
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Directed cis-hydrosilylation of borylalkynes to borylsilylalkenes†
Kinga
Stefanowska
 a,
Tomasz
Sokolnicki
ab,
Jędrzej
Walkowiak
a,
Agnieszka
Czapik
b and
Adrian
Franczyk
*a
a,
Tomasz
Sokolnicki
ab,
Jędrzej
Walkowiak
a,
Agnieszka
Czapik
b and
Adrian
Franczyk
*a
aCenter for Advanced Technology, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 10, Poznań 61-614, Poland. E-mail: adrian.franczyk@amu.edu.pl
bFaculty of Chemistry, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 8, Poznań 61-614, Poland
First published on 13th October 2022
Abstract
Hydrosilylation of borylalkynes to borylsilylalkenes (with a different arrangement of substituents) has been successfully developed. The cis-addition of SiH group to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds was directed by using a specific catalyst. The obtained products are crucial synthons for the introduction of the C
C bonds was directed by using a specific catalyst. The obtained products are crucial synthons for the introduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in organic synthesis.
C bonds in organic synthesis.
Borylsilylalkenes are useful intermediates in organic synthesis because of their ability to achieve straightforward modification through a series of chemical transformations. The development of new and more efficient methodologies for the regio- and stereodefined synthesis of these compounds is one of the most challenging goals in modern organic and organometallic chemistry.
Such molecules combine the synthetic potential of both boryl and silyl groups bonded to sp2-carbon and their different reactivities.1 They can be converted into other functional groups (e.g., halogen, aryl, alkenyl, hydroxyl, etc.) via demetallation processes (e.g., palladium-catalyzed coupling reactions at the boryl (Suzuki–Miyaura)2 or silyl (Hiyama)3 fragment), or become reagents for the synthesis of saturated functional molecules.4 They are considered precursors for the synthesis of di-, tri-, and tetrasubstituted alkenes present in many important pharmaceuticals (e.g., anticancer drugs), bioactive natural compounds,5 liquid crystals,6 and dipeptide mimetics used in conformation studies of cyclic bioactive peptides.7
Methods for the synthesis of borylsilylalkenes include hydro-,8 mono-,9 di-,10 and carboboration11 of silylalkynes, silaboration of alkynes,12 silylative coupling of vinylboranes with vinylsilanes,13 hydrosilylation of borylalkynes,8e,14 and others.15 The detailed literature screening of the protocols for the synthesis of the analog isomers of borylsilylalkenes described in this work is presented in ESI† (Table S6, page 7 and Scheme S1, page 9).
In light of the constant very high interest in the synthesis of stereodefined functionalized alkenes and conjugated systems obtained by the transformation of boryl or silyl groups, herein we decided to test the hydrosilylation of borylalkynes. Hydrosilylation of the carbon–carbon triple bond (CC) is a straightforward, 100% atom efficient, industrially applied, repeatable method, which is easy to scale up and is based on commercially available reagents and catalysts.16 Most importantly it gives the unique opportunity to obtain all possible isomers of cis-, and trans-addition of the SiH group to the CC bond. The R3Si group can be bonded to both α and β-sp-carbons in terminal and internal alkynes. Different products can be obtained with the same set of reagents, but only by changing the type of catalyst and the reaction conditions. The versatility of this method is also a big synthetic challenge aimed at carrying out the process providing one product exclusively in each case.
Based on our experience17 and the literature screening,18 we tested a wide spectrum of catalytic systems (based on Ru, Rh, Ir, Pt) which allow cis- or trans-addition of the SiH bond to the terminal and internal CC. Catalysts dedicated to trans-additions (e.g., RuCHPh(Cl)2(PCy3)2, [Cp*Ru(MeCN)3]PF6, [(coe)2IrCl]219 in the case of boryl derivatives (2a–f) led to the complex post-reaction mixtures containing predominantly cis-addition products. On the other hand, through careful catalyst selection, reaction optimization, and isolation of the resulting products, we achieved a family of borylsilylalkenes by the cis-hydrosilylations of the terminal and internal (boryl,silyl,aryl,alkyl)borylalkynes (2a–f) with a wide spectrum of silanes (1a–g) (Table 1).
