
Static discrete disorder in the crystal structure of iododiflunisal: on the importance of hydrogen bond, halogen bond and π-stacking interactions†
Rafael
Barbas
 a,
Mercè
Font-Bardia
b,
Alfredo
Ballesteros
c,
Gemma
Arsequell
d,
Rafel
Prohens
*a and
Antonio
Frontera
*e
a,
Mercè
Font-Bardia
b,
Alfredo
Ballesteros
c,
Gemma
Arsequell
d,
Rafel
Prohens
*a and
Antonio
Frontera
*e
aUnitat de Polimorfisme i Calorimetria, Centres Científics i Tecnològics, Universitat de Barcelona, Baldiri Reixac 10, 08028 Barcelona, Spain. E-mail: rafel@ccit.ub.edu
bUnitat de Difracció de Raigs X, Centres Científics i Tecnològics, Universitat de Barcelona, Spain
cDepartamento de Química Orgánica e Inorgánica, Instituto de Química Organometálica “Enrique Moles”, Universidad de Oviedo, Julián Clavería, 8, 33006 Oviedo, Spain
dInstitut de Química Avançada de Catalunya (I.Q.A.C.-C.S.I.C.), E-08034, Barcelona, Spain
eDepartament de Química, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma, Spain
First published on 25th March 2022
Abstract
This manuscript reports a combined computational/crystallographic analysis of iododiflunisal (IDIF), a difluorophenyl derivative of salicylic acid (2′,4′-difluoro-4-hydroxy-5-iodo-[1,1′]-biphenyl-3-carboxylic acid). This drug is used to target transthyretin related amyloidosis. In the solid state it shows static discrete disorder and forms the typical R22(8) centrosymmetric dimer that is common in carboxylic acids (via double OH⋯O H-bonds). Parallel face-to-face stacking interactions are also observed in its crystal packing where these R22(8) centrosymmetric dimers are propagated forming infinite 1D columns. Moreover, the presence of iodine, which exhibits a region of large and positive electrostatic potential (σ-hole) along the C–Ha bond and a belt of negative electrostatic potential (σ-lumps) facilitates the formation of halogen bonds (HaBs) and halogen⋯halogen contacts that are also relevant in the solid state. The crystalline disorder was analyzed by means of Hirshfeld surfaces, and hydrogen, halogen and π–π bonding assemblies were analyzed using density functional theory (DFT) calculations, molecular electrostatic potential (MEP) surfaces, the quantum theory of “atom-in-molecules” (QTAIM) and the noncovalent interaction plot (NCIplot).
Introduction
A profound understanding of noncovalent interactions is needed to succeed in the competitive field of supramolecular chemistry and crystal engineering.1 Compared to the ubiquitous hydrogen bonding (HB)2 and π-stacking,3 halogen bonding (HaB) is attracting substantial interest by the research community4,5 due to its high directionality.6–10 Therefore, the synthesis and X-ray characterization of halogenated compounds is of interest to further comprehend the behavior and directing role of HaBs. On the other hand, although disordered crystal structures are frequently found (∼20%) in the Cambridge Structural Database (CSD),11 with relevant examples in pharmaceutical compounds such as carbamazepine12 and caffeine13 among many others, many questions about crystalline disorder are still open, particularly related to crystal structure prediction of pharmaceutical compounds,14 which makes relevant the continuous study of such phenomenon.The small-molecule compound iododiflunisal (IDIF), a iodinated analog of the non-steroidal anti-inflammatory drug (NSAID) diflunisal, is a kinetic stabilizer of the thyroid hormone transporter protein transthyretin (TTR). IDIF was discovered in the framework of a drug discovery project focused in TTR related amyloidosis.15–17 IDIF binds selectively to the unoccupied thyroxine binding sites of the tetramer and kinetically stabilizes the tetrameric form of TTR, preventing the fibrillogenesis. The protein TTR has also a neuroprotective role in Alzheimer's disease (AD) phatogenesis.18 TTR is the main amyloid-β (Aβ) binding protein in the CSF19,20 and this TTR/Aβ binding is believed to naturally prevent Aβ aggregation and toxicity. Tetramer stabilizer IDIF enhances the TTR/Aβ interaction, behaving as small-molecule chaperone.21,22 Importantly, in vivo administration of IDIF to a AD transgenic mice, resulted in decreased brain amyloid burden23,24 and in amelioration of the cognitive functions that are impaired in this AD-like neuropathology.23 Moreover, the formation of TTR-IDIF complexes enhances blood brain barrier (BBB) permeability.25 Imaging studies with the small-molecule chaperone IDIF, a molecule in the discovery phase22,26 are in progress to confirm TTR as a novel target for AD, in the search of effective disease-modifying drugs for AD.
Although the crystal structure of the IDIF/TTR complex has been reported,27 the crystal structure of IDIF remained elusive until now. Thus and with the aim to extend the knowledge about this important drug compound we have analyzed its novel crystal structure from a crystallographic and computational point of view with an emphasis in its static disorder.
Our study reveals the existence of π-stacking, hydrogen and halogen bonds (H⋯O and I⋯O) in the IDIF structure. Moreover, it also exhibits “like⋯like” I⋯I contacts, which have been characterized by means of several computational tools, including molecular electrostatic potential (MEP), the quantum theory of atoms-in-molecules (QTAIM) and the noncovalent interaction plot (NCIPlot). Moreover, the energetic features of π-stacking, H-bonded and Ha-bonded dimers have been evaluated using DFT calculations in relation to the observed disorder, as detailed in the next sections, evidencing the key role of π-stacking in the solid state of IDIF.
