
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriple dehydrofluorination as a route to amidine-functionalized, aromatic phosphorus heterocycles†‡
Nathan T.
Coles
 *ab,
Lucie J.
Groth
a,
Lea
Dettling
a,
Daniel S.
Frost
a,
Massimo
Rigo
a,
Samuel E.
Neale
*c and
Christian
Müller
*a
*ab,
Lucie J.
Groth
a,
Lea
Dettling
a,
Daniel S.
Frost
a,
Massimo
Rigo
a,
Samuel E.
Neale
*c and
Christian
Müller
*a
aInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany. E-mail: c.mueller@fu-berlin.de
bSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: Nathan.Coles@nottingham.ac.uk
cDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: sen36@bath.ac.uk
First published on 3rd November 2022
Abstract
An unexpected route to hitherto unknown amidine-functionalized phosphinines has been developed that is rapid and simple. Starting from primary amines and CF3-substituted λ3,σ2-phosphinines, a cascade of dehydrofluorination reactions leads selectively to ortho-amidinephosphinines. DFT calculations reveal that this unusual transformation can take place via a series of nucleophilic attacks at the electrophilic, low-coordinate phosphorus atom.
λ3,σ2-Phosphinines, also known as phosphabenzenes, are aromatic phosphorus heterocycles which are currently undergoing an intriguing renaissance in the fields of coordination chemistry, homogeneous catalysis, activation of small molecules and photoluminescent molecular materials.1 Our group and others have previously developed a number of (donor-)functionalized λ3- and λ5-phosphinines, which are of particular relevance for their use in such research fields and a brief selection (I–VI) is depicted in Fig. 1.2–7 In this respect, the specific functionalization of phosphinines is of particular importance in order to modify their stereoelectronic properties.
 | ||
| Fig. 1 Selected functionalized λ3- and λ5-phosphinines and brief summary of this work. | ||
During the course of our investigations on [4+2]-cycloaddition reactions on phosphinines, we found not only a synthetic access to hitherto unknown CF3-substituted phosphinines, but also an unexpected series of dehydrofluorination reactions in the presence of primary amines, that yielded novel amidine-substituted phosphinines selectively (Fig. 1).
We have previously synthesized 1-phosphabarrelene 2b from phosphinine 1b in the presence of hexafluoro-2-butyne, which has now been expanded to 1a/2a (Scheme 1).8 Gratifyingly, we were able to characterise the known phosphinine 1a9 and the novel phosphabarrelene 2b also crystallographically (Fig. S92 and S93, ESI‡).
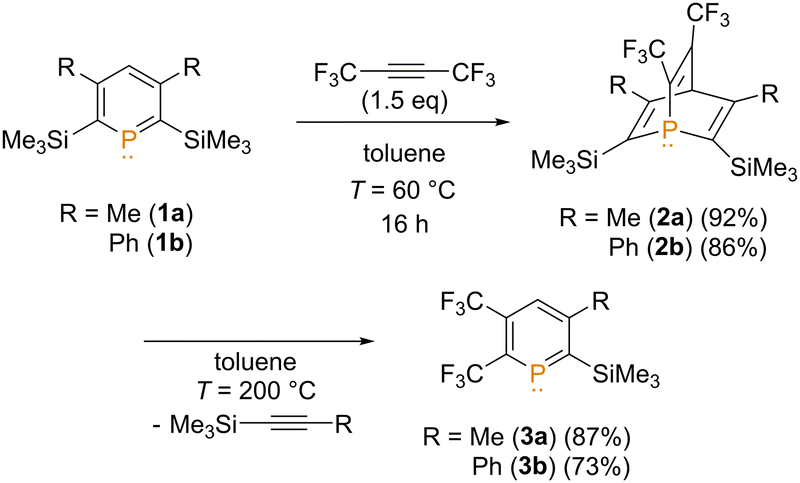 | ||
| Scheme 1 Synthesis of phosphinines 3a/bvia selective cycloaddition–cycloreversion reaction. | ||
Interestingly, we noticed that upon heating a toluene solution of 2a/b in a high-pressure reaction vessel to T = 200 °C, a retro-Diels–Alder reaction occurs, leading selectively to trifluoromethyl substituted phosphinines 3a/b. For 3b, crystals suitable for an X-ray crystallographic analysis, were obtained by storing the oily sample at T = −20 °C. The molecular structure of 3b (Fig S94, ESI‡) in the solid state confirms the presence of the retro-Diels–Alder product. Notably, this reactivity has never been observed in the chemistry of phosphinines and phosphabarrelenes before. It offers the exceptional possibility to introduce electron withdrawing CF3 groups to a phosphinine core in a facile manner.10 Consequently, we were interested in exploring the effect of the CF3 substituents on the energy levels of the frontier molecular orbitals (MOs) of 3a/b first.
