
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphaacene as a structural analogue of thienoacenes for organic semiconductors†
Kyohei Matsuo *a,
Rina Okumuraa,
Hironobu Hayashia,
Naoki Aratania,
Seihou Jinnaib,
Yutaka Ieb,
Akinori Saekic and
Hiroko Yamada*a
*a,
Rina Okumuraa,
Hironobu Hayashia,
Naoki Aratania,
Seihou Jinnaib,
Yutaka Ieb,
Akinori Saekic and
Hiroko Yamada*a
aDivision of Materials Science, Nara Institution of Science and Technology (NAIST), 8916-5 Takayama-cho, Ikoma, Nara, 630-0192, Japan. E-mail: kmatsuo@ms.naist.jp; hyamada@ms.naist.jp
bThe Institute of Scientific and Industrial Research (SANKEN), Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka, 565-0871, Japan
First published on 14th November 2022
Abstract
An air-stable λ3-phosphinine-containing polycyclic aromatic compound without steric protection was synthesized and its charge transport properties were evaluated, which revealed moderate hole mobility. This research is the first experimental demonstration of the organic electronic applications of low-coordinate phosphorus compounds.
Sulfur atoms play an important role in the development of high-performance organic field-effect transistor (OFET) materials.1 The introduction of sulfur atoms, whose valence orbitals (3s and 3p orbitals) are spatially larger than those of the carbon atom (2s and 2p orbitals),2 into π-conjugated frameworks is believed to enhance the charge carrier mobility by increasing the intermolecular orbital overlap.1a On the other hand, organic semiconductors incorporating phosphorus atoms, which also have spatially larger valence orbitals, are popular as electron transporting and host materials for organic light-emitting diodes (OLEDs).3 However, they are very rarely used as OFET materials because of their low charge carrier mobilities.4 The most important factor determining the difference in the charge carrier mobility of organic semiconductors is the coordination number of the heavy elements. A divalent sulfur atom is usually two-coordinate and sterically exposed when embedded at the edge of π-conjugated systems; hence, its valence orbitals can participate in intermolecular orbital overlap. In contrast, the typical coordination numbers of trivalent and pentavalent phosphorus atoms are three or more. One or more substituents act as a steric hindrance on the phosphorus atom and inhibit the formation of conduction channels through its valence orbitals. If this explanation is correct, it can be theorized that as long as the coordination number of the phosphorus atom is reduced to two, it would function similarly to a sulfur atom in enhancing the charge carrier mobility.
λ3-Phosphinine, one of the stable low-coordinate phosphorus compounds, and its derivatives have been extensively studied as ligands in the field of organometallic chemistry because of their higher stability compared to electronically localized phosphaalkenes and unique electron-accepting character.5 Although many derivatives have been synthesized, there are only a few reports on the optoelectronic properties of π-conjugated systems incorporating λ3-phosphinines,6 as well as their applications in organic semiconductors. In terms of π-electrons, λ3-phosphinine has an isoelectronic π-system with thiophene. Hence, it can probably be used as a building block for high-mobility organic semiconductors (Fig. 1a). In other words, heteroacenes incorporating the λ3-phosphinine unit, namely phosphaacenes, are expected to be used as alternatives to typical thienoacenes. Although a few theoretical studies have suggested that the introduction of sp2-phosphorus atoms into π-conjugated frameworks improves the charge carrier mobility of pentacene,7 the synthesis of the proposed molecules is not feasible.