Experimental
Materials
IDIF was synthesized in our laboratory following previously described procedures28 by direct iodination of the drug diflunisal using Barluenga's reagent.29 Single crystals suitable for SXCRD analysis were obtained as follows. IDIF (20 mg, 0.053 mmol) was mixed and dissolved in xylene (2.0 mL) at 70 °C. Single crystals were observed after overnight.X-ray crystallographic analysis
Single crystal X-ray diffraction (SCXRD) intensity data of the IDIF was collected using a D8 Venture system equipped with a multilayer monochromator and a Mo microfocus (λ = 0.71073 Å). Frames were integrated with the Bruker SAINT software package using a SAINT algorithm. Data were corrected for absorption effects using the multi-scan method (SADABS).30,31 The structures were solved and refined using the Bruker SHELXTL soft-ware package, a computer program for automatic solution of crystal structures and refined by full-matrix least-squares method with ShelXle Version 4.8.0, a Qt graphical user interface for SHELXL computer program.32Computational details
The calculations of the non-covalent interactions were carried out using the Gaussian-16 (ref. 33) and the PBE0-D3/def2-TZVP level of theory.34,35 To evaluate the interactions in the solid state, the crystallographic coordinates have been used. The interaction energies have been computed by calculating the difference between the energies of isolated monomers and their assembly. The interaction energies were calculated with correction for the basis set superposition error (BSSE) by using the Boys–Bernardi counterpoise technique.36 The Bader's “Atoms in molecules” theory (QTAIM)37 has been used to study the interactions discussed herein by means of the AIMAll calculation package.38 The molecular electrostatic potential surfaces have been computed using the Gaussian-16 software.In order to assess the nature of interactions in terms of being attractive or repulsive and revealed them in real space, we have used NCIPLOT index, which is a method for plotting non-covalent interaction regions,39 based on the NCI (non-covalent interactions) visualization index derived from the electronic density.40 The reduced density gradient (RDG), coming from the density and its first derivative, is plotted as a function of the density (mapped as isosurfaces) over the molecule of interest. The sign of the second Hessian eigenvalue times the electron density [i.e. sign(λ2)ρ in atomic units] enables the identification of attractive/stabilizing (blue-green coloured isosurfaces) or repulsive (yellow-red coloured isosurfaces) interactions using 3D-plots. For the plots shown in Fig. 5 and 6 the NCIplot index parameters are: RGD = 0.5; ρ cut off = 0.04 a.u.; color range: −0.04 a.u. ≤ sign(λ2)ρ ≤ 0.04 a.u.
Results and discussion
Structural description of IDIF
IDIF crystallizes in the monoclinic P21/n space group and the crystal structure has one molecule in the asymmetric unit (Z′ = 1, Z = 4). The molecule of IDIF shows at 296 K 50% static disorder in the difluorophenyl ring, corresponding to the two possible conformations as the consequence of the C6–C8 bond rotation. Both conformations show a ring torsion angle of 139.5° (C5–C6–C8–C13). The asymmetric unit with ORTEP representation is shown in Fig. 1 and the crystallographic data and structural refinements details are summarized in Table 1.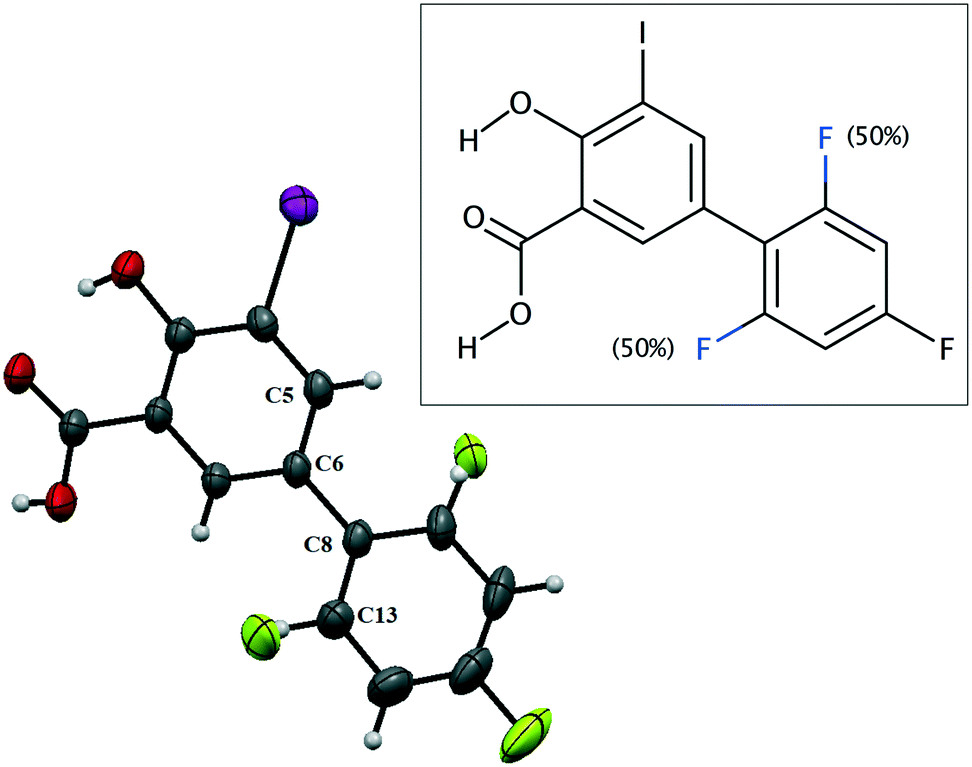 | ||
| Fig. 1 ORTEP representation of the asymmetric unit and chemical structure of IDIF showing the disordered atoms. | ||
| Crystal data | IDIF |
|---|---|
| Empirical formula | C13H7F2IO3 |
| Formula weight | 376.09 |
| Temperature (K) | 296(2) |
| System | Monoclinic |
| Space group | P21/n |
| a (Å) | 16.9777(10) |
| b (Å) | 4.0424(2) |
| c (Å) | 18.0441(11) |
| α (°) | 90 |
| β (°) | 93.412(2) |
| γ (°) | 90 |
| Vol (Å3) | 1236.18(12) |
| Z | 4 |
| Density (calc.) (Mg m−3) | 2.021 |
| Final R indices | R 1 = 0.0352 |
| [I > 2σ(I)] | wR2 = 0.0908 |
| CCDC | 2125800 |
