
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-crystal synthesis of 2- and 4-mercaptopyridines with thiourea and its analogue, trithiocyanuric acid†
Kinga
Wzgarda-Raj
,
Olga
Książkiewicz
and
Marcin
Palusiak
 *
*
Department of Physical Chemistry, Faculty of Chemistry, University of Lodz, Pomorska 163/165, Lodz, 90-236, Poland. E-mail: marcin.palusiak@chemia.uni.lodz.pl
First published on 20th June 2022
Abstract
This article describes the synthesis of four new co-crystals obtained via cross-crystallisation of 2- and 4-mercaptopyridines with thiourea and trithiocyanuric acid and examines their crystal structures. The resulting structures are the first four reported co-crystals of mercaptopyridines. In one case, the synthesis resulted in the production of a ternary co-crystal of unexpected composition instead of the desired product and the correct co-crystal needed to be obtained under dark room conditions; interestingly, when separated from solution, all four co-crystal samples were fully light resistant. The crystal state of the four co-crystals is stabilized by several N–H⋯S type hydrogen bonds. Additionally, in the case of thiourea co-crystals, additional short contacts of S⋯S type are also observed. This relatively complicated network of intermolecular interactions is fully characterized herein.
Introduction
The organosulfur compounds possess a wide spectrum of applications in industry, pharmacology, biology, agrochemistry and synthetic organic chemistry, among others, and their chemistry and application have played significant roles in recent scientific advances. For example, mercaptopyridine derivatives are known for their antimicrobial, antifungal, and alkylation properties1–4 and are used as effective ligands in the syntheses of transition metal complexes.5 In contrast, thiourea demonstrates broad antiviral6 and antibacterial7 activities and has been reported to exhibit anticancer activity and inhibit HIV reverse transcriptase activity.8 Moreover, one form of trithiocyanuric acid, which can be considered a result of cyclic condensation of three thiourea molecules, has been used in plating processes and anticorrosion agents,9 and to remove the palladium formed during the production of drugs.10,11 More information about the application, co-crystal synthesis and interactions of these three chemical species, as well as various theoretical studies, can be found in our earlier works.12–17These previous papers also include a search for thiourea and trithiocyanuric acid crystals in the Crystal Structure Database (CSD).18 The search revealed only a few depositions of para- and ortho-mercaptopyridines, two of which were crystals of pure 4-mercaptopyridine (refcode: AKOVOL;19 AKOVOL01;20 AKOVOL02;21 AKOVOL03;21 AKOVOL04 (ref. 22)) and 2-mercaptopyridine (refcode: PYRIDS;23 PYRIDS01;24 PYRIDS02;25 PYRIDS03;26 PYRIDS11 (ref. 25)). Apart from these two entries, only three metal organic crystals containing 2-mercaptopyridine could be found (refcode: DAHTIP;27 JEHTIC;28 VIJNEH;29 VIJNEH01 (ref. 30)). Clearly knowledge regarding the crystallography of mercaptopyridines is still in its infant stage. The present paper provides the first report of four co-crystal structures containing mercaptopyridines.
This paper reports the continuation of our studies on co-crystal synthesis with the use of thiourea and its analogue, trithiocyanuric acid, as components.12,14–16 It employs a cross-crystallization approach (see graphical abstract) to obtain samples of four new co-crystals (I–IV) of sufficient quality to allow X-ray single-crystal analysis (Scheme 1).
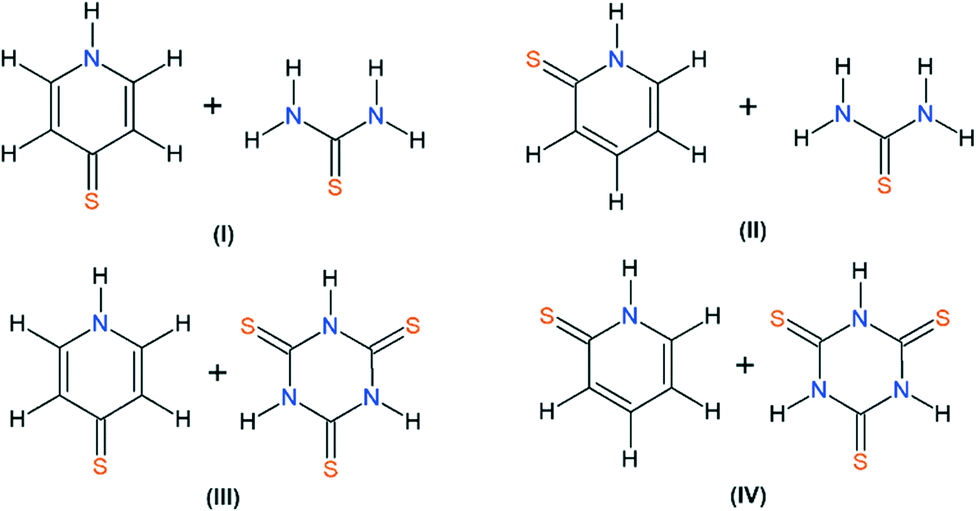 | ||
| Scheme 1 The molecular formulas of I–IV co-crystals (I – 4-mercaptopyridine and thiourea; II – 2-mercaptopyridine and thiourea; III – 4-mercaptopyridine and trithiocyanuric acid; IV – 2-mercaptopyridine and trithiocyanuric acid). | ||
Results and discussion
