
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isostructural σ-hydrocarbyl phospholide complexes of uranium, neptunium, and plutonium†‡
Michaela
Černá
a,
John A.
Seed
 a,
Sara
Garrido Fernandez
a,
Michael T.
Janicke
b,
Brian L.
Scott
c,
George F. S.
Whitehead
a,
Andrew J.
Gaunt
b and
Conrad A. P.
Goodwin
*ab
a,
Sara
Garrido Fernandez
a,
Michael T.
Janicke
b,
Brian L.
Scott
c,
George F. S.
Whitehead
a,
Andrew J.
Gaunt
b and
Conrad A. P.
Goodwin
*ab
aCentre for Radiochemistry Research, Department of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: conrad.goodwin@manchester.ac.uk
bChemistry Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
cMaterials Physics & Applications Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
First published on 28th October 2022
Abstract
σ-Hydrocarbyl complexes of the form [M(η5-PC4Me4)2(μ-η1:η6-CH2Ph)2K(η6-arene)] (M = La, Ce, Pr, U, Np, Pu; arene = benzene or toluene) were synthesised in one-pot reactions from [MI3(THF)4], or [U(BH4)3(toluene)] (M = U). All complexes were examined by multinuclear (1H, 13C{1H}, 31P{1H}) NMR and UV-vis-NIR spectroscopy, as well as single-crystal X-ray diffraction from which molecular metal–phosphorus bonds for Np and Pu, and a σ-hydrocarbyl metal–carbon bond for Pu, have been structurally authenticated.
σ-Hydrocarbyl complexes are an important class of molecule for their numerous roles in both catalysis and fundamental bond formation reactions across the periodic table. While the close energetic matching and spatial overlap of metal and carbon orbitals affords facile redox chemistry in noble metal catalytic processes, poor matching in rare-earth and actinide hydrocarbyl complexes results in highly polar linkages amenable to protonolysis and bond-metathesis chemistry.1
Protonolysis reactions using basic hydrocarbyl groups in UIV and ThIV molecules have proven invaluable for the isolation of diverse metal-element linkages such as imido-, phosphido-, phosphinidene-, alkylidene-, hydrides, and others.2 Thus, their utility in facilitating the comparison of AnIV (An = Th, U) covalency through the examination of metal-element multiple bonds is patent. Hydrocarbyls also serve as precursors, via hydrogenolysis with H2,3 or with silanes,4 to molecular metal-hydrides for which no transuranium examples have been reported. Attempts to extend tetravalent metal-hydrocarbyl chemistry to the next actinide element, Np, have proven non-trivial, in part because reduction of NpIV precursors to NpIII is facile due to the readily accessible NpIV/III redox couple (ca. +0.147 to +0.218 V vs. NHE).5 This complication is likely to extend to many of the transuranium elements for which the trivalent oxidation state is the most prevalent. Conversely, the increasing accessibility of AnIII with higher Z elements can enable direct comparison to LnIII bonding, and hence insight into the comparative role of 4f or 5f electrons in bonding. Trivalent σ-hydrocarbyl complexes, which can be isolated using the small (milligram) reaction scales commonly employed for transuranium research, can help in providing access to new 4f vs. 5f bonding comparisons.
Here we show that a per-alkylated phospholide, TMP (TMP = {PC4Me4}) allows the one-pot synthesis and isolation of trivalent dibenzyl-ate complexes of the form [M(TMP)2{(μ:η1:η6-C7H7)2K(η6-arene)}] (arene = toluene or benzene) of U, Np, and Pu, along with isoradial lanthanide congeners La, Ce, and Pr. The Np and Pu complexes herein represent unique examples of M–P bonds for these elements, and complexation with a carbocyclic ligand other than cyclopentadienide or cyclooctatetraenide. The Pu complex is a rare example of a structurally characterised Pu σ-hydrocarbyl.