Crystalline disorder is defined as dynamic when the crystal constituents are in motion or as static if the constituents are still but occupying either a continuum of different positions (continuous disorder) or a discrete number of possible positions (discrete disorder).41 This phenomenon occurs when four conditions are fulfilled: the non-equivalent molecules of the disordered structure must show similar shape, charge distribution, a similar number and strength of intra and intermolecular interactions and similar energy.14 Thus, taking into account the domain-based statistical nature of disorder and since the symmetry of the P21/n space group transforms the only molecule of the asymmetric unit (existing in two different conformations) into 4-membered chains in the unit cell with a very similar volume occupied by the difluorophenyl ring in both conformations, it can be interpreted that the IDIF molecule is able to pack in the crystal structure with alternating domains consisting of two types of chains in which fluorine atoms are close to each other. Thus, each domain shows a very similar environment but with some important differences, which we have analyzed by means of Hirshfeld surfaces calculations.42 First, we manually removed the disorder from the cif file in order to generate two hypothetical structures in which only one of the two possible conformations exist. Then, we determined the Hirshfeld surface and the associated fingerprint plot43,44 of each structure by using the Crystal Explorer software.45 The most relevant difference between both structures is that in one case the fluorine atom F1 is in syn orientation with respect to the iodine atom (IDIF_Syn, Fig. 2) and in the other case the fluorine atom F1′ is in anti orientation with respect to the iodine atom (IDIF_Anti, Fig. 3). This has a relevant impact in the Hirshfeld surface since in the first case (IDIF_Syn) it is observed a F⋯F distance (2.34 Å) shorter than in the second case (IDIF_Anti, 2.57 Å). Both short contacts are shown in the Hirshfeld surface as a red area near the fluorine atoms together with sharp spikes of different shape in the fingerprint plots. On the other hand, since the R22(8) supramolecular synthon formed by self-interacting carboxylic acid groups are not altered by the static disorder the two corresponding spikes remain exactly the same in both cases. In order to have a clearer picture of the observed disorder we analyzed the crystal structures contained in the CCDC of the parent compound diflunisal (a non-steroidal antiinflamatory drug), which has the same chemical structure but lacking the iodine substituent. There are thirteen structures available at the CCDC (see ESI† for details), including anhydrous forms, inclusion complexes, salts, solvates and cocrystals. Remarkably, only in five cases (two cocrystals, two salts and one anhydrous structure solved from PXRD data) disorder in the fluorine atom was not observed. The propensity of fluorine to exhibit disorder has been recently demonstrated46 by solid-state NMR and DFT calculations. A likely explanation for this common behaviour is the small volum occupied by the fluorine atom, its modest Lewis base ability and absence of σ-hole (in contrast to the rest of halogen atoms) that decreases its ability to form strong noncovalent interactions, thus increasing the probability of mobility and disorder in X-ray structures.
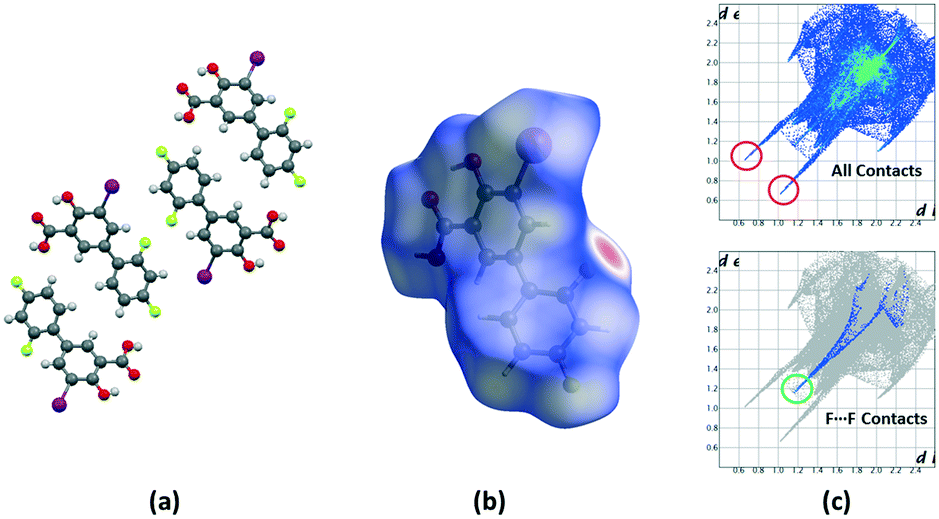 | ||
| Fig. 2 (a) Unit cell representation of the IDIF_Syn domain, (b) Hirshfeld surface mapped with dnorm and (c) fingerprint plots computed from Hirshfeld surfaces. Strong H⋯O contacts are highlighted in red and F⋯F contacts in green. | ||
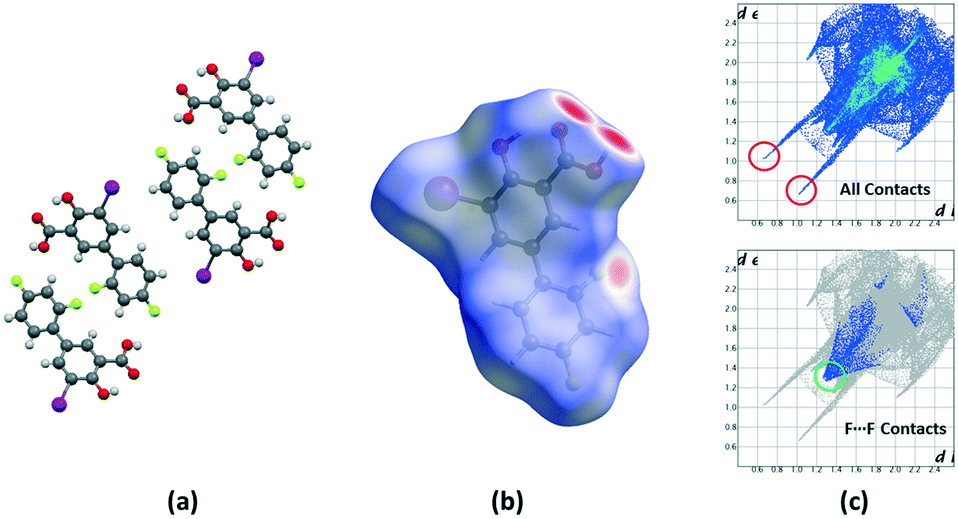 | ||
| Fig. 3 (a) Unit cell representation of the IDIF_Anti domain, (b) Hirshfeld surface mapped with dnorm and (c) fingerprint plots computed from Hirshfeld surfaces. Strong H⋯O contacts are highlighted in red and F⋯F contacts in green. | ||
DFT calculations
First, the MEP surface of IDIF in the syn conformation (IDIF_Syn) was computed and represented in Fig. 4 to investigate the nucleophilic and electrophilic regions. It is also convenient to analyze the anisotropy of the iodine atom and the size and intensity of the iodine's σ-hole. It can be observed that the MEP minimum and maximum are located at the O and H-atoms of the carboxylic group (−25 and +55 kcal mol−1, respectively). Therefore, the most favored assembly from an electrostatic point of view is the formation of COOH⋯COOH dimers, as observed experimentally. The MEP at the phenolic O-atom is also large and negative (−23 kcal mol−1, see Fig. 4). Detail of the MEP surface at the iodine atom is