
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stability of alkyl carbocations†
Thomas
Hansen
 *ab,
Pascal
Vermeeren
a,
F. Matthias
Bickelhaupt
*ac and
Trevor A.
Hamlin
*a
*ab,
Pascal
Vermeeren
a,
F. Matthias
Bickelhaupt
*ac and
Trevor A.
Hamlin
*a
aDepartment of Theoretical Chemistry, Amsterdam Institute of Molecular and Life Sciences (AIMMS), Amsterdam Center for Mul-tiscale Modeling (ACMM), Vrije Universiteit Amsterdam, De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands. E-mail: t.hansen@vu.nl; t.a.hamlin@vu.nl
bDepartament de Química Inorgànica i Orgànica (Secció de Química Orgànica) & Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, 08028 Barcelona, Spain. E-mail: f.m.bickelhaupt@vu.nl
cInstitute for Molecules and Materials (IMM), Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
First published on 6th October 2022
Abstract
The traditional and widespread rationale behind the stability trend of alkyl-substituted carbocations is incomplete. Through state-of-the-art quantum chemical analyses, we quantitatively established a generally overlooked driving force behind the stability of carbocations, namely, that the parent substrates are substantially destabilized by the introduction of substituents, often playing a dominant role in solution. This stems from the repulsion between the substituents and the C–X bond.
Carbocations are ubiquitous reactive intermediates in synthetic chemistry and are involved in many fundamental organic transformations.1,2 These reactive intermediates generally form through a heterolytic C–X bond dissociation, yielding a carbocation C+ and an anionic X−. The heterolytic bond dissociation energy (BDE), for simple alkyl halides MemH3−mC–X, decreases upon increasing methyl substitution along the series of H3C–X (methyl, 0°), MeH2C–X (primary, 1°), Me2HC–X (secondary, 2°), Me3C–X (tertiary, 3°).3 In other words, it is easier to form more substituted carbocations, making them more likely to participate as an intermediate in a chemical reaction.
Currently, the reduced heterolytic bond dissociation energy of the C–X bond is often ascribed to the stabilization of the carbocation, which increases along methyl, primary, secondary, and tertiary substituted carbocations.4 This stability trend stems from the stabilizing interactions (hyperconjugation and inductive effects)1a,5 between the electron-depleted carbon center and the methyl groups. The true definition of structural stability is less straightforward than stated above since, strictly speaking, one cannot directly compare the structural stabilities of non-isomeric species.6,7 Furthermore, this trend also has been attributed to the relief of steric repulsion, going from the substrate to the carbocation, between the substituents of the C–X bond for more substituted systems (also known as B-strain; back-strain).8
Typically, the thermodynamic stability of organic molecules is quantified using hypothetical reactions (e.g., isodesmic or homodesmotic), which are specifically designed to isolate a desired effect.9 Recently, in contrast to common textbook knowledge, we found using isodesmic reactions that methyl substitution destabilizes simple organic radicals.10 In our present study, we partition the energy of the system using the isodesmic reaction shown in eqn (1), which allows us to investigate the effect of the number of methyl groups on the thermodynamic stability of the system. We use the non-substituted H3C–X as our reference compound. This approach has proven useful for quantifying the stability of organic molecules, however, it does not directly permit one to uncover the true origin of the stability trend.