We found that the reactions of ethynylborane (2a) with triethylsilane (1a), 1,1,1,3,5,5,5-heptamethyltrisiloxane (1c), and benzyldimethylsilane (1f) in the presence of PtO2/XPhos (I) led to the formation of products 3 with a selectivity higher than 94%. In the case of the reaction with triphenylsilane (1b), the best results were obtained for Karstedt's catalyst (II) (Pt2(dvs)3). In all tests, product 3 was formed, from the cis-addition in which SiR3 was added to the β-sp-carbon. In the literature, there is only one example of the hydrosilylation of ethynyl MIDA boronate ester (MIDA-N-methylimidodiacetic boronic acid ester) with benzyldimethylsilane (1f) used in excess (1.5 equiv.) in the presence of PtCl2/XPhos. As a result, a product of cis-hydrosilylation was obtained selectively with a 91% yield.14 Therefore, we recreated previously reported conditions for the reaction of borane (2a) with HSiMe2Bn. Resulting from this, a complex mixture of products was observed (ESI,† Table S1, entry 25, page 2). This confirmed that the system proposed by us is much more efficient.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 1 | 2 | [M] | [1]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[2]:[M] :[2]:[M] |
t [h] | Temp. [°C] | Solvent/atmosphere | Selectivity of 3/4/5 [%] NMR (GC-MS) | Isolated yield of 3 or 4 [%] |
| Complete conversion of 1a–g for all tests was confirmed by 1H NMR and GC-MS. Reaction conditions: m1a = 0.0414 g, m1b = 0.0934 g, m1c = 0.0802 g, m1d = 0.04 g, m1e = 0.05 g, m1f = 0.055 g, m1g = 0.064 g, 2 ml of solvent.a m1a/VDCM = 0.116 g ml−1. For more detailed information please see the ESI. XPhos = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl, dvs = 1,3-divinyl-1,1,3,3-tetramethyldisiloxane, Cp = cyclopentadienyl, Cy = cyclohexyl. | |||||||||
| 1 | 1a | 2a | I | 1:1:10−2 |
48 | 60 | THF/argon | aa, 96/4/0 (97/3/0) | 93 |
| 2 | 100 | THF/argon | aa, 100/0/0 (93/7/0) | ||||||
| 3a | V | 1:1:10−2 |
24 | rt | DCM/argon | aa, 100/0/0 (100/0/0) | |||
| 4 | IV | 1:1:2 × 10−2 |
48 | rt | DCM/argon | aa, 10/81/8 (11/80/9) | 57 | ||
| 5 | 1b | II | 1:1:4 × 10−4 |
48 | 100 | Toluene/air | ba, 92/8/0 (92/8/0) | 81 | |
| 6 | IV | 1:1:2 × 10−2 |
24 | rt | DCM/argon | ba, 0/100/0 (0/100/0) | 72 | ||
| 7 | 1c | I | 1:1:10−2 |
24 | 100 | THF/argon | ca, 94/6/0 (94/6/0) | 91 | |
| 8 | 1f | I | 1:1:10−2 |
24 | 100 | THF/argon | fa, 95/5/0 (97/3/0) | 86 | |
| 9 | 1a | 2b | II | 1:1:10−2 |
48 | rt | Toluene/air | ab, 100/0/0 (99/0/1) | 90 |
| 10 | III | 1:1:10−2 |
120 | rt | Toluene/air | ab, 100/0/0 (100/0/0) | |||
| 11 | 1b | III | 1:1:10−2 |
24 | 120 | Toluene/air | bb, 100/0/0 (100/0/0) | 98 | |
| 12 | 1c | III | 1:1:10−2 |
24 | 60 | Toluene/air | cb, 96/0/4 (96/0/4) | 90 | |
| 13 | 1f | III | 1:1:10−2 |
24 | 100 | Toluene/air | fb, 100/0/0 (93/0/7) | 91 | |
| 14 | 1a | 2c | III | 1:1:10−2 |
24 | 60 | Toluene/air | ac, 0/100/0 (3/97/0) | 89 |
| 15 | 1b | II | 1:1:4 × 10−4 |
24 | 100 | Toluene/air | bc, 0/100/0 | 93 | |
| 16 | 1c | III | 1:1:10−2 |
24 | 60 | Toluene/air | cc, 5/95/0 | 89 | |
| 17 | 1d | III | 1:1:10−2 |
24 | 60 | Toluene/air | dc, 0/100/0 | 91 | |