In general, the synthesis of the reported co-crystals was relatively uncomplicated and spontaneous, with the important exception of III, as will be discussed in this section. Briefly, for each of the desired co-crystals, the components were dissolved in a water-ethanol mixture, and the solvents were removed by slow evaporation. From the remaining solid-state material, single crystals were selected for X-ray analysis. The obtained co-crystals were of good quality, and high-quality experimental data could be obtained, confirming the successful co-crystallization of I, II and IV. See Table 1 (with standard X-ray experimental data).| Structure | I | II | III | IV |
|---|---|---|---|---|
| Empirical formula | C5H5NS· CH4N2S | C5H5NS· CH4N2S | C5H5NS· C3H3N3S3 | C5H5NS· C3H3N3S3 |
| Formula weight | 187.28 | 187.28 | 288.42 | 288.42 |
| Temperature [K] | 298 | 298 | 298 | 298 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Triclinic |
| Space group | Pbca | P21/c | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a [Å] | 11.4353(2) | 10.5170(2) | 4.1140(1) | 4.1956(1) |
| b [Å] | 8.3568(1) | 10.8672(1) | 12.3715(1) | 10.848(3) |
| c [Å] | 18.7182(2) | 8.4287(1) | 23.8214(2) | 13.5301(4) |
| α [°] | 90 | 90 | 90 | 99(2) |
| β [°] | 90 | 107(2) | 90(1) | 93(2) |
| γ [°] | 90 | 90 | 90 | 93(2) |
| Volume [Å3] | 1788.76(4) | 916.59(2) | 1212.40(3) | 605.53(3) |
| Z | 8 | 4 | 4 | 2 |
| ρ calc [g cm−3] | 1.391 | 1.357 | 1.580 | 1.582 |
| μ [mm1] | 4.922 | 4.802 | 7.026 | 7.034 |
| F(000) | 784.0 | 392.0 | 592.0 | 296.0 |
| Crystal size [mm3] | 0.262 × 0.069 × 0.048 | 0.355 × 0.316 × 0.143 | 0.056 × 0.033 × 0.027 | 0.520 × 0.050 × 0.045 |
| Radiation | Cu Kα | Cu Kα | Cu Kα | Cu Kα |
| 2Θ range for data collection [°] | 9.45 to 136.978 | 8.836 to 136.988 | 7.422 to 137 | 6.632 to 136.962 |
| Index ranges | −13 ≤ h ≤ 13 | −12 ≤ h ≤ 12 | −4 ≤ h ≤ 4 | −4 ≤ h ≤ 5 |
| −8 ≤ k ≤ 9 | −13 ≤ k ≤ 11 | −14 ≤ k ≤ 14 | −13 ≤ k ≤ 12 | |
| −22 ≤ l ≤ 22 | −10 ≤ l ≤ 6 | −28 ≤ l ≤ 28 | −16 ≤ l ≤ 16 | |
| Reflections collected | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 805 805 |
5349 | 52585 |
5954 |
| Independent reflections | 1630 | 1685 | 2210 | 2201 |
| R int = 0.0601 | R int = 0.0308 | R int = 0.0524 | R int = 0.0365 | |
| R sigma = 0.0251 | R sigma = 0.0306 | R sigma = 0.0161 | R sigma = 0.0398 | |
| Data/restraints/parameters | 1630/0/120 | 1685/0/121 | 2210/0/162 | 2201/0/162 |
| Goodness-of-fit on F2 | 1.076 | 1.089 | 1.054 | 1.061 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0412 | R 1 = 0.0383 | R 1 = 0.0258 | R 1 = 0.0312 |
| wR 2 = 0.1197 | wR 2 = 0.1085 | wR 2 = 0.0622 | wR 2 = 0.0809 | |
| Final R indexes [all data] | R 1 = 0.0440 | R 1 = 0.0412 | R 1 = 0.0282 | R 1 = 0.0350 |
| wR 2 = 0.1229 | wR 2 = 0.1115 | wR 2 = 0.0633 | wR 2 = 0.0832 | |
| Largest diff. peak/hole [e Å−3] | 0.37/−0.34 | 0.34/−0.21 | 0.26/−0.21 | 0.30/−0.27 |
| CCDC deposit number | 2165358 | 2165361 | 2165360 | 2165359 |
The only exception was noted for III, for which a very interesting and relatively rare ternary co-crystal was repeatedly obtained. This molecule consisted of two trithiocyanuric acid molecules, two bis(pyridin-4-yl) sulfide molecules and 1,4-bis(pyridin-4-yl)tetrasulfane, namely, 1,3,5-triazinane-2,4,6-trithione–4-(pyridin-4-ylsulfan-yl)pyridine–1,4-bis(pyridin-4-yl)tetrasulfane in a 2:2:1 stoichiometry. This single crystal structure has been published elsewhere.12
Although the obtained ternary co-crystal itself was a very interesting result, it did not suit our series of desired co-crystals. Any attempt to obtain the crystal sample of III was unsuccessful and always resulted in the production of a ternary co-crystal. Finally, we learned that hydroxyl radicals may be generated from mercaptopyridine N-oxides by photolysis, leading inter alia to the formation of n-sulfides.31 Since our attempts to create III resulted in both the formation of n-sulfides (in reported ternary co-crystals n was equal to 1 and 4) and α-sulfur, it was probable that our substrates, mixed in solution, undergo such decomposition. Therefore, the entire experiment was repeated using only a faint red light source in the laboratory, finally allowing the desired co-crystal of III to be obtained. Interestingly, after the solvents evaporated, the obtained crystalline material was stable under standard conditions, including daylight. The crystallization process applied in our experiments is detailed in the experimental section.