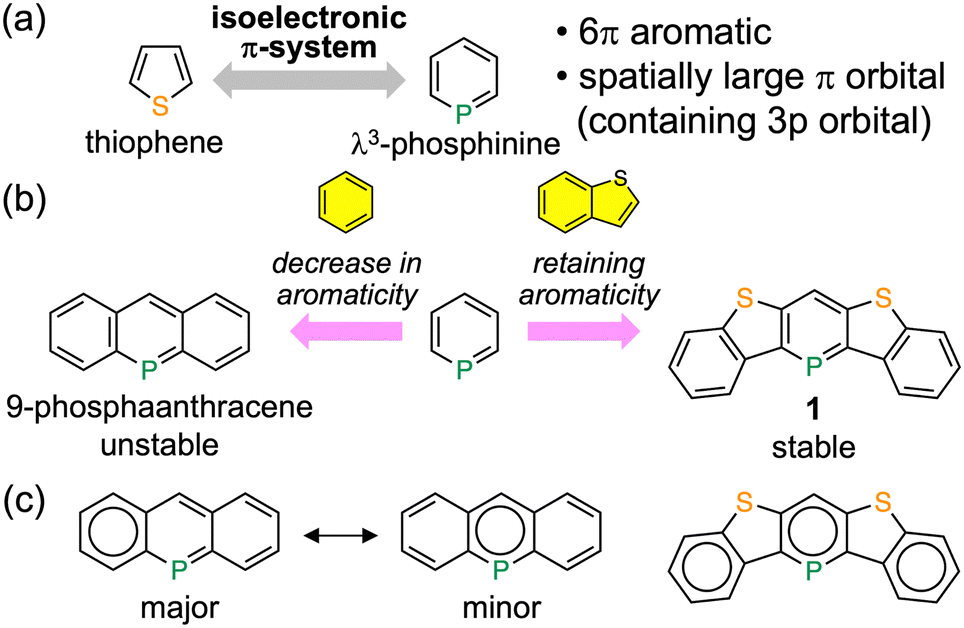 | ||
| Fig. 1 (a) Comparison between thiophene and λ3-phosphinine. (b) Molecular structures and (c) the Clar structures of 9-phosphaanthracene and benzo[b]thiophene-annelated λ3-phosphinine. | ||
Herein, we describe the synthesis and characterization of a new λ3-phosphinine-containing polycyclic aromatic compound, 1 (Fig. 1b). Phosphaacene 1 is stable without any substituents for steric protection. This is contrary to the fact that the introduction of phenyl or more bulky substituents near the phosphorus atom is necessary to stabilize the benzene-annelated λ3-phosphinines, such as 9-phosphaanthracene and 5-phosphaphenanthrene.8 Three Clar sextets can be drawn, including the λ3-phosphinine ring within 1; however, only one sextet is located on the benzene ring compared to the λ3-phosphinine ring within 9-phosphaanthracene (Fig. 1c). Therefore, the fusion of benzo[b]thiophene units retains a high degree of local aromaticity in the central λ3-phosphinine moiety, leading to higher stability.9 A highly planar and rigid structure with no extra substituents is ideal for effective intermolecular interactions in the solid state; hence, 1 is a promising molecule for use in OFET materials. We now report the successful fabrication of the first OFET device using a λ3-phosphinine-based compound.
The synthesis of phosphaacene 1 is illustrated in Scheme 1. The bromination of bis(benzo[b]thiophen-2-yl)methane 2 with N-bromosuccinimide (NBS) resulted in the formation of bis(3-bromobenzo[b]thiophen-2-yl)methane 3 with 87% yield. The halogen–lithium exchange reaction of 3 with four equivalents of sec-butyllithium, followed by treatment with dichloro(dimethylamino)phosphine and heating at 60 °C, successfully produced the desired compound 1 with a yield of 53%.8f The reference compound, thienoacene 4, was prepared with a 33% yield by the direct Bradsher reaction of 2 with dichloromethyl methyl ether and titanium(IV) chloride.10 As expected, the newly synthesized phosphaacene 1 is stable enough to allow handling under atmospheric conditions and can be fully characterized by 1H, 13C, and 31P NMR spectroscopies and high-resolution mass spectrometry. In the 1H NMR spectrum, one characteristic doublet signal, assignable to the proton at the 4-position of the λ3-phosphinine moiety, is observed at 8.50 ppm with a coupling constant (4JPH) of 4.0 Hz.11 This downfield-shifted signal suggests a large extent of aromaticity in the λ3-phosphinine moiety. Moreover, a 31P NMR chemical shift appears in the low-field region at 167 ppm, comparable to those of aryl-substituted λ3-phosphinine derivatives, which is characteristic of aromatic phosphorus heterocycles.6,12 In addition, thermogravimetric analysis (TGA) was performed on 1 and 4 under the flow of N2 gas to evaluate their thermal stability (Fig. S1, ESI†). While the TGA profile of 4 exhibited a typical weight loss curve with a 5% weight loss at a temperature (Td5) of 253 °C, the weight of 1 increased to 10% from approximately 190 °C and then decreased slowly from 300 °C. Although no products were properly identified, this unusual weight increase upon heating can be attributed to the reactivity of trivalent organophosphorus compounds toward oxygen. Nevertheless, it was confirmed that 1 is thermally stable at temperatures as high as 190 °C under oxygen-free conditions, which is sufficient for device fabrication. Indeed, 1 could be purified by temperature-gradient vacuum sublimation at 150 °C/10−3 Pa.