| H3C–X + MemH3−mC+ → H3C+ + MemH3−mC–X | (1) |
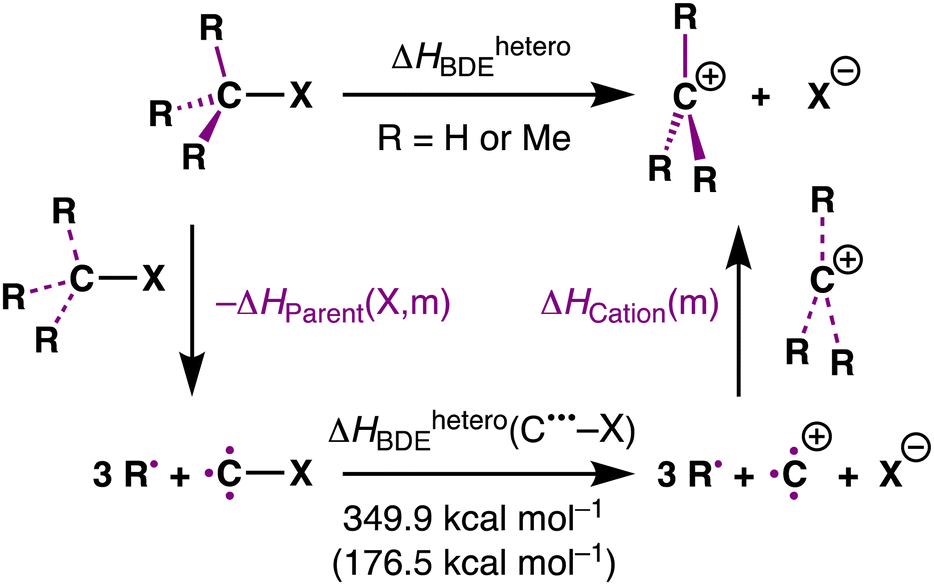
Herein, we reveal the exact source of the stability trend of carbocations by using partial reactions in a thermodynamic cycle quantitatively decomposing the effect of methyl groups on the carbocation species and the parent substrate, as shown in Scheme 1 (purple bonds). We have chosen to study the archetypal MemH3−mC–X model systems with m = 0, 1, 2, 3 and X = F, Cl, Br, I, H, CH3. The activation strain model (ASM)11 was employed to provide quantitative insight into the driving processes for the carbocation stability.
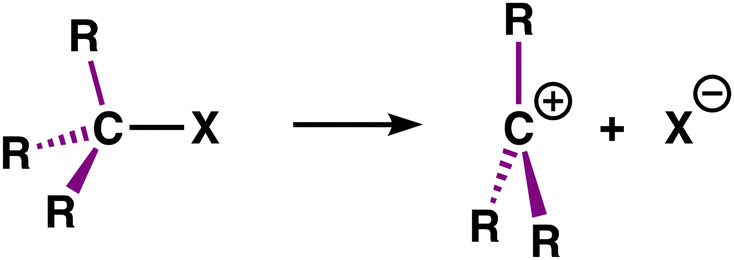 | ||
| Scheme 1 Computationally studied heterolytic bond dissociation of the C–X bond (R = H or Me; X = F, Cl, Br, I, H, CH3). | ||
Here, we focus on the MemH3−mC–I systems as a representative example. However, all model systems we have studied, that is, MemH3−mC–X (m = 0–3; X = F, Cl, Br, H, CH3), possess similar trends and can be found in Table S1 and Fig. S1 (ESI†). Table 1 presents our computed MemH3−mC–I bond lengths (r (C–I)) and heterolytic bond dissociation enthalpies (ΔHheteroBDE). The dissociation of the leaving-group atom, X−, from the tetrahedral substrate leads to a MemH3−mC+ carbocation. The latter adopts a trigonal planar geometry to optimize the C+˙˙˙-substituent bonding overlap between the singly-occupied 2p atomic orbitals of C+ and the E-symmetric SOMO of the substituents R3˙˙˙ (where a dot ˙ represents an unpaired electron; see Fig. S2 for structures, ESI†). The trigonal planar geometry also minimizes steric (Pauli) repulsion between the substituents. Note that, as reported earlier, the ethyl cation adopts a “non-classical” bridged carbocation structure in which the positive charge is delocalized over both carbons (see Fig. S2, ESI†).12
|
|
||||
|---|---|---|---|---|
| System | r (C–X) | ΔHheteroBDE | ΔHParent(X, m) | ΔHCation(m) |
| a Computed at ZORA-(U)M06-2X/QZ4P and values in parentheses at COSMO(H2O)-ZORA-(U)M06-2X/QZ4P, at 298.15 K and 1 atm. | ||||
| H3C–I | 2.132 (2.136) | 210.7 (66.7) | –321.5 (–320.8) | –453.0 (–440.2) |
| MeH2C–I | 2.150 (2.157) | 170.1 (40.1) | –311.5 (–310.3) | –483.5 (–456.3) |
| Me2HC–I | 2.170 (2.181) | 152.5 (29.8) | –302.7 (–301.1) | –492.4 (–457.7) |
| Me3C–I | 2.194 (2.209) | 137.2 (19.2) | –294.5 (–292.4) | –499.5 (–459.4) |
