
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of iminosugars from aminopolyols via selective oxidation using galactose oxidase†
Kathryn
Yeow
 a,
Marianne B.
Haarr
*a,
Jimmy
Muldoon
b and
Elaine
O'Reilly
*a
a,
Marianne B.
Haarr
*a,
Jimmy
Muldoon
b and
Elaine
O'Reilly
*a
aSchool of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: elaine.oreilly@ucd.ie; marianne.haarr@ucd.ie
bMass Spectrometry Facility, School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland
First published on 7th November 2022
Abstract
Minimally protected aminopolyols are novel substrates for the galactose oxidase variant F2. Site-selective oxidation proceeds at the terminal primary alcohol, followed by spontaneous cyclisation to afford stable hemiaminal/hemiacetal anomers of the piperidine and azepane scaffolds, with isolated yields of up to 94%. Simultaneous deprotection and reduction occured readily to afford valuable and biologically relevant iminosugars.
Iminosugars are polyhydroxylated alkaloids and sugar mimics, containing a nitrogen atom in place of the endocyclic oxygen (Scheme 1). These naturally occurring compounds are of pharmaceutical importance because they interact with and inhibit carbohydrate processing enzymes. Known mechanisms of action include mimicking the oxocarbenium-ion transition state of glycoside hydrolysis,1 chaperoning misfolded proteins,2 and allosteric chaperoning.3 Therapeutic applications are wide-ranging and include the treatment of type II diabetes (miglitol),4 Gaucher's disease (miglustat)5 and viral infections.6,7 Whilst the drug-like properties of iminosugars are advantageous, such as chemical and metabolic stability, and good oral bioavailability, first-generation candidates suffer from lack of selectivity, potency and give rise to undesirable side effects.8,9 The absence of efficient syntheses for preparing structurally diverse derivatives has been highlighted as a limitation to the advancement of second-generation iminosugars as therapeutics.8 A robust biocatalytic route for the preparation of iminosugar compounds has not yet been developed and has the potential to greatly simplify and streamline their synthesis.
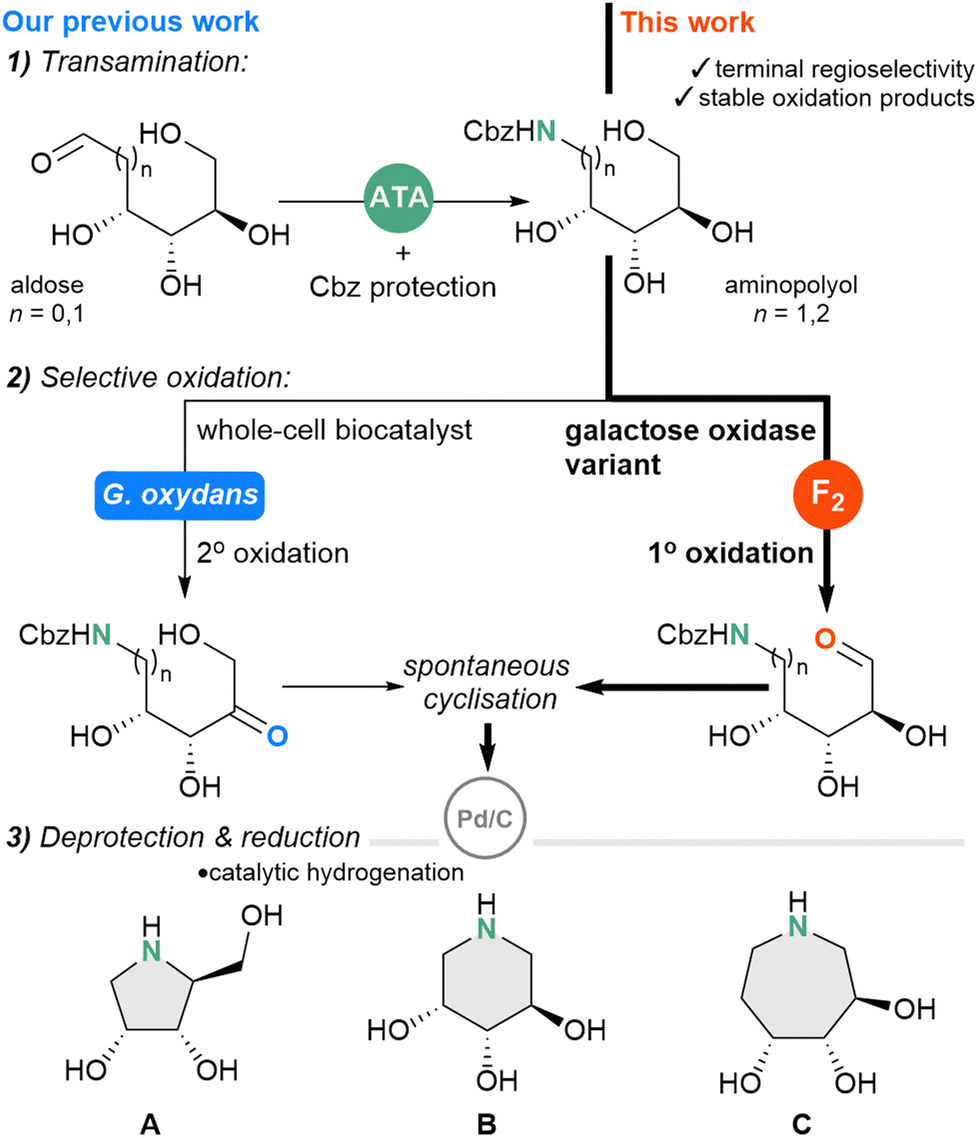 | ||
| Scheme 1 Biocatalytic routes to pyrrolidine-(A), piperidine-(B) and azepane-(C) type iminosugars from monosaccharides. Previous work includes transamination12 and G. oxydans mediated secondary oxidation20 ATA = amine transaminase, F2 = galactose oxidase variant F2. | ||
