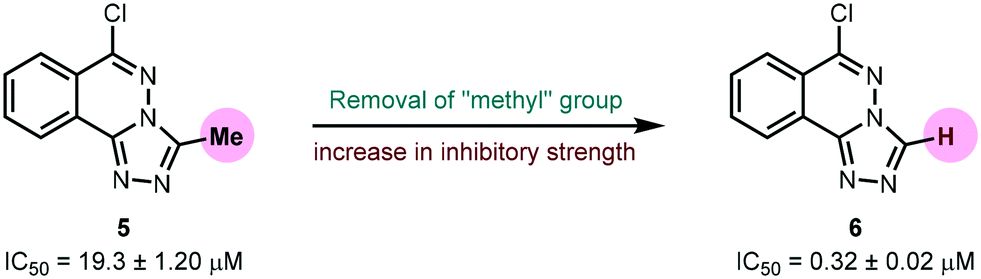DOI:
10.1039/D2CE00351A
(Paper)
CrystEngComm, 2022,
24, 5324-5339
Centroid⋯centroid and hydrogen bond interactions as robust supramolecular units for crystal engineering: X-ray crystallographic, computational and urease inhibitory investigations of 1,2,4-triazolo[3,4-a]phthalazines†
Received
12th March 2022
, Accepted 29th June 2022
First published on 29th June 2022
Abstract
Recognizing the conspicuous role of organic molecular crystals in pharmaceutical industry, we herein report the crystal engineering of two readily accessible 1,2,4-triazolo[3,4-a]phthalazine compounds (5 and 6) involving noncovalent interactions namely hydrogen bonding and aromatic interactions. The synthesis of both compounds was achieved via a facile multi-step protocol in good yield and structures were established using NMR spectroscopy and X-ray crystallographic analysis. The supramolecular assembly of phthalazine derivatives showed a dominated network of hydrogen bonding (C–H⋯N, C–H⋯Cl) and π⋯π interactions. The nature and strength of noncovalent interactions were further visualized using Hirshfeld molecular surface analysis, crystal voids, intermolecular interaction energies and energy frameworks. The structure-directing character of these interactions was confirmed theoretically by density functional theory calculations including molecular electrostatic potential (MEP) surfaces, atoms-in-molecules analysis and NCI plot computational tools. The medicinal potential of phthalazines 5 and 6 was assessed against urease enzyme where compound 6 displayed a remarkable inhibition profile with an IC50 value of 0.32 ± 0.02 μM, ∼70-fold higher activity compared to thiourea (standard inhibitor). Molecular docking analysis of both compounds revealed several vital interactions with key amino acid residues inside the active pocket of urease whereas kinetics studies depicted the competitive mode of compound 6. Furthermore, HYDE assessment and SeeSAR analysis revealed that both compounds show significant docking scores with effective binding affinities. The molecular dynamics simulations were also performed for compound 6 and the results suggested the stability of compound 6 + protein complex over 30 ns time period. Finally, pharmacokinetic properties predicted the drug-likeness and blood–brain barrier permeation of the tested compounds.
1. Introduction
The dominant use of robust supramolecular synthons in crystal engineering has emerged as a leading factor in regulating the solid-state topology and function of macromolecules.1–4 Crystal engineering involving noncovalent interactions such as inter- and intramolecular hydrogen bonding, dispersion interactions, hydrophobic interactions, halogen bonding, C–H⋯π, cation⋯π, N–H⋯π, π⋯π, S–H⋯π, lone pair⋯π, and salt bridge⋯π has gained significant interest in various fields, namely chemistry, biology, materials science and pharmaceuticals.1–5 In particular, hydrogen bonding has remained at the forefront with widespread applications among all noncovalent interactions.6,7 In contrast, a weak non-traditional H-bond of type C–H⋯X demonstrates applications in the broad field of host–guest chemistry and anion recognition. C–H⋯Cl hydrogen bonding is much less frequent as compared to commonly occurring C–H⋯N/O hydrogen bonds, however, well documented in recent years.8–10 In addition, π–π stacking interactions of aromatic systems are ubiquitous in many areas of natural biological and chemical processes. These interactions control significantly the stereoselectivity of organic synthesis,11 host–guest complexes,6 and catalyst design.12
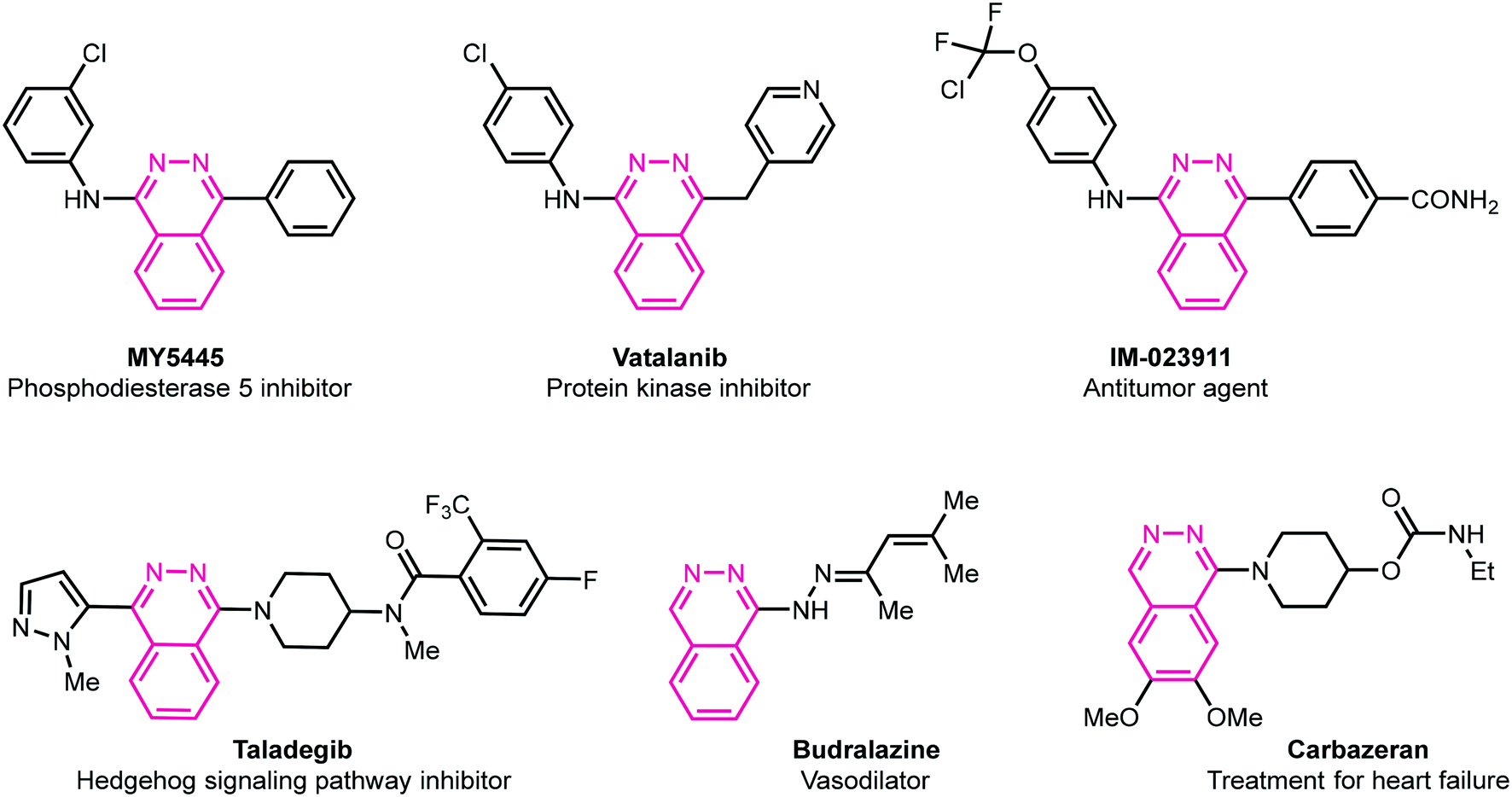
Heterocycles are fundamental chemical entities comprising 87% of known small molecule therapeutics.13–17 Among them, nitrogen-containing heterocyclic molecules are of particular importance due to their widespread occurrence in several natural products, drug molecules, and bioactive designed pharmacophores.13–17 In this family, phthalazine derivatives have been identified as potent leads demonstrating a wide range of therapeutic properties. For instance, these molecules in conjunction with other pharmacophores display anticonvulsant,18 anti-inflammatory,19 vasorelaxant,20 anticancer,21 antibacterial,22 antihypertensive,23 and carbonic anhydrase inhibitory activities.24 These privileged pharmacophores are also present in several marketed drugs such as azelastine (allergic rhinitis),25 hydralazine (antihypertension),26 zaleplon (insomnia),27 budralazine (vasodilator),28 and MY5445 (phosphodiesterase inhibitor).29 Moreover, [1,2,4]triazolo[3,4-a]phthalazines have been reported as inhibitors of bromodomains30 in addition to anticancer31 and positive inotropic activities.32Fig. 1 demonstrates some selective examples of drug molecules encompassing phthalazine nucleus.33
 |
| | Fig. 1 Representative examples of biologically active drug molecules highlighting phthalazine moiety. | |
To explore the noncovalent features of heterocyclic entities and their medicinal chemistry aspects further, and in continuation of our previous work on noncovalent interactions,34 we herein evaluate two readily accessible phthalazine molecules for their utility in supramolecular chemistry, crystal engineering, and drug discovery (anti-urease inhibition). Compared to previous work where we used sulfonamides (H-bonding interactions dominate), this work takes advantage of an extended π-system (three fused rings) to provoke the formation of π-stacking assemblies which control the solid-state architecture of the compounds. A multi-step synthetic approach was utilized to deliver 6-chloro-3-methyl-[1,2,4]triazolo[3,4-a]phthalazine 5 and 6-chloro-[1,2,4]triazolo[3,4-a]phthalazine 6. The supramolecular synthons formed through hydrogen bonding and π-cloud interactions have been explored which were rationalized by computational methods such as molecular electrostatic potential (MEP) surfaces, quantum theory of atoms in molecules (QTAIM) and non-covalent interaction plot (NCIPlot) and Hirshfeld surface analyses. DFT calculations redisclose that the π-stacking interactions are stronger than the CH⋯N and CH⋯Cl H-bonds in compounds 5 and 6. In vitro urease inhibitory potential and mechanism of inhibition were determined and binding modes of the compounds with active site amino acid residues were further rationalized with molecular docking analysis. Further insights were obtained from HYDE assessment, SeeSAR analysis, and molecular dynamics simulations. All the results were in good agreement to those observed in in vitro experiments.
2. Experimental
2.1. Preparation of 2,3-dihydrophthalazine-1,4-dione 2
To a stirred solution of phthalic anhydride 1 (5.92 g, 40 mmol, 1.0 equiv.) in acetic acid (22 mL) was slowly added hydrazine hydrate (2.80 mL, 44 mmol, 1.10 equiv.). The mixture was stirred at 120 °C for 4 h. After completion of reaction, the reaction mixture was cooled to room temperature and filtered. The precipitates were washed with petroleum ether (2 × 10 mL) and dried under vacuum. 2,3-Dihydrophthalazine-1,4-dione 2 was obtained as a white solid (5.95 g, 92%).321H NMR (400 MHz, DMSO-d6) δH: 11.56 (br s, 2H, NH), 8.09–8.05 (m, 2H, ArH), 7.89–7.85 (m, 2H, ArH); 13C{1H} NMR (101 MHz, DMSO-d6) δC: 154.7, 132.7, 127.2, 125.2.
2.2. Preparation of 1,4-dichlorophthalazine 3
A mixture of 2,3-dihydrophthalazine-1,4-dione 2 (4.86 g, 30 mmol) and phosphorus oxychloride (POCl3) (25 mL) was stirred at 110 °C for 12 h. After completion of reaction, the mixture was cooled to room temperature, and slowly added onto crushed ice with stirring for 15 min. The precipitated solid product was filtered off, washed with water, and dried under vacuum. 1,4-Dichlorophthalazine 3 was obtained as a white solid (5.30 g, 89%).321H NMR (400 MHz, DMSO-d6) δH: 8.37–8.33 (m, 2H, ArH), 8.29–8.24 (m, 2H, ArH); 13C{1H} NMR (101 MHz, DMSO-d6) δC: 154.7, 135.7, 126.7, 125.7.
2.3. Preparation of 6-chloro-3-methyl-[1,2,4]triazolo[3,4-a]phthalazine 5
A mixture of 1,4-dichlorophthalazine 3 (1.98 g, 10.0 mmol) and hydrazine hydrate (3.73 mL, 76.5 mmol) in ethanol (50 mL) was stirred at 80 °C for 30 min. After cooling to room temperature, the solid product was filtered off, washed with Et2O (2 × 30 mL) and dried under vacuum. 1-Chloro-4-hydrazineylphthalazine 4 was obtained as a yellow solid (1.25 g, 64%) and used as such without further purification.32
1-Chloro-4-hydrazineylphthalazine 4 (1.25 g, 6.45 mmol) was added to a solution of triethylamine (0.90 mL, 6.45 mmol) and acetyl chloride (0.55 mL, 7.74 mmol) in 1,4-dioxane. The reaction mixture was stirred at 110 °C. After the completion of reaction (monitored by TLC; 30% EtOAc in hexanes), the solvent was removed in vacuo, and the solid obtained was partitioned between DCM (130 mL) and H2O (100 mL). The combined organics were dried over MgSO4, filtered, and concentrated in vacuo to give 5 as a light yellow solid (770 mg, 55%).321H NMR (400 MHz, CDCl3) δH: 8.67 (d, 1H, J = 7.9 Hz, ArH), 8.26 (d, 1H, J = 8.2 Hz, ArH), 8.01–7.97 (m, 1H, ArH), 7.88–7.84 (m, 1H, ArH), 2.81 (s, 3H, CH3); 13C{1H} NMR (101 MHz, CDCl3) δC: 149.9, 147.9, 142.5, 134.9, 131.3, 127.6, 124.2, 123.7, 122.2, 10.0.