As expected, the C–I bond, indeed, significantly weakens as the degree of methyl substitution increases, going from ΔHheteroBDE = 210.7 to 170.1 to 152.5 to 137.2 kcal mol−1, along m = 0, 1, 2, and 3 (see Table 1). At the same time, the C–I bond also becomes longer along this series from 2.132 to 2.150 to 2.170 to 2.194 Å. Our computed bond dissociation energies at ZORA-M06-2X/QZ4P are in good agreement with experiments (see Table S2, ESI†).3 Moreover, the same general conclusions are obtained at ZORA-BLYP-D3(BJ)/QZ4P and ZORA-BP86-D3(BJ)/QZ4P, see Fig. S3, S4 and Tables S3, S4 (ESI†).
To analyze how the heterolytic MemH3−mC–X bond dissociation enthalpy depends on both the bonding of the substituents in the carbocation MemH3−mC+ and in its parent substrate MemH3−mC–X, we have decomposed the heterolytic bond dissociation enthalpy of the system into three terms: ΔHParent(X, m), ΔHheteroBDE(C˙˙˙–X), and ΔHCation(m) (eqn (2)), associated with the three partial reactions of the thermochemical cycle shown in Table 1. The ΔHParent(X, m) is the overall bond enthalpy as the three separate substituents, i.e., MemH3−m˙˙˙ combine with C˙˙˙–X to form the parent substrate MemH3−mC–X. The ΔHheteroBDE(C˙˙˙–X) is the heterolytic C–X bond dissociation enthalpy of the completely unsubstituted C˙˙˙–X species into C+˙˙˙ and X−. The ΔHCation(m) is the overall bond enthalpy as the three separate substituents MemH3−m˙˙˙ combine with C+˙˙˙ to form the carbocation MemH3−mC+. Thus, we have the following relationship of eqn (2):
| ΔHheteroBDE = ΔHheteroBDE(C˙˙˙–X) + ΔHCation(m) − ΔHParent(X, m) | (2) |
| ΔΔHCation(m) = ΔHCation(m) − ΔHCation(m = 0) | (3) |
| ΔΔHParent(X, m) = ΔHParent(X, m) − ΔHParent(X, m = 0) | (4) |
| ΔΔHheteroBDE = ΔΔHCation(m) − ΔΔHParent(X, m) | (5) |
Several trends emerge from our decomposition of ΔHheteroBDE using the thermochemical cycle in Table 1 (Fig. 1). As discussed previously, the MemH3−mC–I bonds become weaker by increasing the number of methyl groups, which is illustrated by the increasingly more negative isodesmic dissociation energy (ΔΔHheteroBDE) in Fig. 1a (black line). We find that the parent substrate MemH3−mC–I is systematically destabilized by the substituents, as seen in Fig. 1a (blue line) by the positive ΔΔHParent(X, m) going from +10.0 to +18.8 to +27.0 kcal mol−1 along m = 1, 2, 3. In contrast, the carbocation is stabilized by the substituents (red line), through, among others, hyperconjugation, as evidenced by the more stabilizing ΔΔHCation(m) going from −30.5 to −39.5 to −46.5 kcal mol−1 along the same series.
