
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fecht's acid revisited: a spirocyclic dicarboxylate for non-aromatic MOFs†
Valentyna D.
Slyusarchuk
 and
Chris S.
Hawes
*
and
Chris S.
Hawes
*
School of Chemical and Physical Sciences, Keele University, Keele ST5 5BG, UK. E-mail: c.s.hawes@keele.ac.uk
First published on 22nd November 2021
Abstract
Spiro[3.3]heptane-2,6-dicarboxylic acid (Fecht's acid, H2SHDC) is examined as a non-aromatic terephthalic acid isostere for the first time. The rigid spirocyclic backbone provides greater steric bulk than conventional aromatic dicarboxylates with consequences for pore chemistry and control of interpenetration, presented here in the structures of two new MOFs. Complex 1 is a three-dimensional rod packed structure consisting of Yb-carboxylate chains bridged by SHDC linkers which, although non-porous, exhibits a surprisingly high thermal stability for a spirocyclic cyclobutane derivative. Complex 2 is a co-ligand complex of SHDC with trans-1,2-bis(4-pyridyl)ethene (bpe) which contains linear solvent channels despite fourfold interpenetration. Although the framework does not retain its structure following evacuation, a clear difference is observed in the extended structure compared to the structurally related terephthalate species. This observation suggests the non-aromatic backbone of Fecht's acid and other rigid aliphatic linkers may prove an effective means to disfavour deleterious close inter-framework contacts which prevail in interpenetrated aromatic MOFs.
Introduction
The successful design and utility of metal–organic frameworks (MOFs) and discrete metallosupramolecular species is contingent on careful ligand design.1 To achieve the properties which are sought after in these materials, especially for applications in small molecule separations,2 catalysis,3 and sensing,4 the pore chemistry must be precisely tailored to match the requirements of the desired guests. Synthetic approaches based on ligand-centred functionality have unlocked countless new applications for porous metallosupramolecular systems. For example, photoswitchable organic spacers can be introduced to modulate structural properties,5 additional metal coordinating functionalities can be installed to bind catalytic metals,6 and responsive chromophores or fluorophores can be built into ligand backbones to report guest uptake or release.7 However, a bottleneck has long existed in this field based on the underlying chemistry of the ligands themselves. To date, the overwhelming majority of ligands studied in MOF chemistry have contained aromatic cores.8 These range from the early named MOFs built from terephthalic acid, trimesic acid, dipyridyl, imidazolate and 4,4′-biphenyldicarboxylic acid, to more exotic functional species based on carbazoles, pyrenes, thiophenes, porphyrins and most other common aromatic moieties.9 Instances of MOF ligands containing aliphatic functionality have until recently been mostly limited to simple cyclohexane or adamantane derivatives or short linear alkyl dicarboxylates,10 while fumarates and oxalates also make effective linkers as conjugated but non-aromatic dicarboxylates.11The dominance of aromatic ligands over aliphatics in MOF chemistry is largely based on two factors. Firstly, aromatic ligands are widely available, with facile synthetic routes and a range of accessible substitution patterns. Secondly, aromatic ligands tend to provide rigid and predictable geometries useful for “reticular” synthesis of families of related MOFs, while the low energy barriers between conformations in aliphatic ligands can make prediction of their extended structures much more difficult. Recent efforts have shown, however, that rigid aliphatic ligands may be a missed opportunity for MOF chemistry, providing access to previously unexplored aliphatic pore chemistries, non-planar geometries, improved optical transparency, and the new application scope that this may provide.12 The evolution in the MOF field can be mapped onto similar considerations in pharmaceutical design, where cubane, bicyclo[2.2.2]octane and bicyclo[1.1.1]pentane have recently been explored as benzene isosteres.13 These fused ring species have the benefit of structural rigidity, negating the tendency for highly flexible aliphatic groups to adopt poorly-defined linker geometries, thus making them well suited to MOF chemistry. Cubane-1,4-dicarboxylate has successfully been incorporated into several MOFs as an isostere for terephthalic acid and demonstrated an inversion in adsorption selectivity for benzene over cyclohexane compared to its aromatic equivalents based on an increased degree of pore contouring caused by its non-planar structure.14 Bicyclo[2.2.2]octane-1,4-dicarboxylate, along with diazabicyclo[2.2.2]octane (dabco), was used to generate a non-aromatic MOF showing complete optical transparency to wavelengths as low as 240 nm.15 Ultrafast rotor dynamics have been observed in MOFs containing bicyclo[1.1.1]pentane-1,3-dicarboxylate, breaking the trend for typically slow rotation observed from aromatic linkers in the solid state.16
While interest in these rigid fused-ring aliphatic systems continues to grow, a structural motif for aliphatic ligands which remains untouched is that of the spirocyclic alkanes. Showing similar rigidity to the parent cycloalkanes, many spirocyclic species also bear the added feature of axial chirality, a highly sought-after property for MOF ligands.17 Despite effective synthetic routes existing for several fully aliphatic spirocyclic dicarboxylates, these have been continually overlooked in the MOF and metallosupramolecular chemistry fields. Here, we introduce spiro[3.3]heptane-2,6-dicarboxylic acid (Fecht's acid, abbreviated here as H2SHDC, and shown in Fig. 1) as a ligand capable of forming rigid aliphatic MOFs with unique structural properties. Having been considered as a chiral auxiliary,18 or purely as a structural oddity, to date there have been no reports describing the coordination chemistry of this molecule.
