
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHetero-Diels–Alder click reaction of dithioesters for a catalyst-free indirect 18F-radiolabelling of peptides†
Timothé
Maujean
 a,
Patrice
Marchand
b,
Patrick
Wagner
a,
Stéphanie
Riché
a,
Frédéric
Boisson
b,
Nicolas
Girard
a,
Dominique
Bonnet
*a and
Mihaela
Gulea
*a
a,
Patrice
Marchand
b,
Patrick
Wagner
a,
Stéphanie
Riché
a,
Frédéric
Boisson
b,
Nicolas
Girard
a,
Dominique
Bonnet
*a and
Mihaela
Gulea
*a
aUniversité de Strasbourg, CNRS, Laboratoire d’Innovation Thérapeutique, LIT UMR 7200, F-67000 Strasbourg, France. E-mail: dominique.bonnet@unistra.fr; gulea@unistra.fr
bUniversité de Strasbourg, CNRS, IPHC UMR 7178, F-67000 Strasbourg, France
First published on 8th September 2022
Abstract
The HDA reaction of dithioesters was developed as a new click-reaction compatible with the indirect 18F-labelling of peptides. It involves dithioester-peptides and a radiofluorinated diene as a novel prosthetic group. The method was applied to a PSMA-ligand for the in vivo detection of LNCap tumors in xenografted mice.
Peptides are present in all living organisms and due to their high binding affinity to specific receptors, they are also widely used for medical applications as therapeutic or targeting agents, as well as diagnostic tools. Hence, the interest in peptide radiolabelling has grown considerably over the past few years, as demonstrated by numerous reviews devoted to this topic.1,2 Fluorine-18, the most used radionuclide in the positron emission tomography (PET) imaging technique, is characterized by a half-life (t1/2) of 109.8 min, thus matching the pharmacokinetics of peptides and explaining its successful use in this field.218F-labelling can be achieved through direct or indirect approaches, however mild reaction conditions are required when a peptide is involved. Despite the interest and the advances in the direct radiofluorination of peptides,3 indirect approaches remain the most used.4 This includes the radiofluorination of a small molecule generating a prosthetic group that subsequently undergoes a fast reaction with a function attached to the tracer. Therefore, click reactions5 including cycloadditions represent a powerful tool for this purpose. The well-known copper-catalyzed reaction of an azide with an alkyne (CuAAC) was widely employed, however with the main drawbacks related to the use of copper (cytotoxicity, undesired oxidative side-reactions, use of large excess of peptide and catalyst, difficulty to remove copper, additional control quality analysis).6 Some metal-free cycloadditions such as SPAAC (strain-promoted azide–alkyne cycloaddition),7 SPSAC (strain-promoted sydnone–alkyne cycloaddition),8 or IEDDA (inverse electron demand Diels–Alder)9 reactions were also reported; however, the accessibility of the involved reagents represents a limitation. Therefore, the introduction of alternative click reactions in the toolbox of peptide radiofluorination still remains a challenging and attractive research area. Some years ago, we developed the use of electron-deficient dithioesters as efficient dienophiles in catalyzed or non-catalyzed hetero-Diels–Alder (HDA) reactions.10 Then, from this work, these reactions were included in the category of click reactions by Barner–Kowollik, Stenzel, and co-workers, thanks to their innovative application in materials science using dithioester-functionalized polymers prepared by RAFT polymerization.11 Based on this knowledge, we report herein the first application of the HDA reaction of dithioesters in a catalyst-free indirect 18F-radiolabelling of peptides using a radiofluorinated diene as a novel prosthetic group (Scheme 1).