2.4. Preparation of 6-chloro-[1,2,4]triazolo[3,4-a]phthalazine 6
1-Chloro-4-hydrazineylphthalazine 4 (1.25 g, 6.45 mmol) was added to a solution of triethylamine (0.90 mL, 6.45 mmol) and triethyl orthoformate (1.28 mL, 7.74 mmol) in 1,4-dioxane. The reaction mixture was stirred at 110 °C. After the completion of reaction (monitored by TLC; 30% EtOAc in hexanes), the solvent was removed in vacuo, and the solid obtained was partitioned between DCM (130 mL) and H2O (100 mL). The combined organics were dried over MgSO4, filtered, and concentrated in vacuo to give 6 as a light yellow solid (982 mg, 75%).321H NMR (400 MHz, DMSO-d6) δH: 9.62 (s, 1H, ArH), 8.57 (d, 1H, J = 7.8 Hz, ArH), 8.30 (d, 1H, J = 8.1 Hz, ArH), 8.17–8.13 (m, 1H, ArH), 8.04–8.00 (m, 1H, ArH); 13C{1H} NMR (101 MHz, DMSO-d6) δC: 149.7, 141.7, 139.8, 135.4, 131.8, 127.5, 123.3, 123.0, 122.2.
2.5. Crystal growth development
Single crystals of compounds 5 and 6 suitable for X-ray diffraction analysis were grown at room temperature from ethyl acetate and hexane solvents using slow evaporation method.
2.6. X-ray structure determination
Diffraction data for the compounds (5 and 6) were collected at 100 K on an Agilent Technologies Supernova system using Cu Kα radiation (λ = 1.54184 Å), processed using CrysAlisPro software. The structures were solved using SHELXT35 and refined using full-matrix least-squares procedures using SHELXL-2019 (ref. 36) within WINGX.37
All non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed in calculated positions with their thermal parameters refined isotropically with Uiso(H) = 1.2 Ueq(C). Molecular plots and packing diagrams were drawn using Mercury38 and additional metrical data were calculated using PLATON.39 Details of the X-ray measurements and crystal data for the compounds are given in Table 1.
Table 1 Crystal data and structure refinement for 5 and 6
| Compound |
5
|
6
|
| Empirical formula |
C10H7ClN4 |
C9H5ClN4 |
| Formula weight |
218.65 |
204.62 |
| Temperature (K) |
100 |
101 |
| Wavelength (Å) |
1.54184 |
1.54184 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/c |
|
a (Å) |
8.8082(1) |
8.7825(4) |
|
b (Å) |
7.3685(1) |
16.4044(7) |
|
c (Å) |
14.2931(2) |
11.3954(4) |
|
α (°) |
90 |
90 |
|
β (°) |
90.213(2) |
96.420(4) |
|
γ (°) |
90 |
90 |
| Volume (Å3) |
927.66(2) |
1631.46(12) |
|
Z
|
4 |
8 |
|
D
calc (Mg m−3) |
1.566 |
1.666 |
| Absorption coefficient (mm−1) |
3.379 |
3.798 |
|
F(000) |
448 |
832 |
| Crystal size (mm) |
0.18 × 0.11 × 0.09 |
0.18 × 0.14 × 0.07 |
| Theta range for data collection (°) |
5.861 to 72.675 |
4.745 to 72.885 |
| Index ranges |
−10 ≦ h ≦ 10 |
−10 ≦ h ≦ 10 |
| −9 ≦ k ≦ 9 |
−19 ≦ k ≦ 20 |
| −17 ≦ l ≦ 17 |
−13 ≦ l ≦ 9 |
| Reflections collected |
16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 468 468 |
6440 |
| Independent reflections |
1833 [R(int) = 0.0309] |
3144 [R(int) = 0.0314] |
| Completeness to theta = 67.684° |
99.1% |
99.9% |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
1833/0/137 |
3144/0/253 |
| Goodness-of-fit on F2 |
1.06 |
1.06 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.027, wR2 = 0.066 |
R
1 = 0.049, wR2 = 0.129 |
|
R indices (all data) |
R
1 = 0.030, wR2 = 0.069 |
R
1 = 0.059, wR2 = 0.141 |
| Largest difference peak and hole (e Å−3) |
0.28 and −0.26 |
0.51 and −0.56 |
| CCDC reference number |
2152622
|
2152626
|
2.7. Theoretical methods
The noncovalent interactions were analyzed energetically using Gaussian-16 (ref. 40) at the PBE0-D3/def2-TZVP level of theory. The binding energies have been corrected using the Boys and Bernardi counterpoise method.41 The Grimme's D3 dispersion correction has also been used in the calculations.42 To evaluate the interactions in the solid state, the crystallographic coordinates were used and only the hydrogen atoms were optimized. This methodology represents a good compromise between the size of the system and the accuracy of the results. It has been used before to analyze similar interactions.43 The interaction energies were estimated by calculating the difference between the energies of the isolated monomers and the ones of their assembly. The QTAIM analysis44 and NCIplot index45 have been computed at the same level of theory by means of the AIMAll program.46
2.8. Urease inhibition assay
Inhibition activity of compounds 5 and 6 against urease was evaluated using the indophenol technique47 with some modifications.48 In 96-well plates, reaction mixtures containing 40 μL buffer (100 mmol L−1 urea, 0.01 mol L−1 K2HPO4, 1 mol L−1 EDTA, and 0.01 mol L−1 LiCl2, pH = 8.2) and 10 μL enzyme (5 U mL−1) were incubated for 30 minutes at 37 °C with 10 μL synthetic compounds (1 mM) and 10 μL urea (1 mM). After 10 min of incubation at 37 °C, the absorbance of phenol reagent (40 μL, 1% w/v phenol, 0.005% w/v sodium nitroprusside) and alkali reagent (40 μL, 0.5% w/v NaOH, 0.1% active chloride NaOCl) added to each well, was measured at 630 nm using a microplate reader (Bio-Tek ELx 800™, Instruments, Inc. USA). All the experiments were performed in triplicates.
| Inhibition percentage = 100 − (Absorbance of test compound/Absorbance of control) × 100 |
The percentage inhibition was calculated using the equation mentioned above.
Different concentrations of synthesized compounds (5 and 6) and thiourea (standard inhibitor) were evaluated under the same experimental parameters to determine IC50 values. The results were analyzed using PRISM 5.0 (GraphPad, San Diego, California, USA).
2.9. Kinetics studies
The type of enzyme inhibition was determined using Michaelis–Menten kinetics assays. To investigate the potential mechanism of action for inhibiting the enzyme, kinetics studies of the potent compound 6 were conducted. The initial velocity of enzyme inhibition was assessed in the absence and presence of four specific concentrations (0, 0.16, 0.32, 0.48 μM) of compound 6 against urease at five different concentrations of substrate, urea (125, 250, 500, 1000, and 1500 μM).
2.10. Docking protocols
2.10.1. Structure selection and preparation.
Molecular docking studies were performed to explore possible interactions of the compounds making complex with the urease enzyme. The RCSB PDB database was used to obtain the crystallographic structure of Jack bean urease (PDB ID: 3LA4).49 Before performing the experiments, the structures of the enzyme and compounds were prepared as follows. The Protonate3D50 algorithm developed within the molecular modeling tool MOE51 was used to protonate the enzyme structure. Amber99 force field was used to optimize the energy of the structure, including all crystallographic solvent molecules. To avoid the collapse of binding pockets during energy minimization calculations, the backbone atoms were constrained with a minor force. The solvent molecules and co-crystallized ligands were then eliminated. MOE was used to remove the crystallographic water molecules and add hypothetical hydrogen atoms to the X-ray structure in standard geometries.
2.10.2. Compounds preparation.
The “wash” module was used to assign protonation and ionization states in the physiological pH range after the 3D structural coordinates of compounds (5 and 6) were created using MOE. For docking studies, the structures of compounds were then energy minimized using the MMFF94x force field.
2.10.3. Docking studies.
Calculations for the docking experiments were conducted with LeadIT from BioSolveIT, GmbH Germany.52 The receptor was uploaded using the LeadIT software's Load or Prepare Receptor tool, with the metal ions selected as part of the receptor. The receptor's binding site was specified by a 9.0 Å spacing of amino acid residues. Compounds were docked using LeadIT's FlexX program. Compounds 5 and 6 were docked into the receptor's active site for this reason, and 50 conformations for each ligand-receptor complex were generated using binding free energies. The default docking parameters were used, and the top 30 docked positions with the highest scores were saved for later study.53 The poses with the lowest free binding energy values were considered as the most stable and had the greatest affinity for interacting with the receptor (Fig. S1†). The 3D possible binding modes of every ligand–protein complex with the lowest binding free energy for interactions were observed using Discovery Studio Visualizer v4.54
2.11. Molecular dynamics simulations
GROMOS96 force field with the 43a1 parameter set was used for the protein modification and protonation of the crystallographic structure of Jack bean urease (PDB ID: 3LA4).49 For the MD simulations, the GROMACS (Groningen Machine for Chemical Simulation) simulation packages, version 5.1.4, were utilized, and the methodology for molecular dynamics simulations was based on previously published methodologies55 with minor changes. The PRODRG servers were used to parameterize compound 6 online.56 For visualization and molecular analysis, MOE and VMD57 were utilized. To neutralize the receptor, the crystallographic structure was solvated (addition a water molecule) and counter ions were added. After that, the system's energy was minimized, and it was equilibrated utilizing two successive NVT (100 ps) and NPT (100 ps) runs, during which the protein's heavy atoms were confined. The generated ensembles were then subjected to 30 ns MD simulations with a time-step of 2 fs for each simulation after minimization. Throughout the simulations, periodic boundary conditions (PBC) were implemented. For simple energy minimizations, the steepest descent method was applied. For temperature (303 K) and pressure coupling (1.01 bar), the Berendsen thermostat and the Parrinello–Rahman barostat were utilized in all NVT and NPT runs, accordingly. For the long-range method, a cut-off radius of 10 Å was used, as well as a smooth particle mesh Ewald (PME) protocol. XMGRACE v5.1.19 was used to plot root mean square deviations, fluctuations, radius of gyration, and distance over time of hydrogen bonds.58
3. Results and discussion
3.1. Synthetic chemistry
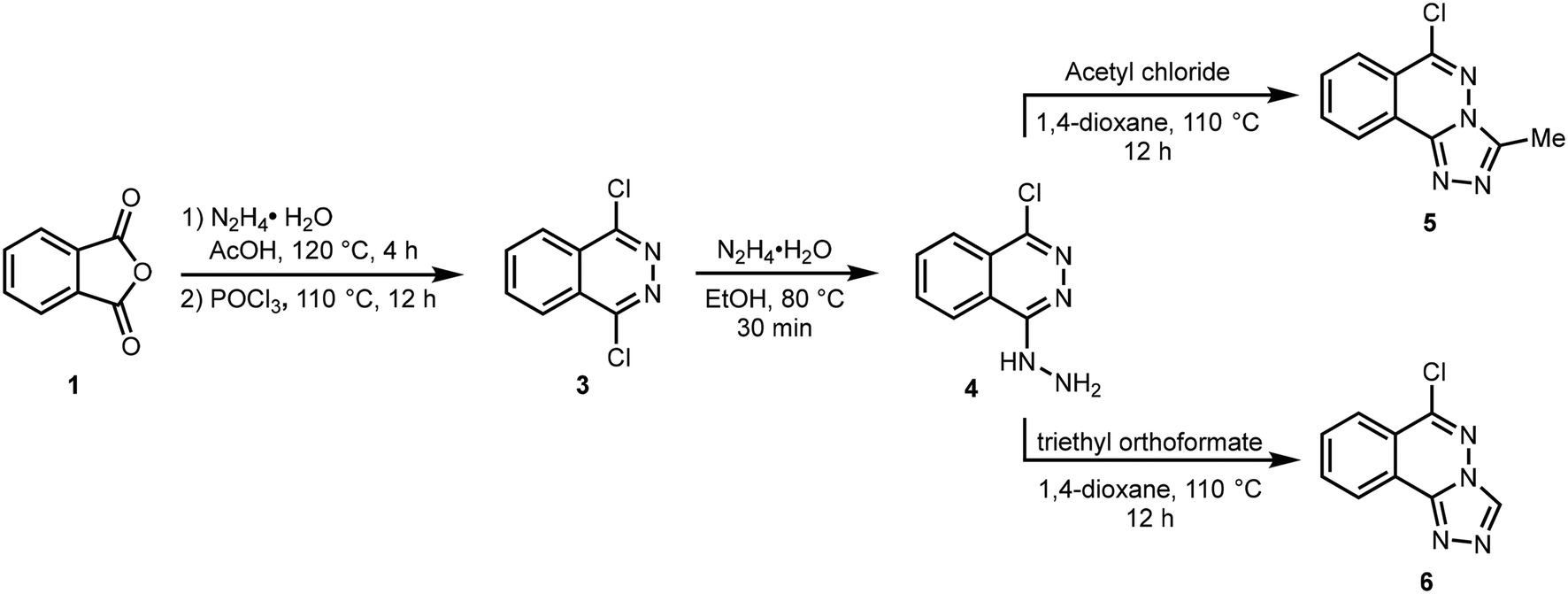
The target compounds 5 and 6 were obtained using the synthetic route depicted in Scheme 1. The reaction of phthalic anhydride 1 with hydrazine hydrate in AcOH produced 2,3-dihydrophthalazine-1,4-dione 2 which was reacted with phosphoryl chloride to afford 1,4-dichlorophthalazine 3. The conversion of phthalic anhydride 1 to 2,3-dihydrophthalazine-1,4-dione 2 was confirmed through NMR analysis where a singlet for two NH protons appeared at 11.56 ppm in 1H NMR spectrum. A characteristic peak due to amide carbonyl (NHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) observed at 154.7 ppm also confirmed the structure of 2. The formation of 1,4-dichlorophthalazine 3 was confirmed by the disappearance of NH protons in 1H NMR spectrum. Subsequent hydrazination in ethanol at 80 °C gave 1-chloro-4-hydrazineylphthalazine 4 which was cyclized to 6-chloro-3-methyl-[1,2,4]triazolo[3,4-a]phthalazine 5 and 6-chloro-[1,2,4]triazolo[3,4-a]phthalazine 6 by reacting with acetyl chloride and triethyl orthoformate in 1,4-dioxane at 110 °C, respectively.32 The structure of compound 5 shows the incorporation of a methyl group at 2.81 and 9.96 ppm in 1H and 13C NMR spectra, respectively, confirming the smooth cyclization of 4 into 5. Similarly, a distinct singlet at 9.62 ppm attributable to Csp2–H of triazole ring confirmed the formation of compound 6. Finally, structures of compounds 5 and 6 were fully established by X-ray crystallography.