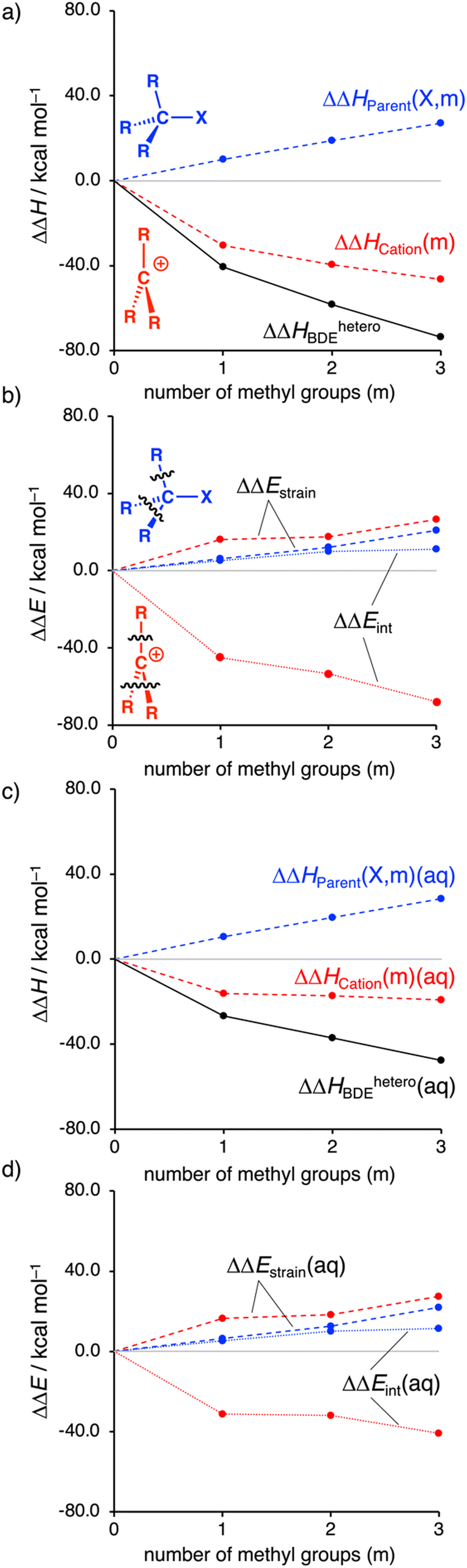 | ||
| Fig. 1 Effect of methyl groups on ΔΔHheteroBDE, ΔΔHParent(X, m), ΔΔHCation(m), and the corresponding activation strain analysis (in kcal mol−1) for R3C–X (R = H or Me; X = I). Computed at ZORA-(U)M06-2X/QZ4P (a and b) and COSMO(H2O)-ZORA-(U)M06-2X/QZ4P (c and d). | ||
To assess the role of solvation, we have also studied the decomposition of ΔHheteroBDE in solution with COSMO. For this purpose, we selected dichloromethane (ε = 9), DMSO (ε = 47), and water (ε = 78), spanning realistic extremes of polarity used in experiments.13 For our discussion, we focus on water solvation and note that in all solvents, the same general trends emerge, see Tables S5, S6 and Fig. S5 (ESI†).
Solvation significantly reduces the stabilizing effects of the methyl groups on the carbocation (Fig. 1c, red line) with ΔΔHCation(m)(aq) of only −16.1, −17.2, −19.2 kcal mol−1 going from m = 1 to 2 to 3, respectively. This stems from the fact that the less substituted carbocation, with its more localized and less shielded positive charge, enters into a more stabilizing interaction with the solvent. In sharp contrast, the observed systematic destabilization of the parent substrate MemH3−mC–I by the substituents is entirely maintained if we go from the gas phase to solution, as reflected by ΔΔHParent(X, m)(aq), which still steeply increases from +10.1, +19.7, +28.4 kcal mol−1 along the same series.
