
Combined computational/experimental investigation of new cocrystals of the drug bosentan†
Rafael
Barbas
 a,
Anna
Portell
a,
Christopher A.
Hunter
b,
Rafel
Prohens
*a and
Antonio
Frontera
*c
a,
Anna
Portell
a,
Christopher A.
Hunter
b,
Rafel
Prohens
*a and
Antonio
Frontera
*c
aUnitat de Polimorfisme i Calorimetria, Centres Científics i Tecnològics, Universitat de Barcelona, Baldiri Reixac 10, 08028 Barcelona, Spain. E-mail: rafel@ccit.ub.edu
bYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK
cDepartament de Química, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma, Spain
First published on 13th June 2022
Abstract
We report the discovery of new cocrystals of bosentan, a drug used in the treatment of pulmonary artery hypertension, with succinic acid, resorcinol, and 4-hydroxybenzoic acid through a combined virtual/experimental cocrystal screening. The X-ray structure of the bosentan/succinic acid cocrystal shows a network of H-bonds involving both components, which have been studied energetically using DFT calculations. The succinic acid molecules interact with one of the pyrazine rings of bosentan establishing an energetically significant lp⋯π interaction. Moreover, they form homodimers stabilized by antiparallel CO⋯CO interactions. This set of interactions has been rationalized using molecular electrostatic potential (MEP) surfaces, the quantum theory of “atoms-in-molecules” (QTAIM), and the noncovalent interaction plot (NCIplot).
1. Introduction
The research on pharmaceutical cocrystals has experienced a great advance in the last decades since it is one of the most successful strategies available for improving the performance of drugs.1 A long list of examples have enriched the corpus of data, which has allowed the rapid development of experimental and computational techniques for the efficient investigation of the multicomponent solid form landscape of pharmaceutical compounds.2 All the available approaches can be considered as complementary tools of the so-called multidisciplinary science named Crystal Engineering, a concept coined by Desiraju, who defined this discipline as the “understanding of intermolecular interactions in the context of crystal packing and the utilization of such understanding in the design of new solids with desired physical and chemical properties”.3 Among these tools, virtual cocrystal screening has emerged as a powerful computational-guided method to select coformers with a high probability of cocrystallization with a particular drug compound. In this sense, Hunter developed a method based on calculated gas phase molecular electrostatic potential surfaces (MEPS),4 which has been validated against experimental data and used successfully in the discovery of new cocrystals of drug compounds with a great ability to establish strong hydrogen bonds like griseofulvin and spironolactone,5 nalidixic acid,6 linezolid,7 betulinic acid,8 1,2,4-thiadiazole9 or sildenafil.10 In fact, hydrogen bonding (HB)11–13 can be considered the most important interaction used in pharmaceutical cocrystallization, an important strategy toward the manufacture of improved formulations of active pharmaceutical ingredients (APIs).14 Moreover, the importance of lone-pair–π (lp–π) interactions, namely, the non-covalent association between a neutral electron-rich molecule and an electron-poor π ring, has been demonstrated in the field of crystal engineering.15 Actually, lp–π interactions have been clearly evidenced in a number of biological systems, exemplified by Z-DNA.16 The stability of the left-handed Z-DNA duplex, regardless of poor base-pair stacking, is ascribable to lp–π interactions between the lp of an O-atom belonging to a 2′-deoxyribose unit and the guanidinium ring.17 However, investigations dealing with lp–π interactions in pharmaceutical cocrystals are scarcely found in the literature, which include interesting examples of lp–π interactions like those recently described in a cocrystal of caffeine and citric acid.18Bosentan (BSN) (4-tert-butyl-N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-pyrimidin-2-ylpyrimidin-4-yl]benzenesulfonamide) is an endothelin receptor antagonist used in the treatment of pulmonary arterial hypertension (Fig. 1).19 The total absence of cocrystals in the literature together with the presence in its structure of a diversity of acceptor functional groups motivated the solid-state investigation reported in this work. Herein, we have used the surface site interaction point (SSIP) molecular descriptors to guide the selection of a very reduced list of coformers, based on the calculation of cocrystallization energies, to experimentally screen for new cocrystals of bosentan. As a result, new cocrystals have been discovered, and in particular the X-ray structure of the cocrystal formed by bosentan and succinic acid has been solved and analyzed in depth. As expected, it discloses the co-existence of several hydrogen bonds (OH⋯O and OH⋯N) between both compounds. But interestingly, it also exhibits a remarkably short O⋯π lp–π interaction between one O-atom of succinic acid and one pyrimidine ring of bosentan, which has been characterized energetically using DFT calculations, the quantum theory of atoms-in-molecules (QTAIM), molecular electrostatic potential (MEP) surfaces, and the noncovalent interaction plot (NCIplot) computational tool.