In our four new co-crystals I–IV, all the components co-crystallise in a 1:1 stoichiometry. Molecular graphs, with atoms represented by thermal ellipsoids, can be found in Fig. 1. As can be seen, in crystal structures I–IV, the mercaptopyridine derivatives adopt an N-protonated form, that is, the sulfur analogue of pyridone tautomers, i.e. pyridinethione. This appears to be the standard arrangement for the crystalline state,12 possibly due to the fact that this tautomeric form is characterized by N–H⋯A type hydrogen bonds, which are stronger than the S–H⋯A type.32,33 However, it must be emphasized that pyridinethione not always is the generally preferred tautomeric form, as the tautomeric preferences of that species depend on a range of factors, including chemical environment (e.g., the type of solvent), the state of matter or even the temperature.34–38
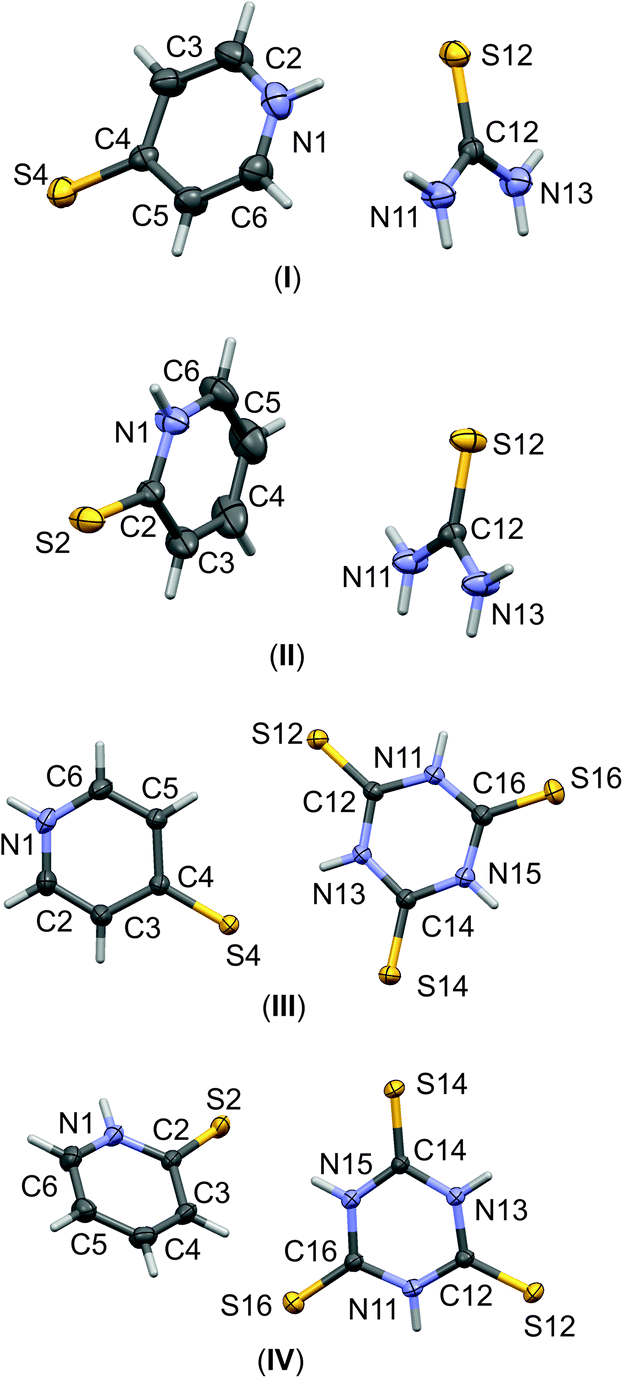 | ||
| Fig. 1 The labelling schemes of molecular components of I–IV co-crystals. The anisotropic displacement parameters of non-hydrogen atoms are drawn as ellipsoids with a 30% probability level. | ||
A similar observation can be made for trithiocyanuric acid, which, out of four possible forms, also favors the non-aromatic tautomeric form with protonated N atoms, i.e., the all-keto 1,3,5-triazi-nane-2,4,6-trithione tautomer. This issue was recently addressed by a combination of structural analysis of experimental data collected in the CSD18 and computational studies for representative molecular models.14,15
Due to the fact that all the components of co-crystals I–IV possess proton-donating amino groups and potential proton-accepting centers, e.g., N and S atoms, it is reasonable to assume that the key effect stabilizing the crystal state would be hydrogen bond formation. In fact, this was found to be the case, and the hydrogen bonding network is relatively expanded. Therefore, to systemize the discussion on the intermolecular interactions present in I–IV, it is divided into two parts. The first discusses the interactions formed by molecules of the same type, and the second examines the H-bridges formed between mercaptopyridines and their co-crystal counterparts. The geometric parameters of all discussed interactions are collected in Table 2.