Note that, in solution, the magnitude of stabilization of the carbocation levels off after the addition of more than one methyl group. This saturation effect finds its origin in the aforementioned interaction between the solvated cation and the substituents. Introducing the first substituent stabilizes the cation, reducing the electron depletion on the carbocation. Intuitively, the next substituent can interact less strongly with the less depleted pertinent carbon center. This effect is also observed in the gas phase, although less apparent.
Next, to further understand the effect of the methyl groups on the carbocation stability, we employ our activation strain model (see ESI† for Computational methods; Fig. 1b and d).9 Note that the computed trends are the same for both ΔH and ΔE (Table S1, ESI†). We continue with the analysis of ΔE. As discussed, the parent substrate is systematically destabilized by adding methyl groups, which can be traced back to both the destabilizing strain (ΔΔEstrain > 0) and less stabilizing interaction energy (ΔΔEint > 0; Fig. 1b). The less stabilizing interaction energy stems from the substituents that engage in steric (Pauli) repulsive interactions (see Fig. S6 and S7, ESI†). The larger methyl groups have more repulsion with the C˙˙˙–X bond and have more mutual repulsion than the smaller hydrogen atoms. This type of repulsion is also known as F-strain (front-strain).14 The destabilizing strain mainly results from the need of the methyl group(s) to deform from their planar methyl radical equilibrium geometry to a pyramidal geometry in the substrate.15
In contrast, the carbocation becomes more stabilized by the introduction of substituents, which stems from the stabilizing interactions between the substituents and the cationic carbon center (Fig. 1b). Our quantitative MO analyses show that this, indeed, finds its origin in, among others, the stabilizing hyperconjugation (Fig. S8 and S9, ESI†). For the carbocations, a systematically slightly more destabilizing strain compared to the parent substrate is found (Fig. 1b; red versus blue dashed line). This can be traced back to the intrinsically shorter carbon–substituent bonds for the carbocations, which require the methyl groups to deform more. The shorter carbon–substituent bonds of the trigonal planar carbocations are a result of the relief of steric repulsion between the substituents.
Note that the jump in both the interaction and strain energy by going from the methyl to the ethyl cation is the direct effect of the “non-classical” bridged carbocation (see Fig. S6 and S7, ESI†). As discussed above, solvation stabilizes the carbocation and thus reduces the electron-accepting capabilities of this species (attenuates hyperconjugative effects), which leads to a substantial reduction of the substituent–cation interaction going from the gas phase to solution (Fig. 1d). While the destabilizing steric repulsive effects in the systems are maintained.
In conclusion, we find that the heterolytic bond strength of C–X bonds decreases as the degree of alkyl substitution increases. We quantitatively established a commonly overlooked driving force behind this trend, namely, the parent substrate is increasingly destabilized by introducing alkyl substituents. This is the result of destabilizing steric repulsion between the alkyl substituents and the C–X bond (also known as F-strain). This trend is reinforced by the stabilization of the carbocation by the alkyl substituents through, among others, hyperconjugation. We found that the destabilization of the parent substrate often plays a dominant role if the species are in solution.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. H. Aue, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 4, 487 Search PubMed; (b) R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905 CrossRef CAS PubMed; (c) D. J. Tantillo, Nat. Prod. Rep., 2011, 28, 1035 RSC; (d) D. J. Tantillo, Chem. Soc. Rev., 2010, 39, 2847 RSC; (e) X. Creary, Chem. Rev., 1991, 91, 1625 CrossRef CAS; (f) M. Saunders and H. A. Jimenez-Vazquez, Chem. Rev., 1991, 91, 375 CrossRef CAS; (g) G. I. Borodkin, I. R. Elanov and V. G. Shubin, Russ. J. Org. Chem., 2021, 57, 301 CrossRef CAS.
