
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled release and characterisation of photocaged molecules using in situ LED illumination in solution NMR spectroscopy†
Jack E.
Bramham
 a,
Matja
Zalar
b and
Alexander P.
Golovanov
*a
a,
Matja
Zalar
b and
Alexander P.
Golovanov
*a
aDepartment of Chemistry, School of Natural Sciences, Faculty of Science and Engineering, The University of Manchester, Manchester, M13 9PL, UK. E-mail: a.golovanov@manchester.ac.uk
bDepartment of Chemical Engineering, School of Natural Sciences, Faculty of Science and Engineering, The University of Manchester, Manchester, M13 9PL, UK
First published on 3rd October 2022
Abstract
Photocaging is an attractive strategy to control molecular behaviour, for example, in chemical synthesis, interaction studies or photodynamic therapies. Here, we demonstrate that in situ illumination by the LED NMRtorch approach enables effective and controlled photocage release with simultaneous monitoring of subsequent reactions by solution NMR spectroscopy.
Photocages, also known as photolabile protecting groups (PPGs), and photolysis have a long history of use for controlling the temporal and spatial availability of molecules throughout biology and chemistry.1–4 Covalent attachment of a photocage results in a chemically or biologically inactive molecule, which can be released and activated through photolysis by illumination at a given wavelength. Thus, photocages can be used as a practical means to control molecular availability, for example, to activate a drug at a particular location in the body during photodynamic therapy,5–7 or control organic synthesis reactions.8,9 Alternatively, photocages may be used as an experimental strategy to initiate and study a reaction or certain molecular behaviour,4,10,11 or to conduct titrations.12 A wide range of photocages with different properties, including targeted chemical moieties and photoactive wavelengths, have been developed13 and applied to an even wider range of target molecules.14–18
To characterise photorelease, ex situ illumination is widely used with subsequent analysis by techniques such as high-performance liquid chromatography (HPLC) or nuclear magnetic resonance (NMR) spectroscopy.19–24 The combination of in situ illumination with continuous NMR monitoring enables further characterisation of photocage behaviour, including kinetics of photorelease. Alternatively, it may be used to initiate reactions in NMR experiments, as pioneered by the Schwalbe group25–31 to characterise protein and nucleotide folding during time-resolved NMR. However, in situ photo-NMR has not been applied to photolysis more widely, perhaps due to the complex laser illumination systems typically used.32,33 Additionally, with the continued expansion of the photosensitivity of photocages beyond the typical ultraviolet (UV) region and into visible and infrared (IR) wavelengths,34,35 an increasing number of expensive wavelength-specific laser light sources may be required.
Recently, inexpensive yet powerful light emitting diodes (LEDs) with a wide range of wavelengths, including UV and IR, have become abundant, resulting in their adoption as a light source for in situ photo-NMR illumination.36–39 Moreover, we have recently demonstrated the NMRtorch approach, which combines an LED-based lighthead inserted into the spectrometer and an etched heavy-walled NMR tube to illuminate NMR samples without using optical fibres.40 The NMRtorch enables high intensity uniform illumination of the NMR sample, allowing, for example, rapid chemical degradation of quinine under UV-A light with simultaneous NMR characterisation.40 While LEDs coupled to an optical fibre have been applied in solid-state NMR spectroscopy to trigger photocage release,41 it has not been assessed if LEDs are powerful enough to perform uncaging in situ in solution NMR spectroscopy within a short timeframe.
Here, we demonstrate that the NMRtorch approach can be used to trigger and simultaneously characterise photocage release by using the model system NPE-ATP (Fig. 1), consisting of the nucleotide adenosine triphosphate (ATP) linked covalently with the 1-(2-nitrophenyl)ethyl (NPE) photocage.1 We show that 365 nm UV-A illumination by the NMRtorch results in almost complete release of 1 mM ATP in seconds. This release can be calibrated, enabling us to perform an ATP titration in a single sample without serial additions, simply by repeated short periods of illumination with controlled timings. Finally, we extend this principle by characterising a model reaction occurring following ATP release, in the form of the phosphorylation of acetate by the enzyme acetate kinase. The LED-based NMRtorch approach may be used to both characterise the structure, behaviour, and kinetics of diverse photocaged systems, and also as an experimental technique to initiate reactions in situ for time-resolved NMR experiments. It may be easily applied in any solution-state spectrometer.