Through a biocatalytic retrosynthetic analysis, we propose a sequential, three-step chemo-enzymatic cascade, whereby monosaccharides undergo transamination, selective oxidation, and reduction, via transaminase (TA), oxidoreductase and catalytic hydrogenation steps, respectively (Scheme 1).
In support of this biomimetic strategy, a biosynthetic gene cluster that initiates 1-deoxynorijirimycin synthesis has been reported and encodes for a TA, alcohol dehydrogenase, and phosphorylase; transforming fructose-6-phosphate into an iminosugar scaffold.10,11 The prospect of accessing aminopolyols from readily available monosaccharides is intriguing, as naturally occurring D-sugars are a major component of plant biomass and considered waste products from industry and agriculture, therefore utilising this feedstock represents a useful valorisation approach. Recently, we reported that TAs can catalyse the direct amination of aldoses to aminopolyols (Scheme 1);12 a study revealing activity towards ketoses was reported shortly after.13
With the first key transaminase step established to afford the aminopolyols from aldoses, we then sought a biocatalytic strategy for the selective oxidation, which must proceed regioselectively at the C5– or C6–OH positions to enable cyclisation. Synthetic approaches for regioselective oxidation of polyol species include the use of transition-state metal (Ru, Pd)14,15 and photoredox catalysis,16 and has even been applied to unprotected carbohydrates, yet scope remains limited to cyclic substrates. The challenge of achieving regioselective oxidation persists for linear polyol species, for which there are fewer variations in the electronic and steric environments between hydroxyl groups. Building on early reports,17–19 we recently employed Gluconobacter oxydans as a whole-cell biocatalyst, which displayed regioselectivity towards the secondary alcohol positions (Scheme 1).20 Since the enzyme(s) responsible for the oxidation was uncharacterised, we next wished to identify specific oxidoreductases that could add to our toolkit of selective aminopolyol oxidation biocatalysts. We reasoned that it would also be valuable to find an enzyme to carry out the primary aminopolyol oxidation to complement the secondary oxidation mediated by G. oxydans, allowing more piperidine-type iminosugars to be derived from the pentose monosaccharides.
