
A new I3O93− group constructed from IO3− and IO55− anion units in Cs3[Ga2O(I3O9)(IO3)4(HIO3)]†
Dandan
Wang
 a,
Xiaoqing
Jiang
a,
Pifu
Gong
b,
Xinyuan
Zhang
*a,
Zheshuai
Lin
b,
Zhanggui
Hu
*a and
Yicheng
Wu
a
a,
Xiaoqing
Jiang
a,
Pifu
Gong
b,
Xinyuan
Zhang
*a,
Zheshuai
Lin
b,
Zhanggui
Hu
*a and
Yicheng
Wu
a
aTianjin Key Laboratory of Functional Crystal Materials, Institute of Functional Crystals, Tianjin University of Technology, Tianjin 300384, China. E-mail: xyzhang@email.tjut.edu.cn
bKey Laboratory of Functional Crystals and Laser Technology, Beijing Center for Crystal Research and Development, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 11th November 2021
Abstract
An alkali metal gallium iodate, Cs3[Ga2O(I3O9)(IO3)4(HIO3)] (CGIO), was synthesized by the traditional hydrothermal method. Interestingly, CGIO revealed a novel I3O93− fundamental building block (FBB) constituted by two IO3 tetrahedrons and one IO5 polyhedron through sharing O atoms. The new I3O93− trimer forms a 0D [Ga2O(I3O9)(IO3)4(HIO3)]3− anion cluster together with the [Ga2O11]16− dimer and isolated IO3− units. The UV-vis-NIR diffuse reflectance spectrum of CGIO shows a wide bandgap of 4.05 eV, and the thermal stability could reach above 400 °C. First-principles calculations explicated that the optical properties of CGIO mainly originated from the iodate anion units with a calculated birefringence of 0.04 at 1064 nm.
Introduction
Inorganic functional materials have become a research hotspot due to their wide applications in the fields of pyroelectricity, birefringence, piezoelectricity, nonlinear optics, and so on.1–4 Recently, metal iodates have drawn lots of attention because of their structural diversity based on I5+ with a stereochemically active lone pair of electrons and constituted by multiple fundamental building blocks (FBBs), such as IO3−, IO43−, IO55− units, etc.5–7 These FBBs could further form polyiodates providing various structural combinations, for instance, the I2O5 dimer in HBa2.5(IO3)6(I2O5),8 the I3O8− trimer in NaI3O8,9 the I3O93− trimer in A2BiI5O15 (A = K+ or Rb+),10 the I4O112− tetramer in HBa(IO3)(I4O11),8 and the I5O143− pentamer in REI5O14 (RE = Y and Gd).11To explore novel inorganic iodates with a large band gap for a wide working spectra region, one of the feasible approaches is to introduce a main group cation with a distorted octahedral structure into the crystal structure. For example, by replacing the V5+ cation with a Ga3+ cation, Pb2GaF2(SeO3)2Cl (ref. 12) achieved an increased band gap from 2.41 eV (Pb2VO2(SeO3)2Cl)13 to 4.32 eV. Via introducing the distorted Ga-centered octahedron, the band gaps of α- and β-Ba2[GaF4(IO3)2](IO3) (4.61 eV and 4.35 eV)14 were widened by 0.29 eV and 0.03 eV, respectively, compared with Ba(IO3)F (4.32 eV).15 Meanwhile, in consideration of the inexistence of d–d or f–f electron transitions for alkaline metals, it is commonly accepted that alkaline metals were also in favour of widening the band gap, e.g., the band gaps within compounds ABi2(IO3)3F5 (A = NH4, K, Rb and Cs) (3.78–3.88 eV)16,17 are larger than that of Bi3OF3(IO3)4 (3.7 eV).18
Therefore, following the above ideas, we found a new gallium iodate compound, Cs3[Ga2O(I3O9)(IO3)4(HIO3)] (CGIO), by using a mild temperature hydrothermal method. CGIO possesses a zero-dimensional (0D) structure consisting of Cs+ cations and novel [Ga2O(I3O9)(IO3)4(HIO3)]3− polyanions, which are constituted by a [Ga2O11]16− dimer, I3O93− trimer, and IO3− triangular pyramids. Different from the previously reported I3O93− group, the newly discovered I3O93− unit in CGIO is constructed from two IO3− units and one IO55− unit. Besides, CGIO also displayed a wide band gap (4.05 eV) and good thermal stability (∼400 °C). First-principles calculations were also performed to investigate the structure–property relationship in detail.