 | ||
| Fig. 1 Uncaging of ATP from NPE-ATP by UV light (hν). The NPE photocage and a H+ are released as by-products. Protons used in NMR analysis highlighted (blue). | ||
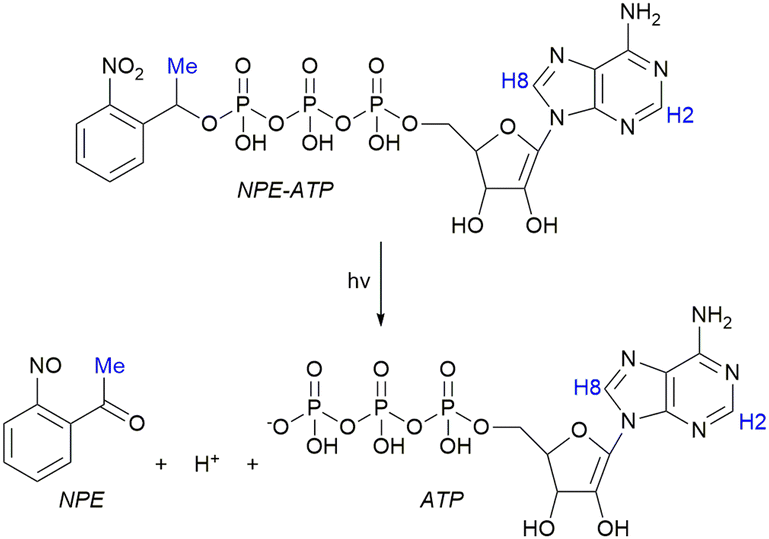
To demonstrate the ability of the NMRtorch to study photocage behaviour, we monitored the photolysis of NPE-ATP by 1H NMR following repeated periods of 1 s illumination, using an NMRtorch with a 365 nm LED array (10 W nominal power consumption) and a quartz NMRtorch tube. Uncaging of 1 mM NPE-ATP was performed in the reaction mixture (200 mM acetate and 5 mM MgCl2 in 100 mM Tris buffer, pH 7.6) to match later experiments with acetate kinase. Photocage release can be followed by NMR using the methyl signal from the NPE photocage, with a decrease in the caged signal (Fig. 2A) and a corresponding increase in free NPE signal (Fig. 2B). Notably, the free NPE appears as a cluster of signals, including some lower intensity ones likely arising from degradation products, in agreement with previous observations of further NPE degradation upon release.42 ATP release can also be directly observed by monitoring the NMR signals of caged and free ATP by 1H (Fig. 2C and Fig. S1, ESI†) or 31P NMR (Fig. S2, ESI†), with H8 and H2 of ATP exhibiting chemical shift and signal splitting perturbations upon release. Additionally, minor background hydrolysis (<5%) of ATP to adenosine diphosphate (ADP) was detected as the shoulder peak of the H8 signal at 8.37 ppm.43 Integration of these signals allows the free ATP concentration after each successive illumination period to be determined (Fig. 2D), with photorelease exhibiting monoexponential behaviour resulting in 90% ATP release by ∼20 s total illumination, and apparent complete release by ∼30 s. This shows that the NMRtorch can be used to characterise release of both the photocage and caged molecule simultaneously.