O) observed at 154.7 ppm also confirmed the structure of 2. The formation of 1,4-dichlorophthalazine 3 was confirmed by the disappearance of NH protons in 1H NMR spectrum. Subsequent hydrazination in ethanol at 80 °C gave 1-chloro-4-hydrazineylphthalazine 4 which was cyclized to 6-chloro-3-methyl-[1,2,4]triazolo[3,4-a]phthalazine 5 and 6-chloro-[1,2,4]triazolo[3,4-a]phthalazine 6 by reacting with acetyl chloride and triethyl orthoformate in 1,4-dioxane at 110 °C, respectively.32 The structure of compound 5 shows the incorporation of a methyl group at 2.81 and 9.96 ppm in 1H and 13C NMR spectra, respectively, confirming the smooth cyclization of 4 into 5. Similarly, a distinct singlet at 9.62 ppm attributable to Csp2–H of triazole ring confirmed the formation of compound 6. Finally, structures of compounds 5 and 6 were fully established by X-ray crystallography.
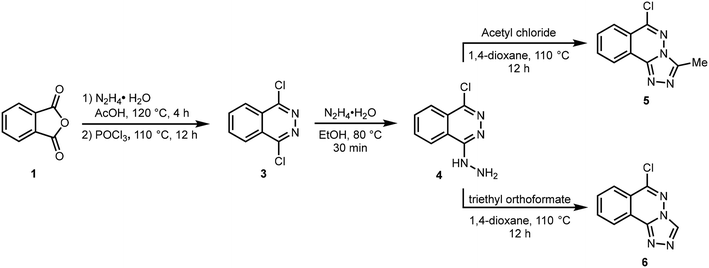 |
| | Scheme 1 Synthetic route to 1,2,4-triazolo[3,4-a]phthalazines (5,6). | |
3.2. X-ray crystallography
A search of the Cambridge structural database version 5.41 November 2019 with 3 updates March 2020 (ref. 59) confirmed the novelty of 6-chloro-triazolophthalazines 5 and 6. A search with the chloro substituent removed gave 24 hits for the triazolophthalazine fragment, all of which had alkyl, alkenyl or aryl substitution in the 3-position. Included in the list is the structure 3-methyl-1,2,4-triazolo[3,4-a]phthalazine monohydrate (PUGHOP),60 a close relative of 5.
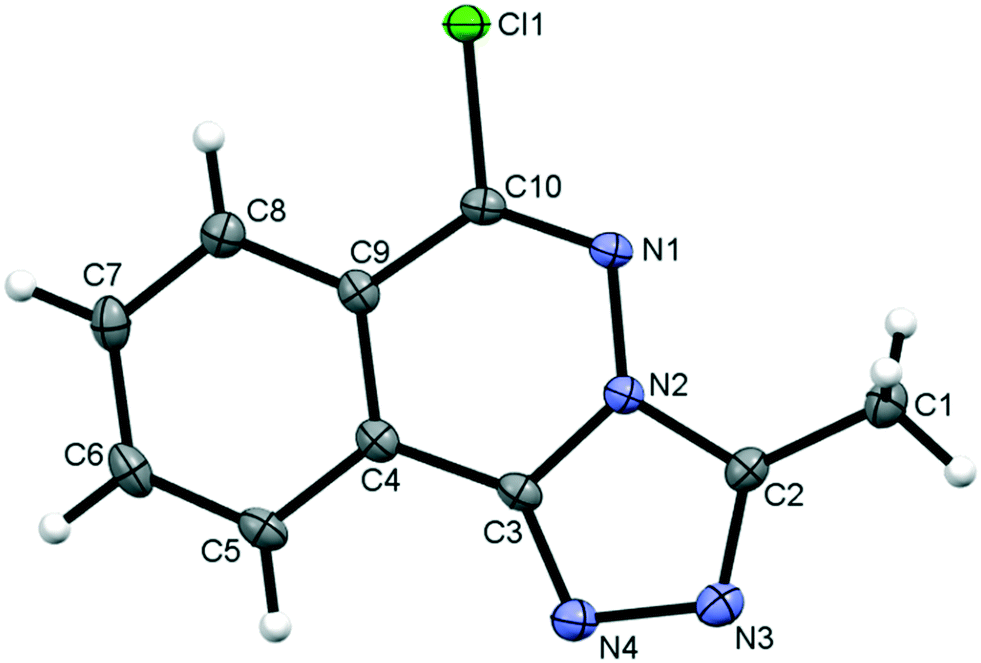
3.2.1. Molecular structure of compound 5.
Compound 5, 6-chloro-3-methyl-[1,2,4]triazolo[3,4-a]phthalazine, is a fusion of a five-membered and two six-membered ring systems, Fig. 2. The combined ring system is planar with an rms deviation of 0.0139 Å for the 13 atoms. The chloro and methyl carbon substituents are also close to the ring plane with deviations of only 0.0782(11) Å and 0.0043(17) Å, respectively. The distance between peri H8 and Cl1 atoms is less than the sum of their van der Waals radii, however, the C8–H8⋯Cl1 angle of 106° falls outside the accepted norms to be considered a weak hydrogen bond.61
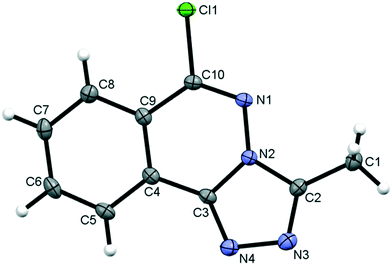 |
| | Fig. 2 The molecular structure of 5 showing the atom numbering scheme with ellipsoids drawn at the 50% probability level. | |
3.2.2. Crystal packing of compound 5.
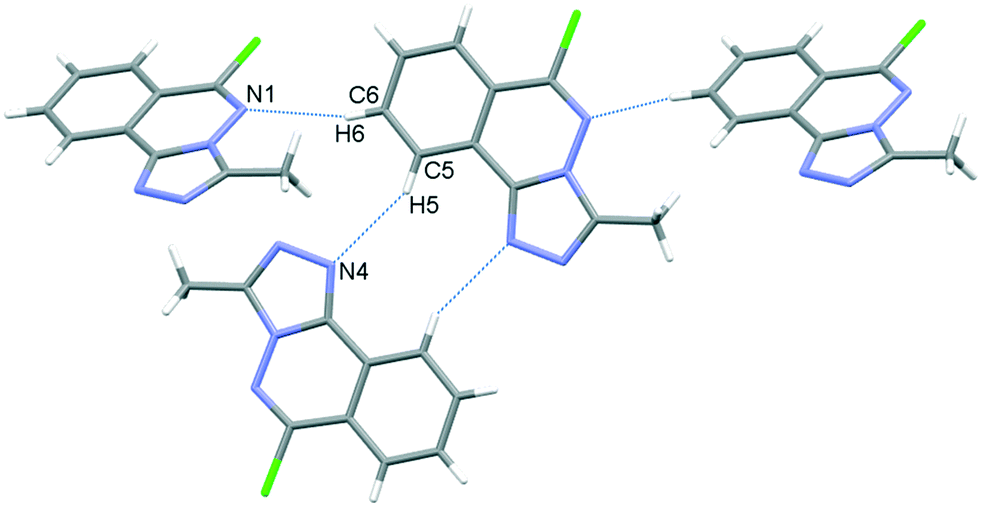
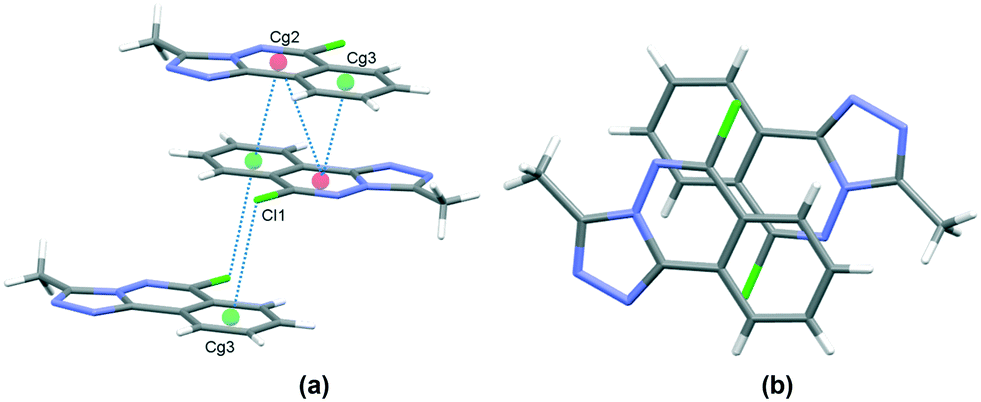
In the crystal, C6–H6⋯N1 hydrogen bonds, link the molecules into chains which propagate by a glide reflection in the (101) plane, Fig. 3, Table 2. An additional C5–H5⋯N4 interaction forms inversion dimers. The electron-rich planar ring system predictably displays π-stacking behaviour. Also revealed is a Cl⋯π close contact.62 These two motifs generate inversion dimer pairs which combine to give stacks in the b axis direction; these are illustrated in Fig. 4 and metrics listed in Table 3.
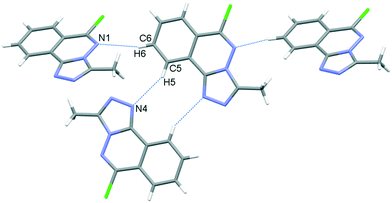 |
| | Fig. 3 C–H⋯N bonding motifs in 5. | |
Table 2 Hydrogen bond distances (Å) and angles (°) for 5
| D–H⋯A |
d(D–H) |
d(H–A) |
d(D–A) |
<(DHA) |
|
Symmetry codes: (i) 1/2 + x, 1/2 − y, −1/2 + z; (ii) 2 − x, 1 − y, 1 − z.
|
| C6–H6⋯N1(i) |
0.93 |
2.55 |
3.4269(19) |
157 |
| C5–H5⋯N4(ii) |
0.93 |
2.67 |
3.5189(19) |
152 |
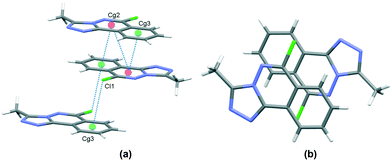 |
| | Fig. 4 Inversion dimer motifs of 5 formed by π⋯π interactions and Cl⋯π close contact, viewed approximately parallel to the (010) plane (a), and stack viewed orthogonal to the (101) plane (b). | |
Table 3 π⋯π and Cl⋯π interactions for 5
| Cg⋯Cg |
Distance Å |
|
Cg2 and Cg3 are the centroids of the N1, N2, C3, C4, C9, C10 ring and C4⋯C9 rings respectively. Symmetry codes: (i) 1 − x, 1 − y, 1 − z; (ii) 1 − x, −y, 1 − z.
|
| Cg2⋯Cg2(i) |
3.6002(7) |
| Cg2⋯Cg3(i) |
3.5492(8) |
| Cl1⋯Cg3(ii) |
3.470 |
3.2.3. Molecular structure of compound 6.
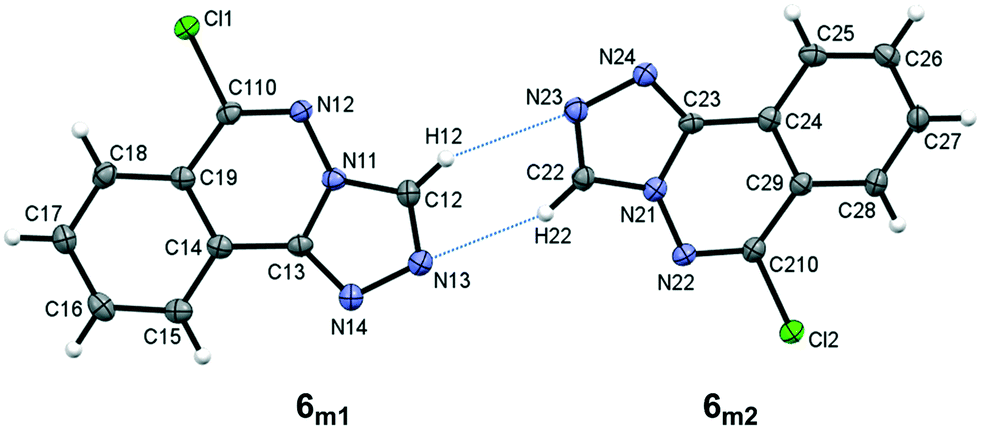
Compound 6, 6-chloro-[1,2,4]triazolo[3,4-a]phthalazine, crystallizes with two unique molecules, (referred to here as 6m1 and 6m2), in the asymmetric unit of the monoclinic unit cell, Fig. 5. They are related by a pseudo 2-fold screw along [101], and linked by C–H⋯N hydrogen bonds, Table 4. Similarities to 5 extend to the significant planarity of the fused ring systems, with an rms deviations of 0.0250 Å for 6m1 and 0.0237 Å for 6m2. The chloro substituents also lie close to the ring planes of both unique molecules with deviations of −0.116(2) Å and 0.117(2), respectively, for 6m1 and 6m2.