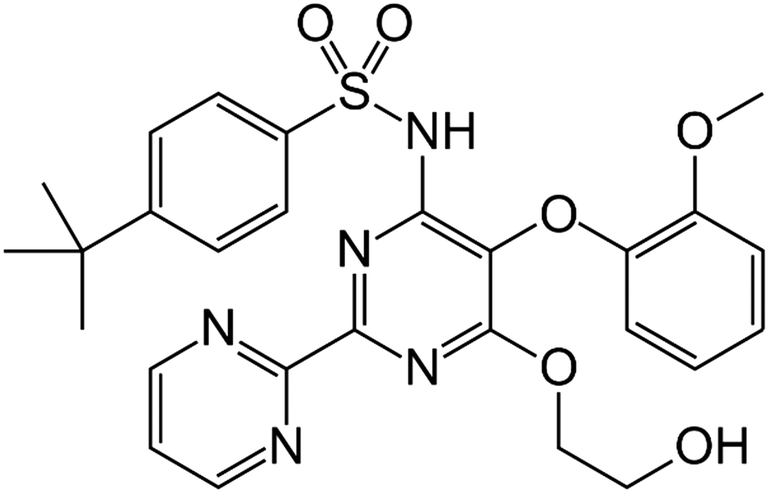 | ||
| Fig. 1 Molecular structure of bosentan. | ||
2. Materials and methods
Bosentan monohydrate was used as the starting material (CCDC refcode: NEQHEY01). The qualitative solubilities of bosentan monohydrate as well as of each coformer were determined experimentally in order to select the solvents to be used in the cocrystal screening, (see the ESI† for further details).2.1 Cocrystal screening
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of bosentan and each coformer and one drop of selected solvents using a grinding mill (Retsch MM 2000). The mixtures were placed in 2 mL stainless steel containers, together with two stainless tungsten grinding balls (3 mm diameter). The experiments were conducted for 15–30 minutes, at a 30 Hz mill frequency. Then, the resulting solids were collected without drying and immediately analyzed by XRPD. Evidence of new solid forms was detected by comparing XRPD patterns of all the known forms of bosentan and the coformers against the resulting solids. RC experiments were conducted with a saturated solution of the most soluble component (bosentan or the coformer) in selected solvents as follows: a small amount of the less soluble component was added to the saturated solution of the most soluble one until it did not dissolve anymore. Then, the mixture was stirred and the final solids filtered and analyzed by XRPD. SMT experiments were performed with suspensions of bosentan and each selected coformer in different molar ratios (40–3000 mg of the final mixture). The mixtures were stirred and the resulting solids filtered and analyzed by XRPD.
:1 mixture of bosentan and each coformer and one drop of selected solvents using a grinding mill (Retsch MM 2000). The mixtures were placed in 2 mL stainless steel containers, together with two stainless tungsten grinding balls (3 mm diameter). The experiments were conducted for 15–30 minutes, at a 30 Hz mill frequency. Then, the resulting solids were collected without drying and immediately analyzed by XRPD. Evidence of new solid forms was detected by comparing XRPD patterns of all the known forms of bosentan and the coformers against the resulting solids. RC experiments were conducted with a saturated solution of the most soluble component (bosentan or the coformer) in selected solvents as follows: a small amount of the less soluble component was added to the saturated solution of the most soluble one until it did not dissolve anymore. Then, the mixture was stirred and the final solids filtered and analyzed by XRPD. SMT experiments were performed with suspensions of bosentan and each selected coformer in different molar ratios (40–3000 mg of the final mixture). The mixtures were stirred and the resulting solids filtered and analyzed by XRPD.
The synthesis of bosentan:resorcinol cocrystal (BSN:RE) bulk powder was conducted by liquid assisted grinding in THF at 25 °C.
The synthesis of bosentan:4-hydroxybenzoic acid cocrystal (BSN:4-HBA) bulk powder was conducted by reaction crystallization (saturated solution of the coformer) in ethyl acetate at 25 °C.
Details of the synthesis and characterization of each form can be found in the ESI† section (see sections 2 and 4). The bosentan:coformer stoichiometry was assessed based on 1H-NMR when the crystal structure is not available.
Single X-ray crystallographic analysis. Single crystal X-ray diffraction intensity data of the bosentan: succinic acid cocrystal crystal were collected using a D8 Venture system equipped with a multilayer monochromator and a Mo microfocus (λ = 0.71073 Å). Frames were integrated with the Bruker SAINT software package using a SAINT algorithm. Data were corrected for absorption effects using the multi-scan method (SADABS).23 The structures were solved and refined using the Bruker SHELXTL software package, a computer program for automatic solution of crystal structures and refined by the full-matrix least-squares method with ShelXle Version 4.8.0, a Qt graphical user interface for the SHELXL computer program.24
Powder X-ray diffraction analysis. Powder X-ray diffraction patterns were obtained using a PANalytical X'Pert PRO MPD diffractometer in transmission configuration using Cu Kα1 radiation (λ = 1.5406 Å) with a focusing elliptic mirror and a PIXcel detector working at a maximum detector's active length of 3.347°. The apparatus was set in a configuration of convergent beam with a focalizing mirror and a transmission geometry, with flat samples sandwiched between low absorbing films measuring from 2 to 40° in 2θ, with a step size of 0.026° 2θ and a total measuring time of 8 to 30 minutes at room temperature (298 K). The powder diffractograms were indexed and the lattice parameters were refined by means of the LeBail method using DAJUST,25,26 and the space groups were determined from the systematic absences. A summary of comparative crystallographic data is given in Table 1.