Our attention turned towards oxidase enzymes of which, many carbohydrate oxidases have been characterised in literature.21
In comparison to the nicotinamide coenzyme requirements of dehydrogenases, oxidases use molecular O2 as the mild oxidant for the regeneration of the reduced enzyme. Galactose oxidase (GOase) from Fusarium graminearum is a well characterised copper-dependent oxidase, and the wild-type enzyme regioselectively oxidises the primary C6–OH of D-galactopyranose. In comparison, other carbohydrate oxidases tend to favour secondary C2–OH oxidation.22 GOase has been the subject of several engineering projects, delivering variants with outcomes such as increased expression and stability in Escherichia coli (E. coli) (M1 variant),23 activity towards non-natural substrates, such as glucose (M3 variant)24 and fructose (R330K variant)25 as well as activity towards aromatic secondary alcohols (M3–5 variant)26 and glycans on glycoproteins (F2 variant) (ESI,† Fig. S2).27 Following these advances, optimising standard operating conditions for GOase has been pursued in a step towards realising its industrial relevance.28 Recently, GOase F2 was evolved further for the desymmetrizing oxidation step in the enzymatic cascade for the synthesis of islatravir.29 For this project, the F2 variant was particularly attractive for its ability to catalyse the terminal selective oxidation of glucosides, showing that activity towards a polyhydroxylated scaffold is retained.30 If the site-selective oxidation of aminopolyols can be performed with the same regioselectivity, the resulting aminoaldehyde products will cyclise to afford the valuable piperidine and azepane ring systems (see Scheme 2) and therefore, the F2 variant was the biocatalyst candidate selected for this study.
 | ||
| Scheme 2 (A) Chemoenzymatic synthesis of piperidine- and azepane-type iminosugars. (B) Substrate scope of the oxidation products and isolated yields, products 2a–e exist predominantly in hemiaminal form, whereas 2f is in hemiacetal form. (C) Iminosugars 3a–f are isolated as the HCl salt. | ||
In view of developing a biocatalytic cascade, it was important that selected sugar starting materials are substrates for the initial transamination. The panel of substrates 1a–f (Scheme 2, ESI† Fig. S1) were derived from D-aldoses arabinose, lyxose, ribose, 2-deoxy-ribose, mannose, and 2-deoxy-galactose and reflect the rich stereochemical landscape that can be accessed through sugar precursors. Aminopolyols were N-Cbz protected to enable isolation and characterisation of the oxidation products.
The enzyme GOase F2 was recombinantly expressed in E. coli BL21(DE3) according to modified protocols31 and purified to homogeneity by streptavidin affinity chromatography, with modest yields of approximately 12 mg per litre of culture. In the initial set of experiments, we screened 1a–f as substrates for the F2-catalysed oxidation on an analytical scale (1 mL) (see ESI,† Section 3). Having established suitable conditions for reaction screening, we opted to monitor biotransformations directly using thin layer chromatography (TLC). TLC analysis indicated conversion of starting material for all the tested substrates after an overnight reaction (see ESI,† Fig. S4). LC/MS analysis confirmed the formation of a mono-oxidation product and there was no evidence of side product formation, such as overoxidation to the carboxylic acid or the geminal diol species that has been previously reported.30
Next, we sought to isolate enough material for full characterisation, as up until this point, neither the site of oxidation nor the form of the oxidised products had been determined. Furthermore, the compounds are not readily accessible commercially or via chemical synthesis, and calibration standards for quantifying reaction conversion were required. For these reasons, biotransformations were scaled up to 10 mL, with substrate loading at 20 mM (0.2 mmol),‡ relatively high compared to what has typically been reported for GOase-catalysed reactions. From the analytical-scale reactions it was evident that the GOase F2 variant precipitates readily from the reaction mixture, and consequently, over time activity likely would diminish. To circumvent a drop off in the reaction progression and to drive the reactions towards completion, we opted to cumulatively add enzyme over the course of the reaction. The progress of the reaction and timeline of sequential additions was informed by TLC analysis. Through these efforts, oxidation products 2a–f were isolated in 48–94% yield, corresponding to up to 53 mg of the D-ribo2c and 2-deoxy-D-galacto2e compounds. It was not possible to achieve complete conversion with all biotransformations within a reasonable time frame, meaning D-lyxo1b and D-manno1f compounds were isolated in yields of approximately 50%. Despite long reaction times, we observed no side product formation, and that the oxidation products were remarkably stable.
Characterisation of the compounds by NMR spectroscopy was initially challenging due to the presence of anomeric mixtures, but analysis of 2D NMR spectra enabled resolution between configurational anomers. The spontaneous cyclisation was supported by the appearance of the characteristic cyclic hemiaminal or hemiacetal peak in the range δ 5.1–5.8 ppm. From this, the ratio of hemiaminal anomers was determined, with product ratios differing across the substrate panel. The absolute stereochemistry of each anomer was not ascertained in this study, with the view that upon deprotection and imine reduction, the anomers converge to a single product.