Experimental section
Reagents
Cs2CO3 (Aladdin, 99.9%), Ga2O3 (Aladdin, 99.99%), and I2O5 (Aladdin, 98%) were purchased from commercial sources without further purification.Synthesis
Cs2CO3 (0.756 g, 2.32 mmol), Ga2O3 (0.0815 g, 0.43 mmol), I2O5 (1.162 g, 3.44 mmol), and 4 mL deionized water were placed into a Teflon liner (30 mL), and sealed in an autoclave and kept at 220 °C for 5 days. After that, the autoclave was cooled down to room temperature at a rate of 3 °C h−1. The crystals of CGIO were washed with deionized water and ethyl alcohol and later dried in air. The photograph of the crystals is illustrated in Fig. 1. | ||
| Fig. 1 X-ray powder diffraction pattern of CGIO and the grown crystals are displayed in the insert picture. | ||
Powder X-ray diffraction
The powder X-ray diffraction (PXRD) data of the target sample was successfully collected on a SmartLab 9 KW X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å) in the 2θ range of 10–70° with a scan step width of 0.01° and a fixed counting time of 1.5 s per step at room temperature. The powder XRD pattern is shown in Fig. 1, indicating that the diffraction peaks of the experiment are in agreement with the calculated data, which clearly demonstrate the phase purity.Energy dispersive X-ray spectroscopy
A field emission scanning electron microscope (FESEM, Quanta FEG 250) made by FEI was applied to analyse the microprobe elementals of CGIO and revealed that the crystal contained Cs, Ga, I, and O elements and the average molar ratio was 6.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.37:15.12:75.12, which is in agreement with that determined from single crystal XRD studies (Fig. S1†).
:3.37:15.12:75.12, which is in agreement with that determined from single crystal XRD studies (Fig. S1†).
Single crystal X-ray determination
A Rigaku XtaLAB PRO single crystal diffractometer with Mo Kα radiation at 299 K was used to collect the single crystal X-ray diffraction data of crystal CGIO, and the data reduction and integration of CGIO were carried out using the SAINT program.19 The crystal structure was figured out by direct methods and refined using least-squares minimization with the ShelXLT20 refinement package through the use of Olex2.21 The potential missing symmetry of the structure was examined by the program PLATON.22 The crystallographic data and structure refinements are shown in Table 1. The atomic coordinates, equivalent isotropic displacement parameters, and selected bond lengths and angles are given in Tables S1–S3 in the ESI.†| a R 1 = Σ||Fo| − |Fc||/Σ|Fo|, ωR2 = [Σω(Fo2 − Fc2)2/Σω(Fo2)2]1/2. | |
|---|---|
| Formula | Cs3[Ga2O(I3O9)(IO3)4(HIO3)] |
| Crystal system | Monoclinic |
| Space group | P21/m |
| a (Å) | 7.9191 (4) |
| b (Å) | 21.1203 (7) |
| c (Å) | 8.6696 (4) |
| α (°) | 90 |
| β (°) | 111.691 (5) |
| γ (°) | 90 |
| Z | 2 |
| ρ calc (g cm−3) | 4.817 |
| μ (mm−1) | 15.259 |
| F(000) | 1704 |
| Reflections collected | 24656 |
| R 1, ωR2 (all data)a | 0.0369/0.0775 |
Thermal analysis
Thermogravimetric (TG) and differential thermal analysis (DTA) were measured in the temperature range of 50–650 °C with a heating rate of 5 °C min−1 under the flow of N2 by using a NETZSCH STA thermal analyzer instrument.UV-vis-NIR diffuse reflectance spectroscopy
The UV-vis-NIR diffuse reflectance spectrum was measured in the range from 200 nm to 1200 nm with a UH4150 UV-vis-NIR spectrophotometer with an integrating sphere at room temperature. The reflectance spectrum was converted to the absorption spectrum by using the Kubelka–Munk function F(R) = (1 − R)2/2R, where R is the reflectance.23,24 The band gap was deduced based on the straightforward extrapolation method.25Infrared spectroscopy
The infrared spectrum was measured in the spectral region from 400 cm−1 to 4000 cm−1 with a Nicolet iS50 FT-IR spectrometer with attenuated total reflection (ATR) at room temperature.Raman spectroscopy