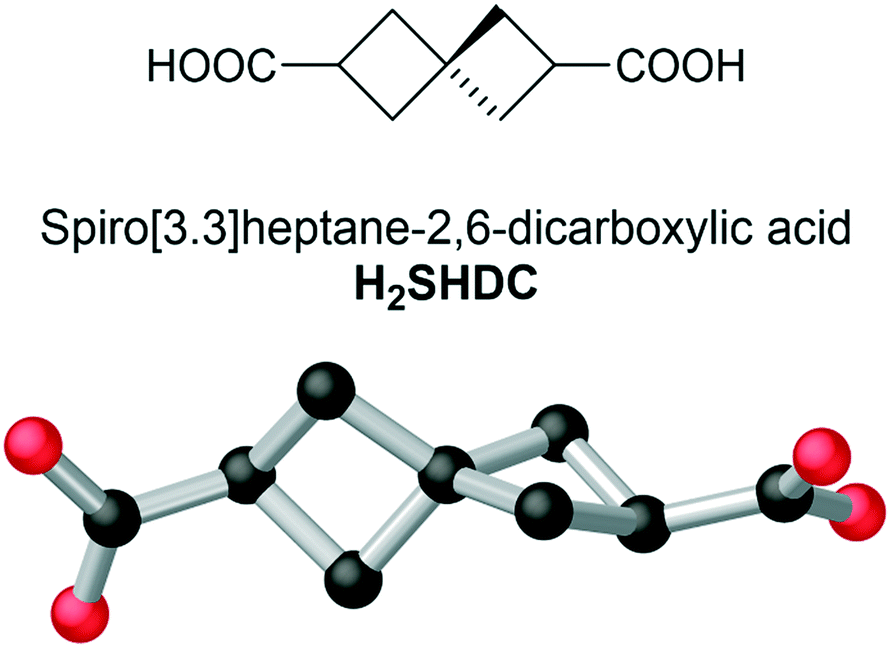 | ||
| Fig. 1 The structure of H2SHDC showing skeletal form (top) and 3-dimensional representation (taken from the crystal structure of complex 1, vide infra) showing the orthogonal orientation of the two cyclobutane rings (bottom). Hydrogen atoms are omitted for clarity. | ||
Results and discussion
Originally reported by Fecht in 1907,19H2SHDC contains of two strained cyclobutane rings with a spiro linkage and two carboxylic acids at the terminal (2,6) positions. These groups are separated at the carboxylate carbon atoms by ca. 6.9 Å, making H2SHDC approximately 20% longer than terephthalic acid. 2,6-disubstituted spiro[3.3]heptanes are axially chiral,20 best visualised here by the ∼90° H–C⋯C–H torsion angle relating the two tertiary C–H groups, which can adopt a left- or right-handed twist. Indeed, it must be stressed that despite generating two new stereocentres, the double decarboxylation step to yield Fecht's acid from the tetracarboxylate precursor generates the product as a mixture of only two enantiomers, rather than giving chemically inequivalent diastereomers. The two possible products following the decarboxylation are related by inversion symmetry in their average conformations (ESI,† Fig. S1). The ligand was prepared as the racemate on the mmol scale in three steps from pentaerythritol tetrabromide using a modification to the procedure reported by Rice and Grogan,21 in an overall 18% yield. Although not our primary focus with this study, it is notable for future applications that a solution of H2SHDC at 40 μM in methanol shows negligible absorbance in the range 235–800 nm (ESI,† Fig. S6). This suggests that SHDC complexes may prove useful for electronic isolation of photophysically-active guests. With H2SHDC in hand, we screened various MOF synthesis conditions with the intention of generating crystalline materials containing the SHDC dianion, to test the influence of its unusual core structure on the resulting framework materials. While our attempts to generate a crystalline homoleptic zinc(II) framework of SHDC akin to MOF-5 were unsuccessful, potentially due to the slightly curved nature of the axially chiral backbone, two new coordination polymers containing SHDC were successfully isolated and analysed.The reaction of H2SHDC with ytterbium(III) chloride hexahydrate in DMF/H2O at 100 °C gave colourless rod-shaped crystals after 24 hours. Analysis by single crystal X-ray diffraction revealed a 3-dimensional polymeric structure consisting of one-dimensional ytterbium carboxylate chains. Each chain is linked to six others by bridging through the fully deprotonated SHDC ligands in a hex-type rod packing topology, as shown in Fig. 2.