| Crystal form | BSN:AS | BSN:RE | BSN:4-HBA |
|---|---|---|---|
| a R-Factor for SXRD and Rwp for XRPD. | |||
| X-ray diffraction analysis | SXRD | XRPD | XRPD |
| T (K) | 300(2) | 298 | 298 |
| System | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/m | P21/m |
| a (Å) | 5.9031(14) | 38.57(1) | 38.43(1) |
| b (Å) | 14.188(4) | 8.030(1) | 9.411(3) |
| c (Å) | 20.665(5) | 22.174(4) | 14.766(6) |
| α (°) | 70.166(10) | 90 | 90 |
| β (°) | 89.202(10) | 96.14(1) | 90.85(3) |
| γ (°) | 83.142(11) | 90 | 90 |
| Vol (Å3) | 1615.8(7) | 6827(20) | 5340(5) |
| Z | 2 | 8 | 4 |
| R (%)a | 7.12 | 8.65 | 7.84 |
2.2 Computational details
The calculations of the non-covalent interactions were carried out using Gaussian-16 (ref. 27) and the PBE0-D3/def2-TZVP level of theory.28,29 To evaluate the interactions in the solid state, the crystallographic coordinates were used. The interaction energies were computed by calculating the difference between the energies of isolated monomers and their assembly. The interaction energies were calculated with correction for the basis set superposition error (BSSE) by using the Boys–Bernardi counterpoise technique.30 Bader's “atoms in molecules” theory (QTAIM)31 were used to study the interactions discussed herein by means of the AIMAll calculation package.32 The molecular electrostatic potential surfaces (isosurface 0.002 a.u.) were computed using the Gaussian-16 software.27In order to assess the nature of interactions in terms of being attractive or repulsive and reveal them in real space, we used the NCIplot index, which is a method for plotting non-covalent interaction regions,33 based on the NCI (non-covalent interactions) visualization index derived from the electronic density.34 The reduced density gradient (RDG), coming from the density and its first derivative, is plotted as a function of the density (mapped as isosurfaces) over the molecule of interest. The sign of the second Hessian eigenvalue times the electron density [i.e. sign(λ2)ρ in atomic units] enables the identification of attractive/stabilizing (blue-green coloured isosurfaces) or repulsive (yellow-red coloured isosurfaces) interactions using 3D-plots. For the plots shown in Fig. 5 and 6, the NCIplot index parameters are as follows: RGD = 0.5; ρ cut off = 0.04 a.u.; color range: −0.04 a.u. ≤ sign(λ2)ρ ≤ 0.04 a.u.
3. Results and discussion
3.1 Experimental
The calculation of the cocrystallization energy (the difference between the calculated energies of the cocrystal and the pure components) for each potential API/coformer combination was conducted as described in detail elsewhere. Briefly, surface site interaction points (SSIPs) for bosentan and a library of potential coformers were extracted from the ab initio molecular electrostatic potential surface (MEPS) of the molecule in the gas phase.22,35 Each SSIP is represented by an interaction parameter, εi (which is positive for a H-bond donor and negative for a H-bond acceptor site). The energy of interaction between each pair of SSIPs, i and j, is given by the product εiεj. The approach assumes that all the interactions between SSIPs are optimised in a crystal, which allows the evaluation of the cocrystal energy without knowing details of the structure.36 The method combines the most positive SSIP with the most negative one, in a decreasing order of strength, giving rise to a hierarchical list of interactions37,38 leading to an estimated energy (E) for each solid (API, conformer, and cocrystal, eqn (1)). Finally, the difference between the energies of the cocrystal and the pure components, ΔE, is calculated to estimate the probability of cocrystal formation (eqn (2)).| E = ∑εiεj | (1) |
| ΔE = −(Ecc − E1 − E2) | (2) |
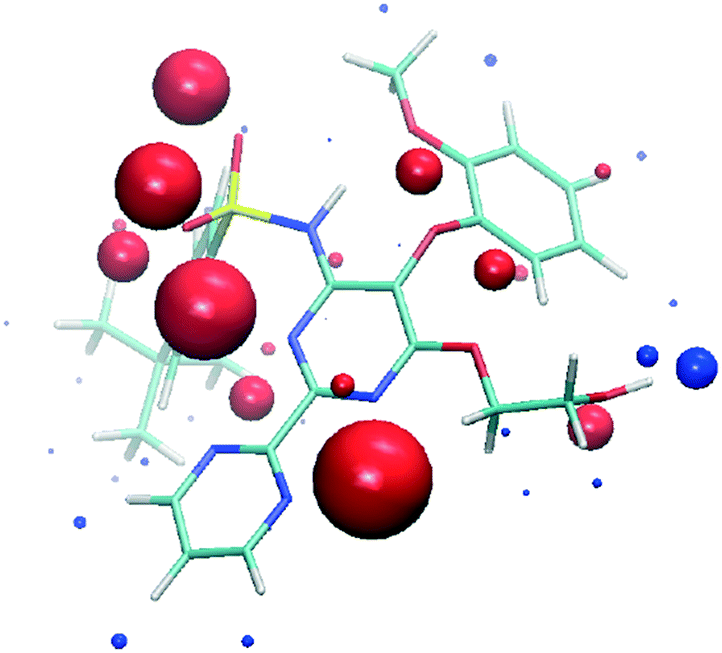 | ||
| Fig. 2 SSIPs calculated for bosentan. Blue spheres correspond to positive interaction parameters (εi) and red spheres correspond to negative values. | ||
The experimental cocrystal screening with a short list of just 10 coformers (resorcinol, tartaric acid, citric acid, malic acid, fumaric acid, 4-hydroxybenzoic acid, inositol, succinic acid, rhamnose, and urea) produced three new cocrystals with succinic acid, resorcinol, and 4-hydroxybenzoic acid. Cocrystals with resorcinol and 4-hydroxybenzoic acid were obtained pure and their diffractograms were indexed (see the ESI† section). Single crystals suitable for SXRD analysis were obtained only for the succinic acid cocrystal (Tables S3 and S4 of the ESI† section contains the crystal structure and refinement data). It crystallizes in the triclinic P space group with one molecule of each component in the asymmetric unit (Z′ = 1, Z = 2). The molecule of bosentan shows at 300 K static disorder (80:20) in the tert-butyl group (C25, C26, and C27 atoms), corresponding to two possible conformations as the consequence of the C21–C24 bond rotation. On the other hand, we analyzed the most relevant intermolecular interactions in the structure by means of Hirshfeld surface calculations39 and the associated fingerprint plot40,41 by using the Crystal Explorer software (Fig. 3).42 In the structure, succinic acid interacts as a H bond donor with the pyrimidine nitrogen (Fig. 3 marked with a black point) and the bosentan alcohol oxygen (Fig. 3 marked with a purple point). Moreover, succinic acid acts as a hydrogen bond acceptor with the bosentan alcohol group (Fig. 3 marked with a red point). Interestingly, in order to efficiently interact with this dual donor/acceptor behavior, succinic acid adopts a gauche conformation, which is present in only 27 out of the 240 crystal structures (11%) in the Cambridge Structural Database (Version 5.41, 2019) (see ESI,† Table S5 for CCDC refcodes). Moreover, a short H⋯H distance (2.13 Å) between methoxy groups was observed (Fig. 3 marked with a green point). Although this intermolecular contact is presumably repulsive based on the hydrogen atom van der Waals diameter (1.1–1.2 Å), a limited number of organic crystal structures have been reported showing H⋯H interatomic distances lower than 2.2 Å,43,44 which can be associated to repulsive forces which preserve the internal equilibrium in the crystal structure.45 All these contacts are shown in the Hirshfeld surface as red areas and with sharp spikes of different shapes in the fingerprint plot. Finally, the crystal structure of the bosentan:succinic acid cocrystal was subjected to a deep computational analysis as described in the following sections.
