
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stars and stripes: hexatopic tris(3,2′:6′,3′′-terpyridine) ligands that unexpectedly form one-dimensional coordination polymers†
Giacomo
Manfroni
,
Alessandro
Prescimone
 ,
Edwin C.
Constable
and
Catherine E.
Housecroft
*
,
Edwin C.
Constable
and
Catherine E.
Housecroft
*
Department of Chemistry, University of Basel, Mattenstrasse 24a, BPR 1096, 4058-Basel, Switzerland. E-mail: catherine.housecroft@unibas.ch
First published on 6th December 2021
Abstract
The hexatopic ligands 1,3,5-tris(4,2′:6′,4′′-terpyridin-4′-yl)benzene (1), 1,3,5-tris(3,2′:6′,3′′-terpyridin-4′-yl)benzene (2), 1,3,5-tris{4-(4,2′:6′,4′′-terpyridin-4′-yl)phenyl}benzene (3), 1,3,5-tris{4-(3,2′:6′,3′′-terpyridin-4′-yl)phenyl}benzene (4) and 1,3,5-trimethyl-2,4,6-tris{4-(3,2′:6′,3′′-terpyridin-4′-yl)phenyl}benzene (5) have been prepared and characterized. The single crystal structure of 1·1.75DMF was determined; 1 exhibits a propeller-shaped geometry with each of the three 4,2′:6′,4′′-tpy domains being crystallographically independent. Packing of molecules of 1 is dominated by face-to-face π-stacking interactions which is consistent with the low solubility of 1 in common organic solvents. Reaction of 5 with [Cu(hfacac)2]·H2O (Hhfacac = 1,1,1,5,5,5-hexafluoropentane-2,4-dione) under conditions of crystal growth by layering resulted in the formation of [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3. Single-crystal X-ray diffraction reveals an unusual 1D-coordination polymer consisting of a series of alternating single and double loops. Each of the three crystallographically independent Cu atoms is octahedrally sited with cis-arrangements two N-donors from two different ligands 1 and, therefore, cis-arrangements of coordinated [hfacac]− ligands; this observation is unusual among compounds in the Cambridge Structural Database containing {Cu(hfacac)2N2} coordination units in which the two N-donors are in a non-chelating ligand.
Introduction
Coordination polymers (CPs)1 have a long history, stretching back at least to the discovery of Prussian blue at the beginning of the 18th century.2 However, it is only during the last three decades that CPs have been the focus of intense research attention.3 The possibility of performing routine single crystal structural determinations is one of the key factors and, since then, widespread potential applications have included catalysis,4,5 gas capture,6,7 molecular separations,8–11 proton conduction12 and sensing.13,14Oligopyridines (N-donors) and polycarboxylates (O-donors) are among the most common classes of organic ligands for CPs. Terpyridine (tpy) ligands are well-established N-donors in coordination chemistry, and have therefore long been successfully exploited for the assembly of discrete and polymeric coordination complexes.15 Of the 48 possible isomeric terpyridines, 2,2′:6′,2′′-tpy is the archetypal. It is a monotopic chelating ligand, although the 2:2′:6′,2′′-tpy domain also exhibits multiple metal-binding modes.16 CPs are formed by combinations of metal nodes and ligand linkers, metal linkers and ligand nodes, or metal linkers and ligand linkers. Thus, if the organic ligand contains a 2:2′:6′,2′′-tpy metal-binding domain, a second-binding domain, such as carboxylate or a heterocycle, is needed to generate an {M(tpy)2} centred ‘expanded ligand’.17
A contrasting approach is to employ oligopyridines with divergent sets of donor atoms. Of all the isomers, however, only the symmetrical 4,2′:6′,4′′-tpy and 3,2′:6′,3′′-tpy have been widely used in the last decade,15,18–22 along with their 4′-functionalized derivatives which are readily accessible using either Wang and Hanan's one-pot synthetic approach23 or the Kröhnke methodology.24 The ligands 4,2′:6′,4′′-tpy and 3,2′:6′,3′′-tpy coordinate through the outer pyridine donors, leaving the central pyridyl-N atom unbound (Scheme 1). CPs with uncoordinated Lewis-base sites within solvent-accessible channels have been considered as potential candidates for small-molecule and ion detection applications,15,22,25–27e.g. through C–H⋯N pyridine hydrogen bond formation.15 These tpy isomers possess different vectorial properties of the N-donor set (Scheme 1). While 4,2′:6′,4′′-tpy offers a fixed V-shaped metal-binding domain, the less explored isomer 3,2′:6′,3′′-tpy has a greater conformational flexibility which arises from rotation about the inter-ring C–C bonds connecting the pyridine rings leading to a more variable and less predictable network assembly.21
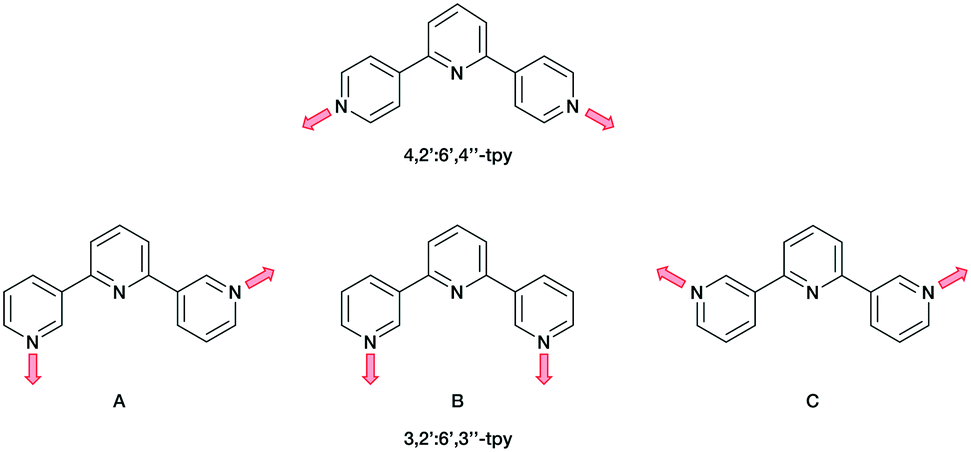 | ||
| Scheme 1 The fixed coordination directionality in 4,2′:6′,4′′-tpy, compared with the three planar conformations A, B and C of 3,2′:6′,3′′-tpy. | ||
Ditopic ligands such as those in Scheme 1 have a limited role as 2-connecting linkers and by connecting multiple tpy domains, the ligands can act as directing nodes in the assembly of multidimensional architectures. Two 4,2′:6′,4′′-tpy or 3,2′:6′,3′′-tpy units, connected by an organic spacer, have the capability to function as 4-connecting nodes (Scheme 2).20–22,28 Substituents, such as n-alkyloxy groups, enhance the solubility of the ligand. However, the chain length and shape have a profound impact in orienting the assembly.20,21,28 Yoshida and co-workers reported the synthesis and structural determination of the first coordination polymer with a bis(4,2′:6′,4′′-tpy) in 2013,29 and further examples followed.20,28 However, the coordination behaviour of bis(3,2′:6′,3′′-tpy) ligands remains little explored.29–31
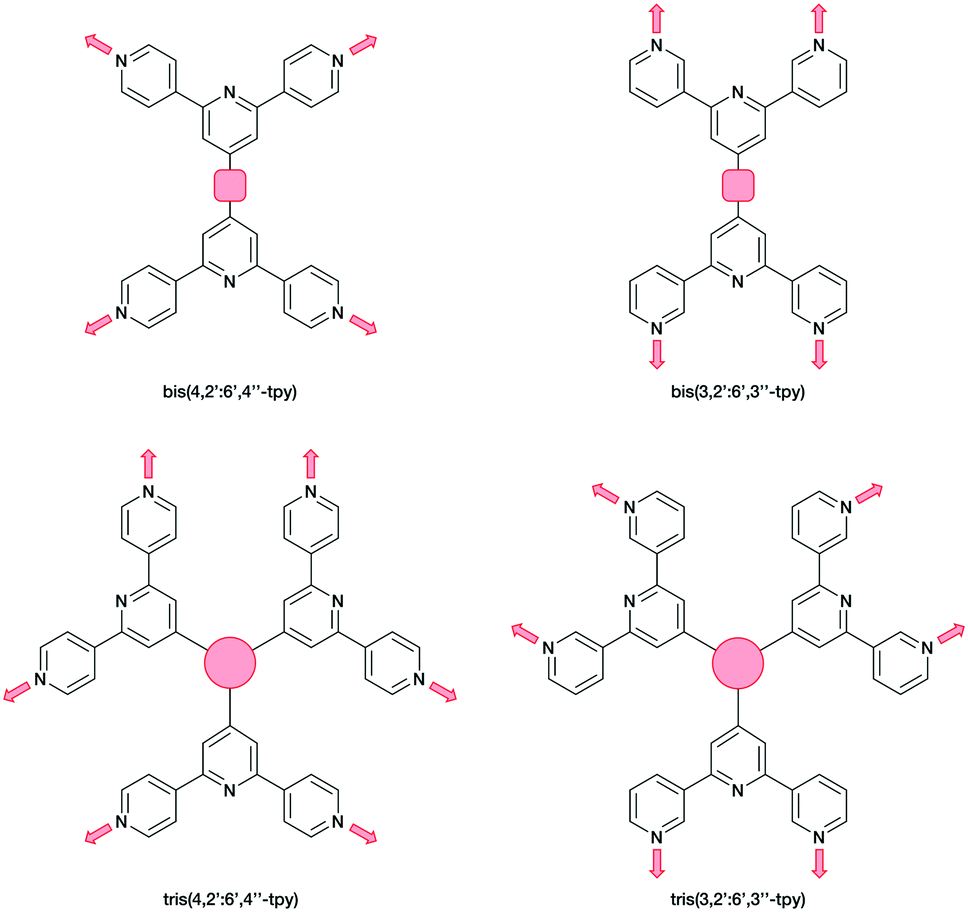 | ||
| Scheme 2 Tetratopic and hexatopic ligands resulting from connecting two and three 4,2′:6′,4′′-tpy or 3,2′:6′,3′′-tpy domains. The red spacer represents any organic linker. Only one of three possible planar conformations of 3,2′:6′,3′′-tpy is shown. | ||
In the present work, we extend our investigations of tetratopic bis(3,2′:6′,3′′-tpy) and bis(4,2′:6′,4′′-tpy) to hexatopic tris(3,2′:6′,3′′-tpy) and tris(4,2′:6′,4′′-tpy) ligands. To date, no tris(4,2′:6′,4′′-tpy) nor tris(3,2′:6′,3′′-tpy) ligands has been reported, despite the fact that they are attractive building blocks for coordination assemblies (Scheme 2). However, this design principle has been exploited with 2,2′:6′,2′′-tpy metal-binding domains and, from the first tris(2,2′:6′,2′′-tpy) in 1992,32 a wide spectrum of poly(2,2′:6′,2′′-tpy) ligands has been used for metallosupramolecular constructs.33,34 This is reflected in a range of complex geometries, such as discrete 2D-35 and 3D-fractals,36 metallodendrimers37 and metallocages38–40 (e.g. molecular polyhedra).41 Some 20 years ago, we introduced the term metallostar to describe such polynuclear assemblies42–46 and the concept of using tritopic tris(2,2′:6′,2′′-tpy) ligands (Scheme 3) for the growth of 2D or 3D CPs was developed by us and others,47,48 and the potential in materials chemistry subsequently demonstrated.49–58 The groups of Nishihara,56,57 Wong54 and Chakraborty51,55 showed a series of coordination nanosheets using different tris(terpyridines) in which durable electrochromism was observed with Fe2+ or Co2+. Moreover, according to Nishihara's group, anion exchange capacities and solvatoluminochromic behaviour were found with Zn(BF4)2 and ZnSO4 respectively.58 With 1,3,5-tris[4-(2,2′:6′,2′′-terpyridin-4′-yl)phenyl]benzene, Higuchi and co-workers described the syntheses of Fe2+, Co2+ and Ni2+-based CPs showing humidity-responsive ionic conduction.53 It should be noted that none of these polymers, containing tris(terpyridines), has been unequivocally characterized by single crystal X-ray diffraction. A search of the Cambridge Structural Database59 (CSD v. 2021.1.0),60 using ConQuest (v. 2021.1.0),60 revealed no CPs assembled with tris 2,2′:6′,2′′, 3,2′:6′,3′′ or 4,2′:6′,4′′ terpyridines. We ignore instances where the tpy unit is one of the less common 45 isomers.