To gain a better understanding of the reaction progress, we determined the reaction conversion after a single addition of enzyme and monitored the reaction progress over time on analytical scale (Fig. 1, Table 1). After 8 h, conversion of substrates 1b–f to oxidised species 2b–f ranged from 33–88%. The initially slow reaction progress of entry 1 can be attributed to the poor solubility of the aminopolyol 1a in buffer at ambient temperature. Despite protein precipitation in the reaction mixture, enzymatic activity was still evident after 24 hours. The results show that aminopolyols 1a–f are good substrates for F2, reaching up to 97% conversion on analytical scale.
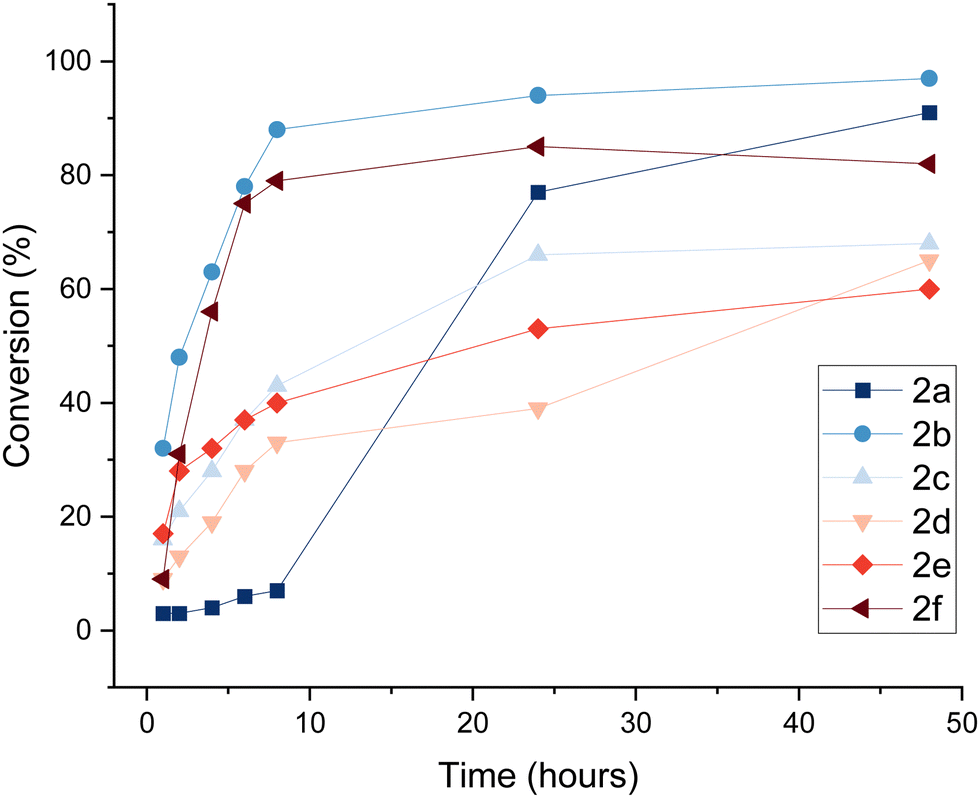 | ||
| Fig. 1 Time course analysis of the GOase F2-catalysed oxidation of N-Cbz-protected aminopolyols. Conversion to products 2a–f was determined by LC/MS analysis, monitoring product formation at timepoints up to 48 hours, and plotted as the average of triplicate reactions (1 mL) (for individual plots with error bars, see ESI,† Section 4). | ||
| Substrate | Absolute configuration | Producta | Conversion [%] | |||
|---|---|---|---|---|---|---|
| 8 h | 24 h | 48 h | ||||
| Reaction conditions: substrate (0.2 mmol, 20 mM), catalase (0.1 mg mL−1), HRP Type I (0.1 mg mL−1), CuSO4 (50 μM), NaPi (100 mM, pH 8), purified GOase F2 (2 mg mL−1) at 20 °C.a A mixture of cyclic hemiaminal and hemiacetal anomers are obtained. | ||||||
| 1 | 1a | D-arabino | 2a | 7 | 77 | 91 |
| 2 | 1b | D-lyxo | 2b | 88 | 94 | 97 |
| 3 | 1c | D-ribo | 2c | 43 | 66 | 68 |
| 4 | 1d | 2-deoxy-D-ribo | 2d | 33 | 39 | 65 |
| 5 | 1e | 2-deoxy-D-galacto | 2e | 40 | 53 | 60 |
| 6 | 1f | D-manno | 2f | 79 | 85 | 82 |
Finally, iminosugars 3a–f were readily obtained from the oxidation products 2a–f by hydrogenation using Pd/C, in which the deprotection and imine reduction steps occur concomitantly (Scheme 2). After 16 h reaction time we observed complete conversion to the desired products 3a–f, regardless of the anomeric hemiaminal/hemiacetal distribution of the precursors 2a–f.