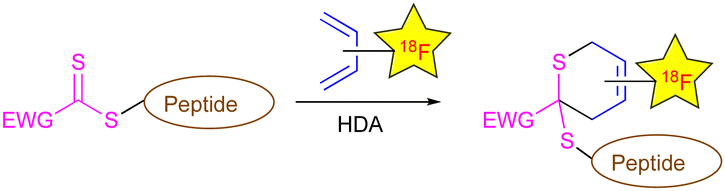 | ||
| Scheme 1 Proposed new catalyst-free indirect 18F-radiolabelling of peptides by HDA reaction; EWG = electron-withdrawing group. | ||
The phosphonodithioformate was selected as the heterodienophile partner as it offers a balanced compromise between reactivity and stability and facile access. Moreover, it enables easy characterization of the substrates and products by 31P NMR spectroscopy. Three phosphonodithioesters (Fig. 1) were prepared by S-alkylation of the diethyl phosphonodithioformate salt12 with the corresponding benzyl bromide derivative (see ESI†). Non-peptidic phosphonodithioester 1 was prepared as a model reagent for kinetic studies. The dithioester functionalized tripeptide 2 containing an AYK–NH2 residue (AYK: alanine–tyrosine–lysine) has been designed as a model peptide for radiofluorination. Starting from a Rink amide resin and using a Fmoc/tBu strategy, it was successfully obtained in a good 81% overall yield (10 steps), demonstrating the chemical stability of the dithioester moiety during cleavage and deprotection steps. This strategy prevented undesired N-thioacylation13 allowing rapid and efficient access to an NH2-containing dithioester-peptide (as its trifluoroacetic acid ammonium salt). Finally, to establish the proof of the applicability of this HDA reaction for in vivo PET imaging, we envisaged the preparation and use of a radiofluorinated PSMA (prostate-specific membrane antigen) ligand.14 To this end, PSMA-phosphonodithioester 3 containing a KuE-residue (KuE: lysine–urea–glutamate) was successfully synthesized in solution with 38% yield over five steps.
 | ||
| Fig. 1 Phosphonodithioesters prepared and used as dienophiles in the HDA reaction in this study. | ||
The choice of the dienic partner was crucial to reach high kinetics, as the cycloaddition reaction time should not exceed one hour, due to the short-lived 18F-radioisotope. Theoretical conversions were plotted for different rate constants under typical conditions of 18F-labelling (with particularly a very low concentration of 5 mM) showing that low or no conversions would be obtained with a rate constant below 1 M−1 s−1 (ESI,† Fig. S1). First, acyclic diene 4 bearing a carbamate moiety was synthesized from the commercially available sorbic alcohol (see ESI†) and reacted with dienophile 1. The reaction was performed in water/isopropanol (70/30) at 60 °C and completed after 24 h, leading to the corresponding cycloadduct in 47% isolated yield (Scheme 2, eqn (1)). The use of water as a co-solvent increased dramatically the rate of this reaction, as more than one week was needed to reach completion when acetonitrile was used. Isopropanol served as a non-toxic co-solvent to keep the substrates soluble during the reaction. The second order rate constant of this reaction was determined by monitoring the absorbance at 530 nm, which corresponds to the n to π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S, and was found to be about 0.023 M−1 s−1 (ESI,† Fig. S2 and S3). This value is not compatible with the kinetic requirement of fluorine-18 chemistry. We thus searched for a much more reactive diene. It is well established that 1,3-dienes need s-trans to s-cis interconversion to react in DA reaction, therefore dienes with locked s-cis conformation are highly reactive.15 In the latter category are included cyclopentadiene derivatives, which are known for their impressive reaction rates but also for their fast dimerization, and exocyclic dienes, which represent a sound alternative as they can react faster than 1,3-butadienes by 100-fold without being prone to dimerization. Thereby, we designed and synthesized the exocyclic diene 6 in five steps starting from cyclohexanone (see ESI†). Its reaction with dithioester 1 was performed under the same conditions to afford the expected cycloadduct 7 in 65% isolated yield (Scheme 2, eqn (2)). The reaction was completed in only 10 min (HPLC monitoring) and the second order rate constant was found to be around 4.3 M−1 s−1 (see ESI,† Fig. S4 and S5), which is 175 times higher than that with the acyclic analogue 4. Gratifyingly, this value is compatible with our objective of 18F-labelling. Both cycloadducts 5 and 7 were obtained as a mixture of four regio/stereoisomers for which the ratio was determined by 31P-NMR spectroscopy (see ESI†). According to the DA reaction rules we assume that the major cycloadduct 7 (around 50% in the mixture) is the endo-regioisomer illustrated in Scheme 2.