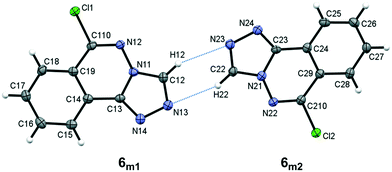 |
| | Fig. 5 The asymmetric unit of 6 showing the atom numbering scheme with ellipsoids drawn at the 50% probability level. Intermolecular C–H⋯N bonds are shown as blue dotted lines. | |
Table 4 Hydrogen bond distances (Å) and angles (°) for 6
|
|
D–H |
H⋯A |
D⋯A |
D–H⋯A |
|
Symmetry codes: (i) 1 − x, 1/2 + y, 3/2 − z; (ii) x, 1 + y, z; (iii) 2 − x, −1/2 + y, 1/2 − z; (iv) x, −1 + y, z.
|
| C12–H12⋯N23 |
0.93 |
2.60 |
3.394(3) |
144 |
| C22–H22⋯N13 |
0.93 |
2.52 |
3.362(3) |
152 |
| C15–H15⋯Cl2(i) |
0.93 |
2.90 |
3.809(3) |
168 |
| C18–H18⋯Cl2(ii) |
0.93 |
2.87 |
3.579(3) |
134 |
| C25–H25⋯Cl1(iii) |
0.93 |
2.98 |
3.872(3) |
162 |
| C28–H28⋯Cl1(iv) |
0.93 |
2.87 |
3.519(3) |
128 |
3.2.4. Crystal packing of compound 6.
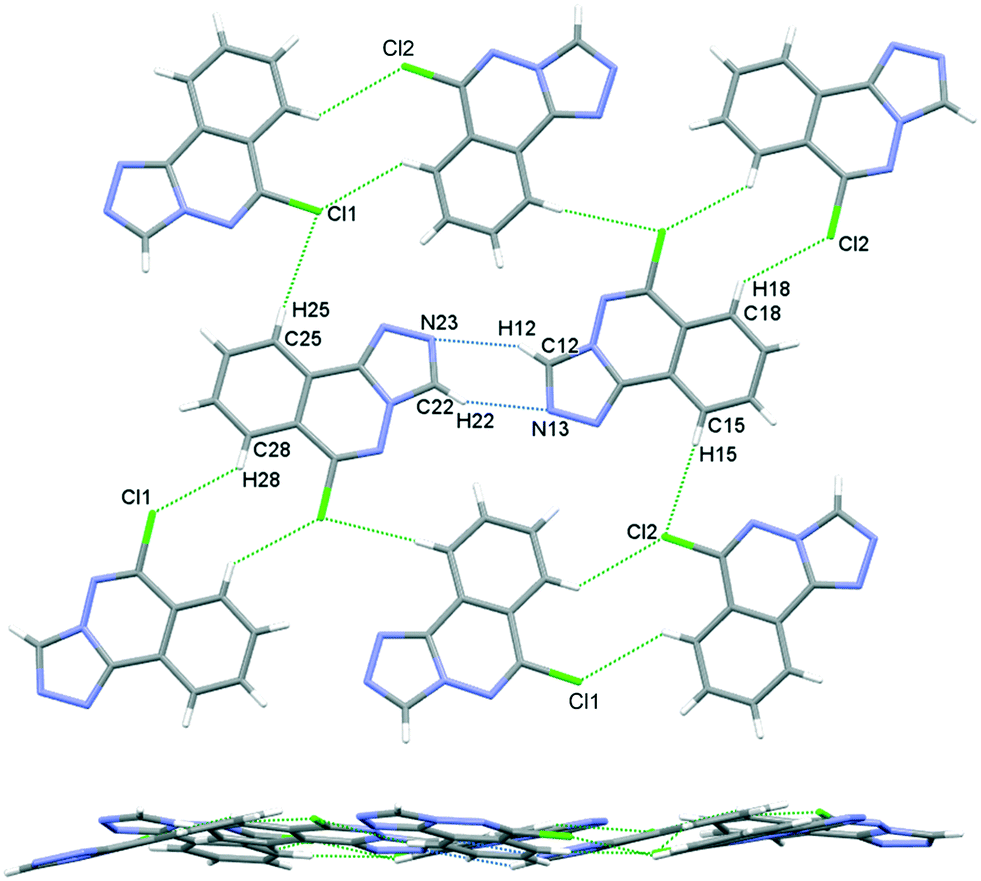
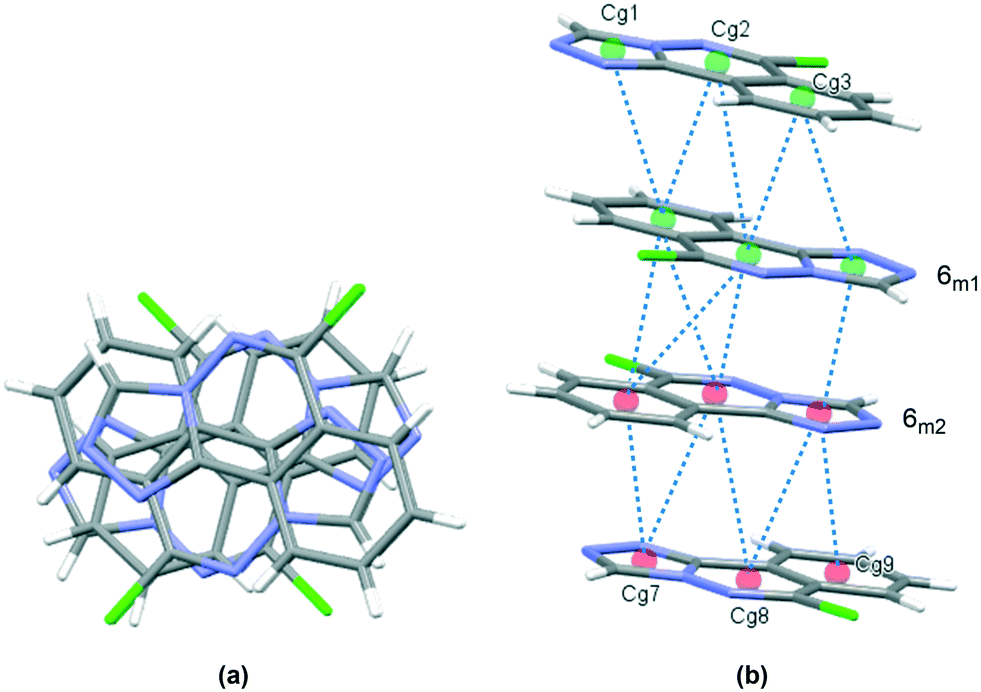
The crystal packing in 6 occurs by stacking of sheets that lie in the (101) plane. The sheets are formed by the intramolecular C–H⋯N hydrogen bonds that link the two molecules in the asymmetric unit, and weak C–H⋯Cl interactions.63 The Cl1 atom acts as a bifurcated acceptor to C25–H25 and C28–H28 donors, and Cl2 relates similarly to C15–H15 and C18–H18, Fig. 6. Completing the packing in 3-dimensions are extensive π⋯π interactions between the electron-rich ring systems, Table 5. Inversion related 6m1 pairs stack with similar 6m2 inversion pairs to generate a 4-layer repeat unit in the [101] direction, Fig. 7.
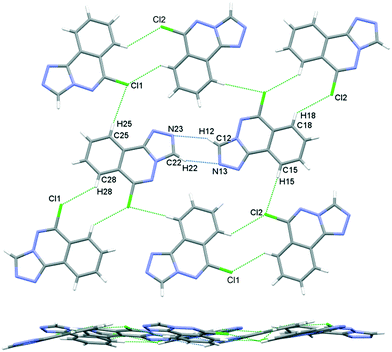 |
| | Fig. 6 Crystal packing in 6, sheets viewed orthogonal to the (101) plane (upper figure), and parallel to the (101) plane (lower figure). | |
Table 5 π⋯π interactions for 6
| Cg⋯Cg |
Distance Å |
|
Cg1, Cg2, Cg3, Cg7, Cg8 and Cg9 are the centroids of the N11, C12, N13, N14, C13; N11, N12, C110, C19, C14, C13; C14⋯C19; N21, C22, N23, N24, C23; N21, N22, C210, C29, C24, C23 and C24⋯C29 rings, respectively. Symmetry codes: (i) 1 − x, 2 − y, 1 − z; (ii) x, 3/2 − y, 1/2 + z; (iii) 1 − x, 2 − y, 1 − z.
|
| Cg1⋯Cg3(i) |
3.8277(14) |
| Cg2⋯Cg2 |
3.7735(14) |
| Cg2⋯Cg3 |
3.4336(13) |
| Cg1⋯Cg7(ii) |
3.4993(14) |
| Cg2⋯Cg8 |
3.9932(13) |
| Cg2⋯Cg9 |
3.8577(14) |
| Cg3⋯Cg8 |
3.7206(14) |
| Cg3⋯Cg9 |
3.6266(15) |
| Cg7⋯Cg8(iii) |
3.4937(14) |
| Cg7⋯Cg9 |
3.5447(14) |
| Cg8⋯Cg8 |
3.6763(14) |
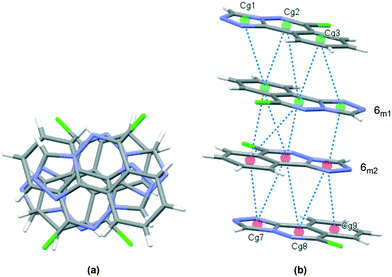 |
| | Fig. 7 Crystal packing in 6, stacks viewed orthogonal to the (101) plane (a), and approximately parallel to the (101) plane (b). π⋯π contacts between ring centroids are shown as blue dotted lines with details given in Table 5. | |
3.3. Hirshfeld surface analysis
In order to visualize the intermolecular interactions in the crystals of compounds 5 and 6, Hirshfeld surface (HS) analyses64 were carried out by using Crystal Explorer 17.5.65 In the HS plotted over dnorm (Fig. 8a and b), the white surface indicates contacts with distances equal to the sum of van der Waals radii, and the red and blue colours indicate distances shorter (in close contact) or longer (distinct contact) than the van der Waals radii, respectively.66 The appearing bright-red spots indicate their roles as the respective donors and/or acceptors; they also appear as blue and red regions corresponding to positive and negative potentials on the HS mapped over electrostatic potential67 as shown in Fig. S1† (for 5). The blue regions indicate the positive electrostatic potential (hydrogen-bond donors), while the red regions indicate the negative electrostatic potential (hydrogen-bond acceptors). The shape-index of the HS is a tool to visualize the π⋯π stacking by the presence of adjacent red and blue triangles; if there are no adjacent red and/or blue triangles, then there are no π⋯π interactions. Fig. S2(a and b)† suggests that there are π⋯π interactions in both compounds.
 |
| | Fig. 8 Views of the three-dimensional Hirshfeld surfaces of compounds 5 (Fig. 8a) and 6 (Fig. 8b) plotted over dnorm in the ranges of −0.1682 to 1.0474 a.u. (for 5) and −0.0860 to 1.0309 a.u (for 6). | |
The overall two-dimensional fingerprint plots, Fig. S3(a and b)† a, and those delineated into H⋯H, H⋯N/N⋯H, H⋯Cl/Cl⋯H, H⋯C/C⋯H, C⋯N/N⋯C, C⋯Cl/Cl⋯C, C⋯C, N⋯N and N⋯Cl/Cl⋯N (for compound 5) and H⋯N/N⋯H, H⋯Cl/Cl⋯H, H⋯H, C⋯C, C⋯N/N⋯C, H⋯C/C⋯H, N⋯Cl/Cl⋯N, C⋯Cl/Cl⋯C and Cl⋯Cl (for compound 6) contacts68 are illustrated in Fig. S3(a)† b–j (for compound 5) and Fig. S3(b)† b–k (for compound 6), together with their relative contributions to the Hirshfeld surfaces. The important interactions H⋯H contribute 25.3% (for 5) and 18.3% (for 6) to the overall crystal packing, which are reflected in Fig. S3(a and b)† b (for 5) and d (for 6) as the widely scattered points of high density due to the large hydrogen contents of the molecules with the tips at de = di = 1.33 Å (for 5) and de = di = 1.13 Å (for 6). The pairs of characteristic wings resulting in the fingerprint plots delineated into H⋯N/N⋯H, Fig. S3(a and b)† c (for 5) and b (for 6) contacts with 25.3% (for 5) and 23.7% (for 6) contributions to the HSs are viewed as pairs of spikes with the tips at de + di = 2.38 Å (for 5) and de + di = 2.52 Å (for 6). The H⋯Cl/Cl⋯H contacts contributing 16.0% (for 5) and 20.8% (for 6) to the overall crystal packing, are reflected in Fig. S3(a and b)† d (for 5) and c (for 6) with the tips at at de + di = 2.98 Å (for 5) and de + di = 2.73 Å (for 6). In the absence of C–H⋯π interactions (for 5 and 6), the H⋯C/C⋯H contacts contributing 12.7% (for 5) and 6.1% (for 6) to the overall crystal packing, are reflected in Fig. S3(a and b)† e (for 5) and g (for 6) with the tips of the wings at de + di = 2.76 Å (for 5) and de + di = 3.17 Å (for 6). Hirshfeld surface representation, crystal voids and interaction energy frameworks are given in supporting information (Fig. S4–S7†).