The introduction of SiMe3-groups at the ortho-position of the aromatic phosphorus heterocycle increases the energy of the occupied MOs.11 Most notably, there is a significant increase in the energy level of the lone-pair (HOMO−1), with respect to the parent phosphinine C5H5P (HOMO−2), while the energy levels of the respective LUMOs do not change significantly (Fig. S97, ESI‡). For 3b, the HOMO−3 represents now the lone-pair at the phosphorus atom, which is at a comparable energy level to the one of the parent phosphinine. In clear contrast, the LUMO of 3b has the character of an empty p-orbital at the phosphorus atom and is stabilized by almost 0.6 eV with respect to the unsubstituted phosphinine, which renders the low-coordinate phosphorus atom much more electrophilic. In this respect, the orientation of the relevant molecular orbitals (HOMO−3 and LUMO) is similar to the situation found in carbenes.
It is well documented that strong nucleophiles, such as organolithium compounds, react at the phosphorus atom to afford 1-R-phosphacyclohexadienyl anions, also known as λ4-phosphinines.12 Due to the energetically low-lying LUMO we anticipated that 3a/b might also react with much less nucleophilic amines, which could lead to interesting follow-up reactions at the phosphorus heterocycle. Initial reactivity studies were performed with 3a and nPrNH2. Using two equivalents of nPrNH2 in DCM, a small new signal at δ(ppm) = 0.3 in the 31P{1H} NMR spectrum was observed (Fig. S1, ESI‡). The signal appears as a quartet with a coupling constant of 3JP–F = 19.0 Hz, which is substantially smaller with respect to the starting material (3JF,P = 53.0 Hz). We tentatively assigned this resonance to compound 4a (Scheme 2).
 | ||
| Scheme 2 Reaction of 3a with nPrNH2 showing the equivalents of amine that has been introduced into the phosphinine. 4a synthesised using 10 equivalents of nPrNH2. 5a/a′ synthesised using an excess of nPrNH2. | ||
The formation of 4a is also corroborated by a large chemical shift difference of δ(ppm) = 245.3, leading to a shift of δ(ppm) = 1.6 in the 31P{1H} NMR spectrum (3a: single resonance at δ(ppm) = 245.6 in CDCl3). The 19F NMR spectrum of 4a (Fig. S2, ESI‡) shows two new signals at δ(ppm) = −65.0 (m) and δ(ppm) = −66.4 (br, s), which had shifted from δ(ppm) = −52.9 and δ(ppm) = −59.4, respectively, compared to 3a. Interestingly, when the solvent was removed and replaced by THF and some additional nPrNH2 and left to stand overnight, a substantial change to the 31P{1H} NMR spectra was noticed (Fig. S3, ESI‡).
Next to 3a and 4a, as well as protodesilylated starting material (3a′, Fig. S3, ESI‡), signals at δ(ppm) = 214.1 (s) (5a) and δ(ppm) = 242.4 (s) (5a′) which could not be assigned initially. The 19F NMR spectrum of the reaction mixture (Fig. S4 and S5, ESI‡) showed two new singlets at δ(ppm) = −61.9 (5a′) and δ(ppm) = −62.2 (5a) alongside a singlet at δ(ppm) = −154.6. Remarkably, the two new singlets in the 19F NMR spectrum were both found in a region similar to that observed for the meta-CF3 of 3a (δ(ppm) = −61.1) and 3a′ (δ(ppm) = −60.9). The 19F data coupled with the observations in the 31P{1H} spectra suggested that a chemical transformation had occurred exclusively at the CF3 moiety adjacent to the phosphorus atom (Scheme 2).