| 18 | 1e | II | 1:1:4 × 10−4 |
24 | 100 | Toluene/air | ec, 0/100/0 | 89 | |
| 19 | 1f | II | 1:1:4×10−4 |
24 | 100 | Toluene/air | fc, 0/100/0 | 91 | |
| 20 | 1a | 2d | II | 1:1:4 × 10−4 |
48 | rt | Toluene/air | ad, 18/82/0 | 76 |
| 21 | 1b | III | 1:1:10−2 |
48 | 120 | Toluene/air | bd, 0/100/0 | 85 | |
| 22 | 1c | IV | 1:1:2 × 10−1 |
24 | rt | Toluene/argon | cd, 84/7/9 (88/7/5) | 79 | |
| 22 | 1e | II | 1:1:4 × 10−4 |
48 | 40 | Toluene/air | ed, (8/85/7) | 88 | |
| 23 | 1f | II | 1:1:4 × 10−4 |
48 | rt | Toluene/air | fd, (3/94/3) | 83 | |
| 24 | IV | 1:1:2 × 10−1 |
24 | rt | 1,4-Dioxane/argon | fd, 89/0/11 (91/0/9) | 86 | ||
| 25 | 1g | IV | 1:1:2 × 10−1 |
24 | rt | Toluene/argon | gd, 91/9/0 (97/0/3) | 91 | |
| 26 | 1b | 2e | III | 1:1:10−2 |
24 | 120 | Toluene/air | be, (14/86/0) | 71 |
| 27 | 1c | IV | 1:1:2 × 10−1 |
24 | rt | Toluene/argon | ce, 100/0/0 (96/2/2) | 89 | |
| 28 | 1f | II | 1:1:4 × 10−4 |
48 | rt | Toluene/air | fe, 3/88/9 (7/85/8) | 73 | |
| 29 | IV | 1:1:2 × 10−1 |
24 | rt | Toluene/argon | fe, (80/10/10) | 70 | ||
| 30 | 1g | IV | 1:1:2 × 10−1 |
24 | rt | Toluene/argon | ge, 100/0/0 (100/0/0) | 92 | |
| 31 | 1a | 2f | II | 1:1:10−2 |
24 | rt | Toluene/air | af, 1/99/0 (3/97/0) | 75 |

Screening different types of catalysts, we found that the reaction of borane 2a with triethylsilane (1a) could also be performed at room temperature if Ru(CO)Cl(H)(PCy3)2 (V) was applied. Unfortunately, the reactions with the remaining silanes were less effective due to the lower conversion of the reagents. Attempts to solve these problems by increasing the reaction temperature resulted in the formation of side products.
On the other hand, the uses of [CpRu(CH3CN)3][PF6] (IV) in the reaction of (2a) with triethyl- (1a) and triphenylsilanes (1b) gave gem-isomers in high yields (products 4aa and 4ba). This proved that, with the same set of substrates and by changing the catalyst and reaction conditions we can direct the cis-hydrosilylation to various products. The attempt to obtain cis-isomer was unsuccessful.
In the next step of the study, hydrosilylation of internal symmetrical 1,2-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyne (2b) was performed. The best results were obtained in the presence of Pt(PPh3)4 (III). The results revealed that targeted product 4 was obtained, which contains two boryl groups and one silyl. Karstedt's (II) catalyst could also be used in the modification of this borane but only when the reactions occurred at room temperature. With elevated temperatures, the formation of side/decomposition products was observed.
The same catalytic systems were used for hydrosilylation of 2-((tert-butyldimethylsilanyl)ethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2c). Both of them were effective, and a higher temperature could be applied. The presence of a SiMe2(t-Bu) group in the structure of the alkyne (2c) directed the addition of SiR3 from silane, to the sp-carbon bonded to the boryl group. The attempt to obtain a second isomer 3 in the presence of [CpRu(CH3CN)3][PF6] (IV) was not successful. The uses of IV gave low conversions or led to complex mixtures of products.