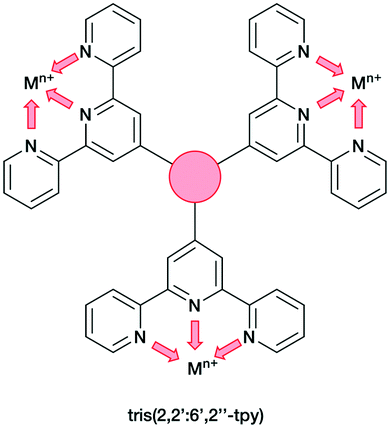 | ||
| Scheme 3 Tris(2,2′:6′,2′′-tpy) as tritopic ligand. Each tpy domain is monotopic, with the three tpy nitrogen atoms binding only one metal ion. | ||
Hence, there is significant scope for investigations of building blocks incorporating three or more 4,2′:6′,4′′-tpy or 3,2′:6′,3′′-tpy domains. Herein, we present the syntheses of a series of star-shaped ligands (Scheme 4). We also describe the self-assembly and single-crystal X-ray characterization of a tris(terpyridine)-based CP to generate an unusual 1D architecture.
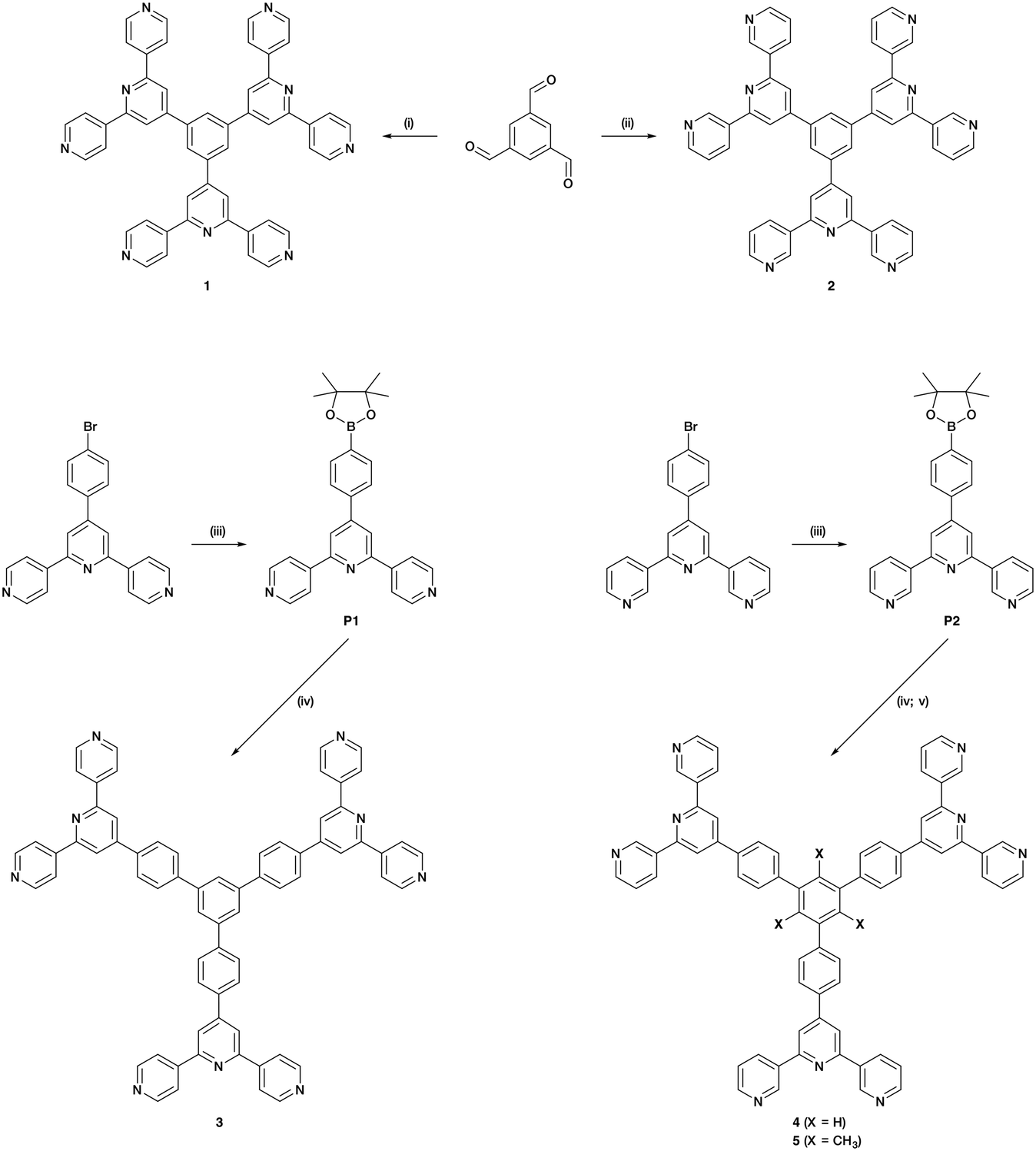 | ||
Scheme 4 Synthetic route to ligands 1–5; reagents conditions: (i) 4-acetylpyridine, KOH, NH3, EtOH, RT, 36 h; (ii) 3-acetylpyridine, KOH, NH3, EtOH, RT, 36 h; (iii) bis(pinacolato)diboron, Pd(dppf)Cl2, AcOK, DMF, 120 °C, 5 h 30 min; (iv) 1,3,5-tribromobenzene, Pd(PPh3)4, aqueous Na2CO3, THF, reflux, 12 h; (v) 2,4,6-tribromomesitylene, Pd(dppf)Cl2, Na2CO3, toluene/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), reflux, 1 h. :1), reflux, 1 h. | ||
Results and discussion
Ligand synthesis and characterization
Compounds 1 and 2 were prepared using Wang and Hanan's one-pot protocol23 (Scheme 4). After purification, 1 and 2 were obtained with yields of 22 and 13%, respectively. The synthetic route to compounds 3 and 4 was similar to the one used to synthesize the related 1,3,5-tris[4-(4′-2,2′:6′,2′′-terpyridyl)phenyl]benzene.61 A Suzuki cross-coupling reaction between 1,3,5-tribromobenzene and 4′-(4-pinacolatoboronphenyl)-4,2′:6′,4′′-terpyridine (P1) or 4′-(4-pinacolatoboronphenyl)-3,2′:6′,3′′-terpyridine (P2) (Scheme 4) was conducted in refluxing THF, with [Pd(PPh3)4] as catalyst and aqueous Na2CO3 as base, affording 3 (37%) and 4 (80%). The compounds were analytically pure and no recrystallization or chromatography was required. However, compounds 3 and 4 were poorly soluble in most common organic solvents which presumably arises from extensive intermolecular stacking interactions. Since ligand solubility is critical for crystallization methods in the assembly of CPs, we moved from ligands 3 and 4 to analogous compounds with solubilizing substituents. Rather than using hindering groups such as long alkyl chains, we opted for methyl groups (ligand 5, Scheme 4). The steric hindrance between adjacent methyl and phenyl groups forces the ligand into a non-planar conformation in the solid state, leading to less efficient π-stacking as is seen in 1,3,5-trimethyl-2,4,6-triphenyl-substituted arenes.62–66 An approach using the Suzuki coupling of 2,4,6-tribromomesitylene and P2 was not successful when utilizing THF and [Pd(PPh3)4], but an improved route to 5 was found using [Pd(dppf)Cl2] and Na2CO3 in refluxing toluene-water. However, some homo-coupling and dehalogenation by-products were identified by MALDI-TOF mass spectrometry. After chromatographic purification eluting with CHCl3, compound 5 was isolated analytically pure in a yield of 33%. Ligands 1–5 and boronic esters P1 and P2 were fully characterized by 1H and 13C{1H} NMR spectroscopies (Fig. S1–S20†), UV-vis (Fig. S21–S25†) and ATR-IR spectroscopies (Fig. S26–S33†), MALDI-TOF mass spectrometry (Fig. S34–S40†), and HR-ESI mass spectrometry (Fig. S41–S43†) or elemental analysis (see ESI† for full details). The data were consistent with the structures shown in Scheme 4. Note that ligands 3–5 with the phenylene spacers are poorly soluble in many organic solvents and, for 3, the absorption spectrum had to be recorded in benzonitrile, this being the only suitable solvent with a high-energy cut-off.Single crystal structure of 1·1.75DMF
Single crystals of 1·1.75DMF grew as a hot DMF solution of 1 was allowed to cool down to 5 °C. Tristerpyridine 1 crystallizes in the orthorhombic space group Pbca and its structure (Fig. 1a) reveals a propeller shaped geometry. The asymmetric unit contains one independent molecule and the three tpy substituents are crystallographically independent. The angles between the plane of the central arene ring and the planes of the pyridine rings containing N2, N5 and N8 (Fig. 1a) are 35.4, 32.8 and 38.0°, respectively. The conformations of the 4,2′:6′,4′-tpy units also differ. For the tpy unit having N1, N2 and N3, the twist angle between the planes of the rings containing N1/N2 and N2/N3 are 9.4 and 10.5°, respectively. In the tpy containing N7, N8 and N9, the angles between the adjacent pyridine rings N7/N8 and N8/N9 are 15.1 and 22.5°, respectively. In contrast, in the tpy possessing N4, N5 and N6, the rings are almost coplanar (angles between N4/N5 and N5/N6 are 6.5 and 1.0°).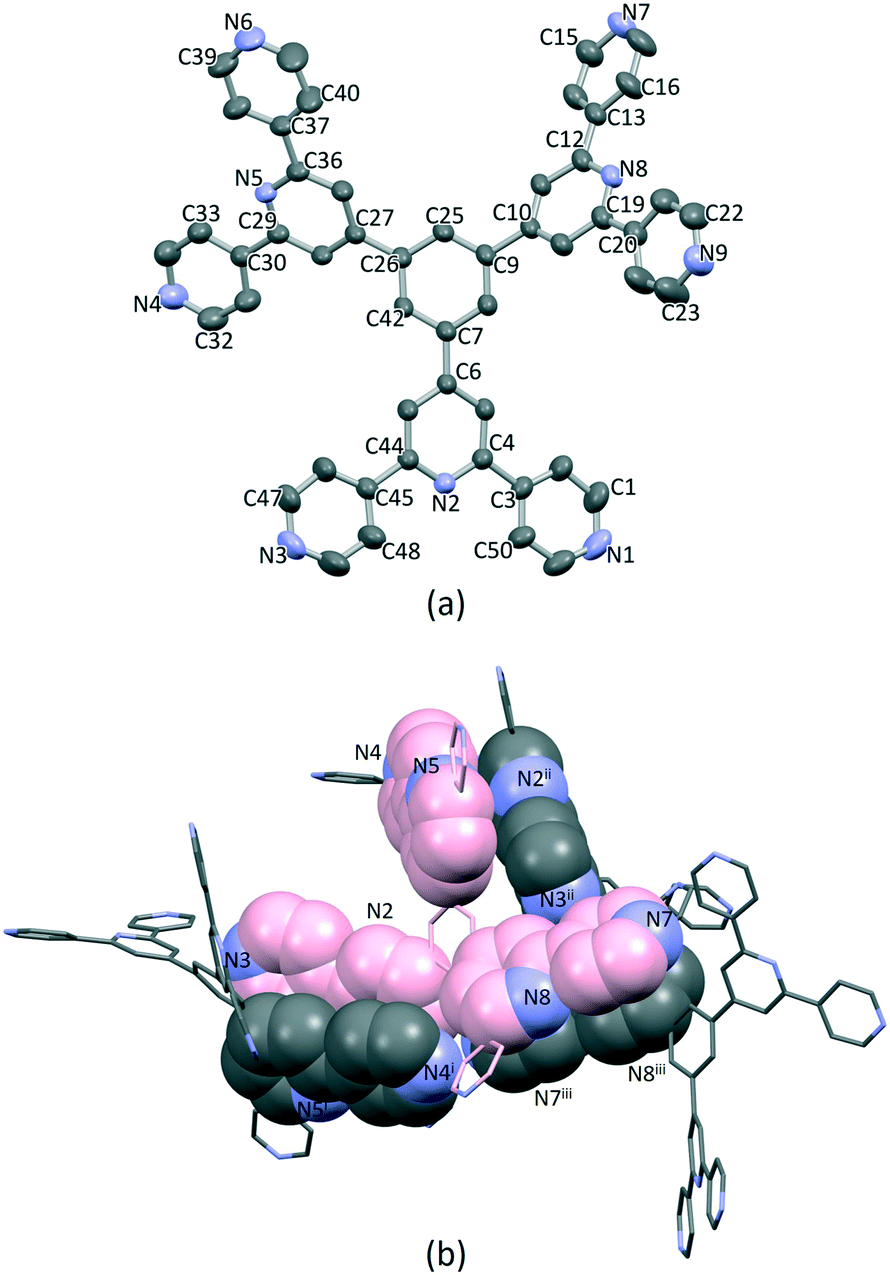 | ||