We have demonstrated that minimally protected aminopolyols, key intermediates for iminosugar synthesis, are novel substrates for the galactose oxidase variant F2. This report represents further progress from our previously reported chemoenzymatic route towards monosaccharide-derived iminosugars.20 Having identified the F2 variant as a suitable oxidase, protein engineering and optimisation studies can now follow to extend the substrate scope, efficiency, stability, and expression of this catalyst. The necessity for sequential addition of enzyme, due to poor stability, enabled enough material to be isolated for characterisation and further analysis, but could potentially be avoided by using immobilised enzyme. Biocatalytic and chemoenzymatic cascades offer enormous advantages over more traditional stepwise synthetic approaches to complex molecules and are being considered as viable industrial approaches.32 Future efforts will focus on eliminating the need for the N-Cbz protecting group on the aminopolyol and identifying and introducing an appropriate imine reductase (IRED) in the last step of the cascade to enable a one-pot fully enzymatic cascade from monosaccharides to iminosugars.
We acknowledge financial support from the Science Foundation Ireland (SFI) Frontiers for the Future Project (19/FFP/6469), SFI grants supporting NMR (12/RI/2341) and mass spectra (18/RI/5702) facilities and equipment and facilities funded by HEA PRTLI Cycle 3. GOase variant F2 was provided by Prof. Sabine Flitsch and substrates 1a, c, d and f were prepared by Dr Justyna Kuska. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission’.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- D. L. Zechel, A. B. Boraston, T. Gloster, C. M. Boraston, J. M. Macdonald, D. M. G. Tilbrook, R. V. Stick and G. J. Davies, J. Am. Chem. Soc., 2003, 125, 14313–14323 CrossRef PubMed.
- C. H. Hill, A. H. Viuff, S. J. Spratley, S. Salamone, S. H. Christensen, R. J. Read, N. W. Moriarty, H. H. Jensen and J. E. Deane, Chem. Sci., 2015, 6, 3075–3086 RSC.
- D. D’Alonzo, M. De Fenza, C. Porto, R. Iacono, M. Huebecker, B. Cobucci-Ponzano, D. A. Priestman, F. Platt, G. Parenti, M. Moracci, G. Palumbo and A. Guaragna, J. Med. Chem., 2017, 60, 9462–9469 CrossRef.
- L. J. Scott and C. M. Spencer, Drugs, 2000, 59, 521–549 CrossRef CAS.
- C. E. M. Hollak, D. Hughes, I. N. Van Schaik, B. Schwierin and B. Bembi, Pharmacoepidemiol. Drug Saf., 2009, 18, 770–777 CrossRef CAS PubMed.
- D. S. Alonzi, K. A. Scott, R. A. Dwek and N. Zitzmann, Biochem. Soc. Trans., 2017, 45, 571–582 CrossRef PubMed.
- K. Prichard, D. Campkin, N. O'Brien, A. Kato, G. W. Fleet and M. I. Simone, Chem. Biol. Drug Des., 2018, 92, 1171–1197 CrossRef.
- G. Horne, F. X. Wilson, J. Tinsley, D. H. Williams and R. Storer, Drug Discovery Today, 2011, 16, 107–118 CrossRef PubMed.
- R. J. Nash, A. Kato, C. Y. Yu and G. W. Fleet, Future Med. Chem., 2011, 3, 1513–1521 CrossRef PubMed.
- L. F. Clark, J. V. Johnson and N. A. Horenstein, ChemBioChem, 2011, 12, 2147–2150 CrossRef.
- K. D. Kang, Y. S. Cho, J. H. Song, Y. S. Park, J. Y. Lee, K. Y. Hwang, S. K. Rhee, J. H. Chung, O. Kwon and S. Il Seong, J. Microbiol., 2011, 49, 431–440 CrossRef.