 | ||
| Fig. 2 Uncaging kinetics of NPE-ATP by UV illumination using the NMRtorch. Appearance of the (A) caged and (B) uncaged methyl group in the NPE caging moiety, and (C) caged and uncaged ATP signals following repeated 1 s illumination periods. (D) Kinetics of ATP release during repeated illumination. Experimental data fitted to an exponential, which then can be used to back-calculate the duration of illumination needed to release a desired concentration of free ATP. | ||
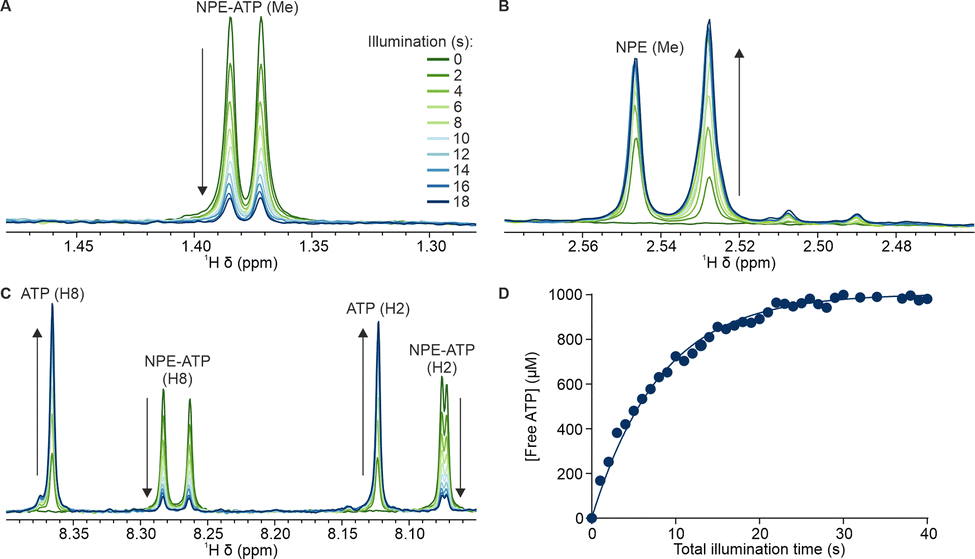
The observed photorelease kinetics can also be used as a calibration curve to determine the illumination time required to release a given concentration of ATP, thus allowing titration experiments to be performed. Such titrations by photorelease may serve as a convenient method to examine the behaviour of a system with increasing concentration of a given molecule in a single sample. Here, we demonstrate a linear titration of free ATP (Fig. 3A), uncaging 100 μM NPE-ATP during each illumination period, with the duration of each illumination increasing exponentially according to the earlier calibration of photorelease kinetics. This illumination strategy successfully resulted in the desired titration, with no additional ATP release or noticeable hydrolysis during the dark periods between illumination events (Fig. 3B). During the illumination however there was ATP hydrolysis noted, reducing the amount of free ATP forming by about 5%, as evidenced by appearance of shoulder peak (H8) at around 8.37 ppm. Such background hydrolysis appears uniform and therefore can be taken into account, if needed, to target specific free ATP values.