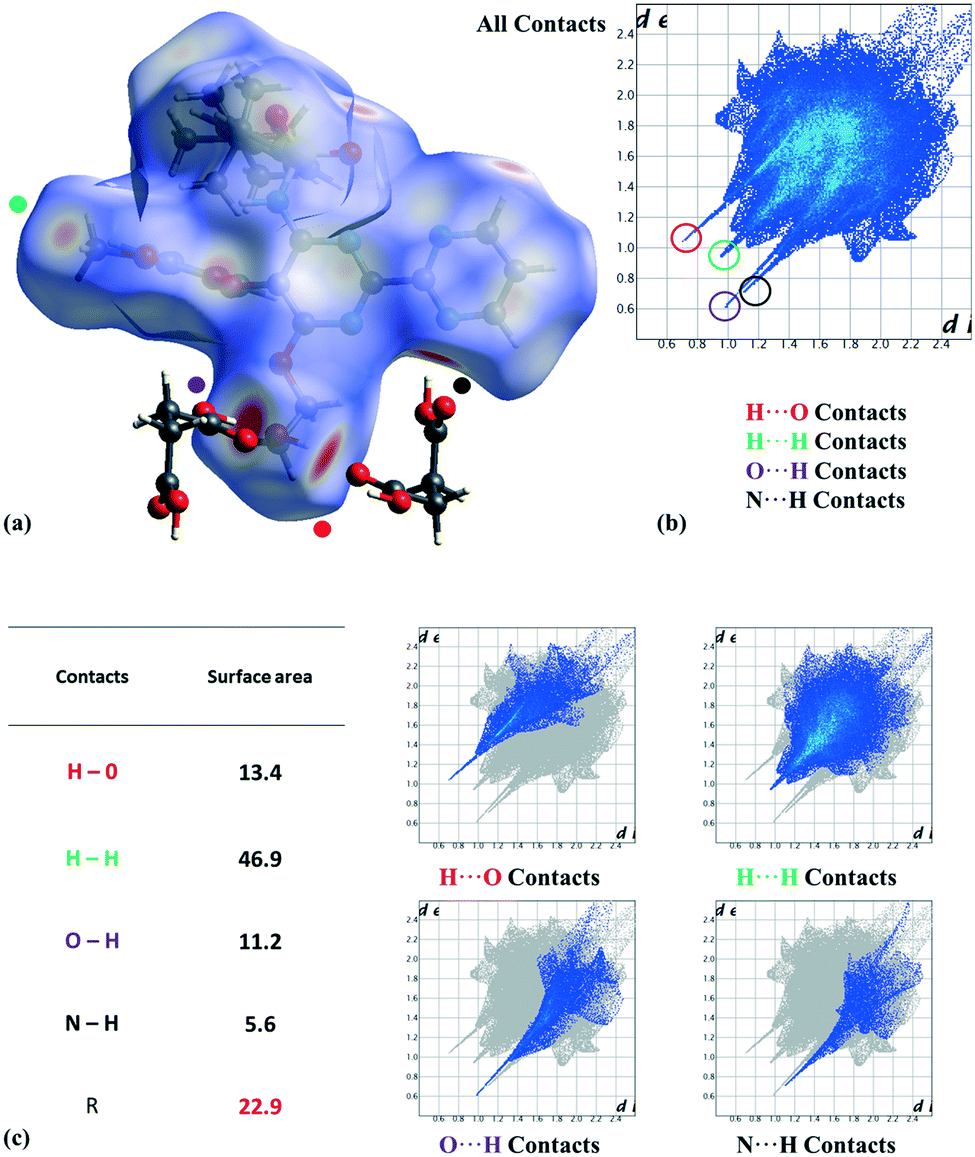 | ||
| Fig. 3 (a) Hirshfeld surface mapped on the bosentan molecule with dnorm. Strong contacts are pointed out and succinic acid is also represented. (b) Fingerprint plot computed from Hirshfeld surfaces. Close contacts are highlighted from inside elements: H⋯O (red), H⋯H (green), O⋯H (purple), and N⋯H (black). (c) Surface area contribution (%) of intermolecular contacts and their associated fingerprint plot regions. | ||
3.2 DFT calculations
The DFT study is basically focused on the analysis of the interactions observed between both coformers in the solid state. Fig. 4 shows a partial view of the X-ray structure of the cocrystal where the bosentan moiety interacts with two succinic acids via H-bonds (OH⋯O and OH⋯N) and another one via a short O⋯π interaction (2.94 Å). In addition, two succinic acids also interact by establishing antiparallel CO⋯CO interactions (C⋯O distance: 3.37 Å).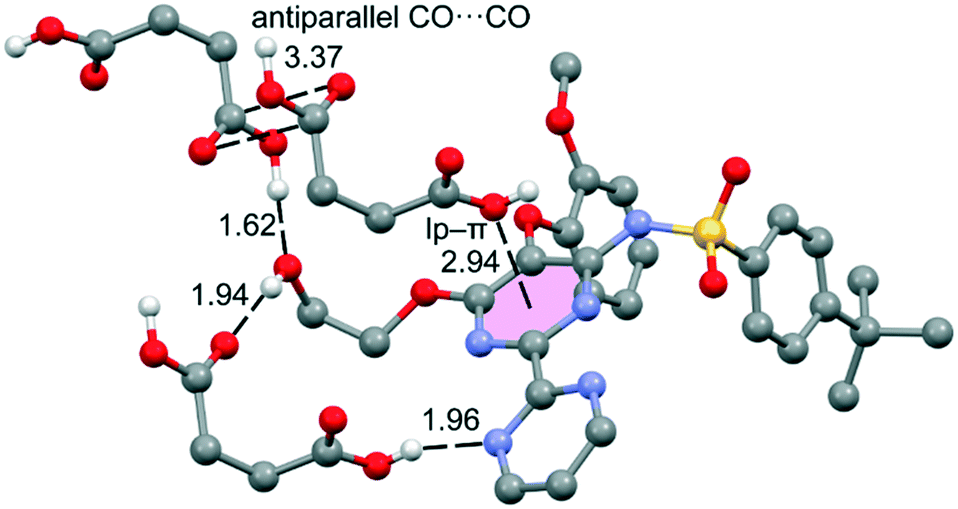 | ||
| Fig. 4 Partial view of the X-ray analysis of the bosentan:succinic acid cocrystal with indication of the H-bonds and lp–π interactions. Distances are in Å. Only acidic H-atoms are shown for clarity. | ||
The MEP surfaces of bosentan and succinic acid, in the conformation they adopt in the crystal structure, were initially computed (see Fig. 5) to investigate the most electron-rich and electron-poor regions of both compounds. It can be observed that the MEP minimum in bosentan (Fig. 5a) is located in the region under the