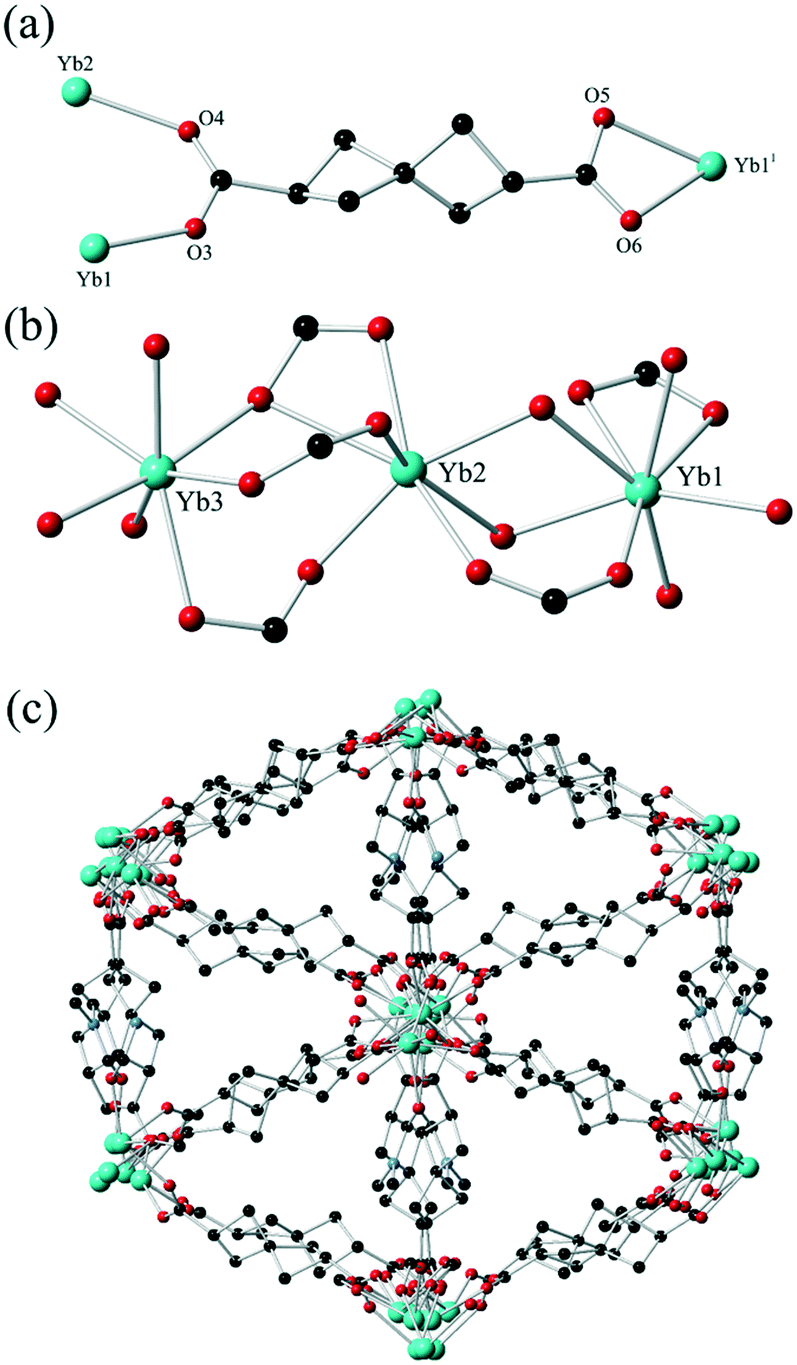 | ||
| Fig. 2 (a) Representative environment for the SHDC ligand in complex 1 with heteroatom labelling scheme. Hydrogen atoms are omitted for clarity. (b) The three unique Yb environments in the structure of 1; (c) The extended structure of complex 1 viewed parallel to the Yb-carboxylate chains showing the hex rod packing motif. Symmetry code used to generate equivalent atoms: 1. 3/2 − x, 1 − y, 3/2 − z. | ||
The asymmetric unit contains three unique Yb sites with charge balance provided by two complete SHDC molecules and five halves of SHDC ligands where the spiro carbon atom resides on a crystallographic inversion centre, which superimposes the two enantiomers of SHDC between the same metal sites. This led to substantial crystallographic disorder on these sites (ESI,† Fig. S3), but nonetheless each SHDC species could be modelled across no more than two overlapped orientations using the two ordered residues as the basis for geometric restraints. One DMF ligand is also present in the asymmetric unit coordinated to Yb3. The SHDC ligands each exhibit μ3 or μ4 bridging modes to the ytterbium ions. As expected, the extent of ring strain is most obvious by examining the angles around the spiro carbon atoms. Internal bond angles in the range 85.8–88.6° are observed on the non-disordered residues, compensated by external C–C–C angles in excess of 120°.
The extended structure of 1 contains isolated pockets of diffuse electron density enclosed by SHDC ligands. While conventional aromatic ligands often form intra-framework π⋯π contacts, in 1 short contacts between ligands are notable by their absence, as adjacent ligands are separated by distances of ca. 5.4–6.5 Å between spiro carbon atoms. The contents of these pockets could not be established crystallographically but the combination of thermogravimetric analysis, elemental analysis and the residual crystallographic electron count are in good agreement with a formulation of 0.5 DMF molecules and 2.5 water molecules per Yb6 formula unit. Thermal analysis showed this solvation was immobile with no substantial mass loss below 250 °C, followed by a loss of 7.8 wt% between 250–350 °C, corresponding to both the lattice solvent and the coordinated DMF ligands (ESI,† Fig. S9). Interestingly, mass loss due to ligand decomposition was only observed with an onset of 425 °C, suggesting that despite the presence of two highly strained cyclobutane rings within the ligand backbone and a particularly distorted spiro carbon atom, the ligand remains remarkably thermally stable when deprotonated. Nonetheless, heating the material to 400 °C with a 30 minute dwell period under a nitrogen flow led to clear discolouration with loss of single crystallinity, indicating that the framework structure itself is not retained under harsh thermal desolvation conditions. The solid was immersed for 3 days in methanol in an attempt to exchange the immobile lattice solvent for a more volatile substitute, however the TGA post-soaking was effectively identical to the pristine material, suggesting no solvent exchange was possible. This suggests that access to these regions is effectively blocked by the larger steric bulk of the SHDC backbone constricting any possible pore windows.