3.4. Theoretical (DFT) study
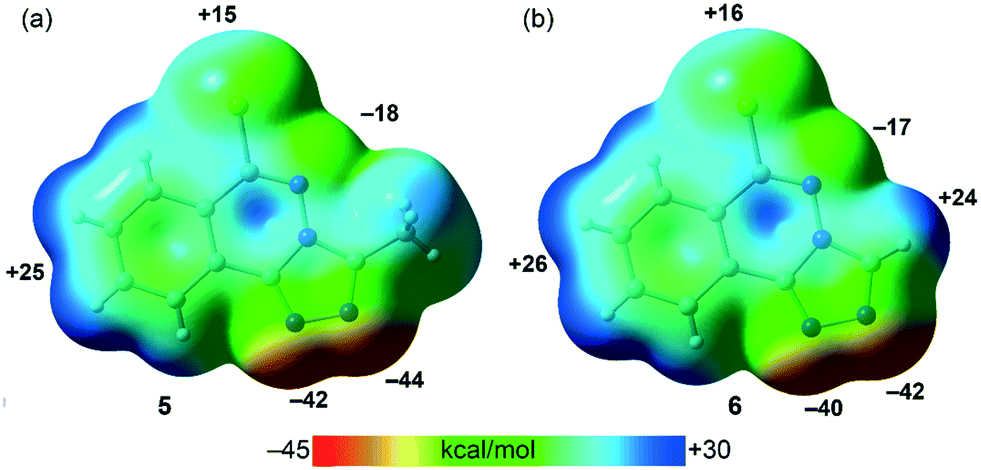
PBE0-D3/def2-TZVP calculations have been used to study the H-bonding and π-stacking interactions described above that govern the crystal packing of compounds 5 and 6 (sections 3.2.2. and 3.2.4.). The molecular electrostatic potential (MEP) surfaces of both compounds have been firstly computed to analyze the most nucleophilic and electrophilic parts of the molecules (Fig. 9). As expected, the most negative MEP values are located at the N-atoms triazole rings, ranging from −40 to −44 kcal mol−1. The MEP values are also negative (−17 and −18 kcal mol−1 for 5 and 6, respectively) at the N-atom of the six-membered ring. The most positive values are located at the aromatic H-atoms (+24 to +26 kcal mol−1) and at the chlorine's σ–hole (+15 kcal mol−1 in 5 and +16 kcal mol−1 in 6).
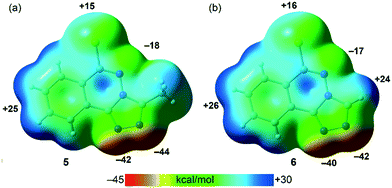 |
| | Fig. 9 MEP surfaces (isosurface 0.001 a.u.) of 5 (a) and 6 (b) at the PBE0-D3/def2-TZVP level of theory. The MEP values at selected points of the surfaces are given in kcal mol−1. | |
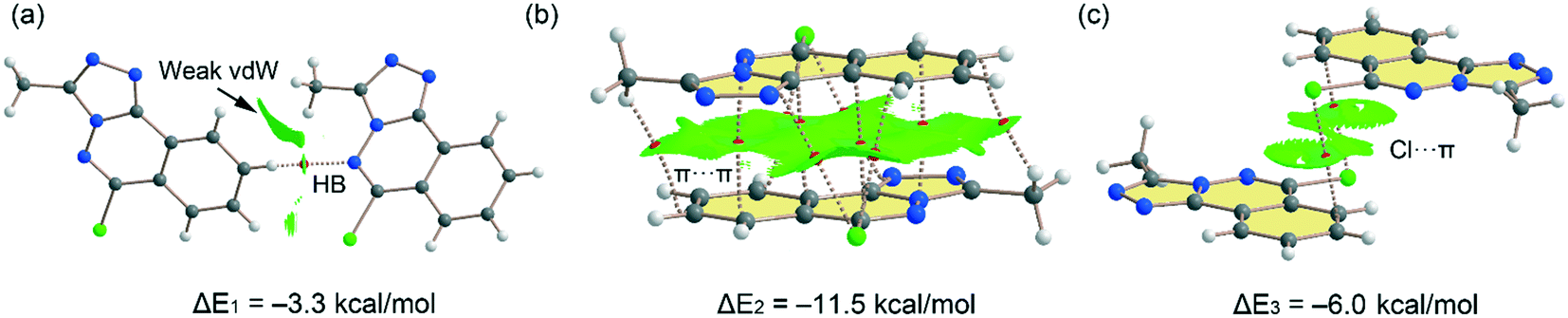
Fig. 10 shows the quantum theory of atoms in molecules (QTAIM) and non-covalent interaction plot (NCIPlot) analyses of three dimers extracted from the solid state of compound 5. Some of these packings have been commented above and highlighted in Fig. 3 and 4. The one in Fig. 10c depicts that a parallel displaced stacking is formed where the Cl-atoms are located over benzene rings. For the H-bonded dimer, the QTAIM analysis shows the formation of bond critical point (CP, red sphere) and bond path connecting the C–H to the N-atom, thus evidencing the H-bond. The interaction is further characterized by a green NCIplot index isosurface, confirming the attractive nature of the interaction. The NCIPlot also reveals the existence of van der Waals interactions between the methyl group of the triazole ring and the aromatic protons (extended green isosurface). The interaction energy is modest (ΔE1 = −3.3 kcal mol−1), due to the low H-bond ability of the C–H group. In contrast, the π-stacked dimer (Fig. 10b) presents a large dimerization energy (ΔE2 = −11.5 kcal mol−1), confirming its relevance in the solid state of compound 5 and that π-stacking forces are more important than H-bonds. The antiparallel orientation of both π-systems also explains the large interaction energy, since this arrangement maximizes the dipole⋯dipole interaction. The π-stacking interaction is characterized by multitude of bond CPs connecting several atoms of the aromatic rings including the Cl and methyl substituents. In fact, the overlap of the π-clouds is large as confirmed by NCIPlot index analysis that shows an extended isosurface embracing both π-systems and substituents. Fig. 10c shows an additional dimer where two symmetrically equivalent Cl⋯π interactions are established. Each one is characterized by a bond CP and bond path connecting the Cl-atom to one C-atom of the benzene ring. The NCIplot isosurface embraces the whole benzene ring, thus evidencing that the whole π-system participates in the interaction. Each Cl⋯π interaction contributes in 3.0 kcal mol−1 to the stabilization of the self-assembled dimer, that is comparable to the C–H⋯N H-bond observed in the H-bonded dimer (Fig. 10a).
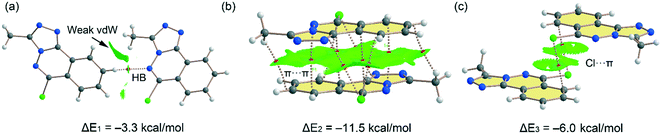 |
| | Fig. 10 (a–c) Combined QTAIM (bond critical points are in red) and NCIplot analyses of three dimers of 5. The dimerization energies are also indicated at the PBE0-D3/def2-TZVP level of theory. Only intermolecular bond CPs and NCIplot isosurfaces are indicated for clarity. The NCIplot parameters are: ρ cut-off = 0.04 a.u.; s = 0.5; color range −0.03 a.u. (blue) ≤ (signλ2) ρ ≤ 0.03 a.u. (red). | |
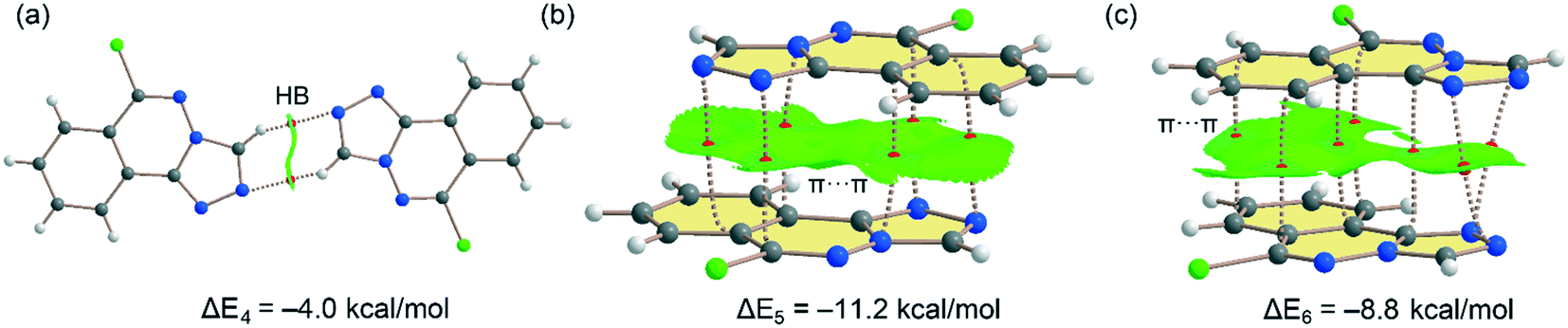
Fig. 11 shows the three dimers studied for compound 6 that have also been described in Fig. 6 and 7 (vide supra). For the H-bonded 6m1⋯6m2 dimer (Fig. 11a), each H-bond is characterized by bond CP and bond path connecting the C–H to the N-atom. The interaction is further characterized by a green NCIplot index isosurface, coincident with the location of the bond CPs. The interaction energy is modest, ΔE4 = −4.0 kcal mol−1 (−2.0 kcal mol−1 each H-bond) in line with compound 5 and the low H-bond ability of the CH group. On the other hand, a large dimerization energy (ΔE5 = −11.2 kcal mol−1) was observed for antiparallel π-stacked dimer (Fig. 11b) which reinforces its critical role in the generation of molecular architectures of compound 6. The interaction energy is like the one obtained for compound 5 and further confirms that π-stacking forces are dominant in the solid state of compounds 5 and 6. The interaction is characterized by six bond CPs interconnecting several C and N-atoms of the aromatic rings. Moreover, the interaction is also characterized by a large NCIPlot green isosurface embracing both π-systems and confirming their large overlap. Fig. 11c shows the combined QTAIM/NCIplot analysis of the other π-stacking mode observed in compound 6 (between molecules 6m1 and 6m2, see Fig. 6). In this case the orientation is not antiparallel and consequently the interaction energy is smaller (ΔE6 = −8.8 kcal mol−1). The interaction is characterized by seven bond CPs interconnecting both molecules and a large and green isosurface, also evidencing a large overlap of the π-system in this π-stacking mode. Finally, it should be mentioned that there are many examples in the literature where extended π-systems exhibit large dimerization energies due to π-stacking interactions, much stronger than H-bonds.69
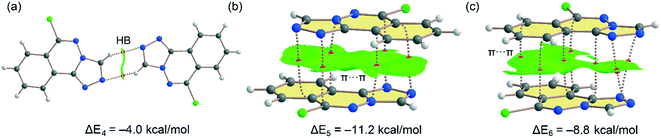 |
| | Fig. 11 (a–c) Combined QTAIM (bond critical points are in red) and NCIplot analyses of three dimers of 6. The dimerization energy is also indicated at the PBE0-D3/def2-TZVP level of theory. Only intermolecular bond CPs and NCIplot isosurfaces are indicated for clarity. The NCIplot parameters are: ρ cut-off = 0.04 a.u.; s = 0.5; color range −0.03 a.u. (blue) ≤ (signλ2) ρ ≤ 0.03 a.u. (red). | |
3.5. Biological screening
Having studied the intermolecular and surface properties of the two phthalazines in solid state we moved on to look for correlations in a biological setting, docking studies in the binding pocket of the metalloenzyme urease (EC 3.5.1.5).
3.5.1. Urease inhibition.
Urease (EC 3.5.1.5), a nickel-containing metalloenzyme responsible for the hydrolysis of urea to ammonia and carbamate, is an important virulent factor in the pathogenesis of several diseases which are detrimental to human and animal health as well as to agriculture.70 With respect to the latter, the increased production of ammonia as a result of high urease activity can lead to severe environmental and economic problems.71 For instance, it causes the ammonia level to increase, thus increasing the pH of the soil and also depriving plants of their essential nutrients.72 A variety of urease inhibitors have been developed to treat various clinical conditions, however, 1,2,4-triazolo[3,4-a]phthalazines have not been reported before. Therefore, in the current study, in vitro evaluation of 6-chloro-3-methyl-[1,2,4]triazolo[3,4-a]phthalazine 5 and 6-chloro-[1,2,4]triazolo[3,4-a]phthalazine 6 against urease enzyme was performed using indophenol method.44 Thiourea was employed as a standard inhibitor showing IC50 value of 22.3 ± 1.06 μM. Both compounds demonstrated remarkable biological efficacy with an IC50 value of 19.3 ± 1.20 and 0.32 ± 0.02 μM, respectively (Table 6). Compound 5 emerged as a slightly better inhibitor than the thiourea standard whereas 6 displayed ∼70-fold higher activity indicating that the presence of a methyl substituent on the triazole ring could be a restraining factor for the low inhibition potential of compound 5 (Fig. 12).
Table 6 Urease inhibitory activity of 1,2,4-triazolo[3,4-a]phthalazines (5 and 6)
| Compound |
Urease inhibition |
| IC50 ± SEM (μM) |
|
5
|
19.3 ± 1.20 |
|
6
|
0.32 ± 0.02 |
| Thiourea |
22.3 ± 1.06 |
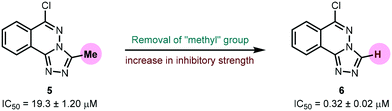 |
| | Fig. 12 Effect of methyl group on urease inhibition. | |
3.5.2. Mechanism of inhibition.
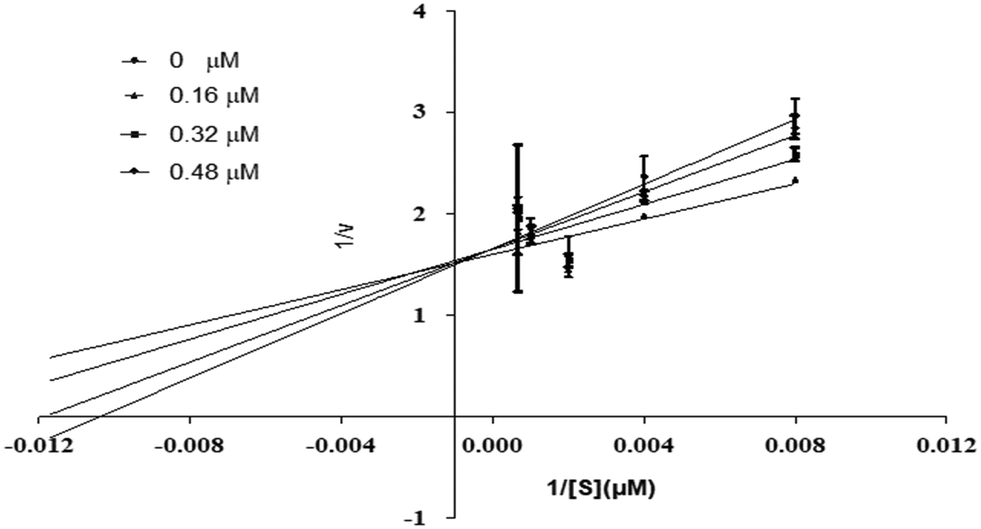
Using urea as a substrate, the mechanism of action of the most potent compound 6 was determined by performing kinetics studies. For determining the type of inhibition and analyzing the effects of the inhibitor on Vmax and Km, a Lineweaver–Burk graph (reciprocal of rate of reaction 1/S and reciprocal of substrate concentration 1/V) was utilized. To estimate the value of Ki, the slope Km/Vmax of each line in the Lineweaver–Burk plot was plotted against various concentrations of substrate and compounds.