The Raman spectrum was investigated in the 0–1000 cm−1 range with an Alpha300 RA at an excitation wavelength of 532 nm at room temperature.Computational method
Utilizing a plane-wave pseudopotential total energy package based on density functional theory (DFT), the electronic band structure and optical properties of CGIO were performed by CASTEP.26,27 The exchange–correlation energy was described by using the functional developed by Perdew–Burke–Ernzerhof (PBE) within the generalized gradient approximation (GGA) form.28,29 The effective interactions between atom cores and valence electrons were modelled using the optimized norm-conserving pseudopotentials30 in the Kleinman–Bylander31 form for all the elements. The valence electron configurations of the atoms were Cs 5s25p66s1, Ga 3d104s24p1, I 5s25p5, O 2s22p4 and H 1s1. Without compromising the computational accuracy, the adoption of a relatively small basis set was allowed. The high kinetic energy cutoff of 880 eV and the k-point meshes of Monkhorst–Pack32 in the Brillouin zones of 3 × 1 × 3 were chosen. The calculation results show that the calculation parameters are accurate enough for this study.Results and discussion
Crystal structure
CGIO crystallizes in the centrosymmetric (CS) space group P21/m (no. 11) with the cell parameters: a = 7.9191 (4) Å, b = 21.1203 (7) Å, c = 8.6696 (4) Å, β = 111.691 (5)° and Z = 2. The crystal structure of CGIO features a novel 0D cluster [Ga2O(I3O9)(IO3)4(HIO3)]3− polyanion, composed of a [Ga2O11]16− dimer, five IO3− triangular pyramids, and one new type of polyiodate anion I3O93−, with alkali metal cations filling the structural void to maintain the charge balance (as shown in Fig. 2). | ||
| Fig. 2 (a) Iodate primitives in the crystal CGIO; (b) [Ga2O(I3O9)(IO3)4(HIO3)]3− polyanion; (c) crystal structure of CGIO along the b axis. | ||
Interestingly, there can be seen as three kinds of I–O polyhedra in CGIO, namely, the IO3 tetrahedron, IO5 polyhedron, and a novel I3O93− anion unit which is composed of two IO3 and one IO5 polyhedron structures by sharing O atoms (Fig. 2a). As shown in Fig. 2b, there exists a neutral molecule HIO3 in CGIO, similar to β-RbIO3(HIO3)2 (ref. 33) and K4[(VO)(IO3)5]2(HIO3)(H2O)2·H2O.34 The bond distances of I–O range from 1.764 (6) Å to 2.469 (4) Å, which are consistent with metal polyiodate compounds reported previously,35,36 such as Cs2I4O11 (1.783 Å to 2.441 Å).37
The Ga atom is octahedrally coordinated by six O atoms from four IO3− units, one IO5− unit, and one terminal O atom, forming the GaO69− group. Two GaO69− groups are bridged by the terminal O atom to form a slightly distorted [Ga2O11]16− dimer group (Fig. 2b), with Ga–O bond lengths ranging from 1.951 (4) Å to 2.008 (3) Å. The magnitude of the out-of-center distortion (Δd) was calculated to be approximately 0.1085.38 One Cs+(1) and two Cs+(2) cations are filled between the two adjacent [Ga2O(I3O9)(IO3)4(HIO3)]3− anion groups to maintain the charge balance, and finally form a 0D cluster Cs3[Ga2O(I3O9)(IO3)4(HIO3)] frame construction (Fig. 2c). It also can be seen that all the 0D clusters in the Cs3[Ga2O(I3O9)(IO3)4(HIO3)] structure are arranged in an anti-parallel manner, leading to the centrosymmetric space group.
The Cs+(1) cation is coordinated with ten O atoms, whereas the Cs+(2) cation is coordinated with eleven O atoms. The bond lengths of Cs–O are in the range of 3.174 (4) Å to 3.554 (4) Å for Cs(1)–O and 3.109 (5) to 3.464 (6) for Cs(2)–O, respectively. Cs(1)O10 and Cs(2)O11 are bridged by O atoms from IO3− and IO55− units to form a [Cs2O19]36− dimer (as shown in Fig. S2†). Bond valence sum (BVS) calculations39,40 verified that the oxidation states of Cs, Ga, I, O and H are 1.062, 3.028, 5.127, 1.950 and 0.953, respectively. It is obvious that the corresponding coordination numbers of atoms are reasonable.