| D–H…A | d(D–H) | d(H…A) | d(D…A) | <(D–H…A) | Symmetry | |
|---|---|---|---|---|---|---|
| Pyridine–pyridine | ||||||
| II | N(1)–H(1)⋯S(2) | 0.83(3) | 2.54(3 | 3.350(2) | 166(2) | 1 − x, −y, 1 − z |
| III | N(1)–H(1)⋯S(4) | 0.84(3) | 2.45(2) | 3.256(2) | 160(2) | 3/2 − x, −1/2 + y, 1/2 − z |
| IV | N(1)–H(1)⋯S(2) | 0.86(3) | 2.45(3) | 3.287(2) | 165(2) | −x, 1 − y, 1 − z |
| Thiourea–thiourea | ||||||
| I | N(11)–H(11A)⋯S(12) | 0.89(3) | 2.53(3) | 3.418(2) | 175(2) | 1 − x, −1/2 + y, 1/2 − z |
| N(13)–H(13A)⋯S(12) | 0.85(2) | 2.69(2) | 3.468(2) | 155(2) | 1 − x, 1/2 + y, 1/2 − z | |
| II | N(11)–H(11A)⋯S(12) | 0.87(2) | 2.52(2) | 3.381(2) | 170(2) | −x, 1 − y, 1 − z |
| N(13)–H(13A)⋯S(12) | 0.83(2) | 2.58(2) | 3.402(2) | 172(3) | −x, 1 − y, −z | |
| Trithiocyanuric acid–trithiocyanuric acid | ||||||
| III | N(11)–H(11)⋯S(12) | 0.80(2) | 2.57(2) | 3.355(2) | 169(2) | 1 − x, −y, −z |
| N(15)–H(15)⋯S(14) | 0.85(2) | 2.50(2) | 3.342(2) | 172(2) | 2 − x, 1 − y, − z | |
| IV | N(11)–H(11)⋯S(12) | 0.81(3) | 2.56(3) | 3.361(2) | 177(2) | 1 − x, 2 − y, −z |
| N(13)–H(13)⋯S(14) | 0.84(2) | 2.45(2) | 3.286(2) | 178(2) | −x, 1 − y, −z | |
| Thiourea–pyridinie | ||||||
| I | N(11)–H(11B)⋯S(4) | 0.72(2) | 2.71(3) | 3.392(2) | 158(2) | x, 1/2 − y, −1/2+ |
| N(13)–H(13B)⋯S(4) | 0.79(3) | 2.72(3) | 3.442(2) | 153(2) | x, 1/2 −y, −1/2 + z | |
| II | N(11)–H(11B)⋯S(2) | 0.84(3) | 2.66(3) | 3.466(2) | 163(2) | x, y, z |
| N(13)–H(13B)⋯S(2) | 0.86(3) | 2.78(3) | 3.574(2) | 155(2) | x, y, z | |
| Trithiocyanuric acid–pyridine | ||||||
| III | N(13)–H(13)⋯S(4) | 0.81(2) | 2.42(2) | 3.216(2) | 168(2) | 1 + x, y, z |
| IV | N(15)–H(15)⋯S(2) | 0.86(3) | 2.34(2) | 3.194(2) | 174(2) | 1 − x, 1 − y, 1 − z |
| Pyridine–thiourea | ||||||
| I | N(1)–H(1)⋯S(12) | 0.85(3) | 2.54(3) | 3.359(2) | 162(2) | 1 − x, 1/2 + y, 1/2 − z |
| Pyridine–trithiocyanuric acid | ||||||
| III | C(5)–H(5)⋯S(12) | 0.93 | 2.92 | 3.832(2) | 169 | x, y, z |
| IV | C(6)–H(6)⋯S(14) | 0.93 | 2.90 | 3.794(2) | 161 | −x + 1, −y + 1, −z + 1 |
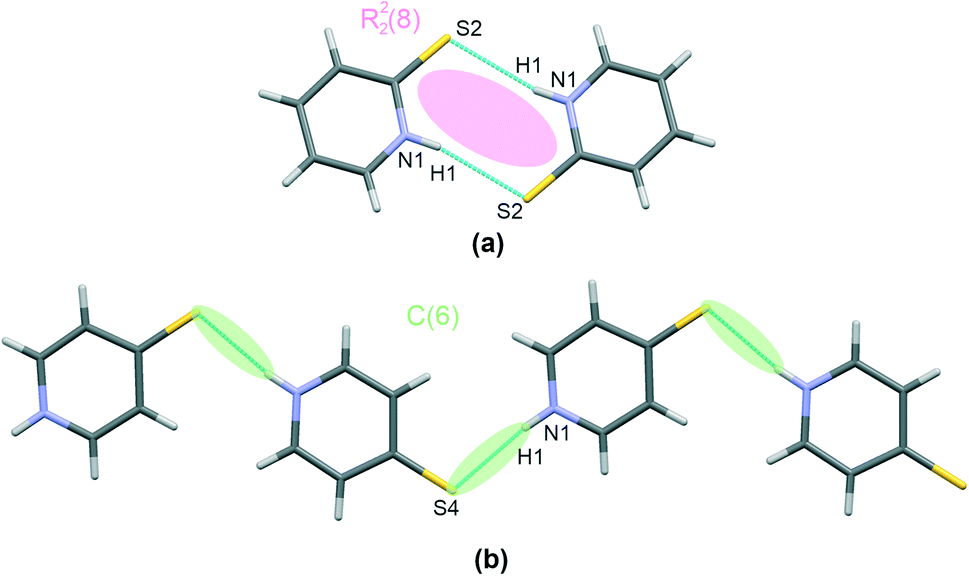
Thiourea, present in co-crystals of I and II, forms chains involving two –NH groups and one S-atom per each molecule, as shown in Fig. 2(a), together with other interactions discussed later. This interaction motif has already been reported16 for other co-crystals containing thiourea, together with a more comprehensive review of crystal structures stabilized using this hydrogen bonding pattern.