Previous reports have shown that salt occlusion and oligomerisation reactions plague trivalent rare earth and f-block phospholide chemistry.6 The facile isolation of [UIII(TMP)(μ-η5:η1-TMP)(BH4)]2 (TMP = {PC4Me4}) by reduction of [UIV(TMP)2(BH4)2] suggested that a suitable tetravalent precursor might allow, through a subsequent reduction step, access to discrete heteroleptic NpIII molecules with which to synthesise σ-hydrocarbyl complexes.7 As [UIV(TMP)3(Cl)] is known,8 and [NpIVCl4(DME)2] is a readily synthesised precursor,9 it seemed reasonable to target [NpIV(TMP)3(Cl)] first to help identify differences in the chemistries of Np/U using a model system. The reaction between [NpIVCl4(DME)2] and 3 equivalents of KTMP in Et2O or THF led to the isolation of several crystals of the NpIII bis-TMP complexes 1 and 2 respectively (Scheme 1). On one occasion, the use of a trivalent precursor, ‘NpIIICl3(THF)n’ prepared in situ,10 afforded several crystals of dinuclear complex, 3. Complexes 1 and 3 could not be separated from impurities such as [K(THF)2(η5-TMP)]n (4) – see ESI‡ for structural details of 1–4, and UV-vis-NIR and multi-nuclear NMR spectroscopy of 2.
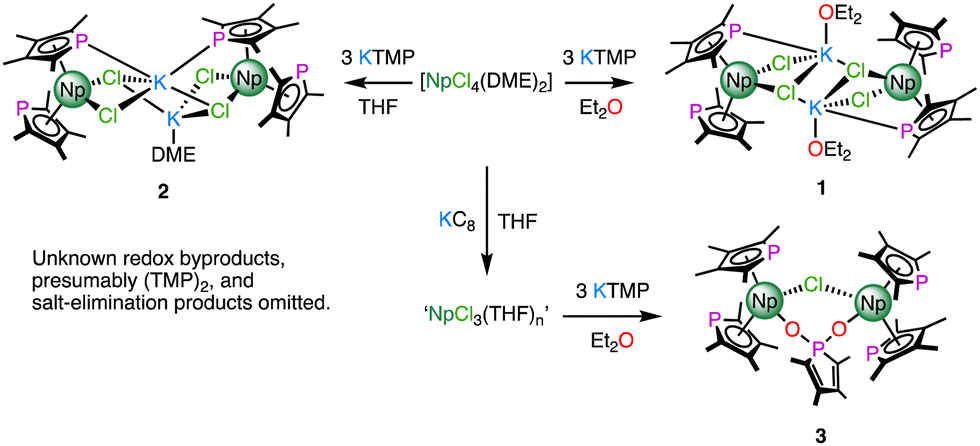 | ||
| Scheme 1 Syntheses of complexes 1–3. | ||
Next, we turned to [MI3(THF)4] (M = La, Ce, Pr, U, Np, Pu) as well-defined trivalent precursors. The micromole-scale (35 mg) reaction in THF between [UI3(THF)4] (prepared in situ from UI3(OEt2)x) and 2 equivalents of KTMP gave a dark green reaction mixture, a small portion of which gave several crystals of [U(TMP)2(I)(THF)] (5). Addition of 2 equivalents of KBn (Bn = benzyl) to the remaining THF solution followed by workup (see ESI‡) gave several crystals of [U(TMP)2(μ:η1:η6-C7H7)2K(η6-C6H6)] (7U).
Separate reactions using [MI3(THF)4] with 2 equivalents of KTMP and 2 equivalents of KBn in THF at −98 °C or room temperature, followed by extraction and crystallisation from boiling toluene gave the isomorphous series [M(TMP)2(μ:η1:η6-C7H7)2K(η6-C7H8)] (6M, M = La, Ce, Pr, Np, Pu; Scheme 2) in low to moderate yield (16%, Pu to 53%, Pr). These metals were chosen based on the similarities of their ionic radii, e.g. UIII and LaIII (1.025 Å vs. 1.032 Å).11 Complex 6U was synthesised on a preparative scale (0.5 mmol) from [U(BH4)3(toluene)]12 in modest yield (29%). Recrystallisation of several mg each of 6U, 6Np, and 6Pu from boiling benzene gave [M(TMP)2(μ:η1:η6-C7H7)2K(η6-C6H6)] (7M), which allowed us to examine the influence of the arene on the structure.