influence of two nitrogen atoms of the 2,2′-bipyrimidinyl moiety and one oxygen atom of the sulphonamide group (−75.0 kcal mol−1). The maximum MEP is located at the NH group (+55.5 kcal mol−1) followed by that at the OH group of the 2-hydroxyethoxy group, which is close in energy (+52.7 kcal mol−1). The MEP in the nucleophilic region between the N-atoms of the 2,2′-bipyrimidinyl moiety (opposite to the MEP minimum) is also large and negative (−48.9 kcal mol−1, see Fig. 5). The MEP values over the pyrimidine rings are very small, indicating the duality of such π-systems. Specifically, it has been previously demonstrated that π-systems with MEP values close to neutral are able to interact favorably with both electron-rich (Lewis bases or anions) and electron-poor (H-bond donors or cations) regions favorably.46 The MEP surface of succinic acid is shown in Fig. 5b. As expected, the MEP maximum is located at the acidic proton (+60.7 kcal mol−1) and the minimum is located at the carbonylic O-atom of the carboxylic group (−35.1 kcal mol−1). The MEP value at the hydroxylic O-atom of the carboxylic group is smaller in absolute value (−18.1 kcal mol−1), thus explaining the formation of the lp–π interaction with the pyrimidine ring. The MEP over the carbon atom of the COOH group is positive (+21.3 kcal mol−1), thus explaining the formation of the antiparallel CO⋯CO interactions detailed in Fig. 4. The MEP surface analyses of both coformers indicate the strong nature of the H-bonds in the solid state.