The studies on the reactivity of 2-phenyl-1-ethynylboronic acid pinacol ester (2d) showed that hydrosilylation was the most efficient when Karstedt's (II) and Pt(PPh3)4 (III) catalysts were applied. With the set of silanes (1a, 1e–f), products 4 were obtained, in which SiR3 was attached to the α-sp-carbon bonded to the boryl group at the same time. The selectivity of these processes was in the range of 80–100%. Exclusive formation of product 4 was observed for HSiPh3 (1b). The cis-additions to 2d leading to products 3 occurred by the use of [CpRu(CH3CN)3][PF6] (IV). These products were formed with a selectivity of around 90%.
Similar results were found for 3-methoxy-1-propyn-1-ylboronic acid pinacol ester (2e). However, in this case, a much higher improvement in selectivity up to 100% was observed for silanes (1c, 1g) with siloxy substituents.
Moreover, the (3,3-dimethyl-1-butynyl)boronic acid diisopropyl ester (2f) bearing t-Bu group and two isopropoxy groups, which are less stable than pinacol derivative, were tested. The reaction catalyzed by Karstedt's (II) catalyst gave product 4 with a high yield.
In the literature, hydrosilylation of 2-(hex-1-yn-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane with an excess of silanes in the presence of [CpRu(CH3CN)3][PF6] (IV) in CH2Cl2 has been reported.8e Moreover, one example of the hydrosilylation of propynyl MIDA ester in the presence of PtCl2/XPhos has also been described.14 However, in both cases, excess of silane (1.5 equiv.) needed to be used since side products were obtained (ESI,† Table S5, entry 17, page 6). The stoichiometric procedures proposed by us herein lead to the selective formation of products of Si–H addition to the CC, which simplify the separation procedures of obtained products and are efficient for a wide spectrum of structurally different terminal and internal borylalkynes and silanes.
It is worth emphasizing that 29 compounds in total were synthesized, 25 of them for the first time (3ca, 3fa, 3ab–cb, 3fb, 4ac–fc, 3cd, 3fd–gd, 4ad–bd, 4ed–fd, 3ce, 3fe–ge, 4be, 4fe, 4af and 6dc). The obtained compounds were fully characterized by 1H, 13C, 29Si, and GC-MS (or ESI MS). For compounds 3ba, 4ba, 3bb, 3fb, and 6dc (which is a product of 4dc condensation), crystal structures were obtained (Scheme 1). The compounds 3aa, 4aa, 3ba, and 4ba had previously been obtained via hydroboration,8b–e dehydrogenative borylation,20 and silylboration21 reactions with lower yields or in traces as side products (ESI,† Table S6, page 7).