With these initial results attempts were made to cleanly synthesize 5a and 5a′. By increasing the equivalents of amine from two to ten the reaction became faster, with ≈80% conversion to 4a by 31P{1H} and 19F NMR spectroscopy in only 30 minutes (Fig. S6 and S7, ESI‡). Leaving this sample for 20 hours led to most of 4a being consumed, with 5a and 5a′ being the major species (Fig. S8 and S9, ESI‡). Visually, the samples became extremely viscous with a large amount of precipitate forming, which was later confirmed as nPrNH3F. Increasing the temperature to T = 60 °C with ten equivalents of amine led to a larger proportion of 5a′ relative to the reaction at room temperature (Fig. S10, ESI‡). Leaving this sample for 2 months at room temperature caused a complete loss of the signal for 5a (Fig. S12, ESI‡). In the 19F NMR spectrum, a signal attributed to 5a′ was observed along with the characteristic signal of FSiMe3 at δ(ppm) = −154.6. This observation indicates that nPrNH3F is generated during the course of the reaction, which over time initiates the protodesilylation reaction. The formation of the 5a/5a′ could be halted by removal of the excess amine after 4a had formed. The 1H NMR spectrum of 4a shows a substantial upfield shift for the proton at the para-position of the heterocyclic ring (δ(ppm) = 0.66 to δ(ppm) = 6.60). Other characteristic signals occur at δ(ppm) = 3.52 ppm and δ(ppm) = 2.24–2.08. The signal at δ(ppm) = 3.52 can be attributed to a proton adjacent to the ortho-CF3 group as it appears as a quartet of doublets, caused by coupling to both 19F and 31P nuclei.
With these findings in mind, an attempt was made to form 5a selectively by using a vast excess of amine. This was implemented to shorten the reaction time and try to prevent the protodesilylation of the SiMe3-group due to the limited solubility of the ammonium salt. Gratifyingly, using 0.1 mL of benzene, to help dissolve 3a (0.13 mmol), with 0.5 mL of nPrNH2 led initially to the formation of the SiMe3-substituted phosphinine, with a conversion of greater than 90%. After filtering the solution to remove the ammonium salt and removal of the volatiles, the residual oil was dissolved in C6D6. A spectroscopic yield of 47% was obtained against an internal standard (PPh3). However, upon filtering, some protodesilylation was observed by means of NMR spectroscopy, most likely due to traces of dissolved nPrNH3F during the isolation of the product.
In order to fully identify the reaction products, the substrate scope of this reaction was further expanded to other amines (Fig. 2 and Scheme S1, ESI‡). We first focused on MeNH2. The reaction was performed by condensing dried MeNH2 directly onto the phosphinine at T = −60 °C and stirring at this temperature for 1 hour. Initially, the SiMe3-substituted phosphinine could again be isolated and characterized. However, upon removal of the volatiles and leaving the residue as an oil for 3 days, a crystalline material formed. When the solid was redissolved, only the protodesilylated product could be observed by means of NMR spectroscopy. Again, we believe that traces of MeNH3F caused the loss of the SiMe3 group.
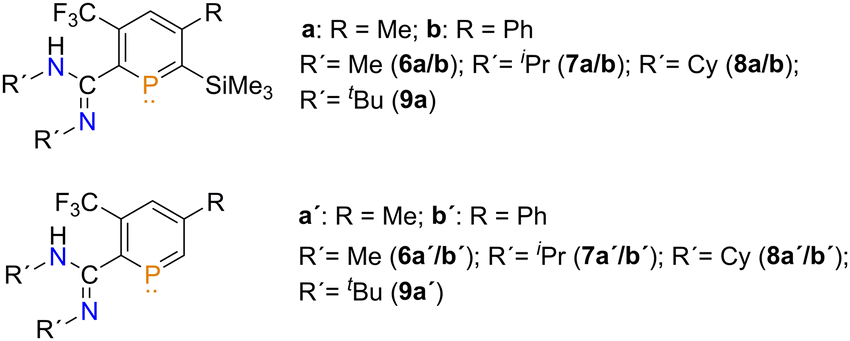 | ||
| Fig. 2 Substrate scope for the novel triple dehydrofluorination reaction. | ||
Pleasingly, slow evaporation of a DCM solution of the 6a′ yielded single crystals suitable for single crystal X-ray diffraction. The molecular structure of 6a′ in the crystal, along with selected bond lengths and angles, is given in Fig. 3.