 | ||
| Scheme 2 Synthesis of complexes 6M (R = Me; M = La, Ce, Pr, U, Np, Pu) and 7M (R = H, M = U, Np, Pu). | ||
All 6M complexes crystallised in the non-centrosymmetric space group Pna21 with one molecule in the asymmetric unit, whereas 7M crystallised in the centrosymmetric space group Pnma with half a molecule in the asymmetric unit. The structure of 7Pu is shown in Fig. 1 (see ESI† for others) which features a bis-η5-TMP wedge-shaped arrangement (Pu1–P1 = 2.9533(13) Å, Pu1⋯TMPcent = 2.5931(15) Å; Pu1–P2 = 2.9816(14) Å, Pu1⋯TMPcent = 2.5821(14) Å; TMPcent⋯Pu1⋯TMPcent = 127.56(5)°). In 7Pu both benzyl groups are bound η1 to the Pu-atom (Pu1–C9 = 2.542(19) Å; Pu1–C16 = 2.614(19) Å; C9–Pu1–C16 = 107.1(4)°), these Pu–C lengths are unremarkable when compared to known UIII mono- and bis-benzyl complexes,13 and accounting for the difference in their between 6-coordinate UIII (1.025 Å) and PuIII (1.00 Å).11 The K-atom is bound η6 to the C6 rings from both benzyl groups (Bncent⋯K1 = 2.875(11) and 2.948(10) Å; Bncent⋯K1⋯Bncent = 113.8(3)°) along with an η6-benzene molecule, and a P–K contact (3.3270(18) Å) with one of the TMP rings. Bond lengths and angles for 6M and 7M complexes are summarised in Table 1 (data for 6Pu is not of sufficient quality for discussion). A homoleptic Pu hydrocarbyl complex has been synthesised previously, [Pu{CH(SiMe3)2}3], though structural data is not available for comparison.14
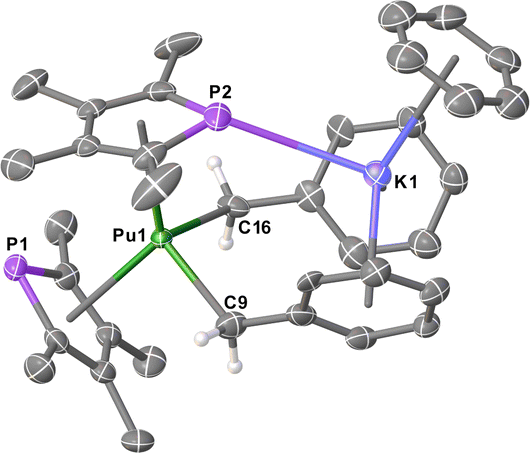 | ||
| Fig. 1 Molecular structure of [Pu(TMP)2Bn2K(benzene)] 7Pu. Ellipsoids set at 50% probability and H-atoms (except on C9 and C16) removed for clarity. | ||
| M–P (Å) | M–CBn (Å) | M⋯TMPcent (Å) | K–P (Å) | TMPcent⋯M⋯TMPcent (°) | |
|---|---|---|---|---|---|
| 6M | |||||
| La | 3.0112(12) | 2.622(8) | 2.650(4) | 3.323(2) | 127.63(12) |
| 3.0407(15) | 2.675(9) | 2.635(4) | |||
| U | 2.9432(11) | 2.577(7) | 2.580(3) | 3.303(2) | 128.33(11) |
| 2.9766(14) | 2.597(7) | 2.564(3) | |||
| Ce | 2.9863(14) | 2.619(8) | 2.623(3) | 3.313(2) | 127.59(11) |
| 3.0197(16) | 2.631(10) | 2.609(4) | |||
| Np | 2.940(2) | 2.519(18) | 2.577(9) | 3.317(4) | 127.8(3) |
| 2.971(3) | 2.573(15) | 2.564(8) | |||
| Pr | 2.9672(9) | 2.592(6) | 2.606(2) | 3.3118(16) | 128.07(8) |
| 3.0034(11) | 2.598(5) | 2.593(2) | |||
| 7M | |||||
| U | 2.9576(11) | 2.556(17) | 2.5915(2) | 3.3275(15) | 127.624(9) |
| 2.9857(11) | 2.654(16) | 2.5816(11) | |||
| Np | 2.9147(11) | 2.479(17) | 2.5547(12) | 3.2805(16) | 127.12(4) |
| 2.9403(12) | 2.603(16) | 2.5484(12) | |||
| Pu | 2.9533(13) | 2.542(19) | 2.5931(15) | 3.3270(18) | 127.56(5) |
| 2.9816(14) | 2.614(19) | 2.5821(14) |
Table 1 shows the arene coordinating to K1 has a small impact on the overall structure. The M–P, K–P, and M⋯TMPcent lengths all shorten somewhat upon switching from toluene to benzene, and the M–CBn bond lengths become less equivalent. For example, the difference between the Np–CBn bonds in 6Np is 0.05(2) Å which is statistically indistinguishable (3σ criteria), but it is 0.12(2) Å for 7Np which shows the two bonds are distinct. This is presumably due to packing differences of the {(η6-Bn)2K(arene)} unit in 6M and 7M. Metrical parameters for the series change broadly as expected with the ionic radii of the metals. Equivalent M–P and M–C bond lengths decrease on average from La to Pr, and from U to Pu. However, within the 7M series, 7Np displays noticeably shorter M–P bonds and M⋯TMPcent distances than 7Pu (though the M–CBn distances overlap slightly within the 3σ criteria). This is not the first time Np complexes have been shown to be outliers.9a As is commonly seen, actinide 6M or 7M have bond lengths that are shorter than corresponding lanthanide complexes by an amount larger than the difference in their ionic radii. For example: the differences between the two unique M–P bond lengths in 6La and 6U are 0.068(16) Å (M–P1) and 0.064(2) Å (M–P2), which is larger than the difference in their radii (0.007 Å). This discrepancy decreases as the series is traversed.9a,15