No fully isostructural Yb-terephthalate species is known for comparison; the various Yb-terephthalates tend to more commonly crystallise with rectangular channels.22 A similar extended structure with a hex rod packing topology is observed in a cerium terephthalate reported by D’Arras et al.23 Interestingly, while no permanent porosity was reported in that structure, the onset of decomposition of the cerium terephthalate species at 360 °C indicates that SHDC may impart a higher thermal stability compared to terephthalate, even despite the high degree of ring strain in the ligand backbone.
To make further comparisons to terephthalate, we turned our attention to transition metal complexes, and in the absence of a homoleptic zinc MOF, explored the use of nitrogen heterocycles as co-ligands. Reaction of H2SHDC with zinc(II) in the presence of trans-1,2-bis(4-pyridyl)ethene (bpe) gave yellow crystals of complex 2, which analysed for the formula poly-[Zn(SHDC)(bpe)]. This structure exhibits a fourfold interpenetrated diamondoid (dia) topology, with tetrahedral zinc(II) nodes connected by both SHDC and bpe linkages as shown in Fig. 3. The asymmetric unit contains a single zinc ion occupying a crystallographic special position, and halves of both ligand types. Similarly to complex 1, the SHDC ligand is disordered across two positions related by a fourfold improper rotation about the spiro carbon atom, again indicating that both enantiomers are superimposed. The bridging distances of 11.8080(7) and 13.4111(9) Å respectively for SHDC and bpe lead to a relatively regular extended network, although the near-linear chains of SHDC-linked zinc ions along the 011 vector (giving a Zn–Zn–Zn angle of 141.54(2)°) leads to slight flattening of the network in the perpendicular direction.
 | ||
| Fig. 3 (a) The coordination environment of the zinc ion in the structure of 2 with labelling scheme for unique heteroatoms. Hydrogen atoms and disorder on the SHDC molecule are omitted for clarity. (b) Extended structure of complex 1 showing the rectangular solvent channels. | ||
Despite the interpenetration, linear solvent channels are present in the extended structure of 2 oriented parallel to the crystallographic b axis. These rectangular channels, of dimensions ca. 8.8 × 9.1 Å interatomic distance, are lined by alternating SHDC and bpe groups which both lends a contouring to the surface and blocks access between adjacent channels. Solvent molecules within these channels could not be modelled crystallographically; the residual electron count from the crystallographic model of 66 electrons per formula unit is marginally higher than that established from bulk-phase methods (1.33 DMF and 1.33 H2O per formula unit, calculated 43 electrons), likely due to solvent loss on air exposure. Indeed, thermogravimetric analysis shows that the pore contents are considerably more mobile in 2 than 1, with onset of desolvation beginning near room temperature but continuing until the first plateau at 170 °C. Complex 2 also shows a lower decomposition temperature with onset of ligand decomposition at 320 °C, most likely involving the bpe co-ligand (ESI,† Fig. S10). The lattice solvent was readily exchanged with methanol to lower the desolvation temperature to 100 °C, while retaining an essentially equivalent volatile fraction by TGA. Capillary PXRD confirmed that the extended structure was conserved during the exchange process (ESI,† Fig. S8).
A terephthalic acid analogue to complex 2 is known, having been reported by Liu and co-workers in 2008.24 In that structure, although an equivalent tetrahedral zinc node is present leading to a dia topology, fivefold interpenetration is observed rather than the fourfold mode observed in 2. The interpenetrated networks associate by π⋯π stacking between the terephthalate and pyridine groups of adjacent networks, and no overall porosity is present. In contrast, the aliphatic nature of the dicarboxylate component in complex 2 prohibits most of the possible interactions between frameworks, with only minor overlap between the π systems of the pyridyl groups. This is a tangible structural outcome from the replacement of an aromatic linker with the aliphatic SHDC spacer, which is a promising starting point for enhancing porosity in similar materials by removing the tendency for aggregation between framework components.