| Fig. 1 (a) Single crystal X-ray structure of 1, H atoms and DMF molecules are omitted for clarity. Thermal ellipsoids are drawn at 40% of probability level. (b) Face-to-face π-stacking between molecules of 1. The carbon atoms of the crystallographic independent molecule are coloured in pink. | ||
Packing of the molecules is dominated by face-to-face π-stacking, which is consistent with the low solubility of 1. For the crystallographic independent molecule, two different intermolecular π-stacking interactions can be classified (Fig. 1b). First, tpy unit with N1, N2 and N3 engages in an interaction with the tpy domain N4i, N5i and N6i (symmetry code i = 1 − x, −1/2 + y, 1/2 − z). A symmetry related interaction occurs between the tpy containing N4, N5 and N6 and that with N1ii, N2ii and N3ii (symmetry code ii = 1 − x, 1/2 + y, 1/2 − z). The centroid⋯centroid separations for pairs of rings containing N2/N4i and N3/N5i are 3.98 and 4.43 Å, and the corresponding angles between the ring planes are 18.2 and 21.6°. The third tpy unit containing N7, N8 and N9 stacks with the domain containing N7iii, N8iii and N9iii across an inversion centre (symmetry code iii = 1 − x, 1 − y, 1 − z). The centroid separation between rings with N7 and N8iii is 4.21 Å. All of these centroid⋯centroid separations are somewhat longer than is typical for efficient π-stacking interactions.67 In addition, the packing of the molecules involves C–H⋯N hydrogen bonds, with H⋯N distances in the range 2.47–2.73 Å (C⋯N range 3.41–3.58 Å, and C–H⋯N angle range 142.3–175.8°). Overall, the packing interactions lead to an intricate 3D-dimensional lattice.
Single-crystal structures of the coordination polymers [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3
Single crystals of [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 were grown under ambient conditions (see Experimental section). Structural analysis revealed the formation of [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 which crystallises in the monoclinic space group P21/n. Numerous crystallization set-ups were attempted with different concentrations of toluene solutions of [Cu(hfacac)2]·H2O and chloroform solutions of 5. The crystals obtained from these attempts had analogous cell parameters to [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 (see Experimental).Single crystals were also grown by layering a 1,2-dichlorobenzene solution of [Cu(hfacac)2]·H2O over a CHCl3 solution of 5 containing anthracene. The latter was added as a potential guest molecule in the assembly. A preliminary structural analysis revealed the formation of solvated [Cu3(hfacac)6(5)]n, which crystallises in the same monoclinic space group (P21/n) as [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 with similar cell dimensions (a = 20.693(4) Å, b = 21.145(4) Å, c = 34.404(7) Å, β = 104.72(3)°). The structure suffered severe disorder, but preliminary data confirmed the assembly of a 1D-coordination polymer that was essentially isostructural with that in [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3.
The structure of the asymmetric unit in [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 is shown in Fig. S44.† The backbone of the coordinated [hfacac]− ligand containing C108 and C109, CF3 groups having C87, C117, C118 and C122, the phenyl spacers containing the atoms C15–C18 and C24–C27, the pyridine ring containing N7, and solvent molecules are disordered and only major occupancies are shown in Fig. S44.† Disordered parts of the [hfacac]− ligands were refined isotropically with geometrical restraints. Fig. 2 shows the repeat unit of the coordination polymer with symmetry-generated atoms; it contains three independent copper atoms and one independent ligand 5.
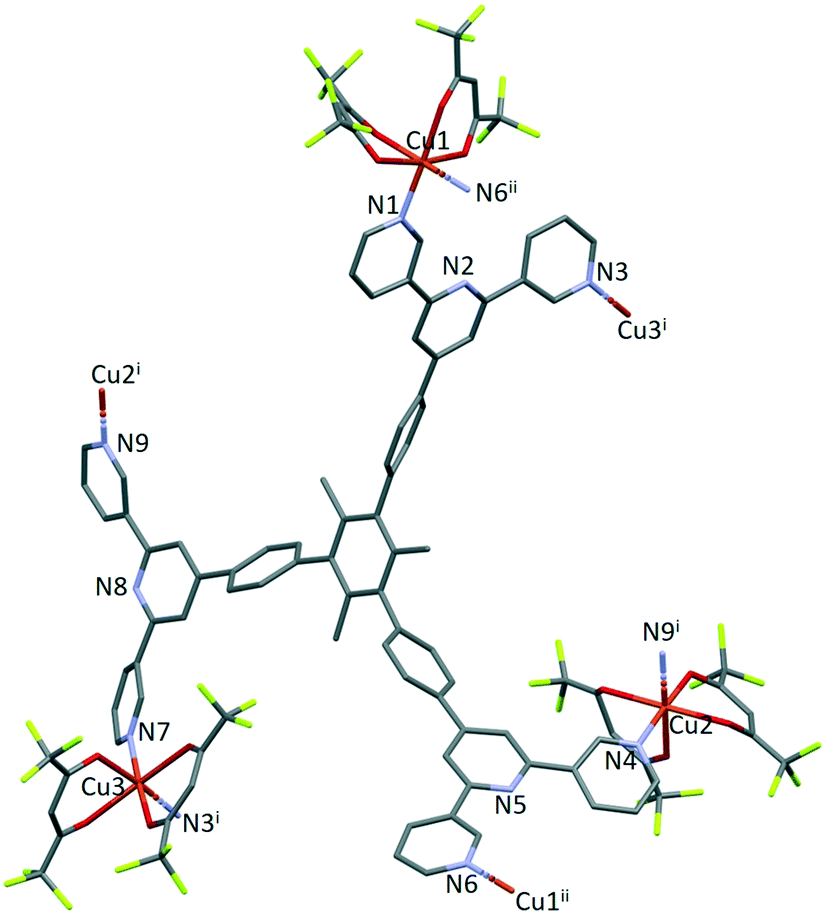 | ||
| Fig. 2 The repeat unit in [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 with symmetry-generated atoms. For clarity, H atoms and solvent are omitted, and only major occupancies are shown. Symmetry codes: i = 1 − x, 1−y, 1 − z; ii = 1 − x, −y, 1 − z. | ||
Each of atoms Cu1, Cu2 and Cu3 is octahedrally coordinated with cis-arrangements of coordinated [hfacac]− ligands. Each Cu atom binds to two pyridine donor atoms of two different ligands 5. The bond lengths and angles for the coordination spheres of Cu1, Cu2 and Cu3 are given in Table 1. A search of the Cambridge Structural Database59 (CSD, v. 2021.1.0 using Conquest v. 2021.1.0)60,68 for structures containing in {Cu(hfacac)2(N1)(N2)} units reveals that in most structures the N–Cu–N angles are close to 180° (Fig. 3.), i.e. a trans-arrangement of N atoms. Most of the cis configurations are associated with chelating ligands. Hence, the cis-arrangement found in [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 is considered to be unusual.