S, and was found to be about 0.023 M−1 s−1 (ESI,† Fig. S2 and S3). This value is not compatible with the kinetic requirement of fluorine-18 chemistry. We thus searched for a much more reactive diene. It is well established that 1,3-dienes need s-trans to s-cis interconversion to react in DA reaction, therefore dienes with locked s-cis conformation are highly reactive.15 In the latter category are included cyclopentadiene derivatives, which are known for their impressive reaction rates but also for their fast dimerization, and exocyclic dienes, which represent a sound alternative as they can react faster than 1,3-butadienes by 100-fold without being prone to dimerization. Thereby, we designed and synthesized the exocyclic diene 6 in five steps starting from cyclohexanone (see ESI†). Its reaction with dithioester 1 was performed under the same conditions to afford the expected cycloadduct 7 in 65% isolated yield (Scheme 2, eqn (2)). The reaction was completed in only 10 min (HPLC monitoring) and the second order rate constant was found to be around 4.3 M−1 s−1 (see ESI,† Fig. S4 and S5), which is 175 times higher than that with the acyclic analogue 4. Gratifyingly, this value is compatible with our objective of 18F-labelling. Both cycloadducts 5 and 7 were obtained as a mixture of four regio/stereoisomers for which the ratio was determined by 31P-NMR spectroscopy (see ESI†). According to the DA reaction rules we assume that the major cycloadduct 7 (around 50% in the mixture) is the endo-regioisomer illustrated in Scheme 2.
 | ||
| Scheme 2 HDA reaction of dithioester 1 with acyclic and exocyclic dienes 4 and 6; one of the four isomers is illustrated for the products. | ||
With these results in hand, we prepared an exocyclic fluorinated diene in order to apply this fast reaction in peptide radiofluorination. Diene F-8 was obtained in seven steps following a similar sequence as that used for diene 6 and additional introduction of the fluorine atom (see ESI†). F-8 was reacted with each phosphonodithioester (1, 2, and 3) under non-radioactive reaction conditions, similar to those used previously (in water/isopropanol, at 60 °C). All reactions showed full conversion after 10 min and led to the expected corresponding cycloadducts F-9 (from 1), F-10 (from 2), and F-11 (from 3) with good isolated yields (Scheme 3) and similar regio/stereoselectivities to those previously obtained for 7 (determined by 31P-NMR spectroscopy, see ESI†). These results demonstrate that the cycloaddition is rapid and fully compatible with the presence of peptide functional groups such as amines, alcohols, or carboxylic acids, and that the size and nature of the attached peptide moiety do not impact the efficiency and selectivity of the reaction.
 | ||
| Scheme 3 HDA reaction of fluorinated diene F-8 with dithioesters 1, 2, and 3; the major of the four isomers is illustrated for the products. | ||
The next step was the radiosynthesis of our [18F]fluoro-cyclic diene [18F]-8. The radiofluorination of the tosylate precursor TsO-8 was performed in classical conditions with [18F]KF/Kryptofix 2.2.2 and we were able to, after a short optimization (ESI,† Fig. S6), reach 90% conversion after just 10 min at 95 °C (Scheme 4). This synthesis was performed on a fully automated apparatus along with the purification of the crude product by C18 HPLC and formulation in ethanol, affording the novel prosthetic group [18F]-8 with a decay corrected isolated radiochemical yield of 70%, a mean molar activity of 150 GBq μmol−1 at the end of synthesis (EOS) and a high radiochemical purity (>98% by HPLC).