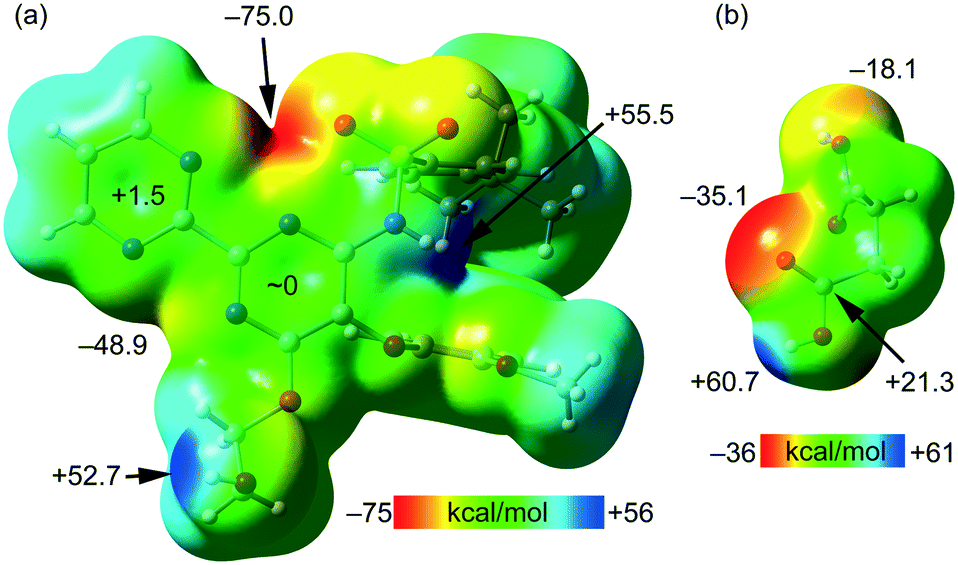 | ||
| Fig. 5 MEP surfaces of bosentan (a) and succinic acid (b) at the PBE0-D3/def2-TZVP level of theory (isosurface 0.002 a.u.). The energies at selected points of the surface are given in kcal mol−1. | ||
Fig. 6a shows one of the binding modes observed in the cocrystal between the succinic acid and bosentan, showing an excellent complementarity, that is, succinic acid establishes two strong H-bonds – one as the donor (OH⋯N) and the other one as the acceptor (OH⋯O) – with bosentan. Moreover, an ancillary CH⋯O H-bond is also established. The dimerization energy is −14.9 kcal mol−1, thus confirming the strong nature of the H-bonds, in line with the MEP analysis. Fig. 6a also shows the combined QTAIM/NCIplot analysis of this dimer, evidencing that each H-bond is characterized by a bond critical point (CP, red sphere) and a bond path interconnecting the H and O,N-atoms. The OH⋯O and NH⋯O interactions are also characterized by blue (strong attractive) NCIplot isosurfaces coincident to the location of the bond CPs. The CH⋯O interaction is characterized by a green NCIplot isosurface indicating that this interaction is weaker. To compare the strengths of the different H-bonds, the methodology proposed by Espinosa et al.47 was used to estimate the dissociation energy of each H-bonding contact. This method uses the potential energy density values (Vr) at the bond CP as the energy predictor (using the equation Edis = −1/2 × Vr). The values are indicated in Fig. 6a (red values) revealing that the NH⋯O interaction is stronger (6.7 kcal mol−1) than the OH⋯O (5.6 kcal mol−1) HB. The strength of the CH⋯O H-bond is weaker (1.1 kcal mol−1) as expected. To total formation energy of the assembly (−14.9 kcal mol−1) is similar to the sum of the HB dissociation energies (13.4 kcal mol−1), thus giving reliability to the energy predictor. The small difference could be due to the small attraction between the OH group of succinic acid and the N-atom of the adjoined pyrimidine ring, as corroborated by the green isosurface located between the H and N atoms (see the small arrow in Fig. 6a). For the other binding mode shown in Fig. 6b, succinic acid and bosentan are simply connected by one H-bond involving the hydroxyl group of bosentan as the acceptor and succinic acid as the donor. In this case, the H-bond is very strong, as confirmed by the dark blue color of the RGD isosurface and the dimerization energy (−8.7 kcal mol−1).
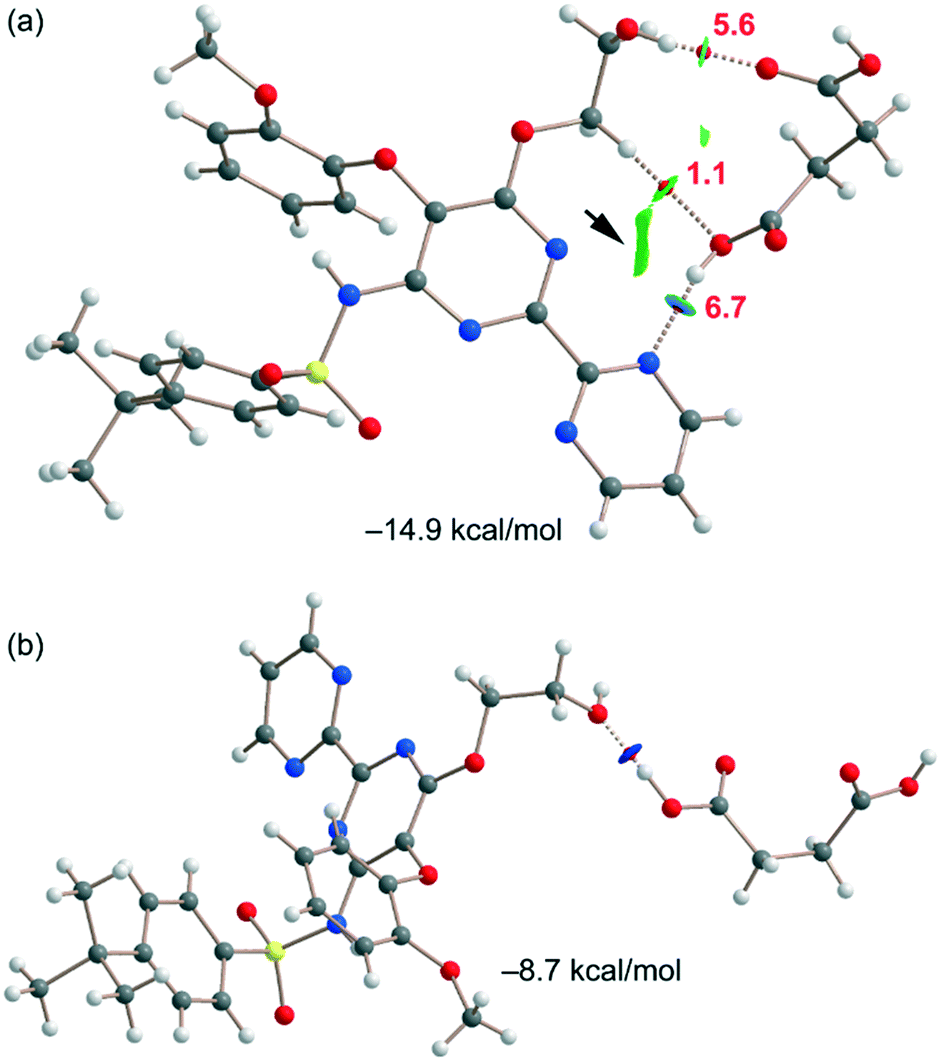 | ||
| Fig. 6 (a and b) QTAIM/NCIplot analysis of intermolecular bond CPs (red spheres), bond paths, and RDG isosurfaces of two heterodimers. | ||