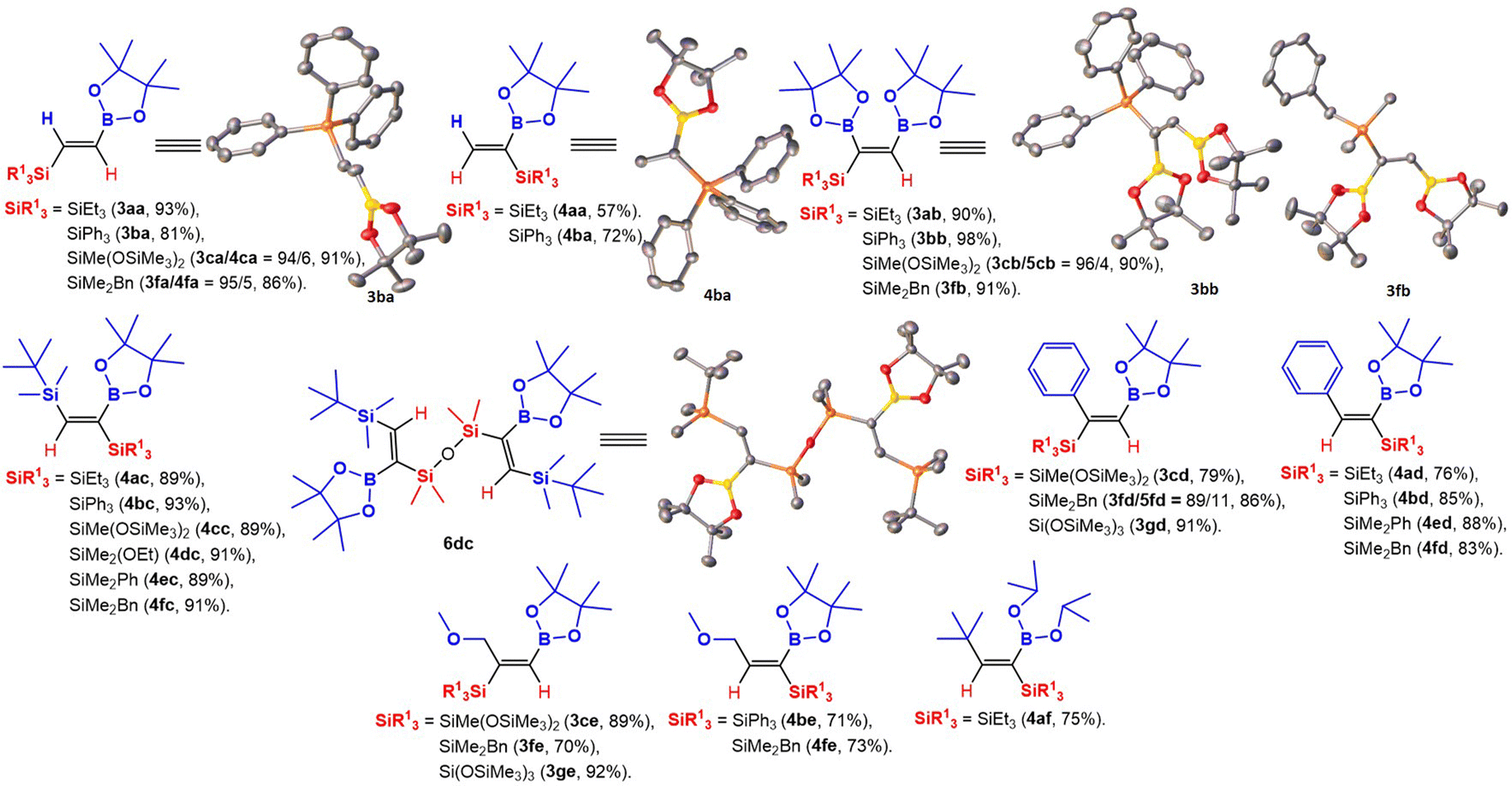 | ||
| Scheme 1 Structural formulas of obtained compounds with indicated isolated yields. Crystal structures of 3ba, 4ba, 3bb, 3fb, and 6dc were confirmed by X-ray. | ||
In summary, we successfully obtained borylsilylalkenes by directed cis-hydrosilylation of a structurally different, wide group of borylalkynes with silanes (1a–f). The processes occurred by cis-addition of the Si–H bond across the carbon–carbon triple bond and led to products 3 or 4 depending on the specific catalyst and reaction conditions used. We proved that, compared to other synthetic methods which can be applied to obtain the targeted products, hydrosilylation is the most powerful method from those previously described and leads to the widest group of products with high yields, which are often not available in different protocols. Moreover, this process is a highly effective method for the synthesis of borylsilylalkenes with a diverse arrangement of substituents, which gives the possibility to use commercially available catalysts and substrates, carrying out the processes with a stoichiometric ratio of reagents. All these features will allow a further straightforward scaling up process. Finally, the obtained products constitute very useful synthons for the synthesis of substituted ethenes or complex materials by widely spread chemical transformation based on the modification of boryl, silyl, or sp2-CH groups which can be performed one after another, based on the different reactivities of the boryl and silyl groups. Such synthons allow the synthesis of new molecules or known systems to be obtained using new approaches.
This work was supported by the National Centre for Research and Development in Poland, Lider Programme No. LIDER/6/0017/L-9/17/NCBR/2018 and the National Science Centre in Poland No. UMO-2018/31/G/ST4/04012. KS gratefully acknowledges the Foundation for Polish Science (FNP) START grant No. 075.2022.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010 RSC.
- J. Jiao, J. K. Nakajima and Y. Nishihara, Org. Lett., 2013, 15, 3294 CrossRef CAS PubMed.
- C. Morrill and N. S. Mani, Org. Lett., 2007, 9, 1505 CrossRef CAS PubMed.
- (a) T. Shimada, K. Mukaide, A. Shinohara, J. W. Han and T. Hayashi, J. Am. Chem. Soc., 2002, 124, 1584 CrossRef CAS PubMed; (b) Y. Sun, J. Guo, X. Shen and Z. Lu, Nat. Commun., 2022, 13, 650 CrossRef CAS PubMed.