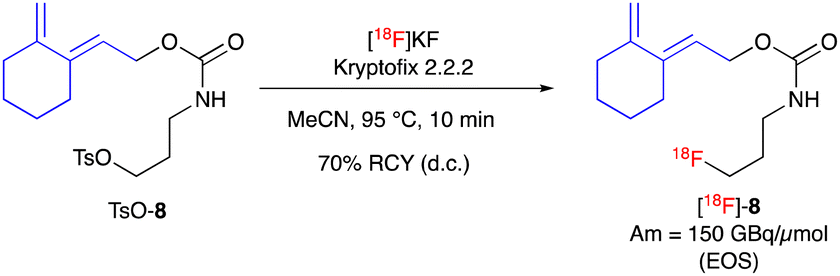 | ||
| Scheme 4 Synthesis of radiofluorinated diene [18F]-8. | ||
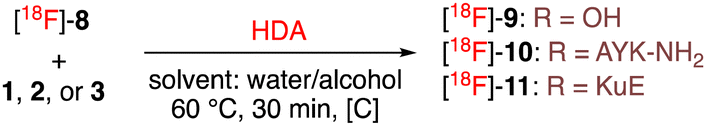
Based on our previous results in non-radioactive reaction conditions the 18F-labelled diene [18F]-8 was subsequently used in the optimization of the HDA reaction with dithioester 1 and 2 by fine-tuning the ratio of water/alcohol and the concentration of the dithioester (Table 1). All reactions were performed using a constant amount of diene in 20 μL of water/alcohol solution (determined using an HPLC calibration curve). With the model dithioester 1, the corresponding cycloadduct [18F]-9 was obtained with high conversion when using a high concentration of 1 (entry 1 vs. entry 2) demonstrating the feasibility of this radiofluorination approach. We then optimized the HDA of dithioester-peptide 2 (entries 3–5). Ethanol was preferred to isopropanol as better tolerated for in vivo injection. The role of water as a co-solvent (entry 4) as well as the importance of using high concentrations (entry 5) were highlighted as the most crucial parameters for this reaction. The optimized conditions were then adapted on a fully automated apparatus to produce using the dithioester 2, the desired radiolabeled peptide [18F]-10 in 54 ± 6% radiochemical yield (from 22–23 GBq of [18F]KF and decay corrected from the start of synthesis, n = 2) after a simple purification on a C18 SEP-PAK cartridge (entry 6). The same conditions and starting radioactivity were used with dithioester 3 (entry 7) to obtain after semi-preparative HPLC purification the radiolabeled PSMA-ligand [18F]-11 in 44 ± 6% radiochemical yield (n = 5). Both automated syntheses were performed within 2 hours (purification and formulation included). Peptides synthesized with this protocol were obtained with high molar activities of 80 ± 20 GBqμmol−1 (n = 3, EOS).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate (nmol); [C] (μM)e | Water/alcohol | Product | Product/[18F]-8f |
| a Manual synthesis with a constant amount of diene, analyzed after 30 min. b Ratio water/isopropanol. c Ratio water/ethanol. d Automated synthesis using 9 to 10 GBq of [18F]-8. e The volume of the solvent was adjusted to the amount of dithioester. f Ratio measured by HPLC on the radioactive channel. | ||||
| 1a | 1 (30); 600 | 70/30b | [18F]-9 | 100/0 |
| 2a | 1 (3); 60 | 70/30b | [18F]-9 | 83/17 |
| 3a | 2 (3); 15 | 80/20c | [18F]-10 | 10/90 |
| 4a | 2 (3); 15 | 60/40c | [18F]-10 | 0/100 |
| 5a | 2 (3); 45 | 70/30c | [18F]-10 | 77/23 |
| 6d | 2 (700); 135 | 70/30c | [18F]-10 | 99/1 |
| 7d | 3 (1500); 290 | 70/30c | [18F]-11 | >99/1 |
Then, radioligand [18F]-11 was evaluated for in vivo PET imaging. [18F]-11 remained intact over a 60 minute incubation in mouse serum at 37 °C, demonstrating the metabolic stability of the newly formed 2-thiopyran heterocyclic linker (see ESI,† Fig. S18). Moreover, the specificity of [18F]-11 was evaluated on LNCap cells in competition with 2-PMPA, a known inhibitor of PSMA receptors.16 The results after 15 min and 60 min of [18F]-11 incubation (with and without 2-PMPA) confirmed that the specificity of the ligand towards the PSMA receptor is preserved after the introduction of the 2-thiopyran linker (ESI,† Fig. S19). In vivo experiments have been performed on mice bearing LNCap tumors. Briefly, male nude mice were injected subcutaneously with LNCap cells at the back of the right shoulder and used after 35 days of tumor growth. The mice were injected with approximately 9 MBq of [18F]-11 and imaged by a 10 min static scan (60 min post-injection) or by a dynamic acquisition of 90 min (18 × 5 min) just after injection (see ESI†). Vascularized tumors with functional sizes below 100 mm3 were efficiently detected as shown in Fig. 2. As expected, the kidneys also exhibited a high uptake of the radiotracer due to the overexpression of the PSMA receptor in this organ. Finally, dynamic acquisition demonstrated an important hepato-biliary excretion of [18F]-11 without accumulation in the liver and a fast blood elimination (see ESI,† Fig. S20).