also included in Fig. 4 (top left) where a reduced MEP scale was used (±19 kcal mol−1). The iodine atom presents a large and intense σ-hole (+19 kcal mol−1) thus anticipating a strong ability to establish halogen bonding interactions. The anisotropic nature of the MEP around the iodine atom anticipates a strong linearity of the halogen bonds. However, the approximation of the electron rich atom to the σ-hole may deviate up to 55° without experimenting the repulsion of the negative belt. Finally, the MEP values over the aromatic rings are negligible, thus favoring the formation of parallel face-to face stacking without electrostatic repulsion. Finally, the MEP at the extension of the C–F bond that participates in the C–F⋯F–C interactions is small (−3.6 kcal mol−1) thus suggesting a small electrostatic repulsion that is likely compensated by other contributions like dispersion or polarization. It must be mentioned that the MEP surface of the IDIF molecule in the anti conformation (IDIF_Anti) is very similar, with identical MEP maximum and minimum values.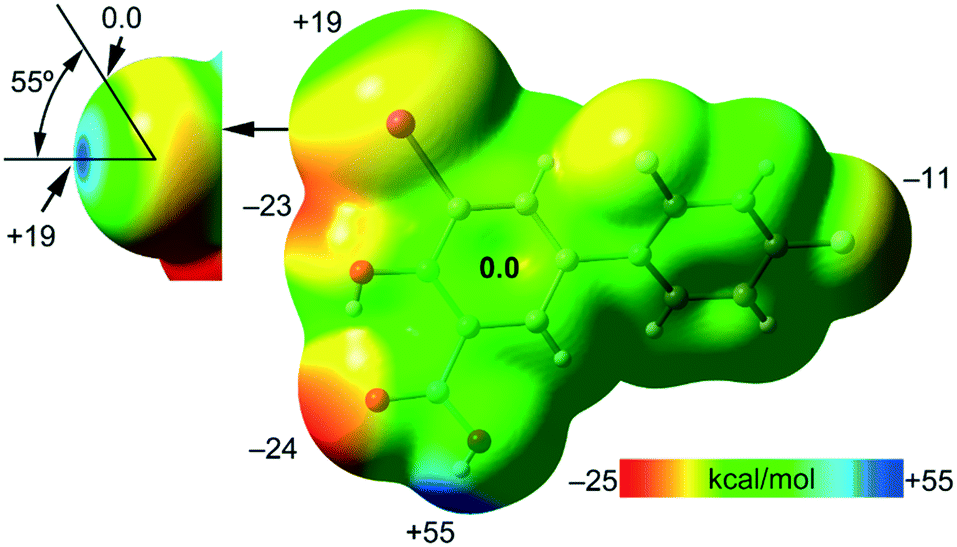 | ||
| Fig. 4 MEP surface of IDIF in the syn conformation at the PBE0-D3/def2-TZVP level of theory. Detail of the MEP around the halogen atom is also shown (top-left) where a reduced ±19 kcal mol−1 energy range has been used. The energies at selected points of the surface are given in kcal mol−1. | ||
The energetic DFT analysis is devoted to analyzing structure-directing interactions, focusing on π–π, HB and HaB interactions. Fig. 5a shows a partial view of the crystal structure of IDIF showing the formation of infinite 1D assemblies where multiple H-bonds (R22(8) synthon) and π–π stacking interactions are established. Fig. 5b and c shows the combined QTAIM/NCIPlot analyses for the R22(8) dimers of both IDIF_Syn and IDIF_Anti structures. They show that each H-bond is characterized by a bond critical point (CP, red sphere) and bond path interconnecting the H and O atoms. The interaction is further characterized by a ring CP (yellow sphere) due to the formation of the R22(8) supramolecular ring. The interactions are characterized by blue (attractive) NCIplot isosurfaces coincident to the location of the bond CPs. The formation energy of the dimer at the PBE0-D3/def2-TZVP level of theory is −10.0 kcal mol−1 and −10.2 kcal mol−1 for both IDIF_Syn and IDIF_Anti, respectively, confirming the relevance of the R22(8) synthon. The QTAIM also shows the existence of an intramolecular H-bond between the phenolic and the carboxylic groups, characterized by the corresponding bond CP, bond path and blue NCIPlot index isosurface. In order to compare the strength of the inter and intramolecular H-bonds, we have evaluated the dissociation energy using the potential energy density (Vr) at the bond CP. This methodology was proposed by Espinosa et al.47 where the dissociation energy is computed using the Edis = −1/2 × Vr equation. The values are indicated in Fig. 5b and c adjacent to the bond CPs in red. It can be observed that the intramolecular OH⋯O(COOH) H-bond is stronger (8.5 kcal mol−1) than the intermolecular one (5.6 kcal mol−1). The strength of the H-bond using the Vr energy predictor (5.6 kcal mol−1) is similar to that computed using the supramolecular approach (half of dimerization energy), giving reliability to the Vr energy predictor. Fig. 5d and e shows the π-stacked dimers of IDIF_Syn and IDIF_Anti extracted from the crystal structure. The QTAIM analysis shows multiple bond and ring CPs and bond paths interconnecting the aromatic rings. Moreover, the QTAIM also reveals the existence of a CH⋯F contact characterized by the corresponding bond CP and bond path in IDIF_Syn and a CH⋯HC vdW contact in IDIF_Anti also characterized by the corresponding bond CP and bond path. The NCIplot analyses for both structures show a very extended green isosurface located between both molecules revealing a strong complementarity. As a consequence, the interaction energy is very large (−11.0 kcal mol−1 and −11.7 kcal mol−1 for IDIF_Syn and IDIF_Anti, respectively), even larger than the R22(8) dimer, thus suggesting that the π-stacking interaction is stronger than the R22(8) H-bonds. This energetic analysis shows that the position of the F1 atom has a little influence on the bonding energies of both synthons.