Compound 6 was subjected to kinetic studies with its various concentrations and that of substrate. There were four concentrations of compound 6 (0, 0.16, 0.32, 0.48 μM) and five concentrations of substrate (125, 250, 500, 1000, and 1500 μM, with 1/S values 0.00067, 0.001, 0.002, 0.004, and 0.008). In the active site of urease, compound 6 competes with substrate (urea) for binding. As demonstrated in Fig. 13, the enzyme's Vmax was unaffected but the urease's Km increased, indicating competitive inhibition. When lines intersect at the y-axis in competitive inhibition, the value of Vmax does not change, but the value of Km increases.
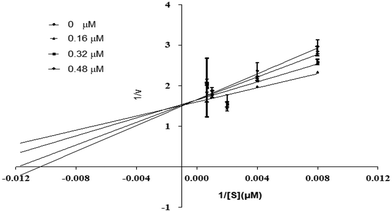 |
| | Fig. 13 Inhibition of urease by compound 6. Lineweaver–Burk graph showing reciprocal rate of reaction 1/V against reciprocal of substrate 1/S. | |
3.5.3. Molecular docking analysis.
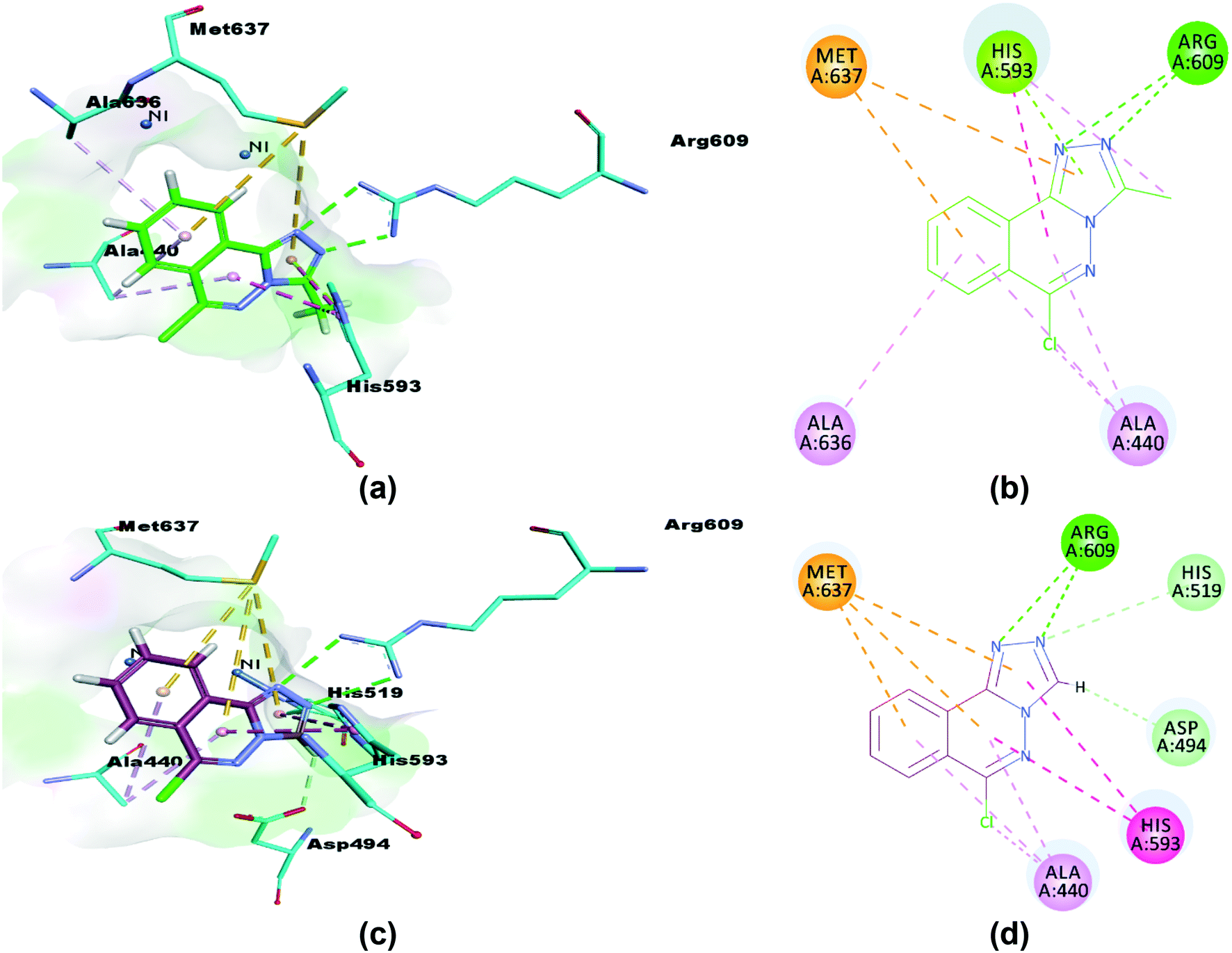
The synthesized compounds (5 and 6) were docked inside the urease binding pocket to investigate their affinities with the receptor's vital residues.49 The analysis revealed short contacts between amino acid residues ARG609, HIS593, MET637, ALA440, and ALA636 inside the active pocket with compound 5. More specifically, the two nitrogen atoms of the triazole ring showed traditional intermolecular N⋯H–N hydrogen bonds with ARG609 in the binding site, Table 7. π–π T-shaped (edge-to-face) and π-lone pair interactions were found between ND1-HIS593 and the phthalazine and triazole rings, respectively. HIS593 also showed an alkyl interaction (a weak C–H⋯N hydrogen bond) with the methyl group (C15) on the triazole ring that defines 5. The close contacts to MET637 are S–π interactions to phthalazine (5.28 Å) and triazole (5.69 Å) rings.73 Completing the pocket⋯5 correspondence were two π–alkyl interactions (C–H⋯π) between the ALA440 residue (in the catalytic region of urease) and phthalazine rings, and similar from the ALA636 residue, Fig. 14(a and b).
Table 7 Hydrogen bonding interactions for 5 and 6
| Compound |
Binding interactions |
| Ligand atoms |
Receptor atoms |
Interaction |
Distance (Å) |
|
NH1: nitrogen atom 1 of the guanidinium group of arginine; NH2: nitrogen atom 2 of the guanidinium group of arginine; NE2: ε-nitrogen of histidine imidazole ring; CE1: ε-carbon of histidine imidazole ring; OD1: carboxylate of aspartate; ND1: the imidazole ring nitrogen of histidine. Numbers automatically generated by docking software 5 N13, N12, C15 & N8 cross-reference to N3, N4, C1 & N1 in Fig. 2; similarly 6 N13, N12 & H19 are N13/23, N14/24 & H12/22 in Fig. 5.
|
|
5
|
N13 |
NH1-ARG609 |
–R609-NH⋯N13-(5 triazole) |
3.46 |
| N12 |
NH2-ARG609 |
–R609-NH⋯N12-(5 triazole) |
3.77 |
| C15 |
ND1-HIS593 |
(5 methyl)-C15H⋯Nδ-H593 |
4.34 |
| N8 |
NE2-HIS593 |
–H593-NH⋯N8-(5 phthalazine) |
2.85 |
|
6
|
N13 |
NH1-ARG609 |
–R609-NH⋯N13-(6 triazole) |
3.12 |
| N12 |
NH2-ARG609 |
–R609-NH⋯N12-(6 triazole) |
3.82 |
| N13 |
CE1-HIS519 |
–H519-CεH⋯N13-(6 triazole) |
2.99 |
| H19 |
OD1-ASP494 |
(6 triazole)-CH⋯O-D494 |
2.67 |
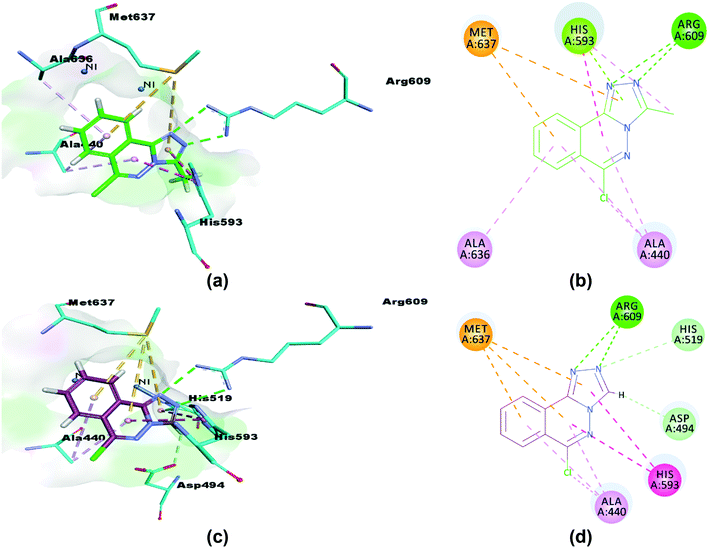 |
| | Fig. 14 3D and 2D interactions of compounds 5 (a and b) and 6 (c and d) with amino acid residues. π–π T-shaped interactions are shown as fuchsia, hydrogen bond as green, S–π as yellow and π–alkyl interactions as light pink dashed lines. | |
Similarly, compound 6 was docked in the catalytic domain of urease with its bi-nickel core. As with 5, ARG609 showed two intermolecular hydrogen bonds with N12 and N13 of the guest triazole ring. A C–H⋯N hydrogen bond interaction was observed between the N13 nitrogen of the triazole ring docked deeply in the active site with the hydrogen of the imidazole ε carbon of HIS519 residue. The HIS593 residue again formed two π–π interactions with triazole (4.71 Å) and phthalazine (4.63 Å) rings, and a short C–H⋯O hydrogen bond was seen between the 6 triazole ring carbon and an oxygen of the ASP494 carboxylate. Analysis of the interactions is completed with a repeat of the two binding motifs observed for 5 with AL440 and MET637 residues, namely the pairs of π–alkyl (phthalazine rings to ALA440) and S–π interactions (MET637 with triazole and phthalazine rings), Fig. 14(c and d). Of note, neither compound 5 nor 6 show any interactions with the metals of the binding pocket. Based on our results, we predict that our compounds are mechanism based inhibitors, which are basically unreactive molecules that inhibit the enzyme.
The binding interactions inside the catalytic domain of the enzyme validated the in vitro results of both compounds demonstrating varied inhibition against urease. Furthermore, both compounds had negative free energy values and were observed to attach with a high affinity.
3.5.4. HYDE affinity calculations.
The HYDE affinity method was used to evaluate the top 30 docked positions.53 The LeadIT software was used to evaluate both ligands. FlexX was used to estimate the docking score and binding free energy for compounds 5 and 6 (Table 8). The results showed that the compounds have a high affinity for the urease active site, as revealed by binding free energies. Both compounds 5 and 6 provided significant scores by FlexX and potent binding free energies after docking studies. The docking score provided the score which compound has depicted within the active pocket of enzyme during the docking studies while the binding free energy is the score of the selected poses and suggests the strength of binding of inhibitor within the active pocket. Compound 6 (without methyl substituent at triazole ring) showed strong docking score as compared to 5. Moreover, the selected pose of 6 for HYDE assessment was 3 whereas, pose 1 was used for 5, which suggests the strength of binding within the active pocket. The selected poses (poses used for the HYDE assessment and calculation) of docked compounds 5 and 6 within the pocket of protein (urease) during HYDE calculation are given in Fig. S9.†
Table 8 Docking scores, binding free energies and their corresponding ranks by Hyde affinity assessment
| Compound |
Docking score by FlexX |
Pose rank |
Binding free energy ΔG (kJ mol−1) |
|
5
|
−14 |
1 |
−18.50 |
|
6
|
−16 |
3 |
−18.63 |
3.5.5. SeeSAR visual drug design.
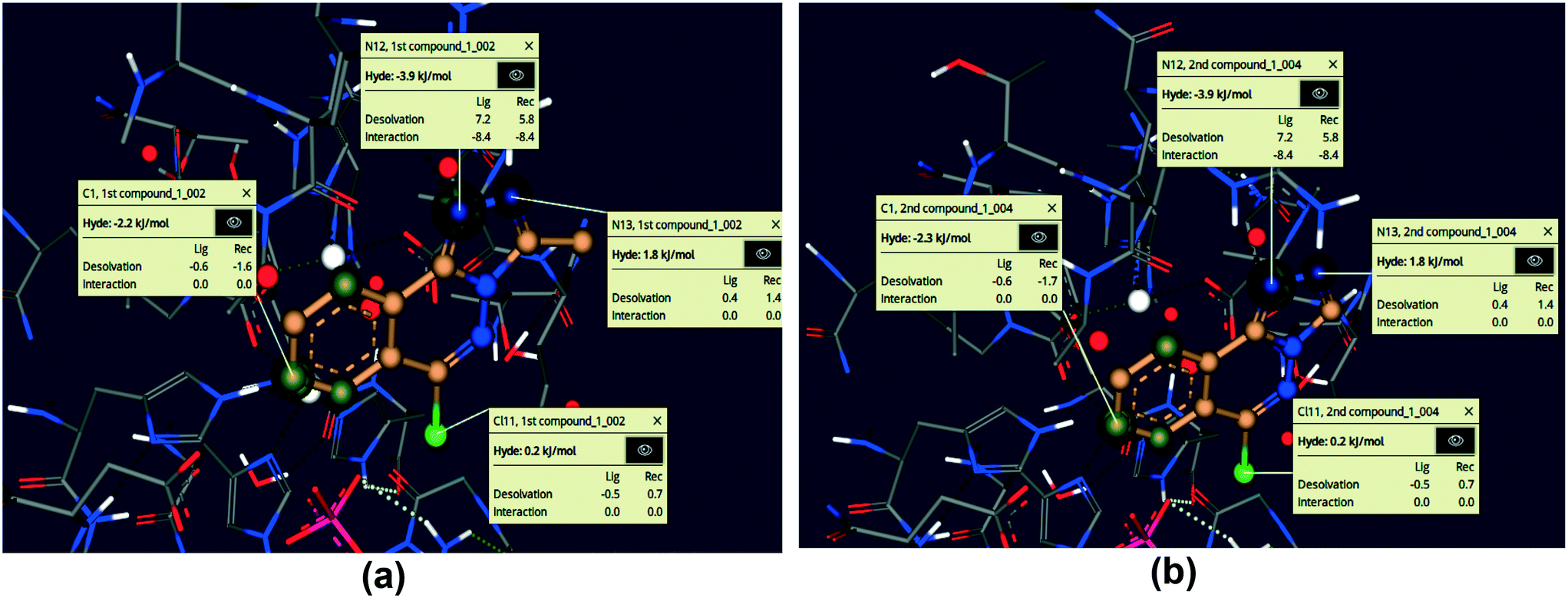
Using the SeeSAR tool of LeadIT software, the visual and analytical modes of the docked posture of compounds 5 and 6 showed explainable, novel, and remarkable conformations.53,74Fig. 15 illustrates the binding and non-binding capability of compounds after iterative and interactive leads adjustment and depicts desolvation and interactions for both compounds. The algorithm estimates visual and interpretable output for explicit hydrogen bonding and dehydration, and it verifies our molecular docking results achieved with FlexX default parameters. The results of Fig. 15 depict the conformation of both inhibitors and their individual atoms within the active pocket of enzyme. The calculations by SeeSAR offers precise results and binding of individual atoms of the inhibitors within the active site of protein.