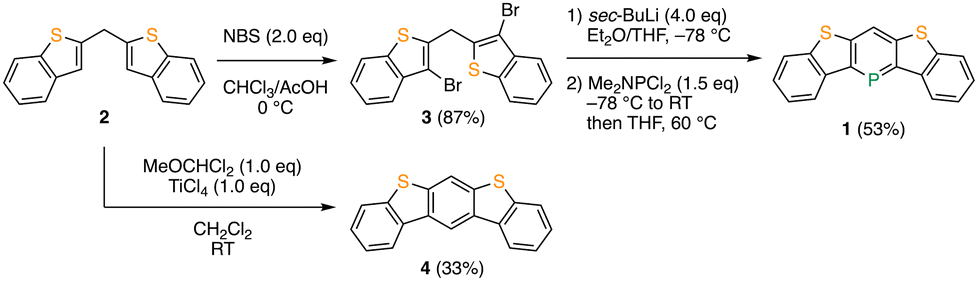 | ||
| Scheme 1 Synthesis of phosphaacene 1 and thienoacene 4. | ||
The molecular structures of 1 and 4 were confirmed by X-ray crystallographic analysis (Fig. 2a and b). Both the five-ring-fused heteroacenes exhibit highly planar structures with small dihedral angles of 2.6° and 7.4° between the two terminal benzene rings for 1 and 4, respectively. The C–C bond lengths of the λ3-phosphinine ring in 1 are in the range of 1.381(4)–1.419(4) Å, and the C–C bond alternation is sufficiently small compared to that of the central benzene ring in 4 (Fig. 2c and d). The C–P bond lengths of 1.737(2) and 1.732(3) Å in 1 are slightly longer than the typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P double bond lengths of 1.60–1.70 Å13 and comparable to those of reported λ3-phosphinine derivatives.5a,12b These results strongly suggest the high extent of aromaticity of the λ3-phosphinine moiety in 1. Notably, 1 shows a 1D-columnar structure with a π–π distance of 3.53 Å in the crystal packing (Fig. 2e). In contrast, 4 forms a typical herringbone structure with a π–π distance of 3.52 Å (Fig. 2f). To estimate the semiconductor characteristics, the charge transfer integrals (t) of the highest occupied molecular orbitals (HOMOs) between adjacent molecules were calculated using the Amsterdam Density Functional (ADF) program for 1 and 4 and are summarized in Table 1.14 The largest transfer integrals in each packing structure calculated for the π-stacking dimer (t1) are 70.1 and 29.4 meV for 1 and 4, respectively. Other calculated t values are comparatively small for both 1 (1.6–7.5 meV) and 4 (2.2–5.1 meV), illustrating quasi-1D conduction channels. Despite the slightly longer π–π distance, the t1 value of 1 is larger than that of 4, possibly because of the introduction of heavier phosphorus atoms.