 | ||
| Fig. 2 Left: Anterior coronal view of a mouse bearing an LNCap tumor 60 min after i.v. injection of 10 MBq of [18F]-11; red arrows point to the tumor, black to the kidneys. Scale bar from 0 to 10% ID g−1 for the left image. Right: Anterior view of whole-body 3D volumic rendering of the same mouse; green arrow for gallbladder and blue for the intestines. | ||
In conclusion, we developed a new fast click-reaction compatible with the indirect radiofluorination of peptides. This consists of a HDA cycloaddition involving a phosphonodithioester-functionalized peptide as the heterodienophile and an 18F-fluorinated highly reactive s-cis constrained exocyclic diene as a novel prosthetic group. It also represents the first example of a direct-electron-demand HDA reaction used in 18F-labelling. Notably, the reaction proceeds free of catalyst and involves partners which are readily available in few steps on a gram scale. This makes this HDA reaction competitive with commonly used cycloadditions for this application. Using this reaction, a model tripeptide containing reactive NH2 and OH functions (from lysine and tyrosine), and a PSMA ligand were efficiently radiolabeled in mild conditions within 2 hours (purification and formulation included) and the conjugates obtained in high molar activities and radiopurities. Finally, the potential of this method to access radiotracers was evaluated through the preparation of PSMA-radiotracer [18F]-11. The latter was found to be stable in mouse serum and the specificity of the ligand towards the PSMA receptor was preserved in the presence of the 3,6-dihydro-2H-thiopyran moiety in the linker structure. This allowed its use for the in vivo detection of small size tumors in xenografted mice. This novel approach paves the way to the rapid and highly chemoselective 18F-radiolabelling of therapeutic peptides for in vivo PET imaging.
Author contributions: T.M., P.M. (Investigation, Conceptualization, Writing original draft); P.W., S.R. (Investigation); F.B. (Resources); N.G. (Supervision); D.B., M.G. (Conceptualization, Project administration, Supervision, Writing original draft, review & editing).
This work is part of the Interdisciplinary Thematic Institutes IMS and InnoVec, ITI 2021-2028 program of the University of Strasbourg, CNRS, and Inserm, supported by IdEx Unistra (ANR-10-IDEX-0002) and SFRI-STRAT’US project (ANR-20-SFRI-0012). T. M. was supported by a fellowship from the ENS Paris-Saclay. We thank the platforms PACSI (GDS 3670) and CYRCé for technical support.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- (a) M. Fani and H. R. Maecke, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39, 11 CrossRef CAS PubMed; (b) C. L. Charron, A. L. Farnsworth, P. D. Roselt, R. J. Hicks and C. A. Hutton, Tetrahedron Lett., 2016, 57, 4119 CrossRef CAS; (c) N. Mohtavinejada, M. S. Ardestani, A. Khalaj, A. Pormohammad, R. Najafi, A. Bitarafan-Rajabi, M. Hajiramezanali and M. Amanlou, Life Sci., 2020, 258, 118206 CrossRef PubMed; (d) P. Kręcisz, K. Czarnecka, L. Kroĺicki, E. Mikiciuk-Olasik and P. Szymanśki, Bioconjugate Chem., 2021, 32, 25 CrossRef PubMed.