 | ||
| Fig. 3 Linear titration of free ATP by controlled UV illumination. (A) Appearance of caged and free ATP signals following illumination with exponentially increasing durations. (B) Concentration of caged and free ATP signals over time, with illumination of variable duration applied at 8 minute intervals, with a following series of NMR spectra acquired then in the dark. | ||
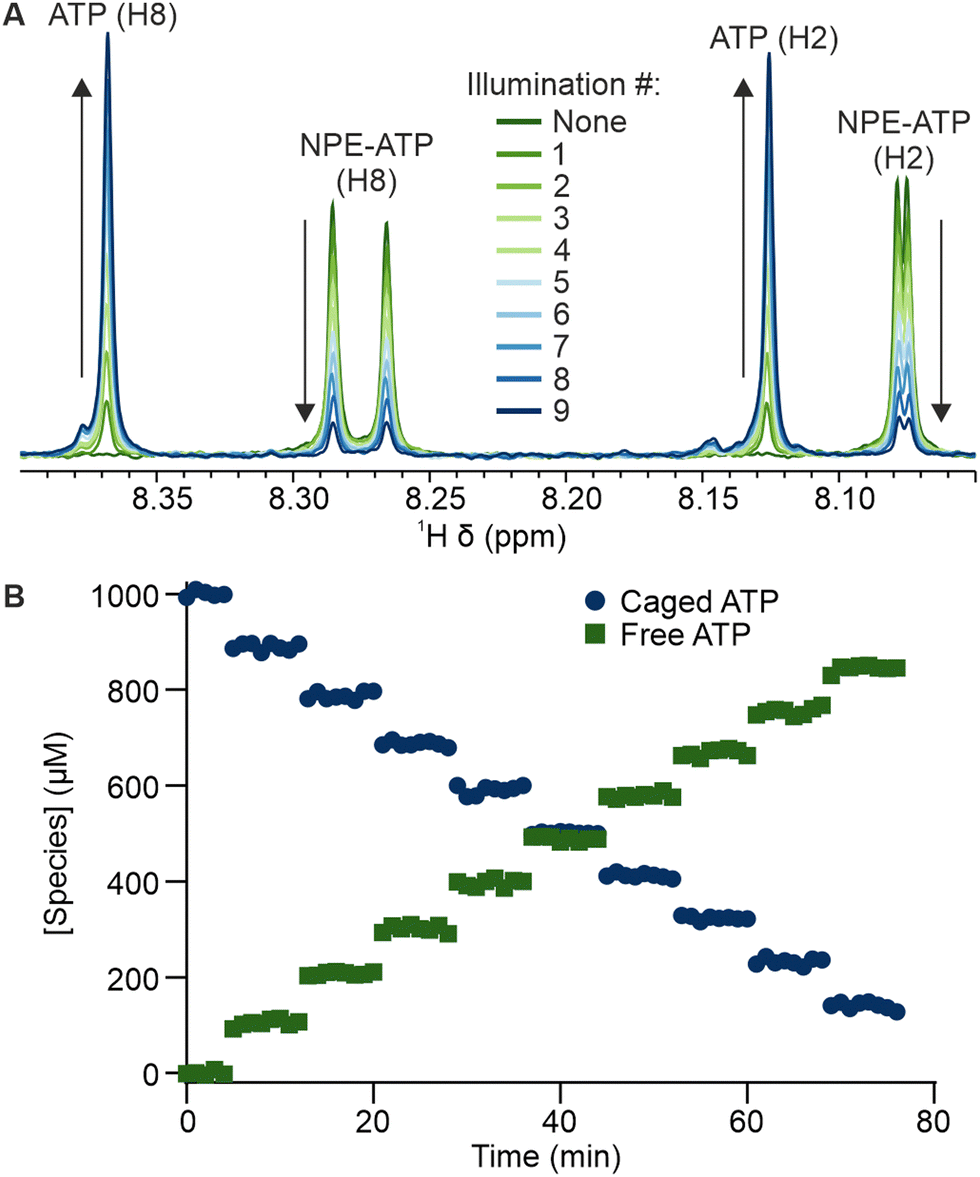
To further demonstrate the potential of such a titration strategy with real time NMR monitoring, we used a model enzyme system, with acetate kinase phosphorylating acetate in the presence of ATP to yield acetyl phosphate and ADP. Here, the behaviour of all components, including photocaged molecules, free substrates and products, can be observed simultaneously by 1H NMR. Before UV illumination, NPE-ATP was enzymatically inactive, with no reaction or ATP hydrolysis occurring. The first release of 100 μM ATP results in initiation of the enzymatic reaction and formation of acetyl phosphate (Fig. S3, ESI†), with spectral deconvolution of the overlapping ADP and ATP H8 signals allowing the phosphorylation reaction to be monitored in real time (Fig. 4). After the initial release, almost all free ATP is converted to ADP within a few minutes, while each successive release of ATP initially perturbs the chemical equilibrium which the enzyme then restores through phosphorylation of acetate.