| Metal atom | Cu–N/Å | N–Cu–N/° | Cu–O/Å | N–Cu–O/° |
|---|---|---|---|---|
| Cu1 | 2.013(5) | 91.1(2) | 2.320(4) | 92.6(2), 90.6(2), 177.7(2), 94.69(19), 90.91(18), 175.1(2), 90.6(2), 93.6(2) |
| 1.993(6) | ||||
| 1.996(4) | ||||
| 1.995(6) | ||||
| 2.238(4) | ||||
| Cu2 | 2.032(5) | 96.6(2) | 1.981(5) | 86.0(2), 92.2(2), 174.0(2), 94.2(2), 176.7(2), 94.4(2), 89.3(2), 90.4(2) |
| 2.222(6) | ||||
| 2.023(6) | 2.001(5) | |||
| 2.290(5) | ||||
| Cu3 | 2.075(9) | 93.5(3) | 1.983(9) | 89.4(4), 95.6(3), 91.1(3), 175.4, 175.1(3), 89.7(3), 94.7(2), 88.2(3) |
| 2.206(7) | ||||
| 2.005(7) | 2.231(5) | |||
| 2.017(8) |
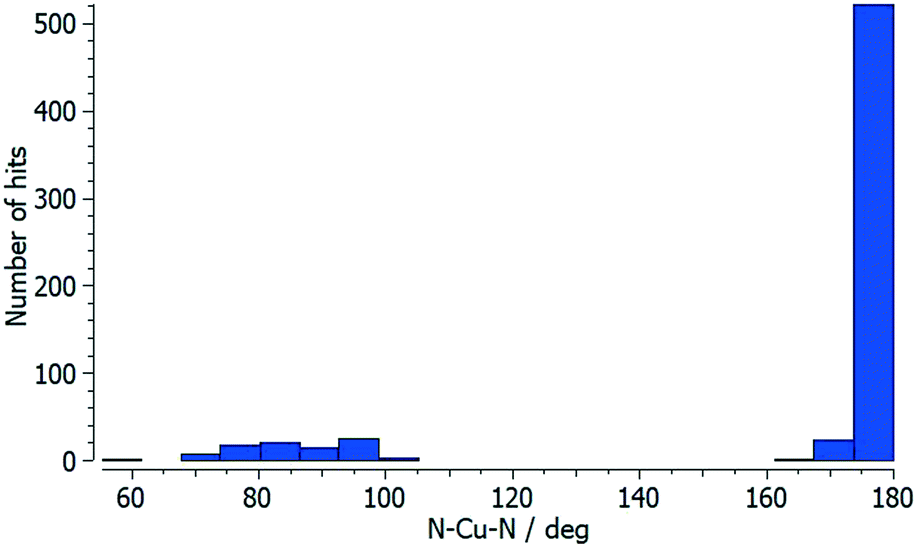 | ||
| Fig. 3 Distribution of the N–Cu–N coordination angle in {Cu(hfacac)2(N1)(N2)} units from a search of the Cambridge Structural Database (v. 2021.1.0) using ConQuest (v. 2021.1.0). | ||
In coordinated ligand 5 (Fig. 2), the three tpy domains are crystallographically independent, and coordination occurs only through the two outer nitrogen atoms. The tpy unit containing N1, N2 and N3 and N4, N5 and N6 display a coordination directionality close to conformation A (Scheme 1), with N1 and N6 pointing out and N4 and N5 oriented towards the centre core of 5. In contrast, the tpy with N7, N8 and N9 adopts conformation C (Scheme 1). Angles between the least squares planes through pairs of adjacent pyridine rings are in the range 7.6–30.7° (Table 2). The larger torsion angles of 60.0, 84.2 and 56.7° between the planes of the arene core and the three arene spacers (Table 2) minimize unfavourable Me⋯H repulsions. Similar twists occur between the arene spacers and the central pyridine of the tpy substituents, although to a lesser extent (35.6, 30.5 and 53.4°), reducing the H⋯H inter-ring repulsions.
| Tpy unit | Angle between planes of connected pyridine rings/° | Angle between the central pyridine ring and arene spacer/° | Angle between arene spacer and arene core/° |
|---|---|---|---|
| N1N2N3 | 7.6, 22.6 | 35.6 | 60.0 |
| N4N5N6 | 17.0, 19.1 | 30.5 | 84.2 |
| N7N8N9 | 30.7, 24.9 | 53.4 | 56.7 |
Ligand 5 presents a 6-connecting building block (Fig. 2) and, combined with the cis-arrangement of the pyridine donors in the Cu(II) coordination sphere, this leads, rather unexpectedly, to a 1D-coordination polymer. There are two building blocks which alternate along the chain. The first is the centrosymmetric metallomacrocyclic unit in Fig. 4a in which Cu1 and Cu1ii bridge pairs of 3,2′:6′,3′′-tpy domains bound through atoms N1 and N6ii, and N6 and N1ii (symmetry code ii = 1 − x, −y, 1 − z).
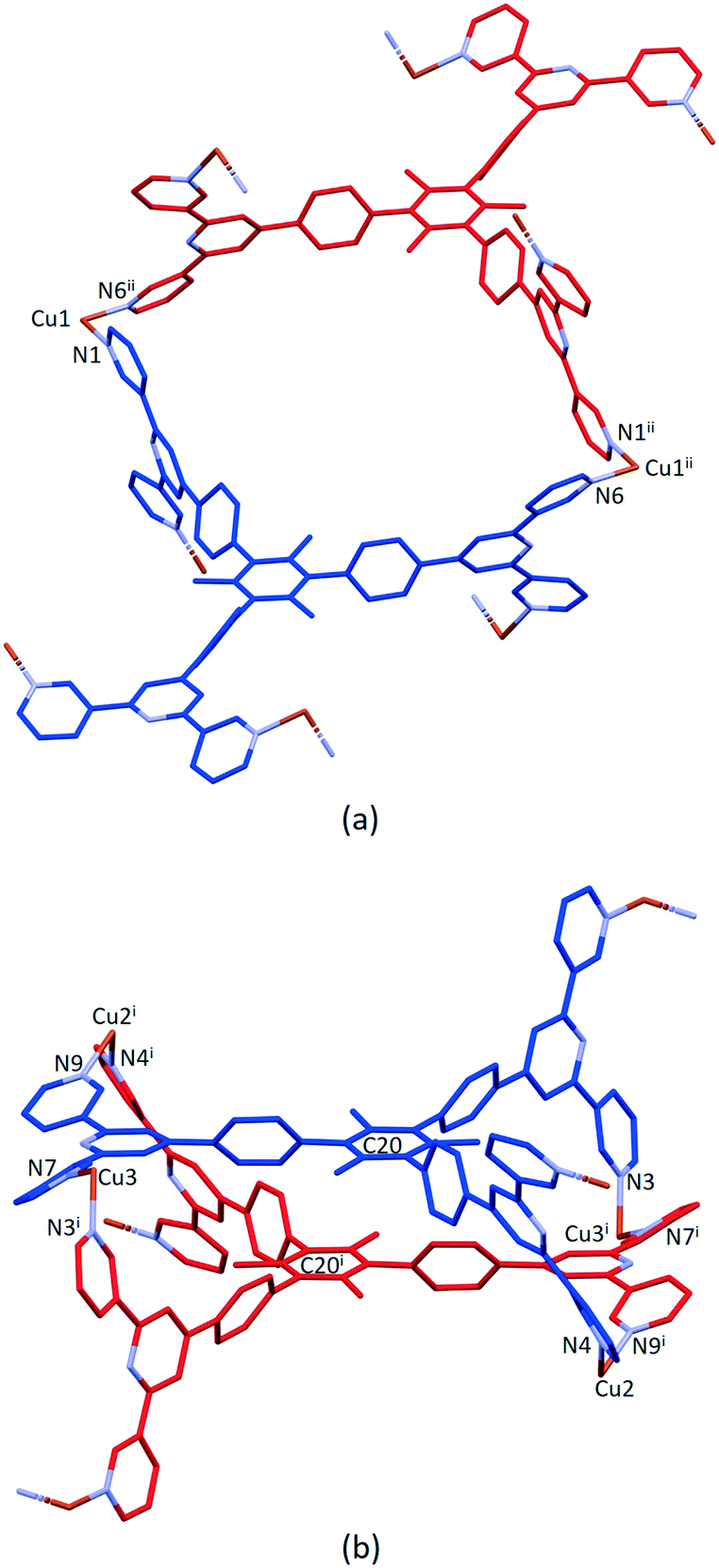 | ||
| Fig. 4 (a) Metallomacrocyclic unit consisting of 5 (blue), Cu1 and the symmetrical equivalents 5ii (red) and Cu1ii. (b) The second motif in the 1D-polymer with symmetry generated copper centres and ligand 5i (red). For clarity, H atoms, coordinated [hfacac]− ligands and solvent are omitted, and only major occupancies are shown. Symmetry codes: i = 1 − x, 1 − y, 1 − z; ii = 1 − x, −y, 1 − z. | ||
The second motif in the 1D-polymer consists of a head-to-tail pairs of two ligands 5 (Fig. 4b). The two tpy ligands are coordinated with Cu2, Cu2i, Cu3 and Cu3i across an inversion centre (symmetry code i = 1 − x, 1 − y, 1 − z). All inwardly oriented nitrogen atoms of 5, which come from all three tpy domains, are involved in the building block. The tpy unit containing N7, N8 and N9 is connected to both the tpy unit with N1i, N2i and N3i and that with N4i, N5i and N6i with atoms Cu3 and Cu2i acting as linkers (Fig. 4b, symmetry code i = 1 − x, 1 − y, 1 − z). Interestingly, there are no π-stacking interactions within the motif, even though the arene cores are coplanar. Centroid⋯centroid separations between for pairs of rings containing C20/C20i is 5.00 Å (Fig. S45†), and it is clear that the steric hindrance caused by the methyl substituents prevents π-interactions. The combination of the two structural motifs in the 1D-polymer (Fig. 5a) leads to a series of connected loops with ligand 5 connecting pairs of Cu atoms (Fig. 5). Atoms Cu1 and Cu1ii lie within a single loop in Fig. 5b, while Cu2, Cu3, Cu2i and Cu3i lie within the double loops in Fig. 5b.