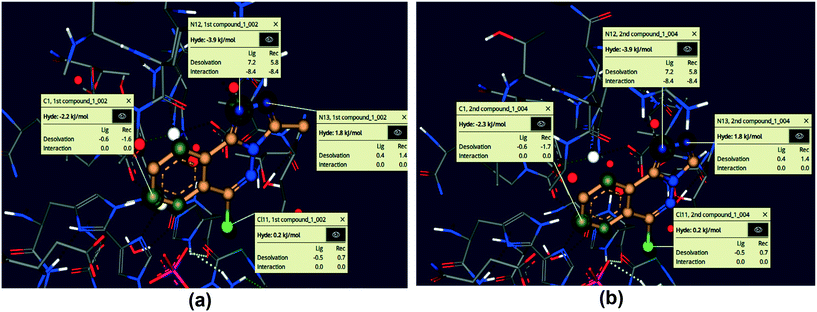 |
| | Fig. 15 Visual and investigative modes of the docked pose of compounds 5 (a) and 6 (b) within the active site of urease (pdb: 3LA4). The green colour represents the favourable and contributing atoms showing interactions inside the active pocket, while light green are chloride atoms. | |
3.5.6. Molecular dynamics simulations.
Molecular dynamics simulations for lead compound 6 were performed. MD simulations of the enzyme in complex with the selected inhibitor 6 were performed in an aqueous environment for 30 ns, with initial conformations derived from the docking pose with the lowest binding free energy. Noncovalent interactions between active site of urease and ligand 6 were observed in a time dependent manner.
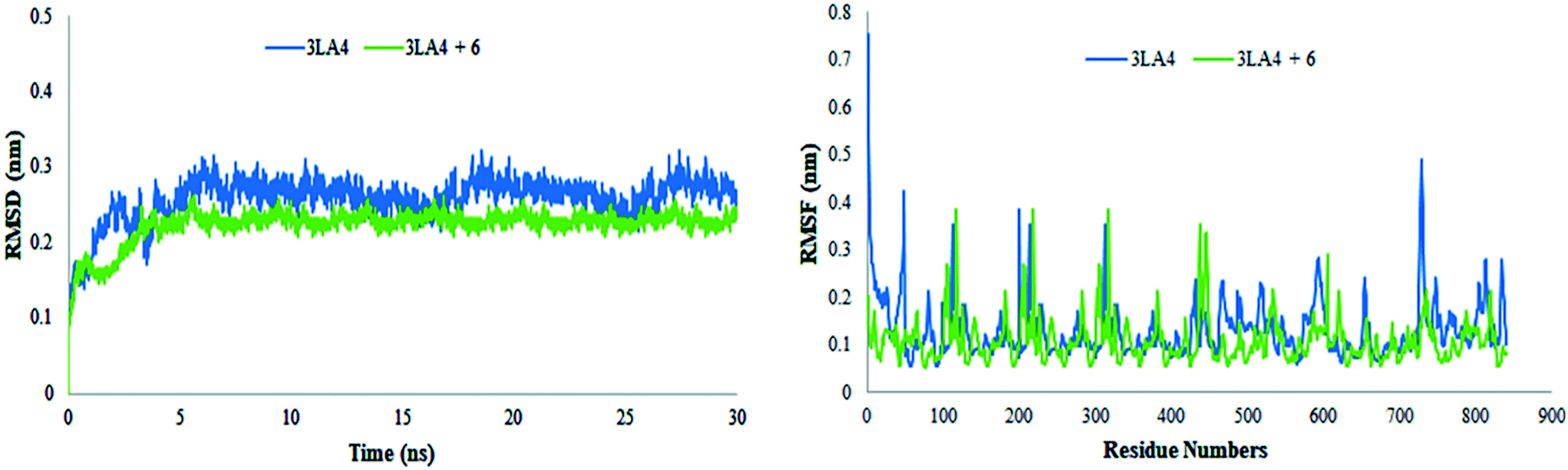
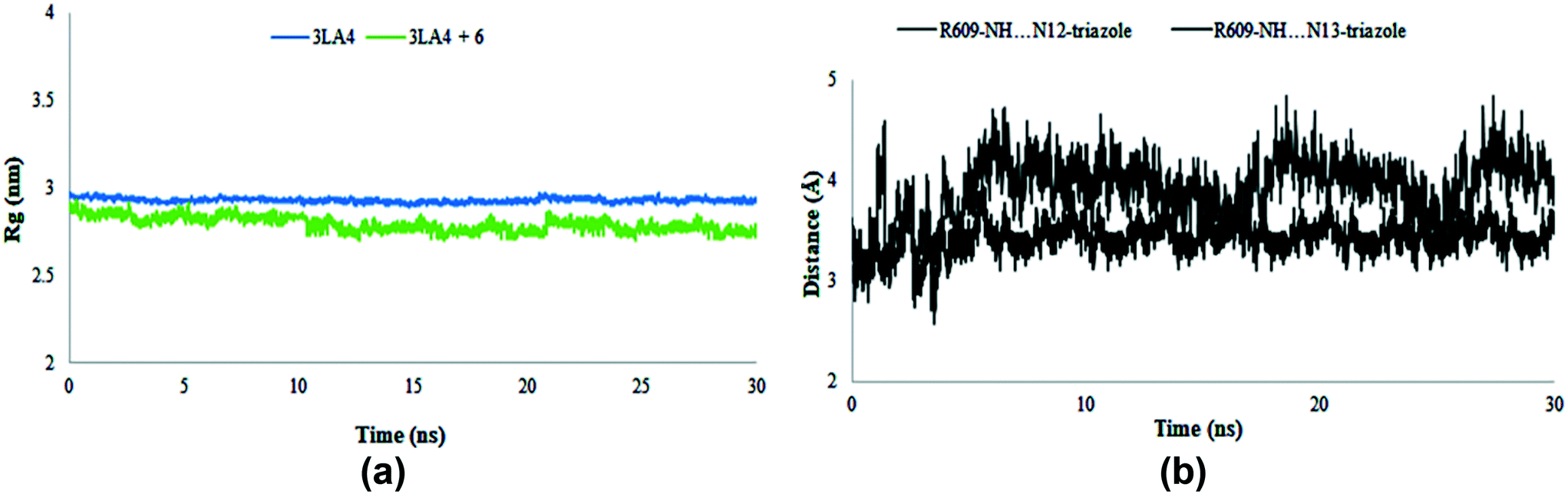
The results of MD simulations are presented as RMSD values, which provided insight into the general stability of the protein and its inhibitor complex. Fig. 16 shows that in the range of 0–0.2 nm, selected ligand 6 exhibited significant stability and minimum variation. The structure of apo protein showed minor alterations, however, the structure of the complex was found stable after 3 ns. When compared to protein alone, the structure of the complex (6 + protein) exhibited a minor change between 16–17.5 ns and 24–26 ns, while the remaining simulations were proven to be stable. There were few variations in protein throughout the simulation (Fig. 16).
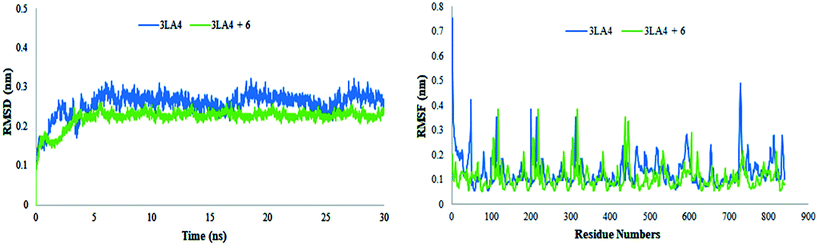 |
| | Fig. 16 Root mean square deviations and fluctuations of amino acid residues of protein (3LA4) during 30 ns simulation time in the absence and presence of compound 6. | |
The value of root mean square variation provides detail on the computations performed to determine the flexibility of the receptor structure in the absence and presence of compound. The systems containing apo and holo proteins revealed a notable pattern of variations, as seen in Fig. 16. The apo protein began at 0.7 nm and fluctuated between 0.1 and 0.4 nm during the simulation time, eventually reaching 0.5 nm. During the simulations, the region with motifs and loops exhibited less fluctuation, but the active site pocket showed great stability. In comparison to protein alone, the results showed greater overall stability of the complex. The stability of internal motion in protein and complex systems was represented by the results of protein structure.
The radius of gyration was evaluated during the MD simulation time frame and gave deep insight about the compactness of the system. Furthermore, it elaborates the folding and unfolding of the structure of protein in the absence and presence of compound 6. The results shown in Fig. 17 suggested the compactness of the system in the presence of selected compound. The average Rg score for urease and its combination with chemical 6 was determined to be 2.9 nm throughout the simulations, indicating the compactness of structures. The distance of intermolecular hydrogen bonds between N12 and N13 of triazole ring and the most crucial amino acid residue ARG609 is shown in Fig. 17b during the whole MD simulation time frame. The results indicated that the distance fluctuated between 3–5 Å during 30 ns within the binding site of protein. These results contribute towards the stability of hydrogen bonds between triazole ring and ARG609 inside the binding site, therefore playing a significant role in the increased affinity of compound 6 towards urease enzyme.
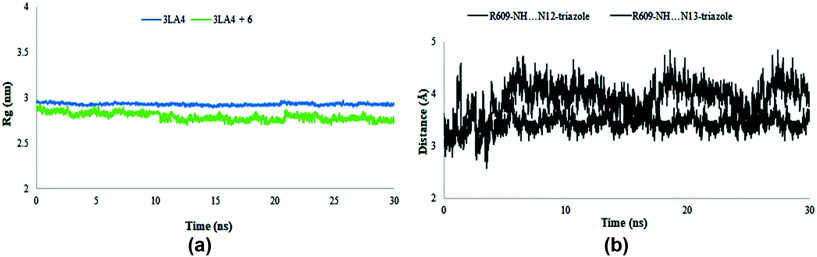 |
| | Fig. 17 Radius of gyration (Rg) of amino acid residues in the presence and absence of compound 6 (a) and distance of hydrogen bonds of N12 and N13 of triazole ring and ARG609 (b) during 30 ns MD simulation run. | |
The noncovalent interactions depicted by the crystal structures of compounds 5 and 6 play important role when docked within the active site of protein. These interactions are responsible for the biological activity against urease enzyme. Furthermore, the results of molecular dynamics studies (RMSD, RMSF, Rg) reveal the stability of the protein ligand complex indicating the strength of noncovalent interactions.
3.5.7. Pharmacokinetic properties.
The impact of different variables on the pharmacokinetic features of drugs (5 and 6) was assessed using previously reported prediction methods.75 These variables include molecular weight, polar surface area, number of atom types (donor/acceptor), molecular refractivity, and lipophilicity, i.e., partition coefficients such as logPo/w, n-octanol, WLOGP, MLOGP, and XLOGP3 that represent solvation free energies and solvent attainable surface area.76 Furthermore, water solubility anticipates compound solubility. These characteristics indicate the drug-likeness and permeability of test compounds across the blood–brain barrier. Table S1† shows that the tested derivatives are safe to be used as drugs and that they can cross the blood–brain barrier.
4. Conclusions
In summary, the present study unravelled the role of noncovalent interactions such as C–H⋯Cl, C–H⋯N, π⋯π, and the Cl⋯π contacts in the generation and stabilization of supramolecular assemblies of two [1,2,4]triazolo[3,4-a]phthalazine compounds which were accessed via a facile multi-step synthetic approach. The crystal structure of compound 5 showed intermolecular C–H⋯N hydrogen bonds, and π stacking interactions. However, two unique molecules of compound 6 demonstrate intermolecular C–H⋯N and weak CH⋯Cl bonding, and extensive π–π interactions within and between pairs of 6m1 and 6m2 molecules of 6. The energetic analysis performed using DFT methods have also demonstrated the dominant role of π-stacking compared to hydrogen bonding interactions. The combined QTAIM/NCIplot analysis also supports the relevance and structure directing role of π-stacking forces in the solid state of compounds 5 and 6. Finally, the urease inhibitory potential of both phthalazine derivatives 5 and 6 was assessed where compound 6 showed ∼70-fold strong inhibition of urease enzyme compared to thiourea (standard inhibitor) and demonstrated an IC50 value of 0.32 ± 0.02 μM. Kinetics studies of 6 suggested a competitive inhibition mechanism while molecular docking analysis revealed various vital interactions of inhibitors with active site amino acid residues augmenting the in vitro urease inhibitory potential. ADME analysis of our compounds demonstrated that these are likely to penetrate the blood brain barrier and influence the relevant biological properties. Taken together, the investigated compounds not only possess crystal engineering properties but also display a remarkable potential to serve as drug candidates for suppressing the effects of ureolytic bacteria.