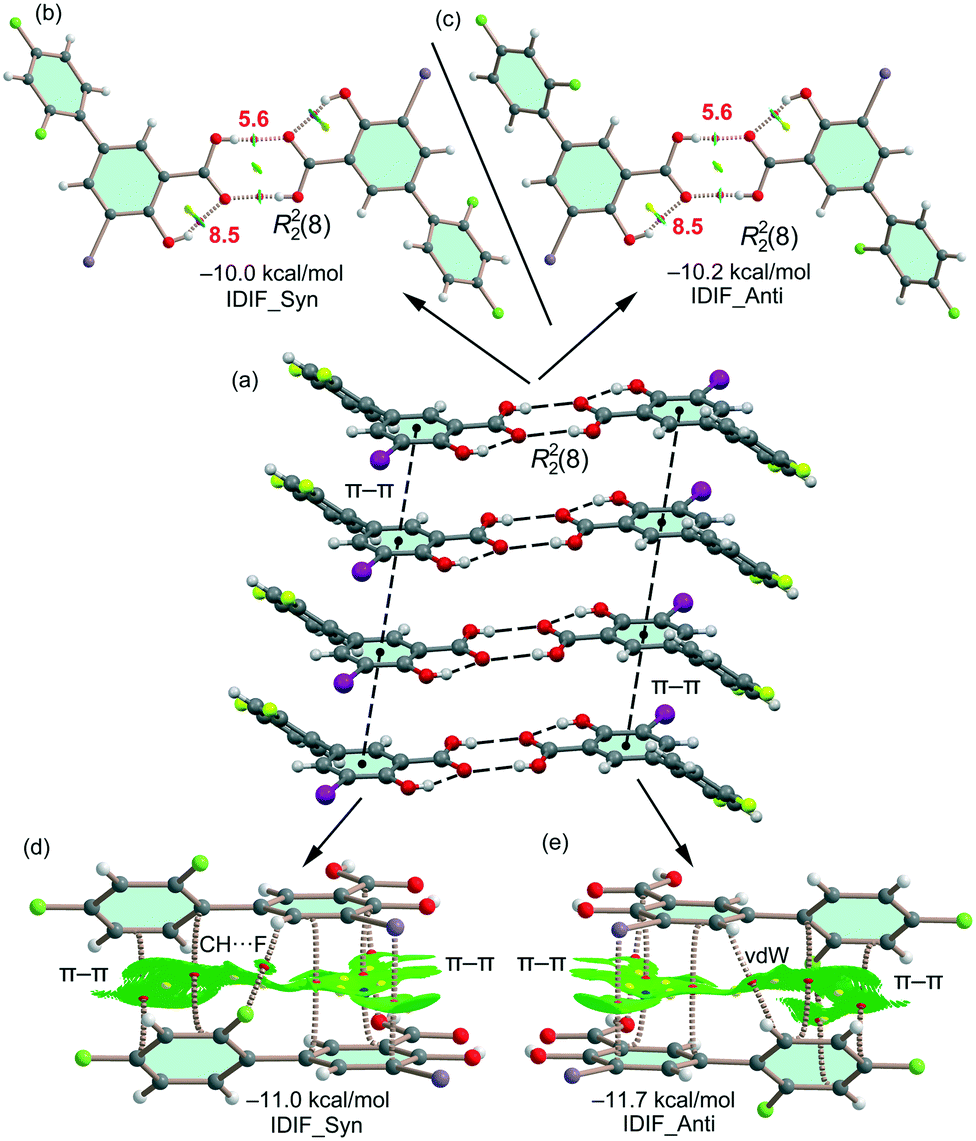 | ||
| Fig. 5 (a) Partial view of the crystal structure of IDIF. Distances in Å. (b–e) QTAIM analysis of intermolecular bond, ring and cage CPs (red, yellow and blue spheres, respectively) and bond paths of the two dimers of IDIF. For the R22(8) dimer, the CP characterizing the intramolecular OH⋯O H-bonds are also shown. The dissociation energies are indicated in red next to the bond CPs. | ||
IDIF also forms supramolecular assemblies in the solid state via multiple I⋯O contacts as depicted in Fig. 6a for the IDIF_Syn structure. The contacts are slightly longer than the sum of the van der Waals radii (∑Rvdw = 3.50 Å). These contacts are in fact revealed by the QTAIM analysis showing the corresponding bond CP and bond path connecting the phenolic O-atom to the I-atom, as observed in the QTAIM/NCIplot analysis of a dimer extracted from the crystal structure (see Fig. 6b). The QTAIM/NCIplot analysis also demonstrates the existence of a I⋯I contact that is expected to be very weak since the I⋯I distance is significantly longer than ∑Rvdw (3.96 Å). The C–I⋯I angle is 120°, therefore this contact cannot be considered as a genuine HaB taking into consideration the IUPAC's definition of HaB, since the negative belt of one iodine is not pointing to the σ-hole of the other iodine. This is further confirmed by the combined QTAIM/MEP plot shown in Fig. 6c, which shows that the bond path connecting both I-atoms does not cross the σ-hole. The dimerization energy is −1.9 kcal mol−1, thus revealing that the HaB interaction is energetically less significant than the H-bond and π-stacking interactions commented above. Unexpectedly, equivalent calculations for the HaB dimer of the IDIF_Anti structure reveal that the interaction energy of the HaB dimer is negligible (0.0 kcal mol−1), thus evidencing that the different location of the F-atom in the IDIF_Anti structure provokes small variation in the charge distribution affecting the delicate balance between the I⋯O and I⋯I interactions.