- (a) O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1 CrossRef CAS PubMed; (b) D. E. Olberg and O. K. Hjelstuen, Curr. Top. Med. Chem., 2010, 10, 1669 CrossRef CAS PubMed; (c) O. Morris, M. Fairclough, J. Grigg, C. Prenant and A. McMahon, J. Labelled Compd. Radiopharm., 2019, 62, 4 CrossRef CAS PubMed; (d) S. Okarvi, Eur. J. Nucl. Med., 2001, 28, 929 CrossRef CAS; (e) S. Richter and F. Wuest, Molecules, 2014, 19, 20536 CrossRef; (f) K. R. Scroggie, M. V. Perkins and J. M. Chalker, Front. Chem., 2021, 9, 687678 CrossRef CAS PubMed.
- (a) D. M. Perrin, Acc. Chem. Res., 2016, 49, 1333 CrossRef CAS PubMed; (b) W. J. McBride, C. A. D’Souza, R. M. Sharkey, H. Karacay, E. A. Rossi, C. H. Chang and D. M. Goldenberg, Bioconjugate Chem., 2010, 21, 1331 CrossRef CAS PubMed; (c) Z. Yuan, M. B. Nodwell, H. Yang, N. Malik, H. Merkens, F. Bénard, R. E. Martin, P. Schaffer and R. Britton, Angew. Chem., Int. Ed., 2018, 57, 12733 CrossRef CAS PubMed; (d) J. Rickmeier and T. Ritter, Angew. Chem., Int. Ed., 2018, 57, 14207 CrossRef CAS; (e) S. Verhoog, C. W. Kee, Y. Wang, T. Khotavivattana, T. C. Wilson, V. Kersemans, S. Smart, M. Tredwell, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2018, 140, 1572 CrossRef CAS PubMed.
- (a) Y. Fu, H. Helbert, N. A. Simeth, S. Crespi, G. B. Spoelstra, J. M. van Dijl, M. van Oosten, L. R. Nazario, D. van der Born, G. Luurtsema, W. Szymanski, P. H. Elsinga and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 10041 CrossRef CAS PubMed; (b) H. S. Krishnan, L. Ma, N. Vasdev and S. H. Liang, Chem. – Eur. J., 2017, 23, 15553 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- (a) J. Marik and J. L. Sutcliffe, Tetrahedron Lett., 2006, 47, 6681 CrossRef CAS; (b) H. S. Gill and J. Marik, Nat. Protoc., 2011, 6, 1718 CrossRef CAS; (c) D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993 CrossRef CAS; (d) S. Li, H. Cai, J. He, H. Chen, S. Lam, C. Tao, Z. Zhu, S. J. Bark and C. Cai, Bioconjugate Chem., 2016, 27, 2315 CrossRef CAS PubMed; (e) C. J. Pickens, S. N. Johnson, M. M. Pressnall, M. A. Leon and C. J. Berkland, Bioconjugate Chem., 2018, 29, 686 CrossRef CAS PubMed.
- (a) L. S. Campbell-Verduyn, L. Mirfeizi, A. K. Schoonen, R. A. Dierckx, P. H. Elsinga and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 11117 CrossRef CAS PubMed; (b) H. L. Evans, R. L. Slade, L. Carroll, G. Smith, Q. D. Nguyen, L. Iddon, N. Kamaly, H. Stockmann, F. J. Leeper, E. O. Aboagye and A. C. Spivey, Chem. Commun., 2012, 48, 991 RSC; (c) K. Sachin, V. H. Jadhav, E. M. Kim, H. L. Kim, S. B. Lee, H. J. Jeong, S. T. Lim, M. H. Sohn and D. W. Kim, Bioconjugate Chem., 2012, 23, 1680 CrossRef CAS PubMed.