 | ||
| Fig. 2 Characteristic patterns of hydrogen bonding found in I–IV. Thiourea motif in I and II – (a), trithiocyanuric acid motif in III – (b), and IV – (c). Atoms are marked with colours, as follows: N – blue, S – yellow, C – grey, H – white. Classification of synthons according to the graph set method is also given. | ||
Regarding the hydrogen bonds present in I and II, the calculated H-bridge structural parameters indicate slightly more effective bonding in the case of structure II than structure I, with shorter distances in I (3.418(2) Å and 3.468(2) Å) than in II (3.381(2) Å and 3.402(2) Å) (Table 2). Similarly, the interacting thiourea molecules are closer to a planar arrangement in II than in I. The appropriate dihedral angles are 18(2)° and 0(2)° for I and II, respectively. This hydrogen bonding motif can be described as a C(6) second order graph and an R22(8) first order graph according to the graph set method.
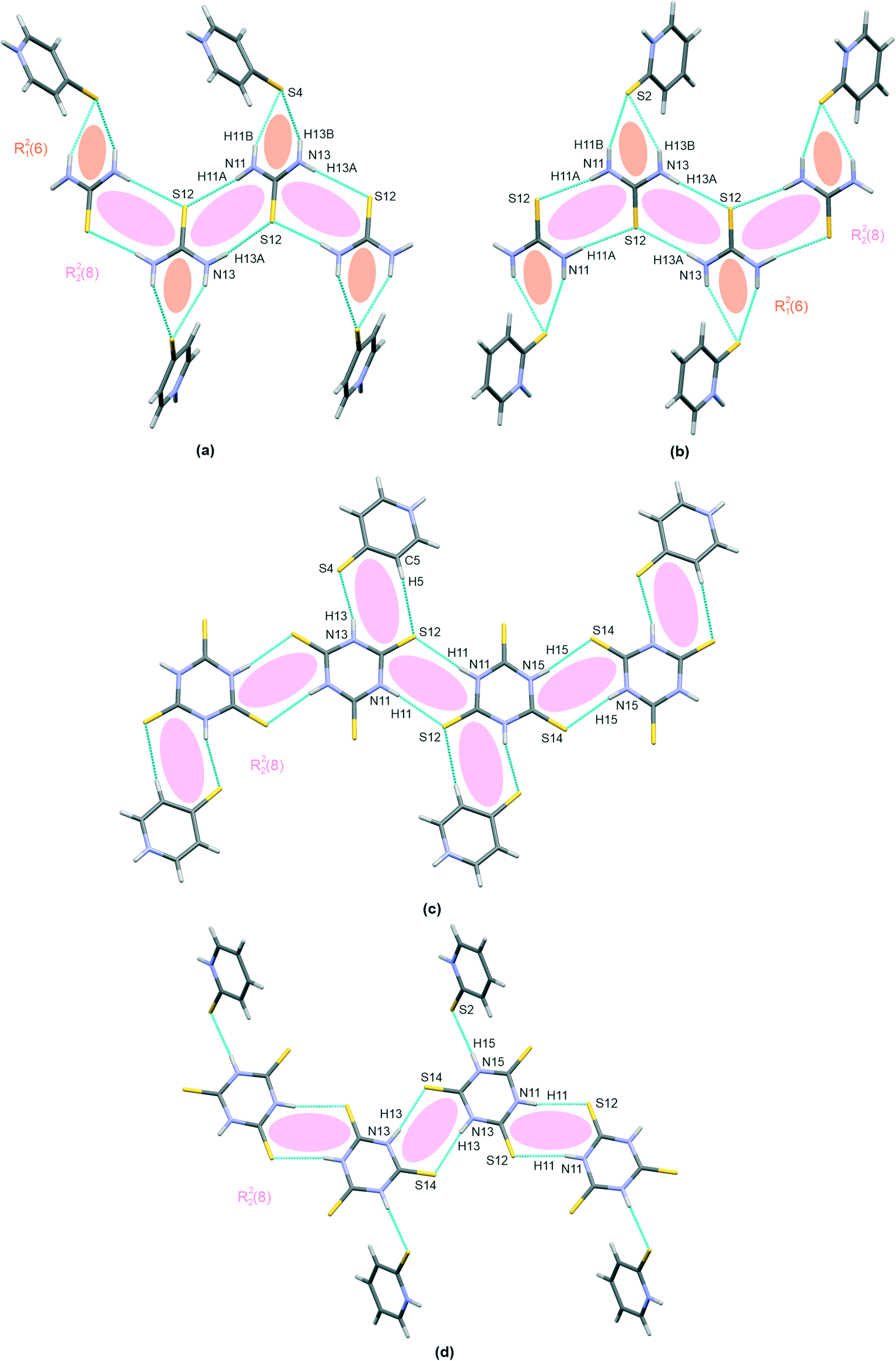
The trithiocyanuric acid present in crystals of III and IV, i.e., the thiourea analogue, also connects into chain-like patterns; however, two different structural types exist, and these are given in Fig. 2(b) and (c). In both cases, the molecules are linked by the same type of hydrogen bonding, i.e., N–H⋯S; however, this motif is straight in III but kinked in IV. Nevertheless, in both cases, when arranged in dimers, the molecules are symmetrically equivalent via an inversion operation located at the centers of R22(8) extra rings.