- (a) J. Bah, V. R. Naidu, J. Teske and J. Franzén, Adv. Synth. Catal., 2015, 357, 148 CrossRef CAS; (b) J. P. Richard, T. L. Amyes and M. M. Toteva, Acc. Chem. Res., 2001, 34, 981 CrossRef CAS PubMed; (c) T. Hansen, L. Lebedel, W. A. Remmerswaal, S. van der Vorm, D. P. A. Wander, M. Somers, H. S. Overkleeft, D. V. Filippov, J. Désiré, A. Mingot, Y. Bleriot, G. A. van der Marel, S. Thibaudeau and J. D. C. Codée, ACS Cent. Sci., 2019, 5, 781 CrossRef CAS PubMed; (d) M. A. Mercadante, C. B. Kelly, T. A. Hamlin, K. R. Delle Chiaie, M. D. Drago, K. K. Duffy, M. T. Dumas, D. C. Fager, B. L. C. Glod, K. E. Hansen, C. R. Hill, R. M. Leising, C. L. Lynes, A. E. MacInnis, M. R. McGohey, S. A. Murray, M. C. Piquette, S. L. Roy, R. M. Smith, K. R. Sullivan, B. H. Truong, K. M. Vailonis, V. Gorbatyuk, N. E. Leadbeater and L. J. Tilley, Chem. Sci., 2014, 5, 3983 RSC; (e) S. van der Vorm, T. Hansen, E. R. van Rijssel, R. Dekkers, J. M. Madern, H. S. Overkleeft, D. V. Filippov, G. A. van der Marel and J. D. C. Codée, Chem. – Eur. J., 2019, 25, 7149 CrossRef CAS PubMed; (f) E. S. Stoyanov, I. V. Stoyanova, F. S. Tham and C. A. Reed, Angew. Chem., Int. Ed., 2012, 51, 9149 CrossRef CAS PubMed; (g) J. M. Madern, T. Hansen, E. R. van Rijssel, H. A. V. Kistemaker, S. van der Vorm, H. S. Overkleeft, G. A. van der Marel, D. V. Filippov and J. D. C. Codée, J. Org. Chem., 2019, 8, 1218 CrossRef PubMed.
- (a) Y. R. Luo and P. D. Pacey, J. Phys. Chem., 1991, 95, 9470 CrossRef CAS; (b) H. M. Rosenstock, R. Buff, M. A. Ferreira, S. G. Lias, A. C. Parr, R. L. Stockbauer and J. L. Holmes, J. Am. Chem. Soc., 1982, 104, 2337 CrossRef CAS; (c) E. M. Arnett and N. J. Pienta, J. Am. Chem. Soc., 1980, 102, 3329–3334 CrossRef CAS.
- (a) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part A: Structure and Mechanisms, Springer, 5 edn, 2007, pp. 300–310 Search PubMed; (b) M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th edn, Wiley, New York, 2013, pp. 234–245 Search PubMed; (c) F. P. Lossing and J. L. Holmes, J. Am. Chem. Soc., 1984, 106, 6917 CrossRef CAS.
- (a) E. S. Stoyanov and G. dos Passos Gomes, J. Phys. Chem. A, 2015, 119, 8619 CrossRef CAS PubMed; (b) I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, e1389 Search PubMed; (c) I. V. Alabugin, K. M. Gilmore and P. W. Peterson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 109 CAS.
- (a) M. L. Coote, A. Pross and L. Radom, Org. Lett., 2003, 5, 4689 CrossRef CAS PubMed; (b) M. Swart, E. Rösler and F. M. Bickelhaupt, Eur. J. Inorg. Chem., 2007, 3646 CrossRef CAS; (c) C. Rüchardt, Angew. Chem., Int. Ed. Engl., 1970, 9, 830 CrossRef; (d) K. B. Clark and D. D. M. Wayner, J. Am. Chem. Soc., 1991, 113, 9363 CrossRef CAS; (e) S. Gronert, J. Org. Chem., 2006, 71, 1209 CrossRef CAS PubMed; (f) S. Gronert, J. Org. Chem., 2006, 71, 7045 CrossRef CAS PubMed; (g) S. Gronert, Org. Lett., 2007, 9, 2211 CrossRef CAS PubMed; (h) M. Swart and F. M. Bickelhaupt, J. Chem. Theory Comput., 2006, 2, 281 CrossRef CAS PubMed; (i) F. M. Bickelhaupt, H. L. Hermann and G. Boche, Angew. Chem., Int. Ed., 2006, 45, 823 CrossRef CAS PubMed.
