. Wawrzak and W. L. Duax, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 874–880 CrossRef CAS PubMed.
- M. Główka, A. Olczak, J. Bojarska, M. Szczesio, W. L. Duax and B. M. Burkhart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 433–441 CrossRef PubMed.
- M. Główka, A. Olczak, J. Bojarska, M. Szczesio, W. L. Duax and B. Burkhart, et al. , Acta Crystallogr., Sect. A: Found. Crystallogr., 2004, 60, 165 Search PubMed.
- M. Główka, A. Olczak, J. Bojarska and M. Szczesio, Pol. J. Chem., 2007, 61, 161–187 Search PubMed.
- J. Bojarska, L. Sieron and W. Maniukiewicz, Struct. Chem., 2018, 29, 1525–1531 CrossRef.
- M. Remko, J. Bojarska, A. Remková and W. Maniukiewicz, Comput. Theor. Chem., 2015, 1062, 50–55 CrossRef CAS.
- M. Remko, J. Bojarska, P. JEzko, W. Maniukiewicz and A. Olczak, J. Mol. Struct., 2013, 1036, 292–297 CrossRef CAS.
- M. Remko, J. Bojarska, P. Jezko, L. Sieron, A. Olczak and W. Maniukiewicz, J. Mol. Struct., 2011, 997, 103–109 CrossRef CAS.
- C. Hamon and B. J. Rawlings, Synth. Commun., 1996, 26(6), 1109–1115 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- CrysAlisPRO, Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe and E. Pidcock, et al. , J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- C. F. MacRae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Version 9.0, Gaussian Inc., Wallingford, CT, 2011 Search PubMed.
- R. G. Parr and W. Wang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1994 Search PubMed.
- R. Neumann, R. H. Nobes and N. C. Handy, Mol. Phys., 1996, 87, 1–36 CrossRef CAS.
- F. M. Bickelhaupt and E. J. Baerends, in Rev. Comput. Chem., ed. K. B. Lipkowitz and D. B. Boyd, Wiley-VCH, New York, 2000, vol. 15, pp. 1–86 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- P. P. Graczyk and M. Mikołajczyk, Topics in Stereochemistry, ed. E. L. Eliel and S. H. Wilen, Wiley, New York, 1994, vol. 21, pp. 159–349 Search PubMed.
- H. Krawczyk, K. Wasek, J. Kedzia, J. Wojciechowski and W. M. Wolf, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o24–o26 CrossRef CAS PubMed.
- F. Biegler-Konig, J. Schonbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
- R. F. W. Bader, Atoms in molecules—a quantum theory, Oxford University Press, Oxford, 1990 Search PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, J. Chem. Theory Comput., 2018, 14, 1614–1623 CrossRef CAS PubMed.
- C. F. MacKenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4(5), 575–587 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- D. Jayatilaka and D. J. Grimwood, Lect. Notes Comput. Sci. (including Subser. Lect. Notes Artif. Intell. Lect. NotesBioinformatics), 2003, vol. 2660, pp. 142–151 Search PubMed.
- A. Daina and V. A. Zoete, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717–42730 CrossRef PubMed.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- R. C. Braga, V. M. Alves, M. F. B. Silva, E. Muratov, D. Fourches, L. M. Liao, A. Tropsha and C. H. Andrade, Mol. Inf., 2015, 34, 698–701 CrossRef CAS PubMed.
- A. A. Lagunin, V. I. Dubovskaja, A. V. Rudik and P. V. Pogodin, et al. , PLoS One, 2018, 13, e0191838–e0191852 CrossRef PubMed.
- A. Avdeef, Anal. Chim. Acta, 1983, 148, 237–244 CrossRef CAS.
- A. Avdeef and J. Comer, J. Pharm. Sci., 1993, 82, 183–190 CrossRef CAS PubMed.
- K. Y. Tam and K. Takacs-Novak, Anal. Chim. Acta, 2001, 434, 157–167 CrossRef CAS.
- A. Avdeef, J. Comer and S. J. Thomson, Anal. Chem., 1993, 65, 42–49 CrossRef CAS.
- H. Putz and K. Brandenburg, Diamond, Crystal and Molecular Structure Visualization, Crystal Impact GbR, Kreuzherrenstr. 102, D-53227 Bonn, Germany Search PubMed.
- A. Osipova, D. S. Yufit and A. de Meijere, Synthesis, 2007, 2007, 131–139 CrossRef.
- W. Huang and F. Meng, Angew. Chem., 2021, 60, 2694–2698 CrossRef CAS PubMed.
- G. Kang, J. Kim, E. Kwon and T. H. Kim, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, o631–o632 CrossRef CAS PubMed.
- L. Qin, L. Ren, S. Wan, G. Liu, X. Luo, Z. Liu, F. Li, Y. Yu, J. Liu and Y. Wei, J. Med. Chem., 2017, 60, 3606–3617 CrossRef CAS PubMed.
- F. Brackman and A. de Meijere, Synthesis, 2005, 12, 2008–2014 Search PubMed.
- Z. Q. Sun, Z. Y. Ding and Z. Y. Shao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o3029–o3030 CrossRef CAS PubMed.