As can be seen in Table 2, the H-bridges in III demonstrate more closely spaced structural parameters than those of IV. For example, the difference between the two extra rings is smaller in III (H⋯S distances of 2.50(2) Å and 2.57(2) Å) compared to 2.45(2) Å and 2.56(3) Å in IV. Also, the whole chain is practically planar in IV, with an interplanar distance of 0.192(2) Å, while III demonstrates a small shift of dimers linked by an H-bridge with a longer hydrogen bond (0.415(2) Å).
Regarding the interactions involving mercaptopyridines, a graphical presentation of contacts corresponding to mercaptopyridine self-assembly hydrogen bonding is given in Fig. 3. Interestingly, only structure I was found to possess (N)H proton-donating and S proton-accepting centers involved in the interaction with the second co–crystal component, that is, the thiourea molecules. Therefore, although 4-mercaptopyridine forms N–H⋯S type hydrogen bonds in this case, this species does not undergo self-assembly in I; it also forms C(6) type chains via N–H⋯S bridges in III (Fig. 2(b)). The same motif, a R22(8) type dimer, is formed by molecules of the second derivative, 2-mercaptopyridine, in II and IV, (Fig. 3(a)). As can be seen from structural parameters collected in Table 2, this hydrogen bonding seems to be slightly more effective in the case of IV than II, with (N)H⋯S distances of 2.45(3) Å and 2.54(3) Å for IV and II.
 | ||
| Fig. 3 I–IV characteristic patterns of the self-assembly of mercaptopyridines via hydrogen bonding. The R22(8) dimer present in II and IV – (a), and the C(6) chain in III – (b). Atoms are marked with colours, as follows: N – blue, S – yellow, C – grey, and H –white. The classification of synthons according to graph set method is also given. | ||
The co-crystal structures I–IV are finally stabilized by a few hetero–molecular N–H⋯S bond linking motifs, as discussed above; all are shown in Fig. 4(a–d). Clearly, I and II demonstrate a characteristic pattern of a bifurcated R21(6) hydrogen bond linking mercaptopyridines with thiourea chains. The co-crystals formed with trithiocyanuric acid, i.e.III and IV, both demonstrate a single N–H⋯S hetero-molecular bond, which is responsible for co-crystal structure stabilization. Additionally, I demonstrates an N–H⋯S hydrogen bond, with 4-mercaptopyridine acting as a proton donor and thiourea as a proton acceptor (pyridine–thiourea dimer in Table 2). Structure III is characterized by two hydrogen bonds, N–H⋯O and C–H⋯N; thus, both molecules in the co-crystal (trithiocyanuric acid and 4-marcaptopyridine) serve simultaneously as a proton donor and an acceptor.
 | ||
| Fig. 4 Selected large patterns of hydrogen bonding stabilizing the co-crystal state in the I–IV characteristic patterns of the self-assembly of mercaptopyridines via the hydrogen bonding. The R21(6)hydrogen bonding linking thiourea chains with mercaptopyridine molecules in I (a) and II (b), and the hydrogen bonding scheme between trithiocyanuric acid chains and mercaptopyridine molecules in III (c) and IV (d). Atoms are marked with colours, as follows: N – blue, S – yellow, C – grey, and H –white. The classification of synthons according to graph set method is also given. | ||
Finally, it is worth mentioning the short S⋯S type bond found in I and II (see Fig. 5). This interaction connects the molecules of both co–crystal components and can potentially be classified as chalcogen bonding. This type of interaction has been noted previously in co-crystals of thiourea. In Table 3 there is a brief comparison of the structural properties selectively collected for such contacts found in thiourea crystal structures deposited at the CSD.18 Additionally, Table S1 (see the ESI† associated with this paper) shows a collection of refcodes together with structural data and literature references that can be found in regard to all urea crystals in which such short S⋯S contacts can be observed.