As both complexes 1 and 2 contained potential porosity within their frameworks, both were analysed for their N2 and CO2 uptake performance. Both compounds exhibit type II adsorption behaviour for nitrogen at 77 K, with the total uptakes minimal suggesting negligible micropore volume. Perhaps unsurprisingly given the lattice solvent could not be removed under accessible conditions, the uptake of CO2 was also low in compound 1, reaching a maximum of 25 cc(STP) g−1 at 100 kPa and 278 K (ESI,† Fig. S11 and S12). The linear shape of the CO2 isotherm suggested minimal penetration of the adsorbate into the micropores of the material itself. For compound 2, expected to show better performance given the facile solvent exchange, the uptake was similarly low (ESI,† Fig. S13 and S14); although CO2 adsorption was marginally higher across the entire pressure range and showed a more typical inflection, the N2 and CO2 loadings are consistent with collapse of the pores during activation rather than micropore filling. Enthalpy of adsorption calculations for CO2 on both materials (ESI,† Fig. S15) show similar zero-loading values of −37 and −33 kJ mol−1 for 1 and 2, respectively, and the expected shallow negative gradients across the loading range. X-ray powder diffraction of the materials post-adsorption showed that complex 1 was unchanged, suggesting that the lattice solvent cannot be effectively displaced without destroying the extended structure, while complex 2 showed conversion into a new, poorly crystalline and presumably close-packed phase during thermal activation.
Experimental
Materials & methods
All starting materials, reagents and solvents were purchased from Sigma-Aldrich, Fluorochem, TCI or Alfa Aesar, were of reagent grade or better and were used as received. NMR spectroscopy was performed on a Bruker Avance III HD 400 spectrometer operating at 400 MHz for 1H NMR and 101 MHz for 13C NMR using deuterated solvents from Cambridge Isotope Laboratories. All spectra were reference to the residual solvent signal and/or TMS. Infrared spectra were measured using a Thermo Scientific Nicolet iS10 instrument with an ATR sampler. Elemental analysis was performed using a Thermo Flash 2000 CHNS analyser, calibrated against sulfanilamide with vanadium pentoxide as a combustion aid. Melting points were measured in air on a Stuart digital melting point apparatus and are uncorrected. High resolution mass spectra were measured at the EPSRC National Mass Spectrometry Facility at Swansea University, United Kingdom. X-ray powder diffraction patterns were measured on a Bruker D8 Advance diffractometer with a Cu-Kα source (λ = 1.54178 Å). Samples were mounted on a zero-background silicon single crystal sample holder. All samples were measured at room temperature in the 2θ range 5–55°, and these data were compared against simulated patterns from the single crystal data collections at 150 K. Gas adsorption data were collected with a Quantachrome Autosorb iQ with three analysis stations. Temperature control was provided by a liquid nitrogen dewar for N2 measurements and a jacketed bath attached to a Julabo recirculating chiller (0.01 K precision) for CO2 measurements. All samples were activated prior to analysis by first exchanging mobile solvent by soaking the samples in methanol for 3 days, refreshing the exchange solvent every 12 hours during this period. The samples were then subjected to initial activation at 100 °C for 4 hours under dynamic vacuum provided by a rotary oil pump, followed by final activation at 100 °C for 10 hours under high vacuum provided by the turbomolecular pump. The sample masses post-activation were 21.7 and 44.5 mg for 1 and 2, respectively.X-ray crystallography
Single crystal XRD data were collected on a Bruker D8 Quest ECO with graphite monochromated Mo Kα (λ = 0.71073 Å). Crystals were mounted on Mitegen micromounts in NVH immersion oil, and all collections were carried out at 150 K using an Oxford cryostream. Data collections and reductions were carried out in the Bruker APEX-3 suite of programs.25 Data were solved using the intrinsic phasing routine in SHELXT26 and refined on F2 in SHELXL27 within OLEX-2 version 1.3.28 In both complexes 1 and 2, substantial disorder was noted on the backbone of the SHDC residues, which was especially manifested by a non-crystallographic S4 improper rotation centred on the spiro carbon which tended to disorder the entire carbon backbone. Where necessary, this was modelled by splitting all backbone carbon atoms across two positions, using SADI and DFIX restraints based on the geometry of a non-disordered residue in the structure of 1. RIGU and ISOR restraints were employed when Uij tensors became unreasonable, and EADP constraints were used where atoms from the two disordered contributors (typically carboxylate atoms) occupied very similar positions. In both compounds, regions of diffuse electron density corresponding to disordered lattice solvent were detected for which no sensible assignment could be made. The scattering contributions of these regions were accounted for using the SMTBX solvent masking routine in Olex2 v1.3.28,29 In both cases, the electron density accounted for was compared with the predicted values from elemental analysis and thermogravimetric analysis. For compound 1 the lattice solvation is estimated by microanalysis at 0.5 DMF and 2.5 H2O molecules per formula unit ([Yb6(SHDC)9(DMF)2]). The expected electron count is 45 e− and a volatile mass of 7.8 wt% (the latter value includes the coordinated DMF molecules), compared to the calculated values of 51 e− and 7.2 wt%. For compound 2, the lattice solvation is estimated by microanalysis at 1.33 DMF and 1.33 H2O molecules per formula unit (calc. 66 e−, 22 wt%, found 43 e−, 15 wt%). CCDC 2106213 and 2106214.Synthesis of H2SHDC, Fecht's acid
H 2 SHDC was prepared according to a modified literature procedure.21 Sodium (575 mg, 25 mmol) was added to n-pentanol (25 mL) under a N2 atmosphere and stirred with gentle heating (55–60 °C) until most of the sodium had reacted. Diethyl malonate (3.80 mL, 24.911 mmol) was added and stirring was continued until all reagents had fully dissolved. Pentaerythritol tetrabromide (2.000 g, 5.158 mmol) was added in one portion and the mixture was heated under reflux under a N2 atmosphere for 48 hours. On completion and cooling to room temperature, any solids present were removed by vacuum filtration and quenched with isopropanol. The pentanol filtrate was concentrated by distillation; to aid this, when near-dryness the solution was cooled and water (5 mL) was added and the solution was reheated to remove the water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pentanol azeotrope. This was repeated 1–2 more times to ensure all pentanol had been removed. The resulting oil was cooled to room temperature and combined with water (5 mL) and extracted into diethyl ether (100 mL) three times. The organic layers were combined, dried over MgSO4, filtered and dried in vacuo to give a viscous oil which consists of a mixture of the ethyl and pentyl esters of the spirocyclic tetracarboxylate.