P double bond lengths of 1.60–1.70 Å13 and comparable to those of reported λ3-phosphinine derivatives.5a,12b These results strongly suggest the high extent of aromaticity of the λ3-phosphinine moiety in 1. Notably, 1 shows a 1D-columnar structure with a π–π distance of 3.53 Å in the crystal packing (Fig. 2e). In contrast, 4 forms a typical herringbone structure with a π–π distance of 3.52 Å (Fig. 2f). To estimate the semiconductor characteristics, the charge transfer integrals (t) of the highest occupied molecular orbitals (HOMOs) between adjacent molecules were calculated using the Amsterdam Density Functional (ADF) program for 1 and 4 and are summarized in Table 1.14 The largest transfer integrals in each packing structure calculated for the π-stacking dimer (t1) are 70.1 and 29.4 meV for 1 and 4, respectively. Other calculated t values are comparatively small for both 1 (1.6–7.5 meV) and 4 (2.2–5.1 meV), illustrating quasi-1D conduction channels. Despite the slightly longer π–π distance, the t1 value of 1 is larger than that of 4, possibly because of the introduction of heavier phosphorus atoms.
 | ||
| Fig. 2 X-Ray crystal structures of (a) 1 and (b) 4 with thermal ellipsoids at a probability level of 50%. Bond lengths [Å] of central rings for (c) 1 and (d) 4. Packing structures of (e) 1 and (f) 4 viewed along the molecular long axis. | ||
| Compounds | t 1 (meV) | t 2 (meV) | t 3 (meV) | t 4 (meV) | t 5 (meV) |
|---|---|---|---|---|---|
| a Calculated at the PBE/TZ2P level of theory. | |||||
| 1 | 70.1 | 5.1 | 7.5 | 1.6 | 2.9 |
| 4 | 29.4 | 2.2 | 5.1 | — | — |
To gain insights into the electronic perturbation of the phosphorus substitution, the UV-Vis absorption and fluorescence spectra in a chloroform solution were measured for 1 and 4 (Fig. S4, ESI†). The UV-Vis absorption spectral shapes of 1 and 4 are similar, with the strongest absorption peaks around 300 nm and weak absorption bands at the longest wavelength. The two absorption peaks of 1 at 402 and 316 nm are red-shifted compared to those of 4 at 352 and 297 nm. The fluorescence wavelength of 1 is also red-shifted from 358 to 433 nm. These bathochromic shifts with phosphorus substitution are attributed to the small energy splitting of the π–π* orbitals containing heavier elements.15 Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements for 1 and 4 in an acetonitrile solution were also conducted (Fig. S5, ESI†). While both 1 and 4 exhibit irreversible oxidation peaks at 0.76 and 1.15 V (vs. Fc/Fc+), respectively, only 1 exhibits an irreversible reduction peak at −1.97 V within the potential window of the solvent. This indicates that oxidation and reduction are more likely to occur in 1 than in 4, for the same reason as that for the change in absorption and fluorescence wavelengths.
To elucidate the electronic properties of 1 and 4, we carried out DFT calculations at the B3LYP/6-311G(d,p) level of theory. The molecular orbital distributions and energy levels are shown in Fig. S7 (ESI†). Although 1 and 4 exhibit very similar orbital distributions for the HOMO and the lowest unoccupied molecular orbital (LUMO), the orbital densities at the phosphorus atom in 1 are spatially expanded compared to those of the corresponding carbon atom in 4, increasing the intermolecular orbital overlap. Moreover, the smaller HOMO–LUMO gap of 1 compared to that of 4 is consistent with the results of the CV measurements. TD-DFT calculations at the same level of theory also support the experimental results from the UV-Vis absorption spectra of 1 and 4 (Fig. S8 and Table S1, ESI†). The lowest singlet excited (S1) state of 1 is attributed to the HOMO → LUMO transition, and its oscillator strength is estimated to be small (0.0121), whereas the most intense absorption band at 316 nm is mainly assigned to the HOMO → LUMO+1 transition with a large oscillator strength of 0.9131. The calculated S1 state of 4 is higher in energy than that of 1, which is consistent with the experimental results. The nucleus-independent chemical shift (NICS) values were calculated at the same level of theory (Fig. S9, ESI†). The NICS(1) value of the λ3-phosphinine moiety (A ring) in 1 was highly negative (−9.7 ppm) and almost comparable to that in 4 (−10.9 ppm), indicating distinct aromaticity. The difference between the NICS(1) values of the B and C rings in 1 and 4 was also small.