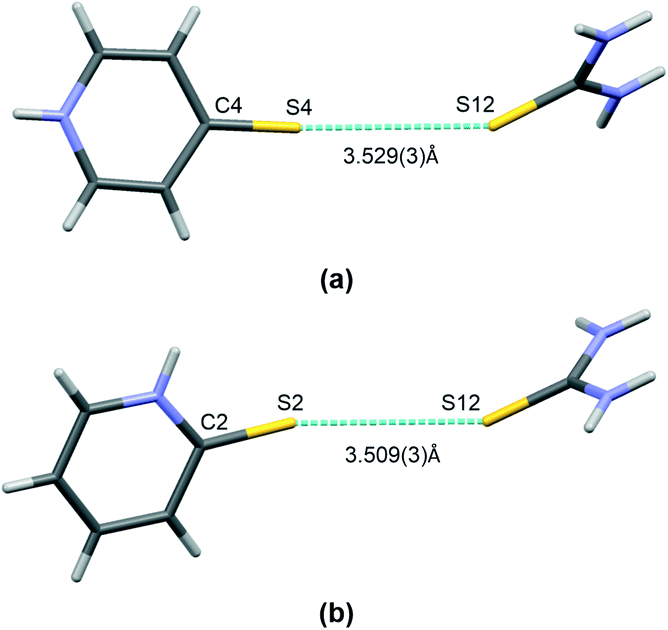 | ||
| Fig. 5 A short contact of S⋯S type found in co-crystals of thiourea: I (a) and II (b). Atoms are marked with colours, as follows: N – blue, S – yellow, C – grey, H –white. | ||
Experimental
Co-crystals synthesis
:1 ratio and dissolved in ethanol. After dissolving the substance in the solvent, the crystallizing dish was covered with a parafilm and allowed to crystallize for more than a week.
:1 ratio and dissolved in ethanol. After dissolving the substance in the solvent, the crystallizing dish was covered with a parafilm and allowed to crystallize for more than a week.
:1 ratio and dissolved in ethanol. Due to poor solubility, the mixture was heated on a magnetic stirrer with a heating plate. After dissolving the substance in the solvent, the crystallizing dish was covered with a parafilm and allowed to crystallize for more than a week. All the above activities were performed under dark room conditions. The crystallization mixture was protected from light and kept at 5 °C.
:1 ratio and dissolved in ethanol. After dissolving the substance in the solvent, the crystallizing dish was covered with a parafilm and allowed to crystallize for more than a week.
Data collection and refinement
X-ray diffraction data were collected on an XtaLAB Synergy, Dualflex, HyPix diffractometer. Integration of the intensities and corrections for Lorentz effects, polarization effects, and analytical absorption were performed with CrysAlis PRO.41 Using Olex2,42 the structure was solved with the SHELXT43 structure solution program using intrinsic phasing and refined with the SHELXL44 refinement package using least squares minimization. The hydrogen atoms of aromatic rings were introduced in the calculated positions with an idealized geometry and constrained using a rigid body model with isotropic displacement parameters equal to 1.2 of the equivalent displacement parameters of their parent atoms. The positions of the hydrogen atoms of the NH– groups were found on a Fourier difference map and refined isotropically without any restrains. The molecular geometries were calculated by the Platon program.45 Atomic coordinates, displacement parameters, and structure factors of the analyzed crystal structures are deposited with the Cambridge Crystallographic Data Centre (CCDC).46Cambridge Structural Database search
A search of the Cambridge Structural Database18 [CSD, Version 5.43, Release of November 2021] was performed. In the case of mercaptopyridines the structure of 4- and 2-mercaptopyridines was the only criterion for the search. The following entries were the result of the search: AKOVOL,19–22 DAHTIP,27 JEHTIC,28 PYRIDS,23–26 and VIJNEH.29,30 When searching S⋯S contacts with thiourea contribution, the main criterion was the structure of thiourea and the presence of the S(thiourea)⋯S(any) contact with a vdW distance limit.Conclusions
This paper describes the results of the cross-crystallization of four new co-crystals composed of 2- and 4-mercaptopyridines with thiourea and its cyclic analogue, trithiocyanuric acid. Our data represents the first accurate description of mercaptopyridine co-crystals. In one case (III), the crystallization procedure led to the production of an unexpected co-crystal; however, this was later found to result from the photoreactivity of the initial compound. The collection of the four obtained co-crystal structures has been characterized by X-ray single crystal analysis. While a relatively complicated network of N–H⋯S hydrogen bonds was found to be responsible for co-crystal state stabilization, additional short S⋯S type contacts seem to support the activity of hydrogen bonds in the case of thiourea co-crystals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MP acknowledges financial support from University of Lodz, Poland (grant number IDUB-B2211102000107.07).Notes and references
- A. Bond and J. Jones, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1536 CrossRef.
- L. W. Scheibel and A. Adler, Mol. Pharmacol., 1980, 18, 320 CAS.
- W. Paulus, Microbicides for the protection of materials: a handbook, Chapman and Hall, London, 1993, p. 294 Search PubMed.
- F. Leonard, F. A. Barkalay, E. V. Brown, F. E. Anderson and D. M. Green, Antibiot. Chemother., 1995, 6, 261 Search PubMed.
- T. Hamagughi, M. Kaneko and I. Ando, Polyhedron, 2013, 50, 215 CrossRef.
- J. Sun, S. Cai, H. Mei, J. Li, N. Yan, Q. Wang, Z. Lin and D. Huo, Chem. Biol. Drug Des., 2010, 76, 245 CAS.
- Z. Zhong, R. Xing, S. Liu, L. Wang, S. Cai and P. Li, Carbohydr. Res., 2008, 343, 566 CrossRef CAS PubMed.
- H. Peng, H. Y. Liang, L. Chen, L. Fu, H. Wang and H. He, Bioorg. Med. Chem. Lett., 2011, 21, 1102 CrossRef CAS.
- W. Chen, S. Hong, H. Q. Luo and N. B. Li, J. Mater. Eng. Perform., 2014, 23, 527 CrossRef CAS.
- S. H. Nile, B. Kumar and S. W. Park, Chem. Biol. Drug Des., 2013, 82, 290 CrossRef CAS PubMed.