 | ||
| Fig. 3 Molecular structure of 6a′. Thermal ellipsoids are represented at 50% probability. Selected bond lengths (Å) and angles (°): P(1)–C(1): 1.728(3); C(1)–C(2): 1.390(4); C(2)–C(3): 1.395(4); C(3)–C(4): 1.395(4); C(4)–C(5): 1.392(4); C(5)–P(1): 1.740(3); C(5)–C(8): 1.508(4); C(8)–N(1): 1.288(4); C(8)–N(2): 1.358(4); C(1)–P(1)–C(5): 101.26(15); C(2)–C(3)–C(4): 124.1(3); N(1)–C(8)–N(2): 121.0(3). | ||
Much to our delight, the crystallographic characterization of 6a′ reveals, that the final product is indeed a protodesilylated, amidine-functionalized phosphinine. The presence of only one CF3 group, the fully planar phosphorus heterocycle and the absence of a SiMe3-group is in full agreement with the NMR spectroscopic data of the product(s) described above and depicted already in Scheme 2. In the solid state, at T = 100 K, the C–N bonds are found to be at lengths expected for both a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond and a C–N single bond, in contrast to what is observed in solution at room temperature where rapid tautomerization of the amidine takes place, as evidenced by the broad signals for the groups bound to the nitrogen atoms. Interestingly, the amidine group orientates itself perpendicular to the plane of the phosphinine ring. This might allow for intermolecular hydrogen bonding in the solid state (Fig. S96, ESI‡).
N double bond and a C–N single bond, in contrast to what is observed in solution at room temperature where rapid tautomerization of the amidine takes place, as evidenced by the broad signals for the groups bound to the nitrogen atoms. Interestingly, the amidine group orientates itself perpendicular to the plane of the phosphinine ring. This might allow for intermolecular hydrogen bonding in the solid state (Fig. S96, ESI‡).
When using aniline, no reactivity towards 3a was observed, even after 6 days. Both iPrNH2 and CyNH2 reacted to give 7a/7a′ and 8a/7a′, respectively. tBuNH2 yielded 9a/9a′, however, in this case an unknown impurity accompanied the desired products, which could not be separated. The reactions were also expanded to phosphinine 3b, resulting in 5b–8b (Fig. 2).
This transformation of an aromatic CF3 group into an amidine, involving the formal loss of three equivalents of HF was thus considered unusual given that CF3-groups in aryl-trifluorides are normally resistant towards chemical degradation. A few methods for chemical modification of such a group have been developed during the last decade,13 with some requiring harsh reaction conditions or the presence of strong Lewis acids.14 CF3 groups of activated aromatic rings, for example, have been shown to degrade in the presence of amines and aqueous NaOH, resulting in amides.15
The first step appears to be the formal addition of the N–H bond across the PC double bond.16 The following steps to the amidine substituent, however, could not be identified experimentally, as no other intermediate species were observed by means of NMR spectroscopy. We therefore employed DFT calculations at the M052X-D3,THF/def2-QZVPP//TPSS/def2-TZVPP level of theory to elucidate the full reaction pathway (see ESI‡ for full computational details17). The calculations focussed on the triple dehydrofluorination of 3a (species I in the DFT profile) to form 6a (VII), with the computed free energy profile outlined in Fig. 4.
 | ||
| Fig. 4 DFT free energy profile of the triple dehydrofluoronation of 3a (labelled I in the profile) with NH2Me, calculated at the M052X-D3, THF/def2-QZVPP//TPSS/def2-TZVPP level. | ||
Initial efforts (not shown in Fig. 4) were dedicated to the description of formal N–H bond activation across the PC bond of the phosphinine and the corresponding product, as the tentative identification of this process was confirmed by NMR spectroscopy. While this process can proceed via an accessible activation barrier of +26.0 kcal mol−1 (see Fig. S98, ESI‡), consistent with the experimental identification of this product, subsequent H–F coupling was disregarded owing to the large computed activation barrier for such a process (+65.2 kcal mol−1).