1H NMR spectroscopy of diamagnetic 6La in C4D8O or C6D6 reveal spectra consistent with C2 symmetry in solution. The TMP 3,4-Me shows a singlet at 1.96 ppm, and the 2,5-Me peak at 2.8 ppm is split (3JHP = 9.6 Hz) due to coupling with 31P (100% I = 1/2). Resonances for the benzyl groups were well resolved as triplets at 6.67 ppm (m) and 6.14 ppm (p), a doublet at 6.40 ppm (o), and a singlet at 1.04 ppm for the benzylic protons. Benzyl CH2 protons were located in the 1H NMR spectra of all paramagnetic 6M complexes (6Ce, 1.22 ppm; 6Pr, 7.48 ppm; 6U, −55.31 ppm; 6Np, −60.30 ppm; 6Pu, −18.75 ppm). In all except 6Pu we could definitively assign all unique CH groups for the benzyl moiety (see ESI‡). The 13C{1H} NMR spectrum of 6La revealed doublets at 15.32 and 16.34 ppm (3JCP = 5.3 and 28.9 Hz) for the TMP methyl groups due to 13C–31P coupling; and the corresponding ring carbons at 135.95 and 142.82 ppm also presented as doublets (3JCP = 2.0 and 41.2 Hz). 13C{1H} NMR spectroscopy was uninformative for all other complexes. The 31P{1H} NMR spectrum of 6La showed a singlet at 98.28 ppm (cf. KTMP 74.32 ppm in C4H8O/C6D6 – see ESI‡), consistent with C2 symmetry in solution. Paramagnetic 6M complexes displayed a broad range of 31P chemical shifts (6Ce, 175.3 ppm; 6Pr, 293.5 ppm; 6U, 738.8 ppm; 6Np, −497.9 ppm). We could not unambiguously locate the TMP 31P peak for 6Pu (see ESI‡).
UV-vis-NIR spectra of 6M (M = La, Ce, Pr) show a broad ligand-to-metal charge-transfer (LMCT) band that tails in from the edge of the spectral range to ca. 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1. This is responsible for the red-orange colour of all of these complexes, and is the only feature for 6La; whereas 6Ce shows a broad feature at ca. 20800 cm−1 (ε = 317 M−1 cm−1) for a symmetry allowed 4f → 5d transition; and 6Pr shows numerous weak 4f → 4f peaks from 6000–20000 cm−1 (ε = 10–52 M−1 cm−1), while a shoulder at 21622 cm−1 (ε = 145 M−1 cm−1) is plausibly due to the 1S0 excitation.16 The spectra of 6Np and 6Pu feature multiple weak peaks between 6000–14000 cm−1 (ε = 16–100 M−1 cm−1) which are characteristic of 5f → 5f transitions. For 6U, this region is broad, mostly featureless, and with modest absorption (ε = 200–550 M−1 cm−1) – see Fig. 2. Comparatively strong features which tail into the visible region at ca. 12000 and 19000 cm−1 for 6U (ε = 550 and 1100 M−1 cm−1 respectively); 16400 cm−1 for 6Np (ε = 300 M−1 cm−1); and ca. 17200 and 22000 cm−1 for 6Pu (ε = 108 and 614 M−1 cm−1 respectively) are responsible for the dark green/brown (6U), apple green (6Np), and golden-brown (6Pu) colours. These excitations are likely too low in energy for 5f → 6d transitions, though have absorptivities somewhat high for typical 5f → 5f transitions. One possible cause for this is an intensity-stealing mechanism by mixing of 5f states with a charge-transfer or intra-ligand transition of different parity which has previously been invoked to explain similar observations.17 It is noteworthy that the transitions between 14000–22000 cm−1 for 6Pu are ca. 5 to 10 times more intense than several other PuIII complexes with reported ε values.9a,18