Time-resolved microwave conductivity (TRMC) measurements were conducted to explore the intrinsic charge carrier mobilities of 1 and 4.16 Photocarriers were generated by excitation at 355 nm. The TRMC profiles for the crystalline powders of 1 and 4, prepared by vacuum sublimation, are shown in Fig. 3. The maximum TRMC signals (ϕΣμmax) are recorded as 1.7 × 10−9 and 1.0 × 10−8 m2 V−1 s−1 for 1 and 4, respectively, indicating a lower charge carrier mobility of 1. This is contrary to the expected values from the calculated transfer integrals. To understand the reason for the low charge carrier mobility of 1, the calculation of the reorganization energy (λ+) for hole transport (Fig. S10 and Table S2, ESI†), which is another factor determining the charge carrier mobility according to the Marcus theory, was also conducted.17 However, no significant differences in λ+ were found between 1 (84 meV) and 4 (88 meV).
 | ||
| Fig. 3 Photoconductivity transients recorded in crystalline powders of 1 and 4 upon excitation at 355 nm at 9.1 × 1015 photons cm−2 under air. | ||
Finally, bottom-gate bottom-contact OFET devices were fabricated from a chloroform solution using the drop casting method. Polarized optical microscopy (POM) images for 1 showed the aggregation of ribbon-like crystals, whereas those for 4 showed a polycrystalline thin film (Fig. S11 and S12, ESI†). The out-of-plane X-ray diffraction (XRD) profile of 1 showed two peaks at 2θ = 6.58 and 7.40°, which are close to the (100) and (002) diffractions, respectively, derived from the single-crystal structure (Fig. S13, ESI†). This result suggests a mixed molecular orientation in thin films, with the short and long axes of the molecule perpendicular to the substrate (Fig. S14, ESI†). On the other hand, the XRD profile of 4 exhibited a peak at 2θ = 6.66°, which corresponds to the (200) diffraction, indicating a dominant orientation with the molecular long axis direction perpendicular to the substrate (Fig. S15 and S16, ESI†). The obtained OFETs based on 1 and 4 exhibited a typical p-type behavior. The transfer and output characteristics are shown in Fig. 4 and Fig. S17 (ESI†). The estimated hole mobility (μh) of 1 is 3.2 × 10−6 cm2 V−1 s−1 with a threshold voltage (Vth) of −51 V and an on/off current ratio (Ion/Ioff) of 103. On the other hand, the μh, Vth, and Ion/Ioff of 4 are 4.7 × 10−4 cm2 V−1 s−1, −32 V, and 103, respectively. Although the inferior performance of 1 compared to that of 4 can be mainly attributed to the poor film morphology observed in the POM images (Fig. S11, ESI†), it is consistent with the results of the TRMC measurements.