:pentanol azeotrope. This was repeated 1–2 more times to ensure all pentanol had been removed. The resulting oil was cooled to room temperature and combined with water (5 mL) and extracted into diethyl ether (100 mL) three times. The organic layers were combined, dried over MgSO4, filtered and dried in vacuo to give a viscous oil which consists of a mixture of the ethyl and pentyl esters of the spirocyclic tetracarboxylate.
This oil was then dissolved in ethanol (40 mL) and KOH (4.5 g, 80 mmol) was added. The mixture was sealed and stirred at room temperature for 72 hours. After this, the yellow solid that had formed was isolated by vacuum filtration and washed with a small amount of ice-cold ethanol. The solid was then dissolved in water (5 mL) and acidified with 5 M HCl to pH ∼3. The resulting solution was poured into a large evaporating dish and left to evaporate for approximately 3 days, until the solid was completely dry. This solid was then heated to 150 °C and the temperature was gradually increased in ∼20 °C increments until a temperature of 215 °C was reached, at which it dwelled for 5 minutes until it was allowed to cool to room temperature. Once cool, the solid was recrystallised from boiling water. Cooling the filtrate yielded colourless crystals. Yield 160 mg, 18% overall, mp 207–209 °C (lit. 212 °CS6). δH (CDCl3, 400 MHz) 11.25 (s, 2H, H1), 3.03 (p, 2H, H2, J = 8.3 Hz), 2.23–2.39 (m, 8H, H3); δC (CDCl3, 100 MHz) 180.61, 37.73, 37.33, 36.84, 32.53. νmax/cm−1 (KBr): 2969 w, 2931 w, 2848 w, 2736 w, 2664 w br, 2554 w br, 1681 s, 1415 m, 1333 w, 1247 s, 1215 s, 1112 w, 931 s, 738 m, 702 m, 541 w, 467 w; m/z (ESMS) 183.0668 ([M − H+], calculated for C9H11O4 183.0657).
Synthesis of poly-[Yb6(SHDC)9(DMF)2]·0.5DMF·2.5H2O complex 1
H 2 SHDC (10 mg, 0.054 mmol) was dissolved in DMF (2 mL) and H2O (1 mL), to which was added YbCl3·6H2O (5 mg, 0.013 mmol). The resulting mixture was sealed in a scintillation vial and heated to 100 °C for 24 hours. Colourless block crystals formed in the vial after 24 hours which were isolated by vacuum filtration from the hot solution. Yield 4.6 mg, 41%; mp >300 °C; νmax/cm−1 (KBr): 2952 w, 2921 w, 1660 w, 1612 m, 1539 s, 1427 s, 1281 m, 1112 w, 859 w, 778 w, 675 w, 566 w, 498 m. Found C, 36.30; H, 3.71; N, 1.52; calculated for C87H104N2O38Yb6·0.5DMF·2.5H2O ([Yb6(SHDC)9(DMF)2]·0.5DMF·2.5H2O), C, 36.58; H, 3.90; N, 1.21%.Synthesis of poly-[Zn(SHDC)(BPE)]·1.33DMF·1.33H2O, complex 2
H 2 SHDC (10 mg, 0.054 mmol) was dissolved in DMF (1 mL) in a scintillation vial, to which was added Zn(NO3)2·6H2O (6 mg, 0.034 mmol) and bpe (9 mg, 0.049 mmol). The resulting mixture was heated in a heat block to 100 °C for 24 hours. Orange needle crystals formed in the vial after 24 hours which were isolated from the hot solution by vacuum filtration. Yield 6 mg, 41%; mp >300 °C.; νmax/cm−1 (KBr): 3421 br, 3063 w, 2995 w, 2920 w, 2845 w, 1668 m, 1611 s, 1567 s, 1506 w, 1429 s, 1387 s, 1274 m, 1207 w, 1097 w, 1069 w, 1025 m, 984 w, 834 m, 772 w, 737 w, 660 w, 552 s, 515 w. Found C, 54.19; H, 5.23; N, 8.91; calculated for C21H20N2O4Zn·1.33DMF·1.33H2O ([Zn(SHDC)(bpe)]·1.33DMF·1.33H2O) C, 54.47; H, 5.85; N, 8.47%.Conclusion
The successful preparation of two new 3-dimensional coordination networks based on Fecht's acid H2SHDC has demonstrated the feasibility of the spiro[3.3]heptane synthon as a 1,4-phenylene isostere in metallosupramolecular systems for the first time. Complex 1 is a 3-dimensional network of ytterbium-carboxylate rod motifs linked by SHDC units, giving a thermally stable but non-porous hex-type rod packed network. Complex 2 incorporates an additional bpe co-ligand, yielding a 3-dimensional dia network with accessible solvent channels. While not stable to evacuation, it is notable that the aliphatic SHDC backbone appears to favour a reduction in inter-framework contacts compared to the close-packed aromatic analogue. The good thermal stability and reduced tendency towards ligand–ligand close contacts in these systems shows that spiroalkanes may be an important general route to improving performance in porous materials development, particularly for applications requiring non-polar void space.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the School of Chemical and Physical Sciences, Keele University, and The Royal Society (research grant RGS\R1\191227) for financial support. We gratefully acknowledge the technical staff of the Lennard-Jones and Central Sciences Laboratories at Keele University for their invaluable work in laboratory and instrument support.References
- D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123–133 CrossRef CAS PubMed; Z. Ji, H. Wang, S. Canossa, S. Wuttke and O. M. Yaghi, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef.