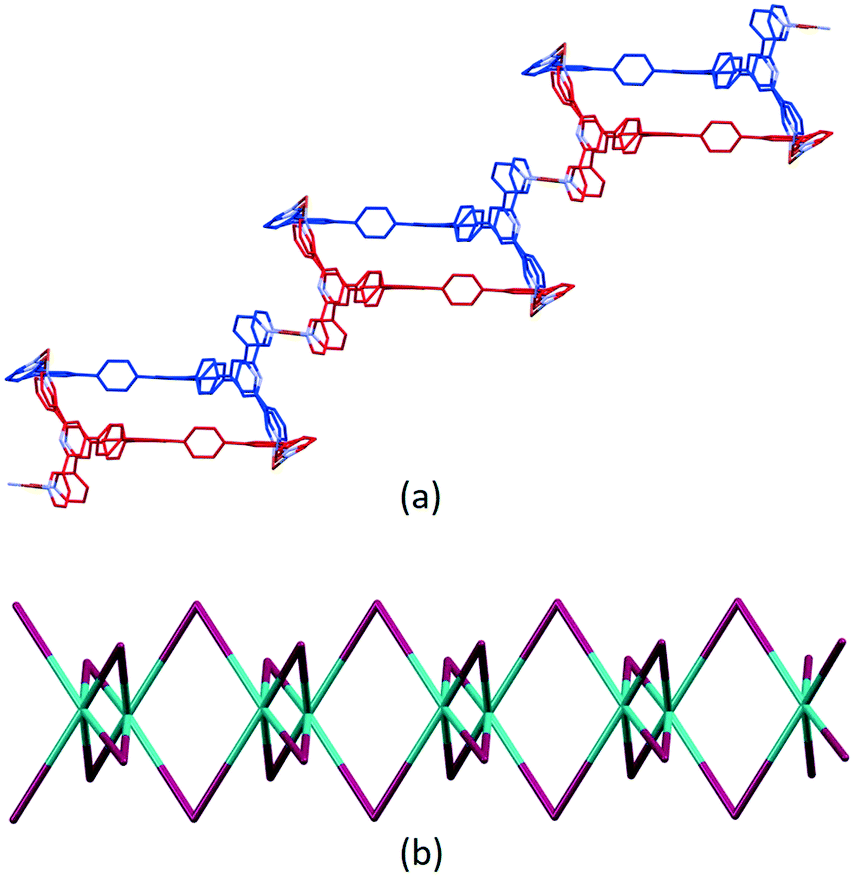 | ||
| Fig. 5 (a) Part of the 1D-coordination polymer. Tristpy building blocks are alternating coloured in blue and red. For clarity, H atoms, coordinated [hfacac]− ligands and solvent are omitted, and only major occupancies are shown. (b) Loops forming the 1D-chain. Magenta: Cu; green: ligand centroids. | ||
The coordination polymer chains follow the crystallographic b-axis and packing interactions between the chains is dominated by short C–H⋯F–C and C–F⋯F–C contacts. However, the disordering of some CF3 groups means that detailed discussion is not meaningful. The nesting of the CP chains shown in Fig. 6 illustrates a zigzag arrangement with the chains slightly offset (Fig. S46†). Channels in the structure are occupied by highly disordered toluene and CHCl3 molecules.
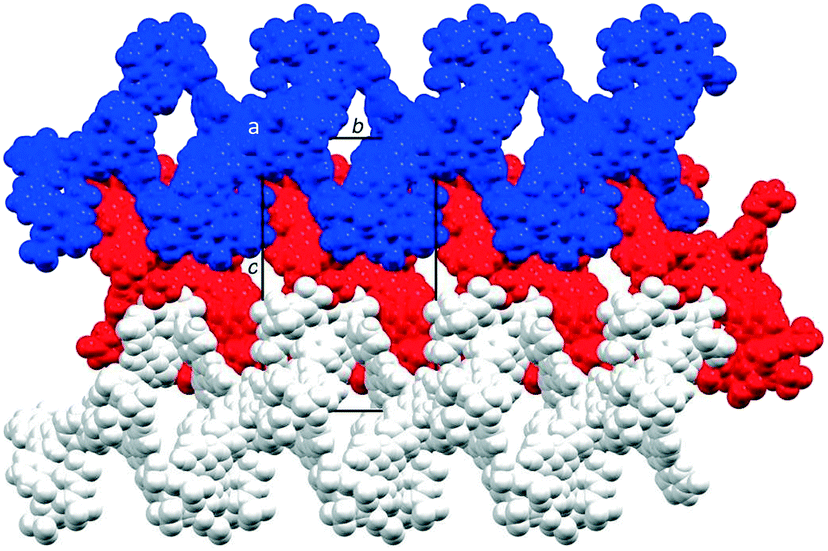 | ||
| Fig. 6 Packing of three 1D ribbons which follow the b-axis, shown in blue, red and white, and viewed down the crystallographic a-axis. For clarity, solvent is omitted and only major occupancies are shown. | ||
PXRD analysis
After four single crystals of [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 had been selected for single-crystal X-ray structure determination, the bulk material was analysed by powder X-ray diffraction (PXRD). The crystals lose solvent rapidly on exposure to air and were, therefore, analysed wet and without washing them. Despite careful handling of the crystals, loss of solvent could not be avoided, and as a consequence, the PXRD spectrum was of poor quality. Confirmation that the single crystal selected was representative of the main phase of the bulk sample came from a comparison of the experimental PXRD pattern (shown in red in Fig. 7) with the pattern predicted from the single crystal structure (black traces in Fig. 7). Minor phases observed in the PXRD spectrum of the bulk material could not be assigned and most likely appear as a consequence of solvent loss during sample preparation and measurement. The differences in intensities (blue traces in Fig. 7) can be justified in terms of differences in the preferred orientations of the crystallites in the bulk powder samples.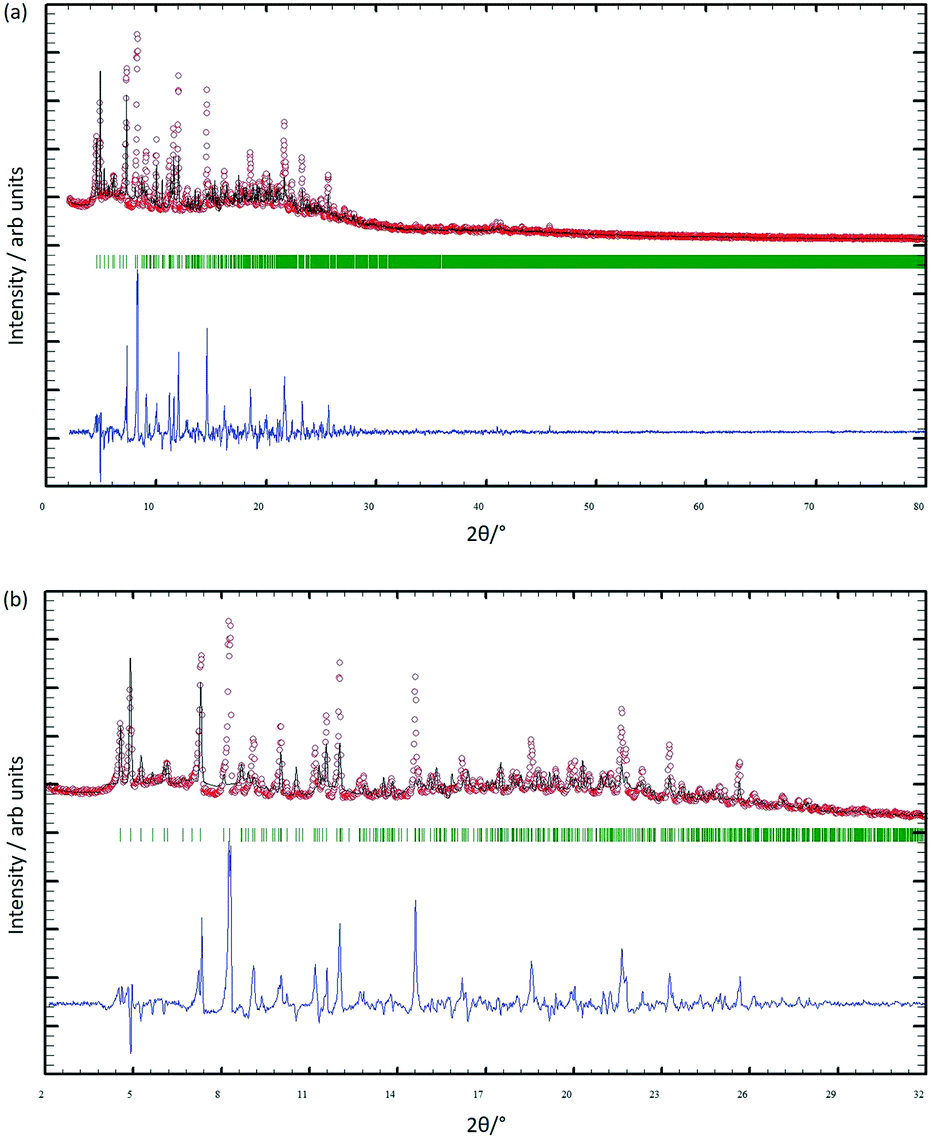 | ||
| Fig. 7 (a) X-ray diffraction (CuKα1 radiation) pattern (red circles) of the bulk crystalline material of [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3, fitting the predicted pattern from the single crystal structure. The black lines are the best fit from Rietveld refinements, and green lines display the Bragg peak positions. The blue plot gives the difference between calculated and experimental points (see text). (b) Expansion in the 2–32° range. | ||
Comments on 1D-coordination polymers containing terpyridine metal-binding domains
The structure of the 1D-chain in [Cu3(hfacac)6(5)]n consists of alternating single and double loops (Fig. 5b) with this motif being a consequence of the 6-connecting node presented by ligand 5. The use of the term ‘node’ in a 1D-chain has been described as ‘subjective’,1 since the chain can be described unambiguously in terms of a combination of metal linkers and ligand linkers. Topologically, the 1D-chains in Scheme 5 are all equivalent, but they are structurally distinct in terms of the organization of the chemical building blocks, with, for example, Scheme 5d and e having enclosed space within the 1D-chain.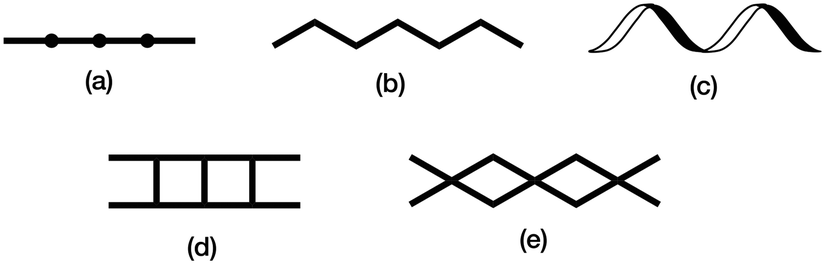 | ||
| Scheme 5 Examples of structure types found in 1D-coordination polymers: (a) linear chain, (b) zigzag chain, (c) helical chain, (d) ladder, (e) series of loops with 4-connecting nodes. | ||
The versatility of terpyridine as a building block for 1D-CPs is worthy of comment. With a 2,2′:6′,2′′-tpy metal-binding domain, {M(tpy)2}n+ units can be functionalized in the 4′-position to provide a linear building block. An example is [Ru(pytpy)2]2+ (pytpy = 4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine), the [PF6]− salt of which reacts with AgNO3 in aqueous MeCN solution to give [{Ru(pytpy)2Ag(NCMe)(NO3)}n][NO3]2n·nH2O·nMeCN containing the 1D-chain shown in Fig. 8a.69 The linearity (Scheme 5a and Fig. 8a) of the chain is a consequence of the vectorial properties of the [Ru(pytpy)2]2+ unit coupled with the N–Ag–N angle of 150.4(1)°. In contrast, ongoing from pytpy to 4-pyridinecarbaldehyde 4′-(2,2′:6′,2′′-terpyridyl)hydrazone (6, Fig. 8b), a significant degree of curvature is introduced into the [Ru(6)2]2+ unit compared to [Ru(pytpy)2]2+. This renders the [Ru(6)2]2+ domain amenable to the formation of 1D-chains comprising a string of loops as seen in [{Fe(NCS)2(Ru(6)2)2}n]4n+ (Fig. 8b).70
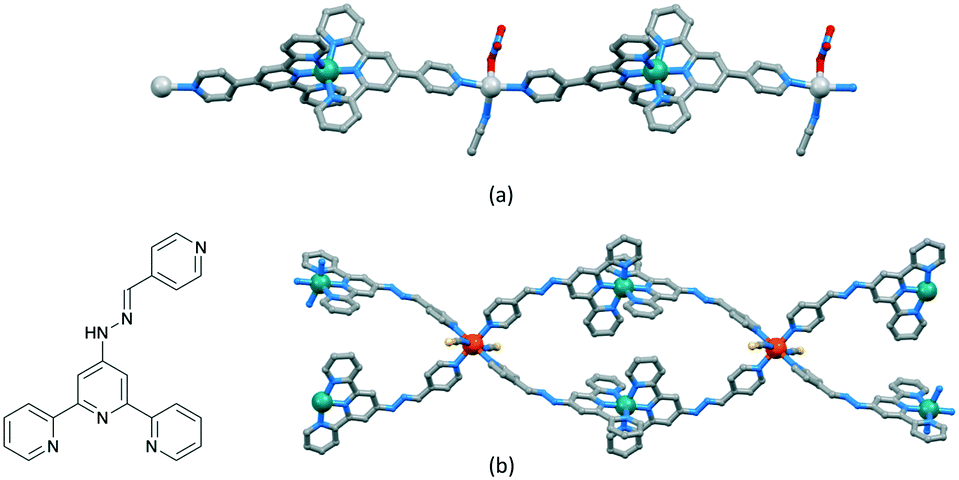 | ||
| Fig. 8 (a) Part of the linear 1D-chain in [{Ru(pytpy)2Ag(NCMe)(NO3)}n][NO3]2n·nH2O·nMeCN (CSD refcode WICSIL). (b) Structure of ligand 6 and part of the chain comprising interconnected loops in [{Fe(NCS)2(Ru(6)2)2}n][Fe2(NCS)6(OEt)2(EtOH)2]n[NCS]2n·4nEtOH·nH2O (CSD refcode TOPLIU). | ||