Fig. 7a shows the last binding mode observed in the solid state of the cocrystal between both coformers. In this case, an interesting O⋯π interaction is formed, as confirmed by the QTAIM/NCIplot analysis performed for the heterodimer. It shows a bond CP and a bond path connecting the O-atom to one N-atom of the pyrimidine ring. The NCIplot isosurface is in this case more informative, showing an extended green isosurface that totally embraces the six-membered ring and confirming the lp–π nature of the interaction, which is moderately strong (−5.2 kcal mol−1) in line with the short lp–π distance. It should be emphasized that the strength of the lp–π interaction is similar to that of the OH⋯O H-bond of the dimer represented in Fig. 6a (−5.6 kcal mol−1), thus revealing that it is an energetically relevant interaction in the solid state. Finally, the succinic acid homodimer exhibiting the antiparallel CO⋯CO interaction has also been analyzed, likewise showing a moderately strong (−4.3 kcal mol−1) dimerization energy. Both the QTAIM and NCIplot analyses confirm the existence and attractive nature of such a contact, which is characterized by a bond CP interconnecting the O-atoms and a green NCIplot isosurface that is located between the antiparallel CO groups.
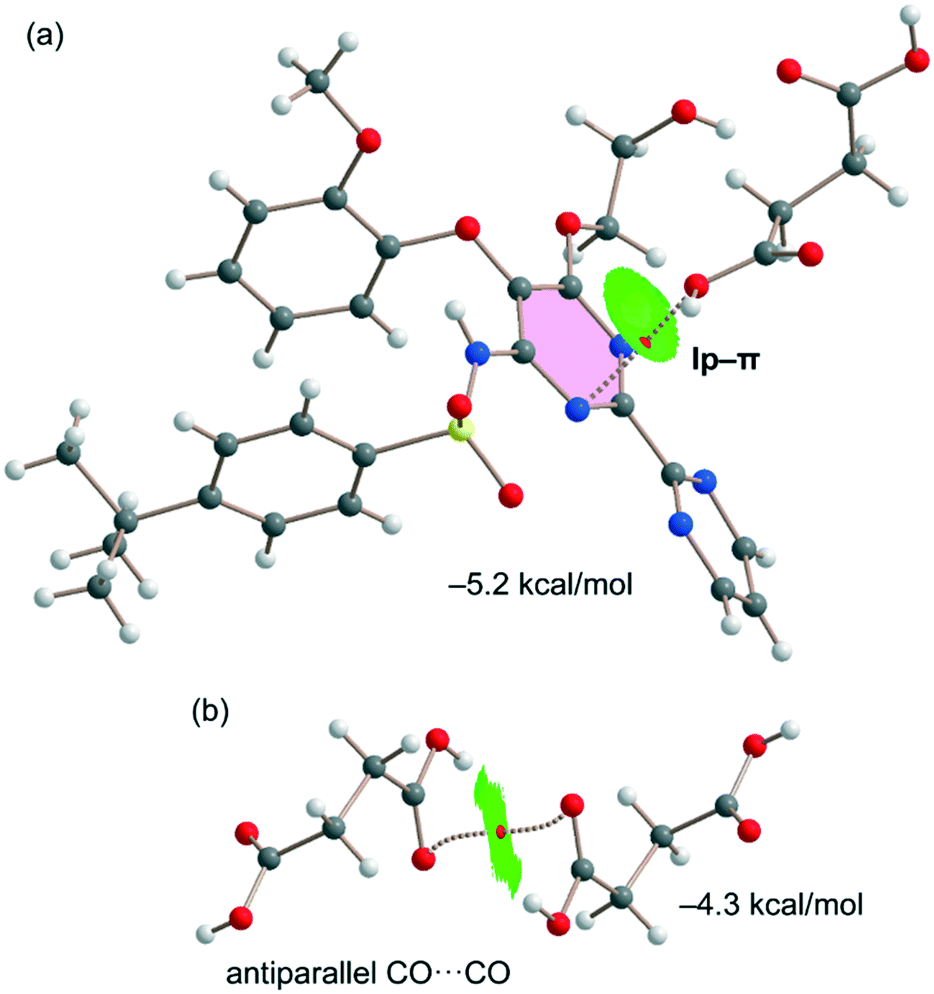 | ||
| Fig. 7 QTAIM/NCIplot analyses of intermolecular bond CPs (red spheres), bond paths, and RDG isosurfaces of one heterodimer (a) and one homodimer (b) observed in the solid state of the cocrystal. | ||
4. Conclusions
New bosentan cocrystals with succinic acid, resorcinol, and 4-hydroxybenzoic acid have been discovered through a combined virtual/experimental cocrystal screen. The X-ray crystal structure of the bosentan:succinic acid cocrystal is reported and described herein, showing interesting binding modes in the solid state. The cocrystal components interact by the formation of strong H-bonds, which were analyzed energetically using density functional theory (DFT) calculations, reduced density gradient isosurfaces, and topological analysis of bond critical points. In addition, energetically relevant O⋯π interactions between succinic acid and the π-system of the pyrazine ring are described. Finally, antiparallel CO⋯CO interactions have been described and evaluated, which are also relevant in the crystal packing of the cocrystal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the MICIU/AEI of Spain (project PID2020-115637GB-I00 FEDER funds) for funding and also Dr. Mercè Font-Bardia (Universitat de Barcelona) for her support concerning SCXRD data. We also thank the CTI (UIB) for computational facilities.Notes and references
- N. Rodriguez-Hornedo, Mol. Pharmaceutics, 2007, 4, 299–300 CrossRef.