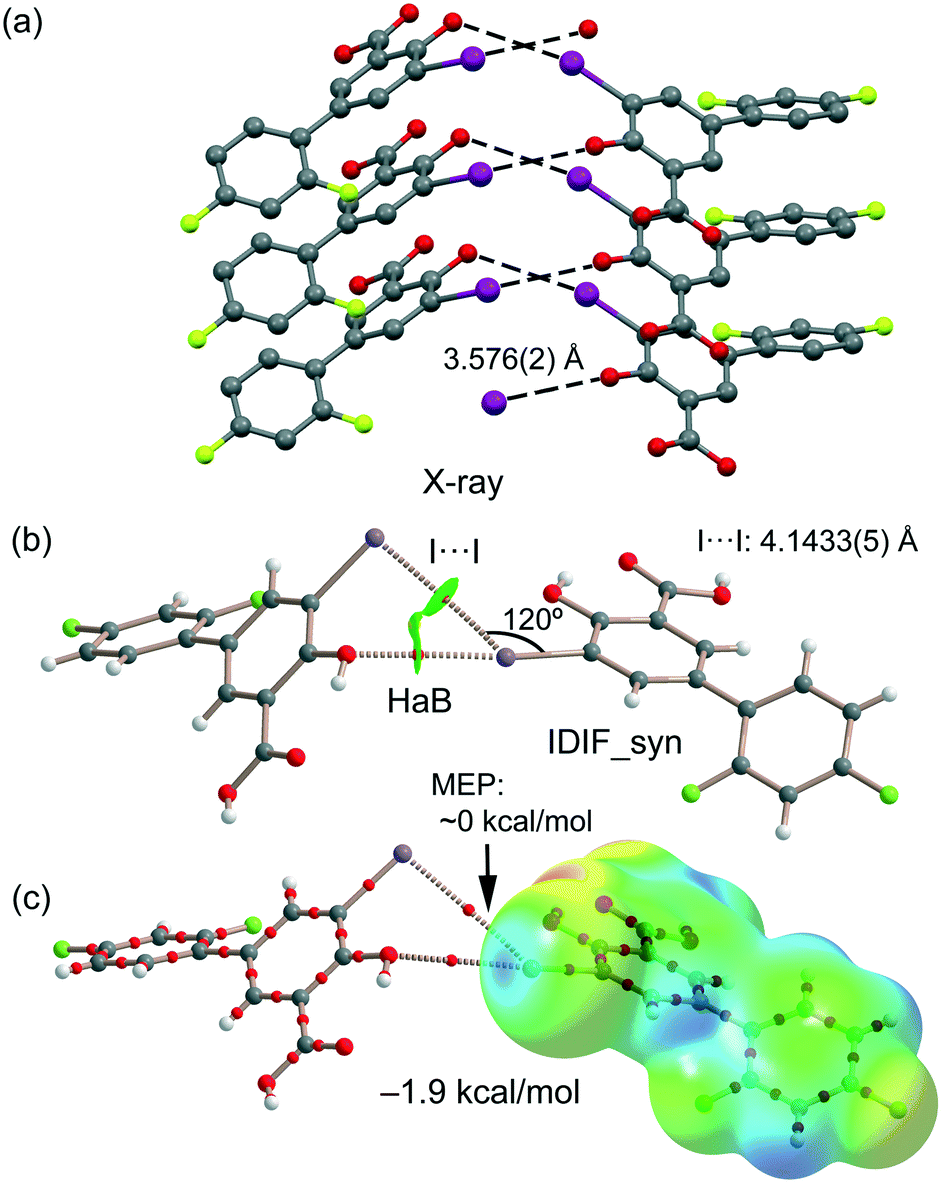 | ||
| Fig. 6 (a) Partial view of the crystal structure of IDIF_Syn. Distances in Å. (b) QTAIM analysis of intermolecular bond and ring CPs (red spheres) and bond paths (dashed lines) of the HaB dimer of IDIF_Syn. (c) QTAIM with the superimposed MEP surface (isosurface 0.001 a.u.) is represented. | ||
As explained above, the most relevant difference of both structures is the orientation of fluorine (F1) that has a major impact on the F⋯F contacts, as revealed by the Hirshfeld surface analysis, and on the shorter F⋯F distance in the IDIF_Syn structure (2.34 Å, see Fig. 2 and 3). It has a minimum impact on the H-bond and π-stacking interactions, as shown in Fig. 5 since the binding energies are almost equivalent for both IDIF_Syn and IDIF_Anti structures. However, it has also an impact on the I⋯O HaB dimer shown in Fig. 6, since the IDIF_Anti presents a negligible binding energy whilst the dimerization energy of IDIF_Syn is −1.9 kcal mol−1. To further analyze the F⋯F contacts, we have computed the dimers of IDIF_Syn and IDIF_Anti where the F⋯F interactions are established, which are shown in Fig. 7, including the QTAIM/NCIplot index analyses for both structures. It can be observed that for both dimers, the QTAIM analysis shows the formation of two H-bonds and one F⋯F contact, each one characterized by a bond CP and bond path. These interactions are also revealed by the NCIPlot index analysis that confirms the attractive nature of the F⋯F contacts. The dimerization energy is larger (in absolute value) for the IDIF_Anti structure that presents one F⋯F and two CH⋯O contacts. The energetic contribution of both CH⋯O interactions has been evaluated using the dissociation energies obtained by means of the Vr predictor, which is −2.0 kcal mol−1. Therefore, the energy of the F⋯F contact can be roughly estimated by difference, that is −1.8 kcal mol−1. The total dimerization energy of the F⋯F dimer observed in IDIF_Syn is smaller (Fig. 7a, −1.7 kcal mol−1). In this dimer, the dissociation energy of each CH⋯I contact has been estimated in 0.6 kcal mol−1 using the Vr predictor (see Fig. 7a), so the total contribution of the H-bonding is −1.2 kcal mol−1, thus evidencing that the energy of the F⋯F contact is approximately −0.5 kcal mol−1. The smaller contribution of the F⋯F interaction in this dimer is most likely related to the directionality of the C–F⋯F–C interaction. In the IDIF_Syn dimer both C–F⋯F angles are equivalent (144.8°) while in the IDIF_Anti dimer are different (146.4° and 101.2°). It has been previously demonstrated that C–X⋯X–C contacts with different angles are energetically stronger than those with equivalent angles.7 Interestingly, the energetic difference between both F⋯F dimers (IDIF_Anti structure is 2.1 kcal mol−1 more favored than IDIF_Syn) is likely compensated by the difference in the formation of the I⋯O dimers (Fig. 6, IDIF_Syn is 1.9 kcal mol−1 more favored), thus providing an energetic explanation for the experimental disorder observed in the solid state.