 | ||
| Fig. 4 UV triggered ATP release and consumption by acetate kinase. (A) Observation of typical ATP conversion to ADP by acetate kinase, here following the second release of 100 μM ATP. (B) Sequential releases of 100 μM ATP, with free ATP and ADP concentrations determined by deconvolution of the overlapping H8 signals at ∼8.37 ppm. The expected values of total released polyphosphate (ATP and ADP) concentration (PP) is shown for comparison. | ||
Together, these results demonstrate that the NMRtorch approach, using LED illumination and an etched sample tube, can be used to characterise photocage behaviour, including release kinetics and subsequent system evolution. Inexpensive LEDs are well suited to the illumination of photocages, particularly with the continued expansion of peak wavelengths available and increases in output power. Additionally, multiple LEDs of different wavelengths may be combined, for example, to study more complex photocage systems.31
Here, the high power UV-A LEDs allowed ∼1 mM ATP to be fully released in 30 seconds (Fig. 2), compared to ∼20 minutes previously demonstrated in solid-state NMR.41 While such release kinetics may be slower than ‘flash’ photolysis achievable by lasers, given the typical timescales of NMR experiments, LED photolysis is still well suited for ‘real time’ NMR monitoring, as individual illumination periods have typical durations similar to relaxation delays used during NMR acquisition. Furthermore, in situ LED illumination by the NMRtorch is markedly easier and more user friendly than complex in situ laser illumination systems. It reduces complications and safety risks associated with use of lasers, and removes the need to work with fragile optical fibres or inserts. Previous studies using lasers and optical fibres, with light distributed in the sample using e.g. quartz rods,33 cone-shaped quartz tips,25 or tapered fibres,44 may suffer from non-uniform light distribution in the sample, which for uncaging experiments would lead to incomplete release in some areas, and subsequent sample inhomogeneity.
Controlled photolysis may also be effectively used in NMR experiments to conduct different titrations. Here, we build upon the previous uses of photocages as reaction initiators in NMR experiments by the Schwalbe group,25–27 by demonstrating the possibility of gradual quantitative uncaging using LEDs with simultaneous NMR monitoring (Fig. 3 and 4). This enables single sample titrations, thereby drastically simplifying experiments by reducing sample preparation and experimental time, and may be applied to a range of typical titrations, including pH, ionic strength, and ligand concentration, using different photocaged molecules. Unlike other single sample NMR ‘titrations’ involving gradients and spatially-selective NMR,45,46 photocage titrations do not suffer from reduced signal arising from the use of smaller ‘slices’ of the sample.
We anticipate that the proposed approach will be especially beneficial for the development and characterisation of new controlled photorelease systems, for example, for photodynamic therapy, or photoactivated chemotherapy, that can be triggered using less harmful visible, IR, or upconverted light.47–49 In principle, any photo-caged system can be studied as long as its characteristic signals can be observed by NMR. NMR combined with ex situ illumination is already used to characterise photocage structure and release,19–24 and in situ illumination with simultaneous NMR monitoring may offer additional insights into kinetics, intermediates, or mechanisms. This approach has advantages compared to other analytical techniques such as HPLC as it provides more atomic-resolution structural details, and removes experimental uncertainties associated with temporal and physical separation between the illumination and detection steps, which is typical for all ex situ approaches. Further information about the photo-caged system, such as determination of quantum yield, can be obtained by calibrating NMRtorch photon flux using reactions with known quantum yields,40 for the same illumination settings, before running the reaction of interest.
In summary, we show that the LED-based NMRtorch enables simple in situ photolysis of photocaged molecules, with simultaneous NMR characterisation of photorelease and subsequent system behaviour. We also demonstrate the potential for single sample NMR titration experiments using photocages. This demonstration may lead to the uptake of in situ photo-NMR as a method to characterise photocage behaviour.
Jack Bramham: investigation, visualisation, writing – original draft. Matja Zalar: resources, writing – review & editing. Alexander P. Golovanov: supervision, writing – review & editing.
This project was supported by EPSRC New Horizons EP/V04835X/1. We acknowledge the use of the Manchester Biomolecular NMR Facility, and are grateful to Dr Matthew Cliff for valuable scientific discussions and NMR support.
Conflicts of interest
A. P. G. is the named author in patent applications GB2008825.8 and PCT/GB2021/051254, International Publication Number WO 2021/250372 A1. J. E. B. and M. Z. declare no competing interests.References
- J. H. Kaplan, B. Forbush, 3rd and J. F. Hoffman, Biochemistry, 1978, 17, 1929–1935 CrossRef CAS PubMed.
- L. H. Clapp and A. M. Gurney, Am. J. Physiol., 1992, 262, H916–920 CrossRef CAS PubMed.