000 cm−1. This is responsible for the red-orange colour of all of these complexes, and is the only feature for 6La; whereas 6Ce shows a broad feature at ca. 20800 cm−1 (ε = 317 M−1 cm−1) for a symmetry allowed 4f → 5d transition; and 6Pr shows numerous weak 4f → 4f peaks from 6000–20000 cm−1 (ε = 10–52 M−1 cm−1), while a shoulder at 21622 cm−1 (ε = 145 M−1 cm−1) is plausibly due to the 1S0 excitation.16 The spectra of 6Np and 6Pu feature multiple weak peaks between 6000–14000 cm−1 (ε = 16–100 M−1 cm−1) which are characteristic of 5f → 5f transitions. For 6U, this region is broad, mostly featureless, and with modest absorption (ε = 200–550 M−1 cm−1) – see Fig. 2. Comparatively strong features which tail into the visible region at ca. 12000 and 19000 cm−1 for 6U (ε = 550 and 1100 M−1 cm−1 respectively); 16400 cm−1 for 6Np (ε = 300 M−1 cm−1); and ca. 17200 and 22000 cm−1 for 6Pu (ε = 108 and 614 M−1 cm−1 respectively) are responsible for the dark green/brown (6U), apple green (6Np), and golden-brown (6Pu) colours. These excitations are likely too low in energy for 5f → 6d transitions, though have absorptivities somewhat high for typical 5f → 5f transitions. One possible cause for this is an intensity-stealing mechanism by mixing of 5f states with a charge-transfer or intra-ligand transition of different parity which has previously been invoked to explain similar observations.17 It is noteworthy that the transitions between 14000–22000 cm−1 for 6Pu are ca. 5 to 10 times more intense than several other PuIII complexes with reported ε values.9a,18
 | ||
| Fig. 2 UV-vis-NIR spectra of [U(TMP)2Bn2K(toluene)] (6U, black line) in THF (0.97 mM), and [Np(TMP)2Bn2K(toluene)] (6Np, red line) in toluene (0.50 mM), and [Pu(TMP)2Bn2K(toluene)] (6Pu, blue line) in toluene (1.00 mM) between 7000–22000 cm−1 (1429–455 nm) at room temperature. | ||
The UIII assignment of 6U was confirmed by SQUID magnetometry (Fig. 3). A powdered sample exhibits slow reduction in μeff (χT) across the entire temperature range from 2.75 μB (0.95 cm3 K mol−1) at 300 K to a minimum of 1.71 μB (0.36 cm3 K mol−1) at 1.8 K, which is consistent with gradual thermal depopulation of excited crystal field states of the split 4I9/2 ground multiplet, and is typical for UIII complexes.19 The magnetisation at 2 K saturates to a value of 0.89 μB mol−1 at 7 T which is also in agreement with a UIII assignment for 6U. Variable field and temperature magnetometry was also performed on 6Ce and 6Pr which displayed μeff values of 2.16 μB and 3.24 μB respectively at 300 K, comparing well with free-ion values for 2F5/2 and 3H4 ground states. Their low temperature μeff values and saturation properties are unremarkable (see ESI‡).20
 | ||
| Fig. 3 Variable-temperature effective magnetic moment (μeff/μB) versus temperature for 6U at 0.5 T and variable-field molar magnetisation for 6U. | ||
We have prepared a range of f-block bis-per-alkylated phospholide complexes from La–Pr, and U–Pu, and shown that this framework is able to support the isolation and characterisation of isomorphous bis-benzyl-potassiate salts. Despite these compounds containing paramagnetic ions (except La), we were able to observe 31P NMR chemical shifts from the phospholide ligands with all metals except Pu. In future, this could be leveraged for in situ monitoring of protonolysis reactions with Np and Pu by NMR spectroscopy – elements for which macroscale chemistry is not routinely possible in most laboratories.