- (a) A. A. Zavitsas, J. Chem. Educ., 2001, 78, 417 CrossRef CAS; (b) J. L. M. Abboud, I. Alkorta, J. Z. Davalos, P. Müller, E. Quintanilla and J. C. Rossier, J. Org. Chem., 2003, 68, 3786 CrossRef CAS PubMed; (c) P. de, A. M. Nicholas and D. R. Arnold, Can. J. Chem., 1984, 62, 1850 CrossRef.
- (a) T. Sorrell, Organic Chemistry, 2 edn, 2006, pp 186–188 Search PubMed; (b) K. Takeuchi, Y. Ohga and T. Kitagawa, J. Org. Chem., 1991, 56, 5007 CrossRef CAS; (c) K. T. Liu, S. J. Hou and M. L. Tsao, J. Org. Chem., 1998, 63, 1360 CrossRef CAS; (d) K. T. Liu, S. J. Hou and M. L. Tsao, J. Chin. Chem. Soc., 2009, 56, 425 CrossRef CAS; (e) K. I. Takeuchi, Y. Ohga, M. Yoshida, K. Ikai, T. Shibata, M. Kato and A. Tsugeno, J. Org. Chem., 1997, 62, 5696 CrossRef CAS; (f) K. Takeuchi, Y. Ohga, T. Ushino and M. Takasuka, J. Phys. Org. Chem., 1997, 10, 717 CrossRef CAS.
- (a) D. A. Ponomarev and V. V. Takhistov, J. Chem. Educ., 1997, 74, 201 CrossRef CAS; (b) R. Fuchs, J. Chem. Educ., 1984, 61, 133 CrossRef CAS; (c) S. E. Wheeler, K. N. Houk, P. v R. Schleyer and W. D. Allen, J. Am. Chem. Soc., 2009, 131, 2547 CrossRef CAS PubMed.
- E. Blokker, W.-J. van Zeist, X. Sun, J. Poater, J. M. van der Schuur, T. A. Hamlin and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2022, 61, e202207477 CrossRef CAS PubMed.
- (a) P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649 CrossRef CAS PubMed; (b) P. Vermeeren, T. A. Hamlin and F. M. Bickelhaupt, Chem. Commun., 2021, 57, 5880 RSC.
- (a) H. S. Andrei, N. Solcà and O. Dopfer, Angew. Chem., Int. Ed., 2007, 47, 395 CrossRef PubMed; (b) B. G. Oliveira, M. L. Vasconcellos and R. R. Olinda, Struct. Chem., 2009, 20, 81 CrossRef CAS.
- (a) T. A. Hamlin, B. van Beek, L. P. Wolters and F. M. Bickelhaupt, Chem. – Eur. J., 2018, 24, 5927 CrossRef CAS PubMed; (b) T. Hansen, J. C. Rooze, F. M. Bickelhaupt and T. A. Hamlin, J. Org. Chem., 2022, 87, 1805 CrossRef CAS PubMed.
- (a) H. C. Brown, J. Am. Chem. Soc., 1945, 67, 374 CrossRef CAS; (b) J. S. Lomas, M. J. D'Souza and D. N. Kevill, J. Am. Chem. Soc., 1995, 117, 5891 CrossRef CAS; (c) K. Takeuchi, Y. Ohga and T. Kitagawa, J. Org. Chem., 1991, 56, 5007 CrossRef CAS.
- W.-J. van Zeist and F. M. Bickelhaupt, Phys. Chem. Chem. Phys., 2009, 11, 10317 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational results; Cartesian coordinates, energies, and the number of imaginary frequencies of all stationary points. See DOI: https://doi.org/10.1039/d2cc04034d |
| This journal is © The Royal Society of Chemistry 2022 |