- S. R. Adams and R. Y. Tsien, Annu. Rev. Physiol., 1993, 55, 755–784 CrossRef CAS PubMed.
- D. C. F. Monteiro, E. Amoah, C. Rogers and A. R. Pearson, Acta Crystallogr., Sect. D: Struct. Biol., 2021, 77, 1218–1232 CrossRef CAS PubMed.
- R. Horbert, B. Pinchuk, P. Davies, D. Alessi and C. Peifer, ACS Chem. Biol., 2015, 10, 2099–2107 CrossRef CAS PubMed.
- J. M. Silva, E. Silva and R. L. Reis, J. Controlled Release, 2019, 298, 154–176 CrossRef CAS PubMed.
- R. Chen, Z. Wang, L. Liu and Z. Pan, Chem. Commun., 2022, 58, 4901–4904 RSC.
- C. G. Bochet and A. Blanc, in Handbook of Synthetic Photochemistry, 2009, ch. 13, pp. 417–447 DOI:10.1002/9783527628193.
- T. Barra, L. Arrue, E. Urzúa and L. Ratjen, J. Phys. Org. Chem., 2019, 32, e3935 CrossRef.
- A. P. Pelliccioli and J. Wirz, Photochem. Photobiol. Sci., 2002, 1, 441–458 CrossRef PubMed.
- I. Josts, S. Niebling, Y. Gao, M. Levantino, H. Tidow and D. Monteiro, IUCrJ, 2018, 5, 667–672 CrossRef CAS PubMed.
- P. Swietach, K. W. Spitzer and R. D. Vaughan-Jones, Biophys. J., 2007, 92, 641–653 CrossRef CAS PubMed.
- P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef CAS PubMed.
- H. M. Bandara, T. P. Walsh and S. C. Burdette, Chemistry, 2011, 17, 3932–3941 CrossRef CAS PubMed.
- J. Cui, R. A. Gropeanu, D. R. Stevens, J. Rettig and A. del Campo, J. Am. Chem. Soc., 2012, 134, 7733–7740 CrossRef CAS PubMed.
- H. K. Agarwal, S. Zhai, D. J. Surmeier and G. C. R. Ellis-Davies, ACS Chem. Neurosci., 2017, 8, 2139–2144 CrossRef CAS PubMed.
- S. Passlick, P. F. Kramer, M. T. Richers, J. T. Williams and G. C. R. Ellis-Davies, PLoS One, 2017, 12, e0187732 CrossRef PubMed.
- N. Ankenbruck, T. Courtney, Y. Naro and A. Deiters, Angew. Chem., Int. Ed., 2018, 57, 2768–2798 CrossRef CAS PubMed.
- C. P. Holmes, J. Org. Chem., 1997, 62, 2370–2380 CrossRef CAS PubMed.
- P. Wang, M. Mondal and Y. Wang, Eur. J. Org. Chem., 2009, 2055–2058 CrossRef CAS.
- C. de Gracia Lux, C. L. McFearin, S. Joshi-Barr, J. Sankaranarayanan, N. Fomina and A. Almutairi, ACS Macro Lett., 2012, 1, 922–926 CrossRef CAS PubMed.
- A. M. Piloto, S. P. G. Costa and M. S. T. Gonçalves, Tetrahedron, 2014, 70, 650–657 CrossRef CAS.
- P. Li and C. M. Dong, ACS Macro Lett., 2017, 6, 292–297 CrossRef CAS PubMed.
- M. Lopez-Corrales, A. Rovira, A. Gandioso, M. Bosch, S. Nonell and V. Marchan, Chemistry, 2020, 26, 16222–16227 CrossRef CAS PubMed.
- T. Kühn and H. Schwalbe, J. Am. Chem. Soc., 2000, 122, 6169–6174 CrossRef.
- J. Wirmer, T. Kuhn and H. Schwalbe, Angew. Chem., Int. Ed., 2001, 40, 4248–4251 CrossRef CAS PubMed.