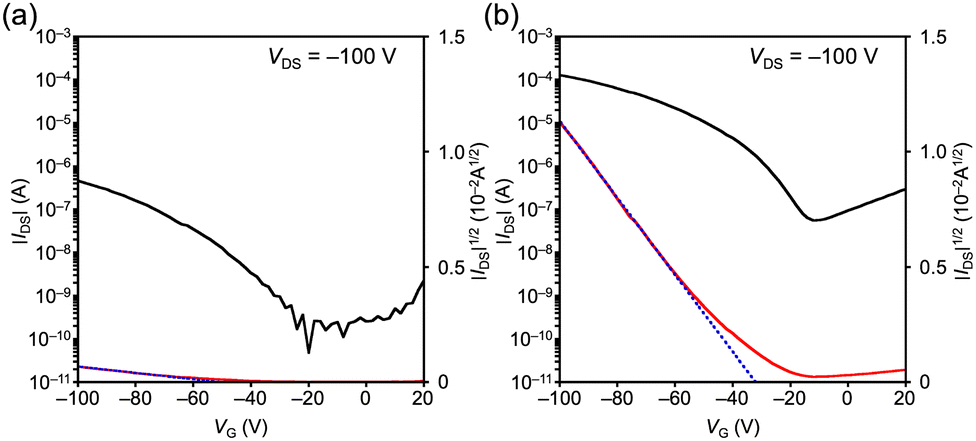 | ||
| Fig. 4 Transfer characteristics of the OFETs based on (a) 1 and (b) 4. | ||
In summary, we synthesized benzo[b]thiophene-annelated λ3-phosphinine 1 to investigate the potential of λ3-phosphinine as a building block for OFET materials. 1 was found to be stable without steric shielding around the CP double bond owing to the high extent of local aromaticity of the λ3-phosphinine moiety, leading to a well-ordered columnar structure in the crystal packing. The large valence orbital of the phosphorus atom was expected to enhance the charge carrier mobility from quantum chemical calculations; however, the actual hole transport properties were inferior to those of the analogue in which a phosphorus atom was replaced by a carbon atom in both the TRMC measurements and OFET characteristics. To the best of our knowledge, this is the first demonstration of the application of low-coordinate phosphorus compounds as organic electronic materials. Although the reason for the intrinsically low carrier mobility is not yet clear and its performance is far inferior to that of reported thienoacenes, the higher electron acceptability of λ3-phosphinine compared to that of thiophene could help address one of the most important current issues: the development of high-mobility n-type organic semiconductors.
This work was partly supported by JSPS KAKENHI grant no. JP20K15261 (KM), JP22K05225 (KM), JP20H02816 (HH), JP20H02711 (NA), JP20H00379 (HY), JP20H05833 (HY), JP20H05841 (YI), and JP20H05836 (AS). We thank Shohei Katao (NAIST) for X-ray crystallographic analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347 CrossRef; (b) W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113 RSC; (c) M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036 CrossRef PubMed.
- J. P. Desclaux, At. Data Nucl. Data Tables, 1973, 12, 311 CrossRef.
- (a) A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876 CrossRef PubMed; (b) D. Joly, P.-A. Bouit and M. Hissler, J. Mater. Chem. C, 2016, 4, 3686 RSC; (c) T. Chatterjee and K.-T. Wong, Adv. Opt. Mater., 2019, 7, 1800565 CrossRef; (d) F. Gao, R. Du, F. Jiao, G. Lu, J. Zhang, C. Han and H. Xu, Adv. Opt. Mater., 2020, 8, 2000052 CrossRef; (e) K. Duan, D. Wang, M. Yang, Z. Liu, C. Wang, T. Tsuboi, C. Deng and Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 30591 CrossRef CAS; (f) J. Bian, S. Chen, L. Qiu, R. Tian, Y. Man, Y. Wang, S. Chen, J. Zhang, C. Duan, C. Han and H. Xu, Adv. Mater., 2022, 34, 2110547 CrossRef CAS PubMed.
- (a) S. Ito, Y. Ueta, T. T. T. Ngo, M. Kobayashi, D. Hashizume, J. Nishida, Y. Yamashita and K. Mikami, J. Am. Chem. Soc., 2013, 135, 17610 CrossRef CAS; (b) Y. Ueta, K. Mikami and S. Ito, Angew. Chem., Int. Ed., 2016, 55, 7525 CrossRef CAS PubMed; (c) S. Ito, Y. Torihata and K. Mikami, ChemistrySelect, 2016, 1, 3310 CrossRef CAS; (d) M. J. Sung, Y. Kim, S. B. Lee, G. B. Lee, T. K. An, H. Cha, S. H. Kim, C. E. Park and Y.-H. Kim, Dyes Pigm., 2016, 125, 316 CrossRef CAS.
- (a) C. Müller and D. Vogt, Dalton Trans., 2007, 5505 RSC; (b) C. Müller and D. Vogt, C. R. Chim., 2010, 13, 1127 CrossRef; (c) C. Müller, L. E. E. Broeckx, I. De Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 187 CrossRef; (d) N. T. Coles, A. S. Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 432, 213729 CrossRef.