Zigzag chains (Scheme 5b) directed by metal paddle-wheel domains such as {Zn2(μ-O2CMe)4} and {Cu2(μ-O2CMe)4} are well represented for a wide range of 4′-substituted 4,2′:6′,4′′-tpy ligands (e.g.Fig. 9a71) and the conformational flexibility of the 3,2′:6′,3′′-tpy domain leads to structural variants on the zigzag backbone.72 Multiply-stranded chains18 which retain the zigzag profile arise when multinuclear metal units can bind to more than two ligands, e.g. [Cd2(OAc)4(4′-(4-PhC6H4)-4,2′:6′,4′′-tpy)]n in which each {Cd2(OAc)4} (a non-paddle wheel motif) binds to four N donors (Fig. 9b73), and [Cu4(μ3-OH)2(OAc)6(4′-(4-MeOC6H4)-3,2′:6′,3′′-tpy)2]n (Fig. 9c74). In contrast, the rotational freedom of the ferrocenyl core in the tetratopic ligand 1,1′-bis(4,2′:6′,4′′-terpyridin-4′-yl)ferrocene allows the assembly of a double-stranded chain in which the ligand ‘folds over’ to produce the two strands (Fig. 9d75). Helical chains are also common. Many are based upon tetrahedral {Zn2X2N2} domains, and are exemplified by [ZnCl2(4,2′:6′,4′′-tpy)]n which was the first CP reported which was directed by a 4,2′:6′,4′′-tpy ligand.76 Ladders (Scheme 5d) in which the uprights and rungs of the ladder are both defined by ligand linkers are less common, and one example is [Cd2(4′-(4-MeOC6H4)-4,2′:6′,4′′-tpy)3(NO3)4]n (Fig. 9e77).
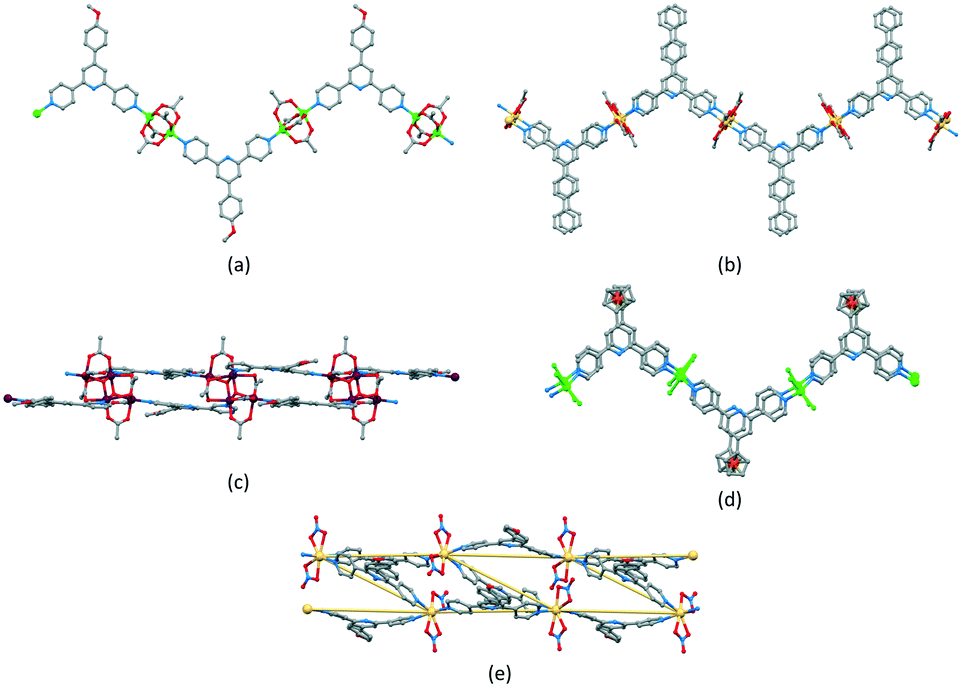 | ||
| Fig. 9 (a) Part of the zigzag chain in [Zn2(OAc)4(4′-(4-MeOC6H4)-4,2′:6′,4′′-tpy)]n (CSD refcode SOXSEF). Parts of the double stranded zigzag chains in (b) [Cd2(OAc)4(4′-(4-PhC6H4)-4,2′:6′,4′′-tpy)]n (refcode RIGJEY), (c) [Cu4(μ3-OH)2(OAc)6(4′-(4-MeOC6H4)-3,2′:6′,3′′-tpy)2]n (refcode KUCHIC), and (d) [Zn2Cl2(1,1′-bis(4,2′:6′,4′′-terpyridin-4′-yl)ferrocene)]n (refcode UMUYUY). (e) Part of the ladder assembly in [Cd2(4′-(4-MeOC6H4)-4,2′:6′,4′′-tpy)3(NO3)4]n (refcode BUDZIL); the Cd⋯Cd vectors which define the ladder are highlighted. | ||
The short survey above is not comprehensive, but illustrates the diversity of 1D-CPs that can be assembled using ligands with 2,2′:6′,2′′-, 3,2′:6′,3′′- and 4,2′:6′,4′′-tpy metal-binding domains. To the best of our knowledge, the looped-structure of the chain in the solvates of [Cu3(hfacac)6(5)]n presented here is unique among terpyridine-based complexes.
Conclusions
We have prepared and characterised five new 1–5 tris(terpyridine) ligands, two with 4,2′:6′,4′′-tpy units and three with 3,2′:6′,3′′-tpy metal-binding domains. The crystal structure of 1 was determined. It possesses a propeller-shaped structure and efficient face-to-face π-stacking interactions predominate in the packing. Reaction of 5 with [Cu(hfacac)2]·H2O under ambient conditions of crystal growth by layering using a combination of CHCl3 and toluene unexpectedly led to the assembly of the 1D-coordination polymer [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3. This possesses an unusual 1D-chain consisting of a series of alternating single and double loops in which the {Cu(hfacac)2(N1)(N2)} units display a cis-arrangement of N atoms. In coordinated ligand 5, two of the crystallographically independent 3,2′:6′,3′′-tpy domains adopt conformation A (Scheme 1), while the third exhibits conformation C. PXRD confirms that the single crystal structure is representative of the bulk material. A preliminary structural analysis has also shown that a switch from toluene to 1,2-dichlorobenzene leads to a reproducible assembly of the 1D-looped-chain. We will now further explore the effects of using different spacers in the hexatopic motifs of tris(4,2′:6′,4′′-tpy) and tris(3,2′:6′,3′′-tpy) building blocks.Experimental
Materials and instrumentation
1,3,5-Tribromobenzene and benzene-1,3,5-tricarbaldehyde were purchased from Acros Organics. 3-Acetylpyridine, 4-acetylpyridine and [Pd(PPh3)4] were bought from Sigma Aldrich and [Cu(hfacac)2]·H2O was purchased from abcr GmbH. Bis(pinacolato)diboron and Pd(dppf)Cl2 were purchased from Fluorochem. All chemicals were used as received.4′-(4-Bromophenyl)-3,2′:6′,3′′-terpyridine and 4′-(4-bromophenyl)-4,2′:6′,4′′-terpyridine were prepared according to the one-pot methods of Hanan23 as reported by Zhang78 but using KOH in place of NaOH. 2,4,6-Tribromomesitylene was synthesized as previously described in literature.79
Analytical thin-layer chromatography was conducted with pre-coated silica gel 60 F254 or with aluminium oxide 60 F254, neutral, aluminium sheets and visualized using ultraviolet light (254 nm). Neutral aluminium oxide 60 F254 (20 × 20 cm; 1.5 mm) sheets were used for the preparative layer chromatography (PLC). Flash column chromatography was performed on a Biotage Selekt system with self-packed silica gel columns (SiliaFlash® P60, 40–63 μm, 230400 mesh from SiliCycle Inc.) or using Biotage Sfär Silica HC D columns (50 g, 20 μm).
1H and 13C{1H} NMR spectra were recorded on a Bruker Avance III-500 spectrometer at 298 K. The 1H and 13C NMR chemical shifts were referenced with respect to the residual solvent peak (δ TMS = 0). MALDI-TOF mass spectra were recorded on a Shimadzu MALDI 8020 without matrix, in some cases using α-cyano-4-hydroxycinnamic acid as matrix. High-resolution electrospray (HR-ESI) mass spectra were measured on a Bruker maXis 4G QTOF instrument. PerkinElmer UATR Two and Cary-5000 instruments were used to record FT-infrared (IR) and UV-vis absorption spectra, respectively. Melting temperatures were determined using a Stuart melting point SMP 30 device. Reactions and procedures under microwave conditions were carried out in a Biotage Initiator 8 reactor. Microwave vials were from Biotage and were selected depending on the required solvent volume. For centrifugation an Eppendorf Centrifuge 5415 R was used with 2, 10 and 50 mL samples.