- P. Wenter, B. Furtig, A. Hainard, H. Schwalbe and S. Pitsch, ChemBioChem, 2006, 7, 417–420 CrossRef CAS PubMed.
- J. Buck, B. Furtig, J. Noeske, J. Wohnert and H. Schwalbe, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15699–15704 CrossRef CAS PubMed.
- B. Furtig, P. Wenter, L. Reymond, C. Richter, S. Pitsch and H. Schwalbe, J. Am. Chem. Soc., 2007, 129, 16222–16229 CrossRef PubMed.
- C. Helmling, D. P. Klotzner, F. Sochor, R. A. Mooney, A. Wacker, R. Landick, B. Furtig, A. Heckel and H. Schwalbe, Nat. Commun., 2018, 9, 944 CrossRef PubMed.
- I. Elamri, C. Abdellaoui, J. K. Bains, K. F. Hohmann, S. L. Gande, E. Stirnal, J. Wachtveitl and H. Schwalbe, J. Am. Chem. Soc., 2021, 143, 10596–10603 CrossRef CAS PubMed.
- C. R. Yonker and S. L. Wallen, Appl. Spectrosc., 2016, 50, 781–784 CrossRef.
- M. Pietrzak, J. Dobkowski, A. Gorski, S. Gawinkowski, M. Kijak, R. Luboradzki, P. E. Hansen and J. Waluk, Phys. Chem. Chem. Phys., 2014, 16, 9128–9137 RSC.
- N. Umeda, H. Takahashi, M. Kamiya, T. Ueno, T. Komatsu, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, ACS Chem. Biol., 2014, 9, 2242–2246 CrossRef CAS PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klan, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- C. Feldmeier, H. Bartling, E. Riedle and R. M. Gschwind, J. Magn. Reson., 2013, 232, 39–44 CrossRef CAS PubMed.
- C. Feldmeier, H. Bartling, K. Magerl and R. M. Gschwind, Angew. Chem., Int. Ed., 2015, 54, 1347–1351 CrossRef CAS PubMed.
- Y. Ji, D. A. DiRocco, J. Kind, C. M. Thiele, R. M. Gschwind and M. Reibarkh, ChemPhotoChem, 2019, 3, 984–992 CrossRef CAS.
- K. L. Skubi, W. B. Swords, H. Hofstetter and T. P. Yoon, ChemPhotoChem, 2020, 4, 685–690 CrossRef CAS PubMed.
- J. E. Bramham and A. P. Golovanov, Commun. Chem., 2022, 5, 90 CrossRef CAS.
- J. de Mos, A. Jakob, J. Becker-Baldus, A. Heckel and C. Glaubitz, Chemistry, 2020, 26, 6789–6792 CrossRef CAS PubMed.
- Y. V. Il'ichev, M. A. Schworer and J. Wirz, J. Am. Chem. Soc., 2004, 126, 4581–4595 CrossRef PubMed.
- Y. Lian, H. Jiang, J. Feng, X. Wang, X. Hou and P. Deng, Talanta, 2016, 150, 485–492 CrossRef CAS PubMed.
- I. Kuprov and P. J. Hore, J. Magn. Reson., 2004, 171, 171–175 CrossRef CAS PubMed.
- M. Wallace, D. J. Adams and J. A. Iggo, Anal. Chem., 2018, 90, 4160–4166 CrossRef CAS PubMed.
- G. Schenck, K. Baj, J. A. Iggo and M. Wallace, Anal. Chem., 2022, 94, 8115–8119 CrossRef CAS PubMed.
- O. Filevich and R. Etchenique, Photochem. Photobiol. Sci., 2013, 12, 1565–1570 CrossRef CAS PubMed.
- Y. H. Chien, Y. L. Chou, S. W. Wang, S. T. Hung, M. C. Liau, Y. J. Chao, C. H. Su and C. S. Yeh, ACS Nano, 2013, 7, 8516–8528 CrossRef CAS PubMed.
- S. H. Askes, A. Bahreman and S. Bonnet, Angew. Chem., Int. Ed., 2014, 53, 1029–1033 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04731d |
| This journal is © The Royal Society of Chemistry 2022 |