- N. Kumar, S. Mukherjee, N. C. Harvey-Reid, A. A. Bezrukov, K. Tan, V. Martins, M. Vandichel, T. Pham, L. M. van Wyk, K. Oyekan, A. Kumar, K. A. Forrest, K. M. Patil, L. J. Barbour, B. Space, Y. Huang, P. E. Kruger and M. Zaworotko, Chem, 2021, 7, 3085–3098 CAS.
- P. Ji, T. Drake, A. Murakami, P. Oliveres, J. H. Skone and W. Lin, J. Am. Chem. Soc., 2018, 140, 10553–10561 CrossRef CAS.
- L. J. Small, M. E. Schindelholz and T. M. Nenoff, Ind. Eng. Chem. Res., 2021, 60, 7998–8006 CrossRef CAS; F.-Y. Yi, D. Chen, M.-K. Wu, L. Han and H.-L. Jiang, ChemPlusChem, 2016, 81, 675–690 CrossRef PubMed.
- C. B. Fan, L. L. Gong, L. Huang, F. Luo, R. Krishna, X. F. Yi, A. M. Zheng, L. Zhang, S. Z. Pu, X. F. Feng, M. B. Luo and G. C. Guo, Angew. Chem., Int. Ed., 2017, 56, 7900–7906 CrossRef CAS.
- R. A. Perlata, M. T. Huxley, Z. Shi, Y.-B. Zhang, C. J. Sumby and C. J. Doonan, Chem. Commun., 2020, 56, 15313–15316 RSC; K. Manna, T. Zhang and W. Lin, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed.
- E. A. Dulgopolova, A. A. Berseneva, M. S. Faillace, O. A. Ejegbavwo, G. A. Leith, S. W. Choi, H. N. Gregory, A. M. Rice, M. D. Smith, M. Chruszsz, G. Garashchuk, K. Mythreye and N. B. Shustova, J. Am. Chem. Soc., 2020, 142, 4769–4783 CrossRef CAS PubMed; J. Cornelio, T.-Y. Zhou, A. Alkaş and S. G. Telfer, J. Am. Chem. Soc., 2018, 140, 15470–15476 CrossRef PubMed; C. S. Hawes, G. M. Ó Máille, K. Byrne, W. Schmitt and T. Gunnlaugsson, Dalton Trans., 2018, 47, 10080–10092 RSC; X.-Y. Liu, X.-M. Yin, S.-L. Yang, L. Zhang, R. Bu and E.-Q. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 20380–20387 CrossRef PubMed.
- J. Gu, M. Wen, X. Liang, Z. Shi, M. V. Kirillova and A. M. Kirillov, Crystals, 2018, 8, 83 CrossRef CAS; B. J. Bucior, A. S. Rosen, M. Haranczyk, Z. Yao, M. E. Ziebel, O. K. Farha, J. T. Hupp, J. I. Siepmann, A. Aspuru-Guzik and R. Q. Snurr, Cryst. Growth Des., 2019, 19, 6682–6697 CrossRef.
- T.-H. Chen, I. Popov, W. Kaveevivitchai and O. Š. Miljanić, Chem. Mater., 2014, 26, 4322–4325 CrossRef CAS; F. P. Kinik, A. Ortega-Guerrero, D. Ongari, C. P. Ireland and B. Smit, Chem. Soc. Rev., 2021, 50, 3143–4177 RSC; C. S. Hawes, Dalton Trans., 2021, 50, 6034–6049 RSC; H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- B. Chen, F. R. Fronczek, B. H. Courtney and F. Zapata, Cryst. Growth Des., 2006, 6, 825–828 CrossRef CAS; J. Wang, Y.-C. Ou, Y. Shen, L. Yun, J.-D. Leng, Z. Lin and M.-L. Tong, Cryst. Growth Des., 2009, 9, 2442–2450 CrossRef; O. Drath, R. W. Gable and C. Boskovic, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 734–741 CrossRef PubMed; K. M. Patil, M. E. Dickinson, T. Tremlett, S. C. Moratti and L. R. Hanton, Cryst. Growth Des., 2016, 16, 1038–1046 CrossRef; I. Boldog, K. V. Domasevitch, J. Sanchiz, P. Mayer and C. Janiak, Dalton Trans., 2014, 43, 12590–12605 RSC; D. Pavlov, T. Sukhikh, E. Filatov and A. Potapov, Molecules, 2019, 24, 2717 CrossRef; M. R. Mani, R. Chellaswamy, Y. N. Marathe and V. K. Pillai, RSC Adv., 2015, 6, 1907–1912 RSC; S. Wang, N. Xharferaj, M. Wahiduzzaman, K. Oyekan, X. Li, K. Wei, B. Zheng, A. Tissot, J. Marrot, W. Shepard, C. Martineau-Corcos, Y. Filinchuk, K. Tan, G. Maurin and C. Serre, J. Am. Chem. Soc., 2019, 141, 17207–17216 CrossRef PubMed.