- V. Kralova, V. Hanusova, P. Stankova, K. Knoppova, K. Canova and L. Skalova, Anti–Cancer Drugs, 2013, 24, 911 CrossRef CAS PubMed.
- K. Wzgarda–Raj, O. Ksiazkiewicz and M. Palusiak, Acta Crystallogr., Sect. C: Struct. Chem., 2021, 77, 479 CrossRef PubMed.
- K. Wzgarda–Raj, M. Nawrot, A. J. Rybarczyk–Pirek and M. Palusiak, Acta Crystallogr., Sect. C: Struct. Chem., 2021, 77, 458 CrossRef PubMed.
- K. Wzgarda–Raj, A. J. Rybarczyk–Pirek, S. Wojtulewski and M. Palusiak, CrystEngComm, 2021, 23(2), 324 RSC.
- K. Wzgarda–Raj, A. J. Rybarczyk–Pirek, S. Wojtulewski and M. Palusiak, Acta Crystallogr., Sect. C: Struct. Chem., 2021, 77, 49 CrossRef PubMed.
- K. Wzgarda–Raj, A. J. Rybarczyk–Pirek, S. Wojtulewski and M. Palusiak, Acta Crystallogr., Sect. C: Struct. Chem., 2020, 76, 170 CrossRef PubMed.
- K. Wzgarda–Raj, A. J. Rybarczyk–Pirek, S. Wojtulewski, E. Pindelska and M. Palusiak, Struct. Chem., 2019, 30, 827 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- M. C. Etter, J. C. MacDonald and R. A. Wanke, J. Phys. Org. Chem., 1992, 5, 191 CrossRef CAS.
- H. T. Flakus, A. Tyl and P. G. Jones, Spectrochim. Acta, Part A, 2002, 58, 299 CrossRef.
- S. Muthu and J. J. Vittal, Cryst. Growth Des., 2004, 4, 1181 CrossRef CAS.
- M. T. Raisanen, N. Runeberg, M. Klinga, M. Nieger, M. Bolte, P. Pyykko, M. Leskela and T. Repo, Inorg. Chem., 2007, 46, 9954 CrossRef PubMed.
- B. R. Penfold, Cryst. Struct. Commun., 1953, 6, 707 CAS.
- U. Ohms, H. Guth, A. Kutoglu, U. Druck and C. Scheringer, Z. Kristallogr., Kristallgeom., Kristallphys., Kristallchem., 1981, 154, 310 Search PubMed.
- U. Ohms, H. Guth, A. Kutoglu and C. Scheringer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 831 CrossRef.
- J. G. Reynolds, S. C. Sendlinger, A. M. Murray, J. C. Huffman and G. Christou, Inorg. Chem., 1995, 34, 5745 CrossRef CAS.
- S. C. Kokkou, V. Schramm and P. Karagiannidis, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1040 CrossRef.
- R. Gericke and J. Wagler, CSD Communication, 2017 Search PubMed.
- L. C. Damude, P. A. W. Dean, V. Manivannan, R. S. Srivastava and J. J. Vittal, Can. J. Chem., 1990, 68, 1323 CrossRef CAS.
- E. Waechtler, R. Gericke, S. Kutter, E. Brendler and J. Wagler, Main Group Met. Chem., 2013, 36, 181 CAS.
- A. J. Vieira, P. J. Telo and R. M. Dias, Methods Enzymol., 1999, 300, 194 CAS.
- G. Gilli and P. Gilli, J. Mol. Struct., 2000, 552, 1 CrossRef CAS.
- M. C. Etter, J. C. MacDonald and A. R. Wanke, J. Phys. Org. Chem., 1992, 5, 191 CrossRef CAS.
- V. Caprio, Pyridines and their Benzo Derivatives: Reactivity of Substituents, Comprehensive Heterocyclic Chemistry III, 2008, vol. 7, p. 101 Search PubMed.
- B. C. Uff, Pyridines and their Benzo Derivatives: Reactivity of Substituents, Comprehensive Heterocyclic Chemistry, 1984, vol. 2, p. 315 Search PubMed.
- P. Beak, J. B. Covington, S. G. Smith, J. M. White and J. M. Zeigler, J. Org. Chem., 1980, 45, 1354 CrossRef CAS.
- S. Stoyanov, T. Stoyanova, P. D. Akrivos, P. Karagiannidis and P. J. Nikolov, Heterocycl. Chem., 1996, 33, 927 CrossRef CAS.
- D. Moran, K. Sukcharoenphon, R. Puchta, H. F. Schaefer, P. V. R. Schleyer and C. D. Hoff, J. Org. Chem., 2002, 67, 9061 CrossRef CAS PubMed.
- A. Okuniewski, D. Rosiak and J. Chojnacki, Acta Crystallogr., Sect. C: Struct. Chem., 2020, 76, 468 CrossRef CAS PubMed.
- P. A. Gaye, A. H. Barry, M. Gaye, A. D. Sarr and A. S. Sall, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, 1252 CrossRef PubMed.
- CrysAlisPRO software system, Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England, 2015 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- The Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK, https://www.ccdc.cam.ac.uk/conts/retrieving.html Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures and crystallographic data. CCDC numbers 2165358–2165361. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00592a |
| This journal is © The Royal Society of Chemistry 2022 |