- R. Cairns, A. Gomm, J. Ryan, T. Clarke, E. Kulcinskaja, K. Butler and E. O’Reilly, ACS Catal., 2019, 9, 1220–1223 CrossRef.
- F. Subrizi, L. Benhamou, J. M. Ward, T. D. Sheppard and H. C. Hailes, Angew. Chem., Int. Ed., 2019, 58, 3854–3858 CrossRef PubMed.
- M. Jäger, M. Hartmann, J. G. De Vries and A. J. Minnaard, Angew. Chem., Int. Ed., 2013, 52, 7809–7812 CrossRef.
- C. K. Hill and J. F. Hartwig, Nat. Chem., 2017, 9, 1213–1221 CrossRef CAS.
- D. J. Gorelik, V. Dimakos, T. Adrianov and M. S. Taylor, Chem. Commun., 2021, 57, 12135–12138 RSC.
- M. Schedel, in Biotechnology: Biotransformations II, ed. D. R. Kelly, WILEY-VCH Verlag GmbH; Weinheim, 2nd edn, 2000, vol. 8b, ch. 7, pp 295–311 Search PubMed.
- M. K. Sethi, A. Kumar, N. Maddur, R. Shukla and L. N. Vemula, J. Mol. Catal. B: Enzym., 2015, 112, 54–58 CrossRef.
- N. G. Oikonomakos, C. Tiraidis, D. D. Leonidas, S. E. Zographos, M. Kristiansen, C. U. Jessen, L. Nørskov-Lauritsen and L. Agius, J. Med. Chem., 2006, 49, 5687–5701 CrossRef PubMed.
- J. Kuska, F. Taday, K. Yeow, J. Ryan and E. O’Reilly, Catal. Sci. Technol., 2021, 11, 4327–4331 RSC.
- S. Savino and M. W. Fraaije, Biotechnol. Adv., 2021, 51, 107634 CrossRef CAS.
- P. Rafighi, P. Bollella, G. Pankratova, C. K. Peterbauer, P. Conghaile, D. Leech, B. Haghighi and L. Gorton, ChemElectroChem, 2019, 6, 801–809 CrossRef CAS.
- L. Sun, I. P. Petrounia, M. Yagasaki, G. Bandara and F. H. Arnold, Protein Eng., Des. Sel., 2001, 14, 699–704 CrossRef CAS.
- L. Sun, T. Bulter, M. Alcalde, I. P. Petrounia, F. H. Arnold, F. H. Arnold, L. Sun, T. Bulter and M. Alcalde, ChemBioChem, 2002, 8, 781–783 CrossRef.
- S. E. Deacon, K. Mahmoud, R. K. Spooner, S. J. Firbank, P. F. Knowles, S. E. V. Phillips and M. J. McPherson, ChemBioChem, 2004, 5, 972–979 CrossRef CAS PubMed.
- F. Escalettes and N. J. Turner, ChemBioChem, 2008, 9, 857–860 CrossRef CAS PubMed.
- J. B. Rannes, A. Ioannou, S. C. Willies, G. Grogan, C. Behrens, S. L. Flitsch and N. J. Turner, J. Am. Chem. Soc., 2011, 133, 8436–8439 CrossRef CAS PubMed.
- A. Toftgaard Pedersen, W. R. Birmingham, G. Rehn, S. J. Charnock, N. J. Turner and J. M. Woodley, Org. Process Res. Dev., 2015, 19, 1580–1589 CrossRef CAS.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS.
- S. C. Cosgrove, A. P. Mattey, M. Riese, M. R. Chapman, W. R. Birmingham, A. J. Blacker, N. Kapur, N. J. Turner and S. L. Flitsch, ACS Catal., 2019, 9, 11658–11662 CrossRef CAS.
- J. Vilím, T. Knaus and F. G. Mutti, Angew. Chem., Int. Ed., 2018, 57, 14240–14244 CrossRef PubMed.
- A. R. Alcántara, P. Domínguez de María, J. A. Littlechild, M. Schürmann, R. A. Sheldon and R. Wohlgemuth, ChemSusChem, 2022, 15, e202102709 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed description of each experiment, protocols, characterisation of compounds, standard curves. See DOI: https://doi.org/10.1039/d2cc04989a |
| ‡ The batch prep reaction of D-lyxo substrate is carried out on 5 mL scale. |
| This journal is © The Royal Society of Chemistry 2022 |