- (a) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed; (b) K. Itami, T. Kamei and J.-I. Yoshida, J. Am. Chem. Soc., 2003, 125, 14670 CrossRef CAS PubMed.
- A. Schultz, S. Diele, S. Laschat and M. Nimtz, Adv. Funct. Mater., 2001, 11, 441 CrossRef CAS.
- S. Oishi, K. Miyamoto, A. Niida, M. Yamamoto, K. Ajito, H. Tamamura, A. Otaka, Y. Kuroda, A. Asai and N. Fujii, Tetrahedron, 2006, 62, 1416 CrossRef CAS.
- (a) M. Magre, B. Maity, A. Falconnet, L. Cavallo and M. Rueping, Angew. Chem., 2019, 58, 7025 CrossRef CAS PubMed; (b) J. Szyling, A. Franczyk, K. Stefanowska, M. Klarek, H. Maciejewski and J. Walkowiak, ChemCatChem, 2018, 10, 531 CrossRef CAS; (c) J. Szyling, A. Franczyk, K. Stefanowska, H. Maciejewski and J. Walkowiak, ACS Sustainable Chem. Eng., 2018, 6, 10980 CrossRef CAS; (d) J. Szyling, A. Franczyk, K. Stefanowska and J. Walkowiak, Adv. Synth. Catal., 2018, 360, 2966 CrossRef CAS; (e) Q. Feng, H. Wu, X. L. Song, L. W. Chung, Y.-D. Wu and J. Sun, J. Am. Chem. Soc., 2020, 142, 13867 CrossRef CAS PubMed; (f) J. R. Lawson, L. C. Wilkins and R. L. Melen, Chem. – Eur. J., 2017, 23, 10997 CrossRef CAS PubMed; (g) Y. Meng, Z. Kong and J. P. Morken, Angew. Chem., Int. Ed., 2020, 59, 8456 CrossRef CAS PubMed; (h) N. Sarkar, S. Bera and S. Nembenna, J. Org. Chem., 2020, 85, 4999 CrossRef CAS PubMed; (i) M. Fleige, J. Mobus, T. Vom Stein, F. Glorius and D. W. Stephan, Chem. Commun., 2016, 52, 10830 RSC; (j) A. Harinath, I. Banerjee, J. Bhattacharjee and T. K. Panda, New J. Chem., 2019, 43, 10531 RSC; (k) C. K. Blasius, V. Vasilenko, R. Matveeva, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2020, 59, 23010 CrossRef CAS PubMed.
- (a) K. Wen, J. Chen, F. Gao, P. S. Bhadury, E. Fan and Z. Sun, Org. Biomol. Chem., 2013, 11, 6350 RSC; (b) Y. M. Chae, J. S. Bae, J. H. Moon, J. Y. Lee and J. Yun, Adv. Synth. Catal., 2014, 356, 843 CrossRef CAS.
- (a) J. Jiao, K. Hyodo, H. Hu, K. Nakajima and Y. Nishihara, J. Org. Chem., 2014, 79, 285 CrossRef CAS PubMed; (b) M. Kidonakis and M. Stratakis, Eur. J. Org. Chem., 2017, 4265 CrossRef CAS; (c) F. Alonso, Y. Moglie, L. Pastor-Perez and A. Sepulveda-Escribano, ChemCatChem, 2014, 6, 857 CrossRef CAS.
- J. R. Lawson, V. Fasano, J. Cid, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2016, 45, 6060 RSC.
- (a) M. Zhao, C.-C. Shan, Z.-L. Wang, C. Yang, Y. Fu and Y.-H. Xu, Org. Lett., 2019, 21, 6016 CrossRef CAS PubMed; (b) M. Suginome, H. Nakamura and Y. Ito, Chem. Commun., 1996, 2777 RSC; (c) T. Ohmura, K. Oshima and M. Suginome, Chem. Commun., 2008, 1416 RSC; (d) T. Ohmura, Y. Takaoka and M. Suginome, Chem. Commun., 2021, 57, 4670 RSC; (e) Y. Gu, Y. Duan, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 2061 CrossRef CAS PubMed.