Transuranium work was conducted at Los Alamos National Laboratory (LANL). AJG and BLS acknowledge the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, Heavy Element Chemistry Program at LANL (DE-AC52-06NA25396). CAPG was sponsored by a Distinguished J. R. Oppenheimer Postdoctoral Fellowship (LANL-LDRD, 20180703PRD1). For work at the University of Manchester (UoM) we thank the Royal Society (URF\R1\211271 to CAPG); and the UoM (Research Bursary to SGF). We acknowledge funding from the EPSRC (EP/K039547/1 and EP/P001386/1 for NMR spectroscopy and X-ray diffraction), and the EPSRC UK National Electron Paramagnetic Resonance Service for SQUID magnetometer access (EP/S033181/1). We thank Prof. Stephen Liddle for support (JAS). Elemental Analyses were performed at the UoM by Mr Martin Jennings and Ms Anne Davies.
Conflicts of interest
There are no conflicts to declare.References
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Prentice Hall, Pearson, 2008 Search PubMed.
- (a) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS; (b) D. S. J. Arney, R. C. Schnabel, B. C. Scott and C. J. Burns, J. Am. Chem. Soc., 1996, 118, 6780–6781 CrossRef CAS; (c) A. J. Wooles, D. P. Mills, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2010, 500–510 RSC; (d) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (e) J. K. Pagano, J. M. Dorhout, K. R. Czerwinski, D. E. Morris, B. L. Scott, R. Waterman and J. L. Kiplinger, Organometallics, 2016, 35, 617–620 CrossRef CAS; (f) E. P. Wildman, G. Balázs, A. J. Wooles, M. Scheer and S. T. Liddle, Nat. Commun., 2016, 7, 12884 CrossRef CAS PubMed.
- P. J. Fagan, J. M. Manriquez, T. J. Marks, C. S. Day, S. H. Vollmer and V. W. Day, Organometallics, 1982, 1, 170–180 CrossRef CAS.
- J. K. Pagano, J. M. Dorhout, R. Waterman, K. R. Czerwinski and J. L. Kiplinger, Chem. Commun., 2015, 51, 17379–17381 RSC.
- (a) S. Kihara, Z. Yoshida, H. Aoyagi, K. Maeda, O. Shirai, Y. Kitatsuji and Y. Yoshida, Pure Appl. Chem., 1999, 71, 1771–1807 CrossRef CAS; (b) A. J. Myers, M. L. Tarlton, S. P. Kelley, W. W. Lukens and J. R. Walensky, Angew. Chem., Int. Ed., 2019, 58, 14891–14895 CrossRef CAS PubMed; (c) D. C. Sonnenberger and J. G. Gaudiello, Inorg. Chem., 1988, 27, 2747–2748 CrossRef CAS.
- (a) F. Nief, P. Riant, L. Ricard, P. Desmurs and D. Baudry-Barbier, Eur. J. Inorg. Chem., 1999, 1041–1045 CrossRef CAS; (b) H.-J. Gosink, F. Nief, L. Ricard and F. Mathey, Inorg. Chem., 2002, 34, 1306–1307 CrossRef; (c) J. Liu, L. E. Nodaraki, P. J. Cobb, M. J. Giansiracusa, F. Ortu, F. Tuna and D. P. Mills, Dalton Trans., 2020, 49, 6504–6511 RSC.
- P. Gradoz, M. Ephritikhine, M. Lance, J. Vigner and M. Nierlich, J. Organomet. Chem., 1994, 481, 69–73 CrossRef CAS.
- A. Elkechai, Y. Mani, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2012, 51, 6943–6952 CrossRef CAS PubMed.
- (a) C. A. P. Goodwin, M. T. Janicke, B. L. Scott and A. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 20680–20696 CrossRef CAS PubMed; (b) S. D. Reilly, J. L. Brown, B. L. Scott and A. J. Gaunt, Dalton Trans., 2014, 43, 1498–1501 RSC.
- S. A. Pattenaude, N. H. Anderson, S. C. Bart, A. J. Gaunt and B. L. Scott, Chem. Commun., 2018, 54, 6113–6116 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- T. V. Fetrow, J. P. Grabow, J. Leddy and S. R. Daly, Inorg. Chem., 2021, 60, 7593–7601 CrossRef CAS PubMed.