- Y. A. Abramov, Computational Pharmaceutical Solid State Chemistry, Wiley, 2016 Search PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- D. Musumeci, C. A. Hunter, R. Prohens, S. Scuderi and J. F. McCabe, Chem. Sci., 2011, 2, 883–890 RSC.
- T. Grecu, R. Prohens, J. F. McCabe, E. J. Carrington, J. S. Wright, L. Brammer and C. A. Hunter, CrystEngComm, 2017, 19, 3592–3599 RSC.
- T. Grecu, H. Adams, C. A. Hunter, J. F. McCabe, A. Portell and R. Prohens, Cryst. Growth Des., 2014, 14, 1749–1755 CrossRef CAS.
- M. Khalaji, M. J. Potrzebowski and M. K. Dudek, Cryst. Growth Des., 2021, 21, 2301–2314 CrossRef CAS.
- M. Nicolov, R. M. Ghiulai, M. Voicu, M. Mioc, A. O. Duse, R. Roman, R. Ambrus, I. Zupko, E. A. Moaca, D. E. Coricovac, C. Farcas, R. M. Racoviceanu, C. Danciu, C.-A. Dehelean and C. Soica, Front. Chem., 2019, 7, 92 CrossRef CAS PubMed.
- A. O. Surov, A. P. Voronin, N. A. Vasilev, A. B. Ilyukhin and G. L. Perlovich, New J. Chem., 2021, 45, 3034–3047 RSC.
- R. Barbas, M. Font-Bardia, A. Paradkar, C. A. Hunter and R. Prohens, Cryst. Growth Des., 2018, 18, 7618–7627 CrossRef CAS.
- A. M. Maharramov, K. T. Mahmudov, M. N. Kopylovich and A. J. L. Pombeiro, Non-Covalent Interactions in the Synthesis and Design of New Compounds, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2016 Search PubMed.
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- J. Chen, B. Sarma, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2011, 11, 887–895 CrossRef CAS.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- M. Egli and R. V. Gessner, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 180–184 CrossRef CAS PubMed.
- S. Sarkhel, A. Rich and M. Egli, J. Am. Chem. Soc., 2003, 125, 8998–8999 CrossRef CAS PubMed.
- P. Verma, A. Srivastava, K. Srivastava, P. Tandon and M. R. Shimpi, Front. Chem., 2021, 9, 708538 CrossRef CAS PubMed.
- L. J. Rubin, D. B. Badesch, R. J. Barst, N. Galiè, C. M. Black, A. Keogh, T. Pulido, A. Frost, S. Roux, I. Leconte, M. Landzberg and G. Simonneau, N. Engl. J. Med., 2002, 346, 896–903 CrossRef CAS PubMed.
- Cresset torchV10lite, https://www.cresset-group.com/products/torch/torchlite/.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and J. D. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. S. Calero, J. Farwer, E. J. Gardiner, C. A. Hunter, M. Mackey, S. Scuderi, S. Thompson and J. G. Vinter, Phys. Chem. Chem. Phys., 2013, 15, 18262–18273 RSC.
- SADABS Bruker AXS; Madison, Wisconsin, USA, 2004; SAINT, Software Users Guide, Version 6.0; Bruker Analytical X–ray Systems, Madison, WI, 1999. Sheldrick, G. M. SADABS v2.03: Area–Detector Absorption Correction. University of Göttingen, Germany, 1999; Saint Version 7.60A (Bruker AXS 2008); SADABS V. 2008–1 (2008).
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Boultif and D. Louër, J. Appl. Crystallogr., 1991, 24, 987–993 CrossRef CAS.
- O. Vallcorba, J. Rius, C. Frontera, I. Peral and C. Miravitlles, J. Appl. Crystallogr., 2012, 45, 844–848 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park, KS, 2013 Search PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- C. A. Hunter, Chem. Sci., 2013, 4, 1687–1700 RSC.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- M. C. Etter, J. Phys. Chem., 1991, 95, 4601–4610 CrossRef CAS.
- C. B. Aakeroy, J. Desper and M. M. Smith, Chem. Commun., 2007, 3936–3938 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. Mckinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. Mckinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- R. Barbas, M. Font-Bardia and R. Prohens, Cryst. Growth Des., 2018, 18, 3740–3746 CrossRef CAS.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- J. D. Dunitz and A. Gavezzotti, Acc. Chem. Res., 1999, 32, 677–684 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Org. Lett., 2003, 5, 2227–2229 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cocrystal screening details, crystal data and structure refinement, characterization of the new solid forms, and CCDC search. CCDC 2168576. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00581f |
| This journal is © The Royal Society of Chemistry 2022 |