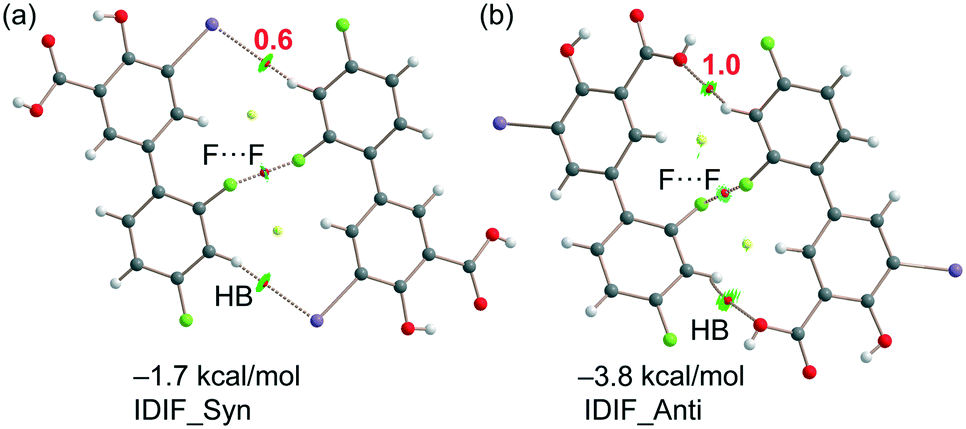 | ||
| Fig. 7 (a) QTAIM analysis of intermolecular bond and ring CPs (red spheres) and bond paths (dashed lines) of the F⋯F dimer of IDIF_Syn. (b) QTAIM analysis of intermolecular bond and ring CPs (red spheres) and bond paths (dashed lines) of the F⋯F dimer of IDIF_Anti. The dissociation energies are indicated in red next to the bond CPs. | ||
Finally and with the aim to compare the overall stability of both crystal domains, we have computed their individual lattice energies by using a supercell of 12 molecules and periodic boundary conditions at the PBE-D/DND level of theory. The computed lattice energies were Elattice = −41.76 kcal mol−1 and −41.59 kcal mol−1 for IDIF_Syn and IDIF_Anti respectively, revealing that they are basically isoenergetic, in line with the experimental findings.
Conclusions
The crystal structure of the small-molecule chaperone IDIF as disease-modifying drug candidate for Alzheimer's disease is reported herein, showing static discrete disorder, which has been analyzed by means of Hirshfeld surface calculations, and with interesting 1D supramolecular assemblies in the solid state. IDIF propagates by means of the formation of multiple H-bonds and π-stacking interactions, that were analyzed energetically using density functional theory (DFT) calculations, showing the dominant role of π-stacking interactions. Moreover, I⋯O halogen bonds are also important in the crystalline domain of the syn conformation of IDIF (IDIF_Syn) in combination with I⋯I contacts. For the latter, we found that the combination of QTAIM/MEP analysis is useful to disclose the nature of these long I⋯I contacts that cannot be classified as genuine HaB, since none of the two iodine atoms is acting as a Lewis acid. In contrast the F⋯F interactions in combination with CH⋯O, I contacts are more important in the crystalline domain of the anti conformation of IDIF (IDIF_Anti), that compensate the energetically relevant I⋯O interactions found in IDIF_Syn. Thus, our computational analysis offers a quantitative explanation of the fourth condition necessary for the presence of static discrete disorder in the IDIF structure, (besides equivalent shape, charge distribution and similar intra and intermolecular interactions), which is the existence of isoenergetic domains of different molecular conformations in the crystal and provides a new example of organic molecule showing this important crystallographic phenomenon.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the MICIU/AEI of Spain (project PID2020-115637GB-I00 FEDER funds). Part of the work was supported by a grant from the Fundació Marató de TV3 (neurodegenerative diseases call, project reference: 20140330-31-32-33-34, https://www.ccma.cat/tv3/marato/en/projectes-financats/2013/212/).Notes and references
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS.
- A. M. Maharramov, K. T. Mahmudov, M. N. Kopylovich and A. J. L. Pombeiro, Non-Covalent Interactions in the Synthesis and Design of New Compounds, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2016 Search PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- Z. T. Li and L. Z. Wu, Hydrogen Bonded Supramolecular Structures, Springer, Berlin/Heidelberg, Germany, 2015 Search PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- P. Metrangolo and G. Resnati, Halogen Bonding: Fundamentals and Applications (Structure and Bonding), Springer, Berlin/Heidelberg, Germany, 2010 Search PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- M. Benito, Y. Roselló, M. Barceló-Oliver, A. Frontera and E. Molins, Int. J. Mol. Sci., 2021, 22, 10663 CrossRef CAS PubMed.