An alternative pathway starting from I was in turn identified in which the addition of MeNH2 occurs via the nitrogen atom at the phosphorus centre, with concomitant H–F coupling. This takes place viaTS(I-II) and a barrier of +31.7 kcal mol−1 to form II (+14.3 kcal mol−1, Fig. 3) indicating that nucleophiles can directly attack the phosphorus LUMO in low-coordinate organophosphorus species under formation of λ4-phosphinines.12 In this initial step the unsaturated CF2 group is formed, where subsequent addition of a second MeNH2 at this unsaturated carbon centre, viaTS(II–III) (+32.1 kcal mol−1) yields III (+26.7 kcal mol−1). From III the phosphinine heterocycle is exergonically re-aromatized to form IV (−6.5 kcal mol−1). Subsequent addition of MeNH2 at the phosphorus centre allows for another process of H–F elimination to yield V (−3.6 kcal mol−1), whereby a repeated process of MeNH2 addition to the unsaturated MeHNFC moiety with concerted proton transfer eliminates MeNH2 from the phosphorus centre and gives the bis-amino-fluoro-substituted species VI (−8.2 kcal mol−1). Dehydrofluorination via an H–F coupling from VI affords the final amidine product, VII (−21.2 kcal mol−1). Most intriguingly, the DFT calculations verify that the unusual transformation of the ortho-CF3-group to an ortho-amidine substituent is feasible owing to the electrophilicity of the low-coordinate phosphorus centre that allows nucleophilic addition of the amine and subsequent elimination of HF.
In summary, a route to hitherto unknown CF3-substituted phosphinines by cycloaddition–cycloreversion reaction on bis-SiMe3 substituted phosphinines has been developed. These compounds undergo a cascade of dehydrofluorination reactions in the presence of primary amines to form novel amidine-functionalized phosphinines. DFT calculations revealed that the CF3 → amidine transformation is driven by a series of nucleophilic additions of the amine to the electrophilic phosphorus centre, which then allows for concerted HF elimination reactions. Phosphinines, decorated with certain functionalities, can thus promote unusual chemical transformations of small molecules. This might also apply for other CF3-substituted organophosphorus compounds containing P–C multiple bonds. Further investigations in this direction and in the coordination chemistry of the here reported novel P,N-hybrid ligands as well as their use in catalytic reactions are currently performed in our laboratories.
C. M. and N. T. C. acknowledge the Deutsche Forschungsgemeinschaft (DFG) for financial support. Dr G. Thiele and C. von Randow are thanked for the supply and handling of dry MeNH2. S. E. N. acknowledges the Anatra HPC service at The University of Bath.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) N. T. Coles, A. S. Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 433, 213729 CrossRef CAS; (b) A. Paderina, R. Ramazanov, R. Valiev, C. Müller and E. Grachova, Inorg. Chem., 2022, 61, 11628 CrossRef PubMed; (c) J. Leitl, M. Marquardt, P. Coburger, D. J. Scott, V. Streitferdt, R. M. Gschwind, C. Müller and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 15407 CrossRef CAS PubMed; (d) X. Chen, Z. Li, F. Yanan and H. Grützmacher, Eur. J. Inorg. Chem., 2016, 633 CrossRef; (e) P. Roesch, J. Nitsch, M. Lutz, J. Wiecko, A. Steffen and C. Müller, Inorg. Chem., 2014, 53, 9855 CrossRef CAS PubMed; (f) J. Moussa, L. M. Chamoreau and H. Amouri, RSC Adv., 2014, 4, 11539 RSC.