- B. Liu and L. Tian, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o4280–o4281 CrossRef CAS.
- N. Rowles, H. G. Heller, D. S. Hughes and M. B. Hursthouse, CSD Communication (Private Communication), 2012 Search PubMed.
- S. M. Wales, E. G. Merisor, H. V. Adcock, C. A. Pearce, I. R. Strutt, W. Lewis, D. Hamza and C. J. Moody, Chem. – Eur. J., 2018, 24, 8233–8239 CrossRef CAS PubMed.
- L. Tian and B. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o4183–o4184 CrossRef CAS.
- H. Y. Bai, X. Fu, J. L. Pan, H. Q. Ma, Z. M. Chen, T. M. Ding and S. Y. Zhang, Adv. Synth. Catal., 2018, 360, 4205–4214 CrossRef CAS.
- R. Hartung, G. Golz, S. Schlaf, G. Silvenoinen, K. Polborn, P. Mayer and H. R. Pfaendler, Synthesis, 2009, 2009, 495–501 CrossRef.
- P. Sivaprakasam, X. Han, R. L. Civiello, S. Jacutin-Porte, K. Kish and M. Pokross, et al. , Bioorg. Med. Chem. Lett., 2015, 25, 1856–1863 CrossRef CAS PubMed.
- P. G. Jones, A. J. Kirby and R. J. Lewis, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 78–81 CrossRef.
- D. V. B. Walji, J. M. D. Storey and W. T. A. Harrison, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1761–o1762 CrossRef CAS.
- G. Bradley, T. J. Ward, J. C. White, J. Coleman, A. Taylor and K. F. Rhodes, J. Med. Chem., 1992, 35, 1515–1520 CrossRef CAS PubMed.
- M. H. Qi, Q. D. Zhang, Y. Liu, F. Z. Ren and G. B. Ren, J. Mol. Struct., 2019, 1178, 242–250 CrossRef CAS.
- H. G. Heller, D. S. Hughes, M. B. Hursthouse and N. G. Rowles, Chem. Commun., 2000, 1397–1398 RSC.
- J. Chen, B. Q. Huang, Z. Q. Wang, X. J. Zhang and M. Yan, Org. Lett., 2019, 21, 9742–9746 CrossRef CAS PubMed.
- M. Viertelhaus, H. C. Holst, J. Volz and R. P. Hummel, J. Mol. Struct., 2013, 1031, 254–262 CrossRef CAS.
- C. Sen, T. Sahoo, H. Singh, E. Suresh and S. C. Ghosh, J. Org. Chem., 2019, 84, 9869–9896 CrossRef CAS PubMed.
- M. W. D. Perry, A. Eriksson and M. R. Landergren, Tetrahedron: Asymmetry, 2017, 28, 1135–1138 CrossRef CAS.
- L. Tian, B. Liu and J. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o273–o274 CrossRef CAS.
- Z. Liu, F. Huang, P. Wu, Q. Wang and Z. Yu, J. Org. Chem., 2018, 83, 5731–5750 CrossRef CAS PubMed.
- A. Guelzim, E. Belloli, S. Yous and C. Vaccher, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 100–101 CrossRef CAS PubMed.
- M. C. Hiller, J. P. Davidson and S. F. Martin, J. Org. Chem., 2011, 66, 1657–1671 CrossRef PubMed.
- R. W. Driver, T. D. W. Claridge, S. Scheiner and M. D. Smith, Chem. – Eur. J., 2016, 22, 16513–16521 CrossRef CAS PubMed.
- R. Perez-Ruiz, J. A. Saez, L. R. Domingo, M. C. Jimenez and M. A. Miranda, Org. Biomol. Chem., 2012, 10, 7928–7932 RSC.
- S. J. Lee, J. Y. Bae and C. W. Cho, Eur. J. Org. Chem., 2015, 2015, 6495–6502 CrossRef CAS.
- J. F. Malone, C. M. Murray, M. H. Charlton, R. Docherty and A. J. Lavery, J. Chem. Soc., Faraday Trans., 1997, 93, 3429–3436 RSC.
- P. Ganguly and G. R. Desiraju, CrystEngComm, 2010, 12, 817–833 RSC.
- A. I. Kitaigorodskii, Organic chemical crystallography, Consultants Bureau, New York, 1961 Search PubMed.
- C. P. Brock, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 807–821 CrossRef CAS PubMed.
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CrossRef CAS PubMed.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- D. T. Manallack, Perspect. Med. Chem., 2017, 1, 25–38 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2019, 47, w357–w364 CrossRef CAS PubMed.
- G. K. Gupta and V. Kumar, Chemical Drug Design, Walter de Gruyter GmbH, Berlin, Germany, 2016 Search PubMed.
- S. Sumanasuriya and J. De Bono, Cold Spring Harbor Perspect. Med., 2018, 8, a030635 CrossRef PubMed.
- H. Suzuki, T. Ueda, T. Ichikawa and H. Ito, Endocr.-Relat. Cancer, 2003, 10, 209–216 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2203890–2203894. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01231f |
| This journal is © The Royal Society of Chemistry 2022 |