- F. Heinen, D. L. Reinhard, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2021, 60, 5069–5073 CrossRef CAS PubMed.
- D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- A. J. Florence, C. K. Leech, N. Shankland, K. Shankland and A. Johnston, CrystEngComm, 2006, 8, 746–747 RSC.
- G. D. Enright, V. V. Terskikh, D. H. Brouwer and J. A. Ripmeester, Cryst. Growth Des., 2007, 7, 1406–1410 CrossRef CAS.
- B. Dittrich, IUCrJ, 2021, 8, 305–318 CrossRef CAS PubMed.
- M. R. Almeida, B. Macedo, I. Cardoso, I. Alves, G. Valencia, G. Arsequell, A. Planas and M. J. Saraiva, Biochem. J., 2004, 381, 351–356 CrossRef CAS PubMed.
- T. Mairal, J. Nieto, M. Pinto, M. R. Almeida, L. Gales, A. Ballesteros, J. Barluenga, J. J. Pérez, J. T. Vázquez, N. B. Centeno, M. J. Saraiva, A. M. Damas, A. Planas, G. Arsequell and G. Valencia, PLoS One, 2009, 4, e4124 CrossRef PubMed.
- L. Gales, S. Macedo-Ribeiro, G. Arsequell, G. Valencia, M. J. Saraiva and A. M. Damas, Biochem. J., 2005, 388, 615–662 CrossRef CAS PubMed.
- T. Gião, J. Saavedra, E. Cotrina, J. Quintana, J. Llop, G. Arsequell and I. Cardoso, Int. J. Mol. Sci., 2020, 21, 2075 CrossRef PubMed.
- A. L. Schwarzman, L. Gregori, M. P. Vitek, S. Lyubski, W. J. Strittmatter, J. J. Enghilde, R. Bhasin, J. Silverman, K. H. Weisgraber, P. K. Coyle, M. G. Zagorki, J. Talafous, M. Eisenberg, A. M. Saunders, A. D. Roses and D. Goldgaber, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8368–8372 CrossRef CAS PubMed.
- K. M. Pate and R. M. Murphy, Isr. J. Chem., 2017, 57, 602–612 CrossRef CAS PubMed.
- C. A. Ribeiro, M. J. Saraiva and I. Cardoso, PLoS One, 2012, 7, e45368 CrossRef CAS PubMed.
- E. Y. Cotrina, A. Gimeno, J. Llop, J. Jiménez-Barbero, J. Quintana, G. Valencia, I. Cardoso, R. Prohens and G. Arsequell, J. Med. Chem., 2020, 63, 3205–3214 CrossRef CAS PubMed.
- C. A. Ribeiro, S. M. Oliveira, L. F. Guido, A. Magalhaes, G. Valencia, G. Arsequell, M. J. Saraiva and I. Cardoso, J. Alzheimer's Dis., 2014, 39, 357–370 CAS.
- L. Rejc, V. Gómez-Vallejo, X. Rios, U. Cossío, Z. Baz, E. Mujica, T. Gião, E. Y. Cotrina, J. Jiménez-Barbero, J. Quintana, G. Arsequell, I. Cardoso and J. Llop, J. Alzheimer's Dis., 2020, 77, 99–112 CAS.
- X. Rios, V. Gómez-Vallejo, A. Martín, U. Cossío, M. A. Morcillo, M. Alemi, I. Cardoso, J. Quintana, J. Jiménez-Barbero, E. Y. Cotrina, G. Valencia, G. Arsequell and J. Llop, Sci. Rep., 2019, 9, 13672 CrossRef PubMed.
- E. Y. Cotrina, L. M. Santos, J. Rivas, D. Blasi, J. P. Leite, M. A. Liz, M. A. Busquets, A. Planas, R. Prohens, A. Gimeno, J. Jiménez-Barbero, L. Gales, J. Llop, J. Quintana, I. Cardoso and G. Arsequell, Eur. J. Med. Chem., 2021, 226, 113847 CrossRef CAS PubMed.
- L. Gales, S. Macedo-Ribeiro, G. Arsequell, G. Valencia, M. J. Saraiva and A. M. Damas, Biochem. J., 2005, 388, 615–621 CrossRef CAS PubMed.
- T. Mairal, J. Nieto, M. Pinto, M. R. Almeida, L. Gales, A. Ballesteros, J. Barluenga, J. J. Pérez, J. T. Vázquez, N. B. Centeno, M. J. Saraiva, A. M. Damas, A. Planas, G. Arsequell and G. Valencia, PLoS One, 2009, 4, e4124 CrossRef PubMed.
- J. Barluenga, J. M. González, M. A. García-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC.
- SADABS Bruker AXS, Madison, Wisconsin, USA, 2004 Search PubMed; SAINT, Software Users Guide, Version 6.0, Bruker Analytical X-ray Systems, Madison, WI, 1999 Search PubMed.
- G. M. Sheldrick, SADABS v2.03, Area-Detector Absorption Correction, University of Göttingen, Germany, 1999 Search PubMed; Saint, Version 7.60A, Bruker AXS 2008, SADABS, V. 2008–1, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park, KS, 2013 Search PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- M. Habgood, R. Grau-Crespo and S. L. Price, Phys. Chem. Chem. Phys., 2011, 13, 9590–9600 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. Mckinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. Mckinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- B. J. Walder and T. M. Alam, J. Am. Chem. Soc., 2021, 143, 11714–11733 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data and structure refinement, characterization of bulk powder, Hirshfeld analysis, list of diflunisal crystal structures in the CCDC. CCDC 2125800. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00202g |
| This journal is © The Royal Society of Chemistry 2022 |