- (a) M. Richard, C. Truillet, V. L. Tran, H. Liu, K. Porte, D. Audisio, M. Roche, B. Jego, S. Cholet, F. Fenaille, B. Kuhnast, F. Taran and S. Specklin, Chem. Commun., 2019, 55, 10400 RSC; (b) M. Kumar Narayanam, B. T. Lai, J. Malette Loredo, J. A. Wilson, A. M. Eliasen, N. A. LaBerge, M. Nason, A. L. Cantu, B. K. Luton, S. Xu, H. D. Agnew and J. M. Murphy, Bioconjugate Chem., 2021, 32, 2073 CrossRef.
- (a) R. Selvaraj, S. Liu, M. Hassink, C. W. Huang, L. P. Yap, J. M. Fox, Z. Li and P. S. Conti, Bioorg. Med. Chem. Lett., 2011, 21, 5011 CrossRef CAS PubMed; (b) S. Liu, M. Hassink, R. Selvaraj, L. P. Yap, R. Park, H. Wang, X. Chen, J. M. Fox, Z. Li and P. S. Conti, Mol. Imaging, 2013, 12, 121 CrossRef CAS.
- (a) M. Heras, M. Gulea and S. Masson, Chem. Commun., 2001, 611 RSC; (b) M. Heras, M. Gulea, S. Masson and C. Philouze, Eur. J. Org. Chem., 2004, 160 CrossRef CAS; (c) R. Bastin, H. Albadri, A.-C. Gaumont and M. Gulea, Org. Lett., 2006, 8, 1033 CrossRef CAS PubMed; (d) H. Dentel, I. Chataigner, F. Le Cavelier and M. Gulea, Tetrahedron Lett., 2010, 51, 6014 CrossRef CAS.
- (a) S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052 RSC; (b) S. Sinnwell, C. V. Synatschke, T. Junkers, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2008, 41, 7904 CrossRef CAS; (c) A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411 CrossRef CAS PubMed; (d) M. Glassner, G. Delaittre, M. Kaupp, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2012, 134, 7274 CrossRef CAS PubMed.
- The procedure was modified from: D. L. Fox, N. R. Whitely, R. J. Cohen and R. N. Salvatore, Synlett, 2003, 2037 CAS.
- M. G. J. ten Cate, H. Rettig, K. Bernhardt and H. G. Börner, Macromolecules, 2005, 38, 10643 CrossRef CAS.
- See as examples of PSMA-labelling: (a) V. I. Böhmer, W. Szymanski, K.-O. van den Berg, C. Mulder, P. Kobauri, H. Helbert, D. van der Born, F. Reebing, A. Huizing, M. Klopstra, D. F. Samplonius, I. F. Antunes, J. W. A. Sijbesma, G. Luurtsema, W. Helfrich, T. J. Visser, B. L. Feringa and P. H. Elsinga, Chem. – Eur. J., 2020, 26, 10871 CrossRef PubMed; (b) C. Barinka, Y. Byun, C. L. Dusich, S. R. Banerjee, Y. Chen, M. Castanares, A. P. Kozikowski, R. C. Mease, M. G. Pomper and J. Lubkowski, J. Med. Chem., 2008, 51, 7737 CrossRef CAS PubMed.
- R. Huisgen, R. Grashey and J. Sauer, Cycloaddition reactions of alkenes, in The Chemistry of alkenes, ed. S. Patai, John Wiley & Sons, 2010, pp. 741–953 Search PubMed.
- P. F. Jackson, D. C. Cole, B. S. Slusher, S. L. Stetz, L. E. Ross, B. A. Donzanti and D. A. Trainor, J. Med. Chem., 1996, 39, 619 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04148k |
| This journal is © The Royal Society of Chemistry 2022 |