- (a) C. Müller, D. Wasserberg, J. J. M. Weemers, E. A. Pidko, S. Hoffmann, M. Lutz, A. L. Spek, S. C. J. Meskers, R. A. J. Janssen, R. A. van Santen and D. Vogt, Chem. – Eur. J., 2007, 13, 4548 CrossRef; (b) N. Nagahora, T. Ogawa, M. Honda, M. Fujii, H. Tokumaru, T. Sasamori, K. Shioji and K. Okuma, Chem. Lett., 2015, 44, 706 CrossRef CAS; (c) N. Nagahora, S. Goto, T. Inatomi, H. Tokumaru, K. Matsubara, K. Shioji and K. Okuma, J. Org. Chem., 2018, 83, 6373 CrossRef CAS; (d) T. Beránek, M. Jakubec, J. Sýkora, I. Císařová, J. Žádný and J. Storch, Org. Lett., 2022, 24, 4756 CrossRef.
- (a) G. Long, X. Yang, W. Chen, M. Zhang, Y. Zhao, Y. Chen and Q. Zhang, Phys. Chem. Chem. Phys., 2016, 18, 3173 RSC; (b) X.-D. Tang, Chem. Phys. Lett., 2017, 684, 402 CrossRef.
- (a) P. de Koe and F. Bickelhaupt, Angew. Chem., Int. Ed. Engl., 1967, 6, 567 CrossRef; (b) P. de Koe, R. van Veen and F. Bickelhaupt, Angew. Chem., Int. Ed. Engl., 1968, 7, 465 CrossRef CAS; (c) P. de Koe and F. Bickelhaupt, Angew. Chem., Int. Ed. Engl., 1967, 7, 889 CrossRef; (d) F. Nief, C. Charrier, F. Mathey and M. Simalty, Tetrahedron Lett., 1980, 21, 1441 CrossRef CAS; (e) P. de Koe and F. Bickelhaupt, Z. Naturforsch. B, 2003, 58, 782 CrossRef CAS; (f) S. Ito, K. Koshino and K. Mikami, Chem. – Asian J., 2018, 13, 830 CrossRef CAS.
- R. E. Messersmith, S. Yadav, M. A. Siegler, H. Ottosson and J. D. Tovar, J. Org. Chem., 2017, 82, 13440 CrossRef CAS.
- (a) M. Ahmed, J. Ashby and O. Meth-Cohn, J. Chem. Soc., Chem. Commun., 1970, 1094 RSC; (b) T. Yamato, N. Sakaue, N. Shinoda and K. Matsuo, J. Chem. Soc., Perkin Trans. 1, 1997, 113 Search PubMed.
- M. Doux, L. Ricard, F. Mathey, P. Le Floch and N. Mézailles, Eur. J. Inorg. Chem., 2003, 687 CrossRef CAS.
- (a) B. Breit, R. Winde, T. Mackewitz, R. Paciello and K. Harms, Chem. – Eur. J., 2001, 7, 3106 CrossRef; (b) L. E. E. Broeckx, S. Güven, F. J. L. Heutz, M. Lutz, D. Vogt and C. Müller, Chem. – Eur. J., 2013, 19, 13087 CrossRef PubMed.
- F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578 CrossRef PubMed.
- ADF2020, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com.
- G. Raabe and J. Michl, Chem. Rev., 1985, 85, 419 CrossRef.
- (a) A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193 CrossRef CAS PubMed; (b) A. Saeki, Polym. J., 2020, 52, 1307 CrossRef CAS PubMed.
- (a) G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 2339 CrossRef CAS PubMed; (b) L. Wang, G. Nan, X. Yang, Q. Peng, Q. Li and Z. Shuai, Chem. Soc. Rev., 2010, 39, 423 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, thermogravimetric analysis, X-ray crystallographic data, photophysical data, electrochemical data, quantum chemical calculations, characterization of thin films, and NMR and HR-MS spectra. CCDC 2194939 and 2194940. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05122b |
| This journal is © The Royal Society of Chemistry 2022 |