Synthesis
600). MALDI-TOF-MS m/z 772.03 [M + H]+ (calc. 772.29). Found C 75.92, H 4.85, N 16.56; required for C51H33N9·1.5DMF: C 75.62, H 4.97, N 16.68.
700). MALDI-TOF-MS m/z 772.33 [M + H]+ (calc. 772.29). Found C 79.02, H 4.31, N 16.58; required for C51H33N9: C 79.36, H 4.31, N 16.33.
:1) and then precipitated at −20 °C to give a colourless solid. The product was isolated by filtration, washed with EtOH and hexane and then dried in vacuo. The filtrate was dried and the yellow residue purified by column chromatography (50 g Biotage Sfär Silica HC D column, Biotage Select, eluent: 30% acetone–70% cyclohexane) to yield a second crop of product (661 mg, 1.52 mmol, 53.7%). M.p. = 229.8–231.5 °C. 1H NMR (500 MHz, CDCl3): δ/ppm 8.80 (d, J = 6.1 Hz, 4H, HA2), 8.09 (d, J = 6.2 Hz, 4H, HA3), 8.06 (s, 2H, HB3), 8.00 (d, J = 8.1 Hz, 2H, HC3), 7.75 (d, J = 8.2 Hz, 2H, HC2), 1.39 (s, 12 H, Hb). 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 155.5 (CB2), 151.2 (CB4), 150.7 (CA2), 146.2 (CA4), 140.6 (CC1), 135.9 (CC3), 130.5 (CC4), 126.6 (CC2), 121.4 (CA3), 119.2 (CB3), 84.3 (Ca), 25.1 (Cb). UV-vis (CHCl3, 2.6 × 10−5 mol dm−3) λ/nm 264 (ε/dm3 mol−1 cm−1 45466). MALDI-TOF-MS m/z 436.19 [M + H]+ (calc. 436.22). Found C 73.68, H 6.08, N 9.83; required for C27H26N3O2B: C 74.49, H 6.02, N 9.65.
712). MALDI-TOF-MS m/z 436.16 [M + H]+ (calc. 436.22). Found C 74.08, H 5.88, N 9.52; required for C27H26N3O2B: C 74.49, H 6.02, N 9.65.
183). MALDI-TOF-MS m/z 1000.30 [M + H]+ (calc. 1000.39). HR-ESI MS m/z 1000.3860 [M + H]+ (calc. 1000.3871).
675). MALDI-TOF-MS m/z 1000.41 [M + H]+ (calc. 1000.39). HR-ESI MS m/z 1000.3859 [M + H]+ (calc. 1000.3871).
400). MALDI-TOF-MS m/z 1042.48 [M + H]+ (calc. 1042.43). HR-ESI MS m/z 1042.4313 [M + H]+ (calc. 1042.4334), 521.7207 [M + 2H]2+ (calc. 521.7206).
In an another attempt, compound 5 (5.2 mg, 0.005 mmol) was dissolved in CHCl3 (6 mL), and then a solution of [Cu(hfacac)2]·H2O (15.3 mg, 0.031 mmol) in toluene (6 mL) was added. A formation of a pale green precipitate was immediately observed. After filtration, the solution was left to evaporate in the air in a test tube with a septum pierced with a syringe-needle. Light green block-like crystals were obtained after 3 months, and cell checks revealed that these were [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 (cell parameters, a = 20.784(6) Å, b = 21.270(3) Å, c = 34.158(9) Å, β = 103.57(2)°).
An additional reaction was carried out to prepare a sample for E.A. and IR spectroscopy. Compound 5 (10.4 mg, 0.010 mmol) was dissolved in CHCl3 (6 mL), and then a solution of [Cu(hfacac)2]·H2O (15.3 mg, 0.031 mmol) in toluene (6 mL) was added. A formation of a pale green precipitate was immediately observed. The solid that formed was collected by centrifugation, washed with CHCl3 and toluene, and then dried in vacuo. The product was isolated as a pale green powder. Yield for [Cu3(hfacac)6(5)]n·0.5nC7H8 (25.0 mg, 0.010 mmol, 99.2%). Found C 50.77, H 2.64, N 5.34; required for C105.5H61Cu3F36N9O12: C 50.26, H 2.44, N 5.00. PXRD showed that the resultant product was amorphous.
Crystallography
Single crystal data for 1·1.75DMF were collected either on a STOE StadiVari diffractometer equipped with a Metaljet D2 source (GaKα radiation) and a Pilatus300K detector. For the CPs, a X06DA-PXIII beamline at the Swiss Light Source (Paul Scherrer Institute, 5232 Villigen, Switzerland) with synchrotron radiation (0.72083 Å) and a PILATUS 2 M-F detector were used. For the former, structures were solved using ShelXT v. 2018/2 (ref. 80) and Olex2,81 and the model was refined with ShelXL v. 2018/3.82 For the latter, data reduction, solution, and refinement used the programs XDS,83 Olex2,81 and ShelXL 2018/3.82 All H atoms were included at geometrically calculated positions and refined using a riding model with Uiso = 1.2 of the parent atom. Structure analysis used CSD Mercury 2020.2.0.68 In [Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3 some CF3 groups of the polymer and solvent molecules were disordered and were refined isotropically with geometrical restraints. A certain amount of the solvent was removed using the solvent mask procedure, and this was added to the formulae and appropriated numbers. In the structural discussions, only the major (or one of the equal) occupancy sites are considered in each disordered entity. A mask was also used to treat the solvent region in 1; 1.75 DMF molecules were found and added to the formulae and numbers.PXRD data were collected at room temperature in transmission mode using a Stoe Stadi P diffractometer, equipped with CuKα1 radiation (Ge(111) monochromator) and a DECTRIS MYTHEN 1K detector. Whole-pattern decomposition (profile matching) analysis of the diffraction patterns was done using the package FULLPROF SUITE (v. September 2020)84,85 using a previously determined instrument resolution function based on a NIST640d standard. The structural models were derived from the single crystal X-ray diffraction data. Refined parameters in Rietveld were scale factor, zero shift, lattice parameters, Cu and halogen atomic positions, background points, and peaks shapes as a Thompson Cox Hastings pseudo-Voigt function. Preferred orientations as a March–Dollase multi-axial phenomenological model were incorporated into the analysis.
1·1.75DMF C51H33N9·C5.25H12.25N1.75O1.75, Mr = 899.78, yellow block, orthorhombic, Pbca, a = 20.3991(5) Å, b = 15.4857(3) Å, c = 30.0422(5) Å, V = 9490.2(3) Å3, T = 150 K, Z = 8, μ(GaKα) = 0.405. Total 59250 reflections, 9577 unique (Rint = 0.0486). Refinement of 5670 reflections (541 parameters) with I > 2σ(I) converged at final R1 = 0.0515 (R1 all data = 0.0746), wR2 = 0.1520 (wR2 all data = 0.1612), F(000) = 3776, gof = 1.177. CCDC 2096132.
[Cu3(hfacac)6(5)]n·2.8nC7H8·0.4nCHCl3: C122H80.1Cl1.2Cu3F36N9O12, Mr = 2781.21, green block, monoclinic, P21/n, a = 19.889(4) Å, b = 21.178(4) Å, c = 34.375(7) Å, β = 104.32(3)°, V = 14029(5) Å3, T = 100 K, Z = 4, μ(synchrotron) = 0.595. Total 531703 reflections, 29721 unique (Rint = 0.0862). Refinement of 20415 reflections (1623 parameters) with I > 2σ(I) converged at final R1 = 0.1912 (R1 all data = 0.2092), wR2 = 0.4998 (wR2 all data = 0.5408), F(000) = 5610, gof = 1.177. CCDC 2096133.
Author contributions
Investigation and methodology, G. M.; crystallography, A. P.; powder diffraction, G. M.; manuscript writing, G. M., C. E. H.; manuscript editing, C. E. H.; E. C. C.; supervision and project administration, C. E. H.; E. C. C.; funding acquisition, C. E. H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Swiss National Science Foundation (grant number 200020_182559) and the University of Basel for financial support. The synchrotron data collection was carried out at the Paul Scherrer Institute (PSI) by Florian Leisinger (University of Basel).References
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, RSC Publishing, Cambridge, 2009 Search PubMed.
- J. E. Berger, Kerrn aller Fridrichs=Städtschen Begebenheiten, Manuskript, Berlin, ca.1730 (Berlin, Staatsbibliothek zu Berlin—Preußischer Kulturbesitz, Handschriftenabteilung, Ms. Boruss. quart. 124).
- E. C. Constable, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Wiley, Chichester, 2012, vol. 6, p. 3073 Search PubMed.
- E. Loukopoulos and G. E. Kostakis, J. Coord. Chem., 2018, 71, 371–410 CrossRef CAS.
- J. Chen, K. Shen and Y. Li, ChemSusChem, 2017, 10, 3165–3187 CrossRef CAS PubMed.
- S. Mukherjee, A. Kumar and M. J. Zaworotko, in Metal-Organic Frameworks (MOFs) for Environmental Applications, ed. S. K. Ghosh, Elsevier, Amsterdam, 2019, pp. 5–61 Search PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. Duan, W. Jin and S. Kitagawa, Coord. Chem. Rev., 2017, 332, 48–74 CrossRef CAS.
- Y. Peng and W. Yang, Adv. Mater. Interfaces, 2020, 7, 1901514 CrossRef CAS.
- R.-B. Lin, S. Xiang, H. Xing, W. Zhou and B. Chen, Coord. Chem. Rev., 2019, 378, 87–103 CrossRef CAS.
- Y. Zhang, X. Cheng, X. Jiang, J. J. Urban, C. H. Lau, S. Liu and L. Shao, Mater. Today, 2020, 36, 40–47 CrossRef CAS.
- Y. Ye, L. Gong, S. Xiang, Z. Zhang and B. Chen, Adv. Mater., 2020, 32, 1907090 CrossRef CAS PubMed.
- J. Deng, F. Wu, P. Yu and L. Mao, Appl. Mater. Today, 2018, 11, 338–351 CrossRef.
- J.-Q. Liu, Z.-D. Luo, Y. Pan, A. K. Singh, M. Trivedi and A. Kumar, Coord. Chem. Rev., 2020, 406, 213145 CrossRef CAS.
- S. M. Elahi, M. Raizada, P. K. Sahu and S. Konar, Chem. – Eur. J., 2021, 27, 5858–5870 CrossRef CAS PubMed.
- E. C. Constable and C. E. Housecroft, Coord. Chem. Rev., 2017, 350, 84–104 CrossRef CAS.
- E. C. Constable, Coord. Chem. Rev., 2008, 252, 842–855 CrossRef CAS.
- C. E. Housecroft, Dalton Trans., 2014, 43, 6594–6604 RSC.
- C. E. Housecroft, CrystEngComm, 2015, 17, 7461–7468 RSC.
- C. E. Housecroft and E. C. Constable, Chimia, 2019, 73, 462–467 CrossRef CAS.
- C. E. Housecroft and E. C. Constable, Chem. Commun., 2020, 56, 10786–10794 RSC.
- C. E. Housecroft and E. C. Constable, Molecules, 2021, 26, 3110 CrossRef CAS PubMed.
- J. Wang and G. S. Hanan, Synlett, 2005, 8, 1251–1254 Search PubMed.
- F. Kröhnke, Synthesis, 1976, 1, 1–24 CrossRef.
- B. Liu, L. Hou, W.-P. Wu, A.-N. Dou and Y.-Y. Wang, Dalton Trans., 2015, 44, 4423–4427 RSC.
- Y. Wu, J. Wu, Z. Luo, J. Wang, Y. Li, Y. Han and J. Liu, RSC Adv., 2017, 7, 10415–10423 RSC.
- F. Yuan, C.-M. Yuan, H.-M. Hu, T.-T. Wang and C.-S. Zhou, J. Solid State Chem., 2018, 258, 588–601 CrossRef CAS.
- C. E. Housecroft and E. C. Constable, J. Inorg. Organomet. Polym. Mater., 2018, 28, 414–427 CrossRef CAS.
- J. Yoshida, S.-I. Nishikiori and H. Yuge, J. Coord. Chem., 2013, 66, 2191–2200 CrossRef CAS.
- Y. M. Klein, E. C. Constable, C. E. Housecroft and A. Prescimone, CrystEngComm, 2015, 17, 2070–2073 RSC.
- G. Manfroni, A. Prescimone, S. R. Batten, Y. M. Klein, D. J. Gawryluk, E. C. Constable and C. E. Housecroft, Crystals, 2019, 9, 529 CrossRef CAS.
- E. C. Constable and A. M. W. C. Thompson, J. Chem. Soc., Chem. Commun., 1992, 617–619 RSC.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- A. Wild, A. Winter, F. Schlütter and U. S. Schubert, Chem. Soc. Rev., 2011, 40, 1459–1511 RSC.
- T. Wu, J. Yuan, B. Song, Y.-S. Chen, M. Chen, X. Xue, Q. Liu, J. Wang, Y.-T. Chan and P. Wang, Chem. Commun., 2017, 53, 6732–6735 RSC.
- X. Lu, X. Li, Y. Cao, A. Schultz, J.-L. Wang, C. N. Moorefield, C. Wesdemiotis, S. Z. D. Cheng and G. R. Newkome, Angew. Chem., Int. Ed., 2013, 52, 7728–7731 CrossRef CAS PubMed.
- J.-L. Wang, X. Li, C. D. Shreiner, X. Lu, C. N. Moorefield, S. R. Tummalapalli, D. A. Medvetz, M. J. Panzner, F. R. Fronczek, C. Wesdemiotis and G. R. Newkome, New J. Chem., 2012, 36, 484–491 RSC.
- T.-Z. Xie, S.-Y. Liao, K. Guo, X. Lu, X. Dong, M. Huang, C. N. Moorefield, S. Z. D. Cheng, X. Liu, C. Wesdemiotis and G. R. Newkome, J. Am. Chem. Soc., 2014, 136, 8165–8168 CrossRef CAS PubMed.
- M. Chen, D. Liu, J. Huang, Y. Li, M. Wang, K. Li, J. Wang, Z. Jiang, X. Li and P. Wang, Inorg. Chem., 2019, 58, 11146–11154 CrossRef CAS PubMed.
- G. Wang, M. Chen, J. Wang, Z. Jiang, D. Liu, D. Lou, H. Zhao, K. Li, S. Li, T. Wu, Z. Jiang, X. Sun and P. Wang, J. Am. Chem. Soc., 2020, 142, 7690–7698 CrossRef CAS PubMed.
- S. Chakraborty, K. J. Endres, R. Bera, L. Wojtas, C. N. Moorefield, M. J. Saunders, N. Das, C. Wesdemiotis and G. R. Newkome, Dalton Trans., 2018, 47, 14189–14194 RSC.
- E. C. Constable, C. E. Housecroft and I. Poleschak, Inorg. Chem. Commun., 1999, 2, 565–568 CrossRef CAS.
- E. C. Constable, O. Eich and C. E. Housecroft, Inorg. Chem. Commun., 1999, 2, 431–433 CrossRef CAS.
- E. C. Constable and P. Harverson, Polyhedron, 1999, 18, 3093–3106 CrossRef CAS.
- E. C. Constable, O. Eich, C. E. Housecroft and D. C. Rees, Inorg. Chim. Acta, 2000, 300–302, 158–168 CrossRef CAS.
- E. C. Constable, O. Eich, D. Fenske, C. E. Housecroft and L. A. Johnston, Chem. – Eur. J., 2000, 6, 4364–4370 CrossRef CAS.
- T. K. Sievers, A. Vergin, H. Möhwald and D. G. Kurth, Langmuir, 2007, 23, 12179–12184 CrossRef CAS PubMed.
- T. Bauer, A. D. Schlüter and J. Sakamoto, Synlett, 2010, 6, 877–880 Search PubMed.
- H. Maeda, R. Sakamoto and H. Nishihara, Coord. Chem. Rev., 2017, 346, 139–149 CrossRef CAS.
- Y. Kuai, T. Yang, F. Yuan, Y. Dong, Q. Song, C. Zhang and W.-Y. Wong, Dyes Pigm., 2021, 194, 109623 CrossRef CAS.
- S. Roy and C. Chakraborty, ACS Appl. Mater. Interfaces, 2020, 12, 35181–35192 CrossRef CAS PubMed.
- C.-W. Hu, T. Sato, J. Zhang, S. Moriyama and M. Higuchi, ACS Appl. Mater. Interfaces, 2014, 6, 9118–9125 CrossRef CAS PubMed.
- R. K. Pandey, M. D. Hossain, T. Sato, U. Rana, S. Moriyama and M. Higuchi, RSC Adv., 2015, 5, 49224–49230 RSC.
- Y. Kuai, W. Li, Y. Dong, W.-Y. Wong, S. Yan, Y. Dai and C. Zhang, Dalton Trans., 2019, 48, 15121–15126 RSC.
- S. Roy and C. Chakraborty, Chem. Commun., 2021, 57, 7565–7568 RSC.
- Y. Liu, R. Sakamoto, C.-L. Ho, H. Nishihara and W.-Y. Wong, J. Mater. Chem. C, 2019, 7, 9159–9166 RSC.
- K. Takada, R. Sakamoto, S.-T. Yi, S. Katagiri, T. Kambe and H. Nishihara, J. Am. Chem. Soc., 2015, 137, 4681–4689 CrossRef CAS PubMed.
- T. Tsukamoto, K. Takada, R. Sakamoto, R. Matsuoka, R. Toyoda, H. Maeda, T. Yagi, M. Nishikawa, N. Shinjo, S. Amano, T. Iokawa, N. Ishibashi, T. Oi, K. Kanayama, R. Kinugawa, Y. Koda, T. Komura, S. Nakajima, R. Fukuyama, N. Fuse, M. Mizui, M. Miyasaki, Y. Yamashita, K. Yamada, W. Zhang, R. Han, W. Liu, T. Tsubomura and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 5359–5366 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B, 2016, 72, 171–179 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B, 2002, 58, 389–397 CrossRef PubMed.
- M. Cavazzini, S. Quici, C. Scalera, F. Puntoriero, G. La Ganga and S. Campagna, Inorg. Chem., 2009, 48, 8578–8592 CrossRef CAS PubMed.
- H. L. Nguyen, C. Gropp, Y. Ma, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 20335–20339 CrossRef CAS PubMed.
- J. N. Moorthy and P. Natarajan, Chem. – Eur. J., 2010, 16, 7796–7802 CrossRef CAS PubMed.
- H. Yamagishi, H. Sato, A. Hori, Y. Sato, R. Matsuda, K. Kato and T. Aida, Science, 2018, 361, 1242–1246 CrossRef CAS PubMed.
- T. Liu, B. Wang, R. He, H. Arman, K. S. Schanze, S. Xiang, D. Li and B. Chen, Can. J. Chem., 2020, 98, 352–357 CrossRef CAS.
- M. F. N. N. Carvalho, M. T. A. Fernanda, M. G. Adelino and J. L. P. Armando, J. Organomet. Chem., 2003, 679, 143–147 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- J. E. Beves, E. C. Constable, C. E. Housecroft, C. J. Kepert and D. J. Price, CrystEngComm, 2007, 9, 456–459 RSC.
- J. E. Beves, E. C. Constable, C. E. Housecroft, C. J. Kepert, M. Neuburger, D. J. Price, S. Schaffner and J. A. Zampese, Dalton Trans., 2008, 6742–6751 RSC.
- Y. M. Klein, E. C. Constable, C. E. Housecroft, J. A. Zampese and A. Crochet, CrystEngComm, 2014, 16, 9915–9929 RSC.
- D. Rocco, S. Novak, A. Prescimone, E. C. Constable and C. E. Housecroft, Chemistry, 2021, 3, 182–198 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, M. Neuburger, J. Schönle, S. Vujovic and J. A. Zampese, Polyhedron, 2013, 60, 120–129 CrossRef CAS.
- D. Rocco, G. Manfroni, A. Prescimone, Y. M. Klein, D. J. Gawryluk, E. C. Constable and C. E. Housecroft, Polymer, 2020, 12, 318 CAS.
- Y. M. Klein, A. Prescimone, E. C. Constable and C. E. Housecroft, Inorg. Chem. Commun., 2016, 70, 118–120 CrossRef CAS.
- M. Barquín, J. Cancela, M. J. González Garmendia, J. Quintanilla and U. Amador, Polyhedron, 1998, 17, 2373–2378 CrossRef.
- Y. M. Klein, E. C. Constable, C. E. Housecroft and A. Prescimone, Inorg. Chem. Commun., 2014, 49, 41–43 CrossRef.
- C.-L. Liu, C.-J. Zheng, X.-K. Liu, Z. Chen, J.-P. Yang, F. Li, X.-M. Ou and X.-H. Zhang, J. Mater. Chem. C, 2015, 3, 1068–1076 RSC.
- J. E. Anthony, S. I. Khan and Y. Rubin, Tetrahedron Lett., 1997, 38, 3499–3502 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A, 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C, 2015, 71, 3–8 CrossRef PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D, 2010, 66, 125–132 CrossRef CAS PubMed.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- T. Roisnel and J. Rodríguez-Carvajal, in In Materials Science Forum. Proceedings of the European Powder Diffraction Conf. (EPDIC 7), Barcelona, Spain, 2001, pp. 118–123 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S20: NMR spectra of compounds 1–5; Fig. S21–S25: absorption spectra; Fig. S26–S33: IR spectra; Fig. S34–S43: mass spectra; Fig. S44–S46: additional structural figures. CCDC 2096132 and 2096133. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01531a |
| This journal is © The Royal Society of Chemistry 2022 |