Author contributions
Sumera Zaib: writing – review & editing original draft, molecular docking analysis, bioactivity, software, funding, Aliya Ibrar: synthesis, formal analysis, investigation, Marriyam Ramay: data curation, bioactivity, Shabab Zahra: data curation, bioactivity, Tuncer Hökelek: data curation, Hirshfeld surface analyses, crystal voids, interaction energy calculations, energy frameworks, Jim Simpson: X-ray crystallography, writing, data curation, software, Christopher John McAdam: X-ray crystallography, Nasser S. Awwad: visualization, formal analysis, resources, funding acquisition, Hala A. Ibrahium: visualization, resources, funding acquisition, Antonio Frontera: DFT analysis, writing, data curation, software, Imtiaz Khan: conceptualization, supervision, synthesis and characterization, writing – review & editing original draft, formal analysis, methodology, investigation, visualization, project administration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for supporting this work through research groups program under grant number R.G.P.2/164/43. We thank the MINIU/AEI from Spain for financial support (project number CTQ2017-85821-R, FEDER funds). We are also grateful to the CTI (UIB) for free allocation of computer time.
References
- M. O. Sinnokrot, E. F. Valeev and C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, 10887–10893 CrossRef CAS PubMed.
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS.
- F. Biedermann and H. J. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS PubMed.
-
Noncovalent forces- challenges and advances in computational chemistry and physics, ed. S. Scheiner, Springer, Dordrecht, 2015 Search PubMed.
-
(a) P. Pal, K. Das, A. Hossain, A. Frontera and S. Mukhopadhyay, New J. Chem., 2020, 44, 7310–7318 RSC;
(b) K. Shivakumar, A. Vidyasagar, A. Naidu, R. G. Gonnade and K. M. Sureshan, CrystEngComm, 2012, 14, 519–524 RSC;
(c) S. Pingali, J. P. Donahue and F. Payton-Stewart, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 388–391 CrossRef CAS PubMed;
(d) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed.
- H. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
-
Non-covalent interaction in the synthesis and design of new compounds, ed. A. M. Maharramov, K. T. Mahmudov, M. N. Kopylovich and A. J. L. Pombeiro, John Wiley & Sons, Inc., Hoboken, NJ, 2016 Search PubMed.
- L. M. Eytel, H. A. Fargher, M. M. Haley and D. W. Johnson, Chem. Commun., 2019, 55, 5195–5206 RSC.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CAS.
-
G. R. Desiraju and T. Steiner, The weak hydrogen bond in structural chemistry and biology, Oxford University Press Inc., New York, 1999 Search PubMed.
- E. H. Krenske and K. N. Houk, Acc. Chem. Res., 2013, 46, 979–989 CrossRef CAS PubMed.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed.
- T. Y. Zhang, Adv. Heterocycl. Chem., 2017, 121, 1–12 CrossRef CAS.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed.
- M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- R. Sivakumar, S. K. Gnanasam, S. Ramachandran and J. T. Leonard, Eur. J. Med. Chem., 2002, 37, 793–801 CrossRef CAS PubMed.
- D. S. Dogruer, E. Kupeli, E. Yesilada and M. F. Sahin, Archiv der Pharmazie - Chemistry in Life Sciences, 2004, 337, 303–310 CrossRef CAS PubMed.
- F. M. Awadallah, W. I. El-Eraky and D. O. Saleh, Eur. J. Med. Chem., 2012, 52, 14–21 CrossRef CAS PubMed.
- N. Curtin, Biochem. Soc. Trans., 2014, 42, 82–88 CrossRef CAS PubMed.
- M. Abbasi, S. M. R. Nazifi, Z. S. Nazifi and A. R. Massah, J. Chem. Sci., 2017, 129, 1257–1266 CrossRef CAS.
- S. Demirayak, A. C. Karaburun and R. Beis, Eur. J. Med. Chem., 2004, 39, 1089–1095 CrossRef CAS PubMed.
- N. Berber, M. Arslan, Ç. Bilen, Z. Sackes, N. Gençer and O. Arslan, Russ. J. Bioorg. Chem., 2015, 41, 414–420 CrossRef CAS.
-
https://go.drugbank.com/drugs/DB00972
.
-
https://go.drugbank.com/drugs/DB01275
.
-
https://go.drugbank.com/drugs/DB00962
.
-
https://drugs.ncats.io/drug/S0177QHV2B
.
- C.-P. Wu, S. Lusvarghi, P.-J. Tseng, S.-H. Hsiao, Y.-H. Huang, T.-H. Hung and S. V. Ambudkar, Am. J. Cancer Res., 2020, 10, 164–178 CAS.
- O. Fedorov, H. Lingard, C. Wells, O. P. Monteiro, S. Picaud, T. Keates, C. Yapp, M. Philpott, S. J. Martin, I. Felletar, B. D. Marsden, P. Filippakopoulos, S. Müller, S. Knapp and P. E. Brennan, J. Med. Chem., 2014, 57, 462–476 CrossRef CAS PubMed.
- Y. Wu, L.-P. Sun, L.-X. Ma, J. Che, M.-X. Song, X. Cui and H.-R. Piao, Chem. Biol. Drug Des., 2013, 81, 591–599 CrossRef CAS PubMed.
-
(a) D.-Q. Xue, X.-Y. Zhang, C.-J. Wang, L.-Y. Ma, N. Zhu, P. He, K.-P. Shao, P.-J. Chen, Y.-F. Gu, X.-S. Zhang, C.-F. Wang, C.-H. Ji, Q.-R. Zhang and H.-M. Liu, Eur. J. Med. Chem., 2014, 85, 235–244 CrossRef CAS PubMed;
(b) L.-X. Maa, B.-R. Cui, Y. Wu, J.-C. Liu, X. Cui, L.-P. Liu and H.-R. Piao, Bioorg. Med. Chem. Lett., 2014, 24, 1737–1741 CrossRef PubMed.
- S. Zaib and I. Khan, Bioorg. Chem., 2020, 105, 104425 CrossRef CAS PubMed.
- H. Andleeb, I. Khan, A. Franconetti, M. N. Tahir, J. Simpson, S. Hameed and A. Frontera, CrystEngComm, 2019, 21, 1780–1793 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- S. B. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–556 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
-
(a) P. Manna, S. K. Seth, M. Mitra, S. Ray Choudhury, A. Bauzá, A. Frontera and S. Mukhopadhyay, Cryst. Growth Des., 2014, 14, 5812–5821 CrossRef CAS;
(b) M. Mirzaei, H. Eshtiagh-Hosseini, Z. Bolouri, Z. Rahmati, A. Esmaeilzadeh, A. Bauza, P. Ballester, M. Barceló-Oliver, J. T. Mague, B. Notash and A. Frontera, Cryst. Growth Des., 2015, 15, 1351–1361 CrossRef CAS.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- J. Contreras-Garcia, E. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
-
T. A. Keith, AIMAll (Version 19.02.13), TK Gristmill Software, Overland Park KS, USA, 2019, https://aim.tkgristmill.com Search PubMed.
- M. W. Weatherburn, Anal. Chem., 1967, 39, 971–974 CrossRef CAS.
- M. K. Rauf, S. Yaseen, A. Badshah, S. Zaib, R. Arshad, Imtiaz-ud-Din, M. N. Tahir and J. Iqbal, J. Biol. Inorg. Chem., 2015, 20, 541–554 CrossRef CAS PubMed.
- A. Balasubramanian and K. Ponnuraj, J. Mol. Biol., 2010, 400, 274–283 CrossRef CAS PubMed.
-
P. Labute, Protonate 3D, Chemical Computing Group, 2007, https://www.chemcomp.com/journal/proton.htm.
- Chemical Computing Group's Molecular Operating Environment (MOE), MOE 2019. 0201, https://www.chemcomp.com/MOEMolecular_Operating_Environment.htm.
- LeadIT version 2.3.2; BioSolveIT GmbH, Sankt Augustin, Germany, 2017, https://www.biosolveit.de/LeadIT.
- N. Schneider, G. Lange, S. Hindle, R. Klein and M. Rarey, J. Comput.-Aided Mol. Des., 2013, 27, 15–29 CrossRef CAS PubMed.
- BIOVIA Discovery Studio Client v19.1.0.18287, Accelrys Discovery Studio, Accelrys Software Inc, San Diego, 2019.
-
(a) R. J. Ferreira, M.-J. Ferreira, U. Daniel and J. V. A. dos Santos, J. Chem. Theory Comput., 2012, 8, 1853–1864 CrossRef CAS PubMed;
(b) B. Özgeris, S. Göksu, L. P. Köse, I. Gülcin, R. E. Salmas, S. Durdagi, F. Tümer and C. T. Supuran, Bioorg. Med. Chem., 2016, 24, 2318–2329 CrossRef PubMed;
(c) B. Mathew, A. Haridas, G. Ucar, I. Baysal, A. A. Adeniyi, M. E. Soliman, M. Joy, G. E. Mathew, B. Lakshmanan and V. Jayaprakash, Int. J. Biol. Macromol., 2016, 91, 680–695 CrossRef CAS PubMed.
- A. W. Schüttelkopf and D. M. F. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
-
P. Turner, XMGRACE, Version 5.1. 19. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR, 2005.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- G. Dutkiewicz, C. S. Chidan Kumar, H. S. Yathirajan, A. N. Mayekar and M. Kubicki, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o2694 CrossRef CAS PubMed.
- C. B. Aakeröy, T. A. Evans, K. R. Seddon and I. Pálinkó, New J. Chem., 1999, 23, 145–152 RSC.
- M. D. Prasanna and T. N. Guru Row, Cryst. Eng., 2000, 3, 135–154 CrossRef CAS.
-
(a) P. K. Thallapally and A. Nangia, CrystEngComm, 2001, 3, 114–119 RSC;
(b) R. Taylor, CrystEngComm, 2014, 16, 6852–6865 RSC.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
-
M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, M. A. Crystal Explorer17, The University of Western Australia, Nedlands, Western Australia, 2017, https://hirshfeldsurface.net Search PubMed.
- P. Venkatesan, S. Thamotharan, A. Ilangovan, H. Liang and T. Sundius, Spectrochim. Acta, Part A, 2016, 153, 625–636 CrossRef CAS PubMed.
-
D. Jayatilaka, D. J. Grimwood, A. Lee, A. Lemay, A. J. Russel, C. Taylor, S. K. Wolff, P. Cassam-Chenai and A. Whitton, TONTO - A System for Computational Chemistry, 2005, Available at: https://hirshfeldsurface.net/.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
-
(a) P. Sharma, T. Baishya, R. M. Gomila, A. Frontera, M. Barcelo-Oliver, A. K. Verma, J. Das and M. K. Bhattacharyya, New J. Chem., 2022, 46, 5296–5311 RSC;
(b) H. Nath, P. Sharma, R. M. Gomila, A. Frontera, M. Barceló-Oliver, A. K. Verma, K. Dutta and M. K. Bhattacharyya, J. Mol. Struct., 2021, 1245, 131038 CrossRef CAS;
(c) S. Chetry, P. Sharma, A. Frontera, U. Saha, A. K. Verma, B. Sarma, P. J. Kalita and M. K. Bhattacharyya, New J. Chem., 2021, 45, 3699–3715 RSC;
(d) P. Sharma, P. Sarma, A. Frontera, S. Hussain, A. K. Verma and M. K. Bhattacharyya, Inorg. Chim. Acta, 2021, 516, 120082 CrossRef CAS.
-
(a) I. Khan, S. Ali, S. Hameed, N. H. Rama, M. T. Hussain, A. Wadood, R. Uddin, Z. Ul-Haq, A. Khan, S. Ali and M. I. Choudhary, Eur. J. Med. Chem., 2010, 45, 5200–5207 CrossRef CAS PubMed;
(b) A. Ibrar, I. Khan and N. Abbas, Archiv der Pharmazie - Chemistry in Life Sciences, 2013, 346, 423–446 CrossRef CAS PubMed.
- Z. Amtul, N. Kausar, Atta-ur-Rahman, S. A. Kazmi, K. M. Khan and M. I. Choudhary, Bioorg. Med. Chem., 2006, 14, 6737–6744 CrossRef CAS PubMed.
- G. Seneviratne, L. H. J. Van Holm and E. M. H. G. S. Ekanayake, Field Crops Res., 2000, 68, 199–203 CrossRef.
- A. L. Ringer, A. Senenko and C. D. Sherrill, Protein Sci., 2007, 16, 2216–2223 CrossRef CAS PubMed.
- I. Reulecke, G. Lange, J. Albrecht, R. Klein and M. Rarey, ChemMedChem, 2008, 3, 885–897 CrossRef CAS PubMed.
-
(a) A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed;
(b) I. Khan, A. Khan, S. A. Halim, M. Khan, S. Zaib, B. Essa, M. Al-Yahyaei, A. Al-Harrasi and A. Ibrar, Int. J. Biol. Macromol., 2021, 167, 233–244 CrossRef CAS PubMed.
- P. Ertl, B. Rohde and B. P. Selzer, J. Med. Chem., 2000, 43, 3714–3717 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and crystal structures of compounds 5 and 6. CCDC 2152622 (5); 2152626 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00351a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 d,
Nasser S.
Awwad
e,
Hala A.
Ibrahium
fg,
Antonio
Frontera
d,
Nasser S.
Awwad
e,
Hala A.
Ibrahium
fg,
Antonio
Frontera