- C. Müller, D. Wasserberg, J. J. M. Weemers, E. A. Pidko, S. Hoffmann, M. Lutz, A. L. Spek, S. C. J. Meskers, R. A. J. Janssen, R. A. van Santen and D. Vogt, Chem. – Eur. J., 2007, 13, 4548 CrossRef PubMed.
- (a) Y. Mao, K. M. H. Lim, Y. Li, R. Ganguly and F. Mathey, Organometallics, 2013, 32, 3562 CrossRef CAS; (b) Y. Hou, Z. Li, Y. Li, P. Liu, C. Y. Su, F. Puschmann and H. Grützmacher, Chem. Sci., 2019, 10, 3168 RSC.
- S. Giese, K. Klimov, A. Mikeházi, Z. Kelemen, D. S. Frost, S. Steinhauer, P. Müller, L. Nyulászi and C. Müller, Angew. Chem., Int. Ed., 2021, 133, 3625 CrossRef.
- R. J. Newland, M. F. Wyatt, R. L. Wingad and S. M. Mansell, Dalton Trans., 2017, 46, 6172 RSC.
- J. Lin, F. Wossidlo, N. T. Coles, M. Weber, S. Steinhauer, T. Böttcher and C. Müller, Chem. – Eur. J., 2022, e202104135 CAS.
- G. Pfeifer, F. Chahdoura, M. Papke, M. Weber, R. Szűcs, B. Geffroy, D. Tondelier, L. Nyulászi, M. Hissler and C. Müller, Chem. – Eur. J., 2020, 26, 10534 CrossRef CAS PubMed.
- M. Rigo, E. R. M. Habraken, K. Bhattacharyya, M. Weber, A. W. Ehlers, N. Mézailles, J. C. Slootweg and C. Müller, Chem. – Eur. J., 2019, 25, 8769 CAS.
- L. Cataldo, S. Choua, T. Berclaz, M. Geoffroy, N. Mézailles, L. Ricard, F. Mathey and P. L. Floch, J. Am. Chem. Soc., 2001, 123, 6654 CrossRef CAS PubMed.
- Only a tetra-CF3-substituted diphosphinine is known so far: Y. Kobayashi, H. Hamana, S. Fujino, A. Ohsawa and I. Kumadaki, J. Am. Chem. Soc., 1980, 102, 252 CrossRef CAS.
- M. H. Habicht, F. Wossidlo, T. Bens, E. A. Pidko and C. Müller, Chem. – Eur. J., 2018, 24, 944 CrossRef CAS PubMed.
- (a) G. Märkl and C. Martin, Angew. Chem., Int. Ed. Engl., 1974, 13, 408 CrossRef; (b) A. Moores, N. Mézailles, L. Ricard and P. Le Floch, Organometallics, 2005, 24, 508 CrossRef CAS.
- (a) Y. Kobayashi and I. Kumadaki, Acc. Chem. Res., 1978, 11, 197 CrossRef CAS; (b) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (c) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915 RSC.
- C. Santamaría, R. Beckhaus, D. Haase, W. Saak and R. Koch, Chem. – Eur. J., 2001, 7, 622 CrossRef.
- G. O′Mahony and A. K. Pitts, Org. Lett., 2010, 12, 2024–2027 CrossRef PubMed.
- See also: (a) B. Schmid, L. M. Venanzi, A. Albinati and F. Mathey, Inorg. Chem., 1991, 30, 4693–4699 CrossRef CAS; (b) I. de Krom, E. A. Pidko, M. Lutz and C. Müller, Chem. – Eur. J., 2013, 19, 7523 CrossRef CAS PubMed.
- Please note that the desilylation process was not modelled, but rather the full dehydrofluorination pathway, both with and in the absence of the ortho-TMS group, where its presence was not shown to impact the kinetics of triple dehydrofluorination. See ESI‡ for details.
Footnotes |
| † Dedicated to Prof. Dr Evamarie Hey-Hawkins on the occasion of her 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2176809–2176812. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05178h |
| This journal is © The Royal Society of Chemistry 2022 |