- (a) J. Walkowiak, B. Marciniec and M. Jankowska–Wajda, J. Organomet. Chem., 2010, 695, 1287 CrossRef CAS; (b) M. Jankowska, B. Marciniec, C. Pietraszuk, J. Cytarska and M. Zaidlewicz, Tetrahedron Lett., 2004, 45, 6615 CrossRef CAS; (c) M. Ludwiczak, J. Szyling, A. Garbicz, T. Sokolnicki, J. Pyziak and J. Walkowiak, Inorg. Chem., 2020, 59, 17555 CrossRef CAS PubMed.
- M. G. McLaughlin, C. A. McAdam and M. J. Cook, Org. Lett., 2015, 17, 10 CrossRef CAS PubMed.
- (a) J. Szyling, J. Walkowiak, T. Sokolnicki, A. Franczyk, K. Stefanowska and M. Klarek, J. Catal., 2019, 376, 219 CrossRef CAS; (b) N. Saito, K. Saito, H. Sato and Y. Sato, Adv. Synth. Catal., 2013, 355, 853 CrossRef CAS; (c) J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 9893 CrossRef CAS PubMed; (d) T. Hata, H. Kitagawa, H. Masai, T. Kurahashi, M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2001, 40, 790 CrossRef CAS; (e) E. K. Edelstein, S. Namirembe and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 5027 CrossRef CAS PubMed; (f) J. Szyling, T. Sokolnicki, A. Franczyk and J. Walkowiak, Catalysts, 2020, 10, 762 CrossRef CAS.
- (a) B. Marciniec, C. Pietraszuk, P. Pawluć and H. Maciejewski, Chem. Rev., 2022, 122, 3996 CrossRef CAS PubMed; (b) B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, Hydrosilylation: A Comprehensive Review on Recent Advances, Springer, 2009, Vol. 1 CrossRef.
- (a) J. Walkowiak, K. Salamon, A. Franczyk, K. Stefanowska, J. Szyling and I. Kownacki, J. Org. Chem., 2019, 84, 2358 CrossRef CAS PubMed; (b) K. Stefanowska, A. Franczyk, J. Szyling, K. Salamon, B. Marciniec and J. Walkowiak, J. Catal., 2017, 356, 206 CrossRef CAS; (c) K. Stefanowska, J. Szyling, J. Walkowiak and A. Franczyk, Inorg. Chem., 2021, 60, 11006 CrossRef CAS PubMed; (d) A. Franczyk, K. Stefanowska, M. Dutkiewicz, D. Frąckowiak and B. Marciniec, Dalton Trans., 2017, 46, 158 RSC; (e) K. Stefanowska, A. Franczyk, J. Szyling, M. Pyziak, P. Pawluć and J. Walkowiak, Chem. – Asian J., 2018, 13, 2101 CrossRef CAS PubMed; (f) K. Stefanowska, A. Franczyk, J. Szyling and J. Walkowiak, ChemCatChem, 2019, 11, 4848 CrossRef CAS; (g) J. Szyling, R. Januszewski, K. Jankowska, J. Walkowiak, I. Kownacki and A. Franczyk, Chem. Commun., 2021, 57, 4504 RSC.
- (a) P. He, M.-Y. Hu, X.-Y. Zhang and S.-F. Zhu, Synthesis, 2021, 49 Search PubMed; (b) J. Walkowiak, J. Szyling, A. Franczyk and R. L. Melen, Chem. Soc. Rev., 2022, 51, 869 RSC.
- (a) S. V. Maifeld, M. N. Tran and D. Lee, Tetrahedron Lett., 2005, 46, 105 CrossRef CAS; (b) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644 CrossRef CAS PubMed; (c) D. C. Apple, K. A. Brady, J. M. Chance, N. E. Heard and T. A. Nile, J. Mol. Catal., 1985, 29, 55 CrossRef CAS.
- N. Kirai, S. Iguchi, T. Ito, J. Takaya and N. Iwasawa, Bull. Chem. Soc. Jpn., 2013, 86, 784 CrossRef CAS.
- T. Kurahashi, T. Hata, H. Masai, H. Kitagawa, M. Shimizu and T. Hiyama, Tetrahedron, 2002, 58, 6381 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2083476, 2107079, 2107080, 2184400 and 2193621. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04318a |
| This journal is © The Royal Society of Chemistry 2022 |