- D. Perales, R. Bhowmick, M. Zeller, P. Miro, B. Vlaisavljevich and S. C. Bart, Chem. Commun., 2022, 58, 9630–9633 RSC.
- B. D. Zwick, A. P. Sattelberger and L. R. Avens, in Transuranium Elements: A Half Century, ed., L. R. Morss and J. Fuger, American Chemical Society, Washington DC, 1992, ch. 25, pp. 239–246 Search PubMed.
- (a) A. J. Gaunt, S. D. Reilly, A. E. Enriquez, B. L. Scott, J. A. Ibers, P. Sekar, K. I. Ingram, N. Kaltsoyannis and M. P. Neu, Inorg. Chem., 2008, 47, 29–41 CrossRef CAS PubMed; (b) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed; (c) M. B. Jones, A. J. Gaunt, J. C. Gordon, N. Kaltsoyannis, M. P. Neu and B. L. Scott, Chem. Sci., 2013, 4, 1189–1203 RSC; (d) J. A. Macor, J. L. Brown, J. N. Cross, S. R. Daly, A. J. Gaunt, G. S. Girolami, M. T. Janicke, S. A. Kozimor, M. P. Neu, A. C. Olson, S. D. Reilly and B. L. Scott, Dalton Trans., 2015, 44, 18923–18936 RSC; (e) C. A. P. Goodwin, A. W. Schlimgen, T. E. Albrecht-Schönzart, E. R. Batista, A. Gaunt, M. T. Janicke, S. A. Kozimor, B. L. Scott, L. M. Stevens, F. D. White and P. Yang, Angew. Chem., Int. Ed., 2021, 60, 9459–9466 CrossRef CAS PubMed; (f) S. E. Gilson and P. C. Burns, Coord. Chem. Rev., 2021, 445, 213994 CrossRef CAS.
- W. A. Hargreaves, J. Condens. Matter Phys., 1992, 4, 6141–6154 CrossRef CAS.
- (a) D. E. Morris, R. E. Da Re, K. C. Jantunen, I. Castro-Rodriguez and J. L. Kiplinger, Organometallics, 2004, 23, 5142–5153 CrossRef CAS; (b) E. M. Matson, W. P. Forrest, P. E. Fanwick and S. C. Bart, Organometallics, 2013, 32, 1484–1492 CrossRef CAS.
- (a) L. R. Avens, S. G. Bott, D. L. Clark, A. P. Sattelberger, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 2248–2256 CrossRef CAS; (b) C. J. Windorff, G. P. Chen, J. N. Cross, W. J. Evans, F. Furche, A. J. Gaunt, M. T. Janicke, S. A. Kozimor and B. L. Scott, J. Am. Chem. Soc., 2017, 139, 3970–3973 CrossRef CAS PubMed; (c) C. J. Windorff, J. M. Sperling, T. E. Albrecht-Schönzart, Z. Bai, W. J. Evans, A. N. Gaiser, A. J. Gaunt, C. A. P. Goodwin, D. E. Hobart, Z. K. Huffman, D. N. Huh, B. E. Klamm, T. N. Poe and E. Warzecha, Inorg. Chem., 2020, 59, 13301–13314 CrossRef CAS PubMed; (d) C. A. P. Goodwin, S. R. Ciccone, S. Bekoe, S. Majumdar, B. L. Scott, J. W. Ziller, A. J. Gaunt, F. Furche and W. J. Evans, Chem. Commun., 2022, 58, 997–1000 RSC.
- (a) F. Moro, D. P. Mills, S. T. Liddle and J. van Slageren, Angew. Chem., Int. Ed., 2013, 52, 3430–3433 CrossRef CAS PubMed; (b) D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed.
- (a) J. A. Seed, L. Vondung, F. Barton, A. J. Wooles, E. Lu, M. Gregson, R. W. Adams and S. T. Liddle, Chem. – Eur. J., 2022, 28, e202200761 CrossRef CAS PubMed; (b) G. Balazs, F. G. N. Cloke, J. C. Green, R. M. Harker, A. Harrison, P. B. Hitchcock, C. N. Jardine and R. Walton, Organometallics, 2007, 26, 3111–3119 CrossRef CAS.
Footnotes |
| † Research data files supporting this work are available from Figshare at DOI: https://doi.org/10.48420/20390763. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2193335–2193346, 2202840. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04803e |
| This journal is © The Royal Society of Chemistry 2022 |