- B. Bozbiyik, J. Lannoeye, D. E. De Vos and J. F. M. Denayer, Phys. Chem. Chem. Phys., 2016, 18, 3294–3301 RSC; F. Jeremias, D. Fröhlich, C. Janiak and S. K. Henninger, RSC Adv., 2014, 4, 24073–24082 RSC; G. Zahn, H. A. Schulze, J. Lippke, S. König, U. Sazama, M. Fröba and P. Behrens, Microporous Mesoporous Mater., 2015, 203, 186–194 CrossRef CAS; C. L. F. Dazem, F. M. A. Noa, J. Nenwa and L. Öhrström, CrystEngComm, 2019, 21, 6156–6164 RSC; K. Zhang, X. Xie, H. Li, J. Gao, L. Nie, Y. Pan, J. Xie, D. Tian, W. Liu, Q. Fan, H. Su, L. Huang and W. Huang, Adv. Mater., 2017, 29, 1701804 CrossRef.
- V. D. Slyusarchuk, P. E. Kruger and C. S. Hawes, ChemPlusChem, 2020, 85, 845–854 CrossRef CAS.
- G. M. Locke, S. S. R. Bernhard and M. O. Senge, Chem. – Eur. J., 2018, 25, 4590–4647 CrossRef CAS; P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC; A. Denisenko, P. Garbuz, S. V. Shishkina, N. M. Voloshchuk and P. K. Mykhailiuk, Angew. Chem., 2020, 59, 20515–20521 CrossRef.
- L. K. Macreadie, E. J. Mensforth, R. Babarao, K. Konstas, S. G. Telfer, C. M. Doherty, J. Tsanaktsidis, S. R. Batten and M. R. Hill, J. Am. Chem. Soc., 2019, 141, 3828–3832 CrossRef CAS PubMed.
- P. J. Llabres-Campaner, J. Pitarch-Jarque, R. Ballesteros-Garrido, B. Abarca, R. Ballesteros and E. García-España, Dalton Trans., 2017, 46, 7397–7402 RSC.
- J. Perego, S. Bracco, M. Negroni, C. X. Bezuidenhout, G. Prando, P. Carretta, A. Comotti and P. Sozzani, Nat. Chem., 2020, 12, 845–851 CrossRef CAS.
- T. Sawano, N. C. Thacker, Z. Lin, A. R. McIsaac and W. Lin, J. Am. Chem. Soc., 2015, 137, 12241–12248 CrossRef CAS PubMed.
- L. A. Hulshof and H. Wynberg, J. Am. Chem. Soc., 1974, 96, 2191–2200 CrossRef CAS; H.-Z. Tang, H. Miura and Y. Kawakami, Enantiomer, 2002, 7, 5–9 CrossRef PubMed.
- H. Fecht, Chem. Ber., 1907, 40, 3883–3891 CrossRef.
- L. A. Hulshof, H. Wynberg, B. van Dijk and J. L. de Boer, J. Am. Chem. Soc., 1976, 98, 2733–2740 CrossRef CAS.
- L. M. Rice and C. H. Grogan, J. Org. Chem., 1961, 26, 54–58 CrossRef CAS.
- Y. Wu, M. I. Breeze, G. J. Clarkson, F. Millange, D. O’Hare and R. I. Walton, Angew. Chem., Int. Ed., 2016, 55, 4992–4996 CrossRef CAS PubMed; J.-J. Wang, Y. Chen, M.-J. Liu, R.-Y. Fan, P.-P. Si, J. Yang, Y.-Y. Pan, Y. Chen, S.-S. Zhao and J. Xu, Polyhedron, 2018, 154, 411–419 CrossRef.
- L. D’Arras, C. Sassoye, L. Rozes, C. Sanchez, J. Marrot, S. Marre and C. Armonier, New J. Chem., 2014, 38, 1477–1483 RSC.
- D. Liu, H. Li, Y. Chen, Y. Zhang and J. Lang, Chin. J. Chem., 2008, 26, 2173–2178 CrossRef CAS.
- Bruker APEX-3, Bruker-AXS Inc., Madison, WI, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallography data, gas adsorption data, NMR spectra, thermogravimetric analysis data and X-ray powder diffraction plots. CCDC 2106213 and 2106214. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01542g |
| This journal is © The Royal Society of Chemistry 2022 |