Notably, the I3O93− in CGIO is totally different from the previously reported ones. In A2BiI5O15 (A = K+ or Rb+), the I3O93− consists of one IO3− unit and two IO43− units via sharing two O atoms in the way shown in Fig. 3a.10 Meanwhile in CGIO, the newly discovered I3O93− unit consists of two IO3− units and one IO55− unit, in which the two IO3− units linked with IO55− by sharing adjacent O atoms in the IO55− group. Their polyhedral structures are exhibited in Fig. 3b.
 | ||
| Fig. 3 The I3O93− type in A2BiI5O15 (A = K+ or Rb+) (a) and crystal CGIO (b). | ||
Thermal analysis
The TG-DTA curves are shown in Fig. S3.†CGIO shows good thermal stability up to ∼400 °C, and it exhibits two main steps of weight losses. The first weight loss is about 41.97% in the temperature range of 100–500 °C caused by the volatilization of one molecule of HIO3, two molecules of I2 and four of molecules O2 (cal. 41.55%), which is also consistent with the endothermic peaks at 419.67 °C in the DTA curve. The second weight loss obtained by the experiment (9.76%) occurred in the temperature region from 500–550 °C which is attributed to the volatilization of 0.5 molecules of I2 and 2 molecules of O2 (cal. 9.77%) which is consistent with the endothermic peaks at 538.91 °C in the DTA curve.UV-vis-NIR diffuse reflectance spectroscopy
The UV-vis-NIR diffuse reflectance spectrum is shown in Fig. 4, and it exhibits a wide experimental band gap of 4.05 eV, comparable with Rb2Ge(IO3)6 (Eg = 4.06 eV)41 and K2Ga(IO3)4(H0.5IO3)2 (Eg = 4.02 eV),42 but much larger than other iodates Ba3Ga2(IO3)12 (Eg = 3.06 eV),43 NH4[MoO3(IO3)] (Eg = 3.26 eV),44 Rb2Ga(IO3)4(H0.5IO3)2 (Eg = 3.67 eV),42 Cs2Ti(IO3)6 (Eg = 3.2 eV),45 Cs2Ge(IO3)6 (Eg = 3.32 eV),46 and CsVO2F(IO3) (Eg = 2.39 eV).47 Compared with K2BiI5O15 (Eg = 3.50 eV) containing different I3O93− groups, the band gap of CGIO (Eg = 4.05 eV) is enhanced to 4.05 eV with the synergistic effect of alkali and main group metals of Cs+ and Ga3+ cations. | ||
| Fig. 4 UV-vis-NIR diffuse reflectance spectrum of CGIO. | ||
Infrared spectroscopy and Raman spectroscopy
As shown in the IR spectrum (Fig. S4†), the characteristic absorption peaks of crystal CGIO in the range from 400–670 cm−1 are denoted as the I–O vibrations;48,49 the peaks at 787 cm−1, 814 cm−1, and 837 cm−1 are attributed to the Ga–O vibrations,14 and the peak at 1057 cm−1 is attributed to the vibrations of the IO3− group;49 the peaks in the region of 1643–3391 cm−1 can be assigned to the characteristic peaks of O–H vibration.42 In the Raman spectrum (Fig. S5†), the peaks in the range of 780–845 cm−1 are attributed to the Ga–O vibrations; the characteristic peaks between 450 cm−1 and 780 cm−1 are considered as the vibrations of I–O; the weak peaks below 300 cm−1 can be denoted as the characteristic peaks of the Ga–O group.43,50Theoretical calculations
First-principles calculations performed by the CASTEP package are carried out to understand the relationship between the structure and properties of CGIO. By utilizing the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) form, the electronic band structure, partial density of states and birefringence are accurately evaluated (Fig. 5 and 6). The calculated electronic band structure is shown in Fig. 5a, revealing that CGIO possesses an indirect band gap of 4.05 eV, which is consistent with the experimental value.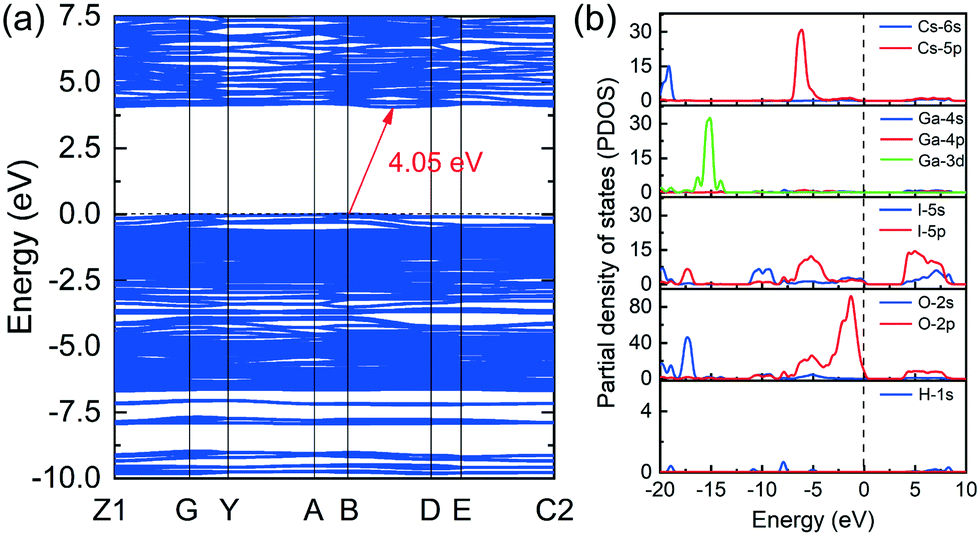 | ||
| Fig. 5 The calculated band structure (a) and the partial density of states (b) of CGIO. | ||
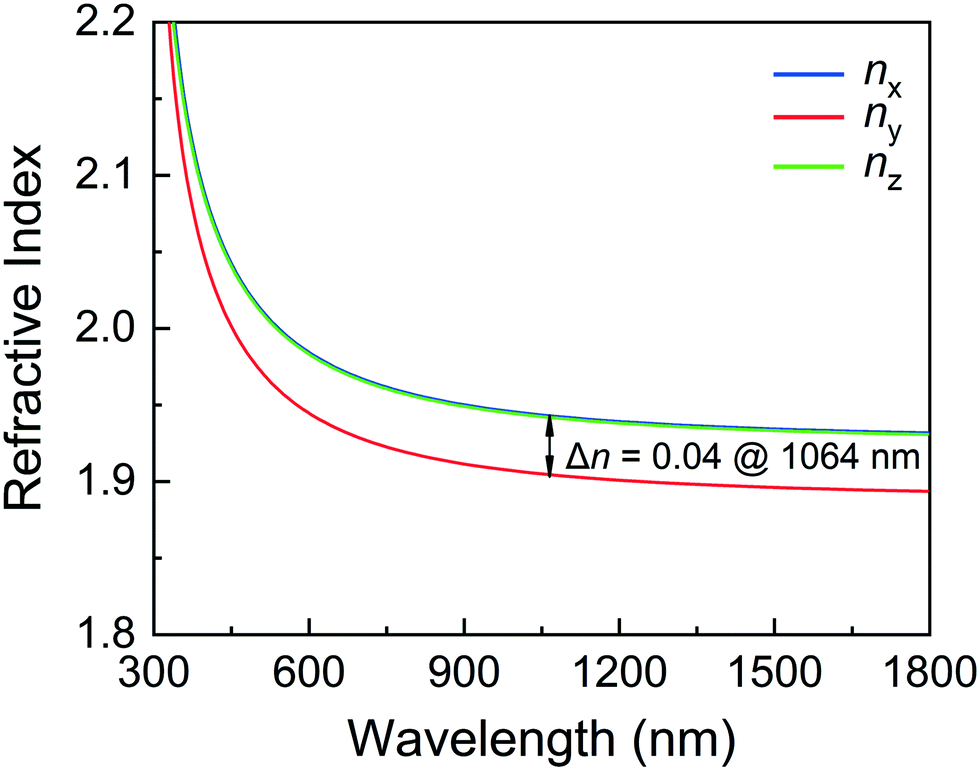 | ||
| Fig. 6 Calculated refractive indices of CGIO. | ||
In order to clearly understand the origin and contribution units of the electronic structure, the partial density of states (PDOS) of the compound was further calculated. The PDOS of CGIO is shown in Fig. 5b and it is obvious that the top states of the valence band (VB) (−5 eV to 0 eV) are mainly attributed to I 5p and O 2p orbitals, while the bottom states of the conduction band (CB) (0 eV to 5 eV) are primarily made up of the unoccupied I 5p and O 2p orbitals. As the optical properties are mainly determined by the electron transitions between the band gaps, it can be deduced that the optical properties of CGIO are mainly determined by IO3− and I3O93− anionic units. The calculated bond orders of the I–O bond by population analysis are 0.27–0.32 e for IO3− and 0.04–0.4 e for IO55−, respectively, indicating that the I–O bonds in IO3− have stronger covalent characteristics than those in IO55−.
As shown in Fig. 6, the refractive indices n and birefringence Δn of CGIO are calculated to evaluate the optical anisotropy. According to the refractive index curves, CGIO is a biaxial crystal with nx > nz > ny and the calculated birefringence (Δn = nx − ny) is 0.04 at 1064 nm.
Conclusions
In conclusion, a new alkali metal gallium iodate crystal, namely, Cs3[Ga2O(I3O9)(IO3)4(HIO3)] (CGIO), with a novel 0D cluster [Ga2O(I3O9)(IO3)4(HIO3)]3− polyanion was obtained by the traditional hydrothermal method. CGIO exhibits a wide band gap of 4.05 eV, and thermogravimetric measurement indicated it possess a high thermal stability above 400 °C. Meanwhile, the theoretical calculations revealed that the birefringence of crystal CGIO can reach 0.04 at 1064 nm and the optical properties originate from the IO3− and I3O93− anionic units. This work would enrich the structural diversity in iodates with wide band gaps by effectively combining different fundamental building blocks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank X. Li for discussion. This work was supported by the National Natural Science Foundation of China (Grant No. 51702232, 51890864, 52002288).Notes and references
- W. W. Zhao, S. L. Pan, L. Y. Dong, Z. H. Yang, X. Y. Dong, Z. H. Chen, M. Zhang and F. F. Zhang, J. Mol. Struct., 2013, 1049, 288–292 CrossRef CAS.
- E. O. Chi, K. M. Ok, Y. Porter and P. S. Halasyamani, Chem. Mater., 2006, 18, 2070–2074 CrossRef CAS.
- M. J. Xia and R. K. Li, Solid State Commun., 2015, 201, 102–106 CrossRef CAS.
- C. Yang, X. Liu, C. Teng, X. Cheng, F. Liang and Q. Wu, Mater. Today Phys., 2021, 19, 100432–100438 CrossRef CAS.
- C. Wu, X. X. Jiang, Z. J. Wang, L. Lin, Z. S. Lin, Z. P. Huang, X. F. Long, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 6, 3464–3468 CrossRef PubMed.
- X. H. Zhang, B. P. Yang, J. Chen, C. L. Hu, Z. Fang, Z. J. Wang and J. G. Mao, Chem. Commun., 2020, 56, 635–638 RSC.
- G. Park, H. R. Byun, J. I. Jang and K. M. Ok, Chem. Mater., 2020, 32, 3621–3630 CrossRef CAS.
- F. F. Mao, C. L. Hu, J. Chen, B. L. Wu and J. G. Mao, Inorg. Chem., 2019, 58, 3982–3989 CrossRef CAS PubMed.
- D. Phanon and I. Gautier-Luneau, Angew. Chem., Int. Ed., 2007, 46, 8488–8491 CrossRef CAS.
- Y. Huang, X. G. Meng, P. F. Gong, L. Yang, Z. S. Lin, X. G. Chen and J. G. Qin, J. Mater. Chem. C, 2014, 2, 4057–4062 RSC.
- J. Chen, C. L. Hu, F. F. Mao, B. P. Yang, X. H. Zhang and J. G. Mao, Angew. Chem., Int. Ed., 2019, 58, 11666–11669 CrossRef CAS.
- F. G. You, F. Liang, Q. Huang, Z. G. Hu, Y. C. Wu and Z. S. Lin, J. Am. Chem. Soc., 2019, 141, 748–752 CrossRef CAS.
- X. L. Cao, F. Kong, C. L. Hu, X. Xu and J. G. Mao, Inorg. Chem., 2014, 53, 8816–8824 CrossRef CAS PubMed.
- J. Chen, C. L. Hu, F. F. Mao, J. H. Feng and J. G. Mao, Angew. Chem., Int. Ed., 2019, 58, 2098–2102 CrossRef CAS PubMed.
- H. X. Fan, G. Peng, C. S. Lin, K. C. Chen, S. D. Yang and N. Ye, Inorg. Chem., 2020, 59, 7376–7379 CrossRef CAS PubMed.
- H. X. Fan, C. S. Lin, K. C. Chen, G. Peng, B. X. Li, G. Zhang, X. F. Long and N. Ye, Angew. Chem., 2020, 59, 5268–5272 CrossRef CAS.
- H. M. Liu, Q. Wu, X. X. Jiang, Z. S. Lin, X. G. Meng, X. G. Chen and J. G. Qin, Angew. Chem., Int. Ed., 2017, 56, 9492–9496 CrossRef CAS.
- M. Zhang, X. Su, M. Mutailipu, Z. H. Yang and S. L. Pan, Chem. Mater., 2017, 29, 945–949 CrossRef CAS.
- SAINT, Version 7.60A, Bruker Analytical X-ray Instruments, Inc, Madison, WI, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- J. Tauc, Mater. Res. Bull., 1970, 5, 721–729 CrossRef CAS.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 593–601 Search PubMed.
- O. Schevciw and W. B. White, Mater. Res. Bull., 1983, 18, 1059–1068 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. RefsonV and M. C. Payne, Z. Kristallogr. Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566–569 CrossRef CAS.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef CAS.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. D. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1227–1230 CrossRef.
- L. Kleinman and D. M. Bylander, Phys. Rev. Lett., 1982, 48, 1425–1428 CrossRef CAS.
- D. J. Chadi, Phys. Rev. B: Solid State, 1977, 16, 1746–1747 CrossRef.
- X. Xu, B. P. Yang, C. Huang and J. G. Mao, Inorg. Chem., 2014, 53, 1756–1763 CrossRef CAS.
- C. F. Sun, C. L. Hu, X. Xu, B. P. Yang and J. G. Mao, J. Am. Chem. Soc., 2011, 133, 5561–5572 CrossRef CAS.
- T. Y. Chang, B. P. Yang, C. L. Hu, D. Yan and J. G. Mao, Cryst. Growth Des., 2017, 17, 4984–4989 CrossRef CAS.
- T. H. Wu, X. X. Jiang, C. Wu, H. Y. Sha, Z. J. Wang, Z. S. Lin, Z. P. Huang, X. F. Long, M. G. Humphrey and C. Zhang, J. Mater. Chem. C, 2021, 2, 8987–8993 RSC.
- K. M. Ok and P. S. Halasyamani, Angew. Chem., Int. Ed., 2004, 43, 5489–5491 CrossRef CAS.
- P. S. Halasyamani, Chem. Mater., 2004, 16, 3586–3592 CrossRef CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- F. F. Mao, C. L. Hu, J. Chen and J. G. Mao, Chem. Mater., 2018, 30, 2443–2452 CrossRef CAS.
- J. H. Zhang, Q. Wu and W. Z. Lai, Dalton Trans., 2020, 49, 2337–2344 RSC.
- L. Xiao, F. G. You, P. F. Gong, Z. G. Hu and Z. S. Lin, CrystEngComm, 2019, 21, 4981–4986 RSC.
- Y. H. Li, G. P. Han, H. W. Yu, H. Li, Z. H. Yang and S. L. Pan, Chem. Mater., 2019, 31, 2992–3000 CrossRef CAS.
- H. Y. Chang, S. H. Kim, K. M. Ok and P. S. Halasyamani, J. Am. Chem. Soc., 2009, 131, 6865–6873 CrossRef CAS PubMed.
- H. M. Liu, X. X. Jiang, X. X. Wang, L. Yang, Z. S. Lin, Z. G. Hu, X. G. Meng, X. G. Chen and J. G. Qin, J. Mater. Chem. C, 2018, 6, 4698–4705 RSC.
- J. Chen, C. L. Hu, X. H. Zhang, B. X. Li, B. P. Yang and J. G. Mao, Angew. Chem., Int. Ed., 2020, 59, 5381–5384 CrossRef CAS PubMed.
- Q. M. Huang, C. L. Hu, B. P. Yang, R. L. Tang, J. Chen, Z. Fang, B. X. Li and J. G. Mao, Chem. Mater., 2020, 32, 6780–6787 CrossRef CAS.
- M. Q. Gai, T. H. Tong, Y. Wang, Z. H. Yang and S. L. Pan, Chem. Mater., 2020, 32, 5723–5728 CrossRef CAS.
- M. J. Bushiri, T. C. Kochuthresia, V. K. Vaidyan and I. Gautier-Luneau, J. Nonlinear Opt. Phys. Mater., 2014, 23, 1450039–1450047 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2107844. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01234g |
| This journal is © The